
Abstract
The activation of the cGAS-STING pathway has tremendous potential to improve anti-tumor immunity by generating type I interferons. In recent decades, we have witnessed that producing dsDNA upon various stimuli is an initiative factor, triggering the cGAS-SING pathway for a defensive host. The understanding of both intracellular cascade reaction and the changes of molecular components gains insight into type I IFNs and adaptive immunity. Based on the immunological study, the STING-cGAS pathway is coupled to cancer biotherapy. The most challenging problem is the limited therapeutic effect. Therefore, people view 5, 6-dimethylxanthenone-4-acetic acid, cyclic dinucleotides and various derivative as cGAS-STING pathway agonists. Even so, these agonists have flaws in decreasing biotherapeutic efficacy. Subsequently, we exploited agonist delivery systems (nanocarriers, microparticles and hydrogels). The article will discuss the activation of the cGAS-STING pathway and underlying mechanisms, with an introduction of cGAS-STING agonists, related clinical trials and agonist delivery systems.
Similar content being viewed by others

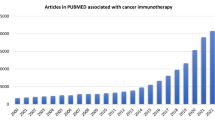
Introduction
Cancer biotherapy replies on stimulating the body’s anti-tumor biological response by initiating the host’s defensive mechanisms and using biological agents. By killing cancer cells and inhibiting its growth, cancer treatment is to regulate the balance between immune responses and tumors to treat cancer, whose mechanism is different from that of chemotherapy, radiotherapy and surgery [1, 2]. Current cancer biotherapy includes cancer vaccine [3], cancer immunotherapy [4], cancer gene therapy [5] and anti-angiogenesis therapy [6]. In the early years, the therapeutic effect was witnessed in tumors with strong immunogenicity, especially melanoma [7]. But now, cancer biotherapy research has been spread into breast cancer [8], lung cancer [9] and liver cancer [10]. Meanwhile, anti-neovascularization drugs, monoclonal antibodies, cancer vaccines, gene therapy drugs and immunomodulators have been used clinically and been studied in clinical trials.
Tumor immune surveillance is illustrated as the immune system’s recognition and the elimination of malignancies. Once tumor-associated antigens are perceived, innate immunity, adaptive immunity and cytokines function together to fight against tumors [11]. In long-term coexistence of the immune system and cancer cells, tumors alter tumor-associated antigens to directly escape immune surveillance. Moreover, tumors remodel their mesenchymal surroundings to support their growth, which is called tumor microenvironment [12]. Under the increasing level of tumor variation, selectively immune pressure and imbalance between immunity and tumors, tumors escape from the surveillance by immunoediting [13]. Innate immunity plays an important role in recognition of exogenous nucleic acids, especially cytoplasmic DNA sensing by the cGAS-STING pathway. Normally, foreign DNA is degraded by nucleases, such as three prime repair exonuclease 1 (TREX1) and ribonuclease H2 (RNase H2) [14, 15]. The destruction of cellular homeostasis would cause cytoplasmic DNA accumulation, such as endogenous retrovirus, DNA damage, genomic instability, damaged mitochondria, dying cells, exosomes, DNA viruses, retroviruses and bacteria [16,17,18]. Under such circumstances, cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS) senses and is activated when binding to double-stranded DNA (dsDNA). In the energy-consuming process, cGAS transforms adenosine 5′-triphosphate (ATP) and guanosine 5′-triphosphate (GTP) into cyclic GMP–AMP (cGAMP). As a secondary messager, cGAMP with other cyclic dinucleotides (CDNs) transmits the signal to the downstream endoplasmic reticulum (ER) protein named stimulator of interferon genes (STING). Then, the signal cascades and culminates in interferon regulatory factor 3 (IRF3) and NF-κB targets in the nucleus, leading to the secretion of type-I interferon (IFN). IFN is important to tumor-specific T cells [19, 20].
cGAS-STING pathway challenges the traditional pathogen-specific structural patterns as an innate immunity’s defensive system. Therefore, to maintain the immune balance, the pathway requires regulatory mechanisms from different levels to guarantee correct immune responses. In the review, we will discuss the knowledge of how DNA is sensed by cGAS-STING pathway, describing why cellular homeostasis happens, what the signal is initiated by, how molecules co-ordinates with each other to execute the command and how the signal is protected from erroneous immune activation. We will also focus on the molecular mechanisms and biological effects of an activated cGAS-STING pathway to enhance cancer biotherapy efficacy. Furthermore, we will review cGAS-STING agonist delivery systems and related drugs.
The molecular mechanisms of cGAS-STING pathway
cGAS activation by pathogens and dsDNAs
As stimuli, endogeneous retrovirus, DNA damage, genomic instability, damaged mitochondria, dying cells, exosomes, DNA viruses, retroviruses and bacteria contribute to the production of dsDNA. Herpes simplex virus-1 (HSV-1), vesicular stomatitis virus (VSV) and porcine circovirus type 2 (PCV2), as DNA viruses, promote IFN by activating cGAS-STING signaling pathway [21,22,23]. Besides, endogeneous retrovirus RNAs triggers a cascade wave of signaling to upregulate INF production by cGAS-STING pathway [24]. For instance, human immunodeficiency virus type 1 (HIV-1) contains single-strand DNAs of stem-loop structures, which activate cGAS in a sequence-dependent way. Mitochondrial DNA is released through outer membrane pores by activating BAX/BAK, followed by the detection of cGAS [25]. Not only in human somatic cells, but also in bacteria, cGAMP signaling activates the phospholipase which destroys bacterial membrane integrity to death before phage reproduction. It provides an evolutionary point to microorganisms against phages [26].
Structurally, how the interaction of dsDNA and cGAS activates cGAS has been studied. As an important functional domain of cGAS, C-terminal nucleotidyltransferase (NTase) domain consists of a catalytic domain and two positive parts. Promoted by zinc ribbon, dsDNA activates cGAS by forming 2:2 cGAS-dsDNA complexes. Each cGAS contains a dsDNA like a ladder network to stabilize the cGAS-dsDNA structure. Then, the stable structure turns on the switch of rearrangement of the catalytic domain to transform GTP and ATP into cyclized cGAMP. The synthesized cGAMP containing two phosphate diester bonds show a higher affinity for STING. Although other nucleic acids (ssDNA, ssRNA and dsRNA) also bind to cGAS, they cannot rearrange the catalytic domain [26, 27]. Another important N-terminal domain is responsible for maintaining the liquid phase of dsDNA and cGAS. By forming droplets of them together, it is facilitated for dimerization and subsequent activation, especially with the help of zinc ions [27, 28]. Notably, such liquid phase is sensitive to the concentration of cGAS and dsDNA. Only when there exists a certain level of dsDNA, cGAS is activated.
Intracellular Cascade reaction of cGAS-STING signal transduction
Activated cGAS produces cGAMP and further binds to STING for activation on the ER membrane. Before STING activation, dimerized STING shapes as V containing two C-terminal domains on the ER surface. Once activated, STING alters into a more closed conformation and transfers to Golgi by ER-Golgi intermediate compartment (ERGIC), which initiates downstream signal cascades [28]. On reaching Golgi, palmitoylation of two cysteine residues is essential for the location of STING on Golgi and allows for both STING oligomerization and TANK Binding Kinase 1 (TBK1) activation [28]. Together with the motif pLxIS in STING, TBK1 is allowed to co-activate IRF3 by phosphorylation. The phosphorylated IRF3 polymerizes and translocates to the nucleus to regulation downstream transcriptional factors of which interferon is the most representative hallmark [29, 30].
cGAS-STING-mediated molecular changes
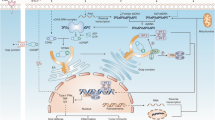
The activated cGAS-STING signal pathway contributes to the increase of IRF3, non-canonical NF-κB and canonical NK-κB, respectively [31]. TBK1 and STING co-phosphate IRF3 (interferon regulatory factor 3). Dimerized IRF3 imports to the nucleus to target corresponding genes. Similarly, like TBK1, mitogen-activated protein kinase kinase kinase 14 (MAP 3 K14/NIK) and IκB kinase (IKK) are other kinases recruited by activated STING. NIK phosphorylates nuclear factor kappa B subunit 2 (NFKB2/p100) combined with RELB. RELB is a proto-oncogene comprising one of the NF-kB subunit. After phosphorylated p100 is degraded by the proteasome to p52, p52 and REBL form into a heterodimer to elicit non-canonical NF-κB signals [32, 33]. Interestingly, the production of canonical NF-κB signals resembles that of non-canonical one. Kinase IKK phosphorylates NFKB inhibitor alpha (NFKBIA/IκB) for the recognition of proteasomal degradation. Thus, heterodimer p65/p50 is separated from IκB/p65/p50 complex to the nucleus, eliciting canonical NF-κB signals [34]. Dimer IRF3, heterodimer p52/RELB and p65/p50 all serve as transcriptional factors. IRF3 regulates the expression of IFNB1 in the nucleus. IFNB1 translation in the cytoplasm results in producing type I IFN, secreting out of cells. Type I IFN stimulates tyrosine kinase-associated receptor, IFNAR1/IFNAR2 heterodimers, which phosphorylates STAT1/STAT2. The process seems like the JAK-STAT signal pathway [35]. The difference is that IRF9 joins phosphorylated dimer STAT/STAT. IRF9/ STAT/STAT as a transcriptional factor transactivates cGAS, developing positive feedback of cGAS-STING signals (shown as Fig. 1) [36].

the cGAS–STING pathway
cGAS-STING signals trigger inflammation and inhibition of cell proliferation and apoptosis. Activation of the cGAS-STING pathway lead to inflammation by rising the concentration of inflammatory cytokines in innate immune responses. We would witness fibrotic changes in chronic diseases [37]. Immunogenicity is enhanced in lymphocytes and other antigen presenting cells (APCs) by high cGAS-STING pathway activity. The mechanism is the activation of NF-κB activation and STING which both activate those immune cells [38]. Sensitive T lymphocytes’ proliferative characteristics are hindered in activated STING cells due to active NF-κB signals [39]. During adaptive immune responses, cGAS-STING signals induce apoptosis for active STING. The phenomenon is often observed in cancerous T cells related to IRF3 and other proapoptotic genes, like p53 [40, 41].
Activated cGAS-STING pathway and cancer biotherapy
Mechanisms of IFNs as immune sensor initiating adaptive immune response
Activated cGAS-STING pathway renders the massive production of type I IFNs. As a ligand, IFN binds to two subunits of the IFN receptor. Human type I IFN is a family of multiple cytokines, containing 13 IFNα subtypes, IFNβ, IFNγ, IFNκ and IFNε [42]. Accumulated evidence shows that T cell infiltration is an indication of the production of type I IFNs. Patients with advanced melanoma, who received oncolytic virotherapy, have increased CD8 + T cells and elevated IFN-γ production [21]. Consistently, clinical data indicate that high levels of extravasating T cells induced by IFN-γ are related to improved survival in patients with colorectal cancer. Those patients with a sound immune system could benefit from biotherapy [43]. On the contrary, tumors are able to abolish IFN formation and reduce T cell recruitment with a lectin secreted by tumors [44]. Therefore, understanding underlying mechanisms among innate immune responses, adaptive immune responses and the cGAS-STING pathway is crucial. Type I IFNs attract cytotoxic T cells to tumor bed [45] and trigger type I T helper cell (Th1) responses [22]. Moreover, type I IFNs make dendritic cells (DCs) mature and present-tumor specific antigens to CD4+ and protective CD8+ T cells [46]. T cell-inflamed tumors contain type I IFN signature. CD8+ T cells show efficient innate priming in triple-negative breast cancer [47]. Overall, these results indicate that type I IFN production is associated with T cell responses against tumor antigens. Mechanically, deficiency in the Ca2+ sensor stromal interaction molecule 1 (STIM1) activates STING spontaneously with subsequently increased type I interferons in immunodeficient mice [48]. In vitro, the expression of Ifnb1, Il6 and interferon-stimulated genes (ISGs) were apparently increased in Stim1−/− murine embryonic fibroblasts (MEFs) compared with those in wild-type (WT) cells. Like murine cells, STIM1-deficient patients exhibited increased type I IFN, pro-inflammatory cytokines and upregulated ISGs. Type I IFN promotes DCs antigen cross-priming in certain mechanisms. It reduces the rate of endosomal-lysosomal acidification to maintain cell-associated antigens in RAB5+ and RAB11+ compartments [30]. Furthermore, IFNα promotes antigen presentation ability of DCs by re-locating MHCI molecules to antigen storage compartments in DCs [29]. In addition, Type I IFN upregulates molecules on the DC surface, such as MHCI, MHCII, CD40, CD80, CD86 [31, 32]. These cluster of differentiation molecules are responsible for providing membrane-associated co-stimulatory signals. Collectively, the results suggest that the innate immune system recognizes cancer cells, leading to the activation of certain signaling pathways and the production of type I IFN, which is crucial for antigen cross-priming.
cGAS-STING signaling pathway initiates type I IFN production and adaptive immunity
Given that IFNs are necessary for the anti-tumor effect, the question that the binding of receptors and ligands causes IFNs production and immune cells’ anti-tumor effect should be understood. Cytosolic DNA activates cGAS further synthesizing cyclic dinucleotides which activates STING. STING gathers around the nucleus and fosters the activation of TBK1. TBK1 phosphorylates IRF3 which transfers signals to downstream type I IFNs gene transcription. Moreover, mutation of STING has been identified in human beings and mice. A heterozygous gain-of-function mutation of STING causes familial chilblain lupus, indicating increased type I IFNs disorders [49]. Patients with STING N154S mutation have STING-associated vasculopathy with onset in infancy (SAVI) whose pathological and pathophysiology changes are pulmonary fibrosis and autoinflammatory disorders. Similarly, STING N153S knock-in mice are vulnerable to develop pulmonary fibrosis after virus infection due to increased type I IFN production [50]. Mechanistic studies found that a low dose of STING agonist ADU-S100 (S100) compatible with checkpoint inhibitors (CPIs) activated tumor-specific CD8 + T cells and enhanced more intensive anti-tumor immunity. TNFα is dispensable for the management of tumor regression [51]. Immunologically, the production of type I IFNs is engaged in the priming of T cells against tumor-associated antigens (TAAs), intratumoral accumulation of dendritic cells and cross-priming of tumor antigen-specific T cells [52,53,54]. Therefore, the cGAS-STING signaling pathway is an important innate defensive sensor that monitors the presence of tumors to mature DCs and promotes T cell priming against TAAs.
The function of the cGAS-STING signaling pathway is identified in several mouse tumor models. In Brca-deficient ovarian cancer models treated with olaparib, the number of intratumoral CD4+ and CD8+ T cells increased, which was accompanied by increased IFNγ and TNFα secreted by themselves. DCs capable of presenting antigens were found to be recruited in the tumor environment (TME). The phenomenon occurred due to the activation of the cGAS-STING pathway and the detection of tumor-derived DNA [55]. Shannon et al. found that mice with ovarian cancer treated with cisplatin had high levels of type I IFN production and inflammation, representing the stimulation of the cGAS-STING pathway. The activated pathway upregulated immunity-recognitive markers. Besides, other non-ovarian solid tumor also showed therapeutic efficacy after intra-tumoral cGAMP administration. Unexpectedly, the cGAS-STING signaling pathway and inflammation have pro-tumor functions [56]. In this study, intra-tumoral injection with STING agonists appeared anti-tumor effect exhibited by T cell responses and regressive tumors in multiple mice tumor models. Meanwhile, experiments invitro suggested that STING agonists administration generated IFNβ production by APCs through the cGAS-STING signaling pathway. STING agonists also contributed to other cytokines production and DCs maturation. Given that 5, 6-dimethylxanthenone-4-acetic acid (DMXAA) capable of disrupting blood vessels induced TNFα by stromal cells in vivo and that its therapeutic efficacy attenuated in TNFR−/− mice, the mechanism of STING agonists therapeutic effect may result from TNFα-mediated angiolysis [57]. In syngeneic ovarian cancer (ID8) and colon cancer (CT26) mice models, poly (ADP-ribose) polymerase inhibitors (PARPi) promoted the levels of phosphorylated Irf3 and STING, which implies active cGAS-STING signaling pathway in vivo. In these PARPi-treated tumor mice models, increased percentages of CD8+ T cells and PD-L1+ cells were observed. These results suggested that PARPi treatment activates the cGAS-STING signaling pathway and rises the levels of type I IFNs and tumor infiltrating lymphocytes (TILs) to trigger an immunogenic response [58].
STING-cGAS signaling pathway applied to Cancer biotherapy
Now that STING-cGAS signaling pathway plays an important role in sensing tumors in innate immunity, two aspects are deserved paying attention to before we develop effective and safe drugs. First, whether tumors could afford STING-cGAS pathway activation by generating T-cell-rich TME should be taken into consideration. After STING adjuvant administration, TME with T cells is related to the activation of the STING-cGAS pathway, indicating that cancer patients react to such biotherapy and show improved prognostic conditions. The failure of generating activated cGAS-STING signals implies a dysfunctional reaction. Therefore, key mlecules in the pathway may serve as predictive biomarkers to direct cancer therapy. Specifically, BATF3-lineage DCs play a central role in anti-tumor immunity [59]. Before pIRF3 translocating to the nucleus, phosphorylation of TBK1 and IRF3 is essential to transfer signals. So BATF3-lineage DCs and pIRF3/pIRF3 are indicators of allowing researches to proceed. Second, therapeutic strategies should conduct clinical trials to prove their bio-therapeutic potentials. Taken one of STING adjuvants for example, 5, 6-dimethylxanthenone-4-acetic acid (DMXAA) is a flavonoid compound used as vascular disrupting agents [60]. Its immunomodulatory function has been proven to have anti-tumor activity in mice [61]. Clinically, DMXAA’s pharmacokinetics showed well-defined efficacy in cancer patients rolled in phase I clinical trial [62]. When DMXAA was combined with carboplatin to conduct a phase II study in untreated patients with advanced non-small cell lung cancer (NSCLC), the therapeutic efficacy of DMXAA was reflected in several parameters, such as tumor response rate, median survival and median time to tumor progression [63, 64]. However, in a randomized phase III placebo-controlled trial, after received DMXAA and platinum-based therapy, patients with advanced NSCLC showed no difference in overall survival (OS), progression-free survival (PFS) and adverse events [65]. Surprisingly, DMXAA only augments murine cGAS-STING signals, instead of that signals in humans [65], which may account for the dysfunction of treating cancer patients in clinical trials. Therefore, DMXAA has exerted concerns about species specificity when people are developing new drugs. Two years later, a newly-designed C7-functionalized DMXAA derivative was synthesized and further evaluated. It shows an affinity to human STING [66]. Together, developing new STING agonists requires human STING to be interacted and activated before clinical trials.
Apart from the stimulus of cGAS-STING agonist, locally increased level of type I IFNs in TME is another challenge to enhance innate immunity. There are two methods: the safety delivery system and targeted radiation. First, developing a rapid and effective delivery system is imperative to increase drug concentrations in the peripheral and central tumor sites. Tumor-targeted mono-antibody (mAb) conjugated with tumor-inhibitory IFNα can induce cancer cell apoptosis. The key technique in the conjugate is Dock-and-Lock method (DNL) to produce 20-2b. 20-2b is an immunocytokine comprising four cytokine groups. Every cytokine group bonds to one anti-CD20 mAb, suggesting that the conjugate has supreme efficacy [67, 68]. IFNAR-deficient tumors are unable to be sensitive to chemotherapy if type I IFN is artificially recharged [69]. Hence, secreting type I IFN in the TME is an essential process for immune priming. Another important point is whether the significant toxicity of delivery systems would produce, suggesting that we need to further observe the effective half-life and dose of drugs in the TME. Nanoparticles (NPs) of chitosan/poly γ-glutamic acid impair the invasion of colorectal cancer cells due to their immunostimulatory characteristics [70]. Castro et al. used NPs carrying IFN-γ to evaluate the therapeutic effect. These IFN-γ-NPs up-regulated costimulatory molecules on cell surfaces and promote to secrete pro-inflammatory cytokines. The mechanism behind T cell proliferation and inhibitory effect against colorectal cancer cells is upregulated CD40 and CD86 molecules [71]. Stephan et al. tested that injecting tumor-reactive T cells do not have a therapeutic effect on pancreatic ductal adenocarcinoma. Subsequently, they used an implantable biopolymer carrier that loaded with CAR T cells and STING agonists to inhibit solid tumors. Consequently, the complex causes the activation of antigen presenting cells, initiation of lymphocyte responses, shrinking of tumors and elimination of metastases [72]. However, the study of these combined agents needs safety evaluation before clinical development.
Targeted radiation is a second method to activate innate immune responses. Irradiated tumors suffer from DNA damage, modulation of signal transduction and changes of TME, exerting anti-tumor responses. Current studies demonstrated that type I IFN-dependent anti-tumor effect after radiation is mediated by the cGAS-STING pathway, rather than the TLR pathway. Administration of cGAMP can reinforce the intensity of the cGAS-STING pathway, consolidating adaptive anti-tumor ability [73]. Hence, radiation is a key initiator for immune priming in the cGAS-STING pathway. The possible mechanism is that radiation-mediated cancer cell death causes tumor-derived DNA to be released. DCs in the TME sense the alteration and subsequent T cell priming coordinates with anti-tumor immune responses. However, the dose of radiotherapy needs to be identified. Exposure to a high dose of ionizing radiation can lead to adverse effects on patients or animals. Low dose may produce inadequate biological responses [74, 75].
Cancer vaccine and cGAS-STING pathway
Cancer vaccines are used to induce immune responses to fight against cancer by stimulating the immune system. The first cancer vaccine is reported in 1983 that Mr. William B. Coley inoculated live bacteria into Soft tissue tumors. He found that some tumors shrunk after inflammation. Researching and developing new cancer vaccines is popular nowadays. In 2006, the FDA approved the first cervical cancer vaccine to lower the risks of HPV16 and HPV18 infection and to prevent cervical cancer. To date, most of the cancer vaccine is under clinic trials. Those trials involve cancer patients who are not bearable during chemotherapy, radiotherapy and operation. Thanks to different antigen constituents, cancer vaccines include cell-based vaccine, virus-based vaccine, DNA-based vaccine, protein- and peptide-based vaccine, anti-idiotype vaccine and carbohydrate-based vaccine. Human tumor antigens have low immunogenicity so that adjuvant is added to support cancer vaccine. Then, cytotoxic T cells are activated and humoral immunity is induced. Current researchers have found correlations between the cGAS-STING pathway and those cancer vaccines. In the part, we would introduce the cGAS-STING pathway agonists.
DMXAA
DMXAA is a flavonoid compound used as tumor vascular disrupting agents (Tumor-VDAs), which is also named as ASA404 and Vadimezan. Tumor-VDAs have two classes of the family: tubulin-binding class and flavonoid class. The flavonoid class is characterized as the induction of inflammatory responses by the innate immune system in the TME. DMXAA is productive to combine with radiation, hyperthermia and chemotherapy reagents. A good illustration of such a combination is DMXAA with carboplatin and paclitaxel in untreated advanced NSCLC. Co-administration was well-tolerated. There is no cardiac adverse events or ophthalmic abnormalities [63]. DMXAA is a STING agonist. A surprising event is that human STING cannot be induced by DMXAA, compared to the fact that mice STING potentiates innate immune responses by DMXAA. Scientists are continuously searching for DMXAA function in the human body and seeking solutions in those disappointing phase III trials [65]. Mechanistically, DMXAA stimulatory function of NF-κB is also detected in monocytes [76], endothelial cells [77] and tumor cells [78]. Increased NF-κB signals give rise to the production of inflammatory cytokines, affecting various immune cells in the TME. The possible mechanism is that DMXAA not only promotes phosphorylated IRF dimers to translocate to nucleus, but also co-stimulates canonical NF-κB targets and non-canonical NF-κB targets simultaneously.
CDNs
CDNs are another kind of cGAS-STING pathway agonist. In the late 1980s, the discovery of them has been found in bacteria as secondary signals. In the pathway, upon interaction with cytosolic DNA, cGAS would synthesize cyclic di-nucleotides to bind to STING for phosphorylation, thus activating STING. Apart from prokaryotic cells, CDNs are thought to participate in innate immune responses of mammalian cells [79]. CDN is a collection of cyclic dinucleotide family, consisting of cyclic di-GMP (c-di-GMP), cyclic di-AMP (c-di-AMP), cyclic AMP-GMP (cGAMP). Amongst themselves, cyclic AMP-GMP contains 3′3’-cGAMP, 2′3’-cGAMP, 3′5’-cGAMP and 2′5’-cGAMP. 2′5’-cGAMP is found to potentiate the activation of human STING due to 2′-5′ linkage [80]. These CDN agonists have anti-cancer potentials. For example, TriVax is a multiple peptide mixture, consisting of CD8 T cell epitope, polyinosinic-polycytidylic acid adjuvant and costimulatory anti-CD40 antibodies. TriVax alone and the combination with c-di-GMP are injected into mice with subcutaneous B16 melanoma cells respectively. C-di-GMP is identified to strengthen TriVax’s function. Co-administration of c-di-GMP slows down tumor growth more intensively. In vitro, adding c-di-GMP to TriVax favors T cells to recognize the B16 tumor cells [81]. Another important case in point is that the anti-tumor efficacy of c-di-GMP is dose- and frequency-dependent. Mice with metastatic breast cancer administrated low dose c-di-GMP are observed elimination of metastases. Meanwhile, 4 T1 tumor cells receiving high dose c-di-GMP are killed. The underlying mechanism is that low dose c-di-GMP promotes myeloid-derived suppressor cells (MDSC) to produce IL-12 for the improvement of T cell responses. High dose c-di-GMP activates caspase-3 for apoptosis. Based on the fact above, low dose c-di-GMP for several times, accompanied with high dose c-di-GMP administration for once, shows no efficacy difference, compared with co-administration of tumor-associated antigen (Mage-b) and c-di-GMP. The explanation is the cross-presentation of TAA by corresponding T cells [82]. Together, enhancing the cGAS-STING pathway by STING adjuvant c-di-GMP is a promising way for cancer biotherapy.
cGAMP is a common CDN molecule, stimulating the immune system and exerting tumor clearance [83, 84]. Notably, cGAMP enhances the cGAS-STING pathway not only by binding to STING, but also by rendering neighboring cells stimuli via transport vesicles and intercellular gap junctions [85, 86]. Furthermore, cGAS-STING signals have been identified in different kinds of tumors, such as B16-BL melanoma, MC38 colon carcinoma and RMA-S lymphoma [87]. In multiple mice models, like 4 T1 breast cancer, squamous cell carcinomas, CT26 colon cancer and B16F10 melanoma, injecting cGAMP generates accumulated macrophages. During the process, those macrophages secrete TNFα and chemokines recruiting T cells, indicating that cGAMP-induced anti-tumor effect is involved with macrophages [83]. Inhalation of nanoparticles with cGAMP in mice with lung matastases is another treatment, which distributes cGAMP in lungs rapidly and promotes APC to produce type I IFNs. Additional fractionated radiation can further strengthen the anti-tumor ability and prolong mice survival [88]. In this study, intra-tumoral injection of cGAMP potentiates CD8+ T cell responses in melanoma and colon cancer mice models respectively. Based on STING agonists promoting immune responses, co-injection of immune checkpoint inhibitors (anti-CTLA-4 and anti-PD-1) further enhances anti-tumor effect [89, 90]. Together, cGAMP, as cGAS-STING pathway agonist, has versatile applications in anti-tumor immunity.
Other agonists
Except for classic CDNs, STING additionally includes ADU-V19, ADU-S100, diABZI STING agonist-1 (Tautomerism), IACS-8779, IACS-8803, E7766, BMS-986301, GSK3745417, IMSA101, MK-1454 and SB 11285. ADU-V19 (RR–S2 cGAMP) is a newly-modified CDN. The novelty is human STING-targeted. As a derivative of STING ligand, ADU-V19 is free from phosphodiesterase by chemical modification and can target human STING. ADU-V19 share a similar function with those common agonists: producing type I IFN, maturating DCs and activating T cells [91]. ADU-S100 (ML RRS2 CDA or MIW815) is another STING agonist, which is similar to ADU-V19. ADU-S100 alone and co-injection of CPIs can both result in anti-tumor effects. The activation of CD8+ T cells and increased survival are observed respectively. What different from ADU-V19 is that ADU-S100 is undergoing phase I or phase II clinical trials (NCT03937141, NCT03172936 and NCT02675439). Tautomerism is a STING against with high selectivity. Subcutaneous injection of 2.5 mg/kg in vivo generates type-I interferon and pro-inflammatory cytokines. Tautomerism’s half-life is 1.4 h and its systemic concentration is higher than the half-maximal effective concentration (EC50) for mouse STING (200 ng/ml). Tautomerism exerts tumor growth inhibition and improves survival. Eight out of ten mice are witnessed tumor-free by the end of the study (day 43) [92]. IACS-8779 (C21H25N9O10P2S2) and IACS-8803 (C20H23FN10O9P2S2) are both STING agonists with robust systemic antitumor efficacy. Mice implanted bilaterally with B16 melanoma cells received intratumoral IACS-8779 injection unilaterally. Scientists observed that superior regression appears on the untreated tumor [93]. The following drugs (E7766, BMS-986301, GSK3745417, IMSA101, MK-1454 and SB 11285) are indicated in the National Cancer Institute (NCI) drug dictionary of National Institutes of Health (NIH) website (https://www.cancer.gov/publications/dictionaries/cancer-drug).
Clinical trials of cGAS-STING pathway agonists
cGAS-STING pathway agonists is a key activator, for the fact that hyperactivity of the pathway is significantly involved in tumor regression, prolonged survival time and enhanced immunity. Therefore, developing drugs targeting the cGAS-STING pathway is worth numerous dedication. Currently, important clinical trials of the cGAS-STING pathway agonists alone and a combination of CPIs are summarized in Table 1 [94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112].
Drug delivery system of STING agonists
Although there exist productive cGAS-STING agonists, they are still suffering their flaws decreasing biotherapeutic efficacy. The hydrophilicity, the vulnerability of enzymatic degradation and the negative charges all refrain from tremendous anti-tumor immunity. Therefore, developing an effective drug delivery system is an essence. The current cGAS-STING agonist delivery system has various forms, ranging from nanocarriers and microparticle to hydrogels. Generally, the smaller delivery vectors more easily transport drugs both locally and distantly. Oppositely, large vectors tend to function in situ after intra-tumoral administration. When choosing delivery vectors, there are two goals needed to be reached. First, at tissue levels, delivery vectors should stick around the cancer lesions to function. Second, at cell levels, drug delivery vectors penetrate the cell membrane arriving at the ER. Meanwhile, they must be protected from endosome decomposition. In the following part, we introduce three kinds of cGAS-STING agonist delivery system which have been widely investigated.
Nanocarriers
Although the activation of the cGAS-STING pathway fights against tumors, delivering agonists targeting tumors still confronts challenges. First, commercially available cGAS-STING agonists are produced soluble, which suffers from clearance easily. In contrast, the phospholipid bilayer of the cell membrane requires fusion substances to be liposoluble. Once soluble drugs are administrated, it is easier to be disseminated throughout the body and to be catabolized for clearance [113]. Even if scientists have tried several methods to overcome it, the results have limitations. A soluble agonist elicits the broadest cytokine response. Cytokine-related toxicity may lead to uncontrollable inflammation and mortal injury [114]. Another limitation is the non-achievement of the therapeutic effect of IT injection. Because non-injected tumor sites are also demonstrated to be regressed due to the activation of distant tumor-specific T cells [57]. Although intramuscular injection of cGAMP inhibits melanoma growth in mice [115], intramuscular injection is not a regular way of delivering drugs.
Next, CDNs are vulnerable to phosphodiesterases (PDEs) of which ecto-nucleotide pyrophosphatase I (ENPP1) has dominant cGAMP-hydrolyzing activity. Li [116] et al. found that ENPP1 locates on the basal lateral surface of the hepatocyte membrane and in the rough ER, instead of in the cytosol. As a Ca2+ store, ER provides an essence of Ca2+ for exerting hydrolytic action. Based on the evidence above, it is suggested that cytosolic CDNs should be transported across the ER membrane in a certain mechanism. Subsequently, scientists synthesized bisphosphothioate analog of 2′3′-cGAMP (2′3′-cGsAsMP) which is hydrolysis-resistant, only to find its increased biostability with higher activity after chemical modification. Apart from PDEs, CD39 and CD73 also contribute to the degradation of ATP. They catalyze ATP hydrolyzed by ENPP1 into AMP, which deepens the immunosuppressive microenvironment [117, 118]. In regards to cancer, ENPP1 overexpression causes different cancer phenotypes. For example, ENPP1-overexpressing breast cancer is involved in the tamoxifen resistance [119]. To address the vulnerability of CDNs, some nucleotide-based ENPP1 inhibitors and non-nucleotide-based ENPP1 inhibitors are developed. However, the high acidity of nucleotide-based ENPP1 inhibitors dampens its oral bioavailability. Off-target biological effects of those drugs would be magnified, because their structure resembles that of natural substrates [120]. These drawbacks of newly-developed ENPP1 inhibitors shed light on CDNs delivery systems using nanocarriers to improve drug efficacy.
Third, the negative charges of CDNs refrain CDNs from permeating through negatively charged cell membrane surface and hydrophobic membrane interior [121]. Since CDNs have more than one anionic phosphate group, such a characteristic limits STING agonists’ entry into the cytoplasm [122]. Cationic liposome with polyethylene glycol (PEG) was explored, because PEGylation enhances nanocarrier’s stability and persistence both in vivo and in vitro [122]. Hence, developing a nanocarrier-based drug delivery system is needed to allow CDNs as potential biotherapeutics penetrating through the cellular surface and arrive at the cytosolic region.
Liposomes is the first nano-biotherapeutics approved by FDA [123]. Liposomes-based delivery encapsulating STING agonists can activate immunologically active cells by cell localization. The function depends on the positively charged spherical vesicle structure exclusively for delivering nucleic acid. The injection of liposomal cGAMP in melanoma tumor-bearing mice are conducted. cGAMP alone cannot be transported into the cytosol due to the presence of two negative charges both on cGAMP surface and on the cell membrane surface. Liposomal cGAMPs are taken up, facilitating the release of cGAMP into the cytosol. Then, the cGAS-STING pathway is activated in the APC (shown as Fig. 2) [122]. On the one hand, liposomes function as vaccine adjuvants. For instance, the study investigated the amplified therapeutic effect of liposomal nanoparticles (NPs) encapsulating cGAMP, compared to that of soluble cGAMP alone. This study demonstrated that cGAMP-delivering NPs reinforce innate immune activation and anti-tumor effect. Particularly, in PD-L1-insensitive models of triple-negative breast cancer (TNBC), cGAMP-NPs induce a variety of inflammatory to fight against cancer, such as proinflammatory cytokines, nitric oxygen, type I IFN and so on. They are more or less involved in promoting M1-like polarization, MHC and costimulatory molecules expression, proliferative inhibition, apoptosis, migration regulation and antigen expression on cell surfaces [113]. Another case in point is that increasing dose of soluble CDNs causes systemic inflammatory toxicity. Hanson et al. found that the administration of CDNs reflects poor lymphatic uptake, because CDNs are eliminated out of tissues by the blood. Thus, scientists used PEGylated liposomal carriers to take place of soluble CDNs, only to find the activation of APCs and active T cell responses [124]. Jointly, a liposomal delivery system containing STING agonists can serve as vaccine adjuvants to enormously improve the anti-tumor effect and immune responses. On the other hand, nanocarriers can overcome the immunosuppressive TME. It is acknowledged that the inhalation of nanocarriers can fight against lung metastases by enhanced immunity. Hence, Liu et al. used liposome encapsulating cGAMP in mice models with lung metastases, indicating pro-inflammatory responses in those metastatic sites. Furthermore, this scientist made a comparison of such a delivery system accompanied by fractionated radiation and the delivery system alone. The results show the immunosuppressive TME exists both in tumors with or without radiation. Adding liposomes loaded cGAMP can control lung metastatic sites and prolong survival time of mice [88]. Therefore, in the immunosuppressive TME, liposomes with CDNs are effective to overcome the disadvantage of the immunosuppressive TME to generate anti-tumor immunity.

liposomal cGAMP structure and therapeutic strategy. a) DOTAP: 1,2-dioleoyl-3-trimethylammonium-propane, DSPE-PEG (2000): 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] polyethylene glycol; b) The injection of liposomal cGAMP in melanoma tumor-bearing mice are conducted. cGAMP alone cannot be transported into the cytosol. Liposomal cGAMPs are taken up, facilitating the release of cGAMP into the cytosol. Then, the cGAS-STING pathway is activated in the APC. Reprinted from [122], copyright (2017) Wiley
Polymeric nanocarriers are another promising nanocarriers to transport STING agonists for cancer biotherapy. They are simple and soft materials for drugs to be built into, because of convenient synthesis and surface structural modification. The feature determinates polymeric nanocarriers’ advantages: improved drug loading efficacy, good biodistribution and biodegradability, controlled pharmacokinetics and safe evaluation [125]. A supreme example is called polymersome. Polymersomes contain two important structures: an aqueous core and a vesicle membrane comprising amphiphilic di-block copolymer chains. The aqueous core is for loading hydrophilic drugs effectively. The vesicle membrane is pH-sensitive and membrane-destabilizing, which mediates the release of intracellular contents and endosomal escape of cGAMP. Polymersomes increase cGAMP activity in monocyte, macrophage and melanoma cell lines. Then, a T-cell- inflamed TME is elicited: increased tumor-infiltrating neutrophils (CD11b+Ly6c+Ly6g+), the polarization from M2 macrophages to M1 macrophages (decreased CD206), overexpression of CD86 in tumor-drainaged lymph nodes, increased CD8+ and CD4+ T cells and increased CD8+/CD4+ T-cell ratio [126]. Another good example is a synthetic polymeric nanoparticle (NP), PC7A NP, developed by Gao et al. The PC7A NP is ultra-pH-sensitive, for the existence of tertiary amines with linear or cyclic side chains. PC7A NPs induce robust antigen-specific CTL, Th1 and Th2 responses. When taking a further look at the underlying mechanisms, Gao et al. identified that promoting antigen delivery and APC cross-presenting both by PC7A NPs jointly stimulate CD8+ T cell responses. In IFNα/βR−/− mice, it is validated that PC7A NPs enhance immune responses by cGAS-STING pathway after APCs are activated in draining lymph nodes. Based on the above information about PC7A NPs, co-administration of a checkpoint inhibitor (anti-PD-1) in multiple tumor models (melanoma, colon cancer and human papilloma virus E6/E7) generates inhibitory effects of tumor growth and prolonged survival time [127].
Microparticles
The micromaterial-based STING agonist delivery system has been developed since 2015. In such a system, the microparticle (MP) is the most widely investigated in the cGAS-STING pathway. For example, microparticles generated from apoptotic cancer cells have been known as a delivery vesicle, carrying chemotherapeutic drugs to inhibit cancer cell growth without any side effect [128]. It is called tumor cell-derived microparticles (T-MP). T-MPs present DNA fragments from cancer cells for APC, stimulating the production of type I IFNs by the activation of the cGAS-STING pathway. Type I IFNs, in turn, make DCs mature by up-regulating CD80, CD86 and MHCII. DCs present tumor antigens to T cells, eliciting anti-tumor effects (shown as Fig. 3) [129, 130]. Besides, acid-sensitive acetylated dextran (Ace-DEX) polymeric MPs are another kind of MP delivery system. Compared to soluble cGAMP, Ace-DEX MPs encapsulating cGAMP increase 1000 times and 50 times the level of type I IFNs in vitro and in vivo respectively, leading to over 100-fold increase in antibody titers. Amazingly, the complex has no toxicity. By overcoming the localization of soluble cGAMP alone, Ace-DEX MPs encapsulating cGAMP potentiate the immunity and expand the germinal center [131]. In another study, Ace-DEX MPs encapsulate cGAMP, imiquimod, murabutide and poly (I:C). Among them, Ace-DEX MPs containing cGAMP are the most effective agent inhibiting tumor growth. The discovery exclusively indicates that NK cells are required for the anti-tumor effect both in TNBC and melanoma mice models [132].

T-MPs function. Upon UV irradiation, tumor cells release MPs containing tumor antigens and DNA fragments. T-MPs present DNA fragments from cancer cells for APC, stimulating the production of type I IFNs by the activation of the cGAS-STING pathway. Type I IFNs, in turn, make DCs mature by up-regulating CD80, CD86 and MHCII. DCs present tumor antigens to T cells, eliciting anti-tumor effects. Reprinted from [129], copyright Taylor & Francis
Hydrogels
Hydrogels are locally-released anti-tumor drug carriers. The unique function is to bind and to retain water, thus forming viscoelastic gels in the aqueous solution [133]. It is reported that hydrogel scaffolds with cross-linking hyaluronic acid (HA) have biological advantages in releasing immunomodulatory drugs, such as biocompatibility and spatiotemporally-controlled drug release. Hydrogels loaded with 2′3′-cdi-AM (PS)2 (Rp,Rp) (“STING-RR”) injecting to intraoperative tumors have therapeutic benefits, however, it is not observed in unresectable tumors. The final result jointly supports that hydrogels loading immunomodulators affect postsurgical resection microenvironment by eliciting immune cells [134]. Linear polyethyleneimine (LPEI)/hyaluronic acid (HA) hydrogels loading cGAMP generate IFNβ and IL-6 in macrophage cells. Notably, the amount of IFNβ is 2.5 times higher than cationic liposomes. The corresponding IFNβ mRNA is elevated in mice spleens [135]. Matrigel is a thermo-responsive hydrogel comprising laminin and collagen IV. Matrigels have a series of advantages: soluble in water, absorption of soluble agents in water, the formation of a gel-like solid at body temperature and rapid degradation. Based on the feature, in the resection cavity, Matrigel incorporating STING ligands cure local tumors in the resection cavity of head and neck squamous cell carcinoma mice models. It is in the host STING expressing cells, not in cancer cells, that the production of type I IFNs contributes to the local control of residual lesions [136]. STINGel is a third peptide hydrogel for CDN intra-tumoral delivery. Multidomain peptides (MDPs) play a crucial role in creating hydrogels. MDPs generate anti-parallel β-sheets of peptide nanofibers in solution when β-sheets electrostatically crosslink with multivalent ions, thus expanding nanofiber networks. It is MDP that controls and extends CDN release. STINGel releases payloads more slowly than common collagen hydrogel does. The duration of releasing high concentration CDNs is simultaneously prolonged. A good thing is that released CDNs fail to recruit infiltrating cells, because immune-cell infiltration is cytotoxic. Meanwhile, cellular necrosis can be witnessed. In mice with oral cancer, the promotion of necrosis and the lack of cytotoxicity both enable cancer-bearing mice models tolerated. The immunotherapeutic mechanisms are the activation of cGAS-STING pathway and the release of danger-associated molecular patterns (DAMPs) from nuclear debris. DAMPs can recruit massive immune cells to the lesion [137].
Conclusions
The cGAS-STING signaling pathway is characterized as the representative production of type I IFNs to elicit anti-tumor immunity. The activation of the cGAS-STING pathway transfer signals from the binding of ligand and receptor to the transcriptional level in the nucleus by phosphorylating second messengers, which renders STING a potential target for cancer biotherapy. The reason for it is that an effective STING- agonist delivery system could substantially enhance anti-tumor immunity. Besides, STING agonists are undergoing basic scientific researches and clinical trials. They have shown promising biological activity. Hence, to optimize the bio-therapeutic efficacy, delivering cGAS-STING pathway agonists to targeted tissues or cells is a method. Pathway agonists penetrate the cell membrane to arrive at Golgi for interaction. However, agonists commercially available have three disadvantages: easily to be cleared, easily to be enzymolyzed by PDEs and the similar charge property of agonists and organelle surfaces. Considering the disadvantages, developing a delivery system to overcome the difficulties and to target cells is imperative. Although studies about delivering agonists to tumor tissues and lymph nodes are investigated, agonists delivery systems are continuously developed for the improvement of cancer biotherapy. In the review, we summarize the cGAS-STING pathway agonists, including DMXAA that experienced failure in phase III clinical trials, CDNs that are widely accepted as common and effective cGAS-STING signal enhancers and other agonists that are investigated at an early stage. Also, we summarize the current drug delivery system, including nanocarriers, microparticles and hydrogels.
It is worth noting that the combination treatment of cGAS-STING agonists and CPIs can synergistically improve cancer biotherapeutic efficacy and decrease the risk of side effects. In mice, the agonist ADU-S100 co-administrated with CPIs is productive to magnify anti-tumor immunity. Moreover, most of the cGAS-STING agonist clinical trials are conducted with CPIs (pembrolizumab, ipilimumab and nivolumab) for cancer biotherapy. However, the underlying mechanism remains to be clarified. Now that cGAS-STING signal cascade transduction changes with the phosphorylation of multiple intracellular molecules after DNA sensing, mediating a series of biological process (inflammation, cell proliferation and apoptosis), clarifying mechanisms and optimizing combinational biotherapeutic strategies are both essential processes to solve drug safety problems for the maximization of drug efficacy. Furthermore, developing a more effective delivery system and designing rational cGAS-STING agonists are coordinatively important for improving cancer therapeutic effect.
In sum, the cGAS-STING pathway shows invaluable potentials in cancer biotherapy. Enhancing the immune ability to fight against cancer by delivering corresponding agonists with appropriate delivery systems can tremendously facilitate the combinational cancer biotherapeutic efficacy.
Availability of data and materials
Not applicable.
Abbreviations
- TREX1:
-
Three prime repair exonuclease 1
- RNase H2:
-
Ribonuclease H2
- cGAS:
-
cyclic guanosine monophosphate-adenosine monophosphate synthase
- dsDNA:
-
double-stranded DNA
- ATP:
-
Adenosine 5´-triphosphate
- GTP:
-
Guanosine 5´-triphosphate
- cGAMP:
-
cyclic GMP-AMP
- CDNs:
-
Cyclic dinucleotides
- ER:
-
Endoplasmic reticulum
- STING:
-
Stimulator of interferon genes
- IRF3:
-
Interferon regulatory factor 3
- IFN:
-
Interferon
- HSV-1:
-
Herpes simplex virus-1
- VSV:
-
Vesicular stomatitis virus
- PCV2:
-
Porcine circovirus type 2
- HIV-1:
-
Immunodeficiency virus type 1
- NTase:
-
Nucleotidyl transferase
- ERGIC:
-
ER-Golgi intermediate compartment
- TBK1:
-
TANK Binding Kinase 1
- MAP 3K14/NIK:
-
Mitogen-activated protein kinase kinase kinase 14
- IKK:
-
IκB kinase
- NFKB2/p100:
-
Nuclear factor kappa B subunit 2
- NFKBIA/IκB:
-
NFKB inhibitor alpha
- APCs:
-
Antigen presenting cells
- Th1:
-
T helper cell
- DCs:
-
Dendritic cells
- STIM1:
-
Stromal interaction molecule 1
- ISGs:
-
Interferon-stimulated genes
- MEFs:
-
Murine embryonic fibroblasts
- WT:
-
Wild-type
- SAVI:
-
STING-associated vasculopathy with onset in infancy
- S100:
-
ADU-S100
- CPIs:
-
Checkpoint inhibitors
- TAAs:
-
Tumor-associated antigens
- TME:
-
Tumor environment
- DMXAA:
-
5, 6-dimethylxanthenone-4-acetic acid
- PARPi:
-
Poly (ADP-ribose) polymerase inhibitors
- TILs:
-
Tumor infiltrating lymphocytes
- NSCLC:
-
Non-small cell lung cancer
- OS:
-
Overall survival
- PFS:
-
Progession-free survival
- mAb:
-
mono-antibody
- DNL:
-
Dock-and-Lock method
- NPs:
-
Nanoparticles
- Tumor-VDAs:
-
Tumor vascular disrupting agents
- c-di-GMP:
-
cyclic di-GMP
- c-di-AMP:
-
cyclic di-AMP
- MDSC:
-
Myeloid-derived suppressor cells
- NCI:
-
National Cancer Institute
- NIH:
-
National Institutes of Health
- PDEs:
-
Phosphodiesterases
- ENPP1:
-
Ecto-nucleotide pyrophosphatase I
- PEG:
-
Polyethylene glycol
- TNBC:
-
Triple-negative breast cancer
- MP:
-
Microparticle
- T-MP:
-
Tumor cell-derived microparticles
- Ace-DEX:
-
Acetalated dextran
- LPEI:
-
Linear polyethyleneimine
- HA:
-
Hyaluronic acid
- MDPs:
-
Multidomain peptides
- DAMPs:
-
Danger-associated molecular patterns
References
Hawkins LK, Lemoine NR, Kirn D. Oncolytic biotherapy: a novel therapeutic plafform. Lancet Oncol. 2002;3:17–26.
Kallies A, Zehn D. Precursor exhausted T cells: key to successful immunotherapy? 2019;20:128–36.
Voelker R. Pursuing an effective ovarian Cancer vaccine. JAMA. 2018;320:858–60.
Goldberg MS. Improving cancer immunotherapy through nanotechnology. Nat Rev Cancer. 2019;19:587–602.
High KA, Roncarolo MG. Gene therapy. N Engl J Med. 2019;381:455–64.
Necitumumab. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012.
Weber RW, O'Day S, Rose M, Deck R, Ames P, Good J, Meyer J, Allen R, Trautvetter S, Timmerman M, et al. Low-dose outpatient chemobiotherapy with temozolomide, granulocyte-macrophage colony stimulating factor, interferon-alpha2b, and recombinant interleukin-2 for the treatment of metastatic melanoma. J Clin Oncol. 2005;23:8992–9000.
Benvenuto M, Focaccetti C, Izzi V, Masuelli L, Modesti A, Bei R. Tumor antigens heterogeneity and immune response-targeting neoantigens in breast cancer. Semin Cancer Biol. 2019;S1044–579X(19)30299–8.
Goetze TO. Immunotherapy: a new era in small-cell lung cancer. Lancet. 2019;394:1884–5.
Keenan BP, Fong L, Kelley RK. Immunotherapy in hepatocellular carcinoma: the complex interface between inflammation, fibrosis, and the immune response. J Immunother Cancer. 2019;7:267.
Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, Blaisdell S, Basham B, Dai J, Grein J, et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell. 2011;20:781–96.
Lambrechts D, Wauters E, Boeckx B, Aibar S. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24:1277–89.
DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405–9.
Coquel F, Neumayer C, Lin YL, Pasero P. SAMHD1 and the innate immune response to cytosolic DNA during DNA replication. Curr Opin Immunol. 2019;56:24–30.
O'Driscoll M. TREX1 DNA exonuclease deficiency, accumulation of single stranded DNA and complex human genetic disorders. DNA Repair (Amst). 2008;7:997–1003.
Genoveso MJ, Hisaoka M, Komatsu T, Wodrich H. Formation of adenovirus DNA replication compartments and viral DNA accumulation sites by host chromatin regulatory proteins including NPM1. FEBS J. 2020;287:205–17.
Takahashi A, Loo TM, Okada R, Kamachi F, Watanabe Y, Wakita M, Watanabe S. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun. 2018;9:1249.
Song X, Ma F, Herrup K. Accumulation of cytoplasmic DNA due to ATM deficiency activates the microglial viral response system with neurotoxic consequences. J Neurosci. 2019;39:6378–94.
Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet. 2019;20:657–74.
Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461–5.
Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell. 2017;170:1109–1119.e1110.
Tough DF. Modulation of T-cell function by type I interferon. Immunol Cell Biol. 2012;90:492–7.
Fan J-B, Miyauchi S, Xu H-Z, Liu D, Kim LJ, Burkart C, Cheng H, Arimoto K-i, Yan M, Zhou Y. Type I interferon regulates a coordinated gene network to enhance cytotoxic T cell–mediated tumor killing. Cancer Discovery. 2020;10:382–93.
Gale M. Interference with virus infection. J Immunol. 2015;195:1909–10.
Hemann EA, Green R, Turnbull JB, Langlois RA, Savan R, Gale M. Interferon-λ modulates dendritic cells to facilitate T cell immunity during infection with influenza a virus. Nat Immunol. 2019;20:1035–45.
Sceneay J, Goreczny GJ, Wilson K, Morrow S, DeCristo MJ, Ubellacker JM, Qin Y, Laszewski T, Stover DG, Barrera V. Interferon signaling is diminished with age and is associated with immune checkpoint blockade efficacy in triple-negative breast cancer. Cancer Discovery. 2019;9:1208–27.
Hutchinson JN, Raj T, Fagerness J, Stahl E, Viloria FT, Gimelbrant A, Seddon J, Daly M, Chess A, Plenge R: Allele-specific methylation occurs at genetic variants associated with complex disease. PloS one 2014;9:e98464.
Srikanth S, Woo JS, Wu B, El-Sherbiny YM, Leung J, Chupradit K, Rice L, Seo GJ, Calmettes G, Ramakrishna C. The Ca 2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol. 2019;20:152–62.
Moffat JM, Cheong WS, Villadangos JA, Mintern JD, Netter HJ. Hepatitis B virus-like particles access major histocompatibility class I and II antigen presentation pathways in primary dendritic cells. Vaccine. 2013;31:2310–6.
Thacker RI, Janssen EM. Cross-presentation of cell-associated antigens by mouse splenic dendritic cell populations. Front Immunol. 2012;3:41.
Todisco E, Gaipa G, Biagi E, Bonamino M, Gramigna R, Introna M, Biondi A. CD40 ligand-stimulated B cell precursor leukemic cells elicit interferon-gamma production by autologous bone marrow T cells in childhood acute lymphoblastic leukemia. Leukemia. 2002;16:2046–54.
Chiriva-Internati M, Liu Y, Weidanz JA, Grizzi F, You H, Zhou W, Bumm K, Barlogie B, Mehta JL, Hermonat PL. Testing recombinant adeno-associated virus-gene loading of dendritic cells for generating potent cytotoxic T lymphocytes against a prototype self-antigen, multiple myeloma HM1.24. Blood. 2003;102:3100–7.
Parvatiyar K, Pindado J, Dev A, Aliyari SR, Zaver SA, Gerami H, Chapon M, Ghaffari AA, Dhingra A, Cheng G. A TRAF3-NIK module differentially regulates DNA vs RNA pathways in innate immune signaling. Nat Commun. 2018;9:2770.
Luo X, Li H, Ma L, Zhou J, Guo X, Woo SL, Pei Y, Knight LR, Deveau M, Chen Y, et al. Expression of STING Is Increased in Liver Tissues From Patients With NAFLD and Promotes Macrophage-Mediated Hepatic Inflammation and Fibrosis in Mice. Gastroenterology. 2018;155:1971–1984.e1974.
Mathur V, Burai R, Vest RT, Bonanno LN, Lehallier B, Zardeneta ME, Mistry KN, Do D, Marsh SE, Abud EM, et al. Activation of the STING-Dependent Type I Interferon Response Reduces Microglial Reactivity and Neuroinflammation. Neuron. 2017;96:1290–1302.e1296.
Xu X, Li M, Wu C, Li D, Jiang Z, Liu C, Cheng B, Mao H, Hu C. The fish-specific protein kinase (PKZ) initiates innate immune responses via IRF3- and ISGF3-like mediated pathways. Front Immunol. 2019;10:582.
Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, Kaufman BA, Park J, Pei L, Baur J, et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab. 2019;30:784–799.e785.
Wang Z, Chen J, Hu J, Zhang H, Xu F, He W, Wang X, Li M, Lu W, Zeng G, et al. cGAS/STING axis mediates a topoisomerase II inhibitor-induced tumor immunogenicity. J Clin Invest. 2019;130:4850–62.
Cerboni S, Jeremiah N, Gentili M. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J Exp Med. 2017;214:1769–85.
Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, Radtke F, Ablasser A. Signalling strength determines proapoptotic functions of STING. Nature Communications. 2017;8:427.
Sze A, Belgnaoui SM, Olagnier D, Lin R, Hiscott J, van Grevenynghe J. Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe. 2013;14:422–34.
Schreiber G, Piehler J. The molecular basis for functional plasticity in type I interferon signaling. Trends Immunol. 2015;36:139–49.
Simpson JA, Al-Attar A, Watson NF, Scholefield JH, Ilyas M, Durrant LG. Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut. 2010;59:926–33.
Gordon-Alonso M, Hirsch T, Wildmann C, van der Bruggen P. Galectin-3 captures interferon-gamma in the tumor matrix reducing chemokine gradient production and T-cell tumor infiltration. Nat Commun. 2017;8:793.
Fan JB, Miyauchi S, Xu HZ, Liu D, Kim LJY, Burkart C, Cheng H, Arimoto KI, Yan M, Zhou Y. Type I interferon regulates a coordinated gene network to enhance cytotoxic T cell-mediated tumor killing; 2020;10:382–93.
Hemann EA, Green R, Turnbull JB. Interferon-lambda modulates dendritic cells to facilitate T cell immunity during infection with influenza a virus. Nat Immunol. 2019;20:1035–45.
Sceneay J, Goreczny GJ, Wilson K, Morrow S, DeCristo MJ, Ubellacker JM, Qin Y, Laszewski T, Stover DG. Interferon signaling is diminished with age and is associated with immune checkpoint blockade efficacy in triple-negative breast Cancer. Cancer Discov. 2019;9:1208–27.
Srikanth S, Woo JS, Wu B, El-Sherbiny YM. The Ca (2+) sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol. 2019;20:152–62.
Konig N, Fiehn C, Wolf C, Schuster M, Cura Costa E, Tungler V, Alvarez HA, Chara O, Engel K, Goldbach-Mansky R, et al. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann Rheum Dis. 2017;76:468–72.
Bennion BG, Ingle H, Ai TL, Miner CA, Platt DJ, Smith AM, Baldridge MT, Miner JJ. A Human Gain-of-Function STING Mutation Causes Immunodeficiency and Gammaherpesvirus-Induced Pulmonary Fibrosis in Mice. J Virol 2019,93:e01806–18.
Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, Hudson TE, Vu UT, Francica BJ, Banda T, et al. Magnitude of Therapeutic STING Activation Determines CD8(+) T Cell-Mediated Anti-tumor Immunity. Cell Rep. 2018;25:3074–3085.e3075.
Laengle J, Stift J, Bilecz A, Wolf B, Beer A, Hegedus B, Stremitzer S, Starlinger P, Tamandl D, Pils D, Bergmann M. DNA damage predicts prognosis and treatment response in colorectal liver metastases superior to immunogenic cell death and T cells. Theranostics. 2018;8:3198–213.
Lorenzi S, Mattei F, Sistigu A, Bracci L, Spadaro F, Sanchez M, Spada M, Belardelli F, Gabriele L, Schiavoni G. Type I IFNs control antigen retention and survival of CD8alpha(+) dendritic cells after uptake of tumor apoptotic cells leading to cross-priming. J Immunol. 2011;186:5142–50.
Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 2014;26:638–52.
Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, Li BB, Xie S, Liu JF, Stover EH, et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018;25:2972–2980.e2975.
Grabosch S, Bulatovic M, Zeng F, Ma T, Zhang L, Ross M, Brozick J, Fang Y, Tseng G, Kim E, et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene. 2019;38:2380–93.
Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11:1018–30.
Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, Yap TA, Mills GB, Peng G. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019;79:311–9.
Corrales L, Matson V, Flood B, Spranger S, Gajewski TF. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017;27:96–108.
Roberts ZJ, Goutagny N, Perera PY, Kato H, Kumar H, Kawai T, Akira S, Savan R, van Echo D, Fitzgerald KA, et al. The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J Exp Med. 2007;204:1559–69.
Baguley BC, Ching LM. Immunomodulatory actions of xanthenone anticancer agents. BioDrugs. 1997;8:119–27.
Jameson MB, Baguley BC, Kestell P, Zhao L, Paxton JW, Thompson PI, Waller S. Pharmacokinetics of 5,6-dimethylxanthenone-4-acetic acid (AS1404), a novel vascular disrupting agent, in phase I clinical trial. Cancer Chemother Pharmacol. 2007;59:681–7.
McKeage MJ, Reck M, Jameson MB, Rosenthal MA, Gibbs D, Mainwaring PN, Freitag L, Sullivan R, Von Pawel J. Phase II study of ASA404 (vadimezan, 5,6-dimethylxanthenone-4-acetic acid/DMXAA) 1800mg/m (2) combined with carboplatin and paclitaxel in previously untreated advanced non-small cell lung cancer. Lung Cancer. 2009;65:192–7.
McKeage MJ, Von Pawel J, Reck M, Jameson MB, Rosenthal MA, Sullivan R, Gibbs D, Mainwaring PN, Serke M, Lafitte JJ, et al. Randomised phase II study of ASA404 combined with carboplatin and paclitaxel in previously untreated advanced non-small cell lung cancer. Br J Cancer. 2008;99:2006–12.
Lara PN Jr, Douillard JY, Nakagawa K, von Pawel J, McKeage MJ, Albert I, Losonczy G, Reck M, Heo DS, Fan X, et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J Clin Oncol. 2011;29:2965–71.
Hwang J, Kang T, Lee J, Choi BS, Han S. Design, synthesis, and biological evaluation of C7-functionalized DMXAA derivatives as potential human-STING agonists. Org Biomol Chem. 2019;17:1869–74.
Rossi EA, Rossi DL, Cardillo TM, Stein R, Goldenberg DM, Chang CH. Preclinical studies on targeted delivery of multiple IFNalpha2b to HLA-DR in diverse hematologic cancers. Blood. 2011;118:1877–84.
Rossi EA, Goldenberg DM, Cardillo TM, Stein R, Chang CH. CD20-targeted tetrameric interferon-alpha, a novel and potent immunocytokine for the therapy of B-cell lymphomas. Blood. 2009;114:3864–71.
Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remedios C, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20:1301–9.
Cardoso AP, Pinto ML, Pinto AT, Oliveira MI, Pinto MT, Goncalves R, Relvas JB, Figueiredo C, Seruca R, Mantovani A, et al. Macrophages stimulate gastric and colorectal cancer invasion through EGFR Y (1086), c-Src, Erk1/2 and Akt phosphorylation and smallGTPase activity. Oncogene. 2014;33:2123–33.
Castro F, Pinto ML. Chitosan/poly (gamma-glutamic acid) nanoparticles incorporating IFN-gamma for immune response modulation in the context of colorectal cancer. Biomater Sci. 2019;7:3386–403.
Smith TT, Moffett HF, Stephan SB, Opel CF, Dumigan AG, Jiang X, Pillarisetty VG, Pillai SPS, Wittrup KD, Stephan MT. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J Clin Invest. 2017;127:2176–91.
Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41:843–52.
Bernard JJ, Gallo RL. Photoimmunology: how ultraviolet radiation affects the immune system. Nat Rev Immunol. 2019;19:688–701.
Fillon M. Lung cancer radiation may increase the risk of major adverse cardiac events. CA Cancer J Clin. 2019;69:435–7.
Philpott M, Baguley BC, Ching L-M. Induction of tumour necrosis factor-α by single and repeated doses of the antitumour agent 5,6-dimethylxanthenone-4-acetic acid. Cancer Chemother Pharmacol. 1995;36:143–8.
Ching LM, Cao Z, Kieda C, Zwain S, Jameson MB, Baguley BC. Induction of endothelial cell apoptosis by the antivascular agent 5,6-dimethylxanthenone-4-acetic acid. Br J Cancer. 2002;86:1937–42.
Woon S, Baguley BC, Palmer BD, Fraser JD, Ching L. Uptake of the Antivascular agent 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) and activation of NF-κB in human tumor cell lines. Oncol Res. 2002;13:95–101.
Danilchanka O, Mekalanos JJ. Cyclic dinucleotides and the innate immune response. Cell. 2013;154:962–70.
Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380–4.
Wang Z, Celis E. STING activator c-di-GMP enhances the anti-tumor effects of peptide vaccines in melanoma-bearing mice. Cancer Immunol Immunother. 2015;64:1057–66.
Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, Zhou J, Hayakawa Y, Karaolis DK, Gravekamp C. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol Res. 2014;2:901–10.
Ohkuri T, Kosaka A, Ishibashi K, Kumai T, Hirata Y, Ohara K, Nagato T, Oikawa K, Aoki N, Harabuchi Y, et al. Intratumoral administration of cGAMP transiently accumulates potent macrophages for anti-tumor immunity at a mouse tumor site. Cancer Immunol Immunother. 2017;66:705–16.
Li T, Cheng H, Yuan H, Xu Q, Shu C, Zhang Y, Xu P, Tan J, Rui Y, Li P, Tan X. Antitumor activity of cGAMP via stimulation of cGAS-cGAMP-STING-IRF3 mediated innate immune response. Sci Rep. 2016;6:19049.
Bridgeman A, Maelfait J, Davenne T, Partridge T, Peng Y, Mayer A, Dong T, Kaever V, Borrow P, Rehwinkel J. Viruses transfer the antiviral second messenger cGAMP between cells. Science. 2015;349:1228–32.
Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, Jacob L, Patwa R, Shah H, Xu K, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533:493–8.
Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity. 2018;49:754–763.e754.
Liu Y, Crowe WN, Wang L, Lu Y, Petty WJ, Habib AA. An inhalable nanoparticulate STING agonist synergizes with radiotherapy to confer long-term control of lung metastases. Nat Commun. 2019;10:5108.
Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A. 2015;112:15408–13.
Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra252.
Kinkead HL, Hopkins A, Lutz E, Wu AA, Yarchoan M, Cruz K, Woolman S, Vithayathil T, Glickman LH, Ndubaku CO, et al. Combining STINGbased neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight. 2018;3:e122857.
Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, Tran JL, Moore P, Lehmann S, Eberl HC, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. 2018;564:439–43.
Ager CR, Zhang H, Wei Z, Jones P, Curran MA, Di Francesco ME. Discovery of IACS-8803 and IACS-8779, potent agonists of stimulator of interferon genes (STING) with robust systemic antitumor efficacy. Bioorg Med Chem Lett. 2019;29:126640.
Glasgow Uo, Institute NC: Dimethylxanthenone Acetic Acid in Treating Patients With Solid Tumors. https://ClinicalTrials.gov/show/NCT00003697 1995.
Research A: Study of AS1404 With Docetaxel in Patients With Hormone Refractory Metastatic Prostate Cancer. https://ClinicalTrials.gov/show/NCT00111618 2005.
Pharmaceuticals N, Novartis: An Open-label, Phase I Trial of Intravenous ASA404 Administered in Combination With Paclitaxel and Carboplatin in Japanese Patients With Non-Small Cell Lung Cancer (NSCLC). https://ClinicalTrials.gov/show/NCT00674102 2008.
Research A: Phase II Study of DMXAA (ASA404) in Combination With Chemotherapy in Patients With Advanced Non-Small Cell Lung Cancer. https://ClinicalTrials.gov/show/NCT00832494; 2004.
Research A: Phase I Safety Study of DMXAA in Refractory Tumors. https://ClinicalTrials.gov/show/NCT00856336; 2003.
UK CR, Auckland CS: Study of DMXAA (Now Known as ASA404) in Solid Tumors. https://ClinicalTrials.gov/show/NCT00863733; 1996.
Research SGfCC: Paclitaxel, Carboplatin, and Dimethylxanthenone Acetic Acid in Treating Patients With Extensive-Stage Small Cell Lung Cancer. https://ClinicalTrials.gov/show/NCT01057342 2010.
Pharmaceuticals N, Novartis: Safety and Tolerability of ASA404 Administered in Combination With Docetaxel in Japanese Patients With Solid Tumors. https://ClinicalTrials.gov/show/NCT01285453; 2009.
Aduro Biotech I, Pharmaceuticals N: Safety and Efficacy of MIW815 (ADU-S100) +/− Ipilimumab in Patients With Advanced/Metastatic Solid Tumors or Lymphomas. https://ClinicalTrials.gov/show/NCT02675439; 2016.
Sharp M, Corp. D: Study of MK-1454 Alone or in Combination With Pembrolizumab (MK-3475) in Participants With Advanced/Metastatic Solid Tumors or Lymphomas (MK-1454-001). https://ClinicalTrials.gov/show/NCT03010176; 2017.
Pharmaceuticals N, Novartis: Study of the Safety and Efficacy of MIW815 With PDR001 to Patients With Advanced/Metastatic Solid Tumors or Lymphomas. https://ClinicalTrials.gov/show/NCT03172936; 2017.
GlaxoSmithKline: Phase 1 First Time in Humans (FTIH), Open Label Study of GSK3745417 Administered to Subjects With Advanced Solid Tumors. https://ClinicalTrials.gov/show/NCT03843359; 2019.
Aduro Biotech I: Efficacy and Safety Trial of ADU-S100 and Pembrolizumab in Head and Neck Cancer. https://ClinicalTrials.gov/show/NCT03937141; 2019.
Squibb B-M: An Investigational Immunotherapy Study of BMS-986301 Alone or in Combination With Nivolumab, and Ipilimumab in Participants With Advanced Solid Cancers. https://ClinicalTrials.gov/show/NCT03956680; 2019.
Inc. IT: Safety and Efficacy Study of IMSA101 in Refractory Malignancies. https://ClinicalTrials.gov/show/NCT04020185; 2019.
Spring Bank Pharmaceuticals I, Medicine PF: Evaluating Safety and Efficacy of SB 11285 Alone and in Combination With Nivolumab in Patients With Advanced Solid Tumors. https://ClinicalTrials.gov/show/NCT04096638; 2019.
Inc. E, Inc. HB: A Study of Stimulator of Interferon Genes (STING) Agonist E7766 in Non-muscle Invasive Bladder Cancer (NMIBC) Including Participants Unresponsive to Bacillus Calmette-Guerin (BCG) Therapy, INPUT-102. https://ClinicalTrials.gov/show/NCT04109092; 2019.
Inc. E, Inc. HB: Study of Intratumorally Administered Stimulator of Interferon Genes (STING) Agonist E7766 in Participants With Advanced Solid Tumors or Lymphomas - INSTAL-101. https://ClinicalTrials.gov/show/NCT04144140; 2020.
Sharp M, Corp. D: Study of Intratumoral (IT) MK-1454 in Combination With Intravenous (IV) Pembrolizumab (MK-3475) Compared to IV Pembrolizumab Alone as the First Line Treatment of Metastatic or Unresectable, Recurrent Head and Neck Squamous Cell Carcinoma (HNSCC) (MK-1454-002). 2020. https://ClinicalTrials.gov/show/NCT04220866.
Cheng N, Watkins-Schulz R, Junkins RD, David CN, Johnson BM, Montgomery SA, Peine KJ, Darr DB, Yuan H, McKinnon KP, et al. A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1-insensitive models of triple-negative breast cancer. JCI Insight. 2018;3:e120638.
Collier MA, Junkins RD, Gallovic MD, Johnson BM, Johnson MM. Acetalated dextran microparticles for Codelivery of STING and TLR7/8 agonists. Mol Pharm. 2018;15:4933–46.
Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, Chen ZJ. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A. 2017;114:1637–42.
Li L, Yin Q, Kuss P, Maliga Z, Millan JL, Wu H, Mitchison TJ. Hydrolysis of 2′3'-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat Chem Biol. 2014;10:1043–8.
Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A, Serra S, Malavasi F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology. 2013;2:e26246.
Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. 2017;276:121–44.
Umar A, Kang H, Timmermans AM, Look MP, Meijer-van Gelder ME, den Bakker MA, Jaitly N, Martens JW, Luider TM, Foekens JA, Pasa-Tolic L. Identification of a putative protein profile associated with tamoxifen therapy resistance in breast cancer. Mol Cell Proteomics. 2009;8:1278–94.
Onyedibe KI, Wang M, Sintim HO. ENPP1, an Old Enzyme with New Functions, and Small Molecule Inhibitors-A STING in the Tale of ENPP1. Molecules. 201924:4192.
Miyabe H, Hyodo M, Nakamura T, Sato Y, Hayakawa Y, Harashima H. A new adjuvant delivery system 'cyclic di-GMP/YSK05 liposome' for cancer immunotherapy. J Control Release. 2014;184:20–7.
Koshy ST, Cheung AS, Gu L, Graveline AR, Mooney DJ. Liposomal Delivery Enhances Immune Activation by STING Agonists for Cancer Immunotherapy. Adv Biosyst. 2017;1(1-2):1600013.
Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20–37.
Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH, Melo MB, Mueller S, Irvine DJ. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. J Clin Invest. 2015;125:2532–46.
Senapati S, Mahanta AK, Kumar S, Maiti P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct Target Ther. 2018;3:7.
Shae D, Becker KW. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat Nanotechnol. 2019;14:269–78.
Luo M, Wang H, Wang Z, Cai H, Lu Z, Li Y, Du M, Huang G, Wang C. A STING-activating nanovaccine for cancer immunotherapy. Nat Nanotechnol. 2017;12:648–54.
Tang K, Zhang Y, Zhang H, Xu P, Liu J, Ma J, Lv M, Li D, Katirai F, Shen GX, et al. Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat Commun. 2012;3:1282.
Zhang H, Huang B. Tumor cell-derived microparticles: a new form of cancer vaccine. Oncoimmunology. 2015;4:e1017704.
Zhang H, Tang K, Zhang Y, Ma R, Ma J, Li Y, Luo S, Liang X, Ji T, Gu Z, et al. Cell-free tumor microparticle vaccines stimulate dendritic cells via cGAS/STING signaling. Cancer Immunol Res. 2015;3:196–205.
Junkins RD, Gallovic MD, Johnson BM, Collier MA, Watkins-Schulz R, Cheng N, David CN, McGee CE, Sempowski GD, Shterev I, et al. A robust microparticle platform for a STING-targeted adjuvant that enhances both humoral and cellular immunity during vaccination. J Control Release. 2018;270:1–13.
Watkins-Schulz R, Tiet P, Gallovic MD, Junkins RD, Batty C, Bachelder EM, Ainslie KM, Ting JPY. A microparticle platform for STING-targeted immunotherapy enhances natural killer cell- and CD8(+) T cell-mediated anti-tumor immunity. Biomaterials. 2019;205:94–105.
Zhu J, Tang X, Jia Y, Ho CT, Huang Q. Applications and delivery mechanisms of hyaluronic acid used for topical/transdermal delivery - a review. Int J Pharm. 2020;578:119127.
Park CG, Hartl CA, Schmid D. Extended release of perioperative immunotherapy prevents tumor recurrence and eliminates metastases. 2018;10:eaar1916.
Lee E, Jang HE, Kang YY, Kim J, Ahn JH, Mok H. Submicron-sized hydrogels incorporating cyclic dinucleotides for selective delivery and elevated cytokine release in macrophages. Acta Biomater. 2016;29:271–81.
Baird JR, Bell RB, Troesch V, Friedman D, Bambina S, Kramer G, Blair TC, Medler T, Wu Y, Sun Z, et al. Evaluation of explant responses to STING ligands: personalized Immunosurgical therapy for head and neck squamous cell carcinoma. Cancer Res. 2018;78:6308–19.
Leach DG, Dharmaraj N, Piotrowski SL, Lopez-Silva TL, Lei YL, Sikora AG, Young S, Hartgerink JD. STINGel: controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy. Biomaterials. 2018;163:67–75.
Acknowledgements
We really appreciate An Biao, Ni Yanghong and Yang Jia to give us guidance about making figures.
Funding
This work is supported by the National Natural Science Foundation Regional Innovation and Development (U19A2003), National Major Scientific and Technological Special Project for “Significant New Drugs Development” (No. 2018ZX09733001), the Excellent Youth Foundation of Sichuan Scientific Committee Grant in China (No. 2019JDJQ008) and the Development Program of China (No. 2016YFA0201402).
Author information
Authors and Affiliations
Contributions
Wei Yuquan and Wei Xiawei offered main direction and significant guidance of this manuscript. Wang Yang and Luo Jingwen drafted the manuscript and illustrated the figures for the manuscript. They contribute equally to the work. Alu Aqu and Han Xuejiao made the figure and the table. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Y., Luo, J., Alu, A. et al. cGAS-STING pathway in cancer biotherapy. Mol Cancer 19, 136 (2020). https://doi.org/10.1186/s12943-020-01247-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-020-01247-w