
Abstract
Haematopoiesis, the process by which a restrained population of stem cells terminally differentiates into specific types of blood cells, depends on the tightly regulated temporospatial activity of several transcription factors (TFs). The deregulation of their activity or expression is a main cause of pathological haematopoiesis, leading to bone marrow failure (BMF), anaemia and leukaemia. TFs can be induced and/or activated by different stimuli, to which they respond by regulating the expression of genes and gene networks. Most TFs are highly pleiotropic; i.e., they are capable of influencing two or more apparently unrelated phenotypic traits, and the action of a single TF in a specific setting often depends on its interaction with other TFs and signalling pathway components. The microphthalmia-associated TF (MiTF) is a prototype TF in multiple situations. MiTF has been described extensively as a key regulator of melanocyte and melanoma development because it acts mainly as an oncogene. Mitf-mutated mice show a plethora of pleiotropic phenotypes, such as microphthalmia, deafness, abnormal pigmentation, retinal degeneration, reduced mast cell numbers and osteopetrosis, revealing a greater requirement for MiTF activity in cells and tissue. A growing amount of evidence has led to the delineation of key roles for MiTF in haematopoiesis and/or in cells of haematopoietic origin, including haematopoietic stem cells, mast cells, NK cells, basophiles, B cells and osteoclasts. This review summarizes several roles of MiTF in cells of the haematopoietic system and how MiTFs can impact BM development.
Similar content being viewed by others

MiTF expression and activity
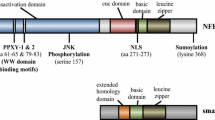
In 1942, Paula Hertwig, considering the progeny of irradiated mice, described animals that shared several pathological defects, including pigmentation loss, microphthalmia, deafness, osteopetrosis and a reduced number of mast cells (MCs) [45] and called them mi/mi (for microphthalmia) mice. The gene encoding the protein whose loss-of-function is critical for the mi/mi phenotype was cloned in 1993 and found to encode a transcription factor (TF) that was called MiTF for Microphthalmia Transcription Factor. MiTF is a member of the basic-helix-loop-helix-leucine zipper (bHLH-ZIP) family [48] characterized by three regions: an HLH and a ZIP motif, which are both involved in protein dimerization and required for the DNA binding mediated by a basic domain, which constitutes the third region [100] (Fig. 1).

(adapted from [1112, 186]. b Different MiTF isoforms. Each isoform is driven by its own promoter and has a partially unique exon, while exons from 2 through 9 are common in all isoforms. c MITF mutations found in melanoma (blue, on top) and in pigment deficiency syndromes (WS2A and TS patients, red, bottom) (adapted from [37]
a Structure of the four MiT family members. AD, acidic domain; bHLH, basic helix-loop-helix; LZ, leucine zipper; Ser, serine-rich region; Gln, glutamine-rich region; Pro, proline-rich segment; Pro + Arg, proline- and arginine-rich region
MiTF forms both homodimers and heterodimers with three other bHLH-ZIP TFs (TFEB, TFEC and TFE3), all members of the MiT family (Fig. 1a), that regulate gene expression by binding to E-box motifs characterized by the core hexanucleotide sequence CA[C/T]GTG [42] or by establishing bridges with and modulating the activity of other TFs. MiTF and TFEC are expressed in a cell-restricted manner [188], while TFEB and TFE3 are ubiquitously expressed [8, 17].
As consequence of different transcription starting sites or alternative splicing of its full-length RNA, MiTF is produced in several isoforms (Fig. 1b). Two isoforms based on alternative promoter usage, the melanocyte-specific (MiTF-M) and heart-specific (MiTF-H) transcripts, were identified in 1994 (Steingrimsson et al. [161]). Other isoforms have been successively identified in different tissues and settings: MiTF-A [6, 185], MiTF-C [31]; MiTF-B [174], MiTF-E [117], MiTF-Mc [168], MiTF-D [167]; MiTF-J [44], and MITF-CM [154]. Finally, MiTF-CX was found to be highly expressed in the cervix during pregnancy [83] and MITF-Mdel, a splice variant of MITF-M, was identified in melanocytes and melanoma cell lines [179].
The existence of multiple isoforms of MiTF, characterized by complex patterns of tissue-specific expression and the propensity to dimerize with other partners, may help explain its pleiotropy and its different biological effects on various cell types.
MiTF expression and/or activity are regulated at both mRNA and protein levels. The major positive regulators of MiTF transcription comprise the SWI/SNF chromatin remodelling complex [175], the WNT/β-catenin pathway (Bellei et al. [10]), SOX10 (sex-determining region Y-box10) [52], and CREB (cAMP response element-binding protein) [143], which can be activated by different signalling pathways. Moreover, MiTF fosters the expression of its own gene by recruiting LEF-1/β-catenin to its promoter [144]. GLI2 (glioma-associated oncogene family member 2), a TF activated by TGFβ [129], DEC1 (differentially expressed in chondrocytes protein 1, which is recruited to the MiTF promoter by HIF1α (hypoxia-inducible factor 1α) [28], and c-MYC [132] are considered negative regulators of MiTF expression. Activated in response to TNF-α exposure, NF-kB can both induce and repress MiTF [64, 71]. Finally, MiTF transcripts can be either stabilized by their association with CRD-BP (coding region determinant-binding protein) (Craig and Spiegelman [21]) or degraded by their interaction with several miRNAs [9].
The transcriptional activity of MiTF is largely regulated post-translationally. Notably, MiTF is phosphorylated by ERK1/2 at Ser73, by p90 ribosomal S6 kinase (p90RSK) at Ser409, by glycogen synthase kinase-3β (GSK3β) at Ser298 and by p38 MAPK at Ser307 [40, 82, 182]. MiTF phosphorylation generally enhances its activity, even though double phosphorylation at Ser73 and Ser409 promotes its proteasome-dependent degradation [182]. SAEI/SAEII- and UBC9-mediated SUMOylation at Lys182 and Lys316 inhibit MiTF [110], which can also lead to its degradation by the proteasome following UBC9-mediated ubiquitylation of Lys201 [183] or cleaved by caspase-3 during the apoptotic process [75].
MITF activity also depends on the availability of cooperating partners that can serve as activators, such as p300/CBP [182], or repressors, such as the member of the histidine triad (HIT) protein family HINT1 (histidine triad nucleotide-binding protein 1) [33].
Diseases associated with MiTF
MiTF is a recognized key TF in melanocyte biology and plays a fundamental role in melanoma, acting essentially as an oncogene. Indeed, in melanoma MiTF is generally overexpressed, with gene amplification observed in approximately 30% of the samples, or overactivated by phosphorylation in a B-RAFV600E/ERK-dependent manner [22, 64, 68].
In humans, germinal MiTF mutations have been mainly identified in the heterozygous state [137], and they are most frequently loss-of-function mutations that result in haploinsufficiency leading to pathologies associated with pigment abnormalities and congenital hearing loss, including Waardenburg syndrome type II (WS2) and Tietz albinism-deafness syndrome (TADS) [170, 177]. MiTF mutations cause type 2A WS2 (WS2A), accounting for 20% of all WS2 cases [137, 130]. Most identified mutations alter exons 7 and 8, encoding the b-HLH-Zip motifs involved in protein dimerization [130]. It has been suggested that MiTF mutations resulting in a truncated protein or in a protein unable to dimerize lead to WS2 through haploinsufficiency, whereas mutations exerting a dominant negative effect result in TADS, mostly characterized by non-truncating mutations in the basic domain [59, 156].
Supporting the possibility that, in contrast to mice, the activity of MiTF is required for normal human development, individuals bearing two inactivated alleles have rarely been identified. A homozygous intronic mutation of the 5′ splice site sequence affecting only the lineage-specific MiTF-M isoform has been associated with a more severe WS2A phenotype [135]. Another homozygous mutation (p.R223H) leads to a classic WS with persistent chronic constipation after the neonatal period, a symptom suggestive of Waardenburg syndrome type 4 (WS4), also known as Waardenburg-Shah syndrome [123, 124]. Finally, compound-heterozygous MiTF mutations have been identified in the severe multisystemic disorder termed COMMAD (coloboma, osteopetrosis, microphthalmia, macrocephaly, albinism, and deafness). These mutations are located in common exons (exons 2–9) and alter all MiTF isoforms, explaining the failure of multiple organ systems, and act in a dominant-negative manner, modifying nuclear localization and DNA-binding proficiency of MiTF homo- and heterodimers [34].
Interestingly, MiTF mutations associated with pigment deficiency syndromes and melanoma have different effects on protein function: mutations associated with WS2A and TADS are located in the bHLH-ZIP domains and prevent MiTF from binding DNA, while mutations found in melanoma cases are mostly located at the amino- or carboxy-termini affecting the transactivation potential of MiTF (Fig. 1c) [37].
MiTF mouse models
Two KO mouse models have been specifically used to study MiTF biology. The first is the mi/mi model (see above). The mi mutant allele (mi-Mitf) encodes a protein that has lost 1 of 4 consecutive arginine residues in the basic domain, which makes mi-Mitf defective in both DNA binding and nuclear localization (Steingrimsson et al. [161]; Morii et al. [101]. The other is the tg/tg mouse model, which does not express any Mitf isoform due to an insertional mutation that destroys the promoter region of Mitf [166]. Notably, the phenotypic abnormalities of the tg/tg mice are relatively mild compared to those observed in the mi/mi mice. Indeed, the transcription of several genes is more profoundly affected in mi/mi than it is in tg/tg cells, indicating that, in addition to the loss of its transactivation ability, mi-Mitf exerts transcription dominant-negative effects sequestering partners outside the nucleus [54, 62].
MiTF and haematopoiesis
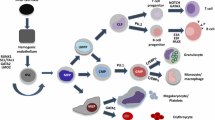
The haematopoietic system is a pyramidal organization with multipotent haematopoietic stem cells (HSCs) at the apex and mature blood cells at the bottom, with haematopoiesis being the process by which the naive population of HSCs are terminally differentiated into various blood cells. HSCs are defined by dormancy, the ability to remain out of the cell cycle for long periods (up to years), self-renewal, the ability to form stem cells, and multipotency, the ability to generate progenitor intermediates, which in turn will differentiate into several blood cell lineages [24, 122]. HSCs develop into multipotent progenitors (MPPs) that differentiate into common lymphoid progenitors (CLPs), precursors of all lymphoid cells, or common myeloid progenitors (CMPs), precursors of all myeloid cells [24, 138] (Fig. 2).

Simplified representation of the haematopoietic hierarchy. Haematopoiesis has a hierarchical organization with HSCs on top and mature blood cells at the bottom. HSCs are the sources of various types of progenitor cells that proliferate extensively, forming more differentiated cells at the expense of their self-renewal capacity
Normal haematopoiesis depends on the tightly regulated temporospatial activity of several TFs. The deregulation of their activity or expression is a main cause of pathological haematopoiesis, leading to bone marrow failure (BMF), anaemia and leukaemia. Although the role of MiTF in haematopoiesis is still unclear in humans, the analysis of the mi/mi and tg/tg mice enabled us to highlight MiTF importance in the haematopoietic context, mainly in the differentiation of some haematopoietic cell lineages: B lymphocytes, natural killer (NK) cells, mast cells (MCs), and osteoclasts (OCs).
B and NK cells develop from CLPs. B lymphocytes constitute a major component of humoral immunity. As pre-B cells, they differentiate in the BM, where they undergo the process of V(D)J recombination that leads to their immunoglobulin heavy and light chain formation, which is the basis of the B cell receptor (BCR). These immature B cells mainly migrate into the spleen, where they become activated B cells when their BCR binds its cognate antigen. Activated B cells undergo somatic hypermutation (SHM) and class switch recombination (CSR) process, which increases the affinity of the antibodies for their antigens and determines the class of specific class of B cells, respectively. Finally, the B cells become either antibody-secreting plasma cells or memory B cells for persistent protection [99, 128].
NK cells are the predominant cytotoxic T lymphocytes involved in innate immunity and mediate antitumour and antiviral responses. They display cytolytic activity that can be initiated through a variety of processes, including degranulation and death receptor ligation, and they produce several inflammatory cytokines, including TNF-α and IFN-γ [1, 79].
MCs and OCs are derived from CMPs. MCs mainly localize in the connective tissue, originate from granulocyte–macrophage progenitors (GMPs) and contribute to allergic responses and host protection against parasitic infection [7, 145]. Their cytoplasm contains large basophilic granules storing mediators of inflammation, such as histamine, heparin and serine proteases, and on their surface they express high-affinity immunoglobulin E (IgE) receptors (FCεRI), whose cross-linking by IgE causes their activation and the subsequent exocytosis of their granule content (degranulation) [70, 72].
OCs are giant, multinucleated, terminally differentiated cells that participate in bone homeostasis through their unique ability for bone resorption. They differentiate from the monocyte/macrophage lineage through two steps: they first proliferate in response to macrophage colony-stimulating factor (M-CSF) and therefore differentiate when the receptor activator of nuclear factor kappa-Β (RANK) on their surface is activated by its ligand RANKL, which is secreted by BM stromal cells and osteoblasts. RANKL/RANK signalling commits the precursors to differentiate into osteoclasts by activating the nuclear factor of activated T cells (NFATc1) to induce osteoclastogenic gene expression [25, 120].
MiTF and HSCs homeostasis
At the top of the haematopoietic process, HSCs play a critical role in maintaining the appropriate number of terminally differentiated and fully functional blood cells during the entire lifespan of the organism. HSC attrition leads to BMF and anaemia and represents a driving force for MDS and leukaemia.
No specific alteration has been described in the HSC compartment in the mi/mi and tg/tg mice. However, a transient induction of MiTF has been recently identified during the awakening of quiescent HSCs in response to haematological stresses, such as blood loss or BM transplant. Following a stress that causes a need for new cells to replenish BM and/or circulating blood, the p38 MAPK pathway is activated in HSCs, with p38α promoting cell cycle entry and progression by stimulating purine metabolism [61]. MiTF was identified among several p38α targets and linked to the increased purine metabolism necessary to support the proliferative activity of awakened HSCs [61, 65, 73, 149]. Indeed, p38-activated MiTF binds to the promoter of the inosine monophosphate dehydrogenase 2 (IMPDH2) gene, which encodes a key rate-limiting enzyme of purine metabolism [61].
Moreover, MiTF enhances HSC homing and long-term engraftment downstream of a signalling cascade initiated by bone morphogenetic protein 4 (BMP4) in R-SMAD-dependent and -independent manner [23, 65]. BMP4 signalling leads to MiTF nuclear translocation, where it upregulates Integrin-α4 (ITGA4) expression, a transmembrane protein critical for HSC homing and retention in the BM [65, 125].
Another example of how MiTF deregulation can impact BM homeostasis was reported in a recent study we published on Fanconi anaemia (FA) [121]. FA, a rare genetic syndrome presenting developmental abnormalities of the skeleton, BM failure, leukaemia predisposition and genetic instability, is due to the loss-of-function of at least one of more than 22 genes that encode proteins constituting a major nuclear pathway involved in DNA repair and replication safeguards and rescue [11, 39, 115]. Interestingly, genome-wide and targeted analyses have shown that MiTF controls the expression of a set of genes involved in DNA replication and genomic stability in melanoma [165] and, in particular, acts as a critical regulator of the FANC pathway, which plays a key role in the proliferation and survival of melanoma, maintaining the high proliferative potential of melanoma cells and contributing to their high resistance to therapeutics [12]. Accordingly, in melanoma cells that constitutionally overexpress MiTF, its siRNA-mediated depletion leads to FANC protein downregulation and the entry of melanoma cells into senescence, at which point they accumulate chromosomal damage and mitotic abnormalities. In this context, MiTF-mediated FANC protein expression appears to be a requirement to cope with the high replication activity of tumour cells. Accordingly, the siRNA-mediated silencing of a FANC protein (FANCA or FANCD2) was sufficient to slow tumour growth, even when MiTF overexpression was maintained [12].
We recently extended the FA-MiTF connection demonstrating that MITF is overexpressed in cells from FA patients, suggesting a regulatory loop in which MiTF induces the FANC proteins that, in turn, downregulate MiTF expression/activity. More importantly, in contrast with the transient p38/MiTF activation observed during stress-induced haematopoiesis, we demonstrated that the p38/MITF axis is constitutionally active and associated with BMF in Fanca−/− mice. Supporting the notion that the unscheduled and constitutional activation of the p38/MiTF axis has pathological consequences, p38 inhibition or siRNA-mediated depletion of MiTF was sufficient to rescue HSCs defects in the Fanca−/− mice [121]. The previous observations shed light on two important physiologic aspects of the biology of both MiTF and FA. They show the key role of MiTF as a biological rheostat, which turns on and off drivers for optimal cell differentiation and functionality, and indicate that the deregulation of its "normal" activity/activation, more than its loss-of-function, leads to pathology. Timely regulated and transient expression of MiTF is important to replenish peripheral blood and BM, whereas its constitutive expression influences BM physiology, affecting HSCs self-renewal and quiescence. Therefore, the attrition of the HSCs pool observed in FA, which has been associated with the inability of cells to recover from DNA damage and p53/p21 axis overactivation, are clearly reinforced by the concomitant loss of the MiTF off switch.
MiTF and lymphoid cells
Lacking B cell precursors within the BM, mi/mi mice rely upon other lymphatic sites, such as the spleen, for B cell development and maturation. This phenotype was initially attributed to the osteopetrotic environment of the BM [140] and the presence of extracellular molecules, including RANKL, stromal-derived factor (SDF-1), B-cell lymphotactin chemokine (BLC) and interferon-β (IFN-β), which affect B cell behaviour [141] and [142]). However, in support of its direct involvement in B cell differentiation, MiTF is highly expressed in naive B cells to repress interferon regulatory factor 4 (IRF-4), a key factor for B cell activation and terminal differentiation in antibody-secreting plasma cells [85, 155]. Accordingly, defective MiTF activity results in spontaneous B cell activation and antibody and autoantibody secretion, while enforced MiTF expression suppresses the expression of IRF-4 and antibody secretion [85]. E2A [41] and Bcl6 [5], which maintain high levels of MiTF in naive B cells, are silenced downstream the antigen-mediated BCR activation through Ca2+ signalling-mediated calmodulin inhibition of E2A [41] and through the upregulation of miR-148a, which targets Bcl6 expression [131], leading MiTF downregulation and B cell terminal differentiation (Fig. 3).

MiTF in B cells. In the normal bone marrow microenvironment, MiTF is highly expressed in naive B cells, where it acts as a repressor of IRF4. After B cell activation, MiTF is downregulated following the inhibition of the E2A factor and the upregulation of miR-148, leading to an increase in IRF4, which is essential for B cell terminal differentiation in antibody-secreting plasma cells. The decreased number of B cells and B cell precursors observed in mi/mi mice can be attributed to both direct and indirect effects of MiTF deficiency in these cells. The lack of MiTF in immature B cells causes an increase in IRF4 expression, leading to diminished proliferative capacity and spontaneous plasma cell differentiation in the absence of external stimuli. Moreover, mi/mi mice are characterized by an osteopetrotic microenvironment, which indirectly leads to the depletion of B cell precursors. In particular, osteoblasts and stromal cells secrete abnormal levels of RANKL to counteract osteoclast dysfunction, thereby activating RANK-expressing cells, which in turn secrete higher levels of IFN-β, which is known to induce cytotoxicity in B cell precursors
Although NK cells differentiate normally in mi/mi mice, mi-Mitf impairs their cytotoxicity, sequestering key TFs or transcription regulators in the cytoplasm [46]. Thus, the expression of perforin, IL-12Rβ2, IL-18Rα, and the receptors for the proinflammatory cytokines IL-12 and IL-18, which mediate IFN-γ expression, is severely impaired in mi/mi NK cells [55, 63]. The relatively mild phenotype observed indicates that MiTF has a redundant function in NK cell physiology than can be performed by other proteins in its absence.
Consequently, MiTF is involved in the immune responses of organisms, acting as an important rheostat regulating immature B cell activation and terminal differentiation, as well as promoting in the optimal cytotoxicity of NK cells.
MiTF and mast cells
Qi and collaborators identified the "pre-basophile and MC progenitors" (pre-BMPs), a subpopulation of the GMPs able to differentiate into basophiles or MCs [134]. A pre-BMP becomes a basophile when MiTF expression is silenced, while it will become an MC when MiTF expression is induced and maintained [145].
MiTF-A, MiTF-E, and MiTF-MC are the major isoforms expressed in MCs [150], which can also express MiTF-H and MiTF-M depending on physiological stimuli that modify both the promoter engagement and splicing [171]. MC proliferation is sustained by MiTF, which is induced by the stem cell factor (SCF)/c-kit signalling pathway, through the downregulation of miR-539 and miR-381 [78]. In turn, induced MiTF fosters c-kit expression. Accordingly, Mitf-mutated MCs respond poorly to SCF [26, 27, 172]. Other external stimuli such as IL-3, IL-4 and aggregated FCεRI proteins as well as activated PI3K pathway, p38α signalling and cytoplasmic adaptor protein SH3-binding protein 2 (3BP2) stabilize/increase MiTF protein synthesis or stability supporting MC differentiation [4, 50, 90, 114].
In MCs, MiTF is negatively regulated by its interaction with HINT [77] or PIAS3 [80]. Downstream IgE/FcεRI signalling activation, lysyl-tRNA synthetase (LysRS) is phosphorylated in a MAPK-dependent manner and translocated into the nucleus, where it produces the diadenosine oligophosphate Ap4A, which binds to HINT, liberating MiTF [77, 136, 184, 16]. The Mitf-PIAS3 interaction is lost as a consequence of MiTF phosphorylation in response to IL-6/IL-6R, SCF/Kit or IgE/FcεRI signalling activation [81, 158, 159].
Separated from its interactors, MITF is free to regulate several genes encoding key proteins involved in MC differentiation and activity, including (a) the granzyme B (GrB), which mediates the cytotoxic activity of MCs [53]; (b) the tryptophan hydroxylase (TPH), the rate-limiting enzyme for serotonin synthesis (Ito et al. [53]); (c) the histidine decarboxylase (Hdc), which regulates histamine synthesis [84], (d) the alpha-melanocyte-stimulating hormone (α-MSH) receptor (MC1R), which controls histamine release [2] and [3], and (e) the haematopoietic PGD2 synthase, which activates the cyclooxygenase pathway [106].
MiTF is also involved in the expression of several proteases stored in the secretory granules, including the endopeptidases MMCP-2, -4, -5, -6, -7, and -9 [32, 60, 101,102,103, 105, 109, 118, 119], the cathepsin G [60], and the transmembrane tryptase (TMT) [104]. MiTF regulates the expression of several adhesion molecules, including the integrin α4 subunit [66], which anchors the MCs to the extracellular matrix in BM and peripheral tissues [38], the plasminogen activator inhibitor-1 (PAI-1), involved in the regulation of extracellular matrix turnover through the inhibition of fibrinolysis [111], and the spermatogenic immunoglobulin superfamily adhesion molecule (SgIGSF) [56], which plays a role in MC migration [57, 107] and degranulation [58].
In quiescent MCs, MiTF interacts in the mitochondria with phosphorylated pyruvate dehydrogenase (PDH), a major regulator of the Krebs cycle that is essential for MC degranulation. In activated MCs, PDH is dephosphorylated and detached from MiTF to participate in degranulation. Thus, MiTF appears to be a negative regulator of PDH activity [151].
MiTF is also involved in the termination of MC-mediated responses. When the stimulus involved in the activation of MCs is resolved, the downregulation of MiTF contributes to the progression of apoptosis and the elimination of exhausted MCs [95, 173].
In summary, MiTF is a key hub in MC physiology during their differentiation and activation, channelling several extracellular signals to target genes. Accordingly, MCs from Mitf-mutant mice appear immature, failing to express genes critical for MC functions [69, 160, 164], (Fig. 4).

MiTF in mast cells. MiTF in mast cells is controlled by both positive and negative regulators: positive regulators can act either at the transcriptional level by increasing MiTF expression and at the post-transcriptional level by modulating MiTF activity and interactions with repressors, while negative regulators preferentially act at the protein level by binding to and inhibiting MiTF activity. Concerning its targets, MiTF controls the expression of proteins that are fundamental for both proliferation (i.e., growth factor receptors) and differentiation of mast cells. The latter include proteins stored in secretory granules, such as proteases, adhesion molecules and metabolic enzymes
MiTF and osteoclasts
MiTF is a major determinant of the behaviour of osteoclasts and it is involved in bone remodelling and BM functionality. The MiTF-A isoform is steadily present in both progenitors and terminally differentiated OCs, whereas MiTF-E, undetectable in the progenitors, is strongly upregulated during osteoclastogenesis [86].
Osteopetrosis, a condition in which the bone hardens, becoming denser, indicating diminished or deficient osteoclast activity, is a major phenotypic characteristic of mi/mi mice [45]. Supporting the key role of MiTFs in the terminal differentiation of OCs, the number of their mononuclear precursors is normal in mi/mi mice [49, 94, 160, 169]. BM cell transplantation from normal donors rescues the osteopetrotic condition of the mi/mi mutant mice, demonstrating the haematopoietic origin of the OCs [178].
Interestingly, only the mi/mi mutation causes osteopetrosis; the tg/tg mice presented only minimal abnormalities during osteoclastogenesis (Steingrimsson et al. [161]). The previous difference in the osteopetrotic phenotype of the mi/mi and the tg/tg mice is due to two reasons: mi-Mitf sequesters partners involved in nuclear gene expression in the cytoplasm, and TFE3, expressed in the OCs, may serve as a backup for MiTF [30, 92, 93, 96, 180]. Accordingly, whereas osteoclasts appear normal in tg/tg and TFE3-null mice, the combined loss of the two genes leads to severe osteopetrosis (Hershey and Fisher 43; Steingrimsson et al. [162]). The MiTF/TFE3 redundancy establishes a critical role for the MiT family in osteoclastogenesis and provides an interpretation of why osteopetrosis in humans has not been related to MiTF genetic alterations.
Osteoclastogenesis depends on M-CSF and RANKL signalling. M-CSF promotes MiTF assembly with TFE3 in a MAPK/ERK signalling-dependent manner [181] or with PU.1 in an NADPH oxidase 2 (Nox2) and ERK signalling-dependent manner [113]. Thus, M-CSF promotes MiTF activity without increasing its protein level. In contrast, the combination of M-CSF and RANKL also increases the intracellular level of MiTF, mainly via the induction of the MiTF-E isoform. Indeed, RANKL induces the expression of the master osteoclast TF NFATc1, which supports MiTF-E expression [152] and in turn amplifies NFATc1-dependent gene transcription [87].
The interaction of RANKL, IL-1, TGFβ and BMPs with their receptors also induces MiTF by activating transforming growth factor β (TGFβ)-activated kinase 1 (TAK1) [74], which regulates the activity and/or expression of p38 MAPK, Smad1/5/8, NF-κB, MiTF, PU.1, c-Fos, and NFATc1 [92, 93, 133]. The intracellular level of MiTF can be elevated by heat shock factor 1 (HSF1), a transcriptional regulator of heat shock and cell stress responses, which enhances osteoclast differentiation [18, 19, 176] or by POH1 (pad one homologue), a deubiquitinating enzyme and component of the 26S proteasome that limits MiTF proteasomal degradation, increasing de facto its level [147, 148]. The transcriptional activity of MiTF is also fostered by p38 MAPK-dependent phosphorylation at its Ser-307, which allows the aggregation of a trimeric complex with the proto-oncogene FUS and the chromatin remodelling ATPase BRG1 [14].
Several factors inhibit MiTF activity in OCs. The Ikaros family protein Eos interacts with both MiTF and PU.1 to repress transcription at specific promoters through the recruitment of the corepressors Sin3A and CtBP [51]. In monocytic precursors, MiTF is excluded from the nucleus by its interaction with the chaperone-like adaptor 14-3-3 proteins and a Ser173 phosphorylated form of Cdc25C-associated kinase 1 (C-TAK) [13]. By inducing protein phosphatase 2A expression, RANK/RANKL signalling promotes pSer173 dephosphorylation, destroying the MiTF/C-TAK/14-3-3 complex and allowing MiTF nuclear translocation [148]. Several other factors bind and inhibit MiTF activity, including the MafB transcription factor [67], PIAS3 [47], the histone deacetylase 7 [127, 163], and inhibitors of differentiation/DNA binding (Ids) and helix-loop-helix (HLH) transcription factors [76]. Finally, at least two miRNAs inhibit MiTF expression in OCs: miR-155, which is induced by the TGFβ1/Smad4 pathway [189] or by interferon-β [187], and miR-340 [91], which interacts with the two binding sites on the 3′ untranslated region (UTR) of the MiTF mRNA, leading to its degradation (Goswami et al. [36]; Zhao et al. [190]).
MiTF regulates OC differentiation, fusion and activity via its interactions with several other TFs, including AP-1, PU.1, eomesodermin (EOMES or Tbr2) [15, 89, 116, 146], TFE3, TFEC, NFATc1, MEF2 or proteins involved in transcriptional regulation as the coactivator P300/CBP [181].
Through previous multiple interactions, MiTF regulates the expression of receptors and membrane-associated proteins involved in osteoclast fusion, such as the OC-associated receptor OSCAR (So et al. [157]); the subunit of the V-ATPase proton pump, V-ATPase d2 [29], the transmembrane 7 superfamily member 4 protein encoded by DC-STAMP [20], or the osteoactivin (OA) encoded by the GPNMB gene [139, 153]. Finally, MiTF participates in the expression of genes encoding proteins involved in the bone resorption activity of mature, terminally differentiated osteoclasts, such as the osteoclast metalloprotein tartrate-resistant acid phosphatase TRAP [88, 93], Partington et al. [126]); the cysteine protease Cathepsin K (CTSK), which plays an essential role in the degradation of protein components of bone matrix [97, 108, 124],and chloride channel 7 (Clcn7) and osteopetrosis-associated transmembrane 1 (Ostm1) proteins, which mutually localize at the membrane to regulate the acidity of the OC extracellular environment [98].
Thus, in OCs, MiTF represents the converging point for the activity of multiple signal pathways, which independently regulate each other or, in cooperative manner, regulate the MiTF expression level and/or activity, highlighting its major role in OC biology (Fig. 5).

MiTF in osteoclast differentiation. MiTF is induced during osteoclastogenesis by both M-CSF, the cytokine responsible for the proliferation of osteoclast progenitors and their commitment towards an osteoclast fate, and RANKL, which induces the differentiation of osteoclasts through RANK signalling. M-CSF induces MiTF through the MAPK/ERK pathway, while RANKL signalling acts through the TAK1/p38 MAPK pathway. In parallel, RANKL also induces the expression of NFATc1, the other transcription factor essential for osteoclast differentiation. The MITF-PU.1 complex interacts with NFATc1 at osteoclast target gene promoters to initiate and maintain the expression of target genes. These genes encode markers of osteoclast differentiation, transmembrane proteins involved in the fusion of osteoclast precursors and enzymes involved in bone resorption
Conclusions
Since its discovery, MiTF has been shown to be a prominent key regulator of many aspects of melanocyte and melanoma biology. In this context, MiTF is unique among TFs for its ability to control a wide range of biological processes, such as cell proliferation, survival, differentiation, metabolism, invasion, senescence and DNA damage responses [35, 64]. Although still unclear, a master role for MiTF in some haematopoietic lineages cannot be ignored, notably in MCs and OCs, and for the homeostasis of the HSCs. On the previous basis, the expression of MiTF should be evaluated in other pathological situations in which BM fails, as well as in leukaemia, where its overexpression or loss-of-function may represent a driving force that allows the selection of clones bearing oncogenic mutations. For instance, subtle alterations in MiTF expression could participate in the emergence of leukemic clones in individuals with clonal haematopoiesis. The pharmacological modulation of the MiTF expression could be useful for care in several pathological setting, as bone marrow failure and bone abnormalities.
References
Abel AM, Yang C, Thakar MS, Malarkannan S. Natural killer cells: development, maturation, and clinical utilization. Front Immunol. 2018;9:1869.
Adachi S, Nakano T, Vliagoftis H, Metcalfe DD. Receptor-mediated modulation of murine mast cell function by alpha-melanocyte stimulating hormone. J Immunol. 1999;163(6):3363–8.
Adachi S, Morii E, Kim DK, Ogihara H, Jippo T, Ito A, Lee YM, Kitamura Y. Involvement of mi-transcription factor in expression of alpha-melanocyte-stimulating hormone receptor in cultured mast cells of mice. J Immunol. 2000;164(2):855–60.
Ainsua-Enrich E, Serrano-Candelas E, Álvarez-Errico D, Picado C, Sayós J, Rivera J, Martín M. The adaptor 3BP2 is required for KIT receptor expression and human mast cell survival. J Immunol. 2015;194(9):4309–18.
Alinikula J, Nera KP, Junttila S, Lassila O. Alternate pathways for Bcl6-mediated regulation of B cell to plasma cell differentiation. Eur J Immunol. 2011;41(8):2404–13.
Amae S, Fuse N, Yasumoto K, Sato S, Yajima I, Yamamoto H, Udono T, Durlu YK, Tamai M, Takahashi K, Shibahara S. Identification of a novel isoform of microphthalmia-associated transcription factor that is enriched in retinal pigment epithelium. Biochem Biophys Res Commun. 1998;247(3):710–5.
Arinobu Y, Iwasaki H, Gurish MF, Mizuno S, Shigematsu H, Ozawa H, Tenen DG, Austen KF, Akashi K. Developmental checkpoints of the basophil/mast cell lineages in adult murine hematopoiesis. Proc Natl Acad Sci U S A. 2005;102(50):18105–10.
Beckmann H, Su LK, Kadesch T. TFE3: a helix-loop-helix protein that activates transcription through the immunoglobulin enhancer muE3 motif. Genes Dev. 1990;4(2):167–79.
Bell RE, Levy C. The three M’s: melanoma, microphthalmia-associated transcription factor and microRNA. Pigment Cell Melanoma Res. 2011;24(6):1088–106.
Bellei B, Pitisci A, Catricalà C, Larue L, Picardo M. Wnt/β-catenin signaling is stimulated by α-melanocyte-stimulating hormone in melanoma and melanocyte cells: implication in cell differentiation. Pigment Cell Melanoma Res. 2011;24(2):309–25.
Bogliolo M, Surrallés J. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev. 2015;33:32–40.
Bourseguin J, Bonet C, Renaud E, Pandiani C, Boncompagni M, Giuliano S, Pawlikowska P, Karmous-Benailly H, Ballotti R, Rosselli F, Bertolotto C. FANCD2 functions as a critical factor downstream of MiTF to maintain the proliferation and survival of melanoma cells. Sci Rep. 2016;6:36539.
Bronisz A, Sharma SM, Hu R, Godlewski J, Tzivion G, Mansky KC, Ostrowski MC. Microphthalmia-associated transcription factor interactions with 14-3-3 modulate differentiation of committed myeloid precursors. Mol Biol Cell. 2006;17(9):3897–906.
Bronisz A, Carey HA, Godlewski J, Sif S, Ostrowski MC, Sharma SM. The multifunctional protein fused in sarcoma (FUS) is a coactivator of microphthalmia-associated transcription factor (MITF). J Biol Chem. 2014;289(1):326–34.
Carey HA, Hildreth BE 3rd, Samuvel DJ, Thies KA, Rosol TJ, Toribio RE, Charles JF, Ostrowski MC, Sharma SM. Eomes partners with PU.1 and MITF to regulate transcription factors critical for osteoclast differentiation. iScience. 2019;11:238–45.
Carmi-Levy I, Motzik A, Ofir-Birin Y, Yagil Z, Yang CM, Kemeny DM, Han JM, Kim S, Kay G, Nechushtan H, Suzuki R, Rivera J, Razin E. Importin beta plays an essential role in the regulation of the LysRS-Ap(4)A pathway in immunologically activated mast cells. Mol Cell Biol. 2011;31(10):2111–21.
Carr CS, Sharp PA. A helix-loop-helix protein related to the immunoglobulin E box-binding proteins. Mol Cell Biol. 1990;10(8):4384–8.
Chai RC, Kouspou MM, Lang BJ, Nguyen CH, van der Kraan AG, Vieusseux JL, Lim RC, Gillespie MT, Benjamin IJ, Quinn JM, Price JT. Molecular stress-inducing compounds increase osteoclast formation in a heat shock factor 1 protein-dependent manner. J Biol Chem. 2014;289(19):13602–14.
Chai RC, McDonald MM, Terry RL, Kovačić N, Down JM, Pettitt JA, Mohanty ST, Shah S, Haffari G, Xu J, Gillespie MT, Rogers MJ, Price JT, Croucher PI, Quinn JMW. Melphalan modifies the bone microenvironment by enhancing osteoclast formation. Oncotarget. 2017;8(40):68047–58.
Courtial N, Smink JJ, Kuvardina ON, Leutz A, Göthert JR, Lausen J. Tal1 regulates osteoclast differentiation through suppression of the master regulator of cell fusion DC-STAMP. FASEB J. 2012;26(2):523–32.
Craig EA, Spiegelman VS. Inhibition of coding region determinant binding protein sensitizes melanoma cells to chemotherapeutic agents. Pigment Cell Melanoma Res. 2012;25(1):83–7.
Cronin JC, Wunderlich J, Loftus SK, Prickett TD, Wei X, Ridd K, Vemula S, Burrell AS, Agrawal NS, Lin JC, Banister CE, Buckhaults P, Rosenberg SA, Bastian BC, Pavan WJ, Samuels Y. Frequent mutations in the MITF pathway in melanoma. Pigment Cell Melanoma Res. 2009;22(4):435–44.
Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–84.
Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10(2):120–36.
Drissi H, Sanjay A. The multifaceted osteoclast; far and beyond bone resorption. J Cell Biochem. 2016;117(8):1753–6.
Ebi Y, Kasugai T, Seino Y, Onoue H, Kanemoto T, Kitamura Y. Mechanism of mast cell deficiency in mutant mice of mi/mi genotype: an analysis by co-culture of mast cells and fibroblasts. Blood. 1990;75(6):1247–51.
Ebi Y, Kanakura Y, Jippo-Kanemoto T, Tsujimura T, Furitsu T, Ikeda H, Adachi S, Kasugai T, Nomura S, Kanayama Y, et al. Low c-kit expression of cultured mast cells of mi/mi genotype may be involved in their defective responses to fibroblasts that express the ligand for c-kit. Blood. 1992;80(6):1454–62.
Feige E, Yokoyama S, Levy C, Khaled M, Igras V, Lin RJ, Lee S, Widlund HR, Granter SR, Kung AL, Fisher DE. Hypoxia-induced transcriptional repression of the melanoma-associated oncogene MITF. Proc Natl Acad Sci U S A. 2011;108(43):E924–33.
Feng H, Cheng T, Steer JH, Joyce DA, Pavlos NJ, Leong C, Kular J, Liu J, Feng X, Zheng MH, Xu J. Myocyte enhancer factor 2 and microphthalmia-associated transcription factor cooperate with NFATc1 to transactivate the V-ATPase d2 promoter during RANKL-induced osteoclastogenesis. J Biol Chem. 2009;284(21):14667–76.
Ferron M, Settembre C, Shimazu J, Lacombe J, Kato S, Rawlings DJ, Ballabio A, Karsenty G. A RANKL-PKCβ-TFEB signaling cascade is necessary for lysosomal biogenesis in osteoclasts. Genes Dev. 2013;27(8):955–69.
Fuse N, Yasumoto KI, Takeda K, Amae S, Yoshizawa M, Udono T, Takahashi K, Tamai M, Tomita Y, Tachibana M, Shibahara S. Molecular cloning of cDNA encoding a novel microphthalmia-associated transcription factor isoform with a distinct amino-terminus. J Biochem. 1999;126(6):1043–51.
Ge Y, Jippo T, Lee YM, Adachi S, Kitamura Y. Independent influence of strain difference and mi transcription factor on the expression of mouse mast cell chymases. Am J Pathol. 2001;158(1):281–92.
Genovese G, Ghosh P, Li H, Rettino A, Sioletic S, Cittadini A, Sgambato A. The tumor suppressor HINT1 regulates MITF and β-catenin transcriptional activity in melanoma cells. Cell Cycle. 2012;11(11):2206–15.
George A, Zand DJ, Hufnagel RB, Sharma R, Sergeev YV, Legare JM, Rice GM, Scott Schwoerer JA, Rius M, Tetri L, Gamm DM, Bharti K, Brooks BP. Biallelic mutations in MITF cause coloboma, osteopetrosis, microphthalmia, macrocephaly, albinism, and deafness. Am J Hum Genet. 2016;99(6):1388–94.
Goding CR, Arnheiter H. MITF-the first 25 years. Genes Dev. 2019;33(15–16):983–1007.
Goswami S, Tarapore RS, Poenitzsch Strong AM, TeSlaa JJ, Grinblat Y, Setaluri V, Spiegelman VS. MicroRNA-340-mediated degradation of microphthalmia-associated transcription factor (MITF) mRNA is inhibited by coding region determinant-binding protein (CRD-BP). J Biol Chem. 2015;290(1):384–95.
Grill C, Bergsteinsdóttir K, Ogmundsdóttir MH, Pogenberg V, Schepsky A, Wilmanns M, Pingault V, Steingrímsson E. MITF mutations associated with pigment deficiency syndromes and melanoma have different effects on protein function. Hum Mol Genet. 2013;22(21):4357–67.
Grodzki AC, Pástor MV, Sousa JF, Oliver C, Jamur MC. Differential expression of integrin subunits on adherent and nonadherent mast cells. Braz J Med Biol Res. 2003;36(8):1101–9.
Gueiderikh A, Rosselli F, Cabral Neto JB. A never-ending story: the steadily growing family of the FA and FA-like genes. Genet Mol Biol. 2017;40(2):398–407.
Hartman ML, Czyz M. MITF in melanoma: mechanisms behind its expression and activity. Cell Mol Life Sci. 2015;72(7):1249–60.
Hauser J, Verma-Gaur J, Wallenius A, Grundström T. Initiation of antigen receptor-dependent differentiation into plasma cells by calmodulin inhibition of E2A. J Immunol. 2009;183(2):1179–87.
Hemesath TJ, Steingrímsson E, McGill G, Hansen MJ, Vaught J, Hodgkinson CA, Arnheiter H, Copeland NG, Jenkins NA, Fisher DE. microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994;8(22):2770–80.
Hershey CL, Fisher DE. Mitf and Tfe3: members of a b-HLH-ZIP transcription factor family essential for osteoclast development and function. Bone. 2004;34(4):689–96.
Hershey CL, Fisher DE. Genomic analysis of the Microphthalmia locus and identification of the MITF-J/Mitf-J isoform. Gene. 2005;347(1):73–82.
Hertwig P. Neue Mutationen und Kopplungsgruppen bei der Hausmaus. Z Indukt Abstammungs- u Vererbungsl. 1942;80:220–46.
Hesslein DG, Lanier LL. Transcriptional control of natural killer cell development and function. Adv Immunol. 2011;109:45–85.
Hikata T, Takaishi H, Takito J, Hakozaki A, Furukawa M, Uchikawa S, Kimura T, Okada Y, Matsumoto M, Yoshimura A, Nishimura R, Reddy SV, Asahara H, Toyama Y. PIAS3 negatively regulates RANKL-mediated osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblasts. Blood. 2009;113(10):2202–12.
Hodgkinson CA, Moore KJ, Nakayama A, Steingrímsson E, Copeland NG, Jenkins NA, Arnheiter H. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell. 1993;74(2):395–404.
Holtrop ME, Cox KA, Eilon G, Simmons HA, Raisz LG. The ultrastructure of osteoclasts in microphthalmic mice. Metab Bone Dis Relat Res. 1981;3(123–129):21.
Hu P, Carlesso N, Xu M, Liu Y, Nebreda AR, Takemoto C, Kapur R. Genetic evidence for critical roles of P38α protein in regulating mast cell differentiation and chemotaxis through distinct mechanisms. J Biol Chem. 2012;287(24):20258–69.
Hu R, Sharma SM, Bronisz A, Srinivasan R, Sankar U, Ostrowski MC. Eos, MITF, and PU.1 recruit corepressors to osteoclast-specific genes in committed myeloid progenitors. Mol Cell Biol. 2007;27(11):4018–27.
Huber WE, Price ER, Widlund HR, Du J, Davis IJ, Wegner M, Fisher DE. A tissue-restricted cAMP transcriptional response: SOX10 modulates alpha-melanocyte-stimulating hormone-triggered expression of microphthalmia-associated transcription factor in melanocytes. J Biol Chem. 2003;278(46):45224–30.
Ito A, Morii E, Maeyama K, Jippo T, Kim DK, Lee YM, Ogihara H, Hashimoto K, Kitamura Y, Nojima H. Systematic method to obtain novel genes that are regulated by mi transcription factor: impaired expression of granzyme B and tryptophan hydroxylase in mi/mi cultured mast cells. Blood. 1998;91(9):3210–21.
Ito A, Morii E, Kim DK, Kataoka TR, Jippo T, Maeyama K, Nojima H, Kitamura Y. Inhibitory effect of the transcription factor encoded by the mi mutant allele in cultured mast cells of mice. Blood. 1999;93(4):1189–96.
Ito A, Kataoka TR, Kim DK, Koma Y, Lee YM, Kitamura Y. Inhibitory effect on natural killer activity of microphthalmia transcription factor encoded by the mutant mi allele of mice. Blood. 2001;97(7):2075–83.
Ito A, Jippo T, Wakayama T, Morii E, Koma Y, Onda H, Nojima H, Iseki S, Kitamura Y. SgIGSF: a new mast-cell adhesion molecule used for attachment to fibroblasts and transcriptionally regulated by MITF. Blood. 2003;101(7):2601–8.
Ito A, Koma Y, Watabe K, Jippo T, Wakayama T, Iseki S, Kitamura Y. Contribution of the SgIGSF adhesion molecule to survival of cultured mast cells in vivo. Biochem Biophys Res Commun. 2004;319(1):200–6.
Ito A, Hagiyama M, Oonuma J, Murakami Y, Yokozaki H, Takaki M. Involvement of the SgIGSF/Necl-2 adhesion molecule in degranulation of mesenteric mast cells. J Neuroimmunol. 2007;184(1–2):209–13.
Izumi K, Kohta T, Kimura Y, Ishida S, Takahashi T, Ishiko A, Kosaki K. Tietz syndrome: unique phenotype specific to mutations of MITF nuclear localization signal. Clin Genet. 2008;74(1):93–5.
Jippo T, Lee YM, Katsu Y, Tsujino K, Morii E, Kim DK, Kim HM, Kitamura Y. Deficient transcription of mouse mast cell protease 4 gene in mutant mice of mi/mi genotype. Blood. 1999;93(6):1942–50.
Karigane D, Kobayashi H, Morikawa T, Ootomo Y, Sakai M, Nagamatsu G, Kubota Y, Goda N, Matsumoto M, Nishimura EK, Soga T, Otsu K, Suematsu M, Okamoto S, Suda T, Takubo K. p38α activates purine metabolism to initiate hematopoietic stem/progenitor cell cycling in response to stress. Cell Stem Cell. 2016;19(2):192–204.
Kataoka TR, Morii E, Oboki K, Kitamura Y. Strain-dependent inhibitory effect of mutant mi-MITF on cytotoxic activities of cultured mast cells and natural killer cells of mice. Lab Invest. 2004;84(3):376–84.
Kataoka TR, Komazawa N, Oboki K, Morii E, Nakano T. Reduced expression of IL-12 receptor beta2 and IL-18 receptor alpha genes in natural killer cells and macrophages derived from B6-mi/mi mice. Lab Invest. 2005;85(1):146–53.
Kawakami A, Fisher DE. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab Invest. 2017;97(6):649–56.
Khurana S, Buckley S, Schouteden S, Ekker S, Petryk A, Delforge M, Zwijsen A, Verfaillie CM. A novel role of BMP4 in adult hematopoietic stem and progenitor cell homing via Smad independent regulation of integrin-α4 expression. Blood. 2013;121(5):781–90.
Kim DK, Morii E, Ogihara H, Hashimoto K, Oritani K, Lee YM, Jippo T, Adachi S, Kanakura Y, Kitamura Y. Impaired expression of integrin alpha-4 subunit in cultured mast cells derived from mutant mice of mi/mi genotype. Blood. 1998;92(6):1973–80.
Kim K, Kim JH, Lee J, Jin HM, Kook H, Kim KK, Lee SY, Kim N. MafB negatively regulates RANKL-mediated osteoclast differentiation. Blood. 2007;109(8):3253–9.
King R, Googe PB, Weilbaecher KN, Mihm MC Jr, Fisher DE. Microphthalmia transcription factor expression in cutaneous benign, malignant melanocytic, and nonmelanocytic tumors. Am J Surg Pathol. 2001;25(1):51–7.
Kitamura Y, Morii E, Jippo T, Ito A. Effect of MITF on mast cell differentiation. Mol Immunol. 2002;38(16–18):1173–6.
Kitamura Y, Oboki K, Ito A. Development of mast cells. Proc Jpn Acad Ser B Phys Biol Sci. 2007;83(6):164–74.
Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, Frederick DT, Barzily-Rokni M, Straussman R, Haq R, Fisher DE, Mesirov JP, Hahn WC, Flaherty KT, Wargo JA, Tamayo P, Garraway LA. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014;4(7):816–27.
Krystel-Whittemore M, Dileepan KN, Wood JG. Mast cell: a multi-functional master cell. Front Immunol. 2016;6:620.
Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma’ayan A. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics. 2010;26(19):2438–44.
Lamothe B, Lai Y, Xie M, Schneider MD, Darnay BG. TAK1 is essential for osteoclast differentiation and is an important modulator of cell death by apoptosis and necroptosis. Mol Cell Biol. 2013;33(3):582–95.
Larribere L, Hilmi C, Khaled M, Gaggioli C, Bille K, Auberger P, Ortonne JP, Ballotti R, Bertolotto C. The cleavage of microphthalmia-associated transcription factor, MITF, by caspases plays an essential role in melanocyte and melanoma cell apoptosis. Genes Dev. 2005;19(17):1980–5.
Lee J, Kim K, Kim JH, Jin HM, Choi HK, Lee SH, Kook H, Kim KK, Yokota Y, Lee SY, Choi Y, Kim N. Id helix-loop-helix proteins negatively regulate TRANCE-mediated osteoclast differentiation. Blood. 2006;107(7):2686–93.
Lee YN, Nechushtan H, Figov N, Razin E. The function of lysyl-tRNA synthetase and Ap4A as signaling regulators of MITF activity in FcepsilonRI-activated mast cells. Immunity. 2004;20(2):145–51.
Lee YN, Brandal S, Noel P, Wentzel E, Mendell JT, McDevitt MA, Kapur R, Carter M, Metcalfe DD, Takemoto CM. KIT signaling regulates MITF expression through miRNAs in normal and malignant mast cell proliferation. Blood. 2011;117(13):3629–40.
Leong JW, Wagner JA, Ireland AR, Fehniger TA. Transcriptional and post-transcriptional regulation of NK cell development and function. Clin Immunol. 2017;177:60–9.
Levy C, Nechushtan H, Razin E. A new role for the STAT3 inhibitor, PIAS3: a repressor of microphthalmia transcription factor. J Biol Chem. 2002;277(3):1962–6.
Levy C, Sonnenblick A, Razin E. Role played by microphthalmia transcription factor phosphorylation and its Zip domain in its transcriptional inhibition by PIAS3. Mol Cell Biol. 2003;23(24):9073–80.
Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006;12(9):406–14.
Li XH, Kishore AH, Dao D, Zheng W, Roman CA, Word RA. A novel isoform of microphthalmia-associated transcription factor inhibits IL-8 gene expression in human cervical stromal cells. Mol Endocrinol. 2010;24(8):1512–28.
Li Y, Liu B, Harmacek L, Long Z, Liang J, Lukin K, Leach SM, O’Connor B, Gerber AN, Hagman J, Roers A, Finkelman FD, Huang H. The transcription factors GATA2 and microphthalmia-associated transcription factor regulate Hdc gene expression in mast cells and are required for IgE/mast cell-mediated anaphylaxis. J Allergy Clin Immunol. 2018;142(4):1173–84.
Lin L, Gerth AJ, Peng SL. Active inhibition of plasma cell development in resting B cells by microphthalmia-associated transcription factor. J Exp Med. 2004;200(1):115–22.
Lu SY, Li M, Lin YL. Mitf induction by RANKL is critical for osteoclastogenesis. Mol Biol Cell. 2010;21(10):1763–71.
Lu SY, Li M, Lin YL. Mitf regulates osteoclastogenesis by modulating NFATc1 activity. Exp Cell Res. 2014;328(1):32–43.
Luchin A, Purdom G, Murphy K, Clark MY, Angel N, Cassady AI, Hume DA, Ostrowski MC. The microphthalmia transcription factor regulates expression of the tartrate-resistant acid phosphatase gene during terminal differentiation of osteoclasts. J Bone Miner Res. 2000;15(3):451–60.
Luchin A, Suchting S, Merson T, Rosol TJ, Hume DA, Cassady AI, Ostrowski MC. Genetic and physical interactions between Microphthalmia transcription factor and PU.1 are necessary for osteoclast gene expression and differentiation. J Biol Chem. 2001;276(39):36703–10.
Ma P, Mali RS, Munugalavadla V, Krishnan S, Ramdas B, Sims E, Martin H, Ghosh J, Li S, Chan RJ, Krystal G, Craig AW, Takemoto C, Kapur R. The PI3K pathway drives the maturation of mast cells via microphthalmia transcription factor. Blood. 2011;118(13):3459–69.
Ma Y, Shan Z, Ma J, Wang Q, Chu J, Xu P, Qin A, Fan S. Validation of downregulated microRNAs during osteoclast formation and osteoporosis progression. Mol Med Rep. 2016;13(3):2273–80.
Mansky KC, Sankar U, Han J, Ostrowski MC. Microphthalmia transcription factor is a target of the p38 MAPK pathway in response to receptor activator of NF-kappa B ligand signaling. J Biol Chem. 2002;277(13):11077–83.
Mansky KC, Sulzbacher S, Purdom G, Nelsen L, Hume DA, Rehli M, Ostrowski MC. The microphthalmia transcription factor and the related helix-loop-helix zipper factors TFE-3 and TFE-C collaborate to activate the tartrate-resistant acid phosphatase promoter. J Leukoc Biol. 2002;71(2):304–10.
Marks SC Jr, Walker DG. The hematogenous origin of osteoclasts: experimental evidence from osteopetrotic (microphthalmic) mice treated with spleen cells from beige mouse donors. Am J Anat. 1981;161(1):1–10.
Martelli F, Ghinassi B, Lorenzini R, Vannucchi AM, Rana RA, Nishikawa M, Partamian S, Migliaccio G, Migliaccio AR. Thrombopoietin inhibits murine mast cell differentiation. Stem Cells. 2008;26(4):912–9.
Martina JA, Diab HI, Li H, Puertollano R. Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell Mol Life Sci. 2014;71(13):2483–97.
Matsumoto M, Kogawa M, Wada S, Takayanagi H, Tsujimoto M, Katayama S, Hisatake K, Nogi Y. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J Biol Chem. 2004;279(44):45969–79.
Meadows NA, Sharma SM, Faulkner GJ, Ostrowski MC, Hume DA, Cassady AI. The expression of Clcn7 and Ostm1 in osteoclasts is coregulated by microphthalmia transcription factor. J Biol Chem. 2007;282(3):1891–904.
Melchers F. Checkpoints that control B cell development. J Clin Invest. 2015;125(6):2203–10.
Moore KJ. Insight into the microphthalmia gene. Trends Genet. 1995;11(11):442–8.
Morii E, Takebayashi K, Motohashi H, Yamamoto M, Nomura S, Kitamura Y. Loss of DNA binding ability of the transcription factor encoded by the mutant mi locus. Biochem Biophys Res Commun. 1994;205(2):1299–304.
Morii E, Tsujimura T, Jippo T, Hashimoto K, Takebayashi K, Tsujino K, Nomura S, Yamamoto M, Kitamura Y. Regulation of mouse mast cell protease 6 gene expression by transcription factor encoded by the mi locus. Blood. 1996;88(7):2488–94.
Morii E, Jippo T, Tsujimura T, Hashimoto K, Kim DK, Lee YM, Ogihara H, Tsujino K, Kim HM, Kitamura Y. Abnormal expression of mouse mast cell protease 5 gene in cultured mast cells derived from mutant mi/mi mice. Blood. 1997;90(8):3057–66.
Morii E, Ogihara H, Oboki K, Kataoka TR, Jippo T, Kitamura Y. Effect of MITF on transcription of transmembrane tryptase gene in cultured mast cells of mice. Biochem Biophys Res Commun. 2001;289(5):1243–6.
Morii E, Oboki K, Kataoka TR, Igarashi K, Kitamura Y. Interaction and cooperation of mi transcription factor (MITF) and myc-associated zinc-finger protein-related factor (MAZR) for transcription of mouse mast cell protease 6 gene. J Biol Chem. 2002;277(10):8566–71.
Morii E, Oboki K. MITF is necessary for generation of prostaglandin D2 in mouse mast cells. J Biol Chem. 2004;279(47):48923–9.
Morii E, Ito A, Jippo T, Koma Y, Oboki K, Wakayama T, Iseki S, Lamoreux ML, Kitamura Y. Number of mast cells in the peritoneal cavity of mice: influence of microphthalmia transcription factor through transcription of newly found mast cell adhesion molecule, spermatogenic immunoglobulin superfamily. Am J Pathol. 2004;165(2):491–9.
Motyckova G, Weilbaecher KN, Horstmann M, Rieman DJ, Fisher DZ, Fisher DE. Linking osteopetrosis and pycnodysostosis: regulation of cathepsin K expression by the microphthalmia transcription factor family. Proc Natl Acad Sci U S A. 2001;98(10):5798–803.
Murakami M, Ikeda T, Ogawa K, Funaba M. Transcriptional activation of mouse mast cell protease-9 by microphthalmia-associated transcription factor. Biochem Biophys Res Commun. 2003;311(1):4–10.
Murakami H, Arnheiter H. Sumoylation modulates transcriptional activity of MITF in a promoter-specific manner. Pigment Cell Res. 2005;18(4):265–77.
Murakami M, Ikeda T, Saito T, Ogawa K, Nishino Y, Nakaya K, Funaba M. Transcriptional regulation of plasminogen activator inhibitor-1 by transforming growth factor-beta, activin A and microphthalmia-associated transcription factor. Cell Signal. 2006;18(2):256–65.
Nabar NR, Kehrl JH. Yale the transcription factor eb links cellular stress to the immune response. J Biol Med. 2017;90(2):301–15.
Nakanishi A, Hie M, Iitsuka N, Tsukamoto I. A crucial role for reactive oxygen species in macrophage colony-stimulating factor-induced RANK expression in osteoclastic differentiation. Int J Mol Med. 2013;31(4):874–80.
Nechushtan H, Zhang Z, Razin E. Microphthalmia (mi) in murine mast cells: regulation of its stimuli-mediated expression on the translational level. Blood. 1997;89(8):2999–3008.
Niraj J, Färkkilä A, D’Andrea AD. The fanconi anemia pathway in cancer. Annu Rev Cancer Biol. 2019;3:457–78.
Nomura S, Sakuma T, Higashibata Y, Oboki K, Sato M. Molecular cause of the severe functional deficiency in osteoclasts by an arginine deletion in the basic domain of Mi transcription factor. J Bone Miner Metab. 2001;19(3):183–7.
Oboki K, Morii E, Kataoka TR, Jippo T, Kitamura Y. Isoforms of mi transcription factor preferentially expressed in cultured mast cells of mice. Biochem Biophys Res Commun. 2002;290(4):1250–4.
Ogihara H, Kanno T, Morii E, Kim DK, Lee YM, Sato M, Kim WY, Nomura S, Ito Y, Kitamura Y. Synergy of PEBP2/CBF with mi transcription factor (MITF) for transactivation of mouse mast cell protease 6 gene. Oncogene. 1999;18(32):4632–9.
Ogihara H, Morii E, Kim DK, Oboki K, Kitamura Y. Inhibitory effect of the transcription factor encoded by the mutant mi microphthalmia allele on transactivation of mouse mast cell protease 7 gene. Blood. 2001;97(3):645–51.
Ono T, Nakashima T. Recent advances in osteoclast biology. Histochem Cell Biol. 2018;149(4):325–41.
Oppezzo A, Bourseguin J, Renaud E, Pawlikowska P, Rosselli F. Microphthalmia transcription factor expression contributes to bone marrow failure in Fanconi anemia. J Clin Invest. 2020;130(3):1377–91.
Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132(4):631–44.
Pang X, Zheng X, Kong X, Chai Y, Wang Y, Qian H, Yang B, Wu C, Chu J, Yang T. A homozygous MITF mutation leads to familial Waardenburg syndrome type 4. Am J Med Genet A. 2019;179(2):243–8.
Pang M, Rodríguez-Gonzalez M, Hernandez M, Recinos CC, Seldeen KL, Troen BR. AP-1 and Mitf interact with NFATc1 to stimulate cathepsin K promoter activity in osteoclast precursors. J Cell Biochem. 2019;120(8):12382–92.
Papayannopoulou T, Priestley GV, Nakamoto B, Zafiropoulos V, Scott LM. Molecular pathways in bone marrow homing: dominant role of alpha(4)beta(1) over beta(2)-integrins and selectins. Blood. 2001;98(8):2403–11.
Partington GA, Fuller K, Chambers TJ, Pondel M. Mitf-PU.1 interactions with the tartrate-resistant acid phosphatase gene promoter during osteoclast differentiation. Bone. 2004;34(2):237–45.
Pham L, Kaiser B, Romsa A, Schwarz T, Gopalakrishnan R, Jensen ED, Mansky KC. HDAC3 and HDAC7 have opposite effects on osteoclast differentiation. J Biol Chem. 2011;286(14):12056–65.
Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol. 2013;131(4):959–71.
Pierrat MJ, Marsaud V, Mauviel A, Javelaud D. Expression of microphthalmia-associated transcription factor (MITF), which is critical for melanoma progression, is inhibited by both transcription factor GLI2 and transforming growth factor-β. J Biol Chem. 2012;287(22):17996–8004.
Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31(4):391–406.
Porstner M, Winkelmann R, Daum P, Schmid J, Pracht K, Côrte-Real J, Schreiber S, Haftmann C, Brandl A, Mashreghi MF, Gelse K, Hauke M, Wirries I, Zwick M, Roth E, Radbruch A, Wittmann J, Jäck HM. miR-148a promotes plasma cell differentiation and targets the germinal center transcription factors Mitf and Bach2. Eur J Immunol. 2015;45(4):1206–15.
Pouryazdanparast P, Brenner A, Haghighat Z, Guitart J, Rademaker A, Gerami P. The role of 8q24 copy number gains and c-MYC expression in amelanotic cutaneous melanoma. Mod Pathol. 2012;25(9):1221–6.
Qi B, Cong Q, Li P, Ma G, Guo X, Yeh J, Xie M, Schneider MD, Liu H, Li B. Ablation of Tak1 in osteoclast progenitor leads to defects in skeletal growth and bone remodeling in mice. Sci Rep. 2014;4:7158.
Qi X, Hong J, Chaves L, Zhuang Y, Chen Y, Wang D, Chabon J, Graham B, Ohmori K, Li Y, Huang H. Antagonistic regulation by the transcription factors C/EBPα and MITF specifies basophil and mast cell fates. Immunity. 2013;39(1):97–110.
Rauschendorf MA, Zimmer AD, Laut A, Demmer P, Rösler B, Happle R, Sartori S, Fischer J. Homozygous intronic MITF mutation causes severe Waardenburg syndrome type 2A. Pigment Cell Melanoma Res. 2019;32(1):85–91.
Razin E, Zhang ZC, Nechushtan H, Frenkel S, Lee YN, Arudchandran R, Rivera J. Suppression of microphthalmia transcriptional activity by its association with protein kinase C-interacting protein 1 in mast cells. J Biol Chem. 1999;274(48):34272–6.
Read AP, Newton VE. Waardenburg syndrome. J Med Genet. 1997;34(8):656–65.
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11.
Ripoll VM, Meadows NA, Raggatt LJ, Chang MK, Pettit AR, Cassady AI, Hume DA. Microphthalmia transcription factor regulates the expression of the novel osteoclast factor GPNMB. Gene. 2008;413(1–2):32–41.
Roundy K, Kollhoff A, Eichwald EJ, Weis JJ, Weis JH. Microphthalmic mice display a B cell deficiency similar to that seen for mast and NK cells. J Immunol. 1999;163(12):6671–8.
Roundy K, Smith R, Weis JJ, Weis JH. Overexpression of RANKL implicates IFN-beta-mediated elimination of B-cell precursors in the osteopetrotic bone of microphthalmic mice. J Bone Miner Res. 2003;18(2):278–88.
Roundy KM, Spangrude G, Weis JJ, Weis JH. Partial rescue of B cells in microphthalmic osteopetrotic marrow by loss of response to type I IFNs. Int Immunol. 2005;17(11):1495–503.
Saha B, Singh SK, Sarkar C, Bera R, Ratha J, Tobin DJ, Bhadra R. Activation of the Mitf promoter by lipid-stimulated activation of p38-stress signalling to CREB. Pigment Cell Res. 2006;19(6):595–605.
Saito H, Yasumoto K, Takeda K, Takahashi K, Fukuzaki A, Orikasa S, Shibahara S. Melanocyte-specific microphthalmia-associated transcription factor isoform activates its own gene promoter through physical interaction with lymphoid-enhancing factor 1. J Biol Chem. 2002;277(32):28787–94.
Sasaki H, Kurotaki D, Tamura T. Regulation of basophil and mast cell development by transcription factors. Allergol Int. 2016;65(2):127–34.
Sato M, Morii E, Takebayashi-Suzuki K, Yasui N, Ochi T, Kitamura Y, Nomura S. Microphthalmia-associated transcription factor interacts with PU.1 and c-Fos: determination of their subcellular localization. Biochem Biophys Res Commun. 1999;254(2):384–7.
Schwarz T, Sohn C, Kaiser B, Jensen ED, Mansky KC. The 19S proteasomal lid subunit POH1 enhances the transcriptional activation by Mitf in osteoclasts. J Cell Biochem. 2010;109(5):967–74.
Schwarz T, Murphy S, Sohn C, Mansky KC. C-TAK1 interacts with microphthalmia-associated transcription factor, Mitf, but not the related family member Tfe3. Biochem Biophys Res Commun. 2010;394(4):890–5.
Seita J, Sahoo D, Rossi DJ, Bhattacharya D, Serwold T, Inlay MA, Ehrlich LI, Fathman JW, Dill DL, Weissman IL. Gene expression commons: an open platform for absolute gene expression profiling. PLoS ONE. 2012;7(7):e40321.
Shahlaee AH, Brandal S, Lee YN, Jie C, Takemoto CM. Distinct and shared transcriptomes are regulated by microphthalmia-associated transcription factor isoforms in mast cells. J Immunol. 2007;178(1):378–88.
Sharkia I, Hadad Erlich T, Landolina N, Assayag M, Motzik A, Rachmin I, Kay G, Porat Z, Tshori S, Berkman N, Levi-Schaffer F, Razin E. Pyruvate dehydrogenase has a major role in mast cell function, and its activity is regulated by mitochondrial microphthalmia transcription factor. J Allergy Clin Immunol. 2017;140(1):204-214.e8.
Sharma SM, Bronisz A, Hu R, Patel K, Mansky KC, Sif S, Ostrowski MC. MITF and PU.1 recruit p38 MAPK and NFATc1 to target genes during osteoclast differentiation. J Biol Chem. 2007;282(21):15921–9.
Sheng MH, Wergedal JE, Mohan S, Lau KH. Osteoactivin is a novel osteoclastic protein and plays a key role in osteoclast differentiation and activity. FEBS Lett. 2008;582(10):1451–8.
Shiohara M, Shigemura T, Suzuki T, Tanaka M, Morii E, Ohtsu H, Shibahara S, Koike K. MITF-CM, a newly identified isoform of microphthalmia-associated transcription factor, is expressed in cultured mast cells. Int J Lab Hematol. 2009;31(2):215–26.
Shukla V, Lu R. IRF4 and IRF8: governing the virtues of B Lymphocytes. Front Biol (Beijing). 2014;9(4):269–82.
Smith SD, Kelley PM, Kenyon JB, Hoover D. Tietz syndrome (hypopigmentation/deafness) caused by mutation of MITF. J Med Genet. 2000;37(6):446–8.
So H, Rho J, Jeong D, Park R, Fisher DE, Ostrowski MC, Choi Y, Kim N. Microphthalmia transcription factor and PU.1 synergistically induce the leukocyte receptor osteoclast-associated receptor gene expression. J Biol Chem. 2003;278(26):24209–16.
Sonnenblick A, Levy C, Razin E. Interplay between MITF, PIAS3, and STAT3 in mast cells and melanocytes. Mol Cell Biol. 2004;24(24):10584–92.
Sonnenblick A, Levy C, Razin E. Immunological trigger of mast cells by monomeric IgE: effect on microphthalmia transcription factor, STAT3 network of interactions. J Immunol. 2005;175(3):1450–5.
Stechschulte DJ, Sharma R, Dileepan KN, Simpson KM, Aggarwal N, Clancy J Jr, Jilka RL. Effect of the mi allele on mast cells, basophils, natural killer cells, and osteoclasts in C57Bl/6J mice. J Cell Physiol. 1987;132(3):565–70.
Steingrímsson E, Moore KJ, Lamoreux ML, Ferré-D’Amaré AR, Burley SK, Zimring DC, Skow LC, Hodgkinson CA, Arnheiter H, Copeland NG, et al. Molecular basis of mouse microphthalmia (mi) mutations helps explain their developmental and phenotypic consequences. Nat Genet. 1994;8(3):256–63.
Steingrímsson E, Tessarollo L, Pathak B, Hou L, Arnheiter H, Copeland NG, Jenkins NA. Mitf and Tfe3, two members of the Mitf-Tfe family of bHLH-Zip transcription factors, have important but functionally redundant roles in osteoclast development. Proc Natl Acad Sci U S A. 2002;99(7):4477–82.
Stemig M, Astelford K, Emery A, Cho JJ, Allen B, Huang TH, Gopalakrishnan R, Mansky KC, Jensen ED. Deletion of histone deacetylase 7 in osteoclasts decreases bone mass in mice by interactions with MITF. PLoS ONE. 2015;10(4):e0123843.
Stevens J, Loutit JF. Mast cells in spotted mutant mice (W Ph mi). Proc R Soc Lond B Biol Sci. 1982;215(1200):405–9.
Strub T, Giuliano S, Ye T, Bonet C, Keime C, Kobi D, Le Gras S, Cormont M, Ballotti R, Bertolotto C, Davidson I. Essential role of microphthalmia transcription factor for DNA replication, mitosis and genomic stability in melanoma. Oncogene. 2011;30(20):2319–32.
Tachibana M, Hara Y, Vyas D, Hodgkinson C, Fex J, Grundfast K, Arnheiter H. Cochlear disorder associated with melanocyte anomaly in mice with a transgenic insertional mutation. Mol Cell Neurosci. 1992;3(5):433–45.
Takeda K, Yasumoto KI, Kawaguchi N, Udono T, Watanabe KI, Saito H, Takahashi K, Noda M, Shibahara S. Mitf-D, a newly identified isoform, expressed in the retinal pigment epithelium and monocyte-lineage cells affected by Mitf mutations. Biochim Biophys Acta. 2002;1574(1):15–23.
Takemoto CM, Yoon YJ, Fisher DE. The identification and functional characterization of a novel mast cell isoform of the microphthalmia-associated transcription factor. J Biol Chem. 2002;277(33):30244–52.
Thesingh CW, Scherft JP. Fusion disability of embryonic osteoclast precursor cells and macrophages in the microphthalmic osteopetrotic mouse. Bone. 1985;6(1):43–52.
Tietz W. A syndrome of deaf-mutism associated with albinism showing dominant autosomal inheritance. Am J Hum Genet. 1963;15(3):259–64.
Tshori S, Sonnenblick A, Yannay-Cohen N, Kay G, Nechushtan H, Razin E. Microphthalmia transcription factor isoforms in mast cells and the heart. Mol Cell Biol. 2007;27(11):3911–9.
Tsujimura T, Morii E, Nozaki M, Hashimoto K, Moriyama Y, Takebayashi K, Kondo T, Kanakura Y, Kitamura Y. Involvement of transcription factor encoded by the mi locus in the expression of c-kit receptor tyrosine kinase in cultured mast cells of mice. Blood. 1996;88(4):1225–33.
Tsujimura T, Hashimoto K, Morii E, Tunio GM, Tsujino K, Kondo T, Kanakura Y, Kitamura Y. Involvement of transcription factor encoded by the mouse mi locus (MITF) in apoptosis of cultured mast cells induced by removal of interleukin-3. Am J Pathol. 1997;151(4):1043–51.
Udono T, Yasumoto K, Takeda K, Amae S, Watanabe K, Saito H, Fuse N, Tachibana M, Takahashi K, Tamai M, Shibahara S. Structural organization of the human microphthalmia-associated transcription factor gene containing four alternative promoters. Biochim Biophys Acta. 2000;1491(1–3):205–19.
Vachtenheim J, Ondrusová L, Borovanský J. SWI/SNF chromatin remodeling complex is critical for the expression of microphthalmia-associated transcription factor in melanoma cells. Biochem Biophys Res Commun. 2010;392(3):454–9.
van der Kraan AG, Chai RC, Singh PP, Lang BJ, Xu J, Gillespie MT, Price JT, Quinn JM. HSP90 inhibitors enhance differentiation and MITF (microphthalmia transcription factor) activity in osteoclast progenitors. Biochem J. 2013;451(2):235–44.
Waardenburg PJ. A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am J Hum Genet. 1951;3(3):195–253.
Walker DG. Bone resorption restored in osteopetrotic mice by transplants of normal bone marrow and spleen cells. Science. 1975;190(4216):784–5.
Wang Y, Radfar S, Liu S, Riker AI, Khong HT. Mitf-Mdel, a novel melanocyte/melanoma-specific isoform of microphthalmia-associated transcription factor-M, as a candidate biomarker for melanoma. BMC Med. 2010;17(8):14.
Weilbaecher KN, Hershey CL, Takemoto CM, Horstmann MA, Hemesath TJ, Tashjian AH, Fisher DE. Age-resolving osteopetrosis: a rat model implicating microphthalmia and the related transcription factor TFE3. J Exp Med. 1998;187(5):775–85.
Weilbaecher KN, Motyckova G, Huber WE, Takemoto CM, Hemesath TJ, Xu Y, Hershey CL, Dowland NR, Wells AG, Fisher DE. Linkage of M-CSF signaling to Mitf, TFE3, and the osteoclast defect in Mitf(mi/mi) mice. Mol Cell. 2001;8(4):749–58.
Wu M, Hemesath TJ, Takemoto CM, Horstmann MA, Wells AG, Price ER, Fisher DZ, Fisher DE. c-Kit triggers dual phosphorylations, which couple activation and degradation of the essential melanocyte factor Mi. Genes Dev. 2000;14(3):301–12.
Xu W, Gong L, Haddad MM, Bischof O, Campisi J, Yeh ET, Medrano EE. Regulation of microphthalmia-associated transcription factor MITF protein levels by association with the ubiquitin-conjugating enzyme hUBC9. Exp Cell Res. 2000;255(2):135–43.
Yannay-Cohen N, Carmi-Levy I, Kay G, Yang CM, Han JM, Kemeny DM, Kim S, Nechushtan H, Razin E. LysRS serves as a key signaling molecule in the immune response by regulating gene expression. Mol Cell. 2009;34(5):603–11.
Yasumoto K, Amae S, Udono T, Fuse N, Takeda K, Shibahara S. A big gene linked to small eyes encodes multiple Mitf isoforms: many promoters make light work. Pigment Cell Res. 1998;11(6):329–36.
Zhang W, Li X, Wang S, Chen Y, Liu H. Regulation of TFEB activity and its potential as a therapeutic target against kidney diseases. Cell Death Discov. 2020;6:32.
Zhang J, Zhao H, Chen J, Xia B, Jin Y, Wei W, Shen J, Huang Y. Interferon-β-induced miR-155 inhibits osteoclast differentiation by targeting SOCS1 and MITF. FEBS Lett. 2012;586(19):3255–62.
Zhao GQ, Zhao Q, Zhou X, Mattei MG, de Crombrugghe B. TFEC, a basic helix-loop-helix protein, forms heterodimers with TFE3 and inhibits TFE3-dependent transcription activation. Mol Cell Biol. 1993;13(8):4505–12.
Zhao H, Zhang J, Shao H, Liu J, Jin M, Chen J, Huang Y. Transforming Growth Factor β1/Smad4 Signaling Affects Osteoclast Differentiation via Regulation of miR-155 Expression. Mol Cells. 2017;40(3):211–21.
Zhao H, Zhang J, Shao H, Liu J, Jin M, Chen J, Huang Y. miRNA-340 inhibits osteoclast differentiation via repression of MITF. Biosci Rep. 2017;37(4):BSR20170302.
Acknowledgments
We apologize to the authors contributing to the studies discussed herein, but not mentioned due to space considerations. We thank X. Renaudin for his critical reading of the manuscript. Research in F.R. lab is funded by La Ligue Contre le Cancer. A.O. is a 4th year PhD student supported by a doctoral fellowship from the «Ministére de l'Enseignement supérieur, de la Recherche et de l'Innovation» and the «Fondation pour La Recherche Médicale» (FDT202001010821).
Author information
Authors and Affiliations
Contributions
AO and FR analyzed the literature, drew the figures and wrote the manuscript. Both authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflicting financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Oppezzo, A., Rosselli, F. The underestimated role of the microphthalmia-associated transcription factor (MiTF) in normal and pathological haematopoiesis. Cell Biosci 11, 18 (2021). https://doi.org/10.1186/s13578-021-00529-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-021-00529-0