
Abstract
Oral–facial–digital syndrome VI (OFD6 OMIM #277170), also called Varadi–Papp syndrome, is a ciliopathy inherited in an autosomal recessive pattern. Recently, mutations in C5orf42 (OMIM #614571) have been associated with OFD6. OFD6 overlaps with Joubert syndrome and mutations in C5orf42 were described in Joubert syndrome 17 (JBTS17, OMIM #614571). Using exome sequencing we report three novel variants and one previously reported variant in the C5orf42 gene in patients with OFD6.
Similar content being viewed by others

Joubert syndrome (JBTS) comprises hypotonia, apnea or hyperpnea in infancy, oculomotor apraxia and variable intellectual impairment.1 The key feature is a complex brain malformation comprising small or absent cerebellar vermis, deepened interpeduncular fossa and elongated superior cerebellar peduncles called the molar tooth sign. Involvement of liver (i.e., congenital liver fibrosis), kidneys (i.e., polycystic kidneys), eyes (i.e., retinopathy), polydactyly or oral–facial abnormalities led to the subclassification of Joubert syndrome and related disorders.2 Oral–facial–digital syndrome VI (OFD6) overlaps with JBTS with respect to key features and also has tongue hamartomas and/or frenulae, upper lip notch, mesoaxial polydactyly of hands or feet, and hypothalamic hamartoma. It has been categorized as Joubert syndrome with oro-facio-digital defects (JS-OFD or JBTS17, OMIM #614615).
Family 1: Two fetuses conceived by a consanguineous (first cousin union) couple from Saudi Arabia. Both were terminated owing to multiple anomalies including post- and preaxial polydactyly on hands with cutaneous syndactyly of hands and feet. On physical examination low-set ears were present, as well as clubfeet. The first fetus also had cleft palate; the second one had anal atresia. Autopsy showed absent cerebellar vermis, a small cerebellum, abnormal gyration, dysplastic corpus callosum and small, cystic kidneys. The second fetus had a single-nucleotide polymorphism microarray showing large areas of homozygosity, but no significant copy-number variation.
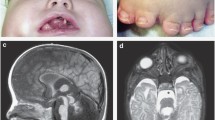
Family 2: The proband in family 2 is a 15-year-old female, who had tongue hamartomata, mesoaxial polydactyly, mild preaxial polydactyly of feet (bifid great toes) and small cerebellar vermis (Figure 1). Other clinical features included bifid epiglottis, esotropia and nystagmus. She had apparent cognitive impairment, unsteady gait and difficulties with articulation. Family history is significant for an older sister who died following surgery for a congenital heart defect associated with a mosaic 45,X/46,XX karyotype. This sister also had preaxial polydactyly of feet (bifid great toes) and absence of the posterior cerebellar vermis, strongly suggesting she was also affected with OFD6.

Fifteen-year old proband of Family 2. (a) Mesoaxial polydactyly hands. (b) Mild preaxial polydactyly feet (bifid great toes). (c) Small cerebellar vermis.
Previous genetic testing included a microarray that showed a 3-MB deletion on the X chromosome (chrX:88,362,258–92,257,523). This deletion includes 15 genes, 3 of which are currently described in OMIM, none are associated with a clinical phenotype to date. None of these were a good candidate for the patient's findings. Sanger sequencing for GLI3 (OMIM #165240) was negative (data not shown).
Family 3: The proband in family 3 was a 1-day-old male infant delivered to a 30-year-old G2P2 Mexican mother. On prenatal ultrasound, the fetus was found to have a large posterior encephalocele, cleft lip and palate and bilateral preaxial polydactyly of the feet. Amniocentesis chromosome analysis was normal and microarray revealed a 169.7-kb loss on 6p21.33 of no known clinical significance. Birth weight was 3,565 grams (50th centile). He expired on the first day of life. On physical examination he had a large posterior encephalocele, cleft lip and palate, micropenis, bilateral undescended testes, postaxial polydactyly of both hands and seven toes bilaterally (with preaxial and postaxial polydactyly) and cutaneous syndactyly of the first and second toes. Autopsy showed occipital encephalocele containing partial herniation of the cerebellum, diffuse polymicrogyria, multifocal heterotopia, absent cerebellar vermis, fused thalami, arrhinencephaly, ventriculomegaly and hypothalamic hamartoma. Family history was significant for consanguinity (first cousin union) and a similarly affected male sibling who died at 5 months of age. Findings in the sibling included macrocephaly, communicating hydrocephalus, Dandy–Walker malformation and absence of cerebellar vermis. Additional features were widely spaced eyes with downslanted palpebral fissures, anteverted nares, soft palate cleft, excess nuchal skin, postaxial polydactyly of hands and bilateral preaxial and postaxial polydactyly of feet.
Exome sequencing on the probands and parents (only the mother in the second family) was performed as part of an effort to expand the mutational spectrum of patients with overlapping features of known GLI3-related phenotypes and oral–facial–digital syndromes with the common attribute of polydactyly. DNA was isolated from whole blood using the salting out method (Qiagen, Germantown, MD, USA) following the manufacturer’s instructions. Exome sequencing was performed as described.3 Filters were implemented using VarSifter software program (Bethesda, MD, USA) for exome and genome sequencing data management.4 Variants were filtered for predicted loss of function and for absence from 938 controls. Sanger sequence analyses of all mutations were performed as described.5 Sequence data were compared with the published C5orf42 sequence (GenBank reference NM_023073.3) using Sequencher 5.0.1 (Gene Codes, Ann Arbor, MI, USA). This study was reviewed and approved by the NHGRI Institutional Review Board. Informed consent was obtained from all families.
Exome sequencing in Family 1 showed a homozygous variant in C5orf42, c.8471-1G>C. This novel splice-site mutation was not present in the Human Gene Mutation Database (HGMD, Version 2014.2) or in the Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP, Version ESP6500SI-V2, Seattle, WA, USA; http://evs.gs.washington.edu/EVS). In addition, it was not present in 938 internal control individuals (ClinSeq data set, NHGRI, Bethesda, MD, USA). Both parents were heterozygous carriers. DNA was unavailable on the affected sibling.
In Family 2 compound heterozygous variants were identified in C5orf42, c.3599C>T, p.Ala1200Val and c.2920+1G>A. The c.3599C>T variant was previously described.6 The c.2920+1G>A variant was novel. Neither variant was present in the ESP dataset or 938 ClinSeq individuals. This patient shares this mutation with her mother. DNA of the father was unavailable. We attempted to amplify a sample of the deceased sister, but DNA appeared to be degraded (data not shown). We hypothesize that her sister had OFD6 owing to her findings in addition to mosaic Turner syndrome.
In Family 3 exome sequencing identified a novel homozygous variant, c.7662_7666del, p.Ser2555Argfs*11. This variant was not present in HGMD, the ESP dataset or 938 ClinSeq individuals. The affected sibling was also homozygous for this mutation and both parents were heterozygous.
C5orf42 was recently identified as the causative gene for this malformation syndrome. Little is known about the function of C5orf42 within the ciliopathy protein network. Exome sequencing in 15 individuals from 11 families with JBTS in Quebec identified 6 distinct mutations in 9 patients of 7 families.7 All patients had cognitive impairment. The majority of patients had oculomotor apraxia and breathing abnormalities; two patients had pre- and postaxial polydactyly and cutaneous syndactyly. Another report described 12 consanguineous Saudi Arabian families with Joubert syndrome.8 In three families, mutations in C5orf42 were identified. Occipital meningocele was present in one patient. Last, Lopez et al. identified 14 homozygous or compound heterozygous mutations in the C5orf42 gene in 12 patients in 9 of 11 families with OFD6. Of note, typical features of ciliopathies, such as polycystic kidney disease and retinal disease were not present.9 The authors concluded that while some patients in their cohort had mutations in TMEM216, C5orf42 was the most commonly mutated gene. In contrast, Romani et al. recently sequenced C5orf42 in 313 patients with JBTS. They identified mutations in 28 (8.9%) of their probands with pure Joubert syndrome. Only 2 out of 17 (11.7%) with features of OFD6 harbored a pathogenic variant in C5orf42. A comparison of mutated versus non-mutated OFD6 patients showed that preaxial and mesoaxial polydacytyly, hypothalamic hamartoma and other congenital defects may predict C5orf42 mutations, whereas tongue hamartomas appear to be more common in patients without an identified mutation.10 All of the probands described in our report had pre- post- or mesoaxial polydactyly which is concordant. The 15-year-old female we describe, however, did have tongue hamartomas in contrast to their conclusion. No clear genotype–phenotype relations have been elucidated.
The local clustering in French Canada led to the hypothesis of a founder effect, but the findings of several different mutations did not support this. With the publication of more patients, it has thus been shown that C5orf42 is likely a common gene in patients with Joubert syndrome with oral–facial findings.9 Our findings with three additional novel mutations support this hypothesis.
One of the patients described had anal atresia. To the best of our knowledge, this finding has not yet been associated with the phenotype. This patient was the offspring of a first-cousin union supported by large areas of homozygosity on microarray. It could be a novel finding, but we suspect it is likely unrelated and an additional finding in this consanguineous family.
In summary, we report three novel mutations in the C5orf42 gene in patients with OFD6. Our findings support recent results that C5orf42 is the major gene mutated in patients with OFD6. Mutations are associated with a wide clinical spectrum comprising overlapping features of JBTS and oral–facial–digital syndromes as well as with Pallister–Hall and Greig Cephalopolysyndactyly syndromes.
The use of next-generation sequencing is slowly, but significantly expanding the phenotypic spectrum of disorders as less typically affected patients are analyzed. This is referred to as phenotypic expansion. This improved understanding of genotype–phenotype relationships facilitates diagnosis, refines our ability to predict phenotype from genotype, and provides insights into the biology of normal and abnormal development.
References
References
Sattar S, Gleeson JG . The ciliopathies in neuronal development: a clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disorders. Dev Med Child Neurol 2011; 53: 793–798.
Brancati F, Dallapiccola B, Valente EM . Joubert Syndrome and related disorders. Orphanet J Rare Dis 2010; 5: 20.
Johnston JJ, Sapp JC, Curry C, Horton M, Leon E, Cusmano-Ozog K et al. Expansion of the TARP syndrome phenotype associated with de novo mutations and mosaicism. Am J Med Genet A 2014; 164A: 120–128.
Teer JK, Green ED, Mullikin JC, Biesecker LG . Varsifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics 2012; 28: 599–600.
Gripp KW, Hopkins E, Johnston JJ, Krause C, Dobyns WB, Biesecker LG . Long-term survival in TARP syndrome and confirmation of RBM10 as the disease-causing gene. Am J Med Genet A 2011; 155A: 2516–2520.
Ohba C, Osaka H, Iai M, Yamashita S, Suzuki Y, Aida N et al. Diagnostic utiliy of whoe exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics 2013; 14: 225–232.
Srour M, Schwartzentruber J, Hamdan FF, Ospina LH, Patry L, Labuda D et al. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am J Hum Genet 2012; 90: 693–700.
Alazami AM, Alshammari MJ, Salih MA, Alzahrani F, Hijazi H, Seidahmed MZ et al. Molecular characterization of Joubert syndrome in Saudi Arabia. Hum Mutat 2012; 33: 1423–1428.
Lopez E, Thauvin-Robinet C, Reversade B, Khartoufi NE, Devisme L, Holder M et al. C5orf42 is the major gene responsible for OFD syndrome type VI. Hum Genet 2014; 133: 367–377.
Romani M, Mancini F, Micalizzi A, Poretti A, Miccinilli E, Accorsi P et al. Oral-facial-digital syndrome type VI: is C5orf42 really the major gene? Hum Genet 2015; 134: 123–126.
Data Citations
Biesecker, Leslie G HGV Database (2015) http://dx.doi.org/10.6084/m9.figshare.hgv.744
Biesecker, Leslie G HGV Database (2015) http://dx.doi.org/10.6084/m9.figshare.hgv.747
Biesecker, Leslie G HGV Database (2015) http://dx.doi.org/10.6084/m9.figshare.hgv.750
Acknowledgements
We thank the families for their participation in this study. This work was supported by funding from the Intramural Research Program of the National Human Genome Research Institute of the US National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Wentzensen, I., Johnston, J., Keppler-Noreuil, K. et al. Exome sequencing identifies novel mutations in C5orf42 in patients with Joubert syndrome with oral–facial–digital anomalies. Hum Genome Var 2, 15045 (2015). https://doi.org/10.1038/hgv.2015.45
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.45
- Springer Nature Limited