
Abstract
Oral-facial-digital syndromes (OFDS) represent a heterogeneous group of rare developmental disorders affecting the mouth, the face and the digits. Additional signs may involve brain, kidneys and other organs thus better defining the different clinical subtypes. With the exception of OFD types I and VIII, which are X-linked, the majority of OFDS is transmitted as an autosomal recessive syndrome. A number of genes have already found to be mutated in OFDS and most of the encoded proteins are predicted or proven to be involved in primary cilia/basal body function. Preliminary data indicate a physical interaction among some of those proteins and future studies will clarify whether all OFDS proteins are part of a network functionally connected to cilia. Mutations in some of the genes can also lead to other types of ciliopathies with partially overlapping phenotypes, such as Joubert syndrome (JS) and Meckel syndrome (MKS), supporting the concept that cilia-related diseases might be a continuous spectrum of the same phenotype with different degrees of severity. To date, seven of the described OFDS still await a molecular definition and two unclassified forms need further clinical and molecular validation. Next-generation sequencing (NGS) approaches are expected to shed light on how many OFDS geneticists should consider while evaluating oral-facial-digital cases. Functional studies will establish whether the non-ciliary functions of the transcripts mutated in OFDS might contribute to any of the phenotypic abnormalities observed in OFDS.
Similar content being viewed by others


Background
The oral-facial-digital syndromes (OFDS) represent a group of rare developmental disorders characterized by abnormalities of the face, oral cavity and digits. Additional signs involving the central nervous system (CNS), and visceral organs, such as the kidney, are also frequently observed. The first case presenting this condition was reported in 1941 [1] and since then a number of different OFDS types with overlapping phenotypes have been described [2, 3] (Table 1). Among the different types, OFD type I is the most frequently observed and can be easily recognized by its typical X-linked dominant male-lethal pattern of inheritance in familial cases. Most of the other OFDS are transmitted as autosomal recessive syndromes or represent sporadic cases. In the last few years, 11 genes responsible for OFDS have been identified allowing a better clinical and genetic definition for this heterogeneous condition. This review will focus on the most recent findings on OFDI, III, IV, VI, IX, XIV and two unclassified OFD subtypes. For all other OFDS please refer to [3]. On the basis of the recent molecular data, we can distinguish (1) two more common types (OFDI and OFDVI), for which the causative genes have been identified; (2) four rare subtypes for which the causative gene has also been identified (OFDIII, OFDIV, OFDIX and OFDXIV), thus allowing molecular diagnosis; (3) two unclassified rare OFD subtypes whose causative genes have been identified but that still require further clinical and molecular validation and (4) additional unclassified OFDS which still await molecular characterization and further definition (Table 1). Table 2 reports a clinical summary of the different OFDS clearly identified to date.
Review
OFD type I syndrome (OFDI)
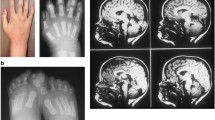
OFDI was described in 1954 [4] and further defined in 1962 [5]. It has an estimated incidence of 1:50,000 live births [6] and it has been reported in different ethnic backgrounds with no evidence of founder effect. It is transmitted as an X-linked dominant condition with male lethality, which usually occurs during the first and second trimester of pregnancy [7–9]. Only a small percentage of cases display familiar inheritance and the majority of mutations are sporadic (~75 %). The clinical spectrum of the disease includes craniofacial, oral and skeletal abnormalities in >80 % of cases (see [7] for details). Renal cystic disease is commonly observed as well as involvement of the CNS, which includes brain developmental anomalies and cognitive defects [10, 11]. Additional findings may include pancreatic, hepatic, and/or ovarian cysts and hearing defects [7, 12]. The gene responsible for OFD type I syndrome was identified on the short arm of the X chromosome [13]. Different mutations have been reported to date, including frameshifts, which represent the majority of mutations (>60 %), splicing, missense, nonsense and genomic rearrangements [7, 10, 11]. Additional file 1: Table S1 summarizes the mutations identified to date. The responsible gene, initially known as CXORF5 and subsequently named OFD1, encodes for the centrosomal/basal body OFD1 protein [14, 15] required for left–right axis specification and for primary cilia formation [16–21]. OFD type I is a male-lethal disorder and male cases with OFD1 mutations associated to a classical OFDI phenotype have never been described. Interestingly, three affected males with “unclassified” X-linked lethal congenital malformation syndrome and a splice mutation in the OFD1 gene have been described. The mother was mildly affected and presented only few accessory oral frenulae and irregular teeth [22]. OFD1 mutations have also been reported in males in X-linked recessive conditions, namely (1) a mental retardation syndrome comprising macrocephaly and ciliary dysfunction [23] mapping to the same locus as Simpson–Golabi–Behemel syndrome type 2 (SGBS2); (2) Joubert syndrome (JS) patients (JBT10) [24–26] and (3) retinitis pigmentosa (RP23) [27]. These findings suggest that mutations in the OFD1 gene may result in a single syndrome spectrum characterized by wide intra- and inter-familial phenotypic variability possibly depending on the contribution of still unknown genetic modifiers.
OFD type III syndrome (OFDIII)
OFDIII was described in 1971 [28]. Affected patients present with orofaciodigital findings similar to those described in the other OFDS, involvement of the CNS and renal disease. The typical manifestation that is only seen, among OFDs, in OFDIII cases is an oculomotor apraxia resulting in “metronome eye movements”. Recent data identified mutations in TMEM231 (Additional file 2: Table S2) in two affected OFDIII siblings during a targeted medical sequencing of 1056 individuals with nephronophthisis-related ciliopathies [29]. The two cases presented with the typical eye movements, lingual hamartomas, postaxial polydactyly and involvement of the CNS (intellectual disabilities, cerebellar vermis hypoplasia and Dandy Walker malformation with cystic dilation of the 4th ventricle). Both cases were born with a normal renal morphology and function but developed end stage renal failure within the third decade of life. In the same report, recurrent TMEM231 mutations were also identified in MKS patients [29]. Functional studies demonstrated that TMEM231 is involved in ciliary functions. Accordingly, mice with mutations in Tmem231 display a clear ciliopathy phenotype including renal cystic disease, malformations of the hepatic ductal plate and skeletal abnormalities [29].
OFD type IV syndrome (OFDIV)
This OFD subtype was originally described in a familial case in which two affected sisters displayed the typical oral-facial-digital findings in addition to severe tibial dysplasia [30, 31]. In 2012, a genome wide homozygosity mapping approach was undertaken on a case born to a consanguineous family and displaying facial dysmorphism with lobulated tongue, polydactyly of all four limbs, renal cystic disease, liver ductal plate proliferation, occipital encephalocele and other brain anomalies. X-rays examination revealed severe tibia hypoplasia and bowing of long bones. Targeted resequencing of candidate genes in homozygosity regions identified a unique nonsense mutation, c.1222C > T (p.Glu408*) in tectonic-3 (TCTN3). Analysis of additional cases led to the identification of three TCTN3 truncating mutations segregating within the affected family members with the expected autosomal recessive inheritance pattern and two compound heterozygous frameshift mutations [32] (Additional file 2: Table S2). All affected cases presented with skeletal dysplasia with long bone bowing and tibia hypoplasia and only two cases displayed associated orofaciodigital findings. Interestingly, in the same study, the authors reported a TCTN3 mutation in a JS case (c.940G > A). The JS mutation involves a nucleotide which is not affected in OFDIV patients but additional studies are required to establish a clear genotype/phenotype correlation. On the basis of these results, the authors concluded that OFDIV phenotypes can include long bone bowing, tibia hypoplasia, cystic kidney, encephalocele and other brain malformations [32].
OFD type VI syndrome (OFDVI)
This form was initially described in 1980 in a Hungarian isolated population presenting with oro-facio-digital findings associated with central and or/cerebellar anomalies [33]. OFD VI is characterized by the presence of a “so-called” molar tooth sign (MTS) on brain MRI associated to one or more of the following: (1) hamartoma(s) of the tongue and/or additional frenula; (2) digital abnormalities (e.g. mesoaxial polydactyly of one or more hands or feet, postaxial and preaxial polydactyly) and (3) hypothalamic hamartoma. Additional oral-facial (e.g. cleft lip and palate) and/or digital signs may also be observed. The presence of the MTS allowed researchers to ascribe OFDVI to the group of Joubert syndrome (JS)-associated disorders. OFDVI differs from pure JS cases for the presence of the oral-facial-digital findings and can be defined as a rare phenotype of JS. Different studies analysed OFDVI patients and identified mutations in TMEM216, OFD1, C5ORF42 and TMEM107 [24, 34–37] (Additional file 2: Table S2). These findings highlight the clinical and genetic overlap among ciliopathies (see below). More recently, a comparison of C5ORF42 mutated versus non-mutated OFDVI cases suggested a major role for this gene in limb development [38]. Interestingly, C5ORF42 and TMEM107 were also found mutated in pure JS cases [38]. The causality of TMEM107 mutations in ciliopathies was confirmed by independent groups [39, 40].
OFD type IX syndrome (OFDIX)
OFD type IX is characterized by retinal colobomata in addition to the typical oro-facio-digital findings. It is inherited as an autosomal recessive trait. Recently, a whole-exome sequencing approach identified mutations in TBC1D32 and SCLT1 in two patients with a severe ciliopathy phenotype [41]. Case 1 was born to healthy consanguineous parents and displayed midline defects including hypertelorism, midline clefts and severe choanal stenosis, left hand postaxial polydactyly and eye abnormalities (right microphthalmia, left anophthalmia, bilateral optic disc coloboma). Brain malformations and cardiac defects were also described. Whole-exome sequencing identified a splicing mutation in TBC1D32 (Additional file 2: Table S2) leading to in-frame truncation of 47 amino acids. Case 2 was also born to healthy consanguineous parents and displayed severe midline cleft lip and palate, microcephaly and choanal atresia. He also presented severe coloboma and congenital heart disease. Brain malformations were also present as well as abnormal inner ear structures. In this case, exon sequencing revealed a splicing mutation in SCLT1 resulting in complete skipping of exon 5 and the introduction of a premature stop codon (Additional file 2: Table S2). Due to the presence of the eye abnormalities, these two cases were classified as OFDIX. TBC1D32 and SCLT1 have both been linked to ciliogenesis. TBC1D32 encodes a ciliary protein predicted to contain a Tre-2, Bub2 and Cdc16 (TBC) domain (TBC1D32). On the other hand, SCLT1 is an important component of the distal appendages, a centrosomal extension that establishes the connection between the mother centriole and the plasma membrane and its deficiency blocks ciliogenesis in the early phases of cilia formation [42].
OFD type XIV syndrome (OFDXIV)
This OFD subtype was defined after the identification of mutations in the C2CD3 gene. The first homozygous nonsense mutation (c.184C > T; p.Arg62*) was identified in a familial case in which the index case presented with classical OFD signs (lingual hamartoma, cleft and lobulated tongue, cleft palate, buccal frenulae, bilateral preaxial polydactyly of feet and postaxial polydactyly of hands) accompanied by microcephaly, micropenis and severe intellectual disabilities. Brain MRI revealed the presence of MTS, the cerebellar anomaly characteristic of JS, and other brain abnormalities (corpus callosum hypoplasia, subarachnoid cysts in the right occipital lobe and the posterior fossa, and incomplete myelination of the white matter). The presence of the MTS is intriguing and suggests a possible link with JS. His younger sister displayed a similar phenotype worsened by the presence of cardiac malformation leading to neonatal death [43]. An additional C2CD3 compound heterozygous mutation (Additional file 2: Table S2) was identified during the screening of 34 OFD cases negative for mutations in known OFD genes in a male foetus exhibiting severe microcephaly, bilateral duplicated hallux and postaxial polydactyly, micropenis, kidney hypoplasia, corpus callosum abnormalities and inferior vermian hypoplasia with posterior cyst [43]. On the basis of these findings and of the peculiar features of microcephaly and cerebral malformations, this OFD subtype was classified as OFDXIV. Functional studies demonstrated that C2Cd3 co-localizes and physically interacts with OFD1 and is involved in centriole elongation, thus defining centriole length regulation as an emerging pathogenetic mechanism in ciliopathies [43].
Unclassified OFDS
A number of OFD subtypes still require molecular definition and characterization of OFD patients negative for mutations in known OFD-associated genes will lead to identification also of unclassified OFD subtypes. This has already happened in the following two examples.
Whole-exome sequencing revealed compound WDPCP heterozygous mutations in a female child with an unclassified form of OFD [44]. This child displayed type A postaxial polydactyly of both hands and 2/3 toe syndactyly, congenital heart defects and tongue hamartomas. The two mutations (Additional file 2: Table S2) were inherited from the asymptomatic father and mother. Interestingly, mutations in the WDPCP transcript, which regulates planar cell polarity and ciliogenesis [45], have also been reported in a patient with Bardet–Biedl syndrome (BBS) [46], one of the first ciliopathy characterized.
Autozygosity mapping identified a minimal interval on chromosome 1 in two multiplex families of Arabian origin displaying oral (tongue lobulation, cleft palate, bifid uvula), facial (frontal bossing, midline lip defects), digital (polydactyly) signs accompanied by additional abnormalities [47]. Exome sequencing analysis identified two homozygous mutations in the DDX59 transcript segregating with the disease in family 1 and 2 (Additional file 2: Table S2). DDX59 is highly enriched in the developing mouse palate and limb buds. Immunofluorescence analysis demonstrated a dynamic nuclear and cytoplasmic localization and normal ciliogenesis pattern in patients’ fibroblasts [47].
In both cases, future studies will clarify whether these conditions represent new OFD subtypes and identification of mutations in additional patients will be necessary to establish these two genes as bona fide ciliopathy genes.
The link between OFDS and other ciliopathies
Mutations in ciliary genes are associated with a wide spectrum of clinical conditions that extends from viable to severe, lethal phenotypes. Oligogenic inheritance may explain this variability implying genetic interaction among different loci to cause/modulate the phenotype. This has already been shown for BBS, nephronophthisis (NPHP) and JS [48–50]. Interestingly, the unclassified OFD case with mutations in WDPCP also carried a deleterious deletion in IQCB1 which is associated to another ciliopathy, Senior–Loken syndrome, type 5 (SLSN5) characterized by early onset retinopathy and renal disease [44]. Mutations in the OFD1 gene can be associated to a very specific phenotype as such in X-linked recessive retinitis pigmentosa (RP23) or more pleiotropic disorders such as in X-linked dominant OFD type 1, and X-linked recessive Joubert syndrome (JBTS10) and a mental retardation syndrome comprising macrocephaly and ciliary dysfunction [23]. It will be interesting to evaluate whether additional mutations in other ciliary transcript may contribute to the phenotypic outcome of OFD1 mutations.
The ciliopathy protein network can be divided in distinct but connected modules: the centrosome/basal body/pericentriolar material, the transition zone, the intraflagellar (IFT) complexes and the BBSome. OFDS genes are often mutated in other ciliopathies, especially Joubert and Meckel Gruber Syndromes. These conditions are mainly due to mutations in genes encoding proteins of the centrosome/basal body/pericentriolar material and transition zone modules (Table 3; Fig. 1) suggesting that these cilia structures have a predominant role in the pathomechanisms underlying OFDS, JS and MKS syndromes.

OFDS proteins map to defined cilia compartments. Left panel, schematic representation of primary cilia. The localization of proteins encoded by OFDS transcripts is depicted. Right panel, the precise cilia localization is defined. The column Cilia/Shh indicate whether a ciliary localization or perturbation of the Shh pathway has been demonstrated (+) or not (−). NK not known
Genes involved in OFDS: the ciliary connection
The gene responsible for OFD type I was identified in 2001 and for a while remained the only OFD gene known. In the last 3–4 years, a number of genes responsible for other OFDS have been identified mainly through next-generation sequencing (NGS) approaches. The majority of OFD genes identified localizes to cilia components (Fig. 1) and/or influence ciliogenesis with the exception of DDX59 and C5ORF52. In this section, the information available on link between genes mutated in OFDS and ciliary functions of ciliary signalling will be reviewed.
Functional studies demonstrated that OFD1 acts at the distal centriole to build distal appendages [18], thus contributing to cilia formation although in a content-specific fashion [16, 19–21]. In addition, in vivo and in vitro studies demonstrated that OFD1-depleted models show defective Shh [16, 20, 21, 51–53] and Wnt [54] signalling. In particular, impairment of Shh signalling from early stages of development may contribute to explain the skeletal malformations observed in OFDI patients.
TCTN3 and TMEM231 are components of a MKS complex localized at the transition zone of primary cilia and physically interact with each other. Functional studies demonstrated that they are both required for ciliogenesis and Shh signalling [29, 32, 55, 56].
TMEM216 localizes at the base of primary cilia and its loss results in defective ciliogenesis and centrosomal docking, with concomitant hyperactivation of RhoA and Dishevelled [35]. No information is available on the role of this transcript in Shh signalling.
TMEM107 and the planar cell polarity WDPCP proteins localize both at the transition zone, contribute to mammalian ciliogenesis [37, 45] and are required for Shh signalling [45, 57].
TBC1D32 is a ciliary protein [58] although the precise localization within cilia is not known. Functional studies demonstrate that TBC1D32 controls ciliary morphology and is required for Shh pathway [59].
SCLT1 and C2CD3 localize at centrioles where C2CD3 co-localizes and physically interacts with OFD1. Both proteins are necessary for ciliogenesis and C2CD3 is also required for Hedgehog signalling in mouse [42, 43, 60].
C5ORF42, also known as NKAPP1, is poorly characterized and no information is available on the role of this transcript in cilia or cilia-mediated signalling [61].
Finally, DDX59 is a member of the DEAD-box-containing RNA helicases with currently unknown function relating to cilia. Functional studies indicated that fibroblasts from affected individuals display a normal ciliogenesis pattern in the presence of reduced Shh signalling [47].
The data summarized above seem to indicate a major role for centrosomal/centriolar function in the pathomechanisms underlying OFD syndrome and components of these cilia-related cellular compartments should be considered candidate genes for the unresolved OFDS. One of the puzzling questions in OFDS as well as in other ciliopathies is how much of the phenotype is due to cilia dysfunction and how much is due to gene functions not related to cilia. Shh impairment which has been demonstrated in more than one OFDS and is linked to the ciliary function of the genes may explain the skeletal and some of the neurological findings observed. However, as we are learning by omics approaches, proteins may display different intracellular localization and functions [62, 63]. OFD1, for example, is localized both to centrosome/basal body and nucleus [14, 15]. Future studies will clarify the contribution of non-ciliary functions of OFD genes to the clinical spectrum of these conditions.
Conclusions and future perspectives
A thorough clinical and molecular characterization of OFD patients will be critical to define how many subtypes do really exist for this pleiotropic condition. NGS-based approaches will define how many genes underlie OFDS and clinical studies will define how many different conditions can be clearly identified. Functional studies will clarify whether ciliopathies can be redefined not according to the genes mutated or the phenotype observed but according to the ciliary structural element functionally compromised. This knowledge may aid in designing the most appropriate approach to slow down disease progression. Finally, it is time for researchers to start studying and defining the non-ciliary functions of the transcripts mutated in OFDS (and other ciliopathies) to understand whether abnormal cilia can justify all the phenotypic abnormalities observed in OFDS.
Abbreviations
- BBS:
-
Bardet–Biedl syndrome
- NGS:
-
next-generation sequencing
- OFD:
-
oral-facial-digital, oro-facio-digital
- OFDS:
-
oral-facial-digital/oro-facio-digital syndromes
- JS:
-
Joubert syndrome
- MKS:
-
Meckel syndrome
- CNS:
-
central nervous system
- Shh:
-
sonic hedgehog
- MTS:
-
molar tooth sign
References
Mohr O. A hereditary lethal syndrome in man. Avh Norske Videnskad Oslo. 1941;14:1–18.
Toriello HV. Oral-facial-digital syndromes, 1992. Clin Dysmorph. 1993;2:95–105.
Gurrieri F, Franco B, Toriello H, Neri G. Oral-facial-digital syndromes: a review and diagnostic guidelines. Am J Med Genet. 2007;143A(24):3314–23.
Papillon-Leage M, Psaume J. Une malformation hereditaire de la muquese buccale: brides et freins anomaux. Rev Stomatol. 1954;55:209–27.
Gorlin RJ, Psaume J. Orofaciodigital dysotosis: a new syndrome. A study of 22 cases. J Pediatr. 1962;61:520–30.
Wahrman J, Berant M, Jacobs J, Aviad I, Ben-Hur N. The oral-facial-digital syndrome: a male-lethal condition in a boy with 47/xxy chromosomes. Pediatrics. 1966;37(5):812–21.
Macca M, Franco B. The molecular basis of oral-facial-digital syndrome, type 1. Am J Med Genet C Semin Med Genet. 2009;151C(4):318–25.
Doege T, Thuline H, Priest J, Norby D, Bryant J. Studies of a family with the oral-facial-digital syndrome. N Engl J Med. 1964;271:1073–80.
Schäfer RW, Kantner G. X-linked dominant inherited diseases with lethality in hemizygous males. Hum Genet. 1983;64:1–23.
Bisschoff IJ, Zeschnigk C, Horn D, Wellek B, Riess A, Wessels M, et al. Novel mutations including deletions of the entire OFD1 gene in 30 families with type 1 orofaciodigital syndrome: a study of the extensive clinical variability. Hum Mutat. 2013;34(1):237–47. doi:10.1002/humu.22224.
Del Giudice E, Macca M, Imperati F, D’Amico A, Parent P, Pasquier L, et al. CNS involvement in OFD1 syndrome: a clinical, molecular, and neuroimaging study. Orphanet J Rare Dis. 2014;9:74. doi:10.1186/1750-1172-9-74.
Chetty-John S, Piwnica-Worms K, Bryant J, Bernardini I, Fischer RE, Heller T, et al. Fibrocystic disease of liver and pancreas; under-recognized features of the X-linked ciliopathy oral-facial-digital syndrome type 1 (OFD I). Am J Med Genet Part A. 2010;152A(10):2640–5. doi:10.1002/ajmg.a.33666.
Ferrante MI, Giorgio G, Feather SA, Bulfone A, Wright V, Ghiani M, et al. Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet. 2001;68(3):569–76.
Romio L, Fry AM, Winyard PJ, Malcolm S, Woolf AS, Feather SA. OFD1 is a centrosomal/basal body protein expressed during mesenchymal-epithelial transition in human nephrogenesis. J Am Soc Nephrol. 2004;15(10):2556–68.
Giorgio G, Alfieri M, Prattichizzo C, Zullo A, Cairo S, Franco B. Functional Characterization of the OFD1 Protein Reveals a Nuclear Localization and Physical Interaction with Subunits of a Chromatin Remodeling Complex. Mol Biol Cell. 2007;18(11):4397–404.
Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, et al. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006;38(1):112–7.
Ferrante MI, Romio L, Castro S, Collins JE, Goulding DA, Stemple DL, et al. Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Hum Mol Genet. 2009;18(2):289–303.
Singla V, Romaguera-Ros M, Garcia-Verdugo JM, Reiter JF. Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev Cell. 2010;18(3):410–24. doi:10.1016/j.devcel.2009.12.022.
Zullo A, Iaconis D, Barra A, Cantone A, Messaddeq N, Capasso G, et al. Kidney-specific inactivation of Ofd1 leads to renal cystic disease associated with upregulation of the mTOR pathway. Hum Mol Genet. 2010;19(14):2792–803. doi:10.1093/hmg/ddq180.
Bimonte S, De Angelis A, Quagliata L, Giusti F, Tammaro R, Dallai R, et al. Ofd1 is required in limb bud patterning and endochondral bone development. Dev Biol. 2011;349(2):179–91. doi:10.1016/j.ydbio.2010.09.020.
D’Angelo A, De Angelis A, Avallone B, Piscopo I, Tammaro R, Studer M, et al. Ofd1 Controls Dorso-Ventral Patterning and Axoneme Elongation during Embryonic Brain Development. PLoS ONE. 2012;7(12):52937. doi:10.1371/journal.pone.0052937.
Tsurusaki Y, Kosho T, Hatasaki K, Narumi Y, Wakui K, Fukushima Y, et al. Exome sequencing in a family with an X-linked lethal malformation syndrome: clinical consequences of hemizygous truncating OFD1 mutations in male patients. Clin Genet. 2013;83(2):135–44. doi:10.1111/j.1399-0004.2012.01885.x.
Budny B, Chen W, Omran H, Fliegauf M, Tzschach A, Wisniewska M, et al. A novel X-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome. Hum Genet. 2006;120(2):171–8.
Coene KL, Roepman R, Doherty D, Afroze B, Kroes HY, Letteboer SJ, et al. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet. 2009;85(4):465–81. doi:10.1016/j.ajhg.2009.09.002.
Field M, Scheffer IE, Gill D, Wilson M, Christie L, Shaw M, et al. Expanding the molecular basis and phenotypic spectrum of X-linked Joubert syndrome associated with OFD1 mutations. Eur J Hum Genet. 2012;20(7):806–9. doi:10.1038/ejhg.2012.9.
Juric-Sekhar G, Adkins J, Doherty D, Hevner RF. Joubert syndrome: brain and spinal cord malformations in genotyped cases and implications for neurodevelopmental functions of primary cilia. Acta Neuropathol. 2012;123(5):695–709. doi:10.1007/s00401-012-0951-2.
Webb TR, Parfitt DA, Gardner JC, Martinez A, Bevilacqua D, Davidson AE, et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum Mol Genet. 2012;21(16):3647–54. doi:10.1093/hmg/dds194.
Sugarman GI, Katakia M, Menkes J. See-saw winking in a familial oral-facial-digital syndrome. Clin Genet. 1971;2(4):248–54.
Roberson EC, Dowdle WE, Ozanturk A, Garcia-Gonzalo FR, Li C, Halbritter J, et al. TMEM231, mutated in orofaciodigital and Meckel syndromes, organizes the ciliary transition zone. J Cell Biol. 2015;209(1):129–42. doi:10.1083/jcb.201411087.
Baraitser M. The orofaciodigital (OFD) syndromes. J Med Genet. 1986;23(2):116–9.
Burn J, Dezateux C, Hall CM, Baraitser M. Orofaciodigital syndrome with mesomelic limb shortening. J Med Genet. 1984;21(3):189–92.
Thomas S, Legendre M, Saunier S, Bessieres B, Alby C, Bonniere M, et al. TCTN3 mutations cause Mohr–Majewski syndrome. Am J Hum Genet. 2012;91(2):372–8. doi:10.1016/j.ajhg.2012.06.017.
Varadi V, Szabo L, Papp Z. Syndrome of polydactyly, cleft lip/palate or lingual lump, and psychomotor retardation in endogamic gypsies. J Med Genet. 1980;17(2):119–22.
Darmency-Stamboul V, Burglen L, Lopez E, Mejean N, Dean J, Franco B, et al. Detailed clinical, genetic and neuroimaging characterization of OFD VI syndrome. Eur J Med Genet. 2013;56(6):301–8. doi:10.1016/j.ejmg.2013.03.004.
Valente EM, Logan CV, Mougou-Zerelli S, Lee JH, Silhavy JL, Brancati F, et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert. Meckel and related syndromes. Nat Genet. 2010;42(7):619–25. doi:10.1038/ng.594.
Lopez E, Thauvin-Robinet C, Reversade B, Khartoufi NE, Devisme L, Holder M, et al. C5orf42 is the major gene responsible for OFD syndrome type VI. Hum Genet. 2014;133(3):367–77. doi:10.1007/s00439-013-1385-1.
Lambacher NJ, Bruel AL, van Dam TJ, Szymanska K, Slaats GG, Kuhns S, et al. TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nat Cell Biol. 2015. doi:10.1038/ncb3273.
Romani M, Mancini F, Micalizzi A, Poretti A, Miccinilli E, Accorsi P, et al. Oral-facial-digital syndrome type VI: is C5orf42 really the major gene? Hum Genet. 2015;134(1):123–6. doi:10.1007/s00439-014-1508-3.
Iglesias A, Anyane-Yeboa K, Wynn J, Wilson A, Truitt Cho M, Guzman E, et al. The usefulness of whole-exome sequencing in routine clinical practice. Genet Med. 2014;16(12):922–31. doi:10.1038/gim.2014.58.
Shaheen R, Almoisheer A, Faqeih E, Babay Z, Monies D, Tassan N, et al. Identification of a novel MKS locus defined by TMEM107 mutation. Hum Mol Genet. 2015;24(18):5211–8. doi:10.1093/hmg/ddv242.
Adly N, Alhashem A, Ammari A, Alkuraya FS. Ciliary genes TBC1D32/C6orf170 and SCLT1 are mutated in patients with OFD type IX. Hum Mutat. 2014;35(1):36–40. doi:10.1002/humu.22477.
Tanos BE, Yang HJ, Soni R, Wang WJ, Macaluso FP, Asara JM, et al. Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev. 2013;27(2):163–8. doi:10.1101/gad.207043.112.
Thauvin-Robinet C, Lee JS, Lopez E, Herranz-Perez V, Shida T, Franco B, et al. The oral-facial-digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nat Genet. 2014;46(8):905–11. doi:10.1038/ng.3031.
Saari J, Lovell MA, Yu HC, Bellus GA. Compound heterozygosity for a frame shift mutation and a likely pathogenic sequence variant in the planar cell polarity-ciliogenesis gene WDPCP in a girl with polysyndactyly, coarctation of the aorta, and tongue hamartomas. Am J Med Genet A. 2015;167A(2):421–7. doi:10.1002/ajmg.a.36852.
Cui C, Chatterjee B, Lozito TP, Zhang Z, Francis RJ, Yagi H, et al. Wdpcp, a PCP protein required for ciliogenesis, regulates directional cell migration and cell polarity by direct modulation of the actin cytoskeleton. PLoS Biol. 2013;11(11):e1001720. doi:10.1371/journal.pbio.1001720.
Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, et al. Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science. 2010;329(5997):1337–40. doi:10.1126/science.1191184.
Shamseldin HE, Rajab A, Alhashem A, Shaheen R, Al-Shidi T, Alamro R, et al. Mutations in DDX59 implicate RNA helicase in the pathogenesis of orofaciodigital syndrome. Am J Hum Genet. 2013;93(3):555–60. doi:10.1016/j.ajhg.2013.07.012.
Novas R, Cardenas-Rodriguez M, Irigoin F, Badano JL. Bardet–Biedl syndrome: is it only cilia dysfunction? FEBS Lett. 2015;589(22):3479–91. doi:10.1016/j.febslet.2015.07.031.
Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013;12(9):894–905. doi:10.1016/S1474-4422(13)70136-4.
Hoefele J, Wolf MT, O’Toole JF, Otto EA, Schultheiss U, Deschenes G, et al. Evidence of oligogenic inheritance in nephronophthisis. J Am Soc Nephrol. 2007;18(10):2789–95. doi:10.1681/ASN.2007020243.
Hunkapiller J, Singla V, Seol A, Reiter JF. The ciliogenic protein Oral-Facial-Digital 1 regulates the neuronal differentiation of embryonic stem cells. Stem Cells Dev. 2011;20(5):831–41. doi:10.1089/scd.2010.0362.
Khonsari RH, Seppala M, Pradel A, Dutel H, Clement G, Lebedev O, et al. The buccohypophyseal canal is an ancestral vertebrate trait maintained by modulation in sonic hedgehog signaling. BMC Biol. 2013;11:27. doi:10.1186/1741-7007-11-27.
Liu YP, Tsai IC, Morleo M, Oh EC, Leitch CC, Massa F, et al. Ciliopathy proteins regulate paracrine signaling by modulating proteasomal degradation of mediators. J Clin Invest. 2014;124(5):2059–70. doi:10.1172/JCI71898.
Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Chen MH, et al. Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol. 2008;10(1):70–6.
Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, et al. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2012;14(1):61–72. doi:10.1038/ncb2410.
Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43(8):776–84. doi:10.1038/ng.891.
Christopher KJ, Wang B, Kong Y, Weatherbee SD. Forward genetics uncovers Transmembrane protein 107 as a novel factor required for ciliogenesis and Sonic hedgehog signaling. Dev Biol. 2012;368(2):382–92. doi:10.1016/j.ydbio.2012.06.008.
Ishikawa H, Thompson J, Yates JR 3rd, Marshall WF. Proteomic analysis of mammalian primary cilia. Curr Biol. 2012;22(5):414–9. doi:10.1016/j.cub.2012.01.031.
Ko HW, Norman RX, Tran J, Fuller KP, Fukuda M, Eggenschwiler JT. Broad-minded links cell cycle-related kinase to cilia assembly and hedgehog signal transduction. Dev Cell. 2010;18(2):237–47. doi:10.1016/j.devcel.2009.12.014.
Hoover AN, Wynkoop A, Zeng H, Jia J, Niswander LA, Liu A. C2cd3 is required for cilia formation and Hedgehog signaling in mouse. Development. 2008;135(24):4049–58. doi:10.1242/dev.029835.
Srour M, Schwartzentruber J, Hamdan FF, Ospina LH, Patry L, Labuda D, et al. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am J Hum Genet. 2012;90(4):693–700. doi:10.1016/j.ajhg.2012.02.011.
Amato R, Morleo M, Giaquinto L, di Bernardo D, Franco B. A system biology approach to dissect the cilia/centrosome complex interactome. BMC Genom. 2014;15:658. doi:10.1186/1471-2164-15-658.
Gupta GD, Coyaud E, Goncalves J, Mojarad BA, Liu Y, Wu Q, et al. A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell. 2015;163(6):1484–99. doi:10.1016/j.cell.2015.10.065.
Thauvin-Robinet C, Cossee M, Cormier-Daire V, Van Maldergem L, Toutain A, Alembik Y, et al. Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral-facial-digital syndrome type 1: a French and Belgian collaborative study. J Med Genet. 2006;43(1):54–61.
Prattichizzo C, Macca M, Novelli V, Giorgio G, Barra A, Franco B. Mutational spectrum of the oral-facial-digital type I syndrome: a study on a large collection of patients. Hum Mutat. 2008;29(10):1237–46.
Poretti A, Vitiello G, Hennekam RC, Arrigoni F, Bertini E, Borgatti R, et al. Delineation and diagnostic criteria of oral-facial-digital syndrome type VI. Orphanet J Rare Dis. 2012;7:4. doi:10.1186/1750-1172-7-4.
Edwards M, Mulcahy D, Turner G. X-linked recessive inheritance of an orofaciodigital syndrome with partial expression in females and survival of affected males. Clin Genet. 1988;34(5):325–32.
Erickson RP, Bodensteiner JB. Oro-facial-digital syndrome IX with severe microcephaly: a new variant in a genetically isolated population. Am J Med Genet A. 2007;143A(24):3309–13. doi:10.1002/ajmg.a.31976.
Figuera LE, Rivas F, Cantu JM. Oral-facial-digital syndrome with fibular aplasia: a new variant. Clin Genet. 1993;44(4):190–2.
Gabrielli O, Ficcadenti A, Fabrizzi G, Perri P, Mercuri A, Coppa GV, et al. Child with oral, facial, digital, and skeletal anomalies and psychomotor delay: a new OFDS form? Am J Med Genet. 1994;53(3):290–3. doi:10.1002/ajmg.1320530315.
Moran-Barroso V, Flores MV, Garcia-Cavazos R, Kofman-Alfaro S, Saavedra-Ontiveros D. Oral-facial-digital (OFD) syndrome with associated features: a new syndrome or genetic heterogeneity and variability? Clin Dysmorphol. 1998;7(1):55–7.
Degner D, Bleich S, Riegel A, Ruther E. Orofaciodigital syndrome—a new variant? Psychiatric, neurologic and neuroradiological findings. Fortschr Neurol Psychiatr. 1999;67(12):525–8. doi:10.1055/s-2007-995229.
Authors’ contributions
CT-R and BF wrote the manuscript. Both authors read and approved the final manuscript.
Acknowledgements
This work was supported by the “Italian Fondazione Telethon” [TGM11CB3 to BF].
Authors’ information
BF is the Principal Investigator at the Telethon Institute of Genetics and Medicine (TIGEM), and also holds an appointment as the Professor of Medical Genetics at the Federico II University, Naples Italy. CT-R is a Professor at the Université de Bourgogne and a hospital practitioner at the Genetics Unit, Hopital d’Enfants, Dijon France. BF and CT-H largely contributed to the characterization of OFD cases.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Franco, B., Thauvin-Robinet, C. Update on oral-facial-digital syndromes (OFDS). Cilia 5, 12 (2016). https://doi.org/10.1186/s13630-016-0034-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13630-016-0034-4