
Abstract
Gradual degeneration and loss of dopaminergic neurons in the substantia nigra, pars compacta and subsequent reduction of dopamine levels in striatum are associated with motor deficits that characterize Parkinson’s disease (PD). In addition, half of the PD patients also exhibit frontostriatal-mediated executive dysfunction, including deficits in attention, short-term working memory, speed of mental processing, and impulsivity. The most commonly used treatments for PD are only partially or transiently effective and are available or applicable to a minority of patients. Because, these therapies neither restore the lost or degenerated dopaminergic neurons, nor prevent or delay the disease progression, the need for more effective therapeutics is critical. In this review, we provide a comprehensive overview of the current understanding of the molecular signaling pathways involved in PD, particularly within the context of how genetic and environmental factors contribute to the initiation and progression of this disease. The involvement of molecular chaperones, autophagy-lysosomal pathways, and proteasome systems in PD are also highlighted. In addition, emerging therapies, including pharmacological manipulations, surgical procedures, stem cell transplantation, gene therapy, as well as complementary, supportive and rehabilitation therapies to prevent or delay the progression of this complex disease are reviewed.
Similar content being viewed by others


Background
Parkinson disease (PD) is second to Alzheimer’s disease as the most common age-related complex, idiopathic neurological disorder [1]. It is characterized by tremor, bradykinesia and muscle rigidity along with impaired gait, and posture [2,3,4]. In addition, about half of the PD patients also exhibit frontostriatal-mediated executive dysfunction, including deficits in attention, speed of mental processing, verbal disturbances, impairment of working memory and impulsivity [5]. Dopaminergic neuronal loss in the substantia nigra pars compacta (SNpc), and depletion of dopamine (DA) levels in the striatum represent the hallmark pathology of PD [6]. Experimental evidence indicates that the prefrontal cortex (PFC), anterior cingulate gyrus, and/or frontostriatal pathways are also affected by PD [7].
Although the exact mechanism of dopaminergic neuronal loss in SNpc is not well understood. Mitochondrial damage, energy failure, oxidative stress, excitotoxicity, protein misfolding and their aggregation, impairment of protein clearance pathways, cell-autonomous mechanisms and “prion-like protein infection” may be involved in the onset and progression of PD [3, 8, 9]. Among them, protein misfolding and its subsequent accumulation in intracellular spaces has become a leading hypothesis for PD [10, 11]. The major misfolded amyloid protein inclusion observed in the intracellular spaces of SNpc neurons in PD is the Lewy bodies (LB) [3, 11, 12], which contain several misfolded amyloid proteins, including alpha-synuclein (SNCA), phosphorylated tau (p-tau), and amyloid beta protein (Aβ) [11, 13]. Several environmental toxins are associated with sporadic PD (SPD), which can be partially mimicked in experimental animal models of PD, such as the use of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and paraquat [14, 15]. Unlike SPD, familial cases are rare, and do not follow the prescribed symptoms of PD, which makes it more difficult to understand the pathogenesis of PD [10, 16].
Although several new treatments for PD have been developed [17], none of them effectively halt the progression of PD. The few symptomatic treatments currently available, are appropriate for only a limited number of patients. Moreover, side-effects, short-life span, and permeability issues are the major problems for use of these drugs against PD. Interestingly, recent developments in stem cell transplantation [18,19,20] and gene therapies [21, 22] have drawn special attention as alternative strategies for treating PD. For example, genetically engineered DA-neurons have shown promising results in mouse models of PD [20, 23]. Similarly, using lentiviral or recombinant adeno-associated viral vectors (rAAV), scientists are able to correct some of the dysfunctional metabolic pathways involved in PD [24]. Further, a very recent development of the gene editing technique, clustered regularly-interspaced short palindromic repeats-associated protein 9 (CRISPR-Cas9), may prove useful for treating PD [22]. Given the pressing need for the development of new, rational therapies for PD, the focus of this review is to provide basic conceptual information on the molecular mechanisms underlying PD, which may assist in the design of more effective drugs or other treatment strategies.
Global scenario and risk factors for PD
Although the symptoms and therapies for PD were first mentioned in the “Indian Ayurveda” (5000 BC) and Chinese medical text, “Nei-Jing” (500 BC), it was James Parkinson, a British physician for whom the disease is named accurately described it as “the shaking palsy” in 1817. Epidemiological studies have revealed that PD is world-wide and affects 1–2% of those older than 65 years, and 4–5% of those aged over 85 years [8, 25]. In US, more than one million cases have been reported [26]. PD is more common in men (about 1.5 times) than in women [8], and a higher incidence of PD has been reported in developed countries [14], due to an increase in the aged population [14, 27]. Aging is the most dominant risk factor for PD. As such, the cases of PD are very low in people under 40 and becomes more prevalent in individuals in their 70s and 80s [10]. People with one or more close relatives who have PD have an increased risk of developing the disease themselves, but the total risk is still just 2–5%, unless the family has a known gene mutation for the disease [26]. Other risk factors exist, including exposure to environmental toxins [14, 27]. However, most scientists agree that PD is not, by itself, a fatal disease, but rather, it causes a worsening of normal functioning with time. Interestingly, the average life expectancy of a PD patient is generally the same as for normal people [28].
Symptoms of PD
The progression of symptoms in PD may take 15 to 20 years or more, but may vary person-to-person [8]. The major symptoms observed in PD patients are categorized into: (i) early symptoms; (ii) primary motor symptoms, (iii) secondary motor symptoms, (iv) primary non-motor symptoms, and (v) secondary non-motor symptoms (Fig. 1).

Different symptoms of PD. The PD symptoms are categorized into five major subtypes: early, primary motor, secondary motor, primary and secondary non-motor symptoms
Early symptoms
The early symptoms are subtle and progress slowly, making them difficult to detect. They include mild tremors, posture difficulty, soft speech, slow handwriting, lack of limb movement, abnormal facial expression, loss of focus in thought and speed, fatigue, irritability or depression, without provocation or cause [2, 29]. Sometimes the person may be stiff, unsteady, or unusually slow, and as the disease progress, the shaking or tremor may appear, which start from one side of the body, they eventually spread bilaterally over time [2, 29]. Generally, family members or close friends, or daily caretakers are more likely to detect the emergence of early symptoms in patients.
Primary motor symptoms
-
(i)
Resting Tremor. Shaking of hands, arms, legs, jaw, head, tongue, lips, chin are the primary motor symptoms observed in PD (Fig. 1). About 70% of people with PD experience “resting tremor” in the early stage of the disease, either in the hand or foot on one side of the body, or less commonly, in the jaw or face [26, 30]. As the disease progresses, both arms may become affected [26, 30]. Typically, the tremor takes the form of a rhythmic, back-and-forth motion at a rate of 4–6 Hz.
-
(ii)
Rigidity. The rigidity or increase in stiffness or tonicity of a muscle is the second most common symptoms noted in PD patient [31]. The person with PD often feels stiff or weak, pain and cramping in muscles and joints. Sometimes muscle rigidity can cause an increase in resistance to the extent that the person feels as if someone else is moving his or her joints [31].
-
(iii)
Slow movement (bradykinesia). Bradykinesia in PD causes unplanned movements, decreases in the extent of movement, or the slowing and loss of spontaneous and automatic movements [4]. Common bradykinesia includes a diminution of their handwriting (micrographia), decreased facial expression, decreased rate of eye blinking, and a soft or lowering of volume in their speech [4]. Sometimes it impairs simple tasks, such as routine movements [4]. Other symptoms include incomplete movement, difficulty initiating movements, and sudden stopping of ongoing movement [4, 29].
-
(iv)
Balance and coordination problems. Impairment of coordination, including losing reflex mechanisms, causes instability or imbalance when the PD patient is standing [31]. In severe cases, PD patients are unable to get up off the ground after falling and have difficulties in making turns or abrupt movements [30].
Secondary motor symptoms
Secondary motor symptoms include stooped posture, a tendency to lean forward, dystonia, fatigue, impaired fine and gross motor coordination, decreased arm swing, akathisia, cramping, drooling, difficulty with swallowing and chewing, and sexual dysfunction [8].
-
(i)
Difficulties in swallowing and chewing. PD patients often have difficulties in swallowing due to losing control of muscle movement around the mouth and throat, which makes it difficult to chew solid foods. This prevents peristaltic movement of GI tract, thus constipation may develop in PD patient [32].
-
(ii)
Muscle cramps and dystonia. A variety of pain, aches, muscle spasms or dystonia have been observed in PD [33]. These muscle cramps can be sustained for prolonged periods and can be very painful. Muscular rigidity is the principal reason for this, which may be exacerbated due to the side-effects of certain medications [34].
-
(iii)
Sexual dysfunction. Sexual dysfunction is one of the major reasons for deterioration of quality of life of a PD patient. Hyper-sexuality, erectile dysfunction, and difficulties in ejaculation are found in some PD male patients. Whereas the loss of lubrication and involuntary urination during sex are common in female PD patients [35]. The tremor, bradykinesia, muscular rigidity, dyskinesia, hyper-salivation, and sweating may be the reasons for sexual dysfunction in PD [36]. In contrast, hyper-sexuality reported in male PD patients may be due to side-effects of medications [36].
-
(iv)
Changes of speech and voice. About 90% of the PD patients have difficulties with voice control and are unable to deliver speech appropriately [37]. They may speak too softly or in a monotone, or may have slurred speech and develop a breathy or hoarse quality [32]. PD patients may hesitate before speaking, slur or repeat their words, or may even speak so fast that is difficult to understand them [32]. Communication difficulties are common during walking or doing any other tasks. Sometimes expression of complicated sentences become difficult for them, along with presence of longer pauses in their conversation [32, 37].
Primary non-motor symptoms
Frequently observed non-motor symptoms in PD patients include depression, insomnia, and cognitive dysfunction.
-
(i)
Depression. Depression is a common problem and an early indicator of PD, which can manifest itself before other symptoms appear [38, 39]. PD patients often experience episodes of sadness and depression, which results in an unpleasant attitude, without any apparent reason, which can reduce the quality of life. The level of depression can be sufficiently severe for some PD patients to have suicidal thoughts and ideations [38].
-
(ii)
Dementia and or cognitive dysfunction. About half of the PD patients have cognitive dysfunction, slowness of thought processing [40]. During their conversation, PD patients have difficulties in finding the right words and in understanding complex sentences [40, 41]. Due to this “tip-of-the tongue” problem, PD patients often have many pauses during conversation, and their audience has a difficult time following their line of thought. This dementia may affect memory, social judgment, language, reasoning, or other mental skills [40, 41].
-
(iii)
Problems in sleep (insomnia). Impairment of sleep is very common, and almost 80% of people with PD have difficulty staying asleep at night, or suffer from some form of restless sleep, nightmares, emotional dreams, drowsiness, or sudden sleep onset during the day [42, 43]. Muscle rigidity, tremors or stiffness at night, or frequent urge to urinate, or experiencing vivid dreams or hallucinations, including violent nightmares may underlie the interference in normal sleep for PD patients [29, 42, 43]. The most common sleep disorders include insomnia, REM sleep behavior disorder, sleep apnea, “sleep attacks, and restless legs syndrome” [43, 44].
Secondary non-motor symptoms
-
(i)
Gesture and emotional changes. Many people with PD have issues in reorganizing words and are unable to deliver their message or express their emotions appropriately [45]. Sometimes their facial expressions do not match the context of their speech or their voice intonation [46]. Furthermore, emotional breakdowns make PD patient fearful, insecure, and uncomfortable. Sometimes they are unable to cope with new environments prefer was not to travel, and to avoid socializing with friends. Also, many PD patients have developed personality problems, with their body gestures, their broken or “flattened voice”, and their disrupted emotional control, leading to misinterpretations about their capabilities, and sometime becoming targets for public ridicule [45, 46].
-
(ii)
Urinary problems and constipation. PD patients often complain about dysfunction in urination and defecation [47]. Movement of smooth muscles in urinary bladder and gastrointestinal (GI) tract are often impaired, which can lead to urination problems and constipation. Constipation can occur because of the slow movement of gastrointestinal tract in PD patients [47].
-
(iii)
Sweating and skin problems. Because of improper function of autonomic nervous system, the PD patient has difficulties in controlling body temperature, which sometimes causes excessive sweating [48]. The face of PD patients become very oily, particularly on the forehead and at the sides of the nose. Sometimes the scalp can become excessively oily, as well, resulting in dandruff, and in some cases, the skin become very dry, rough, and wrinkled [48].
-
(iv)
Blood pressure. PD patients also suffer from increased incidences of cardiovascular diseases [49]. For example, when a PD patient stands up from a lying-down position, his or her blood pressure decreases suddenly, causing dizziness, lightheadedness, and, in extreme cases, loss of balance or fainting [49]. The effects of some medications can be another reason for the sudden dropping of blood pressure [49].
-
(v)
Pain. PD patients often complain of pain in muscles and joints, which may be due to muscle rigidity and abnormal postures [50]. Treatment with dopaminergic agonists can cause aggravate the pain in muscles and joints, along with unexplained burning and stabbing sensations [51].
Causes of motor impairment in PD
-
(i)
Role of dopamine. The principal brain area affected by PD is the substantia nigra, pars compacta (SNpc), a vital part of the basal ganglia [52]. This area is predominantly composed of neurons which secrete DA, an essential brain monoamine, which functions primarily as an inhibitory neurotransmitter. In healthy brain, DA regulates the excitability of striatal neurons, which are involved in controlling the balance of body movement. In PD, DA-neurons of SNpc degenerate, and DA levels are diminished [52, 53]. Inadequate DA levels cause less inhibition of the activity of striatal neurons, allowing them to fire excessively. This makes it difficult for PD patients to control their movements, leading to tremor, rigidity, and bradykinesia, the hallmarks of PD-associated motor symptoms [3] (Fig. 2).
-
(ii)
Role of serotonin. Other than DA, serotonin (5-HT) also plays an important role in PD development, especially in several motor and non-motor symptoms, including tremor, cognition, depression, and psychosis, as well as L-DOPA-induced dyskinesia [54]. A reduction of 5-HT levels in the PFC has been observed up to 18 weeks following an acute injection of MPTP in mice [55]. Similarly, a decline in 5-HT transporter (SERT) levels has been reported in the cortex and anterior cingulate following unilateral striatal lesions in the macaque monkey [56]. In addition, a reduction of SERT-immunoreactive axons in the PFC reduced 5-HT-imunoreactivity in median raphe neurons, or reduced PFC SERT binding capacity have also been observed in brains of PD patients [57, 58]. Furthermore, there is ~25% loss of serotonergic receptor (HT1A) at median raphe nucleus in PD patients, and this is correlated with the severity of resting tremor [59], which suggests that 5-HT projections in midbrain is more relevant for initiation of PD tremor than loss of nigrostriatal DA-projections. Recently, we have shown that 5-HT turnover in the PFC may play a pivotal role in executive dysfunction in MPTP-model of PD [60]. Similarly, a strong relation between decline of 5-HT and depression have been found by several investigators in PD [61], however, the importance of 5-HT and it relationship with the progression of PD warrants further attention.
-
(iii)
Role of acetylcholine. Acetylcholine (ACh), which plays significant role in cognition, is downregulated in several neurological diseases, including PD and AD [62]. Within the basal forebrain subventicular region, there is a broad band of cell clusters, commonly known as nucleus basalis of Meynert (nbM), which are predominantly cholinergic in nature. Different patterns of neuronal loss have been observed in the nbM of patients with PD, LBD, AD, or other forms of dementia, which strongly supports the idea of an involvement of the cholinergic system in PD [62, 63]. Importantly, the presence of LB and neuronal loss were found the nbM of postmortem brain tissue of PD patients with cognitive decline, which suggests that the cholinergic system is also involved in the cognitive dysfunction observed in PD [61].
-
(iv)
Role of GABA/Ca2 + system. The gamma amino butyric acid (GABA) is an inhibitory neurotransmitter, which controls the calcium (Ca++) influx directly via GABAergic receptors and, indirectly, via astrocytes network [64]. The Ca++/GABA mechanism stabilizes neuronal activity both at the cellular and systemic levels. In case of PD, due to mitochondrial damage, Ca++-buffering system become impair, which causes Ca++-excitotoxicity leading to neuronal loss in the SNpc [65], whereas the Ca++-buffering is controlled by GABA activity [66]. It has been observed that ~80% of newly diagnosed PD patients have abnormal olfaction, which is due to damage of the DA-neurons in the olfactory bulbs [67]. The function of the DA-neurons both, in the midbrain and in the olfactory system are controlled by glial cell-derived neurotrophic factor (GDNF), which is also regulated by the Ca++/GABA system. Moreover, GDNF function as a chemo-attractant for GABAergic cells and a strong chemo-attractant for axons of DA. The neuroprotective effects of GDNF was observed in PD animal models when administered in GABAergic neurons in the striatum, but not in the SNpc [68], suggesting collapsing of GABA/Ca++ system are involved in DA-neuronal death in PD [69].

Neuronal circuits and neurotransmission mechanisms of control in the brains of normal individuals and those with Parkinson’s disease. a: Neuronal circuit in basal ganglia in normal brain. b: Degeneration of substantia nigra pars compacta (SNpc) impairs cortico-striatal circuit in PD brain. Decrease in DA levels in the SNpc and striatum causes loss of control of striatal neuronal firing, leading to withdrawal of inhibitory effects on globus pallidus as well as thalamus, therefore, the thalamus becomes over-excitable, which activates the motor cortex excessively. This ultimately leads to impairment of motor coordination and causes Parkinsonism
Molecular mechanisms of PD
PD is a multifactorial disease (Fig. 3), where both genetic and non-genetic, such as environmental factors, are involved [16, 25, 27]. The most salient mechanisms involved in the development of PD include the accumulation of misfolded proteins aggregates, failure of protein clearance pathways, mitochondrial damage, oxidative stress, excitotoxicity, neuroinflammation, and genetic mutations [6, 13, 70].

Schematic diagram showing the involvement of different factors and signaling pathways for degeneration of DA-neurons in PD
The role of aggregation of misfolded proteins in PD
-
(i)
Aggregation of alpha-synuclein (SNCA). One of the hallmark pathologies of PD is the intracellular accumulation of LB in DA neurons of the SNpc [70], which contain misfolded aggregates of SNCA and other associated proteins [13]. Interestingly, several molecular, genetic and biochemical studies evidenced that a mixture of multiple misfolded protein aggregates, such as p-tau, Aβ, and SNCA, are frequently seen in human post-mortem brains of patients who were neuropathologically diagnosed as having mixed dementia with Lewy bodies (DLB) and PD with dementia (PDD) [67]. Gomperts and colleagues examined the brains of several PD patients and found a mixture of amyloid deposition in their brain, which was linked to cognitive declines without dementia, suggesting amyloid contributes to cognitive, but not motor decline over time [68]. Similarly, Hepp and colleagues found that the load and extent of Aβ pathology contribute to cognitive impairments in PDD and LBD [69].
The oligomers, proto-fibrils, and fibrils of SNCA or other misfolded amyloid proteins can make a pore in the membrane, causing neuronal death via oxidative stress, energy failure, excitotoxicity, and neuroinflammation [13, 71] (Fig. 4 ). For example, a single intracerebroventricular (i.c.v.) infusion of SNCA oligomers (α-SYOs) in mice causes the development of late motor and non-motor symptoms, such as deficits in the pole and rotarod tests, along with reduced TH and DA content in the caudate putamen [72]. Similarly, mutations of SNCA gene (e.g. A53T, A30P, E46K and H50Q) cause familial PD with its early onset, rapid progression, and a high association of dementia [73]. Overexpression of SNCA in animal and cell culture models showed an accumulation of SNCA aggregates in mitochondria, marked deficits in mitochondrial motility, and decreased mitochondrial membrane potential [74]. The SNCA knockout mice showed mitochondrial lipid abnormalities and impairment of electron transport chain [75], and the mice became less sensitive to mitochondrial toxins [76]. Furthermore, A53T, a transgenic mouse model of PD develops neuronal mitochondria degeneration with accumulation of SNCA-containing mitochondria and marked reduction of complex IV activity [77]. Mitochondrial DNA damage, respiratory chain dysfunction, oxidative stress, along with SNCA inclusion have also been reported to observe in human DA-neurons in the PD brain [78].
-
(ii)
Tau. Hyper-phosphorylation of tau (p-tau) can cause an accumulation of paired helical filaments of tau, known as neurofibrillary tangles (NFT), a hallmark pathology of different neurodegenerative diseases, including AD, frontotemporal dementia with parkinsonism (FTDP), and progressive supra-nuclear palsy (PSP) [79]. The FTDP is linked to chromosome 17 (FTDP-17), with p-tau accumulation occurring in cortex and SNpc areas [79]. The p-tau can also be co-localized with LB, which is often associated with the development of sporadic PD [80]. Similarly, in the case of FTDP, a mutation of gene coding for microtubule associated protein (MAPT) causes an increase in the accumulation of p-tau [80]. The p-tau also has been linked to the LRRK2 gene mutations [16]. Although NFTs are associated most closely with AD, they can co-localize with SNCA in LB and play an important role in destabilization of DA-neuronal architecture, which ultimately leads to rapid degeneration and death of DA neurons [79, 81, 82].

Schematic diagram showing the steps that cause an accumulation of SNCA. Natural SNCA becomes misfolded under stress and is deposited as oligomers, small aggregates, or fibrils, which play a significant role in DA-neuronal loss in PD
Role of gene mutations in PD
A plethora of recent studies, including the discovery of gene mutations in familial or inherited forms of PD, demonstrate that 5–10% of late-onset forms of PD are linked to genetic factors [16, 24] (Table 1). The most common PD-related genes are SNCA, parkin, DJ-1, PINK1 [16]. Experimental and clinical evidence suggest that there are five different chromosomes (5, 6, 8, 9, and 17), which are linked to an increase in susceptibility to develop PD. For example, the parkin gene is located on chromosome 6, which contains genes that are associated with early-onset of PD [16]. Further, some of the PD patients who do not respond to L-DOPA treatment have specific genes located on chromosome 9 [26]. Similarly, the late-onset PD is related to chromosome 17 (FTDP-17), adjacent to the gene for tau [83]. In addition, gene encoding ubiquitin carboxyl-terminal hydroxylase (UCH-L1), and genes on chromosomes X, 1, 2, and 4, also have influential roles in the etiology of PD in some families [16].
-
(i)
Parkin. Parkin is an important protein associated with protein clearance pathways, such as ubiquitin-proteasome system, which can help degrade misfolded proteins in the cell (Table 1). Parkin acts as an E3 ubiquitin ligase, which can bind covalently with ubiquitin on various misfolded protein substrates to aid in their degradation [84]. Parkin also has been co-localized with SNCA, and can form LB inclusions [85]. In contrast, parkin mutations can cause aggregation of misfolded amyloid proteins within SNpc [85]. Parkin-deficient mice and parkin mutations in idiopathic PD patients show loss of neurons in the locus coeruleus of the midbrain, [85]. Furthermore, in the case of autosomal recessive juvenile Parkinsonism, parkin mutations can cause a significant decline of ubiquitin-ligase enzymatic activity in the SNpc [86, 87], which can decrease the proteasomal degradation process significantly. In addition, parkin is also involved in the regulation of the release of DA from SNpc [6].
-
(ii)
DJ-1 (PARK7). DJ-1, a dimer consists of 189 amino acids, is localized in the cytoplasm, nucleus, and mitochondria, and has been linked to early-onset of PD [88] (Table 1). It is neuroprotective, including regulation of activity of certain cell survival-related genes (PI3 K/Akt pathway), transcriptional regulation, anti-oxidant, chaperone and protease activity, as evidenced by several in vitro studies [89]. DJ-1-deficient mice show locomotor deficits, decreased activity in the D2 type of DA receptors, and enhances sensitivity to MPTP [68]. Similarly, DJ-1 deletions and point mutations cause development of autosomal recessive PD [15]. In addition, DJ-1 has been co-localized with SNCA, p-tau, indicating DJ-1, which may play a key role in synucleinopathies and tauopathies [69]. Furthermore, DJ-1 can bind to several chaperones, including HSP70, carboxy-terminus of HSP70-interacting protein (CHIP), and mitochondrial HSP70/mortalin/Grp75, and can help in the degradation of misfolded SNCA [70]. DJ-1 also modulates the expression of the human TH gene by sequestering the transcriptional repressor, poly-pyrimidine tract-binding protein-associated splicing factor (PSF) from the human TH gene promoter, in order to maintain TH levels in DA neurons of SNpc [88].
-
(iii)
PINK1 (PARK6). PTEN-induced putative kinase-1 (PINK1), is a 63 kDa serine/threonine-protein kinase, which is localized in the mitochondria and protect neurons from stress-induced mitochondrial damage [90]. In vitro studies suggest that PINK1 can act as a cell-survival factor. The PINK1 gene mutation has been observed in several families with PD, in which it causes an increase in cell vulnerability [90, 91]; (Table 1). Mutations of PINK1 gene are also linked to mitochondrial dysfunctions and degeneration of SNpc neurons, which ultimately leads to the development of PD [90].
-
(iv)
LRRK2/PARK8 (dardarin). Leucine-rich repeat kinase 2 (LRRK2), is a 268 kDa multi-domain protein, which is encoded by the PARK8 gene. Several point mutations on the PARK8 gene have been linked with late-onset of PD [16] (Table 1). Post-mortem tissue from PD patients show several point mutations in PARK8, with significant DA neurodegeneration, with or without the presence of LB aggregation. In addition, the p-tau pathology observed in post-mortem brains of PD patients may be linked to mutations of the LRRK2 gene [16].
-
(v)
PARK3, PARK9, PARK10, and PARK11. Familial PD is also related to mutation of the PARK 3-, 9-, 10- and 11- genes (Table 1). For example, the onset of late-stage SPD is linked with a mutation of the PARK3 gene. A few cases have linked PARK9 mutation with PD in one Jordanian family, whereas, PARK10 has been linked to PD in Icelandic families [16] (Table 1).
-
(vi)
Glucocerebrosidase (GBA) gene mutation. GBA is considered one of the most common genetic risk factors associated with Parkinsonism. Recently, Velayati and colleagues reported that the mutations of GBA gene are associated with not only the development of PD, but also for LBD [92]. The GBA mutations are associated with alterations in lipid levels, leading to lysosomal storage disease, which can induce synucleinopathies, and also autophagy-lysosomal dysfunction [92]. Similarly, we also found a mutation of GM1 synthase or the upregulation of ganglioside-3 synthase (GD3S) are associated with decreases in the neuroprotective ganglioside (GM1) and increases in toxic gangliosides (GD3 and GT3 series), which can induce neurodegeneration in SNpc in MPTP-lesion mice [93].
-
(vii)
Mutation of mitochondrial DNA (mtDNA). The mitochondria is a target organelle in PD, and an increase in age-related mtDNA mutations has been observed in PD brain tissue [94]. A group of researchers have developed mitochondrial gene-replacement therapy to replace the mutated human mitochondrial genes as a potential treatment for PD and other sporadic neurodegenerative diseases [95]. This form of therapy could slow down the progression of the type of PD that is closely related to mitochondrial dysfunction.
-
(viii)
Other gene mutations: Mutations of autophagy-related genes, which encode vacuolar protein sorting protein-associated protein 35 (VPS35), are also associated with a rare form of autosomal dominant PD [96]. Similarly, several other genes, including TMEM, COMT, IF4G1E, GRIN2A, GSTP1, TNF-α, COX-2, SLC6A3, ADH1C, rs356219, SREBF1 and SREBF2, HLA-DRB5, BST1, GAK, ACMSD, STK39, MCCC1, SYT1, CCDC62/HIP1R are also involved in development of PD.
PD caused by impairment of protein degradation pathways
-
(i)
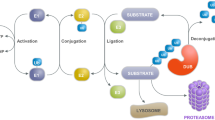
Ubiquitin-Proteasome System (UPS). UPS is the most efficient disposal system of cell, and is mainly responsible for degradation of short polypeptides into small intracellular and plasma-membrane proteins in normal cells [97]. It is also responsible for degradation of misfolded or damaged proteins in the cytosol, nucleus, or endoplasmic reticulum [98]. Impairment or failure of this critical cellular system has been observed in the pathogenesis of PD, leading to aggregation of misfolded amyloid proteins, such as LB, and an increase in neurodegeneration in the SNpc [99, 100]. In the case of PD, several other proteins, such as parkin and UCH-L1, along with UPS, are involved in the degradation of misfolded SNCA. Experimental evidence suggests that UCH-L1 is involved in the production of ubiquitin, which has been co-localized with LB [101] (Fig. 5). Role of UPS in LB degradation in PD can be best studied by inhibiting the UPS system.
For example, the inhibition of proteasome system by using lactacystin resulted in a deposition of LB and degeneration of DA neurons in the fetal rat ventral mesencephalic cells [102, 103]. A retrograde patients of DA neurodegeneration has also been observed in rodent brains, following intrastriatal administration of lactacystin [104]. Similarly, inactivation of ubiquitin hydrolases with ubiquitin aldehyde produce toxic effects in primary neuronal cultures [97, 102]. Furthermore, low levels of proteasome inhibition (100 nM MG115) in human neuroblastoma cells (SH-SY5Y) for several weeks showed mitochondrial degeneration, elevated levels of protein oxidation and aggregates [105, 106], resembling sporadic PD, which strongly supports the role of UPS dysfunction in PD pathogenesis. Furthermore, subcutaneous injections of either the naturally occurring proteasome inhibitor epoxomicin (1.5 mg/kg) or the synthetic proteasome inhibitor PSI (peptidyl aldehyde, selective inhibitor of the chymotrypsin-like activity of the proteasome, 3 or 6 mg/kg) over a period of 2 weeks in adult Sprague-Dawley rats induces progressive motor dysfunction, along with loss of DA nerve terminals in the striatum and a progressive reduction of the DA transporter ligand [107]. To prove the link between the UPS and PD, researchers also developed genetic models of PD, such as parkin-mutated mice, although this mouse model lacks overt signs of parkinsonism [108]. Similarly, inactivation of UCHL-1 in mice did not produce DA neurodegeneration, but did result in axonal dystrophy syndrome or motor ataxia [109]. Interestingly, parkin mutations in Drosophila exhibit selective DA-neuronal death, as well as locomotion deficits, mimicking those of PD patient [110, 111].
-
(ii)
Molecular chaperones (Heat shock proteins, HSP). The molecular chaperone, is one of the most efficient, highly conserved cellular defense mechanisms involved in protein folding, refolding of partially misfolded proteins, and protein degradation [112, 113]. Major HSPs involved in PD are HSP 26, 40, 60, 70, 90 and 100. Some of the HSPs are localized in synapses and axons, and their levels are down-regulated in PD [114] as well as other neurodegenerative diseases [113]. Importantly, HSPs can bind to aggregated SNCA or tau oligomers or pre-fibrillar structures, and interfere by forming low MW soluble oligomers or higher order insoluble structures [115, 116] which reduce their toxicity (Table 2). HSPs also play pivotal roles in the regulation and precise functioning of ubiquitin proteasome and the autophagy-lysosomal pathways [113, 116, 117].
In drosophila and yeast models of PD, HSP70 co-expression prevents DA cell death by decreasing the SNCA toxicity [118], whereas mutations of ATPase domain in HSP70 (K71S) increase toxicity [119]. Similarly, over-expression of HSP70 decreases MPTP- or rotenone-induced neurotoxicity in rat brain slices [120] and also in cultured SK-N-SH or PC12 cells [121]. Furthermore, a reduction of total and detergent-insoluble fractions of misfolded SNCA aggregates were observed in an in vitro model of PD, which co-express different yeast HSPs (HSP104, HSP40, HSP27, or HSP70) [122], suggesting molecular chaperones become dysregulated in PD.
-
(iii)
Autophagy lysosomal pathway (ALP). Because they are too large to pass through the narrow proteasome barrel, large protein debris, such oligomers and fibrils of SNCA, cannot be degraded through UPS [123, 124]. Autophagy, which includes macro-, micro-, and chaperone-mediated autophagy (CMA), are the specialized mechanisms serve as alternative protein clearance machineries present in every cell for degrading LBs in PD [123, 125, 126] (Fig. 6 ). Micro-autophagy is mainly involved in degradation of small cytosolic proteins, even under resting conditions, and macro-autophagy is responsible for degradation of large aggregates. CMA is more specific, performing its activity by interacting with heat-shock cognate protein (HSC70), which specifically bind to small soluble proteins to be degraded via specific pentapeptide targeting motif (KFERQ). The HSC70 docks the t0-be-degraded proteins to the lysosomal membrane receptor, lysosome-associated membrane protein 2 (LAMP2A), and then transport them into the lysosomes, where they are degraded by lysozymes [127]. Experimental evidence suggests that the down-regulation of autophagy-related genes, Atg5 or Atg7, in the CNS leads to aggregation of poly-ubiquitinated protein debris in neurodegenerated tissue in mice [128, 129].
It has been shown that the SNCA is selectively translocated into the lysosomes for degradation by the CMA [123]. Therefore, dysfunction of CMA decreases the efficiency of SNCA degradation, causing excess accumulation of this protein, which impairs neuronal activity significantly. Further, PD brain is particularly vulnerable to dysfunction of autophagy-lysosomal pathway (ALP), which may be due to the failure of autophagosome formation or its inability to bind with lysosomes, due to deficiency of lysozymes, or dysfunction of HSC70 or LAMP2A [6, 130, 131]. Substantial evidence from human post-mortem studies reveals that autophagy mechanisms become impaired in PD brain. For example, accumulation of autophagy vacuoles [132] and levels of the ALP markers, microtubule-associated protein 1 light chain 3 (LC3) [133] have been reported to increase in the SNpc area of postmortem PD brain and temporal cortex of patients with DLB [134] in comparison to age-matched controls, suggesting dysfunction of autophagy is linked with PD progression. Furthermore, decreased levels of LAMP1, LAMP2A, and HSC70 have been observed in the SN of PD patients, suggesting CMA dysregulation [135, 136]. Recent transcriptome studies with postmortem tissue have revealed that several autophagy-related downstream mechanisms, such as mTOR and PI3K/AKT signaling, were also severely affected in PD brain [137, 138].

Role of protein clearance pathways in PD. Different protein clearance pathways, including molecular chaperones (HSPs), ALP (including macro-autophagy, micro-autophagy and chaperone-mediated autophagy), and the ubiquitin-proteasomal system in degradation of misfolded proteins, such as SNCA and LB have been associated with PD

Role of autophagy-lysosomal pathway in degradation of misfolded protein aggregates in PD. Insoluble, larger and smaller SNCA/LB aggregates are degraded by macro-autophagy and micro-autophagy, respectively, whereas soluble, small misfolded SNCA and or LB are degraded by CMA
Role of environmental toxins on PD
Recent yield-boosting advances in the agricultural and fertilizer industries have led farmers to use various types of the pesticides, sometimes indiscriminately, for their crop production. Exposure of those environmental toxins (herbicides, pesticides, fungicides, insecticides etc.) has contributed to the development of SPD [14, 27] (Table 3). Importantly, farmers and people living in rural areas are vulnerable to PD, due to exposure of those toxins, either through direct contact or through drinking water. Many people are also exposed to bacterial toxins, viruses or illegal street drugs, such as the synthetic heroin (MPTP or 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine), leading to SPD [5, 128].
Exposure of MPTP into the cell produces MPP+, the actual toxic metabolite, which can pass through DAT and thus, attack DA-neurons in SNpc and induce parkinsonism [139]; (Fig. 7). Because of this capability, currently MPTP is widely used to produce a severe, permanent parkinsonian syndromes in animal models of PD [140]. There are structural similarities between MPTP and different environmental toxins, such as paraquat, maneb, zineb, nabam, thiram, ziram, and rotenone (Table 3), and many of these environmental toxins may produce Parkinsonism in animals [14, 141].

Mechanistic details of MPTP-induced DA-neuronal loss in PD. After crossing blood brain barrier, MPTP enters glial cells, where it is converted to MPP+. This MPP+ then enters neurons and damage mitochondria, which causes energy failure, oxidative stress, glutamate and Ca++ excitotoxicty, increased aggregation of misfolded SNCA, and DA-neuronal loss
For example, in one of our experiments, mice that were injected with MPTP (25 mg/kg BW) for 5 days, showed an 80% loss of TH-immunoreactivity in SNpc 5 weeks later, indicating a substantial loss of DA-neurons in that area. We also observed TH-positive DA-fibers were more sparse in the striatum (Fig. 8), which suggests that, due to loss of DA neurons in SNPc, the DA fibers become diminished in the striatum [3]. Similarly, injecting 6-hydroxydopamine (6-OHDA), into the striatum can also produce PD-like symptoms in rodents [142]. Similarly, rotenone is another well-known component of pesticides that causes degeneration of DA neurons in SNpc, due to energy failure much like what happens with MPTP exposure [143]. Several studies have shown that the most environmental toxins can inhibit the complex-I activity and interfere the mitochondrial electron transport system, which can ultimately increase free radical production, leading to oxidative stress [140].

Brain areas affected by PD. Substantia nigra in mouse brain (a and b); TH+ DA-neurons in SN (c; 40 x); in control (d) and MPTP-treated mouse brain (e). TH+ fibers in control (f, h) and MPTP-treated (g, i) mouse striatum. Note: The loss of DA-neurons in SN (e), along with loss of TH+ fibers in striatum, have been observed after MPTP treatment (g & i)
Although several investigators have developed many animal models of PD using environmental toxins, none of them demonstrated the primary PD symptoms, like resting tremor or bradykinesia. They also fail to accurately recapitulate the mechanisms of DA neuronal death (Table 4) (Fig. 9).

Different ETs-associated with PD. Chemical structure of different pesticides, herbicides, fungicides, and insecticides which may produce Parkinson-like symptoms in animal models
Moreover, these toxins can cause acute or rapid cell death, unlike progressive neurodegeneration noted in PD [14]. In addition, most therapeutics used to protect against the neurodegeneration caused by these environmental toxins in animal models are unable to translate into effective human therapies. Therefore, the selection of a toxin to produce an animal model of PD is a challenging task (Table 4).
Role of mitochondrial damage and oxidative stress in PD
One of the most promising theories in PD research, as well as other age-related neurodegenerative diseases, is the oxidative stress theory [144]. This theory posits that the mitochondria is the “hot-spot” for degenerative processes. In PD, the abnormal activity of complex-I in mitochondria has been observed, which directly interferes with cellular ATP production, leading to cell death [145]. In addition, the brain monoamines, such as DA and 5-HT, generally act as antioxidants [146]. However, breakdown of DA by monoamine oxidase-B (MAO-B), and combined with ground state O2, leads to the formation of ROS [147] (Fig. 10 ). Researchers have found increased oxidative stress markers and related changes (including free radical damage to DNA, proteins, and fats) in PD patients [147]. In addition, increased levels of the apoptotic marker protein, “Bax” has been observed in DA-neurons of the SNpc in MPTP-treated mice [94]. Recently, investigators have developed hybrid cells, called “cybrid”, to check the role of mitochondria in development of PD [148]. They have placed mitochondrial DNA from PD patients into neuroblastoma cells and found these cybrids develop LB, just like those in the DA-neurons of PD patients. Similarly, certain gene mutations that are involved in cell-survival mechanisms may lead to impairment of mitochondrial activity and ATP production. These findings provide strong support for the idea that mitochondrial defects play a key role in the development of sporadic PD.

Oxidative stress theory in PD. With the help of MAO-B, the DA is converted to DOPAC and produces hydrogen peroxide (H2O2). The H2O2 is then converted to other ROS by Fenton reaction
Role of excitotoxicity in PD
DA is an inhibitory neurotransmitter, which normally maintains the excitation status of the subthalamic nuclei (STN) at basal levels. However, in the case of PD, due to deficiency of DA neurons, the STN become over activated, leading to excessive production of neurotransmitter glutamate [149]. Excessive glutamate binds to its ionotropic receptors (NMDA or AMPA) and open the voltage-gated calcium (Ca++) channels (VGCC), which causes Ca++ excitotoxicity. Excess Ca++ load can damage the mitochondria and produce ROS, leading to oxidative stress [150, 151]. In addition, environmental toxins, can cause increased production of glutamate, leading to Ca++ excitotoxicity which makes DA neurons vulnerable to neurodegeneration [14, 94, 141, 151].
Neuroinflammation involved in PD
A cascade of events are involved in neuroinflammation processes in PD, including activation of microglia and an increase secretion of cytokines [152]. For example, researchers have found strong links between pro-inflammatory cytokines and degeneration of DA neurons, following sub-chronic administration of MPTP in animals [153]. Several clinical studies have shown that the level of inflammatory enzymes, such as cyclo-oxygenase-2 (COX-2), is increased several times in DA-neurons of the postmortem PD brain and in a mouse models of PD [154] (Fig. 11 ).

Mechanism of neuroinflammation in PD. T-lymphocytes and complementary systems can activate microglia to secrete several cytokines, which causes DA-neuronal injury. Similarly, aggregated SNCA can also activate astrocytes, which causes oxidative stress, leading to neuronal injury
Prion hypothesis
During the past few decades, scientists have postulated several mechanisms for the onset and progression of PD. Recently, the “prion hypothesis” is considered one of the most intriguing theories behind its onset. This theory posits that SNCA spreads throughout the CNS, similar to “prion proteins” and infect adjacent new, healthy neurons and that this cycle continues until most of the CNS neurons are infected. Therefore, “prion-like infection” of SNCA may be responsible for the progression and neurodegeneration of some types of PD [11]. According to Braak’s hypothesis, SNCA, bacteria, or viruses can travel via the olfactory tract and into the Vegas nerve to the medala and spread throughout the CNS, which can responsible initiate sporadic PD [155]. Recently, Chandra and colleagues discovered that enteroendocrine cells (EECs) in gastro-intestinal track, possess many neuron-like properties and that SNCA is expressed in the EEC lines, as well as in native EECs of mouse and human intestines. These cells directly connect to SNCA-containing nerves, forming a neural circuit between the gut and the nervous system (gut-brain interaction hypothesis) [156]. Moreover, abundant clinical and pathological evidence have localized misfolded SNCA in EECs, before it appears in the brain. These phenomena suggest that PD pathogenesis may originate in the gut and spread to the CNS via cell-to-cell “prion-like propagation” [156]. Although the “prion hypothesis” provides useful insights into the progression of PD, the presence of SNCA is not always necessary for the emergence of PD pathology or parkinsonism. Therefore, even though SNCA can infect healthy cells like a prion, the “prion hypothesis” of PD remains controversial [157].
Diagnosis of PD
Most clinicians face difficulties in diagnosing PD accurately, because some of the symptoms can emerge during normal aging. Moreover, only a few tests are available, which can help to diagnose PD. Therefore, most PD patients are diagnosed on the basis of their medical history and certain neurological examinations [2]. The presence of LB is one of the most definitive ways to diagnose PD [158], which can be done by microscopic examination of postmortem brain tissues. However, LB can also be found in brains of patients who lack other symptoms without parkinsonism [159]. For example, more than 8% people over 50 years, 13% of people over 70, and 16% of over 80 years of age show LB in their brains, in the absence of any other symptoms of PD [160]. Therefore, the presence of LB in the brain tissue is not the sole indicator of PD, but an accurate diagnosis requires the presence of at least two of the three major motor signs, such as resting tremor, rigidity, and bradykinesia [161] (Table 5). The examination of different reflexes and limb movements are also considered as standard tests for the diagnosis of PD [161]. These can be measured by different means: (i) bradykinesia can be tested by measuring the capability to clamp finger and thumb together, or tap foot up and down; (ii) tremor index can be determined by simple inspection; (iii) muscular rigidity can be tested by moving the neck, upper limbs, and lower limbs; (iv) postural instability can be tested by “pull test”. Overall, the progression of PD can be diagnosed and categorized by different stages, as described by Hoehn and Yahr scale [162] (Table 5).
Treatment of PD
Like other neurodegenerative diseases, PD can cause socioeconomic and emotional breakdown to the immediate family members, caretakers and friends of the patient. Unfortunately, effective therapeutics are not currently available, but early diagnosis and appropriate palliative treatment can provide for a more productive and longer life for most PD patients. Currently, several therapies are available to slow down the disease progression, albeit modestly, or provide transient relief of the severe symptoms of PD. Physicians most commonly use either (i) drug treatment or (ii) surgery to treat PD, however, some accessory therapies are also available.
Drug treatments in PD
Although several generic drugs are available to reduce PD pathogenesis, the most critical factor is the selection of right dose. Therefore, during recommended course of treatment, a physician always assesses the effect of drug on the daily life of the patient, because drugs may take time to affect the patient’s body. Recently microinjection or infusion techniques allow several neuro-active substances to be injected into the damaged brain areas of PD [163]. Currently, the available medications for PD are divided into two categories: (1) dopaminergic drugs; (2) non-dopaminergic drugs (Fig. 12).

Possible therapies for PD. Currently different therapies available for treating PD include pharmacological manipulations, surgical treatments, stem cell and gene therapies, rehabilitation therapies and other complementary and supportive therapies
Dopaminergic drugs
-
(i)
Levodopa (L-DOPA). Physicians usually prescribe DA-drugs to the PD patients in an effect to restore DA levels. As DA, itself, cannot cross the blood brain barrier, therefore, DA precursors, such as levodopa (L-3, 4-dihydroxyphenylalanine/L-DOPA) are commonly prescribed [164]. L-DOPA is very effective in reducing the “resting-tremors” and other primary symptoms [165], but L-DOPA is unable to preserve or replace degenerated DA-neurons or to stop further progression of PD [166]. Furthermore, it may cause nausea, vomiting, low blood pressure, restlessness, drowsiness or sudden onset of sleep (Table 6). In addition, because the conversion of L-DOPA to DA occurs very fast, decreasing its potency when it reaches the target area, physicians often prescribe carbidopa, which prolongs the therapeutic effect when given in conjugation with L-DOPA [167].
-
(ii)
MAO-B inhibitors. Decrease DA levels in PD may be due to its rapid breakdown by the catalytic enzyme monoamine oxidase-B (MAO-B), whose level is increased in the PD brain [17] (Fig. 12). Therefore, inhibition of MAO-B is a good strategy to maintain DA levels in PD brain [168]. Selegiline, (L-deprenyl) and rasagiline are well-tolerated and the most commonly used MAO-B inhibitors [169], which, when administered with L-DOPA, and can sustain the response of L-DOPA even up to a year or more. Although several side effects have been reported with the use of these drugs (Table 5), they have promising effects on restoration of cell functioning or slowing down the loss of DA-neurons in PD [169].
-
(iii)
COMT inhibitors. MAO can breakdown the DA to dihydroxy phenyl acetate, which is further catalyzed by the enzyme, catechol-O-methyl transferase (COMT), to form homovanillic acid (Fig. 13).
-
(iv)
As COMT is responsible for breakdown of DA indirectly, therefore, inhibition of COMT could be another way to restore DA and treat PD [170]. The common COMT inhibitors are entacapone and tolcapone, which prolong the effects of L-DOPA by preventing the breakdown of DA [171]. These drugs also can reduce the sensitivity of L-DOPA in PD patient and produce fewer side-effects (Table 5) [171].
-
(v)
Dopamine agonists. These drugs can increase DA levels in the brain and are most effective during the early stages of PD. They can also be combined with L-DOPA in late-stages of PD to increase the life span of L-DOPA [172]. The common DA agonists used to treat PD patients are pramipexole and ropinirole which are generally less effective than L-DOPA for controlling rigidity and bradykinesia [173]. Unfortunately, these drugs have several potential side effects that are similar to L-DOPA (Table 6) [172, 173].

DA-biosynthesis and degradation. TH: Tyrosine hydroxylase, ALAAD: Aromatic L-amino acid decarboxylase, MAO: Mono amine oxidase, COMT: Catechol O-methyl transferase
Non-dopaminergic drugs
Non-dopaminergic drugs include anti-cholinergic compounds, norepinephrine (NE), serotonergic receptor- and muscarinic-receptor-related compounds, and antiviral drugs.
-
(i)
Anticholinergic drugs. One of most important excitatory neurotransmitters in the brain is ACh, which has been reported to decrease in several areas of the brain parts in PD patients. In PD, DA levels are diminished, causing less inhibitory activity in the brain, allowing ACh-induced excitation to continue to the point of over-excitation. Therefore, anticholinergic drugs may be effective [174]. Although anti-ACh drugs can help to reduce tremors and muscle stiffness in PD, only about 50% of the patients get any relief, and this is only for brief periods, whereas only 30% of patients showing any symptomatic improvements [175]. Moreover, the anti-ACh drugs have several side effects (Table 6) [175].
Other drugs
The non-motor symptoms, including depression and anxiety, can be treated with anti-depressants. Benzodiazepine is one of the most commonly used drugs to treat anxiety in PD patients [176], but it has some side effects. Similarly, clozapine is prescribed to control dyskinesia in PD, but this can cause agranulocytosis and other side effects [177].
Immunotherapies
There are mainly two immunotherapeutic strategies available: active and passive, which have tested in animal models and human PD patients for targeting SNCA. Masliah and colleagues used a transgenic mouse overexpressing human wild type SNCA. These mice exhibit SNCA accumulation in neurons and glia of the neocortex, hippocampus, and SNpc [178]. Mice that were immunized with recombinant human SNCA showed a decreased in accumulation of SNCA inclusions in temporal cortex, and preserved synaptophysin-positive nerve terminals, as well as reduced neurodegeneration [179, 180] (Table 7).
Surgical treatments
Most of the anti-PD drugs have several side-effects and are only transiently effective in a certain population of patients (Table 6). Additionally, they are unable to stop further DA-neuronal loss. Therefore, when there is no adequate relief after medication, clinicians resort to surgical treatments to reduce motor symptoms, especially during the advanced stages of PD. Currently, there are two commonly used surgical treatments for PD: (i) deep brain stimulation (DBS) and (ii) surgical lesions, such as a pallidotomy and/ or a thalamotomy [181].
-
(i)
Deep brain stimulation (DBS). Several basal ganglia nuclei become inactive or dysfunctional in PD. Surgical implantation of very fine electrodes in these areas can be used to keep them functionally active, a process called deep brain stimulation (DBS). The thalamus, globus pallidus interna (Gpi), or STN are target regions for DBS [182], where the electrodes are implanted, in one or both the hemispheres (Fig. 14).
In DBS, devices containing two batteries, which generate finely tuned electrical currents for stimulating those deep brain areas, are implanted in both sides in the chest under the collar bone. The electrical pulses are generated by these batteries, which can be programmed precisely according to the specific needs of the PD patient. At 3–5 year’ intervals, the implanted batteries can be checked or replaced or recharged, accordingly. The DBS can reduce many primary motor symptoms of PD, and also decrease the need for L-DOPA to reduce dyskinesias [183]. In addition, the electrodes can be programmed to be turned on or off, as needed, by using a hand-held device [184]. However, the greatest disadvantage of using DBS, is that it requires surgical implantation of the device, which can cause potential complications, including stroke or hemorrhage, risk of infection, speech, or balance problems. Moreover, DBS is not effective in “atypical” parkinsonian syndromes, such as multiple system atrophy, progressive supra-nuclear palsy, or posttraumatic parkinsonism [184]. In addition, DBS is not used to treat the early stages or for treating mild symptoms of PD, or not suitable for treating the cognitive, psychological, or any other non-motor symptoms [183].
-
(ii)
Pallidotomy and thalamotomy. The parts of the brain which control our voluntary movements include the globus pallidus (GP), a part of basal ganglia which has strong connections to the striatum and the thalamus. In pallidotomy, the surgeon selectively destroys a part of the GP (Fig. 15). Therefore, the synaptic connections with thalamus or striatum are altered in a way which decreases tremor, rigidity, bradykinesia, and posture abnormalities in PD patients [185].
This surgical method can also reduce the amount of L-DOPA that the PD patient requires, which can decrease drug-induced dyskinesia and dystonia. Similarly, destruction of the thalamus, known as thalamotomy, can interrupt the connections between the basal ganglia and motor cortex, in ways that can restore neurotransmitter balance (e.g. glutamate excitation) and reduce symptoms, such as tremor [185]. Thalamotomy is used mainly for controlling tremor, and it is not very effective for bradykinesia, rigidity, or dyskinesias (Fig. 15).

Schematic diagram show the process of DBS. In DBS, STN or thalamus or the globus pallidus interna (Gpi) (in this case STN) are stimulated by an implanted apparatus contains batteries that produce electrical stimulation (like a pace-maker). Stimulating the STN can activate the GPi, which can strongly inhibit the thalamus (right side circuitry) which can activate the motor cortex; in turn, allowing more control into the movement of limbs

Schematic diagram showing pallidotomy (a), and thalamotomy (c) and the basal ganglia circuitory during pallidotomy (b) and thallatomy (d). In case of pallidotomy, the globus pallidus (GP) is surgically destroyed. In the case of a thalamotomy, both thalami are destroyed surgically, which causes a loos of thalamic excitation to the motor cortex, which can decrease Parkinson-like symptoms. Scematic diagram of basal ganglia circuitory in normal brain is shown in “e”
Cell transplantation therapy
Transplantation of neuronal stem cells into the brains of PD patients is considered to be one of the most promising approaches for treating this disease [186]. Over the last two decades, several investigators transplanted DA cells, such as adrenal medullary dopaminergic cells, into the striata in animal models of PD, [5, 6]. Recently, by manipulating several growth factors (such as FGF-2b, FGF8, SHH), researchers are able to generate DA-neurons from rodent embryonic stem cells (Fig. 16), and transplant them into the striata of animal models of PD. Interestingly, these transplanted neurons survive and integrate into the existing brain circuitry in animal model of PD and reverse the behavioral deficits [187, 188]. By over-expressing Nurr1, (a transcription factor for development of DA neuron) in embryonic stem cells, researchers can generate even more DA neurons [18, 19, 189, 190] to transplant into the brains of PD animals.

Different steps of generation of DA-neurons from stem cells for treating PD. Stem cells are obtained from different sources and converted to induced pluoripotent stem cells (iPSCs) using growth factors, such as Fgf2, Shh, Klf4 and c-Myc. The iPSCs is then converted to induced neuronal stem cells (iNSCs). These cells are then converted to DA-neurons by treating different growth factors. These DA-neurons are then injected to the brain of mouse model of PD to supply DA, which ultimately leads to the recovery of motor and cognitive deficits
When human fetal-derived dopaminergic tissues are grafted into the striatum of the PD patients, an increase in DA levels is observed, suggesting that the implanted stem cells are able to survived and differentiate into DA neurons [191]. Further, when researchers implanted porcine-derived DA-producing cells into the brain of a PD patient, they observed modest clinical improvements, suggesting that DA-producing xenografts can survive in human brain [192]. Similarly, the greatest clinical benefits have been observed in allogeneic human fetal ventral mesencephalic (FVM) tissue transplantation in PD patient [193, 194]. These cells survive and make appropriate synaptic connections, while and increasing DA levels within the host cells. Similarly, high-quality DA-cells, such as porcine fetal mesencephalic cells, human retinal pigmented epithelial cells, and progenitor cells, have also been used for clinical or preclinical testing as a therapy for PD [190, 195]. However, further research is needed to evaluate the safety and efficacy of stem cell therapy before it can be approved as a treatment of human PD patients [196].
Gene therapy for PD
Although most cases of PD are sporadic in nature, recent studies have confirmed that several genes are linked with the development of PD [16, 24, 197]. Therefore, gene therapy approaches may offer another promising means of treating PD [24].
-
(i)
Viral vectors-mediated gene delivery. Currently, several viral vectors, such as lentivirus (LV), non-lentivirus, adeno-virus and recombinant adeno-associated virus (rAAV), containing target genes, are injected to the animal brain either by direct stereotaxic administration or via systemic injections. These viruses can integrate with the host cell and induce certain gene expression, promote DA-cell survival, as well as prevent degeneration of DA-neurons, which, ultimately, increases DA levels [24]. Similarly, the use of AAV2 to deliver gene for NGF, BDNF, GDNF, can increase regeneration of DA-neurons in SNpc and boost DA levels in striatum [198, 199]. For example, animals that were injected with the constitutively expressing GDNF vectors showed a long-term and stable improvement for GDNF levels [200]. Similarly, when AAV2-GDNF vectors were injected into the putamen of a MPTP-treated monkey, an enhancement of the locomotor activities and increased the DA-terminals were observed in the putamen [201]. Furthermore, delivering glutamic acid decarboxylase gene (GAD, the rate limiting enzyme for GABA synthesis) using an AAV (AAV-GAD) into the STN can increase GABA, which helps to balance the neuronal firing in the PD brain, resulting in a normalization of inhibitory signaling [202]. A clinical trial with AAV-GAD gene therapy into the STN has been shown to be safe and well tolerated by patients with advanced PD [203].
-
(ii)
AADC-TH-GCH Gene Therapy. Chemical synthesis of DA from L-DOPA requires three-enzyme systems, involving aromatic amino acid decarboxylase (AADC), TH, and guanosine triphosphate cyclohydrolase (GTC). The TH and GCH catalyze the dietary tyrosine and convert it to L-DOPA, whereas AADC turns the L-DOPA to DA. Therefore, delivery of this triple gene therapy (AADC-TH-GCH) could be helpful to in maintaining basal DA levels in advanced PD [204] .
-
(iii)
RNA interference-based therapy. Interference RNA (RNAi) is another powerful gene silencing approach, which could be used to inhibit SNCA, PINK, or parkin genes in PD. Recently, the polyethylene glycol-polyethyleneimine (PEG/PEI) siSNCA complex has been transfected into PC12 cells and a significantly decreased SNCA-mRNA expression, preventing MPTP-induced apoptosis [205].
-
(iv)
CRISPR-Cas9 gene editing system. CRISPR/Cas-9 system is a powerful gene editing tool, including adding, disrupting, or changing the sequence of specific genes [21, 206, 207], which may be applied for PD-related gene therapy (Fig. 17). Using CRISPR/Cas9-mediated genome editing, Basu and colleagues developed a stable cell line that expresses SNCA tagged with a nano-Luc luciferase reporter. They observed an endogenous monitoring of SNCA transcription, which can make an efficient drug screening tool for therapeutic interventions in PD [208]. Although these gene therapy techniques look very promising, further studies are needed before their safe applications in PD patients can be initiated.

Schematic diagram of basics of rAAV-gene therapy. Left: The gene of interest is packaged within a rAAV vector. When the virus infects the host cell, it injects its DNA-containing gene of interest. This foreign DNA then crosses the nuclear membrane and binds with host DNA. Using protein machinery, the nucleus can make DNA and protein using the inserted DNA, replacing mutated or abnormal genes from host cell. Right: CRISPR-Cas9 system can be used to correct defect gene in PD and other genetic diseases. In presence of guide RNA (g-RNA) CRISPR-Cas9 enzyme can breakdown the DNA double strands in the locus where mutated or faulty genes are located. Then using DNA repair system, the normal DNA can be inserted in the cut site to get normal gene expression
Supplementation of neurotrophic factors
One of the primary reasons for neuronal cell death in PD is the depletion of neuronal growth factors, such as decrease BDNF, NGF, GDNF, and FGF-2b [199, 209]. It has been found that treatments with GDNF for 3–6 months can protect DA-neurons and promote their survival in animal models of PD [198]. Importantly, treatment with GDNF protects damaged DA neurons in the SNpc from their further degeneration in PD models [198]. Similarly, combined therapy of GDNF and neurotropin-4 on cultured DA neurons decreased oxidative stress and protected these neurons from further neurodegeneration [198]. Further, when BDNF was administered to rats in an aged rodent model of PD, an increase in the number of DA-receptors in the striatum was observed [210]. Other growth factors, such as FGF-2, released from activated astrocytes have also been shown to stimulate the survival of DA neurons [211], suggesting neurotrophins may be able to rescue DA-neurons in PD.
Complementary and supportive therapies
To overcome some of the motor, as well as non-motor symptoms, several complementary and supportive therapies are available, including physical, occupational, and speech therapies.
-
(i)
Diet. Diets containing fresh vegetables, fruits, green tea, and caffeine provide good sources of antioxidants, which may be beneficial for delaying or preventing oxidative stress-induced cell death in PD [212,213,214]. A high protein diet may aggravate the disease process, including an increase in constipation, and it may also reduce the effectiveness of L-DOPA [212]. Similarly, drinking plenty of liquids can reduce the chance of constipation. Several natural polyphenols (e.g. curcumin) can also prevent cell death in PD by interacting with amyloid proteins, including Aβ, p-tau, and SNCA [215, 216], as well as preventing further deterioration of DA-neurons [113, 216, 217]. Similarly, pleotropic actions of sodium benzoate, the principal ingredient of cinnamon also preserves DA neurons in the SNpc of animal models of PD [218, 219] (Table 8 )
-
(ii)
Aerobic exercise. A plethora of evidence suggests that controlled aerobic exercise may improve gait, posture, mobility, and flexibility of PD patients [220, 221]. Sometimes, voluntary physical exercise, or physiotherapy, can strengthen muscle activity, decrease muscular tonicity and rigidity, improve balance, minimize gait problems, and strengthen certain muscles in PD patients [222]. Even in severe cases of the disease, regular walking, gardening, and other physical activities have been shown to be beneficial, and may improve the ability of the brain to increase DA synthesis. Moreover, experimental results indicate that daily aerobic physical exercise improves brain functioning by increasing neurogenesis, and secretion of neurotropic factors, such as BDNF, NGF, GDNF, FGF, which also help to maintain the emotional balance of people with PD [223].
Rehabilitational therapy
Some of the primary symptoms of PD, especially gait, tremors, and rigidity can be reduced by the use of rehabilitational therapy [224]. Clinicians, who focus on treatments for non-motor symptoms, such as cognitive impairments, urinary tract dysfunction, sleep disorders, and micrographia, often utilize rehabilitational therapy [224]. For example, to reduce or to prevent muscle hypo-tonicity, PD patients need to be involved in their daily activities, such as moving from side to side, lifting their toes, breaking down their actions into individual steps, and keeping busy with house-hold work for at least few hours a day [225]. Similarly, walking, turning, or standing in different postures can help the patient maintain their balance and reduce the risk of falling. Patients with PD should take small steps when turning, but large, exaggerated steps when walking forward [225]. In addition, cognitive and other emotional dysfunction can be reduced by engaging in hobbies requiring focused attending, such as carpentry, fishing, playing cards, practicing “Yoga” or deep breathing and relaxation exercises [225]. These activities may help to control tremor, as well as reduce anxiety and depression. Speech therapy can help rectify the monotone voice and loss of volume which is often observed in PD patients, and it is also critical for evaluating and monitoring the ability to swallow [37].
Monitoring PD progression
Using recent advanced and cutting-edge, noninvasive technology, the brains of PD patients can be more readily imaged for searching potential biomarkers or monitoring disease progression.
-
(i)
Neuroimaging. The lack of easy access to the human brain is the major limitation for studying PD. Novel imaging techniques, such as PET biomarkers, including [18F]-DOPA can be used for estimating DA, [18F] dG for mitochondrial bioenergetics, [18F] BMS for mitochondrial complex-1, [11C] (R)-PK11195 for microglial activation. Similarly, SPECT imaging with 123Iflupane and βCIT can be used for measuring DAT, urinary salsolinol, and 8-hydroxy, 2-deoxyguanosine for neuronal loss [226, 227]. Similarly, terminal DOPA-decarboxylase (DDC) activity of PD brain can be measured with 18F–DOPA-PET, whereas the availability of presynaptic DAT can be assessed with tropane-based PET and SPECT tracers. Furthermore, vesicle monoamine transporter density in DA-terminals can be examined with 11C–dihydrotetrabenazine (DHTBZ)-PET [228]. Several investigators have used these techniques to study PD both in animals and human. For example, the striatal 18F–DOPA uptake has been shown to correlate with DA-neuronal numbers in the SNpc area of post-mortem PD brain and in MPTP-lesioned monkeys [229]. Similarly, 18F–DOPA-PET has been employed to study a series of asymptomatic heterozygote parkin mutation carriers and 11C–PK11195-PET has been used to study microglial activation in both the SNpc and pallidum of a PD patient [228]. In addition, brain regional N-acetyl-aspartate can be used for the in vivo assessment of neuronal loss in PD using MRS and MRI. Furthermore, fMRI and MRS can also be used to monitor the blood flow or metabolic status in the PD brain [230], and using fMRI, it is possible to detect PD and other synucleinopathies, such as LBD. These methods can also be helpful for studying the status of physiological activation of specific motor, cognitive, or mood swings after drug treatments.
-
(ii)
Biomarkers. As PD is a disease with multiple etiologies and heterogeneous clinical symptoms, it is critical to develop several biomarkers to understand the disease more fully and to devise effective therapies. Although radioactive nucleotides are the most feasible biomarkers for screening of PD [231], fluid biomarkers, neuromelanin antibodies, candidate blood-based biomarker testing (e.g. SNCA, DJ-1, uric acid, epidermal growth factor, ApoA1) [232], gene expression profiling, metabolomics, protein profiling (e.g. Aβ and tau) and inflammatory markers (e.g. IL-6) from blood and CSF samples, can be used to predict PD. Because of the high cost of the procedures and the difficulty in administering these techniques, these methods are not always practical for screening PD patients in the general population [233]. In addition, investigating redox status and mitochondrial health could be used as early biomarkers for prediction of the onset of this disease [12]. Further, abnormal motor physiology, cognitive dysfunction, REM sleep disorders, autonomic dysfunction, loss of olfaction, and speech disturbances can, collectively be used to screen for PD [8]. Overall, expanding our knowledge of PD, combined with analyses of the pre-clinical and clinical symptoms, along with the development of more accurate biochemical and molecular markers, should translate into more effective and efficient ways to predict the onset and progression of this disease.
Future directions for PD research and therapeutic developments
From the extensive animal research, we have addressed several critical questions in this complex field. However, no animal model is perfect in reproducing the behavioral deficits and neuropathological changes observed in human PD patients. Therefore, to mimic human PD symptoms and to address specific questions in this field several strategies can be taken, such as (i) development of a novel animal model, which can be produced by combination of two or more animal models, such as transgenic (for genetic effects), as well as sporadic (for toxin effects); (ii) using a range of non-DA drugs, including α2-adrenergic antagonists, serotonergic, and adenosine A2a antagonists, which may offer beneficial effects in late-stage developments of motor symptoms in PD; (iii) development of novel formulae for levodopa/carbidopa drugs (e.g. use of IPX066, XP21279, and Opicapone), MAO-inhibitors (e.g. use of safinamide:100–200 mg/day), which has an immediate and long-term clinical benefit on both early and advanced PD patients, without side effects, such as dyskinesia or depression; (iv) targeting ALP and UPS by novel pharmacological molecules may be an attractive strategy for PD therapy; (v) development of transplantation therapies using novel DA neurons from induced neuronal stem cells (iNSCs) or from induced pluripotent stem cells; (vi) micro-RNA or Si-RNA approach to inhibit mRNA of misfolded protein aggregates; (vii) application of novel gene editing techniques (e.g. CRISP-Cas9) for correction of mutated genes involved in PD. New drugs are constantly being developed tackle PD, and some of them are already improving quality of life in PD patients. There are several issues that need to be addressed before effective gene therapy can be safely used to treat PD. Some of these concerns involve the identification of the exact genes to be used for rectification, appropriate selection of novel vectors, development of safe marker genes, and modulators of precise gene expression in the CNS [24].
Conclusions
The cases of PD are steadily increasing, due to the increase in the aged population. The financial and emotional impact of PD on public health and on those of the family and friends of those suffering from this disease is staggering. Therefore, prognosis or early detection will be critical for screening those who are susceptible for getting this disease. Several treatments are available, but none of them are particularly effective, in terms of reducing dopaminergic neuronal loss and restoring DA levels in the striatum. Moreover, some of the drugs have serious side-effects, and are expensive. Recently, scientists have introduced some promising alternative strategies, such as stem cell transplantation and gene therapy to treat this disease. However, most of these new treatment strategies are still under investigation, with most of these only being tested in animal models, so issues of safety and efficacy need to be adequately addressed before they can be used in clinical trials for PD. Nonetheless, the new therapeutic approaches described in this review offers significant hope that effective treatments for PD are in the near horizon.
Abbreviations
- Ach:
-
Acetylcholine
- ALP:
-
Autophagy lysosomal pathway
- AMPA:
-
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- Atg:
-
Autophagy protein
- BDNF:
-
Brain derived neurotropic factor
- CHIP:
-
Carboxy-terminus of HSP70-interacting protein
- CMA:
-
Chaperone-mediated autophagy
- COMT:
-
Catechol-O-methyl transferase
- COX-2:
-
Cyclo-oxygenase-2
- CRISPR:
-
Cas9-clustered regularly-interspaced short palindromic repeats-CRISPR-associated protein 9
- CSF:
-
Cerebrospinal fluid
- CT:
-
Computarized tomography
- DA:
-
Dopamine
- DAT:
-
Dopamine transporter
- DBS:
-
Deep brain stimulation
- FGF:
-
Fibroblast growth factor
- fMRI:
-
Functional magnetic resonance imaging
- FTDP:
-
Frontotemporal dementia with parkinsonism
- GABA:
-
Gamma amino butyric acid
- GAD:
-
Glutamic acid decarboxylase
- GDNF:
-
Glial derived neurotropic factor
- GI:
-
Gastrointestinal
- Gpi:
-
globus pallidus interna
- H2O2 :
-
Hydrogen peroxide
- HSP:
-
Heat shock protein
- LAMP2A:
-
Lysosome-associated membrane protein 2
- L-DOPA:
-
Levo-3,4-dihydroxyphenylalanine
- LRRK2:
-
Leucine-rich repeat kinase-2
- LRRK2:
-
l-leucine-rich repeat kinase 2
- MAO-B:
-
Monoamine oxidase-B
- MPP:
-
1-methyl-4-phenylpyridinium
- MPTP:
-
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MRI:
-
Magnetic resonance image
- MRS:
-
Magnetic resonance spectroscopy
- mtDNA:
-
Mitochondrial DNA
- NE:
-
Norepinephrine
- NFT:
-
Neurofibrillary tangles
- NGF:
-
Nerve growth factor
- NMDA:
-
N-methyl-D-aspartate
- OH:
-
Hydroxyl radical
- OHDA:
-
Hydroxydopamine
- PD:
-
Parkinson’s disease
- PET:
-
Positron emission tomography
- PFC:
-
Prefrontal cortex
- PINK-1:
-
PTEN-induced putative kinase-1
- PSF:
-
Poly-pyrimidine tract-binding protein-associated splicing factor
- PTEN:
-
Phosphatase and tensin homolog
- rAAV:
-
Recombinant adeno-associated viral vectors
- REM:
-
Rapid eye movement
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- SHH:
-
Sonic hedgehog
- SNpc:
-
Substantia nigra pars compacta
- SPECT:
-
Single photon emission computed tomography
- STN:
-
Subthalamic nucleus
- TH:
-
Tyrosine hydroxylase gene
- TPM:
-
Two-photon microscopy
- UCHL-1:
-
Ubiquitin carboxyl-terminal esterase L-1
- UPS:
-
Ubiquitin-Proteasome System
- VGCC:
-
Voltage-gated calcium channel
- β-CIT:
-
2β-carbomethoxy-3β-[4-iodophenyl)tropane -(4-iodophenyl)tropane]
References
Wright Willis A, Evanoff BA, Lian M, Criswell SR, Racette BA. Geographic and ethnic variation in Parkinson disease: a population-based study of US Medicare beneficiaries. Neuroepidemiology. 2010;34(3):143–51.
Jankovic J. Parkinson's disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79(4):368–76.
Alexander GE. Biology of Parkinson's disease: pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin Neurosci. 2004;6(3):259–80.
Berardelli A, Rothwell JC, Thompson PD, Hallett M. Pathophysiology of bradykinesia in Parkinson's disease. Brain. 2001;124(Pt 11):2131–46.
Solari N, Bonito-Oliva A, Fisone G, Brambilla R. Understanding cognitive deficits in Parkinson's disease: lessons from preclinical animal models. Learn Mem. 2013;20(10):592–600.
Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39(6):889–909.
Shepherd GM. Corticostriatal connectivity and its role in disease. Nat Rev Neurosci. 2013;14(4):278–91.
Davie CA. A review of Parkinson’s disease. Br Med Bull. 2008;86:109–27.
Michel PP, Hirsch EC, Hunot S. Understanding dopaminergic cell death pathways in Parkinson disease. Neuron. 2016;90(4):675–91.
Martin I, Dawson VL, Dawson TM. Recent advances in the genetics of Parkinson's disease. Annu Rev Genomics Hum Genet. 2011;12:301–25.
Chauhan A, Jeans AF. Is Parkinson's disease truly a prion-like disorder? An appraisal of current evidence. Neurol Res Int. 2015;2015:345285.
Berg D. Biomarkers for the early detection of Parkinson's and Alzheimer’s disease. Neurodegener Dis. 2008;5(3–4):133–6.
Kim WS, Kagedal K, Halliday GM. Alpha-synuclein biology in Lewy body diseases. Alzheimers Res Ther. 2014;6(5):73.
Bove J, Prou D, Perier C, Przedborski S. Toxin-induced models of Parkinson's disease. NeuroRx. 2005;2(3):484–94.
Bus JS, Gibson JE. Paraquat: model for oxidant-initiated toxicity. Environ Health Perspect. 1984;55:37–46.
Klein C, Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2(1):a008888.
George JL, Mok S, Moses D, Wilkins S, Bush AI, Cherny RA, Finkelstein DI. Targeting the progression of Parkinson's disease. Curr Neuropharmacol. 2009;7(1):9–36.
Roybon L, Christophersen NS, Brundin P, Li JY. Stem cell therapy for Parkinson's disease: where do we stand? Cell Tissue Res. 2004;318(1):261–73.
Zhang SC, Li XJ, Johnson MA, Pankratz MT. Human embryonic stem cells for brain repair? Philos Trans R Soc Lond Ser B Biol Sci. 2008;363(1489):87–99.
Ziavra D, Makri G, Giompres P, Taraviras S, Thomaidou D, Matsas R, Mitsacos A, Kouvelas ED. Neural stem cells transplanted in a mouse model of Parkinson's disease differentiate to neuronal phenotypes and reduce rotational deficit. CNS Neurol Disord Drug Targets. 2012;11(7):829–35.
Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347–55.
Tu Z, Yang W, Yan S, Guo X, Li XJ. CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases. Mol Neurodegener. 2015;10:35.
Han F, Baremberg D, Gao J, Duan J, Lu X, Zhang N, Chen Q. Development of stem cell-based therapy for Parkinson's disease. Transl Neurodegeneration. 2015;4:16.
Coune PG, Schneider BL, Aebischer P. Parkinson’s disease: gene therapies. Cold Spring Harb Med. 2012;2(4):a009431.
Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease. J Clin Invest. 2005;115(6):1449–57.
Goldenberg MM. Medical management of Parkinson’s disease. P & T : Peer-Reviewed J Formulary Manag. 2008;33(10):590–606.
Cannon JR, Greenamyre JT. The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol Sci. 2011;124(2):225–50.
Ishihara LS, Cheesbrough A, Brayne C, Schrag A. Estimated life expectancy of Parkinson’s patients compared with the UK population. J Neurol Neurosurg Psychiatry. 2007;78(12):1304–9.
Santens P, Boon P, Van Roost D, Caemaert J. The pathophysiology of motor symptoms in Parkinson’s disease. Acta Neurol Belg. 2003;103(3):129–34.
Bhidayasiri R: Differential diagnosis of common tremor syndromes. Postgrad Med J 2005, 81(962):756-762.
Berardelli A, Sabra AF, Hallett M. Physiological mechanisms of rigidity in Parkinson's disease. J Neurol Neurosurg Psychiatry. 1983;46(1):45–53.
Tjaden K. Speech and swallowing in Parkinson's disease. Top Geriatr Rehabil. 2008;24(2):115–26.
Tolosa E, Compta Y. Dystonia in Parkinson’s disease. J Neurol. 2006;253(Suppl 7):VII7–13.
Thanvi B, Lo N, Robinson T. Levodopa-induced dyskinesia in Parkinson's disease: clinical features, pathogenesis, prevention and treatment. Postgrad Med J. 2007;83(980):384–8.
Bronner G, Vodusek DB. Management of sexual dysfunction in Parkinson's disease. Ther Adv Neurol Disord. 2011;4(6):375–83.
Truong DD, Bhidayasiri R, Wolters E. Management of non-motor symptoms in advanced Parkinson disease. J Neurol Sci. 2008;266(1–2):216–28.
Ramig LO, Fox C, Sapir S. Speech treatment for Parkinson’s disease. Expert Rev Neurother. 2008;8(2):297–309.
Kano O, Ikeda K, Cridebring D, Takazawa T, Yoshii Y, Iwasaki Y. Neurobiology of depression and anxiety in Parkinson's disease. Park Dis. 2011;2011:143547.
Aarsland D, Pahlhagen S, Ballard CG, Ehrt U, Svenningsson P. Depression in Parkinson disease--epidemiology, mechanisms and management. Nat Rev Neurol. 2012;8(1):35–47.
Caballol N, Marti MJ, Tolosa E. Cognitive dysfunction and dementia in Parkinson disease. Mov Disord. 2007;22(Suppl 17):S358–66.
Meireles J, Massano J. Cognitive impairment and dementia in Parkinson's disease: clinical features, diagnosis, and management. Front Neurol. 2012;3:88.
Comella CL. Sleep disorders in Parkinson's disease. Curr Treat Options Neurol. 2008;10(3):215–21.
Larsen JP, Tandberg E. Sleep disorders in patients with Parkinson’s disease: epidemiology and management. CNS Drugs. 2001;15(4):267–75.
Suzuki K, Miyamoto M, Miyamoto T, Iwanami M, Hirata K. Sleep disturbances associated with Parkinson’s disease. Park Dis. 2011;2011:219056.
Blonder LX, Slevin JT. Emotional dysfunction in Parkinson’s disease. Behav Neurol. 2011;24(3):201–17.
Bowers D, Miller K, Mikos A, Kirsch-Darrow L, Springer U, Fernandez H, Foote K, Okun M. Startling facts about emotion in Parkinson's disease: blunted reactivity to aversive stimuli. Brain. 2006;129(Pt 12):3356–65.
Blackett H, Walker R, Wood B. Urinary dysfunction in Parkinson's disease: a review. Parkinsonism Relat Disord. 2009;15(2):81–7.
Swinn L, Schrag A, Viswanathan R, Bloem BR, Lees A, Quinn N. Sweating dysfunction in Parkinson’s disease. Mov Disord. 2003;18(12):1459–63.
Tsukamoto T, Kitano Y, Kuno S. Blood pressure fluctuation and hypertension in patients with Parkinson’s disease. Brain Behav. 2013;3(6):710–4.
Ford B. Pain in Parkinson’s disease. Mov Disord. 2010;25(Suppl 1):S98–103.
Sophie M, Ford B. Management of pain in Parkinson’s disease. CNS Drugs. 2012;26(11):937–48.
German DC, Manaye K, Smith WK, Woodward DJ, Saper CB. Midbrain dopaminergic cell loss in Parkinson’s disease: computer visualization. Ann Neurol. 1989;26(4):507–14.
German DC, Manaye KF, Sonsalla PK, Brooks BA. Midbrain dopaminergic cell loss in Parkinson's disease and MPTP-induced parkinsonism: sparing of calbindin-D28k-containing cells. Ann N Y Acad Sci. 1992;648:42–62.
Huot P, Sgambato-Faure V, Fox SH, McCreary AC. Serotonergic approaches in Parkinson’s disease: translational perspectives, an update. ACS Chem Neurosci. 2017;8(5):973–86.
Ansah TA, Ferguson MC, Nayyar T, Deutch AY. Age- and duration-dependent effects of MPTP on cortical serotonin systems. Neurosci Lett. 2011;504(2):160–4.
Sanchez MG, Morissette M, Di Paolo T. Estradiol and brain serotonin reuptake transporter in long-term ovariectomized parkinsonian monkeys. Prog Neuro-Psychopharmacol Biol Psychiatry. 2013;45:170–7.
Guttman M, Boileau I, Warsh J, Saint-Cyr JA, Ginovart N, McCluskey T, Houle S, Wilson A, Mundo E, Rusjan P, et al. Brain serotonin transporter binding in non-depressed patients with Parkinson's disease. Eur J Neurol. 2007;14(5):523–8.
Haapaniemi TH, Ahonen A, Torniainen P, Sotaniemi KA, Myllyla VV. [123I]beta-CIT SPECT demonstrates decreased brain dopamine and serotonin transporter levels in untreated parkinsonian patients. Mov Disord. 2001;16(1):124–30.
Doder M, Rabiner EA, Turjanski N, Lees AJ, Brooks DJ. Study CWP: tremor in Parkinson’s disease and serotonergic dysfunction: an 11C-WAY 100635 PET study. Neurology. 2003;60(4):601–5.
Maiti P, Gregg LC, McDonald MP. MPTP-induced executive dysfunction is associated with altered prefrontal serotonergic function. Behav Brain Res. 2016;298(Pt B):192–201.
Tan SK, Hartung H, Sharp T, Temel Y. Serotonin-dependent depression in Parkinson's disease: a role for the subthalamic nucleus? Neuropharmacology. 2011;61(3):387–99.
Tagliavini F, Pilleri G. Basal nucleus of Meynert. a neuropathological study in Alzheimer's disease, simple senile dementia, Pick's disease and Huntington's chorea. J Neurol Sci. 1983;62(1–3):243–60.
Liu AK, Chang RC, Pearce RK, Gentleman SM. Nucleus basalis of Meynert revisited: anatomy, history and differential involvement in Alzheimer's and Parkinson's disease. Acta Neuropathol. 2015;129(4):527–40.
Allaman I, Belanger M, Magistretti PJ. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 2011;34(2):76–87.
Hurley MJ, Brandon B, Gentleman SM, Dexter DT. Parkinson's disease is associated with altered expression of CaV1 channels and calcium-binding proteins. Brain. 2013;136(Pt 7):2077–97.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–34.
Stefanis L. Alpha-Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2(2):a009399.
Ibanez CF, Andressoo JO. Biology of GDNF and its receptors - relevance for disorders of the central nervous system. Neurobiol Dis. 2017;97(Pt B):80–9.
Blaszczyk JW. Parkinson’s disease and neurodegeneration: GABA-collapse hypothesis. Front Neurosci. 2016;10:269.
Cardinale A, Chiesa R, Sierks M. Protein misfolding and neurodegenerative diseases. Int J Cell Biol. 2014;2014:217371.
Marques O, Outeiro TF. Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis. 2012;3:e350.
Fortuna JTS, Gralle M, Beckman D, Neves FS, Diniz LP, Frost PS, Barros-Aragao F, Santos LE, Goncalves RA, Romao L, et al. Brain infusion of alpha-synuclein oligomers induces motor and non-motor Parkinson’s disease-like symptoms in mice. Behav Brain Res. 2017;333:150–60.
Oczkowska A, Kozubski W, Lianeri M, Dorszewska J. Mutations in PRKN and SNCA genes important for the progress of Parkinson’s disease. Curr Genomics. 2013;14(8):502–17.
Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int J Biochem Cell Biol. 2009;41(10):2015–24.
Ellis CE, Murphy EJ, Mitchell DC, Golovko MY, Scaglia F, Barcelo-Coblijn GC, Nussbaum RL. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol Cell Biol. 2005;25(22):10190–201.
Klivenyi P, Siwek D, Gardian G, Yang L, Starkov A, Cleren C, Ferrante RJ, Kowall NW, Abeliovich A, Beal MF. Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol Dis. 2006;21(3):541–8.
Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson's disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26(1):41–50.
Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283(14):9089–100.
Schraen-Maschke S, Sergeant N, Dhaenens CM, Bombois S, Deramecourt V, Caillet-Boudin ML, Pasquier F, Maurage CA, Sablonniere B, Vanmechelen E, et al. Tau as a biomarker of neurodegenerative diseases. Biomark Med. 2008;2(4):363–84.
Arima K, Hirai S, Sunohara N, Aoto K, Izumiyama Y, Ueda K, Ikeda K, Kawai M. Cellular co-localization of phosphorylated tau- and NACP/alpha-synuclein-epitopes in lewy bodies in sporadic Parkinson's disease and in dementia with Lewy bodies. Brain Res. 1999;843(1–2):53–61.
Hepp DH, Vergoossen DL, Huisman E, Lemstra AW, Netherlands Brain B, Berendse HW, Rozemuller AJ, Foncke EM, van de Berg WD. Distribution and load of amyloid-beta pathology in Parkinson disease and dementia with Lewy bodies. J Neuropathol Exp Neurol. 2016;75(10):936–45.
Walker L, McAleese KE, Thomas AJ, Johnson M, Martin-Ruiz C, Parker C, Colloby SJ, Jellinger K, Attems J. Neuropathologically mixed Alzheimer’s and Lewy body disease: burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol. 2015;129(5):729–48.
Vandrovcova J, Anaya F, Kay V, Lees A, Hardy J, de Silva R. Disentangling the role of the tau gene locus in sporadic tauopathies. Curr Alzheimer Res. 2010;7(8):726–34.
Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20(9):1726–37.
Schlossmacher MG, Frosch MP, Gai WP, Medina M, Sharma N, Forno L, Ochiishi T, Shimura H, Sharon R, Hattori N, et al. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am J Pathol. 2002;160(5):1655–67.
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25(3):302–5.
Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson’s disease. Science. 2001;293(5528):263–9.
Ariga H, Takahashi-Niki K, Kato I, Maita H, Niki T, Iguchi-Ariga SM. Neuroprotective function of DJ-1 in Parkinson’s disease. Oxidative Med Cell Longev. 2013;2013:683920.
Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A. 2004;101(24):9103–8.
Wu HY, Chen SF, Hsieh JY, Chou F, Wang YH, Lin WT, Lee PY, Yu YJ, Lin LY, Lin TS, et al. Structural basis of antizyme-mediated regulation of polyamine homeostasis. Proc Natl Acad Sci U S A. 2015;112(36):11229–34.
Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson’s disease. J Park Dis. 2013;3(4):461–91.
Velayati A, Yu WH, Sidransky E. The role of glucocerebrosidase mutations in Parkinson disease and Lewy body disorders. Curr Neurol Neurosci Rep. 2010;10(3):190–8.
Akkhawattanangkul Y, Maiti P, Xue Y, Aryal D, Wetsel WC, Hamilton D, Fowler SC, McDonald MP. Targeted deletion of GD3 synthase protects against MPTP-induced neurodegeneration. Genes Brain Behav. 2017;16(5):522–36.
Perier C, Vila M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2(2):a009332.
Richter G, Sonnenschein A, Grunewald T, Reichmann H, Janetzky B. Novel mitochondrial DNA mutations in Parkinson’s disease. J Neural Transm. 2002;109(5–6):721–9.
Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol. 2013;9(8):445–54.
McNaught KS, Olanow CW, Halliwell B, Isacson O, Jenner P. Failure of the ubiquitin-proteasome system in Parkinson's disease. Nat Rev Neurosci. 2001;2(8):589–94.
Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. 2015;47:e147.
Larsen KE, Sulzer D. Autophagy in neurons: a review. Histol Histopathol. 2002;17(3):897–908.
Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443(7113):780–6.
Pukass K, Richter-Landsberg C. Inhibition of UCH-L1 in oligodendroglial cells results in microtubule stabilization and prevents alpha-synuclein aggregate formation by activating the autophagic pathway: implications for multiple system atrophy. Front Cell Neurosci. 2015;9:163.
McNaught KS, Mytilineou C, Jnobaptiste R, Yabut J, Shashidharan P, Jennert P, Olanow CW. Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J Neurochem. 2002;81(2):301–6.
Rideout HJ, Lang-Rollin IC, Savalle M, Stefanis L. Dopaminergic neurons in rat ventral midbrain cultures undergo selective apoptosis and form inclusions, but do not up-regulate iHSP70, following proteasomal inhibition. J Neurochem. 2005;93(5):1304–13.
Fornai F, Lenzi P, Gesi M, Ferrucci M, Lazzeri G, Busceti CL, Ruffoli R, Soldani P, Ruggieri S, Alessandri MG, et al. Fine structure and biochemical mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. J Neurosci. 2003;23(26):8955–66.
Ding Q, Dimayuga E, Martin S, Bruce-Keller AJ, Nukala V, Cuervo AM, Keller JN. Characterization of chronic low-level proteasome inhibition on neural homeostasis. J Neurochem. 2003;86(2):489–97.
Sullivan PG, Dragicevic NB, Deng JH, Bai Y, Dimayuga E, Ding Q, Chen Q, Bruce-Keller AJ, Keller JN. Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J Biol Chem. 2004;279(20):20699–707.
McNaught KS, Perl DP, Brownell AL, Olanow CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson's disease. Ann Neurol. 2004;56(1):149–62.
Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278(44):43628–35.
Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, Harada T, Ichihara N, Wakana S, Kikuchi T, et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet. 1999;23(1):47–51.
Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100(7):4078–83.
Cha GH, Kim S, Park J, Lee E, Kim M, Lee SB, Kim JM, Chung J, Cho KS. Parkin negatively regulates JNK pathway in the dopaminergic neurons of drosophila. Proc Natl Acad Sci U S A. 2005;102(29):10345–50.
Dokladny K, Myers OB, Moseley PL. Heat shock response and autophagy--cooperation and control. Autophagy. 2015;11(2):200–13.
Maiti P, Manna J, Veleri S, Frautschy S. Molecular chaperone dysfunction in neurodegenerative diseases and effects of curcumin. Biomed Res Int. 2014;2014:495091.
Karunanithi S, Brown IR. Heat shock response and homeostatic plasticity. Front Cell Neurosci. 2015;9:68.
Bercovich B, Stancovski I, Mayer A, Blumenfeld N, Laszlo A, Schwartz AL, Ciechanover A. Ubiquitin-dependent degradation of certain protein substrates in vitro requires the molecular chaperone Hsc70. J Biol Chem. 1997;272(14):9002–10.
Wyttenbach A. Role of heat shock proteins during polyglutamine neurodegeneration: mechanisms and hypothesis. J Mol Neurosci. 2004;23(1–2):69–96.
Kaushik S, Cuervo AM. Chaperone-mediated autophagy. Methods Mol Biol. 2008;445:227–44.
Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a drosophila model for Parkinson’s disease. Science. 2002;295(5556):865–8.
Flower TR, Chesnokova LS, Froelich CA, Dixon C, Witt SN. Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson’s disease. J Mol Biol. 2005;351(5):1081–100.
Tantucci M, Mariucci G, Taha E, Spaccatini C, Tozzi A, Luchetti E, Calabresi P, Ambrosini MV. Induction of heat shock protein 70 reduces the alteration of striatal electrical activity caused by mitochondrial impairment. Neuroscience. 2009;163(3):735–40.
Fan GH, Zhou HY, Yang H, Chen SD. Heat shock proteins reduce alpha-synuclein aggregation induced by MPP+ in SK-N-SH cells. FEBS Lett. 2006;580(13):3091–8.
Klucken J, Shin Y, Hyman BT, McLean PJ. A single amino acid substitution differentiates Hsp70-dependent effects on alpha-synuclein degradation and toxicity. Biochem Biophys Res Commun. 2004;325(1):367–73.
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305(5688):1292–5.
Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6(4):463–77.
Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278(27):25009–13.
Bandyopadhyay U, Cuervo AM. Chaperone-mediated autophagy in aging and neurodegeneration: lessons from alpha-synuclein. Exp Gerontol. 2007;42(1–2):120–8.
Crotzer VL, Blum JS. Autophagy and intracellular surveillance: modulating MHC class II antigen presentation with stress. Proc Natl Acad Sci U S A. 2005;102(22):7779–80.
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–6.
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–4.
Jankovic J. Motor fluctuations and dyskinesias in Parkinson's disease: clinical manifestations. Mov Disord. 2005;20(Suppl 11):S11–6.
Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain. 2008;131(Pt 8):1969–78.
Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12(1):25–31.
Toulorge D, Schapira AH, Hajj R. Molecular changes in the postmortem parkinsonian brain. J Neurochem. 2016;139(Suppl 1):27–58.
Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K. Alteration of autophagosomal proteins (LC3, GABARAP and GATE-16) in Lewy body disease. Neurobiol Dis. 2011;43(3):690–7.
Murphy KE, Gysbers AM, Abbott SK, Spiro AS, Furuta A, Cooper A, Garner B, Kabuta T, Halliday GM. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson's disease. Mov Disord. 2015;30(12):1639–47.
Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, Schapira AH. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol. 2010;67(12):1464–72.
Dijkstra AA, Ingrassia A, de Menezes RX, van Kesteren RE, Rozemuller AJ, Heutink P, van de Berg WD. Evidence for immune response, axonal dysfunction and reduced endocytosis in the substantia Nigra in early stage Parkinson’s disease. PLoS One. 2015;10(6):e0128651.
Mutez E, Nkiliza A, Belarbi K, de Broucker A, Vanbesien-Mailliot C, Bleuse S, Duflot A, Comptdaer T, Semaille P, Blervaque R, et al. Involvement of the immune system, endocytosis and EIF2 signaling in both genetically determined and sporadic forms of Parkinson’s disease. Neurobiol Dis. 2014;63:165–70.
Watanabe Y, Himeda T, Araki T. Mechanisms of MPTP toxicity and their implications for therapy of Parkinson’s disease. Med Sci Monit. 2005;11(1):RA17–23.
Jackson-Lewis V, Przedborski S. Protocol for the MPTP mouse model of Parkinson's disease. Nat Protoc. 2007;2(1):141–51.
Bjorling-Poulsen M, Andersen HR, Grandjean P. Potential developmental neurotoxicity of pesticides used in Europe. Environ Health. 2008;7:50.
Haik KL, Shear DA, Hargrove C, Patton J, Mazei-Robison M, Sandstrom MI, Dunbar GL. 7-nitroindazole attenuates 6-hydroxydopamine-induced spatial learning deficits and dopamine neuron loss in a presymptomatic animal model of Parkinson's disease. Exp Clin Psychopharmacol. 2008;16(2):178–89.
Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT. A highly reproducible rotenone model of Parkinson's disease. Neurobiol Dis. 2009;34(2):279–90.
Wang X, Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci. 2010;2:12.
Parker WD Jr, Parks JK, Swerdlow RH. Complex I deficiency in Parkinson's disease frontal cortex. Brain Res. 2008;1189:215–8.
Anderson RF, Harris TA. Dopamine and uric acid act as antioxidants in the repair of DNA radicals: implications in Parkinson’s disease. Free Radic Res. 2003;37(10):1131–6.
Lotharius J, Brundin P. Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. 2002;3(12):932–42.
Wilkins HM, Carl SM, Swerdlow RH. Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol. 2014;2C:619–31.
Rodriguez MC, Obeso JA, Olanow CW. Subthalamic nucleus-mediated excitotoxicity in Parkinson's disease: a target for neuroprotection. Ann Neurol. 1998;44(3 Suppl 1):S175–88.
Mark LP, Prost RW, Ulmer JL, Smith MM, Daniels DL, Strottmann JM, Brown WD, Hacein-Bey L. Pictorial review of glutamate excitotoxicity: fundamental concepts for neuroimaging. AJNR Am J Neuroradiol. 2001;22(10):1813–24.
Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. 2009;30(4):379–87.
Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129(2):154–69.
Meredith GE, Rademacher DJ. MPTP mouse models of Parkinson's disease: an update. J Park Dis. 2011;1(1):19–33.
Bartels AL, Leenders KL. Cyclooxygenase and neuroinflammation in Parkinson's disease neurodegeneration. Curr Neuropharmacol. 2010;8(1):62–8.
Rietdijk CD, Perez-Pardo P, Garssen J, van Wezel RJ, Kraneveld AD. Exploring Braak's hypothesis of Parkinson's disease. Front Neurol. 2017;8:37.
Chandra R, Hiniker A, Kuo YM, Nussbaum RL, Liddle RA. alpha-Synuclein in gut endocrine cells and its implications for Parkinson’s disease. JCI Insight. 2017, 2(12). [Epub ahead of print].
Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72(1):57–71.
Gibb WR, Lees AJ. The significance of the Lewy body in the diagnosis of idiopathic Parkinson's disease. Neuropathol Appl Neurobiol. 1989;15(1):27–44.
Colosimo C, Hughes AJ, Kilford L, Lees AJ. Lewy body cortical involvement may not always predict dementia in Parkinson's disease. J Neurol Neurosurg Psychiatry. 2003;74(7):852–6.
Gibb WR, Mountjoy CQ, Mann DM, Lees AJ. A pathological study of the association between Lewy body disease and Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1989;52(6):701–8.
Massano J, Bhatia KP. Clinical approach to Parkinson's disease: features, diagnosis, and principles of management. Cold Spring Harb Perspect Med. 2012;2(6):a008870.
Goetz CG, Poewe W, Rascol O, Sampaio C, Stebbins GT, Counsell C, Giladi N, Holloway RG, Moore CG, Wenning GK, et al. Movement Disorder Society task force report on the Hoehn and Yahr staging scale: status and recommendations. Mov Disord. 2004;19(9):1020–8.
Barua NU, Gill SS. Convection-enhanced drug delivery: prospects for glioblastoma treatment. CNS Oncol. 2014;3(5):313–6.
Jeon MY, Lee WY, Kang HY, Chung EJ. The effects of L-3,4-dihydroxyphenylalanine and dopamine agonists on dopamine neurons in the progressive hemiparkinsonian rat models. Neurol Res. 2007;29(3):289–95.
Barbeau A. L-dopa therapy in Parkinson's disease: a critical review of nine years' experience. Can Med Assoc J. 1969;101(13):59–68.
Foster HD, Hoffer A. The two faces of L-DOPA: benefits and adverse side effects in the treatment of encephalitis lethargica, Parkinson's disease, multiple sclerosis and amyotrophic lateral sclerosis. Med Hypotheses. 2004;62(2):177–81.
Cenci MA. Presynaptic mechanisms of l-DOPA-induced dyskinesia: the findings, the debate, and the therapeutic implications. Front Neurol. 2014;5:242.
Krishna R, Ali M, Moustafa AA. Effects of combined MAO-B inhibitors and levodopa vs. monotherapy in Parkinson’s disease. Front Aging Neurosci. 2014;6:180.
Riederer P, Lachenmayer L, Laux G. Clinical applications of MAO-inhibitors. Curr Med Chem. 2004;11(15):2033–43.
Korczyn AD. Drug treatment of Parkinson's disease. Dialogues Clin Neurosci. 2004;6(3):315–22.
Antonini A, Abbruzzese G, Barone P, Bonuccelli U, Lopiano L, Onofrj M, Zappia M, Quattrone A. COMT inhibition with tolcapone in the treatment algorithm of patients with Parkinson's disease (PD): relevance for motor and non-motor features. Neuropsychiatr Dis Treat. 2008;4(1):1–9.
Brooks DJ. Dopamine agonists: their role in the treatment of Parkinson's disease. J Neurol Neurosurg Psychiatry. 2000;68(6):685–9.
Tintner R, Jankovic J. Dopamine agonists in Parkinson's disease. Expert Opin Investig Drugs. 2003;12(11):1803–20.
Katzenschlager R, Sampaio C, Costa J, Lees A. Anticholinergics for symptomatic management of Parkinson's disease. Cochrane Database Syst Rev. 2003;2:CD003735.
Jankovic J, Aguilar LG. Current approaches to the treatment of Parkinson’s disease. Neuropsychiatr Dis Treat. 2008;4(4):743–57.
Chen JJ, Marsh L. Anxiety in Parkinson’s disease: identification and management. Ther Adv Neurol Disord. 2014;7(1):52–9.
Durif F, Debilly B, Galitzky M, Morand D, Viallet F, Borg M, Thobois S, Broussolle E, Rascol O. Clozapine improves dyskinesias in Parkinson disease: a double-blind, placebo-controlled study. Neurology. 2004;62(3):381–8.
Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68(5):568–78.
Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron. 2005;46(6):857–68.
George S, Brundin P. Immunotherapy in Parkinson’s disease: micromanaging alpha-Synuclein aggregation. J Park Dis. 2015;5(3):413–24.
Groiss SJ, Wojtecki L, Sudmeyer M, Schnitzler A. Deep brain stimulation in Parkinson’s disease. Ther Adv Neurol Disord. 2009;2(6):20–8.
Bronstein JM, Tagliati M, Alterman RL, Lozano AM, Volkmann J, Stefani A, Horak FB, Okun MS, Foote KD, Krack P, et al. Deep brain stimulation for Parkinson disease: an expert consensus and review of key issues. Arch Neurol. 2011;68(2):165.
Deuschl G, Paschen S, Witt K. Clinical outcome of deep brain stimulation for Parkinson’s disease. Handb Clin Neurol. 2013;116:107–28.
de Souza RM, Moro E, Lang AE, Schapira AH. Timing of deep brain stimulation in Parkinson disease: a need for reappraisal? Ann Neurol. 2013;73(5):565–75.
Iacono RP, Henderson JM, Lonser RR. Combined stereotactic thalamotomy and posteroventral pallidotomy for Parkinson’s disease. J Image Guid Surg. 1995;1(3):133–40.
Goodarzi P, Aghayan HR, Larijani B, Soleimani M, Dehpour AR, Sahebjam M, Ghaderi F, Arjmand B. Stem cell-based approach for the treatment of Parkinson’s disease. Med J Islam Repub Iran. 2015;29:168.
Kim JH, Auerbach JM, Rodriguez-Gomez JA, Velasco I, Gavin D, Lumelsky N, Lee SH, Nguyen J, Sanchez-Pernaute R, Bankiewicz K, et al. Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson’s disease. Nature. 2002;418(6893):50–6.
Kim HJ. Stem cell potential in Parkinson’s disease and molecular factors for the generation of dopamine neurons. Biochim Biophys Acta. 2011;1812(1):1–11.
Oh SM, Chang MY, Song JJ, Rhee YH, Joe EH, Lee HS, Yi SH, Lee SH. Combined Nurr1 and Foxa2 roles in the therapy of Parkinson’s disease. EMBO Mol Med. 2015;7(5):510–25.
Fu MH, Li CL, Lin HL, Chen PC, Calkins MJ, Chang YF, Cheng PH, Yang SH. Stem cell transplantation therapy in Parkinson’s disease. SpringerPlus. 2015;4:597.
Lindvall O, Bjorklund A. Cell therapeutics in Parkinson’s disease. Neurotherapeutics. 2011;8(4):539–48.
Fitzpatrick KM, Raschke J, Emborg ME. Cell-based therapies for Parkinson's disease: past, present, and future. Antioxid Redox Signal. 2009;11(9):2189–208.
Freed CR, Breeze RE, Rosenberg NL, Schneck SA, Kriek E, Qi JX, Lone T, Zhang YB, Snyder JA, Wells TH, et al. Survival of implanted fetal dopamine cells and neurologic improvement 12 to 46 months after transplantation for Parkinson's disease. N Engl J Med. 1992;327(22):1549–55.
Hauser RA, Freeman TB, Snow BJ, Nauert M, Gauger L, Kordower JH, Olanow CW. Long-term evaluation of bilateral fetal nigral transplantation in Parkinson disease. Arch Neurol. 1999;56(2):179–87.
Isacson O, Deacon TW, Pakzaban P, Galpern WR, Dinsmore J, Burns LH. Transplanted xenogeneic neural cells in neurodegenerative disease models exhibit remarkable axonal target specificity and distinct growth patterns of glial and axonal fibres. Nat Med. 1995;1(11):1189–94.
Politis M, Lindvall O. Clinical application of stem cell therapy in Parkinson’s disease. BMC Med. 2012;10:1.
Belin AC, Westerlund M. Parkinson's disease: a genetic perspective. FEBS J. 2008;275(7):1377–83.
d’Anglemont de Tassigny X, Pascual A, Lopez-Barneo J. GDNF-based therapies, GDNF-producing interneurons, and trophic support of the dopaminergic nigrostriatal pathway. Implications for Parkinson’s disease. Front Neuroanat. 2015;9:10.
Allen SJ, Watson JJ, Shoemark DK, Barua NU, Patel NK. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol Ther. 2013;138(2):155–75.
Kirik D, Cederfjall E, Halliday G, Petersen A. Gene therapy for Parkinson's disease: disease modification by GDNF family of ligands. Neurobiol Dis. 2017;97(Pt B):179–88.
Eberling JL, Kells AP, Pivirotto P, Beyer J, Bringas J, Federoff HJ, Forsayeth J, Bankiewicz KS. Functional effects of AAV2-GDNF on the dopaminergic nigrostriatal pathway in parkinsonian rhesus monkeys. Hum Gene Ther. 2009;20(5):511–8.
LeWitt PA, Rezai AR, Leehey MA, Ojemann SG, Flaherty AW, Eskandar EN, Kostyk SK, Thomas K, Sarkar A, Siddiqui MS, et al. AAV2-GAD gene therapy for advanced Parkinson's disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 2011;10(4):309–19.
Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, Bland RJ, Young D, Strybing K, Eidelberg D, et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial. Lancet. 2007;369(9579):2097–105.
Jarraya B, Boulet S, Ralph GS, Jan C, Bonvento G, Azzouz M, Miskin JE, Shin M, Delzescaux T, Drouot X, et al. Dopamine gene therapy for Parkinson’s disease in a nonhuman primate without associated dyskinesia. Sci Transl Med. 2009;1(2):2ra4.
Liu YY, Yang XY, Li Z, Liu ZL, Cheng D, Wang Y, Wen XJ, Hu JY, Liu J, Wang LM, et al. Characterization of polyethylene glycol-polyethyleneimine as a vector for alpha-synuclein siRNA delivery to PC12 cells for Parkinson's disease. CNS Neurosci Ther. 2014;20(1):76–85.
Kolli N, Lu M, Maiti P, Rossignol J, Dunbar GL. Application of the gene editing tool, CRISPR-Cas9, for treating neurodegenerative diseases. Neurochem Int. 2017. [Epub ahead of print].
Kolli N, Lu M, Maiti P, Rossignol J, Dunbar GL. CRISPR-Cas9 Mediated Gene-Silencing of the Mutant Huntingtin Gene in an In Vitro Model of Huntington’s Disease. Int J Mol Sci. 2017, 18(4).
Basu S, Adams L, Guhathakurta S, Kim YS. A novel tool for monitoring endogenous alpha-synuclein transcription by NanoLuciferase tag insertion at the 3'end using CRISPR-Cas9 genome editing technique. Sci Rep. 2017;8:45883.
Rangasamy SB, Soderstrom K, Bakay RA, Kordower JH. Neurotrophic factor therapy for Parkinson’s disease. Prog Brain Res. 2010;184:237–64.
Razgado-Hernandez LF, Espadas-Alvarez AJ, Reyna-Velazquez P, Sierra-Sanchez A, Anaya-Martinez V, Jimenez-Estrada I, Bannon MJ, Martinez-Fong D, Aceves-Ruiz J. The transfection of BDNF to dopamine neurons potentiates the effect of dopamine d3 receptor agonist recovering the striatal innervation, dendritic spines and motor behavior in an aged rat model of Parkinson's disease. PLoS One. 2015;10(2):e0117391.
Yang F, Liu Y, Tu J, Wan J, Zhang J, Wu B, Chen S, Zhou J, Mu Y, Wang L. Activated astrocytes enhance the dopaminergic differentiation of stem cells and promote brain repair through bFGF. Nat Commun. 2014;5:5627.
Seidl SE, Santiago JA, Bilyk H, Potashkin JA. The emerging role of nutrition in Parkinson’s disease. Front Aging Neurosci. 2014;6:36.
Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201–4.
Johnson JB, Summer W, Cutler RG, Martin B, Hyun DH, Dixit VD, Pearson M, Nassar M, Telljohann R, Maudsley S, et al. Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic Biol Med. 2007;42(5):665–74.
Mythri RB, Bharath MM. Curcumin: a potential neuroprotective agent in Parkinson's disease. Curr Pharm Des. 2012;18(1):91–9.
Hu S, Maiti P, Ma Q, Zuo X, Jones MR, Cole GM, Frautschy SA. Clinical development of curcumin in neurodegenerative disease. Expert Rev Neurother. 2015;15(6):629–37.
Pan J, Li H, Ma JF, Tan YY, Xiao Q, Ding JQ, Chen SD. Curcumin inhibition of JNKs prevents dopaminergic neuronal loss in a mouse model of Parkinson's disease through suppressing mitochondria dysfunction. Transl Neurodegeneration. 2012;1(1):16.
Pahan P, Pahan K. Can cinnamon bring aroma in Parkinson's disease treatment? Neural Regen Res. 2015;10(1):30–2.
Khasnavis S, Pahan K. Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson's disease. J Neuroimmune Pharmacol. 2014;9(4):569–81.
Petzinger GM, Fisher BE, McEwen S, Beeler JA, Walsh JP, Jakowec MW. Exercise-enhanced neuroplasticity targeting motor and cognitive circuitry in Parkinson's disease. Lancet Neurol. 2013;12(7):716–26.
Shu HF, Yang T, Yu SX, Huang HD, Jiang LL, Gu JW, Kuang YQ. Aerobic exercise for Parkinson's disease: a systematic review and meta-analysis of randomized controlled trials. PLoS One. 2014;9(7):e100503.
Salgado S, Williams N, Kotian R, Salgado M. An evidence-based exercise regimen for patients with mild to moderate Parkinson's disease. Brain Sci. 2013;3(1):87–100.
Morgan JA, Corrigan F, Baune BT. Effects of physical exercise on central nervous system functions: a review of brain region specific adaptations. J Mol Psychiatry. 2015;3(1):3.
de Dreu MJ, van der Wilk AS, Poppe E, Kwakkel G, van Wegen EE. Rehabilitation, exercise therapy and music in patients with Parkinson's disease: a meta-analysis of the effects of music-based movement therapy on walking ability, balance and quality of life. Parkinsonism Relat Disord. 2012;18(Suppl 1):S114–9.
Morris ME, Martin CL, Schenkman ML. Striding out with Parkinson disease: evidence-based physical therapy for gait disorders. Phys Ther. 2010;90(2):280–8.
Antonini A, DeNotaris R. PET and SPECT functional imaging in Parkinson's disease. Sleep Med. 2004;5(2):201–6.
Wang L, Zhang Q, Li H, Zhang H. SPECT molecular imaging in Parkinson's disease. J Biomed Biotechnol. 2012;2012:412486.
Brooks DJ. Neuroimaging in Parkinson’s disease. NeuroRx. 2004;1(2):243–54.
Brooks DJ. Imaging approaches to Parkinson disease. J Nuclear Med. 2010;51(4):596–609.
Pyatigorskaya N, Gallea C, Garcia-Lorenzo D, Vidailhet M, Lehericy S. A review of the use of magnetic resonance imaging in Parkinson's disease. Ther Adv Neurol Disord. 2014;7(4):206–20.
Haas BR, Stewart TH, Zhang J. Premotor biomarkers for Parkinson's disease - a promising direction of research. Transl Neurodegeneration. 2012;1(1):11.
Wu Y, Le W, Jankovic J. Preclinical biomarkers of Parkinson disease. Arch Neurol. 2011;68(1):22–30.
Sharma S, Moon CS, Khogali A, Haidous A, Chabenne A, Ojo C, Jelebinkov M, Kurdi Y, Ebadi M. Biomarkers in Parkinson's disease (recent update). Neurochem Int. 2013;63(3):201–29.
Polymeropoulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, Di Iorio G, et al. Mapping of a gene for Parkinson's disease to chromosome 4q21-q23. Science. 1996;274(5290):1197–9. Epub 1996/11/15. PubMed PMID: 8895469
Fujioka S, Ogaki K, Tacik PM, Uitti RJ, Ross OA, Wszolek ZK. Update on novel familial forms of Parkinson's disease and multiple system atrophy. Parkinsonism Relat Disord. 2014;20(Suppl 1):S29–34. Epub 2013/11/23. doi: 10.1016/S1353-8020(13)70010-5. PubMed PMID: 24262183; PubMed Central PMCID: PMCPMC4215194
Si X, Pu J, Zhang B. Structure, distribution, and genetic profile of alpha-Synuclein and their potential clinical application in Parkinson’s disease. J Mov Dis. 2017;10(2):69–79. Epub 2017/05/10. doi: 10.14802/jmd.16061. PubMed PMID: 28479587; PubMed Central PMCID: PMCPMC5435834
Campelo C, Silva RH. Genetic variants in SNCA and the risk of sporadic Parkinson’s disease and clinical outcomes: a review. Park Dis. 2017;2017:4318416. Epub 2017/08/07. doi: 10.1155/2017/4318416. PubMed PMID: 28781905; PubMed Central PMCID: PMCPMC5525082
Davis AA, Andruska KM, Benitez BA, Racette BA, Perlmutter JS, Cruchaga C. Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression. Neurobiol Aging. 2016;37:209 e1–7. Epub 2015/11/26. doi: 10.1016/j.neurobiolaging.2015.09.014. PubMed PMID: 26601739; PubMed Central PMCID: PMCPMC4688052
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392(6676):605–608. Epub 1998/04/29. doi: 10.1038/33416. PubMed PMID: 9560156.
Bouhouche A, Tibar H, Ben El Haj R, El Bayad K, Razine R, Tazrout S, et al. LRRK2 G2019S mutation: prevalence and clinical features in Moroccans with Parkinson’s disease. Park Dis. 2017;2017:2412486. Epub 2017/05/04. doi: 10.1155/2017/2412486. PubMed PMID: 28465860; PubMed Central PMCID: PMCPMC5390546
Gasser T, Muller-Myhsok B, Wszolek ZK, Oehlmann R, Calne DB, Bonifati V, et al. A susceptibility locus for Parkinson’s disease maps to chromosome 2p13. Nat Genet 1998;18(3):262–265. Epub 1998/03/21. doi: 10.1038/ng0398-262. PubMed PMID: 9500549.
Lahut S, Gispert S, Omur O, Depboylu C, Seidel K, Dominguez-Bautista JA, et al. Blood RNA biomarkers in prodromal PARK4 and rapid eye movement sleep behavior disorder show role of complexin 1 loss for risk of Parkinson’s disease. Dis Model Mech. 2017;10(5):619–31. Epub 2017/01/22. doi: 10.1242/dmm.028035. PubMed PMID: 28108469; PubMed Central PMCID: PMCPMC5451169
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997;276(5321):2045–2047. Epub 1997/06/27. PubMed PMID: 9197268.
Mouton-Liger F, Jacoupy M, Corvol JC, Corti O. PINK1/Parkin-dependent mitochondrial surveillance: from pleiotropy to Parkinson’s disease. Front Mol Neurosci. 2017;10:120. Epub 2017/05/17. doi: 10.3389/fnmol.2017.00120. PubMed PMID: 28507507; PubMed Central PMCID: PMCPMC5410576
Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998;395(6701):451–452. Epub 1998/10/17. doi: 10.1038/26652. PubMed PMID: 9774100.
Wilkinson KD, Lee KM, Deshpande S, Duerksen-Hughes P, Boss JM, Pohl J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science. 1989;246(4930):670–3. Epub 1989/11/03. PubMed PMID: 2530630
Lowe J, McDermott H, Landon M, Mayer RJ, Wilkinson KD. Ubiquitin carboxyl-terminal hydrolase (PGP 9.5) is selectively present in ubiquitinated inclusion bodies characteristic of human neurodegenerative diseases. J Pathol 1990;161(2):153–160. Epub 1990/06/01. doi: 10.1002/path.1711610210. PubMed PMID: 2166150.
Chen S, Annesley SJ, Jasim RAF, Musco VJ, Sanislav O, Fisher PR. The Parkinson’s disease-associated protein DJ-1 plays a positive nonmitochondrial role in endocytosis in Dictyostelium cells. Dis Model Mech 2017. Epub 2017/08/19. doi: 10.1242/dmm.028084. PubMed PMID: 28819044.
Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004;44(4):595–600. Epub 2004/11/16. doi: 10.1016/j.neuron.2004.10.023. PubMed PMID: 15541308.
Galter D, Westerlund M, Carmine A, Lindqvist E, Sydow O, Olson L. LRRK2 expression linked to dopamine-innervated areas. Ann Neurol. 2006;59(4):714–9. Epub 2006/03/15. doi: 10.1002/ana.20808. PubMed PMID: 16532471
Xiong Y, Neifert S, Karuppagounder SS, Stankowski JN, Lee BD, Grima JC, et al. Overexpression of Parkinson’s disease-associated mutation LRRK2 G2019S in mouse forebrain induces behavioral deficits and alpha-Synuclein pathology. eNeuro. 2017;4(2) Epub 2017/03/23. doi: 10.1523/ENEURO.0004-17.2017. PubMed PMID: 28321439; PubMed Central PMCID: PMCPMC5355896.
Hampshire DJ, Roberts E, Crow Y, Bond J, Mubaidin A, Wriekat AL, et al. Kufor-Rakeb syndrome, pallido-pyramidal degeneration with supranuclear upgaze paresis and dementia, maps to 1p36. J Med Genet. 2001;38(10):680–2. Epub 2001/10/05. PubMed PMID: 11584046; PubMed Central PMCID: PMCPMC1734748
Najim Al-din AS, Wriekat A, Mubaidin A, Dasouki M, Hiari M. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol Scand. 1994;89(5):347–52. Epub 1994/05/01. PubMed PMID: 8085432
Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. 2007;68(19):1557–62. Epub 2007/05/09. doi: 10.1212/01.wnl.0000260963.08711.08. PubMed PMID: 17485642
Hicks AA, Petursson H, Jonsson T, Stefansson H, Johannsdottir HS, Sainz J, et al. A susceptibility gene for late-onset idiopathic Parkinson’s disease. Ann Neurol. 2002;52(5):549–55. Epub 2002/10/29. doi: 10.1002/ana.10324. PubMed PMID: 12402251
Pankratz N, Nichols WC, Uniacke SK, Halter C, Rudolph A, Shults C, et al. Significant linkage of Parkinson disease to chromosome 2q36-37. Am J Hum Genet. 2003;72(4):1053–7. Epub 2003/03/15. doi: 10.1086/374383. PubMed PMID: 12638082; PubMed Central PMCID: PMCPMC1180337
Pankratz N, Nichols WC, Uniacke SK, Halter C, Rudolph A, Shults C, et al. Genome screen to identify susceptibility genes for Parkinson disease in a sample without parkin mutations. Am J Hum Genet. 2002;71(1):124–35. Epub 2002/06/12. doi: 10.1086/341282. PubMed PMID: 12058349; PubMed Central PMCID: PMCPMC384969
Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum Mol Genet. 2005;14(15):2099–111. doi: 10.1093/hmg/ddi215. Epub 2005/06/18. PubMed PMID: 15961413
He YC, Huang P, Li QQ, Sun Q, Li DH, Wang T, et al. Mutation analysis of HTRA2 gene in Chinese familial essential tremor and familial Parkinson’s disease. Park Dis. 2017;2017:3217474. Epub 2017/03/01. doi: 10.1155/2017/3217474. PubMed PMID: 28243480; PubMed Central PMCID: PMCPMC5294371
Wang KS, Mullersman JE, Liu XF. Family-based association analysis of the MAPT gene in Parkinson disease. J Appl Genet. 2010;51(4):509–14. Epub 2010/11/11. PubMed PMID: 21063069
O’Regan G, de Souza RM, Balestrino R, Schapira AH. Glucocerebrosidase mutations in Parkinson disease. J Park Dis. 2017;7(3):411–22. Epub 2017/06/10. doi: 10.3233/JPD-171092. PubMed PMID: 28598856
Gubellini P, Kachidian P. Animal models of Parkinson’s disease: an updated overview. Rev Neurol. 2015;171(11):750–61. 10.1016/j.neurol.2015.07.011. PubMed PMID: 26343921
Ebrahimi-Fakhari D, Wahlster L, McLean PJ. Molecular chaperones in Parkinson’s disease--present and future. J Park Dis. 2011;1(4):299–320. PubMed PMID: 22279517; PubMed Central PMCID: PMC3264060
Zourlidou A, Payne Smith MD, Latchman DS. HSP27 but not HSP70 has a potent protective effect against alpha-synuclein-induced cell death in mammalian neuronal cells. J Neurochem. 2004;88(6):1439–48. PubMed PMID: 15009645
Friesen EL, De Snoo ML, Rajendran L, Kalia LV, Kalia SK. Chaperone-based therapies for disease modification in Parkinson’s disease. Park Dis. 2017;2017:5015307. 10.1155/2017/5015307. Epub 2017/09/16. PubMed PMID: 28913005; PubMed Central PMCID: PMCPMC5585656
Feng M, Zhang L, Liu Z, Zhou P, Lu X. The expression and release of Hsp60 in 6-OHDA induced in vivo and in vitro models of Parkinson's disease. Neurochem Res. 2013;38(10):2180–9. 10.1007/s11064-013-1127-8. PubMed PMID: 23943523
Witt SN. Hsp70 molecular chaperones and Parkinson's disease. Biopolymers. 2010;93(3):218–28. 10.1002/bip.21302. PubMed PMID: 19768775
Nillegoda NB, Kirstein J, Szlachcic A, Berynskyy M, Stank A, Stengel F, et al. Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature. 2015;524(7564):247–51. 10.1038/nature14884. Epub 2015/08/08. PubMed PMID: 26245380; PubMed Central PMCID: PMCPMC4830470
Uryu K, Richter-Landsberg C, Welch W, Sun E, Goldbaum O, Norris EH, et al. Convergence of heat shock protein 90 with ubiquitin in filamentous alpha-synuclein inclusions of alpha-synucleinopathies. Am J Pathol. 2006;168(3):947–61. PubMed PMID: 16507910; PubMed Central PMCID: PMC1606542
Jackrel ME, Shorter J. Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease-linked proteins. Dis Model Mech. 2014;7(10):1175–84. 10.1242/dmm.016113. Epub 2014/07/27. PubMed PMID: 25062688; PubMed Central PMCID: PMCPMC4174528
Huang D, Xu J, Wang J, Tong J, Bai X, Li H, et al. Dynamic changes in the nigrostriatal pathway in the MPTP mouse model of Parkinson’s disease. Park Dis. 2017;2017:9349487. 10.1155/2017/9349487. Epub 2017/08/24. PubMed PMID: 28831326; PubMed Central PMCID: PMCPMC5555011
Schmidt WJ, Alam M. Controversies on new animal models of Parkinson’s disease pro and con: the rotenone model of Parkinson’s disease (PD). J Neural Transm Suppl. 2006;70:273–6.
Vaccari C, El Dib R, de Camargo JLV. Paraquat and Parkinson’s disease: a systematic review protocol according to the OHAT approach for hazard identification. Syst Rev. 2017;6(1):98. 10.1186/s13643-017-0491-x. Epub 2017/05/17. PubMed PMID: 28506248; PubMed Central PMCID: PMCPMC5433017
Richter F, Gabby L, McDowell KA, Mulligan CK, De La Rosa K, Sioshansi PC, et al. Effects of decreased dopamine transporter levels on nigrostriatal neurons and paraquat/maneb toxicity in mice. Neurobiol Aging. 2017;51:54–66. 10.1016/j.neurobiolaging.2016.11.015. Epub 2016/12/31.PubMed PMID: 28038352; PubMed Central PMCID: PMCPMC5292275
Atanasov AG, Tam S, Rocken JM, Baker ME, Odermatt A. Inhibition of 11 beta-hydroxysteroid dehydrogenase type 2 by dithiocarbamates. Biochem Biophys Res Commun. 2003;308(2):257–62. PubMed PMID: 12901862
Takahashi RN, Rogerio R, Zanin M. Maneb enhances MPTP neurotoxicity in mice. Res Commun Chem Pathol Pharmacol. 1989;66(1):167–70. PubMed PMID: 2616897
Jia Z, Misra HP. Exposure to mixtures of endosulfan and zineb induces apoptotic and necrotic cell death in SH-SY5Y neuroblastoma cells, in vitro. J Appl Toxicol JAT. 2007;27(5):434–46. 10.1002/jat.1218. PubMed PMID: 17309119
Goldman SM. Environmental toxins and Parkinson's disease. Annu Rev Pharmacol Toxicol. 2014;54:141–64. 10.1146/annurev-pharmtox-011613-135937. PubMed PMID: 24050700
Rizzati V, Briand O, Guillou H, Gamet-Payrastre L. Effects of pesticide mixtures in human and animal models: an update of the recent literature. Chem Biol Interact. 2016;254:231–46. 10.1016/j.cbi.2016.06.003. Epub 2016/06/18. PubMed PMID: 27312199
Chou AP, Maidment N, Klintenberg R, Casida JE, Li S, Fitzmaurice AG, et al. Ziram causes dopaminergic cell damage by inhibiting E1 ligase of the proteasome. J Biol Chem. 2008;283(50):34696–703. 10.1074/jbc.M802210200. PubMed PMID: 18818210; PubMed Central PMCID: PMC2596383
Lee CC, Peters PJ. Neurotoxicity and behavioral effects of thiram in rats. Environ Health Perspect. 1976;17:35–43. PubMed PMID: 1026416; PubMed Central PMCID: PMC1475264
Periquet A, Derache R. Toxicity of the pesticide nabam as a function of dietary protein content in the rat. Toxicol Eur Res Recherche Europeenne Toxicologie. 1978;1(1):27–37. PubMed PMID: 741467
Ungerstedt U, Ljungberg T, Steg G. Behavioral, physiological, and neurochemical changes after 6-hydroxydopamine-induced degeneration of the nigro-striatal dopamine neurons. Adv Neurol. 1974;5:421–6. Epub 1974/01/01. PubMed PMID: 4531217
Anderson G, Noorian AR, Taylor G, Anitha M, Bernhard D, Srinivasan S, et al. Loss of enteric dopaminergic neurons and associated changes in colon motility in an MPTP mouse model of Parkinson’s disease. Exp Neurol. 2007;207(1):4–12. 10.1016/j.expneurol.2007.05.010. Epub 2007/06/26. PubMed PMID: 17586496; PubMed Central PMCID: PMCPMC2277100
Bezard E, Dovero S, Bioulac B, Gross CE. Kinetics of nigral degeneration in a chronic model of MPTP-treated mice. Neurosci Lett. 1997;234(1):47–50. Epub 1997/11/05. PubMed PMID: 9347943
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3(12):1301–6. 10.1038/81834. Epub 2000/12/02. PubMed PMID: 11100151
Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem. 2002;277(3):1641–4. 10.1074/jbc.C100560200. Epub 2001/11/15. PubMed PMID: 11707429
Thiruchelvam M, Richfield EK, Baggs RB, Tank AW, Cory-Slechta DA. The nigrostriatal dopaminergic system as a preferential target of repeated exposures to combined paraquat and maneb: implications for Parkinson's disease. J Neurosci. 2000;20(24):9207–14. Epub 2000/01/11. PubMed PMID: 11124998
Gonzalez-Reyes LE, Verbitsky M, Blesa J, Jackson-Lewis V, Paredes D, Tillack K, et al. Sonic hedgehog maintains cellular and neurochemical homeostasis in the adult nigrostriatal circuit. Neuron. 2012;75(2):306–19. 10.1016/j.neuron.2012.05.018. Epub 2012/07/31. PubMed PMID: 22841315; PubMed Central PMCID: PMCPMC3408586
Jankovic J, Chen S, Le WD. The role of Nurr1 in the development of dopaminergic neurons and Parkinson's disease. Prog Neurobiol. 2005;77(1–2):128–38. 10.1016/j.pneurobio.2005.09.001. Epub 2005/10/26. PubMed PMID: 16243425
Hwang DY, Fleming SM, Ardayfio P, Moran-Gates T, Kim H, Tarazi FI, et al. 3,4-dihydroxyphenylalanine reverses the motor deficits in Pitx3-deficient aphakia mice: behavioral characterization of a novel genetic model of Parkinson's disease. J Neurosci. 2005;25(8):2132–7. 10.1523/JNEUROSCI.3718-04.2005. Epub 2005/02/25. PubMed PMID: 15728853
Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A. 2007;104(4):1325–30. 10.1073/pnas.0605208103. Epub 2007/01/18. PubMed PMID: 17227870; PubMed Central PMCID: PMCPMC1783140
Ekstrand MI, Galter D. The MitoPark mouse - an animal model of Parkinson's disease with impaired respiratory chain function in dopamine neurons. Parkinsonism Relat Disord. 2009;15(Suppl 3):S185–8. 10.1016/S1353-8020(09)70811-9. Epub 2010/01/30. PubMed PMID: 20082987
Yacoubian TA, Standaert DG. Targets for neuroprotection in Parkinson’s disease. Biochim Biophys Acta. 2009;1792(7):676–87. 10.1016/j.bbadis.2008.09.009. Epub 2008/10/22. PubMed PMID: 18930814; PubMed Central PMCID: PMCPMC2740981
Dawson TM, Dawson VL. The role of parkin in familial and sporadic Parkinson’s disease. Mov Disord. 2010;25(Suppl 1):S32–9. 10.1002/mds.22798. Epub 2010/02/27. PubMed PMID: 20187240; PubMed Central PMCID: PMCPMC4115293
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–7. 10.1016/j.neuron.2004.11.005. Epub 2004/11/16. PubMed PMID: 15541309
Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, et al. A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet. 2005;365(9457):415–6. 10.1016/S0140-6736(05)17830-1. Epub 2005/02/01. PubMed PMID: 15680457
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158–60. 10.1126/science.1096284. Epub 2004/04/17. PubMed PMID: 15087508
Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A. 2008;105(32):11364–9. 10.1073/pnas.0802076105. Epub 2008/08/09. PubMed PMID: 18687901; PubMed Central PMCID: PMCPMC2516271
Hedrich K, Djarmati A, Schafer N, Hering R, Wellenbrock C, Weiss PH, et al. DJ-1 (PARK7) mutations are less frequent than Parkin (PARK2) mutations in early-onset Parkinson disease. Neurology. 2004;62(3):389–94. Epub 2004/02/12. PubMed PMID: 14872018
Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299(5604):256–9. 10.1126/science.1077209. Epub 2002/11/26. PubMed PMID: 12446870
Hashimoto M, Rockenstein E, Masliah E. Transgenic models of alpha-synuclein pathology: past, present, and future. Ann N Y Acad Sci. 2003;991:171–88. Epub 2003/07/09. PubMed PMID: 12846986
Koob AO, Ubhi K, Paulsson JF, Kelly J, Rockenstein E, Mante M, et al. Lovastatin ameliorates alpha-synuclein accumulation and oxidation in transgenic mouse models of alpha-synucleinopathies. Exp Neurol. 2010;221(2):267–74. 10.1016/j.expneurol.2009.11.015. Epub 2009/12/01. PubMed PMID: 19944097; PubMed Central PMCID: PMCPMC2812599
Winner B, Rockenstein E, Lie DC, Aigner R, Mante M, Bogdahn U, et al. Mutant alpha-synuclein exacerbates age-related decrease of neurogenesis. Neurobiol Aging. 2008;29(6):913–25. 10.1016/j.neurobiolaging.2006.12.016. Epub 2007/02/06. PubMed PMID: 17275140; PubMed Central PMCID: PMCPMC2896275
van der Putten H, Wiederhold KH, Probst A, Barbieri S, Mistl C, Danner S, et al. Neuropathology in mice expressing human alpha-synuclein. J Neurosci. 2000;20(16):6021–9. Epub 2000/08/10. PubMed PMID: 10934251
Frasier M, Walzer M, McCarthy L, Magnuson D, Lee JM, Haas C, et al. Tau phosphorylation increases in symptomatic mice overexpressing A30P alpha-synuclein. Exp Neurol. 2005;192(2):274–87. 10.1016/j.expneurol.2004.07.016. Epub 2005/03/10. PubMed PMID: 15755545
Freichel C, Neumann M, Ballard T, Muller V, Woolley M, Ozmen L, et al. Age-dependent cognitive decline and amygdala pathology in alpha-synuclein transgenic mice. Neurobiol Aging. 2007;28(9):1421–35. 10.1016/j.neurobiolaging.2006.06.013. Epub 2006/07/29. PubMed PMID: 16872721
Fernagut PO, Hutson CB, Fleming SM, Tetreaut NA, Salcedo J, Masliah E, et al. Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha-synuclein over-expression. Synapse. 2007;61(12):991–1001. 10.1002/syn.20456. Epub 2007/09/20. PubMed PMID: 17879265; PubMed Central PMCID: PMCPMC3097512
Zhou W, Milder JB, Freed CR. Transgenic mice overexpressing tyrosine-to-cysteine mutant human alpha-synuclein: a progressive neurodegenerative model of diffuse Lewy body disease. J Biol Chem. 2008;283(15):9863–70. 10.1074/jbc.M710232200. Epub 2008/02/02. PubMed PMID: 18238775
Ikeda M, Kawarabayashi T, Harigaya Y, Sasaki A, Yamada S, Matsubara E, et al. Motor impairment and aberrant production of neurochemicals in human alpha-synuclein A30P+A53T transgenic mice with alpha-synuclein pathology. Brain Res. 2009;1250:232–41. 10.1016/j.brainres.2008.10.011. Epub 2008/11/11. PubMed PMID: 18992718
Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, et al. Human alpha-synuclein-harboring familial Parkinson's disease-linked ala-53 --> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A. 2002;99(13):8968–73. 10.1073/pnas.132197599. Epub 2002/06/27. PubMed PMID: 12084935; PubMed Central PMCID: PMCPMC124407
Miller RM, Kiser GL, Kaysser-Kranich T, Casaceli C, Colla E, Lee MK, et al. Wild-type and mutant alpha-synuclein induce a multi-component gene expression profile consistent with shared pathophysiology in different transgenic mouse models of PD. Exp Neurol. 2007;204(1):421–32. 10.1016/j.expneurol.2006.12.005. Epub 2007/01/27. PubMed PMID: 17254569
Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci. 2008;28(30):7687–98. 10.1523/JNEUROSCI.0143-07.2008. Epub 2008/07/25. PubMed PMID: 18650345; PubMed Central PMCID: PMCPMC2702093
Graham DR, Sidhu A. Mice expressing the A53T mutant form of human alpha-synuclein exhibit hyperactivity and reduced anxiety-like behavior. J Neurosci Res. 2010;88(8):1777–83. 10.1002/jnr.22331. Epub 2010/01/16. PubMed PMID: 20077428; PubMed Central PMCID: PMCPMC2861296
Cabin DE, Gispert-Sanchez S, Murphy D, Auburger G, Myers RR, Nussbaum RL. Exacerbated synucleinopathy in mice expressing A53T SNCA on a SNCA null background. Neurobiol Aging. 2005;26(1):25–35. 10.1016/j.neurobiolaging.2004.02.026. Epub 2004/12/09. PubMed PMID: 15585343
Gureviciene I, Gurevicius K, Tanila H. Role of alpha-synuclein in synaptic glutamate release. Neurobiol Dis. 2007;28(1):83–9. 10.1016/j.nbd.2007.06.016. Epub 2007/08/11. PubMed PMID: 17689254
Gomez-Isla T, Irizarry MC, Mariash A, Cheung B, Soto O, Schrump S, et al. Motor dysfunction and gliosis with preserved dopaminergic markers in human alpha-synuclein A30P transgenic mice. Neurobiol Aging. 2003;24(2):245–58. Epub 2002/12/25. PubMed PMID: 12498958
Nieto M, Gil-Bea FJ, Dalfo E, Cuadrado M, Cabodevilla F, Sanchez B, et al. Increased sensitivity to MPTP in human alpha-synuclein A30P transgenic mice. Neurobiol Aging. 2006;27(6):848–56. 10.1016/j.neurobiolaging.2005.04.010. Epub 2005/07/12. PubMed PMID: 16006012
Chen L, Thiruchelvam MJ, Madura K, Richfield EK. Proteasome dysfunction in aged human alpha-synuclein transgenic mice. Neurobiol Dis. 2006;23(1):120–6. 10.1016/j.nbd.2006.02.004. Epub 2006/05/23. PubMed PMID: 16713278
Lim Y, Kehm VM, Li C, Trojanowski JQ, Lee VM. Forebrain overexpression of alpha-synuclein leads to early postnatal hippocampal neuron loss and synaptic disruption. Exp Neurol. 2010;221(1):86–97. 10.1016/j.expneurol.2009.10.005. Epub 2009/10/17. PubMed PMID: 19833127; PubMed Central PMCID: PMCPMC2812632
Marxreiter F, Nuber S, Kandasamy M, Klucken J, Aigner R, Burgmayer R, et al. Changes in adult olfactory bulb neurogenesis in mice expressing the A30P mutant form of alpha-synuclein. Eur J Neurosci. 2009;29(5):879–90. 10.1111/j.1460-9568.2009.06641.x. Epub 2009/03/18. PubMed PMID: 19291219
Plaas M, Karis A, Innos J, Rebane E, Baekelandt V, Vaarmann A, et al. Alpha-synuclein A30P point-mutation generates age-dependent nigrostriatal deficiency in mice. J Physiol Pharmacol. 2008;59(2):205–16. Epub 2008/07/16. PubMed PMID: 18622040
Kuo YM, Li Z, Jiao Y, Gaborit N, Pani AK, Orrison BM, et al. Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated alpha-synuclein gene mutations precede central nervous system changes. Hum Mol Genet. 2010;19(9):1633–50. 10.1093/hmg/ddq038. Epub 2010/01/29. PubMed PMID: 20106867; PubMed Central PMCID: PMCPMC2850613
Oertel WH. Recent advances in treating Parkinson’s disease. F1000Res. 2017;6:260. 10.12688/f1000research.10100.1. Epub 2017/03/31. PubMed PMID: 28357055; PubMed Central PMCID: PMCPMC5357034
Pan J, Cai H. Opioid system in L-DOPA-induced dyskinesia. Transl Neurodegeneration. 2017;6:1. 10.1186/s40035-017-0071-y. Epub 2017/01/21. PubMed PMID: 28105331; PubMed Central PMCID: PMCPMC5240307
Cereda E, Cilia R, Canesi M, Tesei S, Mariani CB, Zecchinelli AL, et al. Efficacy of rasagiline and selegiline in Parkinson’s disease: a head-to-head 3-year retrospective case-control study. J Neurol. 2017;264(6):1254–63. 10.1007/s00415-017-8523-y. Epub 2017/05/28. PubMed PMID: 28550482; PubMed Central PMCID: PMCPMC5570795
Mo JJ, Liu LY, Peng WB, Rao J, Liu Z, Cui LL. The effectiveness of creatine treatment for Parkinson’s disease: an updated meta-analysis of randomized controlled trials. BMC Neurol. 2017;17(1):105. 10.1186/s12883-017-0885-3. Epub 2017/06/05. PubMed PMID: 28577542; PubMed Central PMCID: PMCPMC5457735
Wang Y, Sun S, Zhu S, Liu C, Liu Y, Di Q, et al. The efficacy and safety of pramipexole ER versus IR in Chinese patients with Parkinson’s disease: a randomized, double-blind, double-dummy, parallel-group study. Transl Neurodegeneration. 2014;3:11. 10.1186/2047-9158-3-11. Epub 2014/08/13. PubMed PMID: 25114789; PubMed Central PMCID: PMCPMC4128609
Park J, Kim Y, Youn J, Lee PH, Sohn YH, Koh SB, et al. Levodopa dose maintenance or reduction in patients with Parkinson's disease transitioning to levodopa/carbidopa/entacapone. Neurol India. 2017;65(4):746–51. 10.4103/neuroindia.NI_597_16. Epub 2017/07/07. PubMed PMID: 28681744
Pahwa R, Tanner CM, Hauser RA, Isaacson SH, Nausieda PA, Truong DD, et al. ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson disease (EASE LID study): a randomized clinical trial. JAMA Neurol. 2017;74(8):941–9. 10.1001/jamaneurol.2017.0943. Epub 2017/06/13. PubMed PMID: 28604926
Moore AH, Bigbee MJ, Boynton GE, Wakeham CM, Rosenheim HM, Staral CJ, et al. Non-steroidal anti-inflammatory drugs in Alzheimer’s disease and Parkinson’s disease: reconsidering the role of Neuroinflammation. Pharmaceuticals (Basel). 2010;3(6):1812–41. 10.3390/ph3061812. Epub 2010/06/02. PubMed PMID: 27713331; PubMed Central PMCID: PMCPMC4033954
Duits JH, Ongering MS, Martens HJM, Schulte PFJ. Pimavanserin: a new treatment for the Parkinson's disease psychosis. Tijdschr Psychiatr. 2017;59(9):528–36. Epub 2017/09/08. PubMed PMID: 28880354
Schneider JS, Seyfried TN, Choi HS, Kidd SK. Intraventricular Sialidase administration enhances GM1 ganglioside expression and is partially neuroprotective in a mouse model of Parkinson’s disease. PLoS One. 2015;10(12):e0143351. 10.1371/journal.pone.0143351. Epub 2015/12/03. PubMed PMID: 26629687; PubMed Central PMCID: PMCPMC4668049
Farah A. Atypicality of atypical antipsychotics. Prim Care Companion J Clin Psychiatry. 2005;7(6):268–74. Epub 2006/02/25. PubMed PMID: 16498489; PubMed Central PMCID: PMCPMC1324958
de la Mata M, Cotan D, Oropesa-Avila M, Villanueva-Paz M, de Lavera I, Alvarez-Cordoba M, et al. Coenzyme Q10 partially restores pathological alterations in a macrophage model of Gaucher disease. Orphanet J Rare Dis. 2017;12(1):23. 10.1186/s13023-017-0574-8. Epub 2017/02/09. PubMed PMID: 28166796; PubMed Central PMCID: PMCPMC5292786
Zhu ZG, Sun MX, Zhang WL, Wang WW, Jin YM, Xie CL. The efficacy and safety of coenzyme Q10 in Parkinson's disease: a meta-analysis of randomized controlled trials. Neurol Sci. 2017;38(2):215–24. 10.1007/s10072-016-2757-9. Epub 2016/11/11. PubMed PMID: 27830343
Di Rocco A, Rogers JD, Brown R, Werner P, Bottiglieri T. S-Adenosyl-methionine improves depression in patients with Parkinson's disease in an open-label clinical trial. Mov Disord. 2000;15(6):1225–9. Epub 2000/12/05. PubMed PMID: 11104210
Grunig D, Felser A, Bouitbir J, Krahenbuhl S. The catechol-O-methyltransferase inhibitors tolcapone and entacapone uncouple and inhibit the mitochondrial respiratory chain in HepaRG cells. Toxicol in Vitro. 2017;42:337–47. 10.1016/j.tiv.2017.05.013. Epub 2017/05/21. PubMed PMID: 28526448
Phan JA, Stokholm K, Zareba-Paslawska J, Jakobsen S, Vang K, Gjedde A, et al. Early synaptic dysfunction induced by alpha-synuclein in a rat model of Parkinson’s disease. Sci Rep. 2017;7(1):6363. 10.1038/s41598-017-06724-9. Epub 2017/07/27. PubMed PMID: 28743955; PubMed Central PMCID: PMCPMC5526979
Magen I, Torres ER, Dinh D, Chung A, Masliah E, Chesselet MF. Social cognition impairments in mice overexpressing alpha-Synuclein under the Thy1 promoter, a model of pre-manifest Parkinson's disease. J Park Dis. 2015;5(3):669–80. doi: 10.3233/JPD-140503. Epub 2015/01/16. PubMed PMID: 25588356.
Tagliafierro L, Chiba-Falek O. Up-regulation of SNCA gene expression: implications to synucleinopathies. Neurogenetics. 2016;17(3):145–57. 10.1007/s10048-016-0478-0. Epub 2016/03/08. PubMed PMID: 26948950; PubMed Central PMCID: PMCPMC4907864
Blumenstock S, Rodrigues EF, Peters F, Blazquez-Llorca L, Schmidt F, Giese A, et al. Seeding and transgenic overexpression of alpha-synuclein triggers dendritic spine pathology in the neocortex. EMBO Mol Med. 2017;9(5):716–31. 10.15252/emmm.201607305. Epub 2017/03/30. PubMed PMID: 28351932; PubMed Central PMCID: PMCPMC5412764
Delenclos M, Carrascal L, Jensen K, Romero-Ramos M. Immunolocalization of human alpha-synuclein in the Thy1-aSyn (“line 61”) transgenic mouse line. Neuroscience. 2014;277:647–64. 10.1016/j.neuroscience.2014.07.042. Epub 2014/08/06. PubMed PMID: 25090921
Valera E, Masliah E. Immunotherapy for neurodegenerative diseases: focus on alpha-synucleinopathies. Pharmacol Ther. 2013;138(3):311–22. 10.1016/j.pharmthera.2013.01.013. Epub 2013/02/07. PubMed PMID: 23384597; PubMed Central PMCID: PMCPMC3925783
Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338(6109):949–53. 10.1126/science.1227157. Epub 2012/11/20. PubMed PMID: 23161999; PubMed Central PMCID: PMCPMC3552321
Shahaduzzaman M, Nash K, Hudson C, Sharif M, Grimmig B, Lin X, et al. Anti-human alpha-synuclein N-terminal peptide antibody protects against dopaminergic cell death and ameliorates behavioral deficits in an AAV-alpha-synuclein rat model of Parkinson's disease. PLoS One. 2015;10(2):e0116841. 10.1371/journal.pone.0116841. Epub 2015/02/07. PubMed PMID: 25658425; PubMed Central PMCID: PMCPMC4319932.
Apaydin H, Ertan S, Ozekmekci S. Broad bean (Vicia Faba)--a natural source of L-dopa--prolongs “on” periods in patients with Parkinson’s disease who have “on-off” fluctuations. Mov Disord. 2000;15(1):164–6. PubMed PMID: 10634260
Rabey JM, Vered Y, Shabtai H, Graff E, Korczyn AD. Improvement of parkinsonian features correlate with high plasma levodopa values after broad bean (Vicia Faba) consumption. J Neurol Neurosurg Psychiatry. 1992;55(8):725–7. PubMed PMID: 1527547; PubMed Central PMCID: PMC489215
Sarria B, Mateos R, Gallardo E, Ramos S, Martin MA, Bravo L, et al. Nitroderivatives of olive oil phenols protect HepG2 cells against oxidative stress. Food Chem Toxicol. 2012;50(10):3752–8. 10.1016/j.fct.2012.07.030. PubMed PMID: 22842124
Amel N, Wafa T, Samia D, Yousra B, Issam C, Cheraif I, et al. Extra virgin olive oil modulates brain docosahexaenoic acid level and oxidative damage caused by 2,4-Dichlorophenoxyacetic acid in rats. J Food Sci Technol. 2016;53(3):1454–64. 10.1007/s13197-015-2150-3. Epub 2016/08/30. PubMed PMID: 27570270; PubMed Central PMCID: PMCPMC4984713
Singh PK, Kotia V, Ghosh D, Mohite GM, Kumar A, Maji SK. Curcumin modulates alpha-synuclein aggregation and toxicity. ACS Chem Neurosci. 2013;4(3):393–407. 10.1021/cn3001203. PubMed PMID: 23509976; PubMed Central PMCID: PMC3605819
Wang XS, Zhang ZR, Zhang MM, Sun MX, Wang WW, Xie CL. Neuroprotective properties of curcumin in toxin-base animal models of Parkinson’s disease: a systematic experiment literatures review. BMC Complement Altern Med. 2017;17(1):412. 10.1186/s12906-017-1922-x. Epub 2017/08/19. PubMed PMID: 28818104; PubMed Central PMCID: PMCPMC5561616
Liu LX, Chen WF, Xie JX, Wong MS. Neuroprotective effects of genistein on dopaminergic neurons in the mice model of Parkinson's disease. Neurosci Res. 2008;60(2):156–61. 10.1016/j.neures.2007.10.005. PubMed PMID: 18054104
Prediger RD. Effects of caffeine in Parkinson's disease: from neuroprotection to the management of motor and non-motor symptoms. J Alzheimers Dis. 2010;20(Suppl 1):S205–20. 10.3233/JAD-2010-091459. PubMed PMID: 20182024
Postuma RB, Lang AE, Munhoz RP, Charland K, Pelletier A, Moscovich M, et al. Caffeine for treatment of Parkinson disease: a randomized controlled trial. Neurology. 2012;79(7):651–8. 10.1212/WNL.0b013e318263570d. PubMed PMID: 22855866; PubMed Central PMCID: PMC3414662
Hu G, Bidel S, Jousilahti P, Antikainen R, Tuomilehto J. Coffee and tea consumption and the risk of Parkinson’s disease. Mov Disord. 2007;22(15):2242–8. 10.1002/mds.21706. Epub 2007/08/23. PubMed PMID: 17712848
Caruana M, Vassallo N. Tea polyphenols in Parkinson's disease. Adv Exp Med Biol. 2015;863:117–37. 10.1007/978-3-319-18365-7_6. PubMed PMID: 26092629
Pan T, Jankovic J, Le W. Potential therapeutic properties of green tea polyphenols in Parkinson's disease. Drugs Aging. 2003;20(10):711–21. PubMed PMID: 12875608
Ferretta A, Gaballo A, Tanzarella P, Piccoli C, Capitanio N, Nico B, et al. Effect of resveratrol on mitochondrial function: implications in parkin-associated familiar Parkinson's disease. Biochim Biophys Acta. 2014;1842(7):902–15. 10.1016/j.bbadis.2014.02.010. Epub 2014/03/04. PubMed PMID: 24582596
da Silva TM, Munhoz RP, Alvarez C, Naliwaiko K, Kiss A, Andreatini R, et al. Depression in Parkinson's disease: a double-blind, randomized, placebo-controlled pilot study of omega-3 fatty-acid supplementation. J Affect Disord. 2008;111(2–3):351–9. 10.1016/j.jad.2008.03.008. PubMed PMID: 18485485
Calon F, Cole G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: evidence from animal studies. Prostaglandins Leukot Essent Fatty Acids. 2007;77(5–6):287–93. 10.1016/j.plefa.2007.10.019. Epub 2007/11/27. PubMed PMID: 18037281
Cole GM, Frautschy SA. DHA may prevent age-related dementia. J Nutr. 2010;140(4):869–74. 10.3945/jn.109.113910. Epub 2010/02/26. PubMed PMID: 20181786; PubMed Central PMCID: PMCPMC2838628
Su HM. Mechanisms of n-3 fatty acid-mediated development and maintenance of learning memory performance. J Nutr Biochem. 2010;21(5):364–73. 10.1016/j.jnutbio.2009.11.003. Epub 2010/03/18. PubMed PMID: 20233652
Acknowledgements
This work was supported by the Field Neurosciences Institute, St. Mary’s of Michigan, and the John G. Kullavi Professorship and Neuroscience Program at Central Michigan University.
Funding
Field Neurosciences Institute, St. Mary’s of Michigan.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Author information
Authors and Affiliations
Contributions
PM design, performed the background studies, collected data, made all the figs. PM, JM wrote the manuscript. GLD edited and approved the manuscript, contributed the discussion, along with financial supports. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors agreed to publish this article.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Maiti, P., Manna, J. & Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson's disease: Targets for potential treatments. Transl Neurodegener 6, 28 (2017). https://doi.org/10.1186/s40035-017-0099-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-017-0099-z