
Abstract
Type I interferon (IFN-I) plays crucial roles in the regulation of inflammation and it is associated with various inflammatory diseases including systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and periodontitis, impacting people's health and quality of life. It is well-established that IFN-Is affect immune responses and inflammatory factors by regulating some signaling. However, currently, there is no comprehensive overview of the crucial regulatory role of IFN-I in distinctive pathways as well as associated inflammatory diseases. This review aims to provide a narrative of the involvement of IFN-I in different signaling pathways, mainly mediating the related key factors with specific targets in the pathways and signaling cascades to influence the progression of inflammatory diseases. As such, we suggested that IFN-Is induce inflammatory regulation through the stimulation of certain factors in signaling pathways, which displays possible efficient treatment methods and provides a reference for the precise control of inflammatory diseases.
Similar content being viewed by others

Introduction
A significant number of individuals globally suffer from various inflammatory illnesses, such as infection, SLE, RA, systemic sclerosis (SSc), juvenile dermatomyositis (JDM), and periodontitis, presenting significant medical and socio-economic challenge [1,2,3]. Dysregulated IFN-Is signaling could cause inflammatory diseases, including autoimmune diseases and chronic inflections [4,5,6]. The regulation of IFN-I during inflammation is a complex process that typically functions as a double-edged sword, capable of inhibiting pro-inflammatory factors or triggering abnormally high levels of inflammation [7, 8].
IFN-Is belong to a class of cytokines known for their pleiotropic effects and three main functions. Firstly, IFN-Is can induce an anti-bacterial state in infected cells, controlling the spread of infectious and inflammatory agents, particularly viral pathogens [7, 9, 10]. Secondly, they regulate the innate immune response by facilitating antigen presentation and natural killer cell function [11], while mediating inflammatory pathways and cytokine [3, 12, 13]. Thirdly, IFN-Is can trigger the adaptive immune system, prompting high-affinity antigen-immune cell responses and the development of immune memory [5, 14].
Recent studies have revealed that IFN-Is also play a crucial role in the development of inflammatory diseases via regulating the associated signaling pathways [15,16,17]. The IFN-Is family acts as key regulatory factors for specific targets in these pathways, mediating signaling to subsequently inhibit or prompt the inflammation and immune responses [12, 18]. IFN-Is can be secreted by cytosolic receptors such as retinoic acid-inducible gene I (RIG-I), and melanoma differentiation-associated gene 5 (MDA5), and can also respond to toll-like receptors (TLRs) signaling macrophages and dendritic cells (DCs) [13, 19]. Additionally, the cytosolic GAMP synthase (cGAS) detects cytoplasmic DNA and stimulates the synthesis of circular GAMP (cGAMP), which uses the stimulator of interferon genes (STING) as a secondary receptor, and further stimulates STING-dependent inflammatory cytokines, including IFN-Is [20,21,22]. Subsequently, IFN-Is can bind to the heterodimeric transmembrane interferon alpha receptor (IFNAR)1 and IFNAR2, resulting in signal transducers and activators of transcription, which drive different signaling pathways through various cascades to regulate the inflammation responses [23, 24].
This review introduces the IFN-Is family involvement in the progression of inflammatory diseases and summarizes their regulatory role as crucial modulators in downstream inflammatory signaling pathways, including the Janus kinase (JAK)/signal transducers and activators of transcription (STAT) pathway, TLRs pathway, nuclear factor-κB (NF-κB) pathway, activation of the phosphoinositide 3-kinase (PI3K)/serine-threonine kinase (AKT) pathway, and mitogen-activated protein kinases (MAPK) pathway. Furthermore, the review discusses the promising potential and underlying challenges of IFN-Is-based therapy and suggests guidance to develop IFN-Is as disease-specific biomarkers and drug modulators in inflammatory diseases.
Type I interferon
IFN-Is plays a critical role in initiating the innate immune response against a wide range of pathogens [25]. The IFN-Is family comprises IFN-α, which has 13 distinct subtypes in humans and 14 in mice, along with IFN-β, IFN-δ, IFN-ε, IFN-κ, IFN-ω, and IFN-ζ. Among these subtypes, humans can express IFN-α, IFN-β, IFN-ε, IFN-κ, and IFN-ω [26].
The structure of interferon α/β
Among the various subtypes of type I IFNs, IFN-α and IFN-β are the most well-understood. The IFN-α protein family consists of multiple subtypes that share 76–99% amino acid identity. These subtypes contain a 23-amino-acid hydrophobic signal peptide and a 166-amino-acid mature peptide sequence. However, IFN-α2 is an exception as it encodes a 165-amino-acid protein due to a deletion at position 44. Additionally, variant polymorphic forms of IFN-α also exist, including IFN-α2a, -2b, and -2c [27].
In contrast to the IFNA genes, most mammalian genomes have not experienced duplication and expansion of the IFNB gene. Instead, these genomes contain a single gene encoding IFN-β. However, in the genomes of ruminants and pigs, evidence suggests the presence of more than one copy of the IFNB gene, indicating gene duplication in these lineages [28]. While most species have only a single IFNB gene, duplication of the IFNB gene has been observed in some members of two of the 25 Caucasian families studied [29]. Human IFN-β is a protein composed of 166 amino acids and exhibits only 25–32% sequence identity to human IFN-α proteins. In contrast, murine IFN-β is composed of 161 amino acids and shares only 19–23% sequence identity with murine IFN-α [30].
The source of Type I interferon
Most cells in the body could produce IFN-Is in response to the stimulation of pattern recognition receptors (PRRs) by pathogens. Upon the activation by pathogens, various innate immune cells, including macrophages and DCs [31, 32], can produce IFN-Is. However, non-immune cells such as fibroblasts and epithelial cells also contribute to the production of IFN-Is [33].
PRRs are located on the cell surface, in the cytosol and endosomal compartments (Table 1), and are responsible for recognizing various pathogen-associated molecular patterns (PAMPs), including nucleic acids and non-nucleic acid PAMPs [31].
RNA receptors RIG-I and MDA5 are the primary receptors responsible for recognizing RNA in the cytosol [31, 34, 35]. Additionally, AT-rich DNA can be transcribed by RNA polymerase III into 5′-PPP-containing RNA, which serves as a RIG-I agonist [36]. Other DNA motifs in the cytosol can be recognized by various receptors, including DNA-dependent activator of IFN-regulatory factors (DAI), DEAD and DEAH box (DEXD/H box) helicases, and cGAS [31, 32, 37, 38], all of which are highly associated with the induction of IFN-Is. Furthermore, the cytosolic molecular sensors NOD-containing protein 1 (NOD1) and NOD2 are specialized in recognizing bacteria and viruses, leading to IFN-Is production [39].
In addition to cytosolic receptors, TLRs also play a role in activating pathways that lead to IFN-Is production. Among cell-surface TLRs, TLR 4 recognizes lipopolysaccharide (LPS) from bacteria and induces IFNβ through Toll-receptor-domain-containing adapter-inducing interferon-β (TRIF)-dependent pathway [40]. In contrast, other cell-surface TLRs signaling is responsible for IFN-Is production in response to viruses through myeloid differentiation primary response 88 (MyD88)-dependent pathway [41].
Endosomal compartments are also involved in IFN-Is production, with TLR 3, TLR 7, TLR 8, and TLR 9 being responsible for recognizing different types of PAMPs to induce IFN-Is. TLR 3 responds to double-stranded RNA [42], while TLR 7 and TLR 8 recognize single-stranded RNA [43]. TLR 9 responds to unmethylated CpG DNA [44].
Type I interferon signaling in inflammation
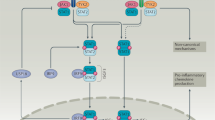
The regulation of IFN-Is in inflammation is a complex process that involves inducing cell-intrinsic antimicrobial states to limit the spread of infectious agents, modulating innate immune responses to inhibit cytokine production, and activating the adaptive immune system, which can lead to either restrained pro-inflammatory pathway or excessive abnormal inflammation [7, 8]. This process is controlled by multiple critical pathways. There is compelling evidence that the IFN-I family serves as mediators for their specific targets in these pathways to regulate cascade reactions, thus subsequently suppressing or promoting sustained inflammation as well as immune activation [45, 46]. This review summarizes IFN-Is that control specific factors to promote or inhibit inflammation through mediating downstream signaling pathways, including the JAK/ STAT pathway, TLRs pathway, NF-κB pathway, PI3K/AKT pathway, and MAPK pathway. (Fig. 1).

Different signaling pathways are involved in the inflammatory regulation of IFN-I. A After being stimulated by bacteria, viruses, PAMP/DAMP, etc. in the external environment, the DNA sensors activate STING, which moves to the Golgi and is phosphorylated by TBK1, allowing for the phosphorylation and activation of IRF 3. Upon binding to their ligands, RIG-I and MDA5 engage MAVS, leading to activation of TBK1 and members of the IKK family of kinases. Similarly, TLRs signal through MyD88 and TRIF adaptor molecules, leading to the activation of TBK1 and members of the IKK family. These kinases trigger the phosphorylation, activation, and dimerization of IRFs and the release of NF-κB. IRFs and NF-κB then migrate into the nucleus where they bind to promoter regions of IFN-I and other target genes, thereby stimulating IFN-I as well as anti-inflammatory and pro-inflammatory cytokine gene transcription. B In the canonical IFN-I signaling pathway, IFN-I binding with IFNAR results in the phosphorylation of JAK1 and TYK2, which then recruit and activate STAT proteins, leading to their trimerization or dimerization and nuclear translocation. Two distinct transcriptional complexes are formed, which regulate the expression of different ISGs' in a sequence-dependent manner. ISGF3, a trimerized complex formed by STAT1, STAT2, and IRF 9, recognizes the ISRE motif and induces a group of gene expression. The other complex formed by STAT1 homodimers binds to the GAS motif and mainly active inflammatory gene expression. B In the uncanonical IFN-I signaling pathway, IFN-I can also induce a set of genes expression independent of STATs, such as MAPKs and PI3K pathways. Additionally, IFN regulates some ISGs’ translation through the mTOR signaling pathway
JAK/STAT pathway
After production, IFN-Is activate a wide range of gene transcription in an autocrine and paracrine manner by triggering the downstream signals [47]. The cognate receptor complex of IFN-Is consists of the ubiquitously expressed transmembrane IFNAR1 and IFNAR2, which initiates signaling cascades upon binding [12, 48]. JAK1 and non-receptor tyrosine kinase 2 (TYK2) are phosphorylated and activated by IFNAR [24], subsequently inducing phosphorylation and dimerization of transcription factors STAT1 and STAT2 [49, 50]. The heterodimer then translocated to the nucleus and recruits IFN-regulatory factor (IRF) 9 to form STAT1-STAT2-IRF 9 tri-complex (IFN-stimulated gene factor 3, ISGF3) [51, 52]. The complex binds to IFN-stimulated response elements (ISRE), a DNA sequence motif, to activate the transcription of a group of genes known as IFN-stimulated genes (ISG) [53, 54]. By regulating IFN-α and IFN-β signaling, ISGs control inflammation. Additionally, IFN-I could also induce STAT1 to form homodimers that are not assembled with IRF 9, subsequently binding to a unique consensus sequence in the ISG promoter called the gamma-activating sequence (GAS) [55]. (Table 2).
Gene activation triggered by IFN-Is
Activation of IFN-I occurs through JAK/STAT signaling pathway, and its role is two-fold, with both pro-inflammatory and anti-inflammatory effect [2, 89]. For instance, research on COVID-19 demonstrated that asymptomatic patients developed a protective IFN-I inflammatory response, whereas severe COVID-19 patients had increased expression of ISGs and excessive inflammation reaction [56]. The angiotensin-converting enzyme inhibitor captopril has been found to reduce circulating and tissue IFN-α levels, along with decreased inflammation of peripheral and central nervous system in lupus-prone mice [57]. Similarly, in a study conducted by Kitamura et al., it was observed that radiation up-regulated the gene expression level of IFN-α in esophageal tissue. It should also be noted that anti-IFN-α neutralizing antibody improved radiation-induced esophageal mucosal inflammation, while IFN-α receptor agonist (RO8191) had the opposite effect, reflecting the pro-inflammatory properties of IFN-α [58]. Additionally, co-expression of the apolipoprotein AI mimetic peptide L37pA with IFN-α resulted in a significant reduction of IFN-α expression, thereby inhibiting inflammatory pathways and responses related to PAMPs and immune cells. This suggests a possible effective treatment for inflammatory processes [59]. Conversely, Antrodia camphorata (A. cinnamomea) was observed to increase the level of IFN-α after dengue virus (DENV) infection, playing an antiviral and anti-inflammatory role [60].
Upon viral infection, IFN-β plays a crucial role. However, under porcine reproductive and respiratory syndrome virus (PRRSV) infection, Paraoxonase-1 (PON1) has been found to inhibit the IFN-β pathway to promote PRRSV replication by interacting with PRRSV nonstructural protein 9 (Nsp9), resulting in an expansion of infection and inflammation [61]. Besides, microRNA (miR)-30a has been identified as a potent negative regulator of IFN-β signaling. It suppresses tripartite motif protein 25 (TRIM25) expression and TRIM25-mediated RIG-I ubiquitination to suppress IFN-β activation and production, leading to enhanced CVB3 replication and inflammation [62]. While IFN-β has been known for its antiviral function, its enhanced response has been found to lead to eosinophilic chronic rhinosinusitis via C–C Motif Chemokine Ligand 11(CCL11) [63]. Interestingly, Cook et al. reported that during acute chikungunya virus (CHIKV) infection, both IFN-α and IFN-β play protective roles, but through different mechanisms: IFN-α restricts CHIKV replication and spread, while IFN-β limits neutrophil-mediated inflammation to prevent CHIKV pathogenesis [64].
IFNAR
IFNARs are essential for the cascade reaction initiated by IFN-I signaling. Inhibition or blocking of causes significant changes in the downstream pro-inflammatory factors and the inflammatory environment [3, 90]. It has been reported that interleukin (IL)-10 levels are significantly reduced in IFNAR knockout (KO) mice during influenza virus infection. The antiviral and anti-inflammatory activities of IFN-I are abolished [66]. In contrast, ultraviolet B (UVB)-irradiated IFNAR KO mice displayed elevated levels of pro-inflammatory cytokines and more severe histological inflammation, suggesting the protective effects of IFN-I [67]. However, IFN-α acts differently under respiratory syncytial virus (RSV) infection. RSV-infected IFNAR-deficient mice showed decreased IFN-α production but demonstrated significantly reduced secretion of pro-inflammatory cytokines and chemokines in the airways. This suggest that IFN-I may contribute to RSV induced inflammation [68]. Studies have shown that using IFNAR1-Ab to bind IFNAR achieves a therapeutic effect by reducing the protein levels of pro-inflammatory cytokines and relieving inflammation and tissue damage [69]. Moreover, scRNA-seq has identified a novel IFN-I signaling-dependent monocyte subpopulation (MO-IFN) that upregulates IFNAR1 expression to increase IFN-I, thereby contributing to monocyte infiltration and the increased inflammation base level [70]. Additionally, Chan et al. has found that activation of the IFN/IFNAR axis increases pro-inflammatory cytokine levels in adipocytes, suggesting further investigation is necessary to understand the roles of adipocyte inflammation in disease pathogenesis [71].
STAT
STAT is a family of transcription factors related to signal transduction and transcriptional activation, which mediates many aspects of cellular immunity and has been identified to significantly regulate IFN-Is signaling [91]. In detail, STAT could combine with IRF to form a complex, and it subsequently binds to the ISRE promoter to induce ISG expression, thereby affecting the regulation of inflammatory factors [49].
In patients with periodontitis, reduced expression of the STAT1 gene leads to impaired downstream of IFN-I signaling, contributing to decreased IFN-I activation and excessive periodontal inflammation [73]. In patients with SLE, glucocorticoid-induced leucine zipper (GILZ) gene directly binds to STAT1, blocking its nuclear translocation and reducing IFN-α-induced gene expression, thereby blocking the pro-inflammatory response of IFN-α [74]. In addition, menthone promotes K48-linked polyubiquitination of TKY2, indirectly inhibiting STAT1 instead of inducing its phosphorylation, significantly restraining local inflammation in collagen II-induced arthritis (CIA) mice [75]. However, during influenza A virus (IAV) infection, STAT1 expression could be hindered by RUNX1, a transcription factor, which subsequently attenuates IFN-β signaling, promoting the expansion of infection and inflammation [76]. Furthermore, hsa_circ_0060450, a circular RNA, functions as a sponge for miR-199a-5p to release its target gene, src homology 2-containing protein tyrosine phosphatase 2 (SHP2), which further targets the inhibition of STAT1, blocking the activation of IFN-I and inhibiting macrophage-mediated inflammation. [77].
STAT2 deficiency may cause failure of feedback from IFN-α signaling, leading to immune dysregulation. Aberrant IFN-α signaling can also switch transcriptional output into a clinically evident inflammatory response [78]. STAT2-dependent IFN-I signaling could accelerate an inflammatory environment due to its release of inflammatory factors by disrupting hypoxia during pathogenic Salmonella typhimurium infection [79]. Additionally, Shen et al. [80] discovered that the capsid protein (Cap) of porcine circovirus 3 (PCV3) could interact with the transactivation domain of STAT2, hindering the the expression of IFN-β and preventing the defense against viral infection and inflammation by binding to ISRE and prevent the ISRE of IRF 9-S2C.
STAT3 indirectly regulates the inflammatory response related to IFN-Is mainly through STAT1 and STAT2. Cannabidiol (CBD) has been found to be able to inhibit Socs3, the main negative regulator gene of STAT3, and downregulated STAT3 blocks the activation of STAT1 transcription factor, inhibiting the IFN-β-dependent pro-inflammatory process [81]. This conclusion was further partially supported by another study, which showed a positive correlation between the expression of STAT3 phosphorylation and IFN-β: the decrease in STAT3 expression suppressed the IFN-β pathway, but resulted in a significant increase in inflammatory cytokines [82].
Toll-like receptor pathway
The canonical IFN-I-JAK/STAT signaling pathway does not operate independently but engages in extensive and critical communication and crosstalk with other signaling pathways, such as PRRs, including TLRs [3, 35]. Downstream of the signaling pathways of host germline-encoded PRRs, which are expressed on the cell membrane or in the cytoplasm of the cells of the innate immune system, IFN-Is can be produced in response to PAMP that includes pathogenic nucleic acids, LPS, and proteins, or in response to host damage-associated molecular patterns (DAMP) [5, 90]. After intracellular TLRs (TLR 3, TLR 7/8, and TLR 9) are activated, IFN-I production is subsequently induced by IRF 3, IRF 7, and IRF 5 [92]. TLRs signaling can be broadly classified into two pathways: the MyD88-dependent and the TRIF-dependent pathway [93]. While other TLRs can activate through the MyD88-dependent pathway [94], only TLR 3 and TLR 4 activate through the TRIF-dependent pathway [95] (Table 3).
TLR
As discussed previously, the activation of TLRs can affect the production of IFN-Is and their interaction with the JAK/STAT signaling pathway, thereby influencing the occurrence and progression of inflammation [136, 137]. TLR 7 signaling has been identified as a prerequisite for human immunodeficiency virus (HIV)-induced IFN-Is production, and antibodies produced during untreated HIV infection may contribute to the sustained high-level IFN-Is response during the infection, suggesting a new mechanism of immune activation through TLRs [96]. Moreover, Yang et al. have demonstrated that upon virus recognition, TLR 3 and TLR 7 activation leads to IFN-β production, which can improve inflammation progression, displaying a protective role in inflammatory regulation [97]. In the MyD88-dependent pathway of TLRs signaling, blocking TLR activation has been considered a potential strategy for addressing excessive inflammation mediated by IFN-Is [98]. For instance, chloroquine loaded by filamentous micelles (CQ-FM), a TLR antagonist, can inhibit TLR activation, leading to a significant reduction in downstream IFN-Is production and decreased inflammation [99]. The TRIF-dependent pathway typically regulates IFN-Is production during bacterial infection. Upon being stimulated by IFN-stimulated genes, such as versican, TLR 3 or TLR 4 could be triggered via LPS to activate the signaling cascade of TRIF adapter, IFN-I as well as IFNAR, allowing IFN-Is to fully exert their anti-inflammatory properties [100]. Interestingly, the current research has found that during Yersinia pestis infection, TLR 7 might have an unconventional signal transduction adapter independent of MyD88, which induces IFN-Is production, inhibiting inflammation caused by the plague [101].
IRF
The IRF family of transcription factors plays a crucial role in IFN-IS induction, with IRF 3 and IRF 7 acting as major mediators downstream of cytoplasmic RNA and DNA receptors, as well as TLRs pathways [138,139,140]. Auger et al. have found that during Streptococcus suis infection, TLR 7 and TLR 9 could recognize bacterial nucleic acids, leading to the activation of IRF 3 and IRF 7, which then induce IFN-β production. The IFN-Is participate in modulating systemic inflammation in host defense, displaying an anti-inflammatory role, when induced relatively mild virulent strains. However, highly virulent strains rapidly induce septic shock and inflammation, which is abnormally regulated by IFN-Is [102].
IRF 3 is expressed ubiquitously and can be activated through phosphorylation to facilitate dimerization, nuclear translocation, the combination with the co-activator cAMP-response element binding protein (CREB)-binding protein (CBP), subsequently binding to canonical ISRE in the promoter of IFN-β and IFN-α [141,142,143,144]. Studies have shown that after the combination of LPS and TLR 4, IRF 3 can be activated via phosphorylation of kinases TANK-binding kinase 1 (TBK1) and inhibitor of NF-κB (IκB) kinase (IKKε), inducing ISG to produce IFN-Is, modulating the process of inflammation [103]. In a separate study, Artusa et al. reported that green coffee and roasted coffee extract can inhibit the effect of IRF 3, thereby inhibiting excessive IFN-β-induced inflammation [104]. Conversely, during viral infection, gastrodin (GTD) can promote the activation of IRF 3 in macrophages to facilitate the production of IFN-Is, resisting inflammation and anti-viral infection [105]. Another significant pathway for the production of IFN-Is through IRF 3 is cGAS/ STING signaling. When cGAS binds to double-stranded DNA (dsDNA), it can be activated and convert adenosine 5'-tri Phosphate (ATP) and guanosine 5'-triphosphate (GTP) to cGAMP, which together with other cyclic dinucleotides (CDNs) signal to STING downstream in the endoplasmic reticulum (ER), subsequently activating IRF 3 in the nucleus, leading to secretion of IFN-Is [145, 146]. Mitochondria can release DNA into the cytoplasm, binding cGAS and promoting the activation of STING, which further activates IRF 3 through phosphorylation by TBK1, contributing to the increased concentrations of IFN-Is as well as inflammatory cytokines in the innate immune response, facilitating the progression of inflammation [106, 147]. Meanwhile, the leaked mitochondrial DNA (mtDNA) could be recognized by TLR9 and trigger MyD88-dependent signaling, promoting pro-inflammatory cytokine expression such as tumor necrosis factor (TNF) as well as IL and IFN-Is secretion through ISG upregulation [148, 149]. This conclusion has been confirmed by demonstrating that oxidized mtDNA drove IFN-Is secretion through the TLR9 pathway in humans with SLE [150, 151]. It is worth noting that during this process, oxidized mtDNA driven by TLR signaling activates the nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing 3 (NLRP3) inflammasome, which in turn facilitates IL-1β maturation in this process, crucially participating in the activation as well as regulation of inflammation [152]. Apart from that, it is currently found that inhibiting mtDNA synthesis through IRF ablation could prevent NLRP3 inflammasome activation and suppress this process of inflammation [153, 154]. DNA polymerase β (Polβ) deficiency can also result in the accumulation of DNA damage in the cell and trigger the leakage of damaged DNA into the cytoplasm, activating STING and facilitating the IRF 3 signaling cascade, promoting the activation of TBK1-phosphorylated IRF 3 to translocate into the nucleus, enhancing the expression of IFN-Is and pro-inflammatory cytokines [107]. Several factors can modulate the level of IFN-Is through IRF 3 via distinct ways, regulating the innate immune response and inhibiting pro-inflammatory signaling. For instance, red rice bran extract (RRBE) can inhibit the phosphorylation of STING, blocking the activation of IRF 3 to hinder initiation of IFN-Is signaling, which function as pro-inflammatory cytokines [108]. Additionally, the E3 ligase RNF 5 can also limit the signaling of IRF 3 through targeting STING, suppressing the production of IFN-Is, which instead promotes viral replication and abnormal inflammation development [109].
In contrast to IRF 3, IRF 7 is usually expressed at very low levels, except in plasmacytoid DCs (pDCs) [155, 156]. IRF 7 can be activated by phosphorylation of TBK1/IKKε and TRIF-dependent pathways downstream of cytoplasmic RNA/DNA sensors, leading to its entry into the nucleus to dimerize with IRF 3, transcriptionally activating and inducing the expression of IFN-α and IFN-β [157, 158]. In addition, IRF-7 is essential for pathways involving MyD88 recruitment, leading to IKKα activation and driving IFN-α and IFN-β expression in response to viruses [159,160,161]. Furthermore, IRF 7 can form a feed-forward loop with IFN-Is, maximizing the expression of IFN-Is and continuously producing a large number of IFN-Is, acting as a positive regulator of IFN-Is [162, 163]. However, IRF7 can also facilitate inflammation and the progress of inflammatory diseases. For instance, gene and protein levels of IRF 7 were significantly enhanced in skin and cultured fibroblasts from patients with SSc, and IRF 7 knockout mice exhibited lower levels of pro-fibrotic factors and less inflammatory response [110]. Additionally, asthmatic patients with higher levels of type 2 innate lymphoid cells (ILC2) in peripheral blood and bronchoalveolar lavage fluid (BALF) to drive inflammation compared had greater IRF 7 expressions in murine lung ILC2s after t stimulation from papain or IL-33 [111]. Furthermore, aryl hydrocarbon receptor-interacting protein (AIP) can inhibit IRF 7 by antagonizing the nuclear localization of IRF 7, hindering the production of IFN-Is induced by IRF 7, reducing the immune response and promotes abnormal inflammation [112]. Nevertheless, IRF7 can also restrain inflammation and suppress the progress of inflammatory diseases in certain conditions. A study has shown that when USP25 was upregulated by virus infection or LPS, IRF 7 could directly bind to two conserved IRF binding sites on the USP25 promoter, driving the transcription of USP25 and promoting the secretion of IFN-Is to adjust the innate immune signal transduction and exhibit an anti-inflammatory effect [114].
RIG-I
Host cells sense invading viruses as well as launch innate immune responses to resist infection, in which detection of viral nucleic acids via RIG-I could produce activated signaling, leading ultimately to the secretion of IFN-Is [164]. In this process, the protein activator of the interferon-induced protein kinase (PACT), also referred to as the protein kinase, interferon-inducible double-stranded RNA-dependent activator (PRKRA), is a crucial component in initiating and maintaining RIG-I-dependent antiviral responses [165]. PACT physically binds to the C-terminal repression domain of RIG-I and then enhances the activation of RIG-I through poly (I:C) of intermediate length [166, 167]. Afterward, RIG-I functions as a virus sensor that triggers the innate antiviral response and could be activated by dsRNA [168]. Then, its N-terminal caspase activation and recruitment domain (CARD) will migrate and link to the CARD on the mitochondrial antiviral signaling protein (MAVS), activating the signal transduction of IFN-Is, and subsequently promoting the innate immune response including inflammatory response [169,170,171]. It is noteworthy that cytoplasmic RIG-I can upregulate the secretion of IRF 3-dependent IFN-Is and reduce the level of MDA5 via combining with c-Cbl-associated protein (CAP), reducing cytotoxicity and alleviating myocarditis [116]. In contrast, Villamayor et al. have revealed a novel interaction between RIG-I and IFN-α-inducible protein 6 (IFI6), which affects RIG-I activation through mediating RNA binding, resulting in negative regulation of innate immunity and excessive inflammation [117]. Moreover, triggered by IFN-Is promoter stimulator-1 (IPS-1) signaling, RIG-I-like receptors (RLRs) could collaborate with TLR 7 to advance pDC recruitment and IFN-α production, restraining the host response to pneumonia viral infection and thus preventing viral bronchiolitis [118]. Apart from that, interestingly, during severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, RIG-I-MAVS siganling could also be blocked by the NSP5 and N proteins of SARS-CoV-2, which could inhibit RIG-I-induced IFN-Is response, resulting in weakening antiviral immunity [119].
MAVS
The protein MAVS is a crucial component of innate immunity, functioning as a central pivot for signal transduction initiated by TLR, RIG-I-like receptors, and MDA5 [172]. Notably, PACT, which is linked to the host antiviral response, promotes the formation of RNA-induced MDA5 oligomers in this process, thereby being beneficial to the initiation of the IFN-Is signaling cascade associated with MAVS [173, 174]. Additionally, MAVS can stimulate the IFN-beta promoter by activating IRF 3, modulating inflammatory signaling related to IFN-Is [172, 175, 176]. However, during infection, a range of factors that target MAVS to affect the control of inflammation by IFN-Is. For instance, TRIM21, a regulator of tissue inflammation and pro-inflammatory cytokine production, interacts with MAVS during coxsackievirus B3 (CVB3) infection to promote upregulation of IFN-β signaling, enhancing host defense against inflammation [124]. Similarly, under the influence of lactic acid bacteria Lactobacillus (LAB), MAVS activates the production of IFN-Is, but directs bacteria-specific immunity [125]. In chemotherapy, mitochondrial RNA (mtRNA) induced by apoptosis damage could activate MDA5, which subsequently upregulates MAVS to promote IFN-Is signaling, suppressing the inflammation caused by the cytoplasmic release of mtRNA in the presence of caspases inhibition [126]. Nevertheless, instead of activating MAVS, sorafenib mainly limits the recruitment of MAVS to negatively regulate IFN-Is signaling. The inhibition of excessive inflammation can prevent the occurrence of inflammatory diseases [127]. Conversely, RING finger protein 114 (RNF114), an E3 ubiquitin ligase, can restrain the production of IFN-Is via interacting with MAVS and inhibiting RIG-I-mediated signaling, promoting viral replication and excessive inflammation [128].
Furthermore, it is worth noting that SARS-CoV-2 could cause coronavirus disease 2019 (COVID-19), and has a non-negligible correlation with RIG-I, MAVS, and MDA5 as well as their involvement in IFN-Is signaling in this process. Han et al. reported that SARS-CoV-2 ORF9b suppressed the components of the cytoplasmic dsRNA sensing pathway transduced via RIG-I/MDA5-MAVS signaling to antagonize the induced activation of IFN-Is, leading to the development of infection and inflammation [120]. Besides, the SARS-CoV-2 membrane (M) protein has also been shown to display a similar role in infections caused by SARS-CoV-2 [121]. Moreover, recent research has confirmed that both SARS-CoV-2 NSP7 and SARS-CoV-2 NSP8 could prevent the formation of the RIG-I/MDA5-MAVS signal body, thereby restraining the induction of IFN-Is, and then facilitating the generation of inflammation as well as virus replication [122, 123].
NF-κB pathway
NF-κB is a group of proteins that function as dimerizing transcription factors to regulate gene expression and various biological processes, including innate and adaptive immunity, as well as inflammation [177]. NF-κB/Rel proteins bind to the inhibitor of NF-κB (IκB) proteins and are thereby inhibited. However, the activation of proinflammatory cytokines, LPS, growth factors, and antigen receptors stimulate an IKK complex (IKKβ, IKKα, and NEMO), which in turn activate IRF, participating in IFN-I transcription and IFN-I production [90]. (Table 4).
NF-κB
As above-mentioned, excessive activation of IFN-β has been demonstrated to lead to abnormal inflammation, tissue damage, or autoimmune disease [1]. However, inhibiting IFN-β to reduce inflammation can be achieved through suppressing NF-κB signaling. For instance, knockdown of the TRIM 72, also known as MG53, can lead to increased ryanodine receptor (RyR)-mediated intracellular calcium oscillations, further activating NF-κB signaling and inhibiting IFN-β induction, thereby suppressing the development of inflammation [178]. Conversely, nc886, a novel inhibitor of IFN-β signaling and inflammation, can restrain NF-κB signaling by suppressing protein kinase R (PKR), thus limiting excessive activation of IFN-β signaling and reducing the inflammatory state [179]. With regard to pro-inflammatory IFN-α downstream of the NF-κB signaling pathway, the Adenosine 2A receptor (A2AR) is primarily transduced through the Protein Kinase A (PKA)/CREB/NF-κB signaling pathway, which increases the level of IFN-α, promotes the viability of T cells and upregulates the secretion of inflammatory factors [180].
TRAF
The TNF receptor-associated factor (TRAF) proteins function as adapter, which transduce activated signals to major signaling pathways and are recruited to activate NF-κB signaling. This process is involved in inducing IFN-I signaling and modulating inflammatory cascades [183]. Recent studies have shown that downregulation of miR-146a inhibits IAV replication by enhancing IFN-β responses in vitro and in vivo through its target gene TRAF6, thereby alleviating infection-induced inflammation [181]. Similarly, USP47, a novel negative immune system regulator, also has been found to display an anti-inflammatory role via targeting TRAF. However, unlike miR-146a: USP47 removes K63-linked polyubiquitin from TRAF, thereby attenuating Sendai virus-induced IFN-β signaling conduction and inhibiting inflammation [182].
PI3K/AKT pathway
The PI3K/AKT pathway is implicated in various human inflammatory and metabolic diseases [184]. This pathway can be induced by IFN-Is via a STAT-independent pathway [185, 186]. In response to IFN-I, the PI3K/AKT pathway displays an important role in mediating gene transcription. IFN-Is cause phosphorylation of insulin receptor substrate 1 (IRS1), which subsequently binds to subunit of PI3K-p85, thus activating PI3K's catalytic subunit p110. Consequently, inflammatory gene transcription is facilitated via phosphorylating protein kinase C (PKC) [24]. Additionally, the PI3K/ AKT signaling cascade dominates the activation of the mammalian target of rapamycin (mTOR), a critical protein mediating mRNA translation, independent of STAT family members [187, 188]. Following IFN-α and IFN-β stimulation, the mTOR pathway kinase-p70 S6K is rapidly phosphorylated and activated, which subsequently inactivates the relative repressor to increase IFN-induced mRNA translation, leading to the development of inflammation[189] (Table 5).
It has been demonstrated that the production of IFN-I in pDCs is dependent on TLR [194, 195] and IRF 7 signal cascades, which modulate the inflammatory state in the physiological process [196, 197]. Guiducci et al. reported that in activated human pDCs, TLR‐mediated IRF 7 nuclear translocation regulates IFN-I, which is controlled by PI3K. This suggests that the production of IFN-I from pDCs relies on PI3K and highlights the potential therapeutic role of PI3K in autoimmune inflammation [190]. A subsequent study has furhter clarified the role of IFN-I in inhibiting inflammation through PI3K. In activated pDCs, inactivation or blockade of PI3K could neutralize IFN-α, inhibiting chemokine cytokines, and leading to the suppression of inflammation in SLE [191].
With regard to AKT, silencing Caveolin-1 (CAV1) could promote AKT-activated IFN to drive inflammatory signaling, inducing downstream IFN-α and IFN-β inflammatory responses [192]. Besides, in the human hepatocyte cells treated with rapamycin (rapa) and AKT inhibitor, it was found that mTOR signaling, rather than AKT signaling, could enhance the antiviral effects of IFN-α against the hepatitis C virus (HCV), contributing to the suppression of relative inflammation [193].
MAPK pathway
MAPK, specially p38 and extracellular signal-regulated kinases (ERK), also play a significant role in IFN-I-modulated gene expression [24]. As reported, the suppression of p38 activity can impede IFNα-induced transcriptional activation of genes through ISRE. This inhibition, however, is not dependent on the phosphorylation of STAT1 or STAT2, nor on the formation of ISGF3 and GAS [198]. Therefore, kinase p38 is essential for IFN-I to mediate relative signaling that is independent of STATs activity, thereby modulating the inflammatory process [199, 200]. In addition to p38, ERK1/2 signaling can also be stimulated by IFN-I [201] and induced by the virus, which further produces IFN-I and facilitates inflammatory signaling [202] (Table 6).
When considering viral infection in MAPK pathway, it is observed that the Zika virus (ZIKV) induces ISG expressing, which subsequently increases the levels of p38 and ERK ½, promoting the secretion of chemokines that facilitate the development of viral infection and inflammation. Meanwhile, ZIKV inhibits the phosphorylation of ribosomal protein S6 (RPS6), leading to reduced IFN-β translation and a consequent increase in of inflammation levels [203]. In addition, it is noteworthy that the myxoma virus could specifically activate ERK1/2 signaling, thereby promoting the increased secretion of IFN-α and IFN-β that resist viral infection and expansion of inflammation [202].
Dysregulation of type I IFN signaling and inflammatory disease
Dysregulated IFN-Is signaling has been implicated in the pathogenesis of various inflammatory diseases, including autoimmune diseases, chronic infections and cancer [4,5,6]. In this context, we mainly focus on dysregulation of IFN-Is in autoimmune diseases and chronic inflammatory diseases.
Functions of type I interferon in autoimmune diseases
Systemic lupus erythematosus (SLE)
SLE is a complex multi-system autoimmune disease and characterized by multiple organ damage [204]. Genetic variants in the IFN-I pathway and regulation of innate immune responses are also important factors in SLE susceptibility [205]. Viruses such as Epstein-Barr virus (EBV) or self-derived nucleic acids, can initiate IFN-I production via activation of intracellular receptors TLR7 and TLR9 [206]. This abnormal production of IFN-I can promote the differentiation of B cells to plasma blasts, leading to inflammation and tissue damage [207, 208]. Additionally, neutrophils may also contribute to the perpetuation of the immune response in SLE through the release of neutrophil extracellular traps (NETs), which could activate pDCs to secrete IFN-Is [209].
Rheumatoid Arthritis (RA)
RA is a chronic autoimmune disorder that can rapidly erode the joint cartilage and bone, leading to joint pain, stiffness, and deformities [210]. Patients with RA have been found to exhibit high levels of IFN-I. even prior to the onset of symptoms [211]. Furthermore, elevated IFN-I signatures have been shown to predict the development of RA in individuals at risk [212]. IRF 5, STAT4, and PTPN22 are the genes that have been identified as increasing the susceptibility to RA, of which are linked to the IFN-Is signaling pathway [213]. Despite the implication of IFN-Is in the pathogenesis of RA, intra-articular injections with IFN-α and intraperitoneal injection with IFN-β have been shown to prevent the occurrence or development of RA in wild-type mice or RA mouse models. This is possibly due to the ability of IFN-Is to inhibit neutrophils recruitment and activation, thereby reducing the release ROS and proteases [214].
Systemic Sclerosis (SSc)
SSc is a condition characterized by fibrosis, dysfunction of internal organs and a vasculopathy [215]. Reports have shown that SSc can occur in patients who have been treated with IFN-α or IFN-β for chronic myelogenous leukemia and hepatitis C [216]. IFN-I signatures have been found in the peripheral blood and affected skin of SSc patients, even in the early stages of the disease [217]. The level of IFN-Is is also linked to severe symptoms in the skin, lung, and skeletal muscle of SSc patients [218]. Additionally, higher IFN-I signatures have been found to be positively correlated with the presence of anti-topoisomerase or anti-U1-RNP antibodies in SSc patients. Conversely, a negative correlation has been observed between higher IFN-I signatures and the presence of anti-RNA polymerase III antibodies in SSc patients [216, 219].
Juvenile dermatomyositis (JDM)
JDM is identified by proximal muscle weakness and characteristic skin rashes. Patients with JDM have overexpressed IFN-I inducible transcripts and activated IFN-I signatures [220]. Similarly, JDM patients have increased serum IFN-α activity, which is associated with high serum muscle-derived enzymes [221]. Furthermore, the expression of IFN-I-inducible genes in muscle biopsy and the levels of proteins induced by IFN-Is, such as myxovirus resistance protein A (MxA), were found to be elevated in JDM patients. This increase in IFN-I signaling may affect both the muscle and skin tissues [222]. Moreover, the activation of TLR7 and IFN-α might lead to the expansion of immature transitional B cell population and skew the cells toward a pro-inflammatory phenotype to promote JDM progression [223].
Functions of type I interferon in chronic infection
Virus infection
IFN-Is have both beneficial and detrimental effects in responding viral infection. On the beneficial side, IFN-Is can protect the host against bacterial assaults. Initial studies found that mice lacking the IFN-I receptor (IFNAR1) displayed susceptibility to various viruses, such as vesicular stomatitis virus, Semliki Forest virus, vaccinia virus and lymphocytic choriomeningitis virus (LCMV) [224, 225]. Moreover, IFN-Is possess the ability to stimulate the production of numerous antiviral proteins, including MX1, PKR, 2′-5′-oligoadenylate synthetase, IFN-induced transmembrane proteins (IFITMs), apolipoprotein B mRNA-editing enzyme catalytic polypeptide 1, and members of the TRIM family, all of which play crucial roles in inhibiting viral replication and promoting viral clearance [226, 227].
Despite the extensive antiviral effects of IFN-I, there are critical considerations. Even though IFN-I signaling can enhance the susceptibility of virally infected cells to apoptosis, thereby controlling viral replication [228], it could also lead to the death of vital cells. In vitro studies have shown that HIV can cause IFN-I-mediated upregulation of TNF-related apoptosis-inducing ligand (TRAIL) expression by pDCs, enabling these cells to induce TRAIL-dependent CD4 + T cell and B cell apoptosis. Nonetheless, by disrupting TRAIL signaling, both T and B cell functions can be restored, including the overall antibody responses to against HIV [229, 230].
However, both excessive anti-inflammation and hyper-inflammation can also lead to the detrimental effect of IFN-I on disease progression. The suppressive effect of IFN-I might contribute to chronic viral infections. Studies have shown that blocking IFN-I signaling, either through the administration of antibodies or receptor deficiency, can enhance the control of chronic infection with LCMV clone 13, mediated by CD4 + T cells [231]. IFN-Is have also been observed to dampen T cell responses by promoting the expression of immunosuppressive genes such as IL-10 and programmed cell death 1 ligand 1 (PDL1) to facilitate persistent virus infection [232]. Meanwhile, intense inflammation can result in excessive inflammation and considerable tissue damage. IFN-Is have the capacity to disrupt the TNF-induced 'cross-tolerance' which protects mice from lethal effects of endotoxins in a living body [233]. IFN-Is can effectively dismantle this TNF-induced cross-tolerance by priming chromatin, thereby facilitating robust transcriptional responses even to weak signals. This process can lead to hyperinflammation through a feedforward mechanism [234]. Moreover, IFN-I has been strongly correlated with the progression of COVID-19. The severity of COVID-19 is often accompanied by IFN-Is response, in addition to the TNF/IL-1β response, indicating that the IFN-I response could aggravate the hyper-inflammatory response by strengthening TNF/IL-1β-driven inflammation, thus influencing the severe progression of COVID-19 [235].
Bacterial infection
IFN-Is exhibits multifaceted effects not only in viral infections but also during bacterial infections. It serves a critical function in adjusting the host's immune response by releasing cytokines such as indoleamine 2,3-dioxygenase, inducible nitric oxide synthase (iNOS), immunoresponsive genes, and guanylate-binding proteins. The primary mechanism is through IFNγ, a part of the type II IFN family, which is indispensable for combating mycobacteria and other intracellular pathogens [236, 237]. However, the effects of IFN-Is are dual-faced, potentially assisting or hindering the host's response to bacterial infections.
IFN-Is are usually needed at the start of bacterial infections. The low level of IFN-Is helps to initiate immune response and protect against the infection. They can inhibit bacterial growth and protect human and mouse cells by depleting l-tryptophan, an essential amino acid needed by bacteria for survival [236]. Furthermore, IFN-Is might protect against Chlamydia pneumoniae infection by working in tandem with IFNγ to suppress bacterial survival [238]. IFN-Is also play a crucial role in inhibiting the replication of L. pneumophila, a common cause of pneumonia in macrophages. They activate macrophages to inhibit bacterial proliferation through reactive oxygen and reactive nitrogen [239]. Moreover, they contribute significantly to recruit protective phagocytic cells and producing chemokines like CXCL10, thereby restoring neutrophil recruitment and facilitating improved bacterial clearance [240].
Although IFN-Is can play a protective role against bacterial infections, they also have detrimental effects, particularly at high concentrations. Overwhelming levels of IFN-Is may inhibit the responsiveness of macrophages to IFNγ activation and stimulate the production of immunosuppressive molecules, potentially decreasing the immune defenses [5]. During infections with L. monocytogenes, macrophage activation by T cell- or NK cell-derived IFNγ is critical for the induction of antimicrobial pathways and elimination of intracellular bacteria [241]. While IFN-Is can significantly inhibit the responsiveness of macrophages to IFNγ, which can be attributed to the downregulation of IFNγ receptor expression on macrophages [242]. And this downregulation occurs due to the silencing of new Ifngr1 transcription by inhibitory transcriptional regulator [243]. Furthermore, during Mycobacterium leprae infections, IFN-Is can hinder macrophages from increasing the production of vitamin D-dependent antimycobacterial peptides and induce IL-10 to cause immunosuppression, which might contribute to the progression of mycobacterial diseases and result in subsequent tissue damage [244].
Additionally, IFN-Is have a detrimental impact by triggering excessive or inappropriate cell apoptosis, which can lead to the loss of essential cells and potentially intensify the severity of infections [245]. For instance, during Listeria monocytogenes infection, IFN-Is can sensitize lymphoid cells to result in large-scale apoptosis of these cells [246]. In infections caused by Tropheryma whipplei, the bacterium responsible for Whipple's disease, IFN-Is might promote macrophage apoptosis and divert macrophages to an alternatively polarized state that is more permissive to the bacteria [247]. IFN-Is can also mediate NLRP3 inflammation during gram-negative bacterial infection by the activation of caspase-11, leading to the production of proinflammatory cytokines IL-1β and IL-18, and inducing cell pyroptosis [248].
Perspectives and conclusion
This review summarizes recent evidence indicating that IFN-Is modulate inflammation via affecting specific key factors in various signaling pathways such as JAK/ STAT pathway, TLRs pathway, NF-κB pathway, PI3K/AKT pathway, and MAPK pathway. IFN-Is' targets have the potential to become a valid approach for future interventions in inflammatory diseases, with implications for the prevention and treatment of abnormal inflammation.
However, some hurdles hamper the therapeutic use of IFN-Is at this phase, mainly due to insufficient understanding of IFN-Is mechanism, lack of sufficient animal experiments and clinical trial evidence, and the difficulties in controlling the precise inflammatory regulation [249, 250]. Further investigation is needed to elucidate the IFN-Is regulatory network specific to the progress of inflammatory diseases. As summarized, the role of IFN-Is in distinct pathways may be influenced by distinct microenvironments, and even in the same cell type, the same IFN-Is signaling may vary according to additional regulation under different conditions, adding difficulty in applying miRNA-based therapeutic approaches in the clinic. It is significant to identify the positive and negative aspects of IFN-Is-regulated signaling, exploit the IFN-Is related pathway to cure persistent inflammatory diseases, and minimize toxicity as well as side effects [89, 251]. Additionally, the main differences of various signaling pathways mediated by IFN-Is in regulating the progression of inflammatory diseases also need to be further clarified, contributing to proposing precision therapies in the future [252]. It is also notable to study whether the related limiting factors and immune activation of all ISGs could achieve balance in the IFN-Is inflammatory regulation, which may improve clinical results such as the treatment of HIV [252, 253]. Moreover, further animal experiments as well as clinical studies need to be carried out [254]. For instance, the use of JAK inhibitors in JDM still needs a large number of clinical trials to solve the existing safety and efficacy issues, even though JAK inhibitors that include Baricitinib, Tofacitinib, and Ruxolitinib have displayed preliminary efficacy of refractory juvenile JDM in several clinical cases and animal experiments [47, 255].
With regards to the development direction of IFN-Is therapy into clinics in the future, it is prospective for inflammatory diseases including new virus infections to obtain ideal IFN-Is-based therapeutic methods [14, 256, 257]. In response to the COVID-19 pandemic, research by Hoagland et al. proposed the utilization of antiviral IFN-Is system as the first line of defense against the pathogenicity of SARS-CoV-2 and supports the application of intranasal IFN-I as an early treatment effective method [258]. Nevertheless, the accurate effect of IFN-Is intervention still needs to be determined. To be specific, timely and potent IFN-Is production (18–24 h post-infection) promotes both innate and acquired immune responses, whereas delayed IFN-Is production (3–4 days post-infection) actually contributes to ineffective anti-infection as well as excessive inflammation [9]. Besides, IFN-Is intervention may have a more pronounced effect on organisms genetically modified to lack innate immune sensors, like TLRs [258]. A complete evaluation of the immune-inflammatory response of IFN-Is against SARS-CoV-2 is crucial for designing harmless and effective vaccines in clinical treatment. In addition, it is necessary to determine the role of IFN-Is treatment in late disease and lethal models to further delineate the nuances of boosting IFN before, during, and/or after SARS-CoV-2 infection [258].
Availability of data and materials
All data relevant to this review is included in the text, references, and figures.
Abbreviations
- LPS:
-
Lipopolysaccharide
- TRIF:
-
Toll-receptor-domain-containing adapter-inducing interferon-β
- SLE:
-
Systemic lupus erythematosus
- JDM:
-
Juvenile dermatomyositis
- SSc:
-
Stemic sclerosis
- MyD88:
-
Myeloid differentiation primary response gene 88
- RIG-I:
-
Retinoic acid-inducible gene I
- TLRs:
-
Toll-like receptors
- PRRs:
-
Pattern recognition receptors
- DEXD/H box:
-
DEAD and DEAH box
- DAI:
-
DNA-dependent activator of IFN-regulatory factors
- MDA5:
-
Melanoma differentiation-associated gene 5
- NF-κB:
-
Nuclear factor-κB
- MAPK:
-
Mitogen-activated protein kinases
- AKT:
-
Serine-threonine kinase
- PI3K:
-
Phosphoinositide 3-kinase
- UVB:
-
Ultraviolet B
- RSV:
-
Respiratory syncytial virus
- MO-IFN:
-
IFN-I signaling-dependent monocyte subpopulation
- JAK:
-
Janus kinase
- GILZ:
-
Glucocorticoid-induced leucine zipper
- CIA:
-
Collagen II-induced arthritis
- IAV:
-
Influenza A virus
- SHP2:
-
Src homology 2-containing protein tyrosine phosphatase 2
- CBP:
-
Co-activator cAMP-response element binding protein (CREB)-binding protein
- GTD:
-
Gastrodin
- CBD:
-
Cannabidiol
- Cap:
-
Capsid protein
- dsDNA:
-
Double-stranded DNA
- ATP:
-
Adenosine 5'-tri Phosphate
- GTP:
-
Guanosine 5'-triphosphate
- CDNs:
-
Cyclic dinucleotides
- ER:
-
Endoplasmic reticulum
- mtDNA:
-
Mitochondrial DNA
- TNF:
-
Tumor necrosis factor
- NLRP3:
-
Nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing 3
- Polβ:
-
Polymerase β
- RRBE:
-
Red rice bran extract
- ILC2:
-
Type 2 innate lymphoid cells
- AIP:
-
Aryl hydrocarbon receptor-interacting protein
- PACT:
-
Protein activator of the interferon-induced protein kinase
- PRKRA:
-
Protein kinase, interferon-inducible double-stranded RNA-dependent activator
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- CVB3:
-
Coxsackievirus B3
- LAB:
-
Lactic acid bacteria Lactobacillus
- mtRNA:
-
Mitochondrial RNA
- RNF114:
-
RING finger protein 114
- COVID-19:
-
Coronavirus disease 2019
- RyR:
-
Ryanodine receptor
- PKR:
-
Protein kinase R
- A2AR:
-
Adenosine 2A receptor
- PKA:
-
Protein Kinase A
- TRAF:
-
TNF receptor-associated factor
- IRS1:
-
Insulin receptor substrate 1
- PKC:
-
Phosphorylating protein kinase C
- mTOR:
-
Mammalian target of rapamycin
- CAV1:
-
Caveolin-1
- rapa:
-
Rapamycin
- HCV:
-
Hepatitis C virus
- ERK:
-
Extracellular signal-regulated kinases
- ZIKV:
-
Zika virus
- RPS6:
-
Ribosomal protein S6
- EBV:
-
Epstein-Barr virus
- NETs:
-
Neutrophil extracellular traps
- MxA:
-
Myxovirus resistance protein A
- CARD:
-
Caspase activation and recruitment domain
- LCMV:
-
Lymphocytic choriomeningitis virus
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- IPS-1:
-
IFN-Is promoter stimulator-1
- RLRs:
-
RIG-I-like receptors
- IFI6:
-
IFN-α-inducible protein 6
- PCV3:
-
Porcine circovirus 3
- HIV:
-
Human immunodeficiency virus
- CQ-FM:
-
Chloroquine loaded by filamentous micelles
- DAMP:
-
Damage-associated molecular patterns
- IKK:
-
Inhibitor of NF-κB (IκB) kinase
- MAVS:
-
Mitochondrial antiviral signaling protein
- DCs:
-
Dendritic cells
- IRF:
-
Interferon regulatory factor
- TBK1:
-
Activation of tank-binding kinase-1
- PDL1:
-
Programmed cell death 1 ligand 1
- iNOS:
-
Inducible nitric oxide synthase
- IFITMs:
-
IFN-induced transmembrane proteins
- GAS:
-
Gamma-activating sequence
- STING:
-
Stimulator of IFN genes
- TYK2:
-
Tyrosine kinase 2
- ISG:
-
IFN-stimulated genes
- ISRE:
-
IFN-stimulated response elements
- cGAMP:
-
Circular GAMP
- cGAS:
-
Cyclic GMP-AMP synthase
- IFNAR:
-
Interferon alpha receptor
- STING:
-
Stimulator of interferon genes
- Lpr:
-
Lymphoproliferation
- BHK-21 cells:
-
Baby hamster Syrian kidney cells
- Ab:
-
Antibody
- PAM:
-
Porcine alveolar macrophages
- PAMPs:
-
Pathogen-associated molecular patterns
- BALF:
-
Bronchoalveolar lavage fluid
- MRSA:
-
Methicillin-resistant Staphylococcus aureus
- NRF2:
-
Nuclear factor erythroid 2-related factor 2
- HS:
-
Hemorrhagic shock
- RA:
-
Rheumatoid arthritis
- PON1:
-
Paraoxonase-1
- CHIKV:
-
Chikungunya virus
- CCL11:
-
C–C Motif Chemokine Ligand 11
- TRIM25:
-
Tripartite motif protein 25
- Nsp9:
-
Nonstructural protein 9
- PRRSV:
-
Syndrome virus
- DENV:
-
Dengue virus
- T1DM:
-
Type 1 diabetes mellitus
- ARDS:
-
Acute respiratory distress syndrome
- ALX/FPR2:
-
Formyl peptide receptor 2
- PBMCs:
-
Peripheral blood mononuclear cells
- BMDM:
-
Bone marrow derived macrophages
- MCT:
-
Mast cell tryptase
- CCI:
-
Controlled cortical impact
- TBI:
-
Traumatic brain injury
- MEFs:
-
Mouse embryonic fibroblasts
- RIPK1:
-
Receptor-interacting serine/threonine-protein kinase 1
- CDT:
-
Cytolethal distending toxi
- A. cinnamomea:
-
Antrodia camphorata
- s.Typhimurium:
-
Serovar Typhimurium
- MCPIP-1:
-
Monocyte chemotactic protein-induced protein 1
- NEMO:
-
NF-Kappa-B Essential Modulator
- PI-IBS:
-
Post-infectious irritable bowel syndrome
- PAH:
-
Pulmonary arterial hypertension
- hBMECs:
-
Human brain microvascular endothelial cell
References
Chen K, Liu J, Cao X. Regulation of type I interferon signaling in immunity and inflammation: a comprehensive review. J Autoimmun. 2017;83:1–11.
Kretschmer S, Lee-Kirsch MA. Type I interferon-mediated autoinflammation and autoimmunity. Curr Opin Immunol. 2017;49:96–102.
LopezdePadilla CM, Niewold TB. The type I interferons: Basic concepts and clinical relevance in immune-mediated inflammatory diseases. Gene. 2016;576(1 Pt 1):14–21.
Fernandez-Ruiz R, Niewold TB. Type I interferons in autoimmunity. J Invest Dermatol. 2022;142(3 Pt B):793–803.
McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15(2):87–103.
Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15(7):405–14.
Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49.
Trinchieri G. Type I interferon: friend or foe? J Exp Med. 2010;207(10):2053–63.
Pasrija R, Naime M. The deregulated immune reaction and cytokines release storm (CRS) in COVID-19 disease. Int Immunopharmacol. 2021;90: 107225.
Duncan CJA, Randall RE, Hambleton S. Genetic lesions of type I interferon signalling in human antiviral immunity. Trends Genet. 2021;37(1):46–58.
Meyer O. Interferons and autoimmune disorders. Joint Bone Spine. 2009;76(5):464–73.
Gallucci S, Meka S, Gamero AM. Abnormalities of the type I interferon signaling pathway in lupus autoimmunity. Cytokine. 2021;146: 155633.
Mayer-Barber KD, Yan B. Clash of the Cytokine Titans: counter-regulation of interleukin-1 and type I interferon-mediated inflammatory responses. Cell Mol Immunol. 2017;14(1):22–35.
Schreiber G. The role of type I interferons in the pathogenesis and treatment of COVID-19. Front Immunol. 2020;11: 595739.
Franco JH, Chattopadhyay S, Pan ZK. How different pathologies are affected by IFIT expression. Viruses. 2023;15(2):342.
Ramaswamy M, Tummala R, Streicher K, Nogueira da Costa A, Brohawn PZ. The pathogenesis, molecular mechanisms, and therapeutic potential of the interferon pathway in systemic lupus erythematosus and other autoimmune diseases. Int J Mol Sci. 2021;22(20):11286.
Zhang E, Fang M, Jones C, Minze LJ, Xing J, Zhang Z. Mechanisms involved in controlling RNA virus-induced intestinal inflammation. Cell Mol Life Sci. 2022;79(6):313.
Kienes I, Weidl T, Mirza N, Chamaillard M, Kufer TA. Role of NLRs in the regulation of type I interferon signaling, host defense and tolerance to inflammation. Int J Mol Sci. 2021;22(3):1301.
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11(5):373–84.
Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–91.
Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339(6121):826–30.
Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341(6148):903–6.
Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6(11):836–48.
Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5(5):375–86.
Kalliolias GD, Ivashkiv LB. Overview of the biology of type I interferons. Arthritis Res Ther. 2010;12(Suppl 1):S1.
Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32.
Sakharkar MK, Chow VT, Kangueane P. Distributions of exons and introns in the human genome. In Silico Biol. 2004;4(4):387–93.
Zhao FR, Wang W, Zheng Q, Zhang YG, Chen J. The regulation of antiviral activity of interferon epsilon. Front Microbiol. 2022;13:1006481.
Krause CD, Pestka S. Cut, copy, move, delete: The study of human interferon genes reveal multiple mechanisms underlying their evolution in amniotes. Cytokine. 2015;76(2):480–95.
Schreiber G, Piehler J. The molecular basis for functional plasticity in type I interferon signaling. Trends Immunol. 2015;36(3):139–49.
Goubau D, Deddouche S. Reis e Sousa C Cytosolic sensing of viruses. Immunity. 2013;38(5):855–69.
Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38(5):870–80.
Perez-Shibayama C, Islander U, Lutge M, Cheng HW, Onder L, Ring SS, et al. Type I interferon signaling in fibroblastic reticular cells prevents exhaustive activation of antiviral CD8(+) T cells. Sci Immunol. 2020;5(51):eabb7066.
Lim YJ, Koo JE, Hong EH, Park ZY, Lim KM, Bae ON, et al. A Src-family-tyrosine kinase, Lyn, is required for efficient IFN-beta expression in pattern recognition receptor, RIG-I, signal pathway by interacting with IPS-1. Cytokine. 2015;72(1):63–70.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–20.
Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138(3):576–91.
Oh S, Lee S. Recent advances in ZBP1-derived PANoptosis against viral infections. Front Immunol. 2023;14:1148727.
Oshiumi H, Sakai K, Matsumoto M, Seya T. DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. Eur J Immunol. 2010;40(4):940–8.
Moreira LO, Zamboni DS. NOD1 and NOD2 signaling in infection and inflammation. Front Immunol. 2012;3:328.
Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9(4):361–8.
Barbalat R, Lau L, Locksley RM, Barton GM. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol. 2009;10(11):1200–7.
Sakaniwa K, Fujimura A, Shibata T, Shigematsu H, Ekimoto T, Yamamoto M, et al. TLR3 forms a laterally aligned multimeric complex along double-stranded RNA for efficient signal transduction. Nat Commun. 2023;14(1):164.
Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, Pitha PM, et al. The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J Biol Chem. 2005;280(17):17005–12.
Moynagh PN. TLR signalling and activation of IRFs: revisiting old friends from the NF-kappaB pathway. Trends Immunol. 2005;26(9):469–76.
Snell LM, McGaha TL, Brooks DG. Type I interferon in chronic virus infection and cancer. Trends Immunol. 2017;38(8):542–57.
Lukhele S, Boukhaled GM, Brooks DG. Type I interferon signaling, regulation and gene stimulation in chronic virus infection. Semin Immunol. 2019;43: 101277.
Ll Wilkinson MG, Deakin CT, Papadopoulou C, Eleftheriou D, Wedderburn LR. JAK inhibitors: a potential treatment for JDM in the context of the role of interferon-driven pathology. Pediatr Rheumatol Online J. 2021;19(1):146.
Ahmed D, Cassol E. Role of cellular metabolism in regulating type I interferon responses: implications for tumour immunology and treatment. Cancer Lett. 2017;409:20–9.
Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–21.
Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5(9):675–87.
Darnell JE Jr. STATs and gene regulation. Science. 1997;277(5332):1630–5.
Fasler-Kan E, Pansky A, Wiederkehr M, Battegay M, Heim MH. Interferon-alpha activates signal transducers and activators of transcription 5 and 6 in Daudi cells. Eur J Biochem. 1998;254(3):514–9.
Wang Y, Nan J, Willard B, Wang X, Yang J, Stark GR. Negative regulation of type I IFN signaling by phosphorylation of STAT2 on T387. EMBO J. 2017;36(2):202–12.
Steen HC, Kotredes KP, Nogusa S, Harris MY, Balachandran S, Gamero AM. Phosphorylation of STAT2 on serine-734 negatively regulates the IFN-alpha-induced antiviral response. J Cell Sci. 2016;129(22):4190–9.
Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–45.
Dean MJ, Ochoa JB, Sanchez-Pino MD, Zabaleta J, Garai J, Del Valle L, et al. Severe COVID-19 is characterized by an impaired type I interferon response and elevated levels of arginase producing granulocytic myeloid derived suppressor cells. Front Immunol. 2021;12: 695972.
Nocito C, Lubinsky C, Hand M, Khan S, Patel T, Seliga A, et al. Centrally acting angiotensin-converting enzyme inhibitor suppresses type I interferon responses and decreases inflammation in the periphery and the CNS in lupus-prone mice. Front Immunol. 2020;11: 573677.
Kitamura H, Tanigawa T, Kuzumoto T, Nadatani Y, Otani K, Fukunaga S, et al. Interferon-alpha exerts proinflammatory properties in experimental radiation-induced esophagitis: Possible involvement of plasmacytoid dendritic cells. Life Sci. 2022;289: 120215.
Fernandez-Sendin M, Di Trani CA, Bella A, Vasquez M, Ardaiz N, Gomar C, et al. Long-term liver expression of an apolipoprotein A-I mimetic peptide attenuates interferon-alpha-induced inflammation and promotes antiviral activity. Front Immunol. 2020;11: 620283.
Chen YJ, Tsao YC, Ho TC, Puc I, Chen CC, Perng GC, et al. Antrodia cinnamomea suppress dengue virus infection through enhancing the secretion of interferon-alpha. Plants. 2022;11(19):2631.
Zhang L, Pan Y, Xu Y, Zhang W, Ma W, Ibrahim YM, et al. Paraoxonase-1 facilitates PRRSV replication by interacting with viral nonstructural protein-9 and inhibiting type I interferon pathway. Viruses. 2022;14(6):1203.
Li J, Xie Y, Li L, Li X, Shen L, Gong J, et al. MicroRNA-30a modulates type I interferon responses to facilitate coxsackievirus B3 replication via targeting tripartite motif protein 25. Front Immunol. 2020;11: 603437.
Jang YJ, Lim JY, Kim S, Lee Y, Kweon MN, Kim JH. Enhanced interferon-beta response contributes to eosinophilic chronic rhinosinusitis. Front Immunol. 2018;9:2330.
Cook LE, Locke MC, Young AR, Monte K, Hedberg ML, Shimak RM, et al. Distinct roles of interferon alpha and beta in controlling chikungunya virus replication and modulating neutrophil-mediated inflammation. J Virol. 2019;94(1):10–128.
Lei Y, Guerra Martinez C, Torres-Odio S, Bell SL, Birdwell CE, Bryant JD, et al. Elevated type I interferon responses potentiate metabolic dysfunction, inflammation, and accelerated aging in mtDNA mutator mice. Sci Adv. 2021;7(22):eabe7548.
Arimori Y, Nakamura R, Yamada H, Shibata K, Maeda N, Kase T, et al. Type I interferon limits influenza virus-induced acute lung injury by regulation of excessive inflammation in mice. Antiviral Res. 2013;99(3):230–7.
Sontheimer C, Liggitt D, Elkon KB. Ultraviolet B irradiation causes stimulator of interferon genes-dependent production of protective type I interferon in mouse skin by recruited inflammatory monocytes. Arthritis Rheumatol. 2017;69(4):826–36.
Ansar M, Qu Y, Ivanciuc T, Garofalo RP, Casola A. Lack of type I interferon signaling ameliorates respiratory syncytial virus-induced lung inflammation and restores antioxidant defenses. Antioxidants. 2021;11(1):67.
Cagliani J, Yang WL, McGinn JT, Wang Z, Wang P. Anti-interferon-alpha receptor 1 antibodies attenuate inflammation and organ injury following hemorrhagic shock. J Trauma Acute Care Surg. 2019;86(5):881–90.
D’Souza SS, Zhang Y, Bailey JT, Fung ITH, Kuentzel ML, Chittur SV, et al. Type I Interferon signaling controls the accumulation and transcriptomes of monocytes in the aged lung. Aging Cell. 2021;20(10): e13470.
Chan CC, Damen M, Moreno-Fernandez ME, Stankiewicz TE, Cappelletti M, Alarcon PC, et al. Type I interferon sensing unlocks dormant adipocyte inflammatory potential. Nat Commun. 2020;11(1):2745.
Minayoshi Y, Maeda H, Yanagisawa H, Hamasaki K, Mizuta Y, Nishida K, et al. Development of Kupffer cell targeting type-I interferon for the treatment of hepatitis via inducing anti-inflammatory and immunomodulatory actions. Drug Deliv. 2018;25(1):1067–77.
Tanaka MH, Giro EM, Cavalcante LB, Pires JR, Apponi LH, Valentini SR, et al. Expression of interferon-gamma, interferon-alpha and related genes in individuals with Down syndrome and periodontitis. Cytokine. 2012;60(3):875–81.
Nataraja C, Flynn J, Dankers W, Northcott M, Zhu W, Sherlock R, et al. GILZ regulates type I interferon release and sequesters STAT1. J Autoimmun. 2022;131: 102858.
Chen X, Wu Q, Cao X, Yang Y, Gong Z, Ren T, et al. Menthone inhibits type-I interferon signaling by promoting Tyk2 ubiquitination to relieve local inflammation of rheumatoid arthritis. Int Immunopharmacol. 2022;112: 109228.
Hu Y, Pan Q, Zhou K, Ling Y, Wang H, Li Y. RUNX1 inhibits the antiviral immune response against influenza A virus through attenuating type I interferon signaling. Virol J. 2022;19(1):39.
Yang L, Han X, Zhang C, Sun C, Huang S, Xiao W, et al. Hsa_circ_0060450 Negatively Regulates Type I Interferon-Induced Inflammation by Serving as miR-199a-5p Sponge in Type 1 Diabetes Mellitus. Front Immunol. 2020;11: 576903.
Gothe F, Stremenova Spegarova J, Hatton CF, Griffin H, Sargent T, Cowley SA, et al. Aberrant inflammatory responses to type I interferon in STAT2 or IRF9 deficiency. J Allergy Clin Immunol. 2022;150(4):955-64 e16.
Wilson RP, Tursi SA, Rapsinski GJ, Medeiros NJ, Le LS, Kotredes KP, et al. STAT2 dependent Type I Interferon response promotes dysbiosis and luminal expansion of the enteric pathogen Salmonella Typhimurium. PLoS Pathog. 2019;15(4): e1007745.
Shen H, Liu X, Zhang P, Wang S, Liu Y, Zhang L, et al. Porcine circovirus 3 Cap inhibits type I interferon signaling through interaction with STAT2. Virus Res. 2020;275: 197804.
Kozela E, Pietr M, Juknat A, Rimmerman N, Levy R, Vogel Z. Cannabinoids Delta(9)-tetrahydrocannabinol and cannabidiol differentially inhibit the lipopolysaccharide-activated NF-kappaB and interferon-beta/STAT proinflammatory pathways in BV-2 microglial cells. J Biol Chem. 2010;285(3):1616–26.
Racicot K, Kwon JY, Aldo P, Abrahams V, El-Guindy A, Romero R, et al. Type I interferon regulates the placental inflammatory response to bacteria and is targeted by virus: mechanism of polymicrobial infection-induced preterm birth. Am J Reprod Immunol. 2016;75(4):451–60.
Febvre-James M, Lecureur V, Augagneur Y, Mayati A, Fardel O. Repression of interferon beta-regulated cytokines by the JAK1/2 inhibitor ruxolitinib in inflammatory human macrophages. Int Immunopharmacol. 2018;54:354–65.
Klopfenstein N, Brandt SL, Castellanos S, Gunzer M, Blackman A, Serezani CH. SOCS-1 inhibition of type I interferon restrains Staphylococcus aureus skin host defense. PLoS Pathog. 2021;17(3): e1009387.
Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369(6504):718–24.
Stefan KL, Fink A, Surana NK, Kasper DL, Dasgupta S. Type I interferon signaling restrains IL-10R+ colonic macrophages and dendritic cells and leads to more severe Salmonella colitis. PLoS ONE. 2017;12(11): e0188600.
Qu B, Cao J, Zhang F, Cui H, Teng J, Li J, et al. Type I interferon inhibition of MicroRNA-146a maturation through up-regulation of monocyte chemotactic protein-induced protein 1 in systemic lupus erythematosus. Arthritis Rheumatol. 2015;67(12):3209–18.
Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, et al. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature. 2015;517(7532):89–93.
Snell LM, Brooks DG. New insights into type I interferon and the immunopathogenesis of persistent viral infections. Curr Opin Immunol. 2015;34:91–8.
Wang L, Ning S. TRIMming type I interferon-mediated innate immune response in antiviral and antitumor defense. Viruses. 2021;13(2):279.
Ihle JN. Cytokine receptor signalling. Nature. 1995;377(6550):591–4.
Honda K, Taniguchi T. Toll-like receptor signaling and IRF transcription factors. IUBMB Life. 2006;58(5–6):290–5.
Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13(5):816–25.
Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med. 2007;13(5):543–51.
Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13(4):539–48.
Veenhuis RT, Freeman ZT, Korleski J, Cohen LK, Massaccesi G, Tomasi A, et al. HIV-antibody complexes enhance production of type I interferon by plasmacytoid dendritic cells. J Clin Invest. 2017;127(12):4352–64.
Yang JY, Kim MS, Kim E, Cheon JH, Lee YS, Kim Y, et al. Enteric viruses ameliorate gut inflammation via toll-like receptor 3 and toll-like receptor 7-mediated interferon-beta production. Immunity. 2016;44(4):889–900.
Sekheri M, Rizo-Tellez SA, Othman A, El Kebir D, Filep JG. Interferon-beta regulates proresolving lipids to promote the resolution of acute airway inflammation. Proc Natl Acad Sci U S A. 2022;119(31): e2201146119.
Allen ME, Golding A, Rus V, Karabin NB, Li S, Lescott CJ, et al. Targeted delivery of chloroquine to antigen-presenting cells enhances inhibition of the type I interferon response. ACS Biomater Sci Eng. 2021;7(12):5666–77.
Chang MY, Kang I, Gale M Jr, Manicone AM, Kinsella MG, Braun KR, et al. Versican is produced by Trif- and type I interferon-dependent signaling in macrophages and contributes to fine control of innate immunity in lungs. Am J Physiol Lung Cell Mol Physiol. 2017;313(6):L1069–86.
Dhariwala MO, Olson RM, Anderson DM. Induction of type I interferon through a noncanonical toll-like receptor 7 pathway during yersinia pestis infection. Infect Immun. 2017;85(11):10–128.
Auger JP, Santinon A, Roy D, Mossman K, Xu J, Segura M, et al. Type I Interferon Induced by Streptococcus suis Serotype 2 is strain-dependent and may be beneficial for host survival. Front Immunol. 2017;8:1039.
Cordoba-David G, Garcia-Gimenez J, Cardoso Castelo-Branco R, Carrasco S, Cannata P, Ortiz A, et al. Crosstalk between TBK1/IKKepsilon and the type I interferon pathway contributes to tubulointerstitial inflammation and kidney tubular injury. Front Pharmacol. 2022;13: 987979.
Artusa V, Ciaramelli C, D’Aloia A, Facchini FA, Gotri N, Bruno A, et al. Green and roasted coffee extracts inhibit interferon-beta release in LPS-stimulated human macrophages. Front Pharmacol. 2022;13: 806010.
Zhou Y, Li M, Lv T, Huang M, Cheng B, Zhang Y, et al. Gastrodin inhibits virus infection by promoting the production of type I interferon. Front Pharmacol. 2020;11: 608707.
Fritsch LE, Ju J, Gudenschwager Basso EK, Soliman E, Paul S, Chen J, et al. Type I interferon response is mediated by NLRX1-cGAS-STING signaling in brain injury. Front Mol Neurosci. 2022;15: 852243.
Huang M, Wu T, Liu R, Wang M, Shi M, Xin J, et al. Polbeta modulates the expression of type I interferon via STING pathway. Biochem Biophys Res Commun. 2022;621:137–43.
Onsa-Ard A, Thongboontho R, Munkong N, Phromnoi K, Ontawong A, Pengnet S, et al. Anti-inflammatory effects of red rice bran extract ameliorate type i interferon production via STING pathway. Foods. 2022;11(11):1622.
Liu Z, Xia L. E3 ligase RNF5 inhibits type I interferon response in herpes simplex virus keratitis through the STING/IRF3 signaling pathway. Front Microbiol. 2022;13: 944101.
Wu M, Skaug B, Bi X, Mills T, Salazar G, Zhou X, et al. Interferon regulatory factor 7 (IRF7) represents a link between inflammation and fibrosis in the pathogenesis of systemic sclerosis. Ann Rheum Dis. 2019;78(11):1583–91.
He J, Yang Q, Xiao Q, Lei A, Li X, Zhou P, et al. IRF-7 is a critical regulator of type 2 innate lymphoid cells in allergic airway inflammation. Cell Rep. 2019;29(9):2718-30 e6.
Zhou Q, Lavorgna A, Bowman M, Hiscott J, Harhaj EW. Aryl hydrocarbon receptor interacting protein targets IRF7 to suppress antiviral signaling and the induction of type I interferon. J Biol Chem. 2015;290(23):14729–39.
Hu R, Xia CQ, Butfiloski E, Clare-Salzler M. Effect of high glucose on cytokine production by human peripheral blood immune cells and type I interferon signaling in monocytes: Implications for the role of hyperglycemia in the diabetes inflammatory process and host defense against infection. Clin Immunol. 2018;195:139–48.
Ren Y, Zhao Y, Lin D, Xu X, Zhu Q, Yao J, et al. The type I interferon-IRF7 axis mediates transcriptional expression of Usp25 gene. J Biol Chem. 2016;291(25):13206–15.
Trevejo-Nunez G, Lin B, Fan L, Aggor FEY, Biswas PS, Chen K, et al. Regnase-1 deficiency restrains klebsiella pneumoniae infection by regulation of a type I interferon response. Bio. 2021;13(1):e0379221.
Valaperti A, Nishii M, Liu Y, Yang H, Naito K, Liu PP, et al. The adapter protein c-Cbl-associated protein (CAP) protects from acute CVB3-mediated myocarditis through stabilization of type I interferon production and reduced cytotoxicity. Basic Res Cardiol. 2014;109(3):411.
Villamayor L, Rivero V, Lopez-Garcia D, Topham DJ, Martinez-Sobrido L, Nogales A, et al. Interferon alpha inducible protein 6 is a negative regulator of innate immune responses by modulating RIG-I activation. Front Immunol. 2023;14:1105309.
Simpson J, Lynch JP, Loh Z, Zhang V, Werder RB, Spann K, et al. The absence of interferon-beta promotor stimulator-1 (IPS-1) predisposes to bronchiolitis and asthma-like pathology in response to pneumoviral infection in mice. Sci Rep. 2017;7(1):2353.
Zheng Y, Deng J, Han L, Zhuang MW, Xu Y, Zhang J, et al. SARS-CoV-2 NSP5 and N protein counteract the RIG-I signaling pathway by suppressing the formation of stress granules. Signal Transduct Target Ther. 2022;7(1):22.
Han L, Zhuang MW, Deng J, Zheng Y, Zhang J, Nan ML, et al. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5-MAVS, TLR3-TRIF, and cGAS-STING signaling pathways. J Med Virol. 2021;93(9):5376–89.
Zheng Y, Zhuang MW, Han L, Zhang J, Nan ML, Zhan P, et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct Target Ther. 2020;5(1):299.
Deng J, Zheng Y, Zheng SN, Nan ML, Han L, Zhang J, et al. SARS-CoV-2 NSP7 inhibits type I and III IFN production by targeting the RIG-I/MDA5, TRIF, and STING signaling pathways. J Med Virol. 2023;95(3): e28561.
Deng J, Zheng SN, Xiao Y, Nan ML, Zhang J, Han L, et al. SARS-CoV-2 NSP8 suppresses type I and III IFN responses by modulating the RIG-I/MDA5, TRIF, and STING signaling pathways. J Med Virol. 2023;95(4): e28680.
Liu H, Li M, Song Y, Xu W. TRIM21 Restricts Coxsackievirus B3 replication, cardiac and pancreatic injury via interacting with MAVS and positively regulating IRF3-mediated type-I interferon production. Front Immunol. 2018;9:2479.
Gutierrez-Merino J, Isla B, Combes T, Martinez-Estrada F, Maluquer De Motes C. Beneficial bacteria activate type-I interferon production via the intracellular cytosolic sensors STING and MAVS. Gut Microbes. 2020;11(4):771–88.
Killarney ST, Washart R, Soderquist RS, Hoj JP, Lebhar J, Lin KH, et al. Executioner caspases restrict mitochondrial RNA-driven Type I IFN induction during chemotherapy-induced apoptosis. Nat Commun. 2023;14(1):1399.
Huang Y, Liang W, Li K, Liao X, Chen J, Qiu X, et al. Sorafenib suppresses the activation of type I interferon pathway induced by RLR-MAVS and cGAS-STING signaling. Biochem Biophys Res Commun. 2022;623:181–8.
Han W, Chen Q, Cui J, Zhao Y, Li M, Li X. E3 ubiquitin ligase RNF114 promotes vesicular stomatitis virus replication via inhibiting type I interferon production. Microb Pathog. 2022;167: 105569.
Pons BJ, Pettes-Duler A, Naylies C, Taieb F, Bouchenot C, Hashim S, et al. Chronic exposure to Cytolethal Distending Toxin (CDT) promotes a cGAS-dependent type I interferon response. Cell Mol Life Sci. 2021;78(17–18):6319–35.
Hsin F, Hsu YC, Tsai YF, Lin SW, Liu HM. The transmembrane serine protease hepsin suppresses type I interferon induction by cleaving STING. Sci Signal. 2021;14(687):eabb4752.
Fischer K, Tschismarov R, Pilz A, Straubinger S, Carotta S, McDowell A, et al. Cutibacterium acnes infection induces type I interferon synthesis through the cGAS-STING pathway. Front Immunol. 2020;11: 571334.
Vail KJ, da Silveira BP, Bell SL, Cohen ND, Bordin AI, Patrick KL, et al. The opportunistic intracellular bacterial pathogen Rhodococcus equi elicits type I interferon by engaging cytosolic DNA sensing in macrophages. PLoS Pathog. 2021;17(9): e1009888.
Wang Y, Karki R, Mall R, Sharma BR, Kalathur RC, Lee S, et al. Molecular mechanism of RIPK1 and caspase-8 in homeostatic type I interferon production and regulation. Cell Rep. 2022;41(1): 111434.
Torre S, Polyak MJ, Langlais D, Fodil N, Kennedy JM, Radovanovic I, et al. USP15 regulates type I interferon response and is required for pathogenesis of neuroinflammation. Nat Immunol. 2017;18(1):54–63.
Li S, Wu Q, Jiang Z, Wu Y, Li Y, Ni B, et al. miR-31-5p regulates type I interferon by targeting SLC15A4 in plasmacytoid dendritic cells of systemic lupus erythematosus. J Inflamm Res. 2022;15:6607–16.
Elkon KB, Wiedeman A. Type I IFN system in the development and manifestations of SLE. Curr Opin Rheumatol. 2012;24(5):499–505.
Severa M, Fitzgerald KA. TLR-mediated activation of type I IFN during antiviral immune responses: fighting the battle to win the war. Curr Top Microbiol Immunol. 2007;316:167–92.
Platanitis E, Decker T. Regulatory networks involving STATs, IRFs, and NFkappaB in inflammation. Front Immunol. 2018;9:2542.
Jefferies CA. Regulating IRFs in IFN driven disease. Front Immunol. 2019;10:325.
Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. 2014;32:461–88.
Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4(5):491–6.
Yanai H, Chiba S, Hangai S, Kometani K, Inoue A, Kimura Y, et al. Revisiting the role of IRF3 in inflammation and immunity by conditional and specifically targeted gene ablation in mice. Proc Natl Acad Sci USA. 2018;115(20):5253–8.
McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci USA. 2004;101(1):233–8.
Petro TM. IFN regulatory factor 3 in health and disease. J Immunol. 2020;205(8):1981–9.
Wang Y, Luo J, Alu A, Han X, Wei Y, Wei X. cGAS-STING pathway in cancer biotherapy. Mol Cancer. 2020;19(1):136.
Srikanth S, Woo JS, Wu B, El-Sherbiny YM, Leung J, Chupradit K, et al. The Ca(2+) sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol. 2019;20(2):152–62.
Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, et al. Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab. 2019;30(4):784-99 e5.
Kaufman BA, Mora AL. IRGM1, a guardian of mitochondrial DAMP-mediated autoinflammation. Nat Immunol. 2021;22(3):272–3.
Riley JS, Tait SW. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020;21(4): e49799.
Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. 2016;213(5):697–713.
Lyu Y, Wang T, Huang S, Zhang Z. Mitochondrial damage-associated molecular patterns and metabolism in the regulation of innate immunity. J Innate Immun. 2023;15(1):665–79.
Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov. 2020;6:36.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–32.
Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560(7717):198–203.
Au WC, Moore PA, LaFleur DW, Tombal B, Pitha PM. Characterization of the interferon regulatory factor-7 and its potential role in the transcription activation of interferon A genes. J Biol Chem. 1998;273(44):29210–7.
Mogensen TH. IRF and STAT transcription factors - from basic biology to roles in infection, protective immunity, and primary immunodeficiencies. Front Immunol. 2018;9:3047.
Ikushima H, Negishi H, Taniguchi T. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol. 2013;78:105–16.
Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434(7034):772–7.
Ning S, Pagano JS, Barber GN. IRF7: activation, regulation, modification and function. Genes Immun. 2011;12(6):399–414.
Qing F, Liu Z. Interferon regulatory factor 7 in inflammation, cancer and infection. Front Immunol. 2023;14:1190841.
Pan H, Yan BS, Rojas M, Shebzukhov YV, Zhou H, Kobzik L, et al. Ipr1 gene mediates innate immunity to tuberculosis. Nature. 2005;434(7034):767–72.
Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17(22):6660–9.
Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998;441(1):106–10.
Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5(7):730–7.
Kok KH, Lui PY, Ng MH, Siu KL, Au SW, Jin DY. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe. 2011;9(4):299–309.
Vaughn LS, Frederick K, Burnett SB, Sharma N, Bragg DC, Camargos S, et al. DYT-PRKRA mutation P222L enhances PACT’s stimulatory activity on type I interferon induction. Biomolecules. 2022;12(5):713.
Chukwurah E, Farabaugh KT, Guan BJ, Ramakrishnan P, Hatzoglou M. A tale of two proteins: PACT and PKR and their roles in inflammation. FEBS J. 2021;288(22):6365–91.
Ho TH, Kew C, Lui PY, Chan CP, Satoh T, Akira S, et al. PACT- and RIG-I-dependent activation of type I interferon production by a defective interfering RNA derived from measles virus vaccine. J Virol. 2016;90(3):1557–68.
Brisse M, Ly H. Comparative structure and function analysis of the RIG-I-Like receptors: RIG-I and MDA5. Front Immunol. 2019;10:1586.
Kell AM, Gale M Jr. RIG-I in RNA virus recognition. Virology. 2015;479–480:110–21.
Xu XX, Wan H, Nie L, Shao T, Xiang LX, Shao JZ. RIG-I: a multifunctional protein beyond a pattern recognition receptor. Protein Cell. 2018;9(3):246–53.
Jacobs JL, Coyne CB. Mechanisms of MAVS regulation at the mitochondrial membrane. J Mol Biol. 2013;425(24):5009–19.
Lui PY, Wong LR, Ho TH, Au SWN, Chan CP, Kok KH, et al. PACT facilitates RNA-induced activation of MDA5 by promoting MDA5 oligomerization. J Immunol. 2017;199(5):1846–55.
Sanchez David RY, Combredet C, Najburg V, Millot GA, Beauclair G, Schwikowski B, et al. LGP2 binds to PACT to regulate RIG-I- and MDA5-mediated antiviral responses. Sci Signal. 2019;12(601):eaar3993.
Mohanty A, Tiwari-Pandey R, Pandey NR. Mitochondria: the indispensable players in innate immunity and guardians of the inflammatory response. J Cell Commun Signal. 2019;13(3):303–18.
Vazquez C, Horner SM. MAVS coordination of antiviral innate immunity. J Virol. 2015;89(14):6974–7.
Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344–62.
Sermersheim M, Kenney AD, Lin PH, McMichael TM, Cai C, Gumpper K, et al. MG53 suppresses interferon-beta and inflammation via regulation of ryanodine receptor-mediated intracellular calcium signaling. Nat Commun. 2020;11(1):3624.
Lee YS, Bao X, Lee HH, Jang JJ, Saruuldalai E, Park G, et al. Nc886, a novel suppressor of the type I interferon response upon pathogen intrusion. Int J Mol Sci. 2021;22(4):2003.
Dong LW, Chen YY, Chen CC, Ma ZC, Fu J, Huang BL, et al. Adenosine 2A receptor contributes to the facilitation of post-infectious irritable bowel syndrome by gammadelta T cells via the PKA/CREB/NF-kappaB signaling pathway. World J Gastroenterol. 2023;29(9):1475–91.
Zhang F, Sun X, Zhu Y, Qin W. Downregulation of miR-146a inhibits influenza A virus replication by enhancing the type I interferon response in vitro and in vivo. Biomed Pharmacother. 2019;111:740–50.
Chen HY, Tang RC, Liang JW, Zhao W, Yu SS, Yao RR, et al. Deubiquitinase USP47 attenuates virus-induced type I interferon signaling. Int Immunopharmacol. 2023;118: 110040.
Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25(51):6717–30.
Hers I, Vincent EE, Tavare JM. Akt signalling in health and disease. Cell Signal. 2011;23(10):1515–27.
Kaur S, Sassano A, Dolniak B, Joshi S, Majchrzak-Kita B, Baker DP, et al. Role of the Akt pathway in mRNA translation of interferon-stimulated genes. Proc Natl Acad Sci USA. 2008;105(12):4808–13.
Uddin S, Yenush L, Sun XJ, Sweet ME, White MF, Platanias LC. Interferon-alpha engages the insulin receptor substrate-1 to associate with the phosphatidylinositol 3’-kinase. J Biol Chem. 1995;270(27):15938–41.
Lekmine F, Sassano A, Uddin S, Smith J, Majchrzak B, Brachmann SM, et al. Interferon-gamma engages the p70 S6 kinase to regulate phosphorylation of the 40S S6 ribosomal protein. Exp Cell Res. 2004;295(1):173–82.
Lekmine F, Uddin S, Sassano A, Parmar S, Brachmann SM, Majchrzak B, et al. Activation of the p70 S6 kinase and phosphorylation of the 4E-BP1 repressor of mRNA translation by type I interferons. J Biol Chem. 2003;278(30):27772–80.
Saleiro D, Platanias LC. Intersection of mTOR and STAT signaling in immunity. Trends Immunol. 2015;36(1):21–9.
Guiducci C, Ghirelli C, Marloie-Provost MA, Matray T, Coffman RL, Liu YJ, et al. PI3K is critical for the nuclear translocation of IRF-7 and type I IFN production by human plasmacytoid predendritic cells in response to TLR activation. J Exp Med. 2008;205(2):315–22.
Ding X, Xiang W, Yi R, Huang X, Lin Q, He X. Neutralizing interferon-alpha blocks inflammation-mediated vascular injury via PI3K and AMPK in systemic lupus erythematosus. Immunology. 2021;164(2):372–85.
Gairhe S, Awad KS, Dougherty EJ, Ferreyra GA, Wang S, Yu ZX, et al. Type I interferon activation and endothelial dysfunction in caveolin-1 insufficiency-associated pulmonary arterial hypertension. Proc Natl Acad Sci U S A. 2021;118(11):e2010206118.
Matsumoto A, Ichikawa T, Nakao K, Miyaaki H, Hirano K, Fujimito M, et al. Interferon-alpha-induced mTOR activation is an anti-hepatitis C virus signal via the phosphatidylinositol 3-kinase-Akt-independent pathway. J Gastroenterol. 2009;44(8):856–63.
Stout-Delgado HW, Yang X, Walker WE, Tesar BM, Goldstein DR. Aging impairs IFN regulatory factor 7 up-regulation in plasmacytoid dendritic cells during TLR9 activation. J Immunol. 2008;181(10):6747–56.
Murayama G, Furusawa N, Chiba A, Yamaji K, Tamura N, Miyake S. Enhanced IFN-alpha production is associated with increased TLR7 retention in the lysosomes of palasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res Ther. 2017;19(1):234.
Bilancio A, Rinaldi B, Oliviero MA, Donniacuo M, Monti MG, Boscaino A, et al. Inhibition of p110delta PI3K prevents inflammatory response and restenosis after artery injury. Biosci Rep. 2017;37(5):BSR20171112.
Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, et al. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J Exp Med. 2005;202(1):135–43.
Katsoulidis E, Li Y, Mears H, Platanias LC. The p38 mitogen-activated protein kinase pathway in interferon signal transduction. J Interferon Cytokine Res. 2005;25(12):749–56.
Mayer IA, Verma A, Grumbach IM, Uddin S, Lekmine F, Ravandi F, et al. The p38 MAPK pathway mediates the growth inhibitory effects of interferon-alpha in BCR-ABL-expressing cells. J Biol Chem. 2001;276(30):28570–7.
Ishida H, Ohkawa K, Hosui A, Hiramatsu N, Kanto T, Ueda K, et al. Involvement of p38 signaling pathway in interferon-alpha-mediated antiviral activity toward hepatitis C virus. Biochem Biophys Res Commun. 2004;321(3):722–7.
David M, Petricoin E 3rd, Benjamin C, Pine R, Weber MJ, Larner AC. Requirement for MAP kinase (ERK2) activity in interferon alpha- and interferon beta-stimulated gene expression through STAT proteins. Science. 1995;269(5231):1721–3.
Wang F, Ma Y, Barrett JW, Gao X, Loh J, Barton E, et al. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat Immunol. 2004;5(12):1266–74.
Wang K, Zou S, Chen H, Higazy D, Gao X, Zhang Y, et al. Zika virus replication on endothelial cells and invasion into the central nervous system by inhibiting interferon beta translation. Virology. 2023;582:23–34.
Accapezzato D, Caccavale R, Paroli MP, Gioia C, Nguyen BL, Spadea L, et al. Advances in the pathogenesis and treatment of systemic lupus erythematosus. Int J Mol Sci. 2023;24(7):6578.
Deng Y, Tsao BP. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat Rev Rheumatol. 2010;6(12):683–92.
Izadi S, Najfizadeh SR, Nejati A, TeimooriRad M, Shahmahmoodi S, Shirazi FG, et al. Potential role of EBV and Toll-like receptor 9 ligand in patients with systemic lupus erythematosus. Immunol Res. 2023;71:698–708.
Dahlgren MW, Plumb AW, Niss K, Lahl K, Brunak S, Johansson-Lindbom B. Type I interferons promote germinal centers through B cell intrinsic signaling and dendritic cell dependent Th1 and Tfh cell lineages. Front Immunol. 2022;13: 932388.
Care MA, Stephenson SJ, Barnes NA, Fan I, Zougman A, El-Sherbiny YM, et al. Network analysis identifies proinflammatory plasma cell polarization for secretion of ISG15 in human autoimmunity. J Immunol. 2016;197(4):1447–59.
Yan M, Gu Y, Sun H, Ge Q. Neutrophil extracellular traps in tumor progression and immunotherapy. Front Immunol. 2023;14:1135086.
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–38.
Barrat FJ, Crow MK, Ivashkiv LB. Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol. 2019;20(12):1574–83.
Lubbers J, Brink M, van de Stadt LA, Vosslamber S, Wesseling JG, van Schaardenburg D, et al. The type I IFN signature as a biomarker of preclinical rheumatoid arthritis. Ann Rheum Dis. 2013;72(5):776–80.
Yarwood A, Huizinga TW, Worthington J. The genetics of rheumatoid arthritis: risk and protection in different stages of the evolution of RA. Rheumatology (Oxford). 2016;55(2):199–209.
Harigai M. Growing evidence of the safety of JAK inhibitors in patients with rheumatoid arthritis. Rheumatology (Oxford). 2019;58(Suppl 1):i34–42.
Zhan Q, Zhang J, Lin Y, Chen W, Fan X, Zhang D. Pathogenesis and treatment of Sjogren’s syndrome: review and update. Front Immunol. 2023;14:1127417.
Skaug B, Assassi S. Type I interferon dysregulation in Systemic Sclerosis. Cytokine. 2020;132: 154635.
Unlu B, Tursen U, Rajabi Z, Jabalameli N, Rajabi F. The immunogenetics of systemic sclerosis. Adv Exp Med Biol. 2022;1367:259–98.
Liu X, Mayes MD, Tan FK, Wu M, Reveille JD, Harper BE, et al. Correlation of interferon-inducible chemokine plasma levels with disease severity in systemic sclerosis. Arthritis Rheum. 2013;65(1):226–35.
Assassi S, Mayes MD, Arnett FC, Gourh P, Agarwal SK, McNearney TA, et al. Systemic sclerosis and lupus: points in an interferon-mediated continuum. Arthritis Rheum. 2010;62(2):589–98.
Ciurtin C. Potential relevance of type I interferon-related biomarkers for the management of polygenic autoimmune rheumatic diseases with childhood onset. Clin Rheumatol. 2023;42:1733–6.
Niewold TB, Kariuki SN, Morgan GA, Shrestha S, Pachman LM. Elevated serum interferon-alpha activity in juvenile dermatomyositis: associations with disease activity at diagnosis and after thirty-six months of therapy. Arthritis Rheum. 2009;60(6):1815–24.
Moneta GM, Pires Marafon D, Marasco E, Rosina S, Verardo M, Fiorillo C, et al. Muscle expression of type I and type II interferons is increased in Juvenile dermatomyositis and related to clinical and histologic features. Arthritis Rheumatol. 2019;71(6):1011–21.
Piper CJM, Wilkinson MGL, Deakin CT, Otto GW, Dowle S, Duurland CL, et al. CD19(+)CD24(hi)CD38(hi) B cells are expanded in Juvenile dermatomyositis and exhibit a pro-inflammatory phenotype after activation through toll-like receptor 7 and interferon-alpha. Front Immunol. 2018;9:1372.
Durbin JE, Fernandez-Sesma A, Lee CK, Rao TD, Frey AB, Moran TM, et al. Type I IFN modulates innate and specific antiviral immunity. J Immunol. 2000;164(8):4220–8.
Koerner I, Kochs G, Kalinke U, Weiss S, Staeheli P. Protective role of beta interferon in host defense against influenza A virus. J Virol. 2007;81(4):2025–30.
McNab FW, Rajsbaum R, Stoye JP, O’Garra A. Tripartite-motif proteins and innate immune regulation. Curr Opin Immunol. 2011;23(1):46–56.
Yan N, Chen ZJ. Intrinsic antiviral immunity. Nat Immunol. 2012;13(3):214–22.
Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. 2006;25(3):373–81.
Wang YJ. Clinical observations on the restoration of kidney function in patients with chronic nephritis treated with “warming kidney” therapy–an analysis of 68 cases. Zhong Xi Yi Jie He Za Zhi. 1985;5(3):158–60.
van Grevenynghe J, Cubas RA, Noto A, DaFonseca S, He Z, Peretz Y, et al. Loss of memory B cells during chronic HIV infection is driven by Foxo3a- and TRAIL-mediated apoptosis. J Clin Invest. 2011;121(10):3877–88.
Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013;340(6129):207–11.
Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340(6129):202–7.
Park SH, Park-Min KH, Chen J, Hu X, Ivashkiv LB. Tumor necrosis factor induces GSK3 kinase-mediated cross-tolerance to endotoxin in macrophages. Nat Immunol. 2011;12(7):607–15.
Park SH, Kang K, Giannopoulou E, Qiao Y, Kang K, Kim G, et al. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat Immunol. 2017;18(10):1104–16.
Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol. 2020;5(49):eabd1554.
Guimaraes ES, Marinho FV, de Queiroz N, Antunes MM, Oliveira SC. Impact of STING inflammatory signaling during intracellular bacterial infections. Cells. 2021;11(1):74.
Sologuren I, Boisson-Dupuis S, Pestano J, Vincent QB, Fernandez-Perez L, Chapgier A, et al. Partial recessive IFN-gammaR1 deficiency: genetic, immunological and clinical features of 14 patients from 11 kindreds. Hum Mol Genet. 2011;20(8):1509–23.
Rackov G, Shokri R, De Mon MA, Martinez AC, Balomenos D. The role of IFN-beta during the course of sepsis progression and its therapeutic potential. Front Immunol. 2017;8:493.
Mraz AL, Weir MH. Knowledge to predict pathogens: legionella pneumophila lifecycle systematic review part II growth within and egress from a host cell. Microorganisms. 2022;10(1):141.
Kelly-Scumpia KM, Scumpia PO, Delano MJ, Weinstein JS, Cuenca AG, Wynn JL, et al. Type I interferon signaling in hematopoietic cells is required for survival in mouse polymicrobial sepsis by regulating CXCL10. J Exp Med. 2010;207(2):319–26.
MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol. 2012;12(5):367–82.
Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med. 2010;207(2):327–37.
Kearney SJ, Delgado C, Eshleman EM, Hill KK, O’Connor BP, Lenz LL. Type I IFNs downregulate myeloid cell IFN-gamma receptor by inducing recruitment of an early growth response 3/NGFI-A binding protein 1 complex that silences ifngr1 transcription. J Immunol. 2013;191(6):3384–92.
Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, et al. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science. 2013;339(6126):1448–53.
Masood E. Chief to leave troubled British biotech. Nature. 1998;393(6683):299.
O’Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, et al. Type I interferon production enhances susceptibility to listeria monocytogenes infection. J Exp Med. 2004;200(4):437–45.
Boumaza A, Mezouar S, Bardou M, Raoult D, Mege JL, Desnues B. Tumor necrosis factor inhibitors exacerbate whipple’s disease by reprogramming macrophage and inducing apoptosis. Front Immunol. 2021;12: 667357.
Karki R, Lee E, Sharma BR, Banoth B, Kanneganti TD. IRF8 regulates gram-negative bacteria-mediated NLRP3 inflammasome activation and cell death. J Immunol. 2020;204(9):2514–22.
Hasselbalch HC, Skov V, Kjaer L, Ellervik C, Poulsen A, Poulsen TD, et al. COVID-19 as a mediator of interferon deficiency and hyperinflammation: rationale for the use of JAK1/2 inhibitors in combination with interferon. Cytokine Growth Factor Rev. 2021;60:28–45.
Ramasamy S, Subbian S. Critical determinants of cytokine storm and type I interferon response in COVID-19 pathogenesis. Clin Microbiol Rev. 2021;34(3):10–128.
Yang L, Wang J, Hui P, Yarovinsky TO, Badeti S, Pham K, et al. Potential role of IFN-alpha in COVID-19 patients and its underlying treatment options. Appl Microbiol Biotechnol. 2021;105(10):4005–15.
Noel N, Jacquelin B, Huot N, Goujard C, Lambotte O, Muller-Trutwin M. Interferon-associated therapies toward HIV control: the back and forth. Cytokine Growth Factor Rev. 2018;40:99–112.
Nganou-Makamdop K, Douek DC. Manipulating the interferon signaling pathway: implications for HIV infection. Virol Sin. 2019;34(2):192–6.
Mary A, Henaut L, Macq PY, Badoux L, Cappe A, Poree T, et al. Rationale for COVID-19 treatment by nebulized interferon-beta-1b-literature review and personal preliminary experience. Front Pharmacol. 2020;11: 592543.
Jorgensen SCJ, Tse CLY, Burry L, Dresser LD. Baricitinib: a review of pharmacology, safety, and emerging clinical experience in COVID-19. Pharmacotherapy. 2020;40(8):843–56.
Wang Y, Wu M, Li Y, Yuen HH, He ML. The effects of SARS-CoV-2 infection on modulating innate immunity and strategies of combating inflammatory response for COVID-19 therapy. J Biomed Sci. 2022;29(1):27.
Yang L, Xie X, Tu Z, Fu J, Xu D, Zhou Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct Target Ther. 2021;6(1):255.
Hoagland DA, Moller R, Uhl SA, Oishi K, Frere J, Golynker I, et al. Leveraging the antiviral type I interferon system as a first line of defense against SARS-CoV-2 pathogenicity. Immunity. 2021;54(3):557-70 e5.
Acknowledgements
The figure was created with BioRender.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
LJ and TLL wrote the manuscript with feedback from all authors. HMC, YQY, EYL and JYL polished the manuscript. WQ and HC gave their comments and suggestions to the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ji, L., Li, T., Chen, H. et al. The crucial regulatory role of type I interferon in inflammatory diseases. Cell Biosci 13, 230 (2023). https://doi.org/10.1186/s13578-023-01188-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-023-01188-z