
Abstract
Chimeric antigen receptor (CAR)-engineered T (CAR-T) cells have obtained prominent achievement in the clinical immunotherapy of hematological malignant tumors, leading to a rapid development of cellular immunotherapy in cancer treatment. Scientists are also aware of the prospective advantages of CAR engineering in cellular immunotherapy. Due to various limitations such as the serious side effects of CAR-T therapy, researchers began to investigate other immune cells for CAR modification. Natural killer (NK) cells are critical innate immune cells with the characteristic of non-specifically recognizing target cells and with the potential to become “off-the-shelf” products. In recent years, many preclinical studies on CAR-engineered NK (CAR-NK) cells have shown their remarkable efficacy in cancer therapy and their superiority over autologous CAR-T cells. In this review, we summarize the generation, mechanisms of anti-tumor activity and unique advantages of CAR-NK cells, and then analyze some challenges and recent clinical trials about CAR-NK cells therapy. We believe that CAR-NK therapy is a promising prospect for cancer immunotherapy in the future.
Similar content being viewed by others
Introduction
Cellular immunotherapy plays an indispensable role in cancer treatment. Chimeric antigen receptor (CAR)–engineered T cells therapy, especially for the treatment of hematological malignant tumors, has become a research hotspot over the past decade. One BCMA-targeted and four CD19-targeted CAR-T products have been approved for marketing by the American Food and Drug Administration (FDA): tisagenlecleucel and axicabtagene ciloleucel for the treatment of relapsed/refractory (R/R) large B-cell lymphoma and pediatric B-cell acute lymphocytic leukemia, brexucabtagene autoleucel for R/R mantle cell lymphoma, lisocabtagene maraleucel for R/R large B-cell lymphoma, and idecabtagene vicleucel for R/R multiple myeloma [1,2,3,4,5]. However, CAR-T therapy still has many unavoidable limitations that hinder further development in clinical treatment (Table 1): (1) The safety of CAR-T cells needs to be solved. Cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) are the major inevitable toxicities that still lack corresponding effective management in most cases [6]. (2) CAR-T therapy requires T cells that can only be derived and engineered from the autologous peripheral blood and then injected back into the patient, so it is time-consuming and expensive for patients. (3) T cells are the main effector cells of graft-versus-host disease (GVHD), so CAR-T therapies have the risk of developing GVHD, which requires higher safety of CAR-T products [7]. (4) Other limitations include the off-targeted toxicity and the ineffective treatment of solid tumors due to the immunosuppressive microenvironment [8,9,10,11]. Given the shortcomings of CAR-T therapy, researchers began to apply the principles of CAR-engineered therapy to other immune cells [12, 13]. Because of the unique biological characteristics, anti-tumor mechanisms, wide range of sources, and higher safety of natural killer (NK) cells, CAR-engineered NK (CAR-NK) cells which are regarded as a promising alternative platform have attracted considerable attention in recent years [13,14,15,16,17]. In this review, we will discuss CAR-NK cells in detail, including the generation, tumor-killing mechanisms and promising advantages of CAR-NK cells, and then analyze some challenges and recent clinical trials about CAR-NK cells therapy.
Generation of CAR-NK cells
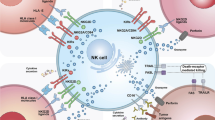
The success of CAR-T therapy in clinical trials has led to the development of CAR-NK cells. Genetic engineering of NK cells with a CAR structure is also similar with the generation of CAR-T cells. The procedure for adoptive CAR-NK therapy in cancer patients is described in the Fig. 1.

Generation of CAR-NK cells for adoptive transfer immunotherapy in cancer patients. A. Evolution of CAR structures from the first generation to the fourth generation according to NK cell intracellular motifs and functions. The new NK-CAR structure contains NKG2D ectodomain with CD3ζ, DAP10 or DAP12 cytoplasmic signal domain. B. The mechanisms of tumor destruction by CAR-NK cells through both CAR-dependent and CAR-independent manners. C. Different cell sources and corresponding procedures for manufacturing CAR-NK cell products
CAR structures
At present, most preclinical studies on CAR-NK cells still follow the CAR structures adopted by CAR-T therapies. The CAR construct is similar to that of the T cell receptor. It consists of several parts including an extracellular antigen binding region, an extracellular hinge domain and a transmembrane region, and an intracellular signal transduction region that transmits activation signals and costimulatory signals to T cells. The extracellular antigen binding region is a single-chain variable fragment (scFv), which is constructed by connecting the heavy chain variable region and the light chain variable region of the monoclonal antibody via a short linker peptide [18,19,20]. The CAR structure has undergone at least five generations of evolution. The first-generation structure that only provides the first activation signal for T cells cannot effectively activate T cells and provide a continuous anti-tumor effect in vivo [21, 22]. In the second- or third-generation structures, one or two costimulatory signal molecules are added to provide the second activation signal. CD28 and 4-1BB (CD137) are two commonly used classical costimulatory domains [23], and others such as CD27, OX40 (CD134), ICOS (CD278), CD40, toll-like receptors, and complement 3a receptor (C3aR) have also been studied [24,25,26,27,28]. In the fourth generation, some structures such as suicide genes and cytokines are integrated to regulate T cells precisely, which is worthwhile exploring to reshape the tumor microenvironment (TME) [29,30,31]. In the new generation of CAR-T cells, endogenous T cell receptors and leukocyte antigen class I (HLA) are knocked out and fusion proteins are integrated for reducing the toxic side effects and re-establishing the immune system after infusion [20].
Previous studies of CAR-NK cells used the first four generations of CAR constructs designed for T cells, such as 4-1BB, CD28 costimulatory domains, and CD3ζ signaling domains. However, recent studies have shown that the domains specific to NK cells can produce stronger activation of NK cells and anti-tumor response [32]. Ye Li and his team demonstrated that CAR constructs specifically designed to enhance NK cell activity have a potent ability to kill tumors both in vitro and in vivo. They finally concluded that CAR-NK cells with scFv (anti-mesothelin)-NKG2D-2B4-CD3ζ (NK-CAR4 structure) have stronger cytotoxicity, expansibility and persistence than scFv-CD28-CD137-CD3ζ (T-CAR structure) [33]. CD3ζ and DNAX-activation proteins (DAP10 and DAP12) are signaling adaptor molecules that contain immunoreceptor tyrosine-based activation motifs (ITAMs). They can initialize the activation of NK cells through phosphorylation mediated by protein tyrosine kinases of the Src family and then up-regulate the phosphorylation of signaling pathways such as the Syk-vav1-Erk and NF-kB pathways, resulting in cytotoxicity (release of cytotoxic granules, including perforin and granzymes) and cytokines production (e.g., TNF-α and IFN-γ) [34,35,36]. As an important activation receptor, NKG2D is a C-type lectin-like family molecule expressed on the surface of almost all NK cells [37]. DAP10 is a downstream signal molecule that transmits signals for the activating receptor NKG2D. The two can combine specifically via the induced fit theory and further induce phosphorylation [38, 39]. Similarly, DAP12 transmits activation signals for activating receptors such as NKG2C and NKp44. The DAP12 domain that contains only one ITAM can provide sufficient and better activation signaling to NK cells compared with a CAR containing the CD3ζ chain with three ITAMs [40]. Another activating receptor, 2B4, belongs to the signaling lymphocytic activation molecule family and contains an immunoreceptor tyrosine-based switch motif that mediates signal transduction associated with the activation of NK cells. And 2B4 was demonstrated to enhance cytokine secretion after linking with NKG2D [41]. The evolution of the CAR structures and the specific NK-CAR were displayed in the Fig. 1A.
CAR vector transfer in NK cells
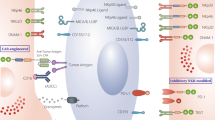
NK cells are more difficult to be successfully engineered with CAR structures compared with T cells, as a result of different sensitivity to the interference of foreign introduced genes [42]. Thus, it leads to lower transduction efficiency than CAR engineered to T cells due to the resistance of inserting foreign genes mediated by their natural defense receptors [43]. Approaches for CAR gene transferred into NK cells can be divided into viral and non-viral ways (Fig. 2). And currently, there is no available gene transfer method that is universally applicable because every method more or less has flaws.

Various approaches of delivering CAR into NK cells. The methods for genetic engineering of NK cells including viral transduction (upper) and non-viral electroporation (lower), as well as their strengths and drawbacks are illustrated
Viral transductions including the retroviral and lentiviral-based vectors enable foreign genes to insert into the targeted cell genome and stably express for a long time. Viral vectors can accommodate complicated and long sequences up to 10kbp, and transfer them into the host integrally [44]. It is also the main method used in a large number of CAR-NK preclinical studies. However, there are likewise some drawbacks of viral transduction. Retroviral vectors are likely to cause insertion mutation and toxic damage to primary NK cells, and can only introduce foreign genes when the host cells are in the stage of proliferation with their nuclear membrane dissolving [45]. Before transduction, NK cells need to be stimulated into an expanded state with some cytokines (e.g., IL-2, IL-21, IL-15, IL-12, IL-18) [46, 47]. In contrast to retroviral vectors, lentiviral vectors can integrate their genetic materials into resting cells but with lower efficiency. Multiple rounds of transfection are often needed to achieve the transduction efficiency required [48, 49]. In order to improve the efficiency, it is often necessary to add auxiliary transfection reagents, such as polybrene, RetroNectin, or Vectofusin-1[50,51,52]. Müller and his coworkers compared two different vectors, lentiviral and alpharetroviral, both combined with two different transduction enhancers (RetroNectin and Vectofusin-1). They concluded that using RD114-TR pseudotyped retroviral particles in combination with Vectofusin-1 is a promising strategy to genetically modify NK cells to achieve highly cytotoxic CD19-CAR-NK cells with high yield [53]. In addition, it can also optimize virus packaging by selecting the best pseudotyping envelope protein, such as using baboon envelop glycoprotein (BaEV-gp) instead of the commonly used vesicular stomatitis virus glycoprotein G (VSV-G). A research group reported that using BaEV pseudotyped lentiviral vectors can obtain a transduction rate of about 83.4% in activated NK-cells, which outperformed VSV-G-, RD114- and measles virus-pseudotyped lentivirus [54]. Another group found that spinfection transduction (centrifugation at a low speed) showed better efficiency (19–73%) with CD19-CAR lentiviral transduced to NK cells from cord blood compared with static transduction (12–30%) [55]. Thus, spinfection may be another method to further optimize viral transduction. Recently, recombinant adeno-associated virus (AAV) has been broadly adopted and been regarded as an alternative gene delivery tool, owing to their high efficiency of transduction and a better safety profile. The combination of CRISPR/Cas9 and AAV resulted in highly efficient and stable CD33-CAR expression applicable to cancer immunotherapy. But AAV also has limitations of constrained packaging capacity and vector production difficulty [56].
Another non-viral strategy of CAR-NK engineering is transfection with electroporation. Electroporation can transport mRNA or DNA into the cell by utilizing short electric pulses to make tiny holes of the cell membrane. But it may damage cells due to membrane leakage [57, 58]. DNA electroporation needs higher transfection conditions and leads to the lower efficiency and viability of cells than mRNA electroporation [59, 60], so it has not been used in clinical trials of CAR-NK cells. Compared with viral transduction, although the efficiency of mRNA electroporation is higher, the expression of the CAR gene cannot be maintained for a long time owing to transient transfection without integrating into the cell genome [61, 62]. And Boissel, L concluded that lentiviral vectors should be used for CAR transduction if primary NK cells are considered. Transfection with mRNA only is suitable for meeting clinical demand in NK-92 cells [55]. Lin Xiao et al. reported that utilizing optimized mRNA electroporation for NK cells expressing NKG2D-DAP12 CAR structures provided almost 99% transfection efficiency, which was maintained for about 5 days [63]. Some researchers combine electroporation with transposons or CRISPR/Cas9-based integration [64,65,66]. The sleeping beauty transposon system for integrating CAR vectors has been tried out in CAR-NK cells [33]. Recently, the new technology of CRISPR/Cas9 can accurately select the site of integration of the CAR gene and perform gene knockout in NK cells, which provides new possibilities for improving CAR-NK cell products [67]. Gurney et al. applied CRISPR/Cas9 genome editing to knock down the CD38 gene during NK expansion with a mean efficiency of 84% and then expressed an affinity optimized CD38-CAR. They found that the cytotoxic potential of CD38 KO-CD38 CAR-NK cells was augmented and proposed a viable immunotherapeutic approach for the treatment of acute myeloid leukemia (AML) [68].
Mechanisms of cancer killing
CAR-independent killing mechanism
The cytotoxicity of NK cells is controlled by a series of active and inhibitory receptors expressed on themselves. When encountering tumor cells or other stress conditions, activating receptors on NK cells, such as NKG2D, NKp30, NKp44 and NKp46, can be stimulated to engage with ligands, thus triggering NK activation and mediating their killing activity [69, 70]. On the contrary, some inhibitory receptors such as killer immunoglobulin-like receptors (KIRs) can inhibit the activity of NK cells when they bind to corresponding ligands. For example, our healthy cells express human leukocyte antigen (HLA) ligands that bind to KIRs so that NK cells do not attack them under normal circumstances [71]. In addition, NK cells exert their non-targeted cytotoxicity in a non-major histocompatibility complex restricted manner. First, NK cells kill target cells through similar cytotoxic mechanisms with CD8+ cytotoxic T cells, including releasing perforin/granzyme, which leads to target cell lysis; upregulating Fas ligand or TRAIL receptor on their surface to induce tumor cell apoptosis; and releasing cytokines and chemokines to recruit and activate other immune effector cells such as macrophages and dendritic cells [72,73,74]. Moreover, CD16 (Fc receptor) expressed on NK cells recognizes the Fc segment of immunoglobulin G that binds to the target cells, thereby mediating the cytotoxicity of NK cells against tumors. It is the antibody-dependent cell-mediated cytotoxicity (ADCC) [75].
CAR-dependent killing mechanism
Although the cytotoxicity of NK cells is non-targeted, NK cells can reach the tumor site where specific antigens are expressed and kill tumors in targeted ways after genetically engineered with the CAR structure. Figure 1B displayed the two kinds of killing mechanisms of CAR-NK cells. Different specific targets of CAR can be selected according to the various tumor types. Table 2 and Table 3 summarize some preclinical researches of the anti-target CAR-NK cell therapy for hematological malignancies and solid tumors, respectively. For the hematological malignant tumors such as lymphomas and leukemias, CD19 is the most commonly used target that is only overexpressed on B-cell malignancies. BCMA, CD138 and CS1 are the targets of multiple myeloma for CAR-NK cells. Others such as CD7, CD123 and CD5 are also the anti-targets for engineered-NK cell therapy (Table 2). For solid tumors, anti-HER2 and anti-EGFR engineered-NK therapy have been reported in several cancers like glioblastoma, breast, and ovarian cancers. Han reported that EGFR-CAR-engineered NK cells displayed enhanced cytolytic capability and production of IFN-γ, which restrained glioblastoma growth and obviously prolonged the survival of tumor-bearing mice [76]. Other antigens including mesothelin, PSCA, GPA7 and EpCAM were explored as CAR-NK cell targets in solid tumors (Table 3).
Advantages of CAR-NK immunotherapy
Abundant sources of NK cells
Compared with CAR-T cells that can only be derived from autologous (patient-derived) peripheral blood, CAR-NK cells have more diverse sources to choose from. Different cell sources and corresponding procedures for manufacturing CAR-NK cell products were summarized in the Fig. 1C. Allogeneic NK cells provide a greater possibility of making “off-the-shelf” products [125], which can not only reduce the long period needed for CAR-T production, but also prevent patients with poor physical condition from being unable to receive CAR-engineered cellular immunotherapy. Because patients whose immunity is already severely damaged may not provide enough sufficient functional lymphocytes. Thus, CAR-NK immunotherapy may be more applicable than CAR-T cells in the future. CAR-NK cells can be obtained from several different sources of NK cells, including peripheral blood-derived NK (PB-NK) cells; cord blood-derived NK (CB-NK) cells; some clonal cell lines such as KHYG-1, NK-92, NKL and YT cells; NK cells derived from human induced pluripotent stem cells (iPSC-NK) or human embryonic stem cells [47, 69, 126, 127].
PB-NK and CB-NK cells are both primary NK cells, which need to be activated and expanded before the cytotoxicity of CAR-NK therapy can be effectively exerted [128]. Many clinical trials used PB-NK cells as they are derived from HLA-mismatched donors with no risk of GVHDs [129]. NK cells can be isolated from peripheral blood mononuclear cells by CD3 depletion or combining consecutive CD56-positive selection of NK cell isolation kits [130,131,132]. Then they can be expanded and activated in NK cell-specific expansion media with cytokines and other stimulated cells suitable for clinical use [133]. It has been proved that a large number of highly active NK cells can still be obtained from frozen CB compared with fresh CB [134, 135]. Therefore, sufficient CB can be obtained at one time from the CB bank to produce large-scale CAR-NK cells, without temporary screening of a single qualified adult donor and leukapheresis as the PB-NK. In addition, the CB bank provides another strength for selecting NK cells of donors with certain HLA types and specific NK cell receptor characteristics. Selecting a donor with HLA-KIR mismatch may enhance the alloreactivity of NK cells [136, 137]. Therefore, compared with PB-NK, CAR-NK cells derived from CB-NK are more likely to serve as “off-the-shelf” products for cellular immunotherapy. However, some researchers found that CB-NK cells have immature phenotypes, exhibiting normal levels of degranulation but lower cytotoxicity, with decreased expression of some molecules such as CD16 compared with PB-NK cells [138,139,140].
NK cell lines are also the main source of CAR-NK cells in preclinical and clinical studies as they are relatively easy to successfully transduce CAR genes [45]. Among these cell lines, NK-92 is the most commonly used and the only one approved by the FDA for clinical trials of CAR-NK-92 therapy [141]. Tang reported a first-in-man clinical trial of CD33-CAR NK-92 cells and showed that this therapy can be safely used in patients with relapsed and refractory AML [142]. There are a series of activated receptors but almost no inhibitory killer receptors expressed on NK-92 cells, and NK-92 cells are unable to mediate ADCC because of shortage of the CD16 receptor [143,144,145]. CAR-engineered NK-92 cells have strong cytotoxicity and can obtain a large number of cells with the same phenotypes in a short time [86, 146]. The disadvantage of CAR-NK92 cells is that they must be irradiated to avoid malignant proliferation before infused into patients [83]. The proliferation of irradiated CAR-NK cells is inhibited in vivo, which leads to a short persistence time [114]. Although it can avoid some side effects of CAR-T therapy such as “off-target effects,” it also reduces its anti-tumor effect, thereby requiring multiple transfusions.
NK cells can also be generated from hematopoietic progenitor cells (HPCs). Peripheral blood apheresis after stimulation, CB, bone marrow, and ESC are the approaches for obtaining CD34+HPC [147, 148]. After induced by the culture medium including a mixture of cytokines such as IL-3, stem cell factor, IL-15, FLT3L and IL-7, CD34+HPC will differentiate into NK cells for adoptive immunotherapy [149]. IPSC-NK is considered to be a potential cell source for “off-the-shelf” CAR-NK products because of its unlimited expansion ability and higher transduction efficiency than primary NK cells [33, 150, 151]. After proper ways of differentiation and expansion, only one CAR-iPSC cell can be used to obtain a large number of CAR-NK cells with uniform phenotypes. That's why it is easier to get a standardized “off-the-shelf” product. However, similar to CB-NK, iPSC-NK cells have lower cytotoxicity due to their immature phenotypes such as low CD16 and KIR expression, and high NKG2A expression compared with PB-NK cells [151,152,153]. Therefore, some problems need to be addressed before iPSCs can be used on a large scale in the future. The first clinical trial about CAR-engineered iPSC-NK product, FT596, was carried out in 2019. These CD19-targeted CAR iPSC-NK cells express a high-affinity, non-cleavable CD16 with an IL-15 receptor fusion protein. Therefore, their anti-tumor ability was enhanced by overcoming the low expression of CD16 and improving the expansion and persistence of CAR-NK cells in vivo. The ongoing phase I trial (NCT04245722) also provides a promising guidance for CAR-iPSC-NK to become “off-the-shelf” products for clinical cellular immunotherapy.
Better safety and multiple available mechanisms of cytotoxicity
In contrast to CAR-T cell-based immunotherapy, CAR-NK cells have superior safety performance mainly reflected in the following aspects. First, allogeneic NK cells do not cause a lethal risk of GVHD that extremely associated with T cells, which has been verified in some animal or human clinical trials of CAR-NK cells [154, 155]. In addition, the cytokines secreted by NK cells are almost GM-CSF and IFN-γ with lower toxicity profiles [156]. They are different from most inflammatory cytokines secreted by T cells such as IL-1, IL-2, IL-6, TNF-a, IL-8, IL-15, MCP1, and IL-10, which are closely related to the side effects of CAR-T therapy [157, 158]. Therefore, CAR-NK cells injected into the patient nearly have no side effects such as CRS or ICANS, making them become a more attractive choice for anti-tumor cellular immunotherapy. In addition, the lifespan of NK cells in the blood cycle is relatively limited [159], further reducing the potential for toxicity to normal tissue, such as B cell aplasia caused by long-term persistence of CAR-T cells in vivo [160]. But it is possible that designed CAR structures will induce unanticipated toxicity due to excessive cytokine production [161]. Considering the potential toxicity caused by long-lived genetically modified CAR-NK cells, suicide genes have been used as safety switches and inducible caspase 9 (iCas9) has been shown to effectively eliminate CAR-NK cells both in vitro and in vivo [162, 163].
As mentioned earlier, NK cells release cytotoxicity depending on their germline-encoded activating and inhibitory receptors. And they do not rely on the existence of tumor-associated antigens on tumor cells. Therefore, even if tumor cells initiate immune escape mechanisms and lead to the down-regulation of the targeted antigens, CAR-NK cells can still reserve the intrinsic killing efficiency mediated by their receptors. Some studies have also attempted to add a fusion protein of CD16 into the CAR structure to enhance ADCC [164, 165]. A high-affinity, non-cleavable CD16 (hnCD16) expressed on NK cells with CAR structures provides the possibility of combining CAR-NK cells with antibodies targeting different antigens. An iPSC-derived CAR-NK cell product, iDuo NK cell, is engineered with three functional elements including a CD19-CAR, a high-affinity and non-cleavable CD16, and a membrane-bound IL-15/IL-15R fusion molecule (IL-15RF). The modified iDuo NK cells exhibited effective elimination of both CD19+ and CD20+ lymphoma cells through a combination of intrinsic cytotoxicity, anti-CD19 CAR mediated killing, and ADCC mediated by hnCD16 engagement with rituximab [166]. Thus, unlike CAR-T cells that only rely on CAR-targeted mechanism, CAR-NK cells keep immune-monitoring and killing tumor cells that express different levels of CAR-targeted antigens through multiple cytotoxic mechanisms.
High feasibility of “off-the-shelf” products
Simplifying the manufacturing process and saving cost are also major difficulties that should be overcome for the clinical widespread of CAR-T therapy. Many studies made efforts to develop efficient and reliable “off-the-shelf” T cell products. However, the requirements of individual specificity for autologous CAR-T cells, demand for facilities for cold-chain transportation, and unavoidable GVHD risks make it difficult to achieve. Unlike CAR-T cells, CAR-NK cells are regarded as an ideal alternative platform for cancer immunotherapy by researchers because of their unique advantages and the potential for the production of “off-the-shelf” immunotherapeutic products. For example, NK-92 cells engineered to express different CARs targeting CD19, CD20, CD38, HER2, PSMA or GD2 were considered as allogeneic “off-the-shelf” immunotherapeutic products and held great promise for the development of effective anti-cancer treatments [114, 144, 167, 168]. Moreover, CAR-NK cells obtained from iPSC are a standardized homogeneous cell population that can be promoted in a clinically scalable manner [151, 169]. With abundant sources, no risk of GVHD response, and no need for patient-specificity, CAR-NK cells are more feasible to steadily acquire “off-the-shelf” products.
CAR-NK cells against solid tumors
Although there are large similarities between CAR-NK and CAR-T cell therapies and they are both known to be effective against hematological malignancies, the inefficiency of CAR-T cells in solid tumors should not be extended to CAR-NK therapy. The natural properties of NK cells and multiple killing mechanisms offer a promising prospect for CAR-NK cells in solid cancer therapy. A growing number of studies have examined the activity of CAR-NK cells against solid tumors, such as glioblastoma, breast, ovarian and pancreatic cancer [170]. Intracranial injections of bispecific EGFR- and EGFRvIII- targeted CAR-NK-92 prolonged the survival of glioblastoma xenograft mouse [171]. Several studies have demonstrated the effectiveness of CAR-NK cell therapies targeting mesothelin, CD24 and glypican-3 in ovarian cancer models [112, 117, 172]. Additionally, the first clinical trial of CAR-NK therapy for solid tumor treatment used MUC-1 CAR-NK cells to target against multiple malignant solid tumors, e.g., glioblastoma, pancreatic, colorectal, breast and ovarian cancer (NCT02839954). Of the eight patients, seven achieved stable disease without serious adverse events. Although CAR-NK therapy has a bright potential for solid tumor, there are still many limitations to overcome. Furthermore, the application of CAR-NK therapy for solid tumors may require additional modifications of the NK cells beyond CAR transduction to improve intratumor trafficking, overcome the immunosuppressive TME and prevent tumor antigens escape.
Current clinical trials of CAR-NK cell immunotherapy
As of June 2022, there have been 32 clinical trials (summarized in Table 4) of CAR-NK therapy registered on the ClinicalTrials.Gov website. Two of them have been completed, nine of them are in early phase I, and 21 of them are in phase I/II. Among these trials, there are some targets for hematological malignant tumors, such as CD19, CD7, BCMA, CD33, CD22. Eleven trials are about solid tumors (including ovarian cancer, prostate cancer, non-small cell lung cancer, pancreatic cancer, and glioblastoma), in which CAR-NK cells target some over-expressed antigens such as MUC1, PSMA, ROBO1, mesothelin, and HER2. According to the data disclosed, PB-NK (8/32) and NK-92 (9/32) are the most common cell sources, and lentiviral transduction is mostly used (not shown in the table). To date, the results of a few clinical trials have been published. Two are small-scale (n = 3) clinical trials, using NK-92 targeting CD33 (NCT02944162) and PB-NK cells targeting NKG2DL (NCT03415100) [63, 142]. Both the small-scale clinical trials demonstrated the advantages of CAR-NK in the treatment of tumors, such as not inducing GVHD or other immune toxicities, providing the potential of “off-the-shelf” products for cancer immunotherapy.
Another one is a promising large-scale (NCT03056339) phase I/II clinical trial, which was published in February 2020. In this large-scale trial, 11 patients with R/R CD19-positive cancers (non-Hodgkin’s lymphoma or chronic lymphocytic leukemia) received an allogeneic CB-derived CAR-NK cell product after lymphodepleting chemotherapy [173]. Anti-CD19 CAR-NK cells were transduced to express IL-15 and an iCaspase-9-based safety switch. Eight of the 11 patients (73%) received treatment had no obvious side effects, such as CRS, ICANS and GVHD, seven patients had complete remission. The corresponding preclinical study reported that CB-derived NK cells were transduced with the genes for CAR-CD19 and IL-15. These cells showed effective cytotoxicity and apparent prolongation of survival with IL-15 production by the transduced CB-NK cells in a xenograft Raji lymphoma murine model [82]. In this clinical trial, expansion was observed as early as 3 days after injection. And CAR-NK cells persisted for at least 12 months, which was also associated with the incorporation of IL-15 in the CAR vector. In summary, the small progress of clinical trials indicates a bright future for the clinical application of CAR-NK products.
Obstacles to the popularization of CAR-NK therapy and corresponding coping strategies
Regardless of the great prospect of CAR-NK cells in tumor immunotherapy, there are still some thorny problems hindering their further approval and promotion. Researchers are constantly exploring how to improve them for their successful popularization in the clinic.
Expansion and purification of NK cells
A large-scale highly cytotoxic NK cells are needed for clinical adoptive therapy. The proportion of NK cells in peripheral blood lymphocytes does not exceed 10–15%, so in vitro culture for expansion is needed. Some cytokines can improve the expansion of NK cells, but it is very difficult to induce a constant proliferation of human NK cells under a kind of cytokine such as IL-2 [174]. Some researchers have shown that IL-2 in combination with other cytokines (anti-CD3 antibody or IL-15) can exert better stimulatory effects on NK cells [175,176,177]. Chronic myelogenous leukemia-derived cell line K562 can stimulate NK cells [178], but the unmodified K562 cell is of limited use. It needs to be modified before it can be used to amplify NK cells. Co-culturing PB-NK cells with K562 feeder cells genetically modified to express IL-15 and the adhesion molecule 4-1BBL is a good method [78]. A manually performed experiment with GMP-compliant media containing IL-2, IL-21 and irradiated autologous feeder cells made an effective cell expansion, which induced an 85-fold NK cell expansion [98]. Recently, Yan Yang et al. applied a new feeder cell system (human B-lymphoblastoid cell-line 721.221) with human B-lymphoblastoid cell-line 721.221 to stimulate NK cells. The results showed that new feeder cells were superior to K562-mIL-21 cells with better proliferation and less apoptosis during expansion [179]. NK cells from human ESC and iPSC have been certified to have functions similar to conventional primary cells. But they are more homogenous and feasible to be genetically modified at a clonal level, and easy to be expanded in a clinical scale [139, 153, 180]. Another trouble is allogenic NK cells may be contaminated with T cells. In a first-in-human trial of adoptive transfer of donor-derived IL-15/4-1BBL-activated NK cells (aNK), researchers found that aNK-DLI contributed to acute GVHD, likely by augmenting the underlying T-cell alloreactivity [181]. Pre-lymphocyte deletion may contribute to purifying NK cells [182]. Moreover, regulatory T cells and myeloid-derived suppressor cells (MDSCs) may influence NK cells therapies [45]. It is necessary that finding a suitable method to expand and purify NK cells to serve the clinic effectively.
Persistence of CAR-NK cells in vivo
NK cells do not go through massive expansions and have a limited lifespan in vivo, which is why they do not cause severe side effects unlike CAR-T cells but result in low persistence. Cytokines such as IL-2 and IL-15 are key molecules involved in NK cells functions, including differentiation, proliferation, activation, and survival [183, 184]. IL-2 diphtheria toxin fusion protein (IL-2DT) may improve the expansion of NK cells in AML patients [185]. However, a high dose of IL-2 can cause serious adverse effects [186]. IL-15 has been demonstrated to stimulate NK cells proliferation and up-regulate activating receptor NKp30 expression on NK cells in vitro [82, 187, 188]. ALT-803, a kind of superagonist, was shown to be well tolerated by patients and could promote CD8+ T and NK cell expansion in vivo [188]. A TriKE (IL-15 trispecific killer engager that targets CD33) was constructed to induce expansion and persistence in vivo with a modified IL-15 cross-link [189, 190]. Currently, TriKE is being evaluated in phase I/II clinical trials in patients with CD33-expressing high-risk myelodysplastic syndromes, such as R/R AML (NCT03214666).
Suppression of TME
After being attacked, tumor cells may directly inhibit the functions of CAR-NK cells or contribute to the generation of a local suppressive TME. This is the reason why CAR-NK cells, with high cytotoxicity against cancer cells, gradually lose their anti-tumor ability in vivo.
Tumor cells can produce some soluble immune factors such as TGF-β, prostaglandin E2 (PGE2), indoleamine 2,3-dioxygenase (IDO), IL-10, and PD-1, which suppress the function of NK cells. Produced by neutrophils, macrophages, and Tregs in the TME, TGF-β may affect the metabolism of NK cells and down-regulate NKG2D and NKp30 expression [191, 192]. Tregs and immunosuppressive MDSCs are actively recruited to the local TME to inhibit the cytotoxicity of CAR-NK cells and promote tumor growth [193]. PGE2 has a negative impact on the differentiation and cytotoxicity of NK cells, and inhibits expression of NKp44 and NKp30 on NK cells. [194, 195].
TME is frequently characterized by hypoxia and it is a common metabolic disturbance. It can greatly influence NK cells by affecting their metabolism and infiltration, cytokines production, and expression of several activating receptors [196,197,198]. Activating receptors such as NKp30, NKp46, NKp44, and NKG2D on NK cells would be down-regulated for tumor growth and metastasis [197]. CD73 induces the expression of arginase, an immunosuppressive metabolite. Arginase is produced under hypoxia conditions and inhibits NK cell functions. The inhibition of CD73 increases the homing of NKG2D-CAR NK cells that target tumor cells expressing NKG2D ligands and improves anti-tumor responses in animal models of lung cancer [199]. In terms of migration, hypoxia may also influence the surface expression of CCR7 and CXCR4 on CD56 bright NK cells, increasing their migration response to CCL19/21 and CXCL12 [198].
Tumor cells can also express ligands for the so-called checkpoint proteins that help tumor cells evade immune surveillance. PD-1 has been verified to be expressed in PB-NK cells from patients with multiple myeloma, and in blood and tumor-associated NK cells from patients with renal and ovarian cancers [200,201,202]. CAR constructions combined with blockers of checkpoint proteins such as anti-PD-1, CTLA-4, LAG3, and TIGIT have more therapeutic benefits compared with using traditional CAR structures [203]. NK-92 cells engineered to express high-affinity CD16, IL-2, and PD-L1-specific CAR structure released high levels of perforin and granzyme and lysed human cancer cell lines including breast, lung, and gastric cancers [204].
Storage, shipping, and recovery of “off-the-shelf” CAR-NK cells
The storage, shipping, and recovery of “off-the-shelf” CAR-NK cells are necessary to facilitate large-scale clinical promotion, but some problems come across during these processes. Compared with T cells, NK cells are more sensitive to the process of freezing and thawing. The survival rate and cytotoxicity of NK cells are significantly down-regulated after thawing [205]. Some researchers found that the influence of frozen NK cells can be improved by culturing with IL-2 [206]. Moreover, cytokine-activated NK cells are very sensitive to lower temperature [207]. Their cytotoxicity should be maintained under body temperature during the process of shipping. Appropriate cell density is also crucial in the process of shipping. High cell concentrations may lead to the loss of cells activity possibly due to the rapid consumption of medium and changes in glucose and pH value. Moreover, the quality of cells is difficult to control in the shipping process. The sensitivity of NK cells to cryopreservation makes them difficult to store and transport, which is a limitation of CAR-NK cell therapy. Strategies for optimal cryopreservation must be explored to make CAR-NK cells therapy available for “off-the-shelf” products.
Combination strategies of CAR-NK cells with other therapies
NK cells express CD16 and exert tumor killing through ADCC. Another strategy for combination therapy is to combine CAR-NK cells with antibodies targeting different tumor antigens. The ADCC-inducing antibodies of anti-CD20, anti-HER2, anti-EGFR and anti-GD2 have promising results on refractory NHL, breast cancer, colorectal cancer and neuroblastoma [208]. FT596 not only targeted CD19+ lymphoma, but also exhibited enhanced killing effect on the CD20+ lymphoma cells when combined with the anti-CD20 antibody rituximab (NCT04245722). IL-2 and IL-15 have been identified as key cytokines that upregulate the activity of NK cells. The antitumor effect of PD-L1 CAR-NK cells, in combination with anti-PD-1 and N-803, an IL-15 superagonist, resulted in superior control of tumor growth in C57BL/6 mice [204]. Therefore, cytokines-based treatment may improve the persistence and cytotoxicity of CAR-NK cells. In addition, chemotherapy not only reduces rejection of infused allogeneic NK cells by the recipient, but may also reprogram the TME to facilitate NK cell infiltration and survival in vivo [209]. The sequential treatment with chemotherapeutic agent followed by CAR-NK cells led to the strongest clinical efficacy of ovarian cancer [210]. Chemotherapy remains an important adjuvant therapeutic approach for future CAR-NK cell therapy.
Blockade of PD-1/PDL1 axis in combination with CAR-T therapy has been proved to be effectively in improving the anti-tumor effect [211, 212]. PD-1 disruption by CRISPR/Cas9 augments anti-CD19 CAR-T cell mediated tumor killing [213]. Similar to T cells, NK cells express some immune checkpoints molecules that inhibit the anti-tumor activity of activated NK cells. Immune checkpoint blockade (ICB) therapies by corresponding antibodies (e.g., anti-PD1/PDL1 and anti-CTLA4) that block inhibitory signals of NK cells activation can also enhance NK cell activity. Combination therapy of anti-PSMA CAR-NK cells and anti-PD-L1 monoclonal antibody enhances the anti-tumor efficacy against prostate cancer [115]. Studies have demonstrated that targeting ICB molecules like PD-1/PDL1 or B7-H3 can enhance the anti-tumor activity of CAR-NK cells [214, 215]. The silencing of immune checkpoint NKG2A enhances NK cell mediated cytotoxicity against multiple myeloma [216]. Therefore, CAR-NK cells in combination with ICB therapy or with knockout of ICB molecules by gene-editing will be a promising approach for improving the efficiency of CAR-NK therapy, especially for solid tumors.
Conclusion and outlook
Currently, CAR-NK cell-based therapy has become a popular research field because of its unique advantages and feasibility of “off-the-shelf” products compared with CAR-T therapy, so it might become an alternative cellular immunotherapy for cancers. Combining CAR engineering with CD16 expression on NK cells also further enhances their anti-tumor ability by utterly utilizing their cytotoxic capacity and ADCC. However, some problems need to be studied and addressed continuously, including how to optimize the gene editing technology of CAR structure specific for NK cells, how to expand and activate NK cells briefly and effectively, and how to improve persistence and repair TME to advance the therapeutic effect of solid tumors. With the approval of more clinical trials and the guidance of more clinical data in the next few years, CAR-NK cells will be gradually optimized to provide efficient and safe “off-the-shelf” products for cancer immunotherapy.
Availability of data and materials
Not applicable.
Abbreviations
- CAR:
-
Chimeric antigen receptor
- NK:
-
Natural killer
- R/R:
-
Relapsed/refractory
- GVHD:
-
Graft-versus-host disease
- CRS:
-
Cytokine release syndrome
- ICANS:
-
Immune effector cell-associated neurotoxicity syndrome
- scFv:
-
Single-chain variable fragment
- ITAMs:
-
Immunoreceptor tyrosine-based activation motifs
- BaEV-gp:
-
Baboon envelop glycoprotein
- VSV-G:
-
Vesicular stomatitis virus glycoprotein G
- KIRs:
-
Killer immunoglobulin-like receptors
- HLA:
-
Human leukocyte antigen
- hnCD16:
-
High-affinity, non-cleavable CD16
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- PB:
-
Peripheral blood
- CB:
-
Cord blood
- iPSCs:
-
Induced pluripotent stem cells
- AML:
-
Acute myeloid leukemia
- HPCs:
-
Hematopoietic progenitor cells
- MDSCs:
-
Myeloid-derived suppressor cells
- TME:
-
Tumor microenvironment
- PGE2:
-
Prostaglandin E2
References
Mullard A. FDA approves first CAR T therapy. Nat Rev Drug Discov. 2017;16(10):669.
Mullard A. Second anticancer CAR T therapy receives FDA approval. Nat Rev Drug Discov. 2017;16(12):818.
Reagan PM, Friedberg JW. Axicabtagene ciloleucel and brexucabtagene autoleucel in relapsed and refractory diffuse large B-cell and mantle cell lymphomas. Future Oncol. 2021;17(11):1269–83.
Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, Mehta A, Purev E, Maloney DG, Andreadis C, Sehgal A, Solomon SR, Ghosh N, Albertson TM, Garcia J, Kostic A, Mallaney M, Ogasawara K, Newhall K, Kim Y, Li D, Siddiqi T. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396(10254):839–52.
Anderson LD. Idecabtagene vicleucel (ide-cel) CAR T-cell therapy for relapsed and refractory multiple myeloma. Future Oncol. 2022;18(3):277–89.
Neelapu SS. Managing the toxicities of CAR T-cell therapy. Hematol Oncol. 2019;37(Suppl 1):48–52.
Yang Y, Jacoby E, Fry TJ. Challenges and opportunities of allogeneic donor-derived CAR T cells. Curr Opin Hematol. 2015;22(6):509–15.
Gross G, Eshhar Z. Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: counteracting off-tumor toxicities for safe CAR T cell therapy. Annu Rev Pharmacol Toxicol. 2016;56:59–83.
Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, Kalos M, June CH. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2(2):112–20.
Bagley SJ, O’Rourke DM. Clinical investigation of CAR T cells for solid tumors: Lessons learned and future directions. Pharmacol Ther. 2020;205: 107419.
Portillo AL, Hogg R, Poznanski SM, Rojas EA, Cashell NJ, Hammill JA, Chew MV, Shenouda MM, Ritchie TM, Cao QT, Hirota JA, Dhesy-Thind S, Bramson JL, Ashkar AA. Expanded human NK cells armed with CAR uncouple potent anti-tumor activity from off-tumor toxicity against solid tumors. iScience. 2021;24(6):102619.
Chan LY, Dass SA, Tye GJ, Imran SAM, Zaman WSWK, Nordin F. CAR-T Cells/-NK cells in cancer immunotherapy and the potential of MSC to enhance its efficacy: a review. Biomedicines. 2022. https://doi.org/10.3390/biomedicines1004.
Ebrahimiyan H, Tamimi A, Shokoohian B, Minaei N, Memarnejadian A, Hossein-Khannazer N, Hassan M, Vosough M. Novel insights in CAR-NK cells beyond CAR-T cell technology; promising advantages. Int Immunopharmacol. 2022;106: 108587.
Basar R, Daher M, Rezvani K. Next-generation cell therapies: the emerging role of CAR-NK cells. Hematol Am Soc Hematol Educ Prog. 2020;2020(1):570–8.
Rafei H, Daher M, Rezvani K. Chimeric antigen receptor (CAR) natural killer (NK)-cell therapy: leveraging the power of innate immunity. Br J Haematol. 2020. https://doi.org/10.1111/bjh.17186.
Liu SZ, Galat V, Galat Y, Lee YKA, Wainwright D, Wu J. NK cell-based cancer immunotherapy: from basic biology to clinical development. J Hematol Oncol. 2021. https://doi.org/10.1186/s13045-020-01014-w.
Chu J, Gao F, Yan M, Zhao S, Yan Z, Shi B, Liu Y. Natural killer cells: a promising immunotherapy for cancer. J Transl Med. 2022;20(1):240.
Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2020;17(3):147–67.
Pfefferle A, Huntington ND. You have got a fast CAR: chimeric antigen receptor NK cells in cancer therapy. Cancers. 2020. https://doi.org/10.3390/cancers12030706.
Huang R, Li X, He Y, Zhu W, Gao L, Liu Y, Gao L, Wen Q, Zhong JF, Zhang C, Zhang X. Recent advances in CAR-T cell engineering. J Hematol Oncol. 2020;13(1):86.
Firor AE, Jares A, Ma Y. From humble beginnings to success in the clinic: chimeric antigen receptor-modified T-cells and implications for immunotherapy. Exp Biol Med (Maywood). 2015;240(8):1087–98.
Qu J, Mei Q, Chen L, Zhou J. Chimeric antigen receptor (CAR)-T-cell therapy in non-small-cell lung cancer (NSCLC): current status and future perspectives. Cancer Immunol Immunother. 2021;70(3):619–31.
Drent E, Poels R, Ruiter R, van de Donk N, Zweegman S, Yuan H, de Bruijn J, Sadelain M, Lokhorst HM, Groen RWJ, Mutis T, Themeli M. Combined CD28 and 4–1BB costimulation potentiates affinity-tuned chimeric antigen receptor-engineered T Cells. Clin Cancer Res. 2019;25(13):4014–25.
Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. 2021;21(3):145–61.
Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol. 2004;172(1):104–13.
Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunology. 2019;8(5): e1049.
Lai P, Chen X, Wang Y, Wang J, Zhang Y, Geng S, Li P, Du X, Weng J, Pei D. C3aR costimulation enhances the antitumor efficacy of CAR-T cell therapy through Th17 expansion and memory T cell induction. J Hematol Oncol. 2022;15(1):68.
Nouri Y, Weinkove R, Perret R. T-cell intrinsic Toll-like receptor signaling: implications for cancer immunotherapy and CAR T-cells. J Immunother Cancer. 2021. https://doi.org/10.1136/jitc-2021-003065.
Lanitis E, Rota G, Kosti P, Ronet C, Spill A, Seijo B, Romero P, Dangaj D, Coukos G, Irving M. Optimized gene engineering of murine CAR-T cells reveals the beneficial effects of IL-15 coexpression. J Exp Med. 2021. https://doi.org/10.1084/jem.20192203.
Morgan MA, Schambach A. Chimeric antigen receptor T Cells: extending translation from liquid to solid tumors. Hum Gene Ther. 2018;29(10):1083–97.
Salaroli A, Spilleboudt C, Bron D, Lewalle P. Chimeric antigen receptor T-cell lymphoma immunotherapy: the next questions. Curr Opin Oncol. 2020;32(5):434–41.
Zenere G, Olwenyi OA, Byrareddy SN, Braun SE. Optimizing intracellular signaling domains for CAR NK cells in HIV immunotherapy: a comprehensive review. Drug Discov Today. 2019;24(4):983–91.
Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23(2):181–92.
Oberschmidt O, Kloess S, Koehl U. Redirected primary human chimeric antigen receptor natural killer cells as an “Off-the-Shelf Immunotherapy” for improvement in cancer treatment. Front Immunol. 2017;8:654.
Wu J, Cherwinski H, Spies T, Phillips JH, Lanier LL. DAP10 and DAP12 form distinct, but functionally cooperative, receptor complexes in natural killer cells. J Exp Med. 2000;192(7):1059–68.
Liu Y, Li R, Chen XX, Zhi Y, Deng R, Zhou EM, Qiao S, Zhang G. Nonmuscle myosin heavy chain IIA recognizes sialic acids on sialylated RNA viruses to suppress proinflammatory responses via the DAP12-Syk pathway. mBio. 2019. https://doi.org/10.1128/mBio.00574-19.
Raulet DH, Gasser S, Gowen BG, Deng WW, Jung HY. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. 2013;31(31):413–41.
Kavazovic I, Lenartic M, Jelencic V, Jurkovic S, Lemmermann NAW, Jonjic S, Polic B, Wensveen FM. NKG2D stimulation of CD8(+) T cells during priming promotes their capacity to produce cytokines in response to viral infection in mice. Eur J Immunol. 2017;47(7):1123–35.
Wang J, Li C-Di, Sun Lin. Recent advances in molecular mechanisms of the NKG2D pathway in hepatocellular carcinoma. Biomolecules. 2020. https://doi.org/10.3390/biom10020301.
Topfer K, Cartellieri M, Michen S, Wiedemuth R, Muller N, Lindemann D, Bachmann M, Fussel M, Schackert G, Temme A. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol. 2015;194(7):3201–12.
Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood. 2010;115(11):2167–76.
Gong Y, Klein Wolterink RGJ, Janssen I, Groot AJ, Bos GMJ, Germeraad WTV. Rosuvastatin enhances VSV-G lentiviral transduction of NK cells via upregulation of the low-density lipoprotein receptor. Mol Ther Methods Clin Dev. 2020;17:634–46.
Sutlu T, Nystrom S, Gilljam M, Stellan B, Applequist SE, Alici E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: implications for gene therapy. Hum Gene Ther. 2012;23(10):1090–100.
Schmidt P, Raftery MJ, Pecher G. Engineering NK cells for CAR therapy-recent advances in gene transfer methodology. Front Immunol. 2020;11: 611163.
Carlsten M, Childs RW. Genetic manipulation of NK cells for cancer immunotherapy: techniques and clinical implications. Front Immunol. 2015;6:266.
Liu M, Meng Y, Zhang L, Han Z, Feng X. High-efficient generation of natural killer cells from peripheral blood with preferable cell vitality and enhanced cytotoxicity by combination of IL-2, IL-15 and IL-18. Biochem Biophys Res Commun. 2021;534:149–56.
Kundu S, Gurney M, O’Dwyer M. Generating natural killer cells for adoptive transfer: expanding horizons. Cytotherapy. 2021. https://doi.org/10.1016/j.jcyt.2020.12.002.
Schott JW, Leon-Rico D, Ferreira CB, Buckland KF, Santilli G, Armant MA, Schambach A, Cavazza A, Thrasher AJ. Enhancing lentiviral and alpharetroviral transduction of human hematopoietic stem cells for clinical application. Mol Ther Methods Clin Dev. 2019;14:134–47.
Gill KP, Denham M. Optimized transgene delivery using third-generation lentiviruses. Curr Protoc Mol Biol. 2020;133(1): e125.
Anastasov N, Hofig I, Mall S, Krackhardt AM, Thirion C. Optimized lentiviral transduction protocols by use of a poloxamer enhancer, spinoculation, and scFv-antibody fusions to VSV-G. Methods Mol Biol. 2016;1448:49–61.
Han M, Yu D, Song Q, Wang J, Dong P, He J. Polybrene: Observations on cochlear hair cell necrosis and minimal lentiviral transduction of cochlear hair cells. Neurosci Lett. 2015;600:164–70.
Radek C, Bernadin O, Drechsel K, Cordes N, Pfeifer R, Strasser P, Mormin M, Gutierrez-Guerrero A, Cosset FL, Kaiser AD, Schaser T, Galy A, Verhoeyen E, Johnston ICD. Vectofusin-1 improves transduction of primary human cells with diverse retroviral and lentiviral pseudotypes, enabling robust automated closed-system manufacturing. Hum Gene Ther. 2019;30(12):1477–93.
Muller S, Bexte T, Gebel V, Kalensee F, Stolzenberg E, Hartmann J, Koehl U, Schambach A, Wels WS, Modlich U, Ullrich E. High cytotoxic efficiency of lentivirally and alpharetrovirally engineered CD19-specific chimeric antigen receptor natural killer cells against acute lymphoblastic leukemia. Front Immunol. 2019;10:3123.
Colamartino ABL, Lemieux W, Bifsha P, Nicoletti S, Chakravarti N, Sanz J, Romero H, Selleri S, Beland K, Guiot M, Tremblay-Laganiere C, Dicaire R, Barreiro L, Lee DA, Verhoeyen E, Haddad E. Efficient and robust NK-Cell transduction With baboon envelope pseudotyped lentivector. Front Immunol. 2019;10:2873.
Boissel L, Betancur M, Lu W, Wels WS, Marino T, Van Etten RA, Klingemann H. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk Lymphoma. 2012;53(5):958–65.
Kararoudi MN, Likhite S, Elmas E, Yamamoto K, Schwartz M, Sorathia K, Pereira MD, Sezgin Y, Devine RD, Lyberger JM, Behbehani GK. Optimization and validation of CAR transduction into human primary NK cells using CRISPR and AAV. Cell Rep Methods. 2022;2(6):100236.
Sherba JJ, Hogquist S, Lin H, Shan JW, Shreiber DI, Zahn JD. The effects of electroporation buffer composition on cell viability and electro-transfection efficiency. Sci Rep. 2020;10(1):3053.
Jakstys B, Jakutaviciute M, Uzdavinyte D, Satkauskiene I, Satkauskas S. Correlation between the loss of intracellular molecules and cell viability after cell electroporation. Bioelectrochemistry. 2020;135: 107550.
Boissel L, Betancur M, Wels WS, Tuncer H, Klingemann H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk Res. 2009;33(9):1255–9.
Bugeon S, de Chevigny A, Boutin C, Core N, Wild S, Bosio A, Cremer H, Beclin C. Direct and efficient transfection of mouse neural stem cells and mature neurons by in vivo mRNA electroporation. Development. 2017;144(21):3968–77.
Diener Y, Jurk M, Kandil B, Choi YH, Wild S, Bissels U, Bosio A. RNA-based, transient modulation of gene expression in human haematopoietic stem and progenitor cells. Sci Rep. 2015;5:17184.
Lehner M, Gotz G, Proff J, Schaft N, Dorrie J, Full F, Ensser A, Muller YA, Cerwenka A, Abken H, Parolini O, Ambros PF, Kovar H, Holter W. Redirecting T cells to Ewing’s sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLoS ONE. 2012;7(2): e31210.
Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, Jin Q, Su L, Liu X, Wang K, Yan G, Dong T, Wu S, Zhou P, Zhang J, Liang W, Ren J, Teng Y, Chen C, Xu XH. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol Ther. 2019;27(6):1114–25.
Yudovich D, Backstrom A, Schmiderer L, Zemaitis K, Subramaniam A, Larsson J. Combined lentiviral- and RNA-mediated CRISPR/Cas9 delivery for efficient and traceable gene editing in human hematopoietic stem and progenitor cells. Sci Rep. 2020;10(1):22393.
Nowakowski A, Andrzejewska A, Janowski M, Walczak P, Lukomska B. Genetic engineering of stem cells for enhanced therapy. Acta Neurobiol Exp. 2013;73(1):1–18.
Gonzalez-Estevez C, Momose T, Gehring WJ, Salo E. Transgenic planarian lines obtained by electroporation using transposon-derived vectors and an eye-specific GFP marker. Proc Natl Acad Sci U S A. 2003;100(24):14046–51.
Subrakova VG, Kulemzin SV, Belovezhets TN, Chikaev AN, Chikaev NA, Koval OA, Gorchakov AA, Taranin AV. shp-2 gene knockout upregulates CAR-driven cytotoxicity of YT NK cells. Vavilovskii Zhurnal Genet Selektsii. 2020;24(1):80–6.
Gurney M, Stikvoort A, Nolan E, Kirkham-McCarthy L, Khoruzhenko S, Shivakumar R, Zweegman S, Van de Donk N, Mutis T, Szegezdi E, Sarkar S, O’Dwyer M. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica. 2020. https://doi.org/10.3324/haematol.2020.271908.
Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: a promising cellular immunotherapy for cancer. EBioMedicine. 2020;59: 102975.
Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, Biassoni R, Moretta L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol. 2001;19:197–223.
Dizaji Asl K, Velaei K, Rafat A, Tayefi Nasrabadi H, Movassaghpour AA, Mahdavi M, Nozad CH. The role of KIR positive NK cells in diseases and its importance in clinical intervention. Int Immunopharmacol. 2021;92: 107361.
Liu S, Galat V, Galat Y, Lee YKA, Wainwright D, Wu J. NK cell-based cancer immunotherapy: from basic biology to clinical development. J Hematol Oncol. 2021;14(1):7.
Sordo-Bahamonde C, Vitale M, Lorenzo-Herrero S, Lopez-Soto A, Gonzalez S. Mechanisms of resistance to NK cell immunotherapy. Cancers. 2020. https://doi.org/10.3390/cancers12040893.
Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8(+) T cells. Nat Rev Immunol. 2011;11(10):645–57.
Sanchez-Martinez D, Allende-Vega N, Orecchioni S, Talarico G, Cornillon A, Vo DN, Rene C, Lu ZY, Krzywinska E, Anel A, Galvez EM, Pardo J, Robert B, Martineau P, Hicheri Y, Bertolini F, Cartron G, Villalba M. Expansion of allogeneic NK cells with efficient antibody-dependent cell cytotoxicity against multiple tumors. Theranostics. 2018;8(14):3856–69.
Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, Victor A, Meisen WH, Kim SH, Grandi P, Wang QE, He X, Nakano I, Chiocca EA, Glorioso Iii JC, Kaur B, Caligiuri MA, Yu J. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep. 2015;5:11483.
Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106(1):376–83.
Shimasaki N, Fujisaki H, Cho D, Masselli M, Lockey T, Eldridge P, Leung W, Campana D. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy. 2012;14(7):830–40.
Oelsner S, Wagner J, Friede ME, Pfirrmann V, Genssler S, Rettinger E, Buchholz CJ, Pfeifer H, Schubert R, Ottmann OG, Ullrich E, Bader P, Wels WS. Chimeric antigen receptor-engineered cytokine-induced killer cells overcome treatment resistance of pre-B-cell acute lymphoblastic leukemia and enhance survival. Int J Cancer. 2016;139(8):1799–809.
Suerth JD, Morgan MA, Kloess S, Heckl D, Neudorfl C, Falk CS, Koehl U, Schambach A. Efficient generation of gene-modified human natural killer cells via alpharetroviral vectors. J Mol Med. 2016;94(1):83–93.
Oelsner S, Friede ME, Zhang C, Wagner J, Badura S, Bader P, Ullrich E, Ottmann OG, Klingemann H, Tonn T, Wels WS. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy. 2017;19(2):235–49.
Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, Orange J, Wan X, Lu X, Reynolds A, Gagea M, Banerjee P, Cai R, Bdaiwi MH, Basar R, Muftuoglu M, Li L, Marin D, Wierda W, Keating M, Champlin R, Shpall E, Rezvani K. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520–31.
Liu Q, Xu Y, Mou J, Tang K, Fu X, Li Y, Xing Y, Rao Q, Xing H, Tian Z, Wang M, Wang J. Irradiated chimeric antigen receptor engineered NK-92MI cells show effective cytotoxicity against CD19(+) malignancy in a mouse model. Cytotherapy. 2020;22(10):552–62.
Jamali A, Hadjati J, Madjd Z, Mirzaei HR, Thalheimer FB, Agarwal S, Bonig H, Ullrich E, Hartmann J. Highly efficient generation of transgenically augmented CAR NK cells overexpressing CXCR4. Front Immunol. 2020;11:2028.
Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, Mendt M, Kerbauy LN, Banerjee PP, Shanley M, Imahashi N, Li L, Lim F, Fathi M, Rezvan A, Mohanty V, Shen Y, Shaim H, Lu J, Ozcan G, Ensley E, Kaplan M, Nandivada V, Bdiwi M, Acharya S, Xi Y, Wan X, Mak D, Liu E, Jiang XR, Ang S, Muniz-Feliciano L, Li Y, Wang J, Kordasti S, Petrov N, Varadarajan N, Marin D, Brunetti L, Skinner RJ, Lyu S, Silva L, Turk R, Schubert MS, Rettig GR, McNeill MS, Kurgan G, Behlke MA, Li H, Fowlkes NW, Chen K, Konopleva M, Champlin RE, Shpall EJ, Rezvani K. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood. 2021;137(5):624–36.
Roex G, Campillo-Davo D, Flumens D, Shaw PAG, Krekelbergh L, De Reu H, Berneman ZN, Lion E, Anguille S. Two for one: targeting BCMA and CD19 in B-cell malignancies with off-the-shelf dual-CAR NK-92 cells. J Transl Med. 2022;20(1):124.
Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, Czuczman M, Cairo MS. Targeting CD20+ aggressive B-cell non-hodgkin lymphoma by Anti-CD20 CAR mRNA-modified expanded natural killer cells in vitro and in NSG mice. Cancer Immunol Res. 2015;3(4):333–44.
Chu Y, Yahr A, Huang B, Ayello J, Barth M, Cairo MS. Romidepsin alone or in combination with anti-CD20 chimeric antigen receptor expanded natural killer cells targeting Burkitt lymphoma in vitro and in immunodeficient mice. Oncoimmunology. 2017;6(9):e1341031.
Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, Zeng T, Huang H, Zhang X, Sun W, Man-Yuen Sze D, Yi Q, Hou J. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol. 2014;8(2):297–310.
Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, Peng Y, Mao H, Yi L, Ghoshal K, He X, Devine SM, Zhang X, Caligiuri MA, Hofmeister CC, Yu J. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28(4):917–27.
Martín EM, Encinas J, García-Ortiz A, Ugalde L, Fernández RA, Leivas A, Paciello ML, Garrido V, Martín-Antonio B, Suñe G, Cedena M-T, Powell DJ, Río P, Martinez-Lopez J, Valeri A. Exploring NKG2D and BCMA-CAR NK-92 for adoptive cellular therapy to multiple myeloma. Clin Lymphoma Myeloma Leuk. 2019;19(10):e24–5.
Ng YY, Du Z, Zhang X, Chng WJ, Wang S. CXCR4 and anti-BCMA CAR co-modified natural killer cells suppress multiple myeloma progression in a xenograft mouse model. Cancer Gene Ther. 2022;29(5):475–83.
Gurney M, Stikvoort A, Nolan E, Kirkham-McCarthy L, Khoruzhenko S, Shivakumar R, Zweegman S, Van de Donk N, Mutis T, Szegezdi E, Sarkar S, O’Dwyer M. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica. 2022;107(2):437–45.
Albinger N, Pfeifer R, Nitsche M, Mertlitz S, Campe J, Stein K, Kreyenberg H, Schubert R, Quadflieg M, Schneider D, Kuhn MWM, Penack O, Zhang C, Moker N, Ullrich E. Primary CD33-targeting CAR-NK cells for the treatment of acute myeloid leukemia. Blood Cancer J. 2022;12(4):61.
Chen KH, Wada M, Pinz KG, Liu H, Lin KW, Jares A, Firor AE, Shuai X, Salman H, Golightly M, Lan F, Senzel L, Leung EL, Jiang X, Ma Y. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia. 2017;31(10):2151–60.
Xu YX, Liu Q, Zhong MJ, Wang ZZ, Chen ZQ, Zhang Y, Xing HY, Tian Z, Tang KJ, Liao XL, Rao Q, Wang M, Wang JX. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J Hematol Oncol. 2019. https://doi.org/10.1186/s13045-019-0732-7.
You FT, Wang YY, Jiang LC, Zhu XJ, Chen D, Yuan L, An GL, Meng HM, Yang L. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am J Cancer Res. 2019;9(1):64.
Kloss S, Oberschmidt O, Morgan M, Dahlke J, Arseniev L, Huppert V, Granzin M, Gardlowski T, Matthies N, Soltenborn S, Schambach A, Koehl U. Optimization of human NK Cell manufacturing: fully automated separation, improved ex vivo expansion using IL-21 with Autologous feeder cells, and generation of anti-CD123-CAR-expressing effector cells. Hum Gene Ther. 2017;28(10):897–913.
Morgan MA, Kloos A, Lenz D, Kattre N, Nowak J, Bentele M, Keisker M, Dahlke J, Zimmermann K, Sauer M, Heuser M, Schambach A. Improved activity against acute myeloid leukemia with chimeric antigen receptor (CAR)-NK-92 cells designed to target CD123. Viruses. 2021. https://doi.org/10.3390/v13071365.
Du Z, Ng YY, Zha S, Wang S. piggyBac system to co-express NKG2D CAR and IL-15 to augment the in vivo persistence and anti-AML activity of human peripheral blood NK cells. Mol Ther Methods Clin Dev. 2021;23:582–96.
Kruschinski A, Moosmann A, Poschke I, Norell H, Chmielewski M, Seliger B, Kiessling R, Blankenstein T, Abken H, Charo J. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc Natl Acad Sci USA. 2008;105(45):17481–6.
Liu H, Yang B, Sun T, Lin L, Hu Y, Deng M, Yang J, Liu T, Li J, Sun S, Jiao S. Specific growth inhibition of ErbB2expressing human breast cancer cells by genetically modified NK92 cells. Oncol Rep. 2015;33(1):95–102.
Schonfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, Nowakowska P, Bonig H, Kohl U, Kloess S, Kohler S, Holtgreve-Grez H, Jauch A, Schmidt M, Schubert R, Kuhlcke K, Seifried E, Klingemann HG, Rieger MA, Tonn T, Grez M, Wels WS. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. 2015;23(2):330–8.
Zhang C, Burger MC, Jennewein L, Genssler S, Schonfeld K, Zeiner P, Hattingen E, Harter PN, Mittelbronn M, Tonn T, Steinbach JP, Wels WS. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. 2016. https://doi.org/10.1093/jnci/djv375.
Wu X, Huang S. HER2-specific chimeric antigen receptor-engineered natural killer cells combined with apatinib for the treatment of gastric cancer. Bull Cancer. 2019;106(11):946–58.
Muller N, Michen S, Tietze S, Topfer K, Schulte A, Lamszus K, Schmitz M, Schackert G, Pastan I, Temme A. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1alpha-secreting glioblastoma. J Immunother. 2015;38(5):197–210.
Chen X, Han J, Chu J, Zhang L, Zhang J, Chen C, Chen L, Wang Y, Wang H, Yi L, Elder JB, Wang QE, He X, Kaur B, Chiocca EA, Yu J. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget. 2016;7(19):27764–77.
Ma R, Lu T, Li Z, Teng KY, Mansour AG, Yu M, Tian L, Xu B, Ma S, Zhang J, Barr T, Peng Y, Caligiuri MA, Yu J. An oncolytic virus expressing IL15/IL15Ralpha combined with off-the-shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res. 2021;81(13):3635–48.
Liu Y, Zhou Y, Huang KH, Fang X, Li Y, Wang F, An L, Chen Q, Zhang Y, Shi A, Yu S, Zhang J. Targeting epidermal growth factor-overexpressing triple-negative breast cancer by natural killer cells expressing a specific chimeric antigen receptor. Cell Prolif. 2020;53(8): e12858.
Esser R, Muller T, Stefes D, Kloess S, Seidel D, Gillies SD, Aperlo-Iffland C, Huston JS, Uherek C, Schonfeld K, Tonn T, Huebener N, Lode HN, Koehl U, Wels WS. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med. 2012;16(3):569–81.
Kailayangiri S, Altvater B, Spurny C, Jamitzky S, Schelhaas S, Jacobs AH, Wiek C, Roellecke K, Hanenberg H, Hartmann W, Wiendl H, Pankratz S, Meltzer J, Farwick N, Greune L, Fluegge M, Rossig C. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology. 2017;6(1): e1250050.
Cao B, Liu M, Wang L, Liang B, Feng Y, Chen X, Shi Y, Zhang J, Ye X, Tian Y, Zhi C, Li J, Lian H, Wu Q, Zhang Z. Use of chimeric antigen receptor NK-92 cells to target mesothelin in ovarian cancer. Biochem Biophys Res Commun. 2020;524(1):96–102.
Cao B, Liu M, Huang J, Zhou J, Li J, Lian H, Huang W, Guo Y, Yang S, Lin L, Cai M, Zhi C, Wu J, Liang L, Hu Y, Hu H, He J, Liang B, Zhao Q, Zhu K. Development of mesothelin-specific CAR NK-92 cells for the treatment of gastric cancer. Int J Biol Sci. 2021;17(14):3850–61.
Montagner IM, Penna A, Fracasso G, Carpanese D, Dalla Pieta A, Barbieri V, Zuccolotto G, Rosato A. Anti-PSMA CAR-engineered NK-92 cells: an off-the-shelf cell therapy for prostate cancer. Cells. 2020. https://doi.org/10.3390/cells9061382.
Wang F, Wu L, Yin L, Shi H, Gu Y, Xing N. Combined treatment with anti-PSMA CAR NK-92 cell and anti-PD-L1 monoclonal antibody enhances the antitumour efficacy against castration-resistant prostate cancer. Clin Transl Med. 2022;12(6): e901.
Yu M, Luo H, Fan M, Wu X, Shi B, Di S, Liu Y, Pan Z, Jiang H, Li Z. Development of GPC3-Specific chimeric antigen receptor-engineered natural killer cells for the treatment of hepatocellular carcinoma. Mol Ther. 2018;26(2):366–78.
Ueda T, Kumagai A, Iriguchi S, Yasui Y, Miyasaka T, Nakagoshi K, Nakane K, Saito K, Takahashi M, Sasaki A, Yoshida S, Takasu N, Seno H, Uemura Y, Tamada K, Nakatsura T, Kaneko S. Non-clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti-glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci. 2020;111(5):1478–90.
Huang Y, Zeng J, Liu T, Xu Q, Song X, Zeng J. DNAM1 and 2B4 costimulatory domains enhance the cytotoxicity of anti-GPC3 chimeric antigen receptor-modified natural killer cells against hepatocellular cancer cells in vitro. Cancer Manag Res. 2020;12:3247–55.
Sahm C, Schonfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother. 2012;61(9):1451–61.
Jan CI, Huang SW, Canoll P, Bruce JN, Lin YC, Pan CM, Lu HM, Chiu SC, Cho DY. Targeting human leukocyte antigen G with chimeric antigen receptors of natural killer cells convert immunosuppression to ablate solid tumors. J Immunother Cancer. 2021. https://doi.org/10.1136/jitc-2021-003050.
Zhang G, Liu R, Zhu X, Wang L, Ma J, Han H, Wang X, Zhang G, He W, Wang W, Liu C, Li S, Sun M, Gao B. Retargeting NK-92 for anti-melanoma activity by a TCR-like single-domain antibody. Immunol Cell Biol. 2013;91(10):615–24.
Liu B, Liu ZZ, Zhou ML, Lin JW, Chen XM, Li Z, Gao WB, Yu ZD, Liu T. Development of cMETspecific chimeric antigen receptorengineered natural killer cells with cytotoxic effects on human liver cancer HepG2 cells. Mol Med Rep. 2019;20(3):2823–31.
Yang S, Cao B, Zhou G, Zhu L, Wang L, Zhang L, Kwok HF, Zhang Z, Zhao Q. Targeting B7–H3 immune checkpoint with chimeric antigen receptor-engineered natural killer cells exhibits potent cytotoxicity against non-small cell lung cancer. Front Pharmacol. 2020;11:1089.
Liu M, Huang W, Guo Y, Zhou Y, Zhi C, Chen J, Li J, He J, Lian H, Zhou J, Ye X, Hu Y, Hu H, Liu Z, Huang J, Lin L, Cai M, Wang X, Huang J, Zhang Z, Zhu K, Zhao Q, Cao B. CAR NK-92 cells targeting DLL3 kill effectively small cell lung cancer cells in vitro and in vivo. J Leukoc Biol. 2022. https://doi.org/10.1002/JLB.5MA0122-467R.
Hoerster K, Uhrberg M, Wiek C, Horn PA, Hanenberg H, Heinrichs S. HLA class I knockout converts allogeneic primary NK cells into suitable effectors for “Off-the-Shelf” immunotherapy. Front Immunol. 2020;11: 586168.
Mehta RS, Rezvani K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front Immunol. 2018;9:283.
Glienke W, Esser R, Priesner C, Suerth JD, Schambach A, Wels WS, Grez M, Kloess S, Arseniev L, Koehl U. Advantages and applications of CAR-expressing natural killer cells. Front Pharmacol. 2015;6:21.
Reina-Ortiz C, Constantinides M, Fayd-Herbe-de-Maudave A, Presumey J, Hernandez J, Cartron G, Giraldos D, Diez R, Izquierdo I, Azaceta G, Palomera L, Marzo I, Naval J, Anel A, Villalba M. Expanded NK cells from umbilical cord blood and adult peripheral blood combined with daratumumab are effective against tumor cells from multiple myeloma patients. Oncoimmunology. 2020;10(1):1853314.
Bjorklund AT, Carlsten M, Sohlberg E, Liu LL, Clancy T, Karimi M, Cooley S, Miller JS, Klimkowska M, Schaffer M, Watz E, Wikstrom K, Blomberg P, Wahlin BE, Palma M, Hansson L, Ljungman P, Hellstrom-Lindberg E, Ljunggren HG, Malmberg KJ. Complete remission with reduction of high-risk clones following haploidentical NK-Cell therapy against MDS and AML. Clin Cancer Res. 2018;24(8):1834–44.
Herrera L, Santos S, Vesga MA, Anguita J, Martin-Ruiz I, Carrascosa T, Juan M, Eguizabal C. Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. Sci Rep. 2019;9(1):18729.
Torelli GF, Rozera C, Santodonato L, Peragine N, D’Agostino G, Montefiore E, Napolitano MR, Monque DM, Carlei D, Mariglia P, Pauselli S, Gozzer M, Bafti MS, Girelli G, Guarini A, Belardelli F, Foa R. A good manufacturing practice method to ex vivo expand natural killer cells for clinical use. Blood Transfus. 2015;13(3):464–71.
Oberschmidt O, Morgan M, Huppert V, Kessler J, Gardlowski T, Matthies N, Aleksandrova K, Arseniev L, Schambach A, Koehl U, Kloess S. Development of automated separation, expansion, and quality control protocols for clinical-scale manufacturing of primary human NK cells and alpharetroviral chimeric antigen receptor engineering. Hum Gene Ther Methods. 2019;30(3):102–20.
Fernandez A, Navarro-Zapata A, Escudero A, Matamala N, Ruz-Caracuel B, Mirones I, Pernas A, Cobo M, Casado G, Lanzarot D, Rodriguez-Antolin C, Vela M, Ferreras C, Mestre C, Viejo A, Leivas A, Martinez J, Fernandez L, Perez-Martinez A. Optimizing the procedure to manufacture clinical-grade NK cells for adoptive immunotherapy. Cancers. 2021. https://doi.org/10.3390/cancers13030577.
Shah N, Martin-Antonio B, Yang H, Ku S, Lee DA, Cooper LJ, Decker WK, Li S, Robinson SN, Sekine T, Parmar S, Gribben J, Wang M, Rezvani K, Yvon E, Najjar A, Burks J, Kaur I, Champlin RE, Bollard CM, Shpall EJ. Antigen presenting cell-mediated expansion of human umbilical cord blood yields log-scale expansion of natural killer cells with anti-myeloma activity. PLoS ONE. 2013;8(10): e76781.
Nham T, Poznanski SM, Fan IY, Vahedi F, Shenouda MM, Lee AJ, Chew MV, Hogg RT, Lee DA, Ashkar AA. Ex Vivo-expanded natural killer cells derived from long-term cryopreserved cord blood are cytotoxic against primary breast cancer cells. J Immunother. 2018;41(2):64–72.
Alkindi S, Dennison D. Umbilical cord blood banking and transplantation: a short review. Sultan Qaboos Univ Med J. 2011;11(4):455–61.
Lupo KB, Matosevic S. Natural killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers. 2019. https://doi.org/10.3390/cancers11060769.
Luevano M, Daryouzeh M, Alnabhan R, Querol S, Khakoo S, Madrigal A, Saudemont A. The unique profile of cord blood natural killer cells balances incomplete maturation and effective killing function upon activation. Hum Immunol. 2012;73(3):248–57.
Sarvaria A, Jawdat D, Madrigal JA, Saudemont A. Umbilical cord blood natural killer cells, their characteristics, and potential clinical applications. Front Immunol. 2017;8:329.
Tanaka H, Kai S, Yamaguchi M, Misawa M, Fujimori Y, Yamamoto M, Hara H. Analysis of natural killer (NK) cell activity and adhesion molecules on NK cells from umbilical cord blood. Eur J Haematol. 2003;71(1):29–38.
Bachiller M, Battram AM, Perez-Amill L, Martin-Antonio B. Natural killer cells in immunotherapy: are we nearly there? Cancers. 2020. https://doi.org/10.3390/cancers12113139.
Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, Yin J, You F, Zhu M, Shen W, Chen G, Zhu X, Wu D, Yu J. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8(6):1083–9.
Maki G, Klingemann HG, Martinson JA, Tam YK. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J Hematother Stem Cell Res. 2001;10(3):369–83.
Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, Wels WS. Chimeric antigen receptor-engineered NK-92 cells: an off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol. 2017;8:533.
Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8(4):652–8.
Zhang J, Zheng H, Diao Y. Natural killer cells and current applications of chimeric antigen receptor-modified NK-92 cells in tumor immunotherapy. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20020317.
Papa L, Djedaini M, Kintali M, Schaniel C, Hoffman R. Ex vivo expansion of adult hematopoietic stem and progenitor cells with valproic acid. Methods Mol Biol. 2021;2185:267–80.
Knorr DA, Bock A, Brentjens RJ, Kaufman DS. Engineered human embryonic stem cell-derived lymphocytes to study in vivo trafficking and immunotherapy. Stem Cells Dev. 2013;22(13):1861–9.
Goldenson BH, Zhu H, Wang YM, Heragu N, Bernareggi D, Ruiz-Cisneros A, Bahena A, Ask EH, Hoel HJ, Malmberg KJ, Kaufman DS. Umbilical cord blood and iPSC-derived natural killer cells demonstrate key differences in cytotoxic activity and KIR profiles. Front Immunol. 2020;11: 561553.
Valamehr B, Robinson M, Abujarour R, Rezner B, Vranceanu F, Le T, Medcalf A, Lee TT, Fitch M, Robbins D, Flynn P. Platform for induction and maintenance of transgene-free hiPSCs resembling ground state pluripotent stem cells. Stem Cell Reports. 2014;2(3):366–81.
Saetersmoen ML, Hammer Q, Valamehr B, Kaufman DS, Malmberg KJ. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin Immunopathol. 2019;41(1):59–68.
Hermanson DL, Bendzick L, Pribyl L, McCullar V, Vogel RI, Miller JS, Geller MA, Kaufman DS. Induced pluripotent stem cell-derived natural killer cells for treatment of ovarian cancer. Stem Cells. 2016;34(1):93–101.
Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJ, Lee DA, Kaufman DS. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med. 2013;2(4):274–83.
Gao F, Ye Y, Gao Y, Huang H, Zhao Y. Influence of KIR and NK cell reconstitution in the outcomes of hematopoietic stem cell transplantation. Front Immunol. 2020;11:2022.
Ruggeri L, Mancusi A, Capanni M, Urbani E, Carotti A, Aloisi T, Stern M, Pende D, Perruccio K, Burchielli E, Topini F, Bianchi E, Aversa F, Martelli MF, Velardi A. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood. 2007;110(1):433–40.
Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology. 2014;3: e28147.
Wei J, Liu Y, Wang C, Zhang Y, Tong C, Dai G, Wang W, Rasko JEJ, Melenhorst JJ, Qian W, Liang A, Han W. The model of cytokine release syndrome in CAR T-cell treatment for B-cell non-Hodgkin lymphoma. Signal Transduct Target Ther. 2020;5(1):134.
Wang Z, Han W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark Res. 2018;6:4.
Zhang Y, Wallace DL, de Lara CM, Ghattas H, Asquith B, Worth A, Griffin GE, Taylor GP, Tough DF, Beverley PC, Macallan DC. In vivo kinetics of human natural killer cells: the effects of ageing and acute and chronic viral infection. Immunology. 2007;121(2):258–65.
Davila ML, Kloss CC, Gunset G, Sadelain M. CD19 CAR-targeted T cells induce long-term remission and B cell Aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLoS ONE. 2013;8(4): e61338.
Daher M, Rezvani K. Outlook for new CAR-based therapies with a focus on CAR NK Cells: what lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov. 2021;11(1):45–58.
Oelsner S, Waldmann A, Billmeier A, Roder J, Lindner A, Ullrich E, Marschalek R, Dotti G, Jung G, Grosse-Hovest L, Oberoi P, Bader P, Wels WS. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int J Cancer. 2019;145(7):1935–45.
Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, Brenner MK. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–83.
Hu Z. Tissue factor as a new target for CAR-NK cell immunotherapy of triple-negative breast cancer. Sci Rep. 2020;10(1):2815.
Franks SE, Wolfson B, Hodge JW. Natural born killers: NK cells in cancer therapy. Cancers. 2020. https://doi.org/10.3390/cancers12082131.
Cichocki F, Goodridge J, Bjordahl R, Mahmood S, Davis ZB, Gaidarova S, Abujarour R, Groff B, Witty AD, Wang H, Tuininga K, Kodal B, Felices M, Bonello GB, Huffman J, Dailey T, Lee TT, Walcheck B, Valamehr B, Miller JS. Dual-antigen targeted off-the-shelf NK cells show durable response and prevent antigen escape in lymphoma and leukemia. Blood. 2022. https://doi.org/10.1182/blood.2021015184.
Mitwasi N, Feldmann A, Arndt C, Koristka S, Berndt N, Jureczek J, Loureiro LR, Bergmann R, Mathe D, Hegedus N, Kovacs T, Zhang C, Oberoi P, Jager E, Seliger B, Rossig C, Temme A, Eitler J, Tonn T, Schmitz M, Hassel JC, Jager D, Wels WS, Bachmann M. “UniCAR”-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci Rep. 2020;10(1):2141.
Suck G, Odendahl M, Nowakowska P, Seidl C, Wels WS, Klingemann HG, Tonn T. NK-92: an “off-the-shelf therapeutic” for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol Immunother. 2016;65(4):485–92.
Arias J, Yu J, Varshney M, Inzunza J, Nalvarte I. HSC and iPS cell-derived CAR-NK cells as reliable cell-based therapy solutions. Stem Cells Transl Med. 2021. https://doi.org/10.1002/sctm.20-0459.
Wrona E, Borowiec M, Potemski P. CAR-NK cells in the treatment of solid tumors. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22115899.
Genssler S, Burger MC, Zhang C, Oelsner S, Mildenberger I, Wagner M, Steinbach JP, Wels WS. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology. 2016;5(4): e1119354.
Klapdor R, Wang S, Morgan M, Dork T, Hacker U, Hillemanns P, Buning H, Schambach A. Characterization of a novel third-generation anti-CD24-CAR against ovarian cancer. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20030660.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, Nassif Kerbauy L, Overman B, Thall P, Kaplan M, Nandivada V, Kaur I, Nunez Cortes A, Cao K, Daher M, Hosing C, Cohen EN, Kebriaei P, Mehta R, Neelapu S, Nieto Y, Wang M, Wierda W, Keating M, Champlin R, Shpall EJ, Rezvani K. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–53.
London L, Perussia B, Trinchieri G. Induction of proliferation in vitro of resting human natural killer cells: IL 2 induces into cell cycle most peripheral blood NK cells, but only a minor subset of low density T cells. J Immunol. 1986;137(12):3845–54.
Alici E, Sutlu T, Bjorkstrand B, Gilljam M, Stellan B, Nahi H, Quezada HC, Gahrton G, Ljunggren HG, Dilber MS. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood. 2008;111(6):3155–62.
Satwani P, van de Ven C, Ayello J, Cairo D, Simpson LL, Baxi L, Cairo MS. Interleukin (IL)-15 in combination with IL-2, fms-like tyrosine kinase-3 ligand and anti-CD3 significantly enhances umbilical cord blood natural killer (NK) cell and NK-cell subset expansion and NK function. Cytotherapy. 2011;13(6):730–8.
Tomchuck SL, Leung WH, Dallas MH. Enhanced cytotoxic function of natural killer and CD3+CD56+ cells in cord blood after culture. Biol Blood Marrow Transplant. 2015;21(1):39–49.
Phillips JH, Lanier LL. A model for the differentiation of human natural killer cells Studies on the in vitro activation of Leu-11+ granular lymphocytes with a natural killer-sensitive tumor cell, K562. J Exp Med. 1985;161(6):1464–82.
Yang Y, Badeti S, Tseng HC, Ma MT, Liu T, Jiang JG, Liu C, Liu D. Superior expansion and cytotoxicity of human primary NK and CAR-NK cells from various sources via enriched metabolic pathways. Mol Ther Methods Clin Dev. 2020;18:428–45.
Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J Immunol. 2005;175(8):5095–103.
Shah NN, Baird K, Delbrook CP, Fleisher TA, Kohler ME, Rampertaap S, Lemberg K, Hurley CK, Kleiner DE, Merchant MS, Pittaluga S, Sabatino M, Stroncek DF, Wayne AS, Zhang H, Fry TJ, Mackall CL. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood. 2015;125(5):784–92.
Klingemann H. Challenges of cancer therapy with natural killer cells. Cytotherapy. 2015;17(3):245–9.
Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, Corcuff E, Mortier E, Jacques Y, Spits H, Di Santo JP. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J Exp Med. 2009;206(1):25–34.
Lehmann C, Zeis M, Uharek L. Activation of natural killer cells with interleukin 2 (IL-2) and IL-12 increases perforin binding and subsequent lysis of tumour cells. Br J Haematol. 2001;114(3):660–5.
Martin-Antonio B, Sune G, Perez-Amill L, Castella M, Urbano-Ispizua A. Natural killer cells: angels and devils for immunotherapy. Int J Mol Sci. 2017. https://doi.org/10.3390/ijms18091868.
Schwartz RN, Stover L, Dutcher JP. Managing toxicities of high-dose interleukin-2. Oncology. 2002;16(11 Suppl 13):11–20.
Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna D, Le C, Defor TE, Burns LJ, Orchard PJ, Blazar BR, Wagner JE, Slungaard A, Weisdorf DJ, Okazaki IJ, McGlave PB. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):3051–7.
Mathios D, Park CK, Marcus WD, Alter S, Rhode PR, Jeng EK, Wong HC, Pardoll DM, Lim M. Therapeutic administration of IL-15 superagonist complex ALT-803 leads to long-term survival and durable antitumor immune response in a murine glioblastoma model. Int J Cancer. 2016;138(1):187–94.
Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, Zhang B, Lenvik AJ, Panoskaltsis-Mortari A, Verneris MR, Tolar J, Cooley S, Weisdorf DJ, Blazar BR, Miller JS. IL15 Trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res. 2016;22(14):3440–50.
Schmohl JU, Felices M, Taras E, Miller JS, Vallera DA. Enhanced ADCC and NK cell activation of an anticarcinoma bispecific antibody by genetic insertion of a modified IL-15 cross-linker. Mol Ther. 2016;24(7):1312–22.
Zaiatz-Bittencourt V, Finlay DK, Gardiner CM. Canonical TGF-beta signaling pathway represses human NK cell metabolism. J Immunol. 2018;200(12):3934–41.
Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, Biassoni R, Bottino C, Moretta L, Moretta A. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci USA. 2003;100(7):4120–5.
Heyman B, Yang Y. Chimeric antigen receptor T cell therapy for solid tumors: current status, obstacles and future strategies. Cancers. 2019. https://doi.org/10.3390/cancers11020191.
Park A, Lee Y, Kim MS, Kang YJ, Park YJ, Jung H, Kim TD, Lee HG, Choi I, Yoon SR. Prostaglandin E2 secreted by thyroid cancer cells contributes to immune escape through the suppression of natural killer (NK) cell cytotoxicity and NK cell differentiation. Front Immunol. 2018;9:1859.
Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, Queirolo P, Vermi W, Facchetti F, Moretta A, Moretta L, Mingari MC, Vitale M. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci U S A. 2009;106(49):20847–52.
Velasquez SY, Killian D, Schulte J, Sticht C, Thiel M, Lindner HA. Short term hypoxia synergizes with interleukin 15 priming in driving glycolytic gene transcription and supports human natural killer cell activities. J Biol Chem. 2016;291(25):12960–77.
Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, Varesio L, Moretta L, Bosco MC, Vitale M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol. 2013;43(10):2756–64.
Parodi M, Raggi F, Cangelosi D, Manzini C, Balsamo M, Blengio F, Eva A, Varesio L, Pietra G, Moretta L, Mingari MC, Vitale M, Bosco MC. Hypoxia modifies the transcriptome of human NK cells, modulates their immunoregulatory profile, and influences NK cell subset migration. Front Immunol. 2018;9:2358.
Wang J, Lupo KB, Chambers AM, Matosevic S. Purinergic targeting enhances immunotherapy of CD73(+) solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J Immunother Cancer. 2018;6(1):136.
Benson DM, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, Baiocchi RA, Zhang J, Jianhua Y, Smith MK, Greenfield CN, Porcu P, Devine SM, Rotem-Yehudar R, Lozanski G, Byrd JC, Caligiuri MA. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010;116(13):2286–94.
MacFarlane AW, Jillab M, Plimack ER, Hudes GR, Uzzo RG, Litwin S, Dulaimi E, Al-Saleem T, Campbell KS. PD-1 expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor resection. Cancer Immunol Res. 2014;2(4):320–31.
Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, Moretta L, Moretta A, Marcenaro E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: a phenotypic and functional characterization. J Allergy Clin Immunol. 2017;139(1):335–46.
Guo Y, Wang Y, Han W. Chimeric Antigen Receptor-Modified T Cells for Solid Tumors: Challenges and Prospects. J Immunol Res. 2016;2016:3850839.
Fabian KP, Padget MR, Donahue RN, Solocinski K, Robbins Y, Allen CT, Lee JH, Rabizadeh S, Soon-Shiong P, Schlom J, Hodge JW. PD-L1 targeting high-affinity NK (t-haNK) cells induce direct antitumor effects and target suppressive MDSC populations. J Immunother Cancer. 2020. https://doi.org/10.1136/jitc-2019-000450.
van Ostaijen-ten DMM, Prins HJ, Boerman GH, Vervat C, Pende D, Putter H, Lankester A, van Tol MJ, Zwaginga JJ, Schilham MW. Preparation of cytokine-activated NK Cells for use in adoptive cell therapy in cancer patients: protocol optimization and therapeutic potential. J Immunother. 2016;39(2):90–100.
Domogala A, Madrigal JA, Saudemont A. Cryopreservation has no effect on function of natural killer cells differentiated in vitro from umbilical cord blood CD34(+) cells. Cytotherapy. 2016;18(6):754–9.
Koehl U, Brehm C, Huenecke S, Zimmermann SY, Kloess S, Bremm M, Ullrich E, Soerensen J, Quaiser A, Erben S, Wunram C, Gardlowski T, Auth E, Tonn T, Seidl C, Meyer-Monard S, Stern M, Passweg J, Klingebiel T, Bader P, Schwabe D, Esser R. Clinical grade purification and expansion of NK cell products for an optimized manufacturing protocol. Front Oncol. 2013;3:118.
Maskalenko NA, Zhigarev D, Campbell KS. Harnessing natural killer cells for cancer immunotherapy: dispatching the first responders. Nat Rev Drug Discov. 2022;21(8):559–77.
Jimenez-Sanchez A, Cybulska P, Mager KL, Koplev S, Cast O, Couturier DL, Memon D, Selenica P, Nikolovski I, Mazaheri Y, Bykov Y, Geyer FC, Macintyre G, Gavarro LM, Drews RM, Gill MB, Papanastasiou AD, Sosa RE, Soslow RA, Walther T, Shen R, Chi DS, Park KJ, Hollmann T, Reis-Filho JS, Markowetz F, Beltrao P, Vargas HA, Zamarin D, Brenton JD, Snyder A, Weigelt B, Sala E, Miller ML. Unraveling tumor-immune heterogeneity in advanced ovarian cancer uncovers immunogenic effect of chemotherapy. Nat Genet. 2020;52(6):582–93.
Klapdor R, Wang S, Hacker U, Buning H, Morgan M, Dork T, Hillemanns P, Schambach A. Improved killing of ovarian cancer stem cells by combining a novel chimeric antigen receptor-based immunotherapy and chemotherapy. Hum Gene Ther. 2017;28(10):886–96.
Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS, Garrison SM, Specht JM, Lee SM, Amezquita RA, Voillet V, Muhunthan V, Yechan-Gunja S, Pillai SPS, Rader C, Houghton AM, Pierce RH, Gottardo R, Maloney DG, Riddell SR. Immunogenic chemotherapy enhances recruitment of CAR-T cells to lung tumors and improves antitumor efficacy when combined with checkpoint blockade. Cancer Cell. 2021;39(2):193–208.
Adusumilli PS, Zauderer MG, Riviere I, Solomon SB, Rusch VW, O’Cearbhaill RE, Zhu A, Cheema W, Chintala NK, Halton E, Pineda J, Perez-Johnston R, Tan KS, Daly B, Araujo Filho JA, Ngai D, McGee E, Vincent A, Diamonte C, Sauter JL, Modi S, Sikder D, Senechal B, Wang X, Travis WD, Gonen M, Rudin CM, Brentjens RJ, Jones DR, Sadelain M. A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with malignant pleural disease, in combination with the anti-PD-1 agent pembrolizumab. Cancer Discov. 2021;11(11):2748–63.
Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. 2019;36(5):471–82.
Robbins Y, Greene S, Friedman J, Clavijo PE, Van Waes C, Fabian KP, Padget MR, Abdul Sater H, Lee JH, Soon-Shiong P, Gulley J, Schlom J, Hodge JW, Allen CT. Tumor control via targeting PD-L1 with chimeric antigen receptor modified NK cells. Elife. 2020. https://doi.org/10.7554/eLife.54854.
Liu J, Yang S, Cao B, Zhou G, Zhang F, Wang Y, Wang R, Zhu L, Meng Y, Hu C, Liang H, Lin X, Zhu K, Chen G, Luo KQ, Di L, Zhao Q. Targeting B7–H3 via chimeric antigen receptor T cells and bispecific killer cell engagers augments antitumor response of cytotoxic lymphocytes. J Hematol Oncol. 2021;14(1):21.
Bexte T, Alzubi J, Reindl LM, Wendel P, Schubert R, Salzmann-Manrique E, von Metzler I, Cathomen T, Ullrich E. CRISPR-Cas9 based gene editing of the immune checkpoint NKG2A enhances NK cell mediated cytotoxicity against multiple myeloma. Oncoimmunology. 2022;11(1):2081415.
Acknowledgements
Thanks to figdraw for providing the elements in the figure.
Funding
This work was supported by National Natural Science Foundation of China (81770182) and Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant (20152507).
Author information
Authors and Affiliations
Contributions
All authors contributed to the manuscript. XW and XJY wrote the paper and XY prepared the figure and tables. YYW and WBW reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, X., Yang, X., Yuan, X. et al. Chimeric antigen receptor-engineered NK cells: new weapons of cancer immunotherapy with great potential. Exp Hematol Oncol 11, 85 (2022). https://doi.org/10.1186/s40164-022-00341-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-022-00341-7