
Abstract
Osteoarthritis (OA) is a common chronic disabling disease that affects hundreds of millions of people around the world. The most important pathological feature is the rupture and loss of articular cartilage, and the characteristics of avascular joint tissues lead to limited repair ability. Currently, there is no effective treatment to prevent cartilage degeneration. Studies on the mechanism of cartilage metabolism revealed that hypoxia-inducible factors (HIFs) are key regulatory genes that maintain the balance of cartilage catabolism−matrix anabolism and are considered to be the major OA regulator and promising OA treatment target. Although the exact mechanism of HIFs in OA needs to be further clarified, many drugs that directly or indirectly act on HIF signaling pathways have been confirmed by animal experiments and regarded as promising treatments for OA. Targeting HIFs will provide a promising strategy for the development of new OA drugs. This article reviews the regulation of HIFs on intra-articular cartilage homeostasis and its influence on the progression of osteoarthritis and summarizes the recent advances in OA therapies targeting the HIF system.
Similar content being viewed by others


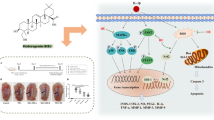
Introduction
In the recent years, as one of the most common chronic diseases of orthopedics, the incidence of osteoarthritis has been increasing year by year with the aging of the population and the increasing proportion of obesity [1]. The most important change in this disease is the destruction of articular cartilage, which is mainly caused by the decomposition of extracellular matrix by degradation enzymes and the death of chondrocytes caused by apoptosis or autophagy. Owing to the lack of blood supply and the relatively closed joint cavity, the articular cartilage itself is in a hypoxic environment. Since there are no capillaries in articular cartilage, the oxygen concentration gradient varies from only 1–10% [2]. The physiological homeostasis of this hypoxic environment is mainly regulated by hypoxia-inducible factors (HIFs), especially HIF-1 and HIF-2 [3]. The hypoxic environment induces chondrocytes to produce a series of hypoxia-related molecules, which are involved in the regulation of osteoarthritis extracellular matrix-degrading enzymes and chondrocyte autophagy and apoptosis. The purpose of this article is to review recent studies on the HIF signaling pathway and its roles in the occurrence and development of osteoarthritis and to explore potential therapies targeting the HIF system.
HIF family
HIFs are heterodimeric transcription factors composed of α (HIF-1α, HIF-2α, and HIF-3α) and β (HIF-1β, HIF-2β, and HIF-3β, also known as ARNT1, ARNT2, and ARNT3) subunits [4,5,6]. HIF-α and HIF-β have the same structural characteristics: both possess basic helix-loop-helix (bHLH) and PAS domains (PAS is named after the three proteins PER, ARNT and SIM all of which have this domain) [7] (Fig. 1A, B). The bHLH-PAS domains mediate heterodimerization and further bind with the hypoxia response elements (HREs) of the target genes [8]. Binding of HIF-1α to HRE causes upregulation of HIF-1α target genes and is precisely regulated by many factors [9, 10]. Besides bHLH and PAS domains, the HIF-1α protein also contains an oxygen-dependent degradation (ODD) domain and two transactivation domains (N-TAD and C-TAD). Interestingly, HIF-1β only has the C-terminal transactivation domain (C-TAD), however, it has two repeat regions PAS domains known as PAS-A and PAS-B. HIF-2β and HIF-3β have similar domain organizations as HIF-1β (Fig. 1A, B).

Schematic diagram of the domain organization of HIFs. A Illustrated functional domain arrangements of HIFs. Domains: bHLH, basic helix-loop-helix (DNA binding and dimerization); PAS, Per/Ahr-ARNT/Sim (dimerization); PAS-A PAS-associated domain A; PAS-B PAS-associated domain B; ODDD: oxygen-dependent degradation domain; N-TAD, N-terminal transactivation domain (transcriptional transactivation); C-TAD, C-terminal transactivation domain (transcriptional transactivation); Factors: PHD1-3, HIF prolyl hydroxylase 1–3; VHL, von Hippel–Lindau tumor suppressor protein. CBP/P300: p300/CREB-binding protein. B 3D structural illustrations of HIF domain organization. Mouse HIF-1α:HIF-1β:HRE (DNA element) are shown as an example to show domain organizations. The structure is retrieved from the Protein Data Bank (ID: 4ZPR). Color schemes are the same as in panel A. Because the structure only contains partial sequences of HIF1α:HIF-1β, only bHLH and PAS domains are shown
Similar to HIF-1α, HIF-2α also interacts with HREs to upregulate transcriptional activity of target genes [11]. The HIF-2α protein shares 48% sequence identity with HIF-1α protein, and has many structural and biochemical similarities with HIF-1α (e.g., heterodimerization and HREs binding). However, when compared with HIF-1α, which is widely expressed, HIF-2α is mainly expressed in the lung, carotid body, and endothelial cells [12]. HIF-2β and HIF-3β also expression in the endothelial tissue and have 70% similarity with HIF-1β and sharing similar structure [13,14,15]. In contrast, HIF-3α is involved in hypoxic downregulation through selective splicing of transcription factors that may act as inhibitors of HIF-1α [16]. HIF-3α is also expressed in a variety of tissues, dimerizes with HIF-1α and binds to HREs [17]. Currently, HIF-1 and HIF-2 have been more widely studied, while HIF-3 and other HIFs have been relatively less studied (Fig. 1A, B).
Regulation of HIFs
HIFs are key heterodimer transcription factors expressed under hypoxic conditions. It mediates adaptive responses from normoxic (~ 21% oxygen) to hypoxic conditions by binding to the promoters of numerous hypoxia-inducible genes, such as those involved in iron metabolism, angiogenesis, and glucose metabolism. It plays important roles for cells and tissues to adapt to low oxygen tension [6, 18, 19].
The regulation of HIF system is mainly through the α subunit HIF-α, whereas β subunit HIF-β is constitutively expressed [20]. Under normoxic condition, the expression of two main HIF-α isoforms (HIF-1α and HIF-2α) is regulated by oxygen-independent mechanisms, the mitogen-activated protein kinase (MAPK) pathway and the growth factor-mediated phosphoinositide 3 kinase (PI3K) pathway [21, 22]. Taking the oxygen-independent mechanism as an example, HIF-α protein degradation is mediated by the ODD domain. HIF-α remains stable even in the absence of a hypoxia signal when the entire ODD region is removed [23]. Hydroxylation of proline residues 402 and 564 in the ODD domain controls the interaction between HIF-α and the von Hippel-Lindau tumor suppressor protein (pVHL) which facilitates the ubiquitination and degradation of HIIF-α [24,25,26,27]. The hydroxylation process is regulated by three conserved HIF prolyl hydroxylases (PHD1, PHD2, PHD3, and also known as EGLN2, EGLN1, EGLN3), and their activity lies in the presence of oxygen, iron, 2-oxoglutarate and ascorbate [18, 28]. Interestingly, it has been shown that PHD2 has remarkable significance in regulating HIF-1α levels by using small interfering RNA (siRNA) techniques [29].
In hypoxia, prolyl hydroxylation of the ODD domain is suppressed, and the interaction between HIF-1α and pVHL is inhibited. As a result, HIF-α degradation is interrupted and its concentration consequently increases. HIF-α is then translocated and accumulated in the nucleus where it binds to HIF-β via bHLHs and PAS domains to form the HIF-α/β dimer complex [6]. Transcriptional coactivators, such as p300/CBP (p300/CREB-binding protein), help the HIF complex couple to HRE elements within the promoter region of HIF target genes, consequently regulating their transcriptional activation [30, 31]. Asparagine hydroxylase, known as FIH-1 (factor inhibiting HIF-1), also mediates the transcriptional activity of HIF-1α [32,33,34,35]. Hydroxylation of Asn803 from FIH-1 blocks the HIF-1α interaction with p300/CBP in its C-TAD under normoxic conditions, so FIH-1 acts as a negative regulator of HIF-1α by interacting with pVHL to suppress transcriptional activity and modulate stabilization (Fig. 2).

Schematic diagram of the HIF-1 pathway. Under normal oxygen conditions, HIF-1α protein is hydroxylated by prolyl 4-hydroxylase (PHD) on proline residues and polyubiquitinated by von Hippel–Lindau protein (pVHL). In this case, it will be degraded by the 26S proteasome system. Under hypoxia, HIF-1α can enter the nucleus and form a transcription complex with HIF-1β subunits and then recruit coactivators (such as CBP/p300) to regulate the transcriptional activity of downstream genes. PHD, molecules containing proline-hydroxylase domain; Ub, ubiquitin; FIH, a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity
The roles of HIF-1α in OA
It is extremely important to maintain chondrocyte metabolism under hypoxic conditions. In healthy cartilage, hypoxic conditions can enhance the expression and activity of HIF-1α [36,37,38], which not only induces the expression of Erythropoietin (EPO), SRY-Box Transcription Factor 9 (SOX9), Collagen Type II Alpha 1 (COL2A1), Vascular endothelial growth factor (VEGF), Nitric Oxide Synthase (NOS) and glucose transporter protein type 1 (GLUT1) to maintain cartilage homeostasis, but also inhibits the expression of Collagen Type I Alpha 1 (COL1A1), Collagen Type I Alpha 2 (COL1A2) and Collagen Type III Alpha 1 (COL3A1) to prevent the degradation of the extracellular matrix (ECM) [39,40,41,42], thereby mediating anti-catabolic reactions and preventing spontaneous and induced destruction of human cartilage. In addition, HIF-1α can also protect chondrocytes from apoptosis by inducing heat shock protein 70 (HSP70) to increase ECM gene expression levels and cell viability [43,44,45,46,47,48,49,50].
Studies have found that HIF-1α levels are significantly related to the severity and progression of osteoarthritis [51, 52]. In the early stage of OA, articular cartilage undergoes metabolic adaptation under harmful stimulating conditions. Chondrocytes dedifferentiate into a hypertrophic phenotype, and ECM is degraded which is characterized by decreased synthesis of Collagen II and newly synthesized Collagen I and Collagen X, accompanied by activation of matrix-degrading enzymes such as MMP13 [53]. During this period, chondrocytes attemp to repair damaged cells and ECM through adaptive changes in metabolism. Owing to the changes in the chondrocyte microenvironment in OA, the activation of AMP-activated protein kinase (AMPK) and inhibition of the mammalian target of rapamycin (mTOR) induced by HIF-1α can also cause chondrocyte autophagy [54]. Osteoarthritis of the temporomandibular joint (TMJ) is one of the common types of OA. In TMJ-OA, the HIF-1-VEGF-Notch signaling pathway accelerates cartilage angiogenesis, thereby accelerating the development of TMJ-OA [55,56,57]. F-box and WD repeat domain-containing 7 (FBW7) can negatively regulate the HIF-1α/VEGF pathway to inhibit angiogenesis, thereby inhibiting the degradation of chondrocytes induced by IL-1β [58]. Studies have found that the expression of HIF-1α and Runx2 in degenerative chondrocytes increases simultaneously. Studies showed that Runx2 protein can induce the expression of HIF-1α at the transcriptional level and accelerate the progression of OA [59]. However, with the development of OA, this metabolic adaptation and the self-repair ability of cartilage decrease, leading to serious tissue damage [60, 61]. The abnormal deposition of ECM can further lead to synovial fibrosis. Among them, TGF-β, procollagen-lysine, 2-oxoglutarate 5-dioxygenase2, COL1A1 and tissue inhibitor of metalloproteinases 1 are involved in osteoarthritis-related fibrosis and are considered fibrosis markers [62,63,64]. Fibroblast-like synovial cells (FLSs) are the main effector cells of synovial fibrosis in knee osteoarthritis [65]. More recently, a form of programmed inflammatory cell death called pyroptosis has been discovered [66]. HIF-1α activates nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3), assembles inflammasome complexes through the adapter protein apoptosis-associated speck-like protein containing a caspase recruitment domain, and drives caspase-1-mediated inflammation to mediate knee synovial fibrosis in OA [67,68,69]. The HIF-1α/NLRP3 inflammasome is one of the inflammasome signal transductions closely related to the KOA process. Inhibiting the activation of inflammasomes improves synovial fibrosis in KOA [70,71,72,73].
In addition, the progression of HIF-1α in OA is also regulated by long noncoding RNAs (lncRNAs) and miRNAs. LncRNAs refer to a subpopulation of noncoding RNAs longer than 200 nucleotides. Previous studies have shown that abnormal expression of lncRNAs plays an important role in the development of OA [74, 75]. For example, lncRNA UFC1 can increase the proliferation of chondrocytes in OA [76], and lncRNA cartilage injury-related (lncRNA-CIR) can promote the degradation of chondrocyte extracellular matrix in the disease [77]. Long noncoding HIF-1α co-activating RNA (LncHIFCAR) positively regulates HIF-1α and HIF-1α target genes (such as VEGF and BNIP3), thereby promoting the hypoxia-induced inflammatory response and matrix synthesis and inducing cell apoptosis [78]. miRNAs are single-stranded small noncoding RNAs abundant in cells. Many miRNAs also plays an important role in hypoxia cartilage homeostasis or the OA stress microenvironment through the HIF-1α pathway [79, 80]. For example, miR-146a upregulates the expression of ULK-1, HIF-1α and ATG-5 by targeting TRAF6/IRAK1, thereby slowing the progression of OA [81]. miR-204 and miR-211 affect nerve growth factor (NGF) expression in a Runx2-dependent manner to regulate homeostasis and OA progression [82]. miR-411 regulates chondrocyte autophagy by targeting HIF-1α [83]. Whereas miR-373 regulates the damage to chondrocytes treated with lipopolysaccharide by targeting HIF-1α [84].
The roles of HIF-2α in OA
HIF-2α and HIF-1α have different functions in cartilage. In the articular cartilage cells of synovial joints, HIF-1α promotes the homeostatic pathway, while HIF-2α promotes the degradation pathway. HIF-2α can target genes related to the hypertrophy and differentiation of chondrocytes, such as Runt-related transcription factor 2 (RUNX2) and COL10A1, and genes related to the degradation of ECM, such as matrix metalloproteinase MMP9, MMP13, MMP3 and A Disintegrin and Metalloproteinase with Thrombospondin motifs 48–51 (ADAMTS48-51) [85]. In addition, proinflammatory factors (such as interleukin IL-1β, IL-6, and tumor necrosis factor TNF-α) can upregulate the expression of HIF-2α in articular chondrocytes by activating Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathways [86, 87], thereby promoting chondrocytes from the prehypertrophic state to the terminal hypertrophic state [87]. Previous studies have found that nicotinamide phosphoribosyltransferase (NAMPT) is the direct target gene of HIF-2α in articular chondrocytes and is upregulated in OA cartilage. Inhibition of NAMPT enzyme activity by injection of the NAMPT inhibitor FK866 (intra-articular or intraperitoneal) can inhibit the destruction of osteoarthritic cartilage caused by intra-articular injection or DMM surgery with Epas1 adenovirus (Ad-Epas1) or Ad-Nampt [88]. Fas (CD95) is a member of the tumor necrosis factor receptor family, containing a death domain to activate apoptosis signals. The binding of Fas ligand (FasL) or an agonistic anti-Fas antibody to the Fas receptor triggers the apoptosis signal. The increase in the chondrocyte apoptosis is also related to the severity of human OA cartilage damage [89, 90]. The combination of FasL or anti-Fas antibodies can induce chondrocyte apoptosis [91, 92]. Researchers have found that HIF-2α promotes Fas-mediated chondrocyte apoptosis by upregulating Fas expression [93]. The accumulation of iron-dependent lipid hydroperoxides causes cell death, known as ferroptosis [94,95,96]. The recent evidence suggests that ferroptosis in chondrocytes is associated with the progression of OA [94,95,96]. These findings indicate that ferroptosis suppression is a new potential choice to prevent the progression of OA. Human lysyl oxidase (LOX) is a hypoxia response gene whose product can catalyze collagen cross-linking, while HIF-2α can upregulate LOX and play a crucial role in osteoarthritis [97, 98]. HIF-2α also causes cartilage destruction by regulating the expression of various catabolic factors, such as VEGF, type X collagen, prostaglandin intra peroxidase synthase 2 (PTGS2) and nitric oxide synthase 2 (NOS2) [87].
When compared with normal cartilage, the expression of various miRNAs in osteoarthritis has also undergone some changes, which indicates that the expression of miRNAs may also be involved in the metabolic balance of cartilage through HIF-2α pathway [99]. There are experiments have shown that miR-365 regulates HIF-2α at the posttranscriptional level and cross-regulates the MAPK-NF-kB signaling pathway to reduce IL-1β-induced chondrocyte catabolism [100] (Fig. 3).

HIF-1α and HIF-2α have different functions in OA. HIF-1α mainly maintains the extracellular matrix synthesis of chondrocytes and chondrocyte differentiation and promotes the balance of articular cartilage autophagy in the body. NF-κB activation promotes the heterodimerization of HIF-2α and RNTL, leading to the activation of the transcription factor HIF-2α, and through the IHH and RUNX2 axes, coactivating MMP13 prompts articular chondrocytes to show a hypertrophic state of differentiation, leading to the occurrence and progression of OA
Targeting HIF-1α for OA therapy
Because PHD can hydroxylate HIF-1α and lead to ubiquitination degradation, inhibiting the hydroxylation of HIF-1α by PHD may have potential therapeutic value. Dimethyloxaloylglycine (DMOG), an analog of 2-oxoglutarate, can competitively bind to PHD and eventually inhibit HIF degradation [101]. In addition, the PHD inhibitors TM6008 and TM6089 are designed based on the active site of the three-dimensional protein structure of human PHD2 and inhibit HIF-1α degradation [102, 103]. Moreover, FK506-binding protein 38 can reduce the stability of PHD2 protein by interacting with the N-terminal domain of PHD2, thereby accumulating HIF-1 and increasing cartilage stability [104].
The NLRP3 inflammasome (HIF-1α/NLRP3 inflammasome) is one of the inflammasome signal transduction pathways closely related to the KOA process, and inhibiting the activation of the NLRP3 inflammasome can improve synovial fibrosis in KOA. Researchers found that Agnuside (AGN), a nontoxic, natural small molecule isolated from the extract of Vitex negundo L., can reduce the fibrosis of experimental KOA by inhibiting the accumulation of HIF-1α and the activation of NLRP3 inflammasomes [105]. Casticin is a compound purified from the Chinese herbal medicine Viticis Fructus. It has the effects of promoting the immune response [106], anti-inflammation [107], antioxidative stress [108] and antifibrosis [109]. Similarly, casticin reduces MIA-induced KOA by inhibiting HIF-1α/NLRP3 inflammasome activation. Therefore, casticin may be a potential treatment strategy for KOA [110].
In the recent years, magnesium-based biomedical devices have shown great potential for translation in orthopedics [111]. The use of magnesium ions (Mg2+) to promote the synthesis of cartilage matrix mediated by HIF-1α is a new treatment option for OA [112]. However, oxidative stress can reduce the expression of HIF-1α and enhance the inflammatory response, which may impair the efficacy of Mg2+ in the treatment of OA. Vitamin C is an effective antioxidant that can enhance the efficacy of Mg2+ in the treatment of OA [113].
In summary, there are many different ways to treat OA through the different HIF-1α pathways. Firstly, cartilage homeostasis can be strengthened through inhibiting degradation of HIF-1α in the early of OA, using compounds such as DMOG, FK506-binding protein 38, PHD inhibitors TM6008 and TM6089. However, with the progression of OA, the improving synovial fibrosis through inhibiting accumulation of HIF-1α become more important using compunds, such as Agnuside and Casticin. Besides, Mg2+ can be used to promote the synthesis of cartilage matrix mediated by hypoxia-inducible factor-1α.
Targeting HIF-2α for OA therapy
HIF-2α is a regulatory factor for the expression of catabolic factors during the development of osteoarthritis. Therefore, HIF-2α inhibitors have potential therapeutic prospects for osteoarthritis. Studies have found that curcumin CMC2.24 regulates chondrocyte apoptosis and ECM homeostasis by inhibiting the NF-κB/HIF-2α pathway, thereby providing a new perspective for the treatment of OA [114]. One of the extracts of Cirsium japonicum var. maackii (CJM), apigenin inhibits HIF-2α through the NF-κB pathway, effectively blocking the expression of Prostaglandin-endoperoxide synthase 2 (COX-2), MMP3 and MMP13, and is worthy of use as a therapeutic drug for OA to block cartilage inflammation [115]. D-mannose inhibits chondrocyte ferroptosis enhanced by HIF-2α and has a chondroprotective effect on the progression of OA [116]. Icariin (ICA) is a typical flavonoid compound extracted from Epimedii Folium that may inhibit inflammatory damage by inhibiting the NF-κB/HIF-2α signaling pathway, thereby increasing chondrocyte viability [117]. Inhibition of syndecan-4 (SDC-4) induces the expression of microRNA-96-5p (miR-96-5p), targets HIF-2α 3′-UTR sequences and inhibits HIF-2α signaling in mouse cartilage tissue and chondrocytes. Therefore, this method may provide a potential new strategy to prevent the progression of osteoarthritis [118]. 4 (2′-Aminoethyl) amino-1,8-dimethylimidazo(1,2-a)quinoxaline (BMS-345541) is a selective inhibitor of the subunits of IκBα kinase (IKK). Intra-articular administration of BMS-345541 may inhibit the development of OA by downregulating NF-κB/HIF-2α signaling [119]. Studies using vectors to deliver siRNA and silence HIF-2α expression can prevent cartilage degradation in mice affected by OA [120]. In brief, there are many HIF-2α inhibitors (CMC2.24, CJM, D-mannose, ICA, SDC-4 and BMS-345541 et. al) have the potential therapeutic prospects in the OA diseases.
Conclusion
HIF-1α α and HIF-2α have different functions in cartilage. The regulation of HIF-1α is crucial in maintaining cartilage homeostasis. It induces the expression of COL2A1, SOX9, GLUT1, EPO, NOS and VEGF to maintain cartilage homeostasis and inhibits the expression of COL1A1, COL1A2 and COL3A1 to prevent ECM degradation. HIF-2α is involved in a pathway that promotes osteoarthritis degradation and regulates the expression of genes related to chondrocyte hypertrophy and differentiation, such as COL10A1 and RUNX2, and the expression of genes related to ECM degradation, such as MMP9, MMP13, MMP3, ADAMTS. Many of those proteins may serve potential targets for novel therapy development. However, many research gaps still exists for further in-depth studies. For example, HIFs pathways may cross-talk with other pathways and regulation of the potential targets may thus result in serious side effects [121]. What’s more, the molecular mechanism of osteoarthritis is very complicated and many of those reported potential therapeutics still need further provement in clinical experiemnts [122]. In conclusion, OA is a dynamic change caused by the imbalance between the anabolic and catabolic of joint tissue. For osteoarthritis, increasing the accumulation and activity of HIF-1α to increase cartilage stability and inhibiting the activity of HIF-2α to reduce ECM degradation are promising therapeutic approaches.
Availability of data and materials
Not applicable.
Abbreviations
- ADAMTS:
-
A disintegrin and metalloproteinase with thrombospondin motifs
- AGN:
-
Agnuside
- AMPK:
-
AMP-activated protein kinase
- bHLH:
-
Basic helix-loop-helix
- CJM:
-
Cirsium japonicum var. maackii
- COL1A1:
-
Collagen Type I Alpha 1
- COL1A2:
-
Collagen Type I Alpha 2
- COL2A1:
-
Collagen Type II Alpha 1
- COL3A1:
-
Collagen Type III Alpha 1
- COL10A1:
-
Collagen Type X Alpha 1
- COX-2:
-
Prostaglandin-endoperoxide synthase 2
- DMOG:
-
Dimethyloxaloylglycine
- ECM:
-
Extracellular matrix
- EPO:
-
Erythropoietin
- FBW7:
-
F-box and WD repeat domain-containing 7
- FIH-1:
-
Factor inhibiting HIF-1
- HIF:
-
Hypoxia-inducible factor
- FasL:
-
Fas ligand
- FLS:
-
Fibroblast-like synovial cell
- HSP70:
-
Heat shock protein 70
- HRE:
-
Hypoxia response elements
- ICA:
-
Icariin
- IKK:
-
IκBα kinase
- IL:
-
Interleukin
- LncHIFCAR:
-
Long noncoding HIF-1α co-activating RNA
- LncRNA:
-
Long noncoding RNAs
- LOX:
-
Lysyl oxidase
- MAPK:
-
Mitogen-activated protein kinase
- MMP:
-
Matrix metalloproteinase
- mTOR:
-
The mammalian target of rapamycin
- NAMPT:
-
Nicotinamide phosphoribosyltransferase
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NGF:
-
Nerve growth factor
- NLRP3:
-
Nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3
- NOS2:
-
Nitric oxide synthase 2
- OA:
-
Osteoarthritis
- ODD:
-
Oxygen-dependent degradation
- OPN:
-
Osteopontin
- PI3K:
-
Phosphoinositide 3 kinase
- PTGS2:
-
Prostaglandin intra peroxidase synthase 2
- PHD:
-
HIF prolyl hydroxylase
- pVHL:
-
Von Hippel−Lindau tumor suppressor protein
- RUNX2:
-
Runt-related transcription factor 2
- SDC-4:
-
Syndecan-4
- SiRNA:
-
Small interfering RNA
- SOX9:
-
SRY-Box Transcription Factor 9
- TAD:
-
Transactivation domain
- TGF-β:
-
Transforming growth factor beta
- TNF:
-
Tumor necrosis factor
- TMJ:
-
Temporomandibular joint
References
James SL, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, Abbastabar H, Abd-Allah F, Abdela J, Abdelalim A. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet. 2018;392(10159):1789–858.
Ströbel S, Loparic M, Wendt D, Schenk AD, Candrian C, Lindberg RLP, Moldovan F, Barbero A, Martin I. Anabolic and catabolic responses of human articular chondrocytes to varying oxygen percentages. Arthritis Res Ther. 2010;12(2):1–15.
Yudoh K, Nakamura H, Masuko-Hongo K, Kato T, Nishioka K. Catabolic stress induces expression of hypoxia-inducible factor (HIF)-1α in articular chondrocytes: involvement of HIF-1α in the pathogenesis of osteoarthritis. Arthritis Res Ther. 2005;7(4):1–11.
Semenza GL. Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev. 1998;8(5):588–94.
Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3’to the human erythropoietin gene. Proc Natl Acad Sci. 1991;88(13):5680–4.
Wang GL, Jiang B-H, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci. 1995;92(12):5510–4.
Jiang B-H, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem. 1996;271(30):17771–8.
Michel G, Minet E, Ernest I, Roland I, Durant F, Remacle J, Michiels C. A model for the complex between the hypoxia-inducible factor-1 (HIF-1) and its consensus DNA sequence. J Biomol Struct Dyn. 2000;18(2):169–79.
Carrero P, Okamoto K, Coumailleau P, O’Brien S, Tanaka H, Poellinger L. Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1α. Mol Cell Biol. 2000;20(1):402–15.
Richard DE, Berra E, Gothié E, Roux D, Pouysségur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1α (HIF-1α) and enhance the transcriptional activity of HIF-1. J Biol Chem. 1999;274(46):32631–7.
Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15(1):551–78.
Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci. 1997;94(9):4273–8.
Labrecque MP, Prefontaine GG, Beischlag TV. The aryl hydrocarbon receptor nuclear translocator (ARNT) family of proteins: transcriptional modifiers with multi-functional protein interfaces. Curr mol med. 2013;13(7):1047–65.
Maltepe E, Keith B, Arsham AM, Brorson JR, Simon MC. The role of ARNT2 in tumor angiogenesis and the neural response to hypoxia. Biochem Biophys Res Commun. 2000;273(1):231–8.
Takahata S, Sogawa K, Kobayashi A, Ema M, Mimura J, Ozaki N, Fujii-Kuriyama Y. Transcriptionally active heterodimer formation of an Arnt-like PAS protein, Arnt3, with HIF-1a, HLF, and clock. Biochem Biophys Res Commun. 1998;248(3):789–94.
Makino Y, Kanopka A, Wilson WJ, Tanaka H, Poellinger L. Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3α locus. J Biol Chem. 2002;277(36):32405–8.
Gu Y-Z, Moran SM, Hogenesch JB, Wartman L, Bradfield CA. Molecular characterization and chromosomal localization of a third α-class hypoxia inducible factor subunit, HIF3α. Gene Expr The J Liver Res. 1998;7(3):205–13.
Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294(5545):1337–40.
Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5(5):343–54.
Berchner-Pfannschmidt U, Frede S, Wotzlaw C, Fandrey J. Imaging of the hypoxia-inducible factor pathway: insights into oxygen sensing. Eur Respir J. 2008;32(1):210–7.
Fukuda R, Hirota K, Fan F, Do Jung Y, Ellis LM, Semenza GL. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem. 2002;277(41):38205–11.
Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1α (HIF-1α) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21(12):3995–4004.
Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1α is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci. 1998;95(14):7987–92.
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–8.
Jaakkola P, Mole DR, Tian Y-M, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–72.
Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J. 2001;20(18):5197–206.
Yu F, White SB, Zhao Q, Lee FS. HIF-1α binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci. 2001;98(17):9630–5.
Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A. C elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43–54.
Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J. 2003;22(16):4082–90.
Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci. 1993;90(9):4304–8.
Kallio PJ, Okamoto K, O’Brien S, Carrero P, Makino Y, Tanaka H, Poellinger L. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-induciblefactor-1α. EMBO J. 1998;17(22):6573–86.
Hewitson KS, McNeill LA, Riordan MV, Tian Y-M, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002;277(29):26351–5.
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16(12):1466–71.
Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain: a hypoxic switch. Science. 2002;295(5556):858–61.
Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15(20):2675–86.
Coimbra IB, Jimenez SA, Hawkins DF, Piera-Velazquez S, Stokes DG. Hypoxia inducible factor-1 alpha expression in human normal and osteoarthritic chondrocytes. Osteoarthritis Cartilage. 2004;12(4):336–45.
Schrobback K, Malda J, Crawford RW, Upton Z, Klein TJ. Effects of oxygen on zonal marker expression in human articular chondrocytes. Tissue Eng Part A. 2011;18(9–10):920–33.
Hong YH, Park CW, Kim HS, Won KC, Kim YW, Lee CK. Effects of hypoxia/ischemia on catabolic mediators of cartilage in a human chondrocyte, SW1353. Biochem Biophys Res Commun. 2013;431(3):478–83.
Xu TX, Zhao SZ, Dong M, Yu XR. Hypoxia responsive miR-210 promotes cell survival and autophagy of endometriotic cells in hypoxia. Eur Rev Med Pharmacol. 2016;20(3):399–406.
Provot S, Schipani E. Fetal growth plate: a developmental model of cellular adaptation to hypoxia. Ann N Y Acad Sci. 2010;1117(1):26–39.
Sanz-Ramos P, Mora G, Vicente-Pascual M, Ochoa I, Alcaine C, Moreno R, Doblaré M, Izal-Azcárate Í. Response of sheep chondrocytes to changes in substrate stiffness from 2 to 20 Pa: effect of cell passaging. Connect Tissue Res. 2013;54(3):159–66.
Duval E, Leclercq S, Elissalde JM, Demoor M, Galéra P, Boumédiene K. Hypoxia-inducible factor 1α inhibits the fibroblast-like markers type I and type III collagen during hypoxia-induced chondrocyte redifferentiation: hypoxia not only induces type II collagen and aggrecan, but it also inhibits type I and type III collagen in the hypoxia-inducible factor 1α–dependent redifferentiation of chondrocytes. Arthritis Rheum Off J Am College Rheum. 2009;60(10):3038–48.
Tsuchida S, Arai Y, Takahashi KA, Kishida T, Kubo T. HIF-1α-induced HSP70 regulates anabolic responses in articular chondrocytes under hypoxic conditions. J Orthop Res. 2014. https://doi.org/10.1002/jor.22623.
Ngarmukos S, Scaramuzza S, Theerawattanapong N, Tanavalee A, Honsawek S. Circulating and synovial fluid heat shock protein 70 are correlated with severity in knee osteoarthritis. Cartilage. 2020;11(3):323–8.
Suzuki T, Segami N, Nishimura M, Hattori H, Nojima T. Analysis of 70Kd heat shock protein expression in patients with internal derangement of the temporomandibular joint. Int J Oral Maxillofac Surg. 2000;29(4):301–4.
Terauchi R, Takahashi KA, Arai Y, Ikeda T, Ohashi S, Imanishi J, Mazda O, Kubo T. Hsp70 prevents nitric oxide-induced apoptosis in articular chondrocytes. Arthritis Rheum. 2003;48(6):1562–8.
Grossin L, Etienne S, Gaborit N, Pinzano A, Cournil-Henrionnet C, Gerard C, Payan E, Netter P, Terlain B, Gillet P. Induction of heat shock protein 70 (Hsp70) by proteasome inhibitor MG 132 protects articular chondrocytes from cellular death in vitro and in vivo. Biorheology. 2004;41(3–4):521–34.
Tonomura H, Takahashi KA, Mazda O, Arai Y, Inoue A, Terauchi R, Shin-Ya M, Kishida T, Imanishi J, Kubo T. Glutamine protects articular chondrocytes from heat stress and NO-induced apoptosis with HSP70 expression. Osteoarthritis Cartilage. 2006;14(6):545–53.
Etienne S, Gaborit N, Henrionnet C, Pinzano A, Galois L, Netter P, Gillet P, Grossin L. Local induction of heat shock protein 70 (Hsp70) by proteasome inhibition confers chondroprotection during surgically induced osteoarthritis in the rat knee. Biomed Mater Eng. 2008;18(4–5):253–60.
Takahashi KA, Tonomura H, Arai Y, Terauchi R, Honjo K, Hiraoka N, Hojo T, Kunitomo T, Kubo T. Hyperthermia for the treatment of articular cartilage with osteoarthritis. Int J Hyperthermia. 2009;25(8):661–7.
Araldi E, Schipani E. Hypoxia, HIFs and bone development. Bone. 2010;47(2):190–6.
Qing L, Lei P, Hao L, Jie X, Long W, Wen T, Hu Y. Expression of hypoxia-inducible factor-1α in synovial fluid and articular cartilage is associated with disease severity in knee osteoarthritis. Exp Ther Med. 2017;13(1):63–8.
Charlier E, Deroyer C, Ciregia F, Malaise O, Neuville S, Plener Z, Malaise M, de Seny D. Chondrocyte dedifferentiation and osteoarthritis (OA). Biochem Pharmacol. 2019;165:49–65.
Chen Y-C, Zhang L, Li E-N, Ding L-X, Zhang G-A, Hou Y, Yuan W. Association of the insulin-like growth factor-1 single nucleotide polymorphisms rs35767, rs2288377, and rs5742612 with osteoporosis risk: a meta-analysis. Medicine. 2017;96(51):e9231.
Shirakura M, Tanimoto K, Eguchi H, Miyauchi M, Nakamura H, Hiyama K, Tanimoto K, Tanaka E, Takata T, Tanne K. Activation of the hypoxia-inducible factor-1 in overloaded temporomandibular joint, and induction of osteoclastogenesis. Biochem Biophys Res Commun. 2010;393(4):800–5.
Milam SB, Schmitz JP. Molecular biology of temporomandibular joint disorders: proposed mechanisms of disease. J Oral Maxillofac Surg. 1995;53(12):1448–54.
Ke J, Liu Y, Long X, Li J, Fang W, Meng Q, Zhang Y. Up-regulation of vascular endothelial growth factor in synovial fibroblasts from human temporomandibular joint by hypoxia. J Oral Pathol Med. 2007;36(5):290–6.
Zhu W, Chang B, Wang X, Zang Y, Zheng Z, Zhao H, Cui Q. FBW7 regulates HIF-1alpha/VEGF pathway in the IL-1beta induced chondrocytes degeneration. Eur Rev Med Pharmacol Sci. 2020;24(11):5914–24.
Kong P, Chen R, Zou F, Wang Y, Liu M, Wang W. HIF-1α repairs degenerative chondrocyte glycolytic metabolism by the transcriptional regulation of Runx2. Eur Rev Med Pharmacol Sci. 2021;25(3):1206–14.
Guilak F, Nims RJ, Dicks A, Wu C-L, Meulenbelt I. Osteoarthritis as a disease of the cartilage pericellular matrix. Matrix Biol. 2018;71:40–50.
Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019;393(10182):1745–59.
Meng X-M, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12(6):325–38.
Van Der Kraan PM. The changing role of TGFβ in healthy, ageing and osteoarthritic joints. Nat Rev Rheumatol. 2017;13(3):155–63.
Remst DFG, Davidson E, Vitters EL, Blom AB, Stoop R, Snabel JM, Bank RA, Berg W, Kraan PMVD. Osteoarthritis-related fibrosis is associated with both elevated pyridinoline cross-link formation and lysyl hydroxylase 2b expression. Osteoarthritis Cartilage. 2013;21(1):157–64.
Remst DF, Blaney Davidson EN, van der Kraan PM. Unravelling osteoarthritis-related synovial fibrosis: a step closer to solving joint stiffness. Rheumatology. 2015;54(11):1954–63.
Walle LV, Lamkanfi M. Pyroptosis. Curr Biol. 2016;26(13):R568–72.
Zhao LR, Xing RL, Wang PM, Zhang NS, Yin SJ, Li XC, Zhang L. NLRP1 and NLRP3 inflammasomes mediate LPS/ATP-induced pyroptosis in knee osteoarthritis. Mol Med Rep. 2018;17(4):5463–9.
Cheng F, Yan F-F, Liu Y-P, Cong Y, Sun K-F, He X-M. Dexmedetomidine inhibits the NF-κB pathway and NLRP3 inflammasome to attenuate papain-induced osteoarthritis in rats. Pharm Biol. 2019;57(1):649.
McAllister M, Chemaly M, Eakin AJ, Gibson DS, McGilligan VE. NLRP3 as a potentially novel biomarker for the management of osteoarthritis. Osteoarthritis Cartilage. 2018;26(5):612–9.
Zhang L, Xing R, Huang Z, Zhang N, Zhang L, Li X, Wang P. Inhibition of synovial macrophage pyroptosis alleviates synovitis and fibrosis in knee osteoarthritis. Mediators Inflamm. 2019;2019:2165918.
Li X, Geng J, Zhao J, Ni Q, Zhao C, Zheng Y, Chen X, Wang L. Trimethylamine N-oxide exacerbates cardiac fibrosis via activating the NLRP3 inflammasome. Front Physiol. 2019;10:866.
Ding S, Wang H, Wang M, Bai L, Yu P, Wu W. Resveratrol alleviates chronic “real-world” ambient particulate matter-induced lung inflammation and fibrosis by inhibiting NLRP3 inflammasome activation in mice. Ecotoxicol Environ Saf. 2019;182:109425.
Wang Y, Li H, Li Y, Zhao Y, Xiong F, Liu Y, Xue H, Yang Z, Ni S, Sahil A. Coriolus versicolor alleviates diabetic cardiomyopathy by inhibiting cardiac fibrosis and NLRP3 inflammasome activation. Phytother Res. 2019;33(10):2737–48.
Regmi S, Fu A, Luo KQ. High shear stresses under exercise condition destroy circulating tumor cells in a microfluidic system. Sci Rep. 2017;7:39975.
Xing D, Liang Jq, Li Y, Lu J, Jia Hb, Xu Ly, Ma XI. Identification of long noncoding RNA associated with osteoarthritis in humans. Orthopaedic surg. 2014;6(4):288–93.
Zhang G, Wu Y, Xu D, Yan X. Long noncoding RNA UFC1 promotes proliferation of chondrocyte in osteoarthritis by acting as a sponge for miR-34a. DNA Cell Biol. 2016;35(11):691–5.
Li Y-F, Li S-H, Liu Y, Luo Y-T. Long noncoding RNA CIR promotes chondrocyte extracellular matrix degradation in osteoarthritis by acting as a sponge for Mir-27b. Cell Physiol Biochem. 2017;43(2):602–10.
Sun J, Song X, Su L, Cao S. Long non-coding RNA LncHIFCAR promotes osteoarthritis development via positively regulating HIF-1α and activating the PI3K/AKT/mTOR pathway. Int J Clin Exp Pathol. 2018;11(6):3000.
Miyaki S, Asahara H. Macro view of microRNA function in osteoarthritis. Nat Rev Rheumatol. 2012;8(9):543–52.
Musumeci G, Castrogiovanni P, Trovato FM, Weinberg AM, Al-Wasiyah MK, Alqahtani MH, Mobasheri A. Biomarkers of chondrocyte apoptosis and autophagy in osteoarthritis. Int J Mol Sci. 2015;16(9):20560–75.
Chen G, Gao X, Wang J, Yang C, Wang Y, Liu Y, Zou W, Liu T. Hypoxia-induced microRNA-146a represses Bcl-2 through Traf6/IRAK1 but not Smad4 to promote chondrocyte autophagy. Biol Chem. 2017;398(4):499–507.
Huang J, Zhao L, Fan Y, Liao L, Ma PX, Xiao G, Chen D. The microRNAs miR-204 and miR-211 maintain joint homeostasis and protect against osteoarthritis progression. Nat Commun. 2019;10(1):2876.
Yang F, Huang R, Ma H, Zhao X, Wang G. miRNA-411 regulates chondrocyte autophagy in osteoarthritis by targeting hypoxia-inducible factor 1 alpha (HIF-1α). Med Sci Monit Int Med J Exp Clin Res. 2020;26:e921155.
Chen G, Liu T, Yu B, Wang B, Peng Q. CircRNA-UBE2G1 regulates LPS-induced osteoarthritis through miR-373/HIF-1a axis. Cell Cycle. 2020;19(13):1696–705.
Fernandez-Torres J, Zamudio-Cuevas Y, Martinez-Nava G, Lopez-Reyes A. Hypoxia-inducible factors (HIFs) in the articular cartilage: a systematic review. Eur Rev Med Pharmacol Sci. 2017;21(12):2800–10.
Ryu JH, Yang S, Shin Y, Rhee J, Chun CH, Chun JS. Interleukin-6 plays an essential role in hypoxia-inducible factor 2α–induced experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum. 2011;63(9):2732–43.
Yang S, Kim J, Ryu J-H, Oh H, Chun C-H, Kim BJ, Min BH, Chun J-S. Hypoxia-inducible factor-2α is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med. 2010;16(6):687–93.
Yang S, Ryu J-H, Oh H, Jeon J, Kwak J-S, Kim J-H, Kim HA, Chun C-H, Chun J-S. NAMPT (visfatin), a direct target of hypoxia-inducible factor-2α, is an essential catabolic regulator of osteoarthritis. Ann Rheum Dis. 2015;74(3):595–602.
Blanco FJ, Guitian R, Vázquez-Martul E, de Toro FJ, Galdo F. Osteoarthritis chondrocytes die by apoptosis: a possible pathway for osteoarthritis pathology. Arthritis Rheum Off J Am Coll Rheumatol. 1998;41(2):284–9.
Heraud F, Heraud A, Harmand M. Apoptosis in normal and osteoarthritic human articular cartilage. Ann Rheum Dis. 2000;59(12):959–65.
Kühn K. D’lima D, Hashimoto S, Lotz M: Cell death in cartilage. Osteoarthritis Cartilage. 2004;12(1):1–16.
Kim H, Blanco F. Cell death and apoptosis in ostearthritic cartilage. Curr Drug Targ. 2007;8(2):333–45.
Ryu J, Shin Y, Huh Y, Yang S, Chun C, Chun J. Hypoxia-inducible factor-2 α regulates Fas-mediated chondrocyte apoptosis during osteoarthritic cartilage destruction. Cell Death Differ. 2012;19(3):440–50.
Yao X, Sun K, Yu S, Luo J, Guo J, Lin J, Wang G, Guo Z, Ye Y, Guo F. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. J Orthop Translat. 2021;27:33–43.
Jing X, Lin J, Du T, Jiang Z, Li T, Wang G, Liu X, Cui X, Sun K. Iron overload is associated with accelerated progression of osteoarthritis: the role of DMT1 mediated iron homeostasis. Front Cell Dev Biol. 2021;8:1699.
Simão M, Gavaia PJ, Camacho A, Porto G, Pinto IJ, Ea HK, Cancela ML. Intracellular iron uptake is favored in Hfe-KO mouse primary chondrocytes mimicking an osteoarthritis-related phenotype. BioFactors. 2019;45(4):583–97.
Kim J-H, Lee G, Won Y, Lee M, Kwak J-S, Chun C-H, Chun J-S. Matrix cross-linking–mediated mechanotransduction promotes posttraumatic osteoarthritis. Proc Natl Acad Sci. 2015;112(30):9424–9.
Wang V, Davis DA, Yarchoan R. Identification of functional hypoxia inducible factor response elements in the human lysyl oxidase gene promoter. Biochem Biophys Res Commun. 2017;490(2):480–5.
Nugent M. MicroRNAs: exploring new horizons in osteoarthritis. Osteoarthritis Cartilage. 2016;24(4):573–80.
Hwang HS, Park SJ, Lee MH, Kim HA. MicroRNA-365 regulates IL-1β-induced catabolic factor expression by targeting HIF-2α in primary chondrocytes. Sci Rep. 2017;7(1):1–13.
Yuan Q, Bleiziffer O, Boos AM, Sun J, Brandl A, Beier JP, Arkudas A, Schmitz M, Kneser U, Horch RE. PHDs inhibitor DMOG promotes the vascularization process in the AV loop by HIF-1a up-regulation and the preliminary discussion on its kinetics in rat. BMC Biotechnol. 2014;14(1):1–10.
Nangaku M, Izuhara Y, Takizawa S, Yamashita T, Fujii-Kuriyama Y, Ohneda O, Yamamoto M, Van Y, Hirayama N, Miyata T. A novel class of prolyl hydroxylase inhibitors induces angiogenesis and exerts organ protection against ischemia. Arterioscler Thromb Vasc Biol. 2007;27(12):2548–54.
Barth S, Nesper J, Hasgall PA, Wirthner R, Nytko KJ, Edlich F, Katschinski DM, Stiehl DP, Wenger RH, Camenisch G. The peptidyl prolyl cis/trans isomerase FKBP38 determines hypoxia-inducible transcription factor prolyl-4-hydroxylase PHD2 protein stability. Mol Cell Biol. 2007;27(10):3758–68.
Fan L, Li J, Yu Z, Dang X, Wang K. The hypoxia-inducible factor pathway, prolyl hydroxylase domain protein inhibitors, and their roles in bone repair and regeneration. Biomed Res Int. 2014;2014:239356.
Zhang L, Li X, Zhang H, Huang Z, Zhang N, Zhang L, Xing R, Wang P. Agnuside alleviates synovitis and fibrosis in knee osteoarthritis through the inhibition of HIF-1α and NLRP3 inflammasome. Mediators Inflamm. 2021;2021:5534614.
Lai K-C, Lu H-F, Chen K-B, Hsueh S-C, Chung J-G, Huang W-W, Chen C-C, Shang H-S. Casticin promotes immune responses, enhances macrophage and NK cell activities, and increases survival rates of leukemia BALB/c mice. Am J Chin Med. 2019;47(01):223–36.
Mu Y, Hao W, Li S. Casticin protects against IL-1β-induced inflammation in human osteoarthritis chondrocytes. Eur J Pharmacol. 2019;842:314–20.
Ma J, Yin G, Lu Z, Xie P, Zhou H, Liu J, Yu L. Casticin prevents DSS induced ulcerative colitis in mice through inhibitions of NF-κB pathway and ROS signaling. Phytother Res. 2018;32(9):1770–83.
Zhou L, Dong X, Wang L, Shan L, Li T, Xu W, Ding Y, Lai M, Lin X, Dai M. Casticin attenuates liver fibrosis and hepatic stellate cell activation by blocking TGF-β/Smad signaling pathway. Oncotarget. 2017;8(34):56267.
Li X, Mei W, Huang Z, Zhang L, Xu B, Shi X, Xiao Y, Ma Z, Liao T, Zhang H. Casticin suppresses monoiodoacetic acid-induced knee osteoarthritis through inhibiting HIF-1α/NLRP3 inflammasome signaling. Int Immunopharmacol. 2020;86:106745.
Sun Y, Wu H, Wang W, Zan R, Peng H, Zhang S, Zhang X. Translational status of biomedical Mg devices in China. Bioact mater. 2019;4:358–65.
Yao H, Xu J, Zheng N, Wang J, Mok S, Lee Y, Shi L, Wang J, Yue J, Yung S. Intra-articular injection of magnesium chloride attenuates osteoarthritis progression in rats. Osteoarthritis Cartilage. 2019;27(12):1811–21.
Yao H, Xu J, Wang J, Zhang Y, Zheng N, Yue J, Mi J, Zheng L, Dai B, Huang W. Combination of magnesium ions and vitamin C alleviates synovitis and osteophyte formation in osteoarthritis of mice. Bioact mater. 2021;6(5):1341–52.
Zhou Y, Ming J, Deng M, Li Y, Li B, Li J, Ma Y, Chen Z, Wang G, Liu S. Chemically modified curcumin (CMC2. 24) alleviates osteoarthritis progression by restoring cartilage homeostasis and inhibiting chondrocyte apoptosis via the NF-κB/HIF-2α axis. J Mol Med. 2020;98(10):1479–91.
Cho C, Kang LJ, Jang D, Jeon J, Lee H, Choi S, Han SJ, Oh E, Nam J, Kim CS. Cirsium japonicum var. maackii and apigenin block Hif-2α-induced osteoarthritic cartilage destruction. J cell mol med. 2019;23(8):5369–79.
Zhou X, Zheng Y, Sun W, Zhang Z, Liu J, Yang W, Yuan W, Yi Y, Wang J, Liu J. D-mannose alleviates osteoarthritis progression by inhibiting chondrocyte ferroptosis in a HIF-2α-dependent manner. Cell Prolif. 2021;54(11):e13134.
Wang P, Meng Q, Wang W, Zhang S, Xiong X, Qin S, Zhang J, Li A, Liu Z. Icariin inhibits the inflammation through down-regulating NF-κB/HIF-2α signal pathways in chondrocytes. Biosci Rep. 2020;40(11):BSR20203107.
Zhou K, He S, Yu H, Pei F, Zhou Z. Inhibition of syndecan-4 reduces cartilage degradation in murine models of osteoarthritis through the downregulation of HIF-2α by miR-96–5p. Lab Invest. 2021. https://doi.org/10.1038/s41374-021-00595-5.
Murahashi Y, Yano F, Kobayashi H, Makii Y, Iba K, Yamashita T, Tanaka S, Saito T. Intra-articular administration of IκBα kinase inhibitor suppresses mouse knee osteoarthritis via downregulation of the NF-κB/HIF-2α axis. Sci Rep. 2018;8(1):1–8.
Pi Y, Zhang X, Shao Z, Zhao F, Hu X, Ao Y. Intra-articular delivery of anti-Hif-2α siRNA by chondrocyte-homing nanoparticles to prevent cartilage degeneration in arthritic mice. Gene Ther. 2015;22(6):439–48.
Zeng CY, Wang XF, Hua FZ. HIF-1alpha in osteoarthritis: from pathogenesis to therapeutic implications. Front Pharmacol. 2022;13:927126.
Jiang Y. Osteoarthritis year in review 2021: biology. Osteoarthritis Cartilage. 2022;30(2):207–15.
Acknowledgements
Not applicable.
Funding
This work is supported by the Science and Technology Innovation Committee of Shenzhen (No. GJHZ20190820115203714, JSGG20191129094218565), the Guangdong Basic and Applied Basic Research Foundation (2021A1515220134), the National Natural Science Foundation of China (No. 31900046, No. 81972085 and No. 82172465) and Guangdong Provincial Key Laboratory of Advanced Biomaterials (2022B1212010003).
Author information
Authors and Affiliations
Contributions
HG and JH contributed equally to this work. HG was a major contributor in drafting of the manuscript. HG, HZ and JH participated in revising the manuscript. YL concepted and prepared figures. DW was a contributor in the constructive discussions. All authors have read and agreed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All of the authors agreed to publish this manuscript.
Competing interests
The authors declare there are no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Guo, H., Huang, J., Liang, Y. et al. Focusing on the hypoxia-inducible factor pathway: role, regulation, and therapy for osteoarthritis. Eur J Med Res 27, 288 (2022). https://doi.org/10.1186/s40001-022-00926-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-022-00926-2