
Abstract
N6-Methyladenosine (m6A) is the most abundant, dynamic, and reversible epigenetic RNA modification that is found in coding and non-coding RNAs. Emerging studies have shown that m6A and its regulators affect multiple steps in RNA metabolism and play broad roles in various cancers. Worldwide, breast cancer is the most prevalent cancer in female. It is a very heterogeneous disease characterized by genetic and epigenetic variations in tumor cells. Increasing evidence has shown that the dysregulation of m6A-related effectors, as methyltransferases, demethylases, and m6A binding proteins, is pivotal in breast cancer pathogenesis. In this review, we have summarized the most up-to-date research on the biological functions of m6A modification in breast cancer and have discussed the potential clinical applications and future directions of m6A modification as a biomarker as well as a therapeutic target of breast cancer.
Similar content being viewed by others
Background
Breast cancer is the most common malignancy and leading cause of cancer-related death in women [1]. In 2018, up to 2.1 million women worldwide were diagnosed with breast cancer, occupying one out of four cancer cases among the female population [2]. At present, approximately 70–80% of non-metastatic breast cancer patients get cured, while advanced (metastatic) breast cancer patients do not attain remission using the currently available treatment regimens [1]. Breast cancer is known to be associated with molecular heterogeneity and exhibits a variety of histological features, prognostic patterns, and responses to treatment [3,4,5]. Thus, it is imperative to understand the underlying molecular mechanism of the development of breast cancer in detail.
Several studies have recently shown the importance of the intricate signaling at genetic, transcriptomic, and epigenetic levels that affects tumorigenesis and progression of breast cancer [6,7,8]. N6-Methyladenosine (m6A) is one of the most common internal epigenetic modifications found in RNA molecules [9]. After its discovery by Desrosiers in the 1970s [10], owing to the limitations in technology, research on m6A modification has slowly gained attention in the past couple of decades. Recently, with the advances in molecular biology and sequencing, the research on m6A modification has made remarkable progress [11,12,13]. To date, m6A modifications have been identified in almost every kind of RNA, including mRNA, tRNA, and non-coding RNA, and they are involved in multiple RNA processing and metabolism activities such as splicing, localization, export, translation, stabilization, and decay [14,15,16,17,18]. Notably, m6A modification sites are evolutionally conserved (mammals, insects, plants, bacteria, yeast and some viruses) and occur within a consensus sequence DRACH (D = G, A, or U; R = G or A; H = A, C, or U) [11, 12]. m6A methylation is not randomly distributed and is commonly detected in the coding sequences and 3′ untranslated regions (3′ UTRs), around the stop codons in mRNAs, or near the last exon in non-coding RNAs [19,20,21]. Deposition of m6A preferentially in the 5′ UTR was also observed in a few cases [22, 23].
It has become clear that the global abundance of m6A and expression levels of its regulators are frequently dysregulated in a variety of cancers, including breast cancer [24, 25]. The functions of m6A are critical for multiple biological processes such as tumor initiation, promotion, and progression in breast cancer. In this review, we first provide a comprehensive elucidation of m6A modification, and then focus on the emerging pathophysiological roles and molecular mechanism of m6A modification in breast cancer. More importantly, we highlight the potential clinical applications and future directions of m6A modification as a biomarker as well as a therapeutic target of breast cancer.
Regulation of m6A modification
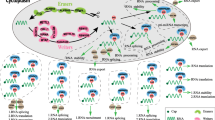
The m6A modification, as its name suggests, involves the transfer of a methyl group to the N-6 position of the adenosine in the nucleic acid [26]. Similar to DNA and histone methylation, m6A modification is a dynamic and reversible biological process that is regulated by methyltransferases (also called “writers”) and demethylases (also called “erasers”). In addition to writers and erasers, “readers” are binding proteins that recognize the chemical signatures important for the regulation of m6A modification (Fig. 1) [27, 28].

The molecular mechanism involved in m6A modification of consensus adenosine (A) bases. This is a dynamic and reversible epigenetic modification that is regulated by “writers” and “erasers.” m6A methylation is primarily catalyzed by the m6A methyltransferase complex comprising METTL3/METTL14/WTAP and other regulatory proteins (RBM15/15B, KIAA1429, Hakai, or ZC3H13). The erasers mainly include FTO, ALKBH5, ALKBH3, and ALKBH1. In addition to writers and erasers, “readers” are binding proteins that recognize m6A marks in the RNA. m6A modification can affect multiple steps in RNA processing, such as RNA splicing, export, translation, stabilization, and decay
m6A writers
Writers of m6A methylation include the multicomponent m6A methyltransferase complex (MTC) comprising methyltransferase-like 3 (METTL3), METTL14, Wilms tumor 1-associated protein (WTAP), and other regulatory proteins, including RNA-binding motif protein 15 (RBM15), RBM15B, Vir-like m6A methyltransferase associated (VIRMA, also termed as KIAA1429 or Virilizer), Cbl proto-oncogene like 1 (CBLL1, also termed as Hakai), and zinc finger CCCH-type containing 13 (ZC3H13) [29]. In the MTC, METTL3 is the active catalyzing enzyme, while METTL14 is responsible for maintaining the catalytic activity of METTL3 and substrate recognition. The heterodimer formed by METTL3 and METTL14 is indispensable for m6A methylation [30, 31]. WTAP helps in binding of this METTL3/METTL14 heterodimer to regulatory proteins and in localization of MTC in nuclear spots, thereby facilitating m6A methylation at selective group of transcripts and regions [32]. Moreover, certain m6A methyltransferases do not exert their function via the MTC. METTL16, METTL5, and zinc finger CCHC-type containing 4 (ZCCHC4) are RNA m6A methyltransferases that directly catalyze m6A modification in RNA molecules [33,34,35].
m6A erasers
Demethylases (“erasers”) are proteins that remove the m6A modification from RNA and include the fat mass and obesity-associated protein (FTO), α-ketoglutarate-dependent dioxygenase alk B homolog 5 (ALKBH5), ALKBH3, and ALKBH1 [36]. All these molecules belong to the α-ketoglutarate-dependent dioxygenase family of proteins and share a common mechanism for demethylation: m6A is oxidized to N6-hydroxymethyladenosine (hm6A) that is converted to N6-formyladenosine (f6A) before finally reverting to adenosine (A), i.e., m6A-hm6A-f6A-A in a step-wise manner [37]. FTO was the first m6A demethylase identified (2011), and it can not only remove methyl group of m6A in RNA, but can also demethylate N6,2-O-dimethyladenosine (m6Am), which is predominantly located in the 5′ UTR [38,39,40]. ALKBH5, primarily localized to the nucleus, was the second m6A demethylase to be identified (2013). It can remove the m6A modification from nuclear RNA (mostly mRNA), thereby affecting mRNA export, splicing, and stability [41, 42].
m6A readers
Readers of m6A methylation constitute m6A-binding proteins that recognize the modified site and induce a series of physiological functions [43]. These proteins can be divided into three categories depending on the mechanism of m6A recognition: direct reader, m6A switch reader, and indirect reader [36]. Direct readers comprise the most-studied category and include YTH domain-containing proteins and eukaryotic translation initiation factor (eIF) 3 [36]. The YTH domain is an RNA-binding domain that interacts with m6A via a “tryptophan cage” [44]. There are five proteins that form the YTH domain-containing (YTHDC) family of proteins, namely, YTHDC1, YTHDC2, and YTHDF1-3 [45]. YTHDC1 and the YTHDF family are primarily localized to the nucleus and cytoplasm, respectively, while YTHDC2 is found in both the nucleus and cytoplasm [14, 46, 47]. They identify specific m6A sites, and accordingly regulate export, degradation as well as translation of m6A-containing mRNAs [48]. Heterogeneous nuclear ribonucleoproteins (hnRNPs) including hnRNPG, hnRNPC, and hnRNPA2B1 and insulin-like growth factor 2 mRNA binding proteins (IGF2BPs) including IGF2BP1, IGF2BP2, and IGF2BP3 can function as m6A switch readers by remodeling specific RNA structure and consequently impacting the binding mode of RNA and protein [36, 49, 50]. Fragile-X mental retardation protein (FMRP) has been recently identified to be an indirect reader since it can regulate m6A-modified mRNA by binding with the YTHDF proteins [51].
m6A sequencing technology
m6A-antibody immunoprecipitation (m6A-IP) and methylated RNA m6A immunoprecipitation sequencing (MeRIP, also called m6A-seq) were used to reveal the landscape of transcriptome-wide m6A sites in 2012 [11, 12]. However, these methods could only detect m6A sites within 100–200 nucleotides long RNA fragments and could not identify m6A sites at base resolution [52]. Thus, to overcome low resolution, a series of new detection methods have been developed. For example, the RNA-antibody photo-crosslinking and immunoprecipitation (CLIP) methods (PA-m6A-seq, miCLIP, and UV-CLIP) are antibody-based methods with better resolution [53]. m6A-REF-seq or MAZTER-seq are antibody-free m6A-seq methods that are based on the RNA m6A methylation-sensitive endoribonuclease MazF. It identifies unknown m6A sites that have been reported to be undetectable by CLIP [54, 55]. Another antibody-free method, termed DART-seq, is based on the fusion construct of m6A binding protein YTH and C-to-U editing enzyme APOBEC1. This technique requires low amounts of RNA and simple library preparation [56]. It is noteworthy that the methods mentioned above mostly detect m6A modification indirectly and may result in inaccuracies [57]. Recently, the Oxford nanopore technology is used to study transcriptome-wide m6A using a direct RNA sequencing protocol, which could prevent bias associated with amplification or reverse transcription [58].
m6A modification in breast cancer
With the elucidation of mechanisms involved in m6A modification, current research has focused on the roles of m6A modification in various diseases. Although studies on the function of m6A in breast cancer are in their early stages, increasing evidence has shown that m6A is essential in many aspects of this tumor, including tumorigenesis, metastasis, prognosis, and therapy resistance. Herein, we review the physiological effects of m6A modification in breast cancer (Table 1) and elaborate its future research trends and potential clinical applications.
Roles of m6A in breast cancer proliferation and apoptosis
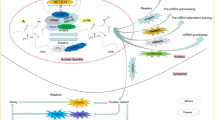
Immortality and evasion of apoptosis are the two hallmarks of cancer [59]. Numerous studies have shown the dysregulation of writers/erasers/readers associated with m6A are responsible for tumorigenesis and progression in breast cancer (Fig. 2a). METTL3, the core component of MTC, enhances cell proliferation via a positive feedback loop of the HBXIP/let-7g/METTL3/HBXIP axis in breast cancer [60]. METTL3 also induces proliferation, inhibits apoptosis, and accelerates tumor growth by targeting Bcl-2 [61]. Overexpression of the METTL3/14 m6A methylation complex results in malignant transformation [62]. METTL14 interacts with and modifies lncRNA-942 by adding m6A to enhance the expression and stability of CYP1B1 and CXCR4, respectively, thereby increasing cell proliferation and colony formation and suppressing cell apoptosis [63]. Interestingly, a similar study showed that the overexpression of METTL14 inhibits cell viability and colony formation in breast cancer [64]. KIAA1429 is an oncogene and it promotes breast cancer cell proliferation and colony formation by stabilizing the CDK1 mRNA (a cell cycle regulator) [65]. Similarly, Lewinska et al. [66] demonstrated that decrease in the m6A signature promotes cell cycle arrest and senescence, thereby exerting anticancer effects.

The pathophysiological roles and molecular mechanism of m6A modification in breast cancer. a m6A and its regulators control RNA fate and metabolism to affect proliferation, apoptosis and cell cycle. b The mechanism of m6A modification involved in breast cancer migration, invasion and metastasis
As an m6A eraser, FTO demethylates the 3′ UTR of the BNIP3 mRNA and induces its decay in an YTHDF2-independent manner, resulting in breast cancer cell proliferation, colony formation, and metastasis [67]. Polymorphisms in FTO are associated with breast cancer, especially estrogen receptor (ER)-positive breast cancer [68, 69]. Estrogen stimulates breast cancer cell proliferation by upregulating FTO and activating PI3K/Akt signaling [69]. Moreover, a recent study has demonstrated that FTO mediates the survival of metabolically adaptable triple-negative breast cancer (TNBC) cells in glutamine-deficient microenvironments [70]. The function of ALKBH5 in breast cancer is controversial. Wu et al. [64] have shown that silencing ALKBH5 leads to inhibition of breast cancer cell viability, colony formation, and migration. However, Fry et al. demonstrated the overexpression of ALKBH5 and METTL3/14 in immortalized human mammary epithelial cells. Depletion of ALKBH5 increases cell proliferation and migration [62].
The expression of eIF3m, one of the 13 subunits of m6A reader eIF3, positively correlates with the development and progression of breast cancer. Downregulation of eIF3m inhibits breast cancer proliferation and increases the rate of apoptosis [71]. Wu et al. have reported high levels of hnRNPC associated with breast cancer proliferation. Downregulation of hnRNPC promotes the formation of endogenous double-stranded RNA and induces immune response that results in antiproliferative activity [72]. hnRNPA2B1 also has a positive role in breast cancer. Knockdown of hnRNPA2B1 decreases breast cancer cell proliferation, increases apoptosis, and prolongs the S phase of cells by inhibiting STAT3/ERK1/2 signaling [73].
Roles of m6A in breast cancer migration, invasion and metastasis
Metastasis is a major cause of cancer-related deaths. Although the survival rate of breast cancer has improved immensely over the past decades, the therapeutic effect of metastatic breast cancer is still not optimistic [74]. Migration and invasion of tumor cells are key processes in cancer metastasis (Fig. 2b) [75]. METTL14 promotes the migration and invasion of breast cancer cells by directly regulating hsa-miR-146a-5p and m6A modification [76]. Similarly, KIAA1429 has also been found to promote breast cancer cell migration and invasion [65]. ERs constitute the most active transcription factors involved in breast cancer. Inhibiting ERα activity is currently used as a strategy for treating patients with ER-positive breast cancer [77, 78]. Hakai is a coregulator of ERα and suppresses breast cancer cell migration by competitively binding to ERα [79]. Although breast cancer stem cells (BCSCs) constitute a minor proportion of breast cancer cells, accumulating evidence has demonstrated the vital role of BCSCs in tumor initiation, progression, and metastasis [80, 81]. Hypoxia stimulates ALKBH5 or ZNF217 that stabilize the NANOG and KLF4 mRNAs and induce the phenotype associated with BCSCs and lung metastasis [82, 83]. IGF2BP binds to lncRNA FGF13-AS1 and Myc to form a positive feedback loop to regulate breast cancer cell stemness [84]. Epithelial–mesenchymal transition (EMT) accelerates the progress of tumor metastasis [85]. Liu et al. [86] demonstrated that hnRNPA2B1 inhibits EMT and metastasis in breast cancer by directly binding to PFN2 mRNA and reducing its stability. Conversely, eIF3m promotes breast cancer cell migration and invasion by activating EMT [71]. eIF3g, another subunit of eIF3, interacts with hnRNPU, HSZFP36, and β-actin in the nucleus and promotes the metastasis of breast cancer to the lymph nodes [87].
Roles of m6A in the clinicopathology and prognosis of breast cancer
A growing number of studies have confirmed the correlation between m6A modification and clinical pathological characteristics and prognosis of breast cancer (Fig. 3). Typically, breast cancer is classified into three major subtypes based on molecular markers: ER or progesterone receptor (PR)-positive (luminal A and luminal B), human epidermal growth factor receptor 2 (HER2)-positive, and TNBC [3, 88]. Different subtypes of breast cancer are associated with distinct etiologies, response to treatment, and prognosis. Wu et al. [64] reported that METTL3, METTL14, FTO, and ALKBH5 are upregulated and WTAP is downregulated in luminal breast cancer patients, while the expression level of FTO is significantly decreased in HER2-positive breast cancer. However, the study by Tan et al. demonstrated overexpression of FTO in hormone receptor-negative and HER2-positive breast cancer. A significant proportion of FTO-positive cells have also been reported in P53-positive or histological grade 3 breast cancer [89]. Overexpression of eIF3m has been observed in TNBC but not in non-TNBC or normal breast tissues and it reduces overall survival (OS), relapse-free survival, and post-progression survival in breast cancer patients [71]. Using the data from The Cancer Genome Atlas-Breast Cancer cohort, Liu et al. revealed that the overexpression of YTHDF1, YTHDF3, and KIAA1429 is predictive of poor prognosis. Especially, YTHDF3 is an independent prognostic factor of OS in breast cancer patients [25].

The roles of m6A modification in the clinicopathology of breast cancer
Zeng et al. performed a case–control study based on Chinese population to determine the correlation between polymorphisms in FTO and risk associated with prognosis of breast cancer patients. Their results showed variants of FTO are concerned with varying susceptibility of breast cancer; however, they cannot predict survival outcomes in patients with this disease [90]. Meanwhile, it is acknowledged that obesity increases the risk of breast cancer substantially, but the molecular mechanism involved remain to be understood [91]. As the name implies, FTO is intimately associated with obesity. Thus, the advent of FTO may well explain the relationship between obesity and breast cancer [69, 92]. In addition, epidemiological studies have found that reproductive history is linked to the development of breast cancer. The risk of breast cancer is significantly less in early pregnancy (before age 20), while the risk transiently increases after parturition [91, 93]. Peri et al. [94] have demonstrated that hnRPA2B1 is overexpressed in the mammary tissues of post-menopausal parous women, suggesting that m6A modification may contribute to the correlation between pregnancy and breast cancer.
Discussion
With the discovery of FTO as an m6A demethylase, research on m6A modification has become the hotspot of epigenetics. Recent reports have demonstrated that m6A-related regulators play essential and diverse biological functions in the development of various types of cancer, including breast cancer, glioblastoma, hepatocellular carcinoma, acute myeloid leukemia, and cervical cancer [24, 95,96,97]. This review summarizes the recent advances in the understanding of the roles, mechanisms, and potential clinical applications of m6A in breast cancer. Notably, the specific mechanism for m6A modification in breast cancer is complex and even inconsistent among studies. For instance, Wu et al. [64] showed that m6A methylation suppresses the growth and metastasis of breast cancer, while Fry et al. [62] reported malignant progression with increasing m6A methylation. This “double-edged sword” phenomenon is also reported in other tumors [98] and may be attributed to differences in the origin of tumor tissues, intratumoral heterogeneity, and ethnicity at the macro level. For example, the polymorphisms rs9939609 and rs1477196 in FTO are implicated in an increased risk of breast cancer among women excluding those from Iran [99]. Moreover, at the molecular level, there are two types of m6A sites in different cell lines: structural m6A sites and dynamic m6A sites. Dynamic m6A sites are cell-specific sites regulated by spatio-temporal regulators [100]. This category of m6A sites can make the gene play diverse roles in different cells that may contribute to the phenomenon.
Studies have shown the importance of m6A regulatory enzymes as novel potential biomarkers for the early diagnosis and prognosis of breast cancer. Different enzymes involved in catalyzing m6A modification correlate with specific molecular subtypes of breast cancer that are classified based on the presence of certain biomarkers (ER, PR, and HER2). For example, eIF3m is overexpressed in TNBC, while it is expressed to the same extent in tumors and corresponding adjacent normal breast tissues in non-TNBC. The upregulation of eIF3m represents poor pathological differentiation, high degree of malignant transformation, and increased rates of lymph node and distant metastases in TNBC. Moreover, elevated expression of eIF3m implies poor survival outcomes for TNBC patients [71]. Therefore, eIF3m may be a reliable biomarker of TNBC. Of interest, we also found that both the m6A writer and eraser genes are aberrantly overexpressed and play oncogenic roles in breast cancer. Thus, global m6A signatures may be unreliable as diagnostic and prognostic biomarkers in patients with breast cancer. To that extent, the m6A profiles of specific transcripts or transcript loci could serve as better biomarkers. However, the techniques currently available for studying transcriptome-wide m6A modification are not precise enough [52]. This has resulted in the difficulty in fully understanding the correlation between m6A-modified RNAs and disease. Additionally, these methods are limited by the requirement of large amounts of RNA, experienced technical skills, and high cost, thereby limiting the feasibility of m6A-seq in large-scale screening [57]. Therefore, novel detection methods with high precision, reduced sample volume, and low cost are warranted. This will help develop m6A profiles/signatures of specific transcripts or transcript loci as early diagnostic and prognostic biomarkers for breast cancer. The improved methods of m6A-seq may enable the use of peripheral blood for screening of cancer in the future.
m6A may also serve as a novel therapeutic target in breast cancer. Targeting dysregulated m6A regulators represents an attractive strategy for cancer therapy. However, only a few studies have focused on the development of potent and specific drugs that target m6A regulators in breast cancer. MO-I-500 is a small-molecule inhibitor of the m6A demethylase activity of FTO and inhibits the survival and/or colony formation of a SUM149 triple-negative inflammatory breast cancer cell line [70]. In addition to small-molecule compounds, PROTAC (proteolysis targeting chimera)-based inhibitors can also be developed to treat breast cancer by selectively degrading dysregulated m6A regulators [101]. Systemic therapies, such as chemotherapy, radiotherapy, endocrine therapy, and targeted therapy, comprise the most important arm of breast cancer treatment [1]. Resistance to these therapies is catastrophic and contributes to failed treatment and/or cancer recurrence [102, 103]. Recent studies have indicated that dysregulation of m6A regulators plays an important role in developing resistance to therapy in cancer [104, 105]. Klinge et al. observed higher RNA and protein levels of hnRNPA2B1 in tamoxifen-resistant breast cancer cells. The upregulation of hnRNPA2B1 alters the expression of multiple miRNAs and reduces the sensitivity of MCF-7 cells to tamoxifen [106], suggesting the importance of hnRNPA2B1 in resistance to endocrine therapy. Future research should focus on abrogating m6A-mediated resistance of breast cancer cells via different treatment regimens.
Immunotherapy is emerging as a new treatment modality in breast cancer, especially metastatic breast cancer [107]. Owing to the unsatisfactory effect of immunotherapy in the early stages of patients with breast cancer, breast cancer has previously been assumed to be unresponsive to the immunotherapy [108]. This could be attributed to the lacunae in the molecular mechanism in breast cancer that has resulted in the slow development of effective immunotherapy in such patients. Recent studies have shown the regulatory effect of m6A RNA modification on host immunity and in enhancing anticancer immunotherapy. Depleting FTO promotes the degradation of downstream genes PD-1, CXCR4, and SOX10 in an m6A-dependent manner, thereby sensitizing patients with melanoma to anti-PD-1 checkpoint blockade therapy [109]. Similarly, Han et al. demonstrated a new mechanism for immune evasion: the m6A reader YTHDF1 binds to and promotes the translation of mRNAs encoding lysosomal proteases that result in the reduction of cross-presentation of tumor antigens in dendritic cells. Silencing YTHDF1 inhibits immune evasion and improves the efficacy of anti-PD-1 therapy [110]. Given the vital roles of m6A modification in breast cancer as well as the promising effect of immunotherapy in other tumors, combining m6A signatures and anticancer immunotherapy may serve as a breakthrough in breast cancer immunotherapy.
Currently, the roles and mechanisms involved in m6A modification in breast cancer remain to be elucidated and several issues need to be addressed in the future. First, high-throughput research on m6A modification should be performed to generate m6A methylation-centric networks in breast cancer. Second, although researchers have noted the potential of m6A as a diagnostic and prognostic marker for breast cancer, no studies that have focused on the sensitivity or specificity of this marker in large patient cohorts. Current m6A sequencing technologies are not sufficient to support large-scale screening. Thus, a novel sequencing technology is indispensable to study the role of m6A in breast cancer. Third, there is preliminary evidence for the potential of m6A as a therapeutic target for breast cancer. Studies have only focused on the molecular mechanisms involved at this stage and a few reports have focused on drug development and pre-clinical/clinical trials. Future experiments should examine the efficacy of m6A-targeted drugs alone or in combination with other treatments for breast cancer.
Conclusions
Taken together, we have discussed the dysregulation of m6A modification in breast cancer to help develop broad clinical applications in the prevention, treatment, and management of breast cancer. Detailed efforts to understand the underlying mechanism of m6A modification in breast cancer, identify and develop diagnostic and prognostic factors, and devise m6A-targeted therapy will help better treat patients with breast cancer in the future. This will also highlight the diverse (undiscovered) aspects of m6A modification and mark the beginning of the era of RNA epigenetics in cancer therapy.
Availability of data and materials
Not applicable.
Abbreviations
- ALKBH5:
-
α-Ketoglutarate-dependent dioxygenase alk B homolog
- BCSC:
-
Breast cancer stem cell
- CBLL1:
-
Cbl proto-oncogene like 1
- CLIP:
-
RNA-antibody photo-crosslinking and immunoprecipitation
- eIF3:
-
Eukaryotic translation initiation factor 3
- EMT:
-
Epithelial–mesenchymal transition
- ER:
-
Estrogen receptor
- f6A:
-
N6-Formyladenosine
- FMRP:
-
Fragile-X mental retardation protein
- FTO:
-
Fat mass and obesity-associated protein
- HER2:
-
Human epidermal growth factor receptor 2
- hm6A:
-
N6-Hydroxymethyladenosine
- hnRNP:
-
Heterogeneous nuclear ribonucleoprotein
- IGF2BP:
-
Insulin-like growth factor 2 mRNA binding protein
- m6A:
-
N6-Methyladenosine
- m6A-IP:
-
M6A-antibody immunoprecipitation
- MeRIP-seq:
-
Methylated RNA immunoprecipitation sequencing
- METTL:
-
Methyltransferase like
- MTC:
-
Methyltransferase complex
- OS:
-
Overall survival
- PR:
-
Progesterone receptor
- PROTAC:
-
Proteolysis targeting chimera
- RBM:
-
RNA-binding motif
- TNBC:
-
Triple-negative breast cancer
- UTR:
-
Untranslated region
- VIRMA:
-
Vir-like m6A methyltransferase associated
- WTAP:
-
Wilms tumor 1-associated protein
- YTHDC:
-
YTH domain-containing
- ZC3H13:
-
Zinc finger CCCH-type containing 13
- ZCCHC4:
-
Zinc finger CCHC-type containing 4
References
Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast cancer. Nat Rev Dis Primers. 2019;5(1):66.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Prat A, Parker JS, Fan C, Perou CM. PAM50 assay and the three-gene model for identifying the major and clinically relevant molecular subtypes of breast cancer. Breast Cancer Res Treat. 2012;135(1):301–6.
Pedrosa R, Mustafa DA, Soffietti R, Kros JM. Breast cancer brain metastasis: molecular mechanisms and directions for treatment. Neuro Oncol. 2018;20(11):1439–49.
Nagini S. Breast cancer: current molecular therapeutic targets and new players. Anticancer Agents Med Chem. 2017;17(2):152–63.
Chen Y, Lin Y, Shu Y, He J, Gao W. Interaction between N(6)-methyladenosine (m(6)A) modification and noncoding RNAs in cancer. Mol Cancer. 2020;19(1):94.
Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13(11):674–90.
Loibl S, Gianni L. HER2-positive breast cancer. Lancet. 2017;389(10087):2415–29.
Zhang Y, Hui ZG, Zhang JH, Yu ZH, Liu XF, Jin J, et al. Survey on the use of radiotherapy to treat early breast cancer following breast-conserving surgery in China. Tumori. 2014;100(5):512–7.
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA. 1974;71(10):3971–5.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149(7):1635–46.
Huang H, Weng H, Chen J. m(6)A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell. 2020;37(3):270–88.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-Methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–99.
Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-Methyladenosine marks primary microRNAs for processing. Nature. 2015;519(7544):482–5.
Haussmann IU, Bodi Z, Sanchez-Moran E, Mongan NP, Archer N, Fray RG, et al. m(6)A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. 2016;540(7632):301–4.
Helm M, Motorin Y. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet. 2017;18(5):275–91.
Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561(7724):556–60.
Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A, et al. A majority of m6A residues are in the last exons, allowing the potential for 3’ UTR regulation. Genes Dev. 2015;29(19):2037–53.
Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vagbo CB, Geula S, et al. m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017;31(10):990–1006.
Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. 2015;347(6225):1002–6.
Yoon KJ, Ringeling FR, Vissers C, Jacob F, Pokrass M, Jimenez-Cyrus D, et al. Temporal control of mammalian cortical neurogenesis by m(6)A methylation. Cell. 2017;171(4):877.e17-889.e17.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5’ UTR m(6)A promotes cap-independent translation. Cell. 2015;163(4):999–1010.
Li Y, Xiao J, Bai J, Tian Y, Qu Y, Chen X, et al. Molecular characterization and clinical relevance of m(6)A regulators across 33 cancer types. Mol Cancer. 2019;18(1):137.
Liu L, Liu X, Dong Z, Li J, Yu Y, Chen X, et al. N6-Methyladenosine-related genomic targets are altered in breast cancer tissue and associated with poor survival. J Cancer. 2019;10(22):5447–59.
Zhao Y, Shi Y, Shen H, Xie W. m(6)A-binding proteins: the emerging crucial performers in epigenetics. J Hematol Oncol. 2020;13(1):35.
Wu R, Jiang D, Wang Y, Wang X. N (6)-methyladenosine (m(6)A) methylation in mRNA with A dynamic and reversible epigenetic modification. Mol Biotechnol. 2016;58(7):450–9.
Zhu ZM, Huo FC, Pei DS. Function and evolution of RNA N6-methyladenosine modification. Int J Biol Sci. 2020;16(11):1929–40.
Shi H, Wei J, He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell. 2019;74(4):640–50.
Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. 2016;63(2):306–17.
Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural basis of N(6)-adenosine methylation by the METTL3–METTL14 complex. Nature. 2016;534(7608):575–8.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–89.
Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y, et al. S-Adenosylmethionine synthesis is regulated by selective N(6)-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep. 2017;21(12):3354–63.
Leismann J, Spagnuolo M, Pradhan M, Wacheul L, Vu MA, Musheev M, et al. The 18S ribosomal RNA m(6) A methyltransferase Mettl5 is required for normal walking behavior in Drosophila. EMBO Rep. 2020;21(7):e49443.
Ren W, Lu J, Huang M, Gao L, Li D, Wang GG, et al. Structure and regulation of ZCCHC4 in m(6)A-methylation of 28S rRNA. Nat Commun. 2019;10(1):5042.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20(10):608–24.
Fu Y, Jia G, Pang X, Wang RN, Wang X, Li CJ, et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat Commun. 2013;4:1798.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, et al. Reversible methylation of m(6)A(m) in the 5’ cap controls mRNA stability. Nature. 2017;541(7637):371–5.
Mauer J, Sindelar M, Despic V, Guez T, Hawley BR, Vasseur JJ, et al. FTO controls reversible m(6)Am RNA methylation during snRNA biogenesis. Nat Chem Biol. 2019;15(4):340–7.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29.
Tang C, Klukovich R, Peng H, Wang Z, Yu T, Zhang Y, et al. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3’-UTR mRNAs in male germ cells. Proc Natl Acad Sci USA. 2018;115(2):E325–33.
Meyer KD, Jaffrey SR. Rethinking m(6)A readers, writers, and erasers. Annu Rev Cell Dev Biol. 2017;33:319–42.
Theler D, Dominguez C, Blatter M, Boudet J, Allain FH. Solution structure of the YTH domain in complex with N6-methyladenosine RNA: a reader of methylated RNA. Nucleic Acids Res. 2014;42(22):13911–9.
Liao S, Sun H, Xu C. YTH domain: a family of N(6)-methyladenosine (m(6)A) readers. Genom Proteom Bioinform. 2018;16(2):99–107.
Hartmann AM, Nayler O, Schwaiger FW, Obermeier A, Stamm S. The interaction and colocalization of Sam68 with the splicing-associated factor YT521-B in nuclear dots is regulated by the Src family kinase p59(fyn). Mol Biol Cell. 1999;10(11):3909–26.
Wojtas MN, Pandey RR, Mendel M, Homolka D, Sachidanandam R, Pillai RS. Regulation of m(6)A transcripts by the 3’→5’ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell. 2017;68(2):374-87.e12.
Patil DP, Pickering BF, Jaffrey SR. Reading m(6)A in the transcriptome: m(6)A-binding proteins. Trends Cell Biol. 2018;28(2):113–27.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-Methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518(7540):560–4.
Wu B, Su S, Patil DP, Liu H, Gan J, Jaffrey SR, et al. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat Commun. 2018;9(1):420.
Zhang F, Kang Y, Wang M, Li Y, Xu T, Yang W, et al. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum Mol Genet. 2018;27(22):3936–50.
Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12(8):767–72.
Haberman N, Huppertz I, Attig J, König J, Wang Z, Hauer C, et al. Insights into the design and interpretation of iCLIP experiments. Genome Biol. 2017;18(1):7.
Garcia-Campos MA, Edelheit S, Toth U, Safra M, Shachar R, Viukov S, et al. Deciphering the “m(6)A code” via antibody-independent quantitative profiling. Cell. 2019;178(3):731-47.e16.
Shi J, Zhang P, Liu L, Min X, Xiao Y. Weighted gene coexpression network analysis identifies a new biomarker of CENPF for prediction disease prognosis and progression in nonmuscle invasive bladder cancer. Mol Genet Genom Med. 2019;7(11):e982.
Meyer KD. DART-seq: an antibody-free method for global m(6)A detection. Nat Methods. 2019;16(12):1275–80.
Capitanchik C, Toolan-Kerr P, Luscombe NM, Ule J. How do you identify m(6) a methylation in transcriptomes at high resolution? A comparison of recent datasets. Front Genet. 2020;11:398.
Liu H, Begik O, Lucas MC, Ramirez JM, Mason CE, Wiener D, et al. Accurate detection of m(6)A RNA modifications in native RNA sequences. Nat Commun. 2019;10(1):4079.
Gordon K, Clouaire T, Bao XX, Kemp SE, Xenophontos M, de Las Heras JI, et al. Immortality, but not oncogenic transformation, of primary human cells leads to epigenetic reprogramming of DNA methylation and gene expression. Nucleic Acids Res. 2014;42(6):3529–41.
Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–9.
Wang H, Xu B, Shi J. N6-Methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene. 2020;722:144076.
Fry NJ, Law BA, Ilkayeva OR, Carraway KR, Holley CL, Mansfield KD. N(6)-Methyladenosine contributes to cellular phenotype in a genetically-defined model of breast cancer progression. Oncotarget. 2018;9(58):31231–43.
Sun T, Wu Z, Wang X, Wang Y, Hu X, Qin W, et al. LNC942 promoting METTL14-mediated m(6)A methylation in breast cancer cell proliferation and progression. Oncogene. 2020;39(31):5358–72.
Wu L, Wu D, Ning J, Liu W, Zhang D. Changes of N6-methyladenosine modulators promote breast cancer progression. BMC Cancer. 2019;19(1):326.
Qian JY, Gao J, Sun X, Cao MD, Shi L, Xia TS, et al. KIAA1429 acts as an oncogenic factor in breast cancer by regulating CDK1 in an N6-methyladenosine-independent manner. Oncogene. 2019;38(33):6123–41.
Lewinska A, Adamczyk-Grochala J, Deregowska A, Wnuk M. Sulforaphane-induced cell cycle arrest and senescence are accompanied by DNA hypomethylation and changes in microRNA profile in breast cancer cells. Theranostics. 2017;7(14):3461–77.
Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18(1):46.
Akbari ME, Gholamalizadeh M, Doaei S, Mirsafa F. FTO gene affects obesity and breast cancer through similar mechanisms: a new insight into the molecular therapeutic targets. Nutr Cancer. 2018;70(1):30–6.
Gholamalizadeh M, Jarrahi AM, Akbari ME, Bourbour F, Mokhtari Z, Salahshoornezhad S, et al. Association between FTO gene polymorphisms and breast cancer: the role of estrogen. Expert Rev Endocrinol Metab. 2020;15(2):115–21.
Singh B, Kinne HE, Milligan RD, Washburn LJ, Olsen M, Lucci A. Important role of FTO in the survival of rare panresistant triple-negative inflammatory breast cancer cells facing a severe metabolic challenge. PLoS ONE. 2016;11(7):e0159072.
Han W, Zhang C, Shi CT, Gao XJ, Zhou MH, Shao QX, et al. Roles of eIF3m in the tumorigenesis of triple negative breast cancer. Cancer Cell Int. 2020;20:141.
Wu Y, Zhao W, Liu Y, Tan X, Li X, Zou Q, et al. Function of HNRNPC in breast cancer cells by controlling the dsRNA-induced interferon response. Embo J. 2018;37(23):e99017.
Hu Y, Sun Z, Deng J, Hu B, Yan W, Wei H, et al. Splicing factor hnRNPA2B1 contributes to tumorigenic potential of breast cancer cells through STAT3 and ERK1/2 signaling pathway. Tumour Biol. 2017;39(3):1010428317694318.
Santa-Maria CA, Gradishar WJ. Changing treatment paradigms in metastatic breast cancer: lessons learned. JAMA Oncol. 2015;1(4):528–34 (quiz 49).
Tang Y, He Y, Zhang P, Wang J, Fan C, Yang L, et al. LncRNAs regulate the cytoskeleton and related Rho/ROCK signaling in cancer metastasis. Mol Cancer. 2018;17(1):77.
Yi D, Wang R, Shi X, Xu L, Yilihamu Y, Sang J. METTL14 promotes the migration and invasion of breast cancer cells by modulating N6-methyladenosine and hsa-miR-146a-5p expression. Oncol Rep. 2020;43(5):1375–86.
Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687–717.
Saha T, Makar S, Swetha R, Gutti G, Singh SK. Estrogen signaling: an emanating therapeutic target for breast cancer treatment. Eur J Med Chem. 2019;177:116–43.
Gong EY, Park E, Lee K. Hakai acts as a coregulator of estrogen receptor alpha in breast cancer cells. Cancer Sci. 2010;101(9):2019–25.
Yousefnia S, Seyed Forootan F, Seyed Forootan S, Nasr Esfahani MH, Gure AO, Ghaedi K. Mechanistic pathways of malignancy in breast cancer stem cells. Front Oncol. 2020;10:452.
Gu HF, Mao XY, Du M. Prevention of breast cancer by dietary polyphenols-role of cancer stem cells. Crit Rev Food Sci Nutr. 2020;60(5):810–25.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA. 2016;113(14):E2047–56.
Zhang C, Zhi WI, Lu H, Samanta D, Chen I, Gabrielson E, et al. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016;7(40):64527–42.
Ma F, Liu X, Zhou S, Li W, Liu C, Chadwick M, et al. Long non-coding RNA FGF13-AS1 inhibits glycolysis and stemness properties of breast cancer cells through FGF13-AS1/IGF2BPs/Myc feedback loop. Cancer Lett. 2019;450:63–75.
Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2020;21(6):341–52.
Liu Y, Li H, Liu F, Gao LB, Han R, Chen C, et al. Heterogeneous nuclear ribonucleoprotein A2/B1 is a negative regulator of human breast cancer metastasis by maintaining the balance of multiple genes and pathways. EBioMedicine. 2020;51:102583.
Zheng Q, Liu H, Ye J, Zhang H, Jia Z, Cao J. Nuclear distribution of eIF3g and its interacting nuclear proteins in breast cancer cells. Mol Med Rep. 2016;13(4):2973–80.
Waks AG, Winer EP. Breast cancer treatment: a review. JAMA. 2019;321(3):288–300.
Tan A, Dang Y, Chen G, Mo Z. Overexpression of the fat mass and obesity associated gene (FTO) in breast cancer and its clinical implications. Int J Clin Exp Pathol. 2015;8(10):13405–10.
Zeng X, Ban Z, Cao J, Zhang W, Chu T, Lei D, et al. Association of FTO mutations with risk and survival of breast cancer in a Chinese population. Dis Mark. 2015;2015:101032.
Jiralerspong S, Goodwin PJ. Obesity and breast cancer prognosis: evidence, challenges, and opportunities. J Clin Oncol. 2016;34(35):4203–16.
Chen J, Du B. Novel positioning from obesity to cancer: FTO, an m(6)A RNA demethylase, regulates tumour progression. J Cancer Res Clin Oncol. 2019;145(1):19–29.
Slepicka PF, Cyrill SL, Dos Santos CO. Pregnancy and breast cancer: pathways to understand risk and prevention. Trends Mol Med. 2019;25(10):866–81.
Peri S, de Cicco RL, Santucci-Pereira J, Slifker M, Ross EA, Russo IH, et al. Defining the genomic signature of the parous breast. BMC Med Genom. 2012;5:46.
Han SH, Choe J. Diverse molecular functions of m(6)A mRNA modification in cancer. Exp Mol Med. 2020;52(5):738–49.
Li F, Yi Y, Miao Y, Long W, Long T, Chen S, et al. N(6)-Methyladenosine modulates nonsense-mediated mRNA decay in human glioblastoma. Cancer Res. 2019;79(22):5785–98.
Zuo X, Chen Z, Gao W, Zhang Y, Wang J, Wang J, et al. M6A-mediated upregulation of LINC00958 increases lipogenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. J Hematol Oncol. 2020;13(1):5.
Hu BB, Wang XY, Gu XY, Zou C, Gao ZJ, Zhang H, et al. N(6)-Methyladenosine (m(6)A) RNA modification in gastrointestinal tract cancers: roles, mechanisms, and applications. Mol Cancer. 2019;18(1):178.
Mojaver M, Mokarian F, Kazemi M, Salehi M. Specific TaqMan allelic discrimination assay for rs1477196 and rs9939609 single nucleotide polymorphisms of FTO gene demonstrated that there is no association between these SNPs and risk of breast cancer in Iranian women. Adv Biomed Res. 2015;4:136.
An S, Huang W, Huang X, Cun Y, Cheng W, Sun X, et al. Integrative network analysis identifies cell-specific trans regulators of m6A. Nucleic Acids Res. 2020;48(4):1715–29.
Paiva SL, Crews CM. Targeted protein degradation: elements of PROTAC design. Curr Opin Chem Biol. 2019;50:111–9.
Szostakowska M, Trebinska-Stryjewska A, Grzybowska EA, Fabisiewicz A. Resistance to endocrine therapy in breast cancer: molecular mechanisms and future goals. Breast Cancer Res Treat. 2019;173(3):489–97.
Echeverria GV, Ge Z, Seth S, Zhang X, Jeter-Jones S, Zhou X, et al. Resistance to neoadjuvant chemotherapy in triple-negative breast cancer mediated by a reversible drug-tolerant state. Sci Transl Med. 2019;11(488):eaav0936.
Taketo K, Konno M, Asai A, Koseki J, Toratani M, Satoh T, et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J Oncol. 2018;52(2):621–9.
Meng Q, Wang S, Zhou S, Liu H, Ma X, Zhou X, et al. Dissecting the m(6)A methylation affection on afatinib resistance in non-small cell lung cancer. Pharmacogenom J. 2020;20(2):227–34.
Klinge CM, Piell KM, Tooley CS, Rouchka EC. HNRNPA2/B1 is upregulated in endocrine-resistant LCC9 breast cancer cells and alters the miRNA transcriptome when overexpressed in MCF-7 cells. Sci Rep. 2019;9(1):9430.
Adams S, Gatti-Mays ME, Kalinsky K, Korde LA, Sharon E, Amiri-Kordestani L, et al. Current landscape of immunotherapy in breast cancer: a review. JAMA Oncol. 2019;5(8):1205–14.
Esteva FJ, Hubbard-Lucey VM, Tang J, Pusztai L. Immunotherapy and targeted therapy combinations in metastatic breast cancer. Lancet Oncol. 2019;20(3):e175–86.
Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10(1):2782.
Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature. 2019;566(7743):270–4.
Acknowledgements
Not applicable.
Funding
This work was supported by the Nature Science Foundation of Zhejiang Province (LQ19H060002 and LQ19H160041) and the Medical and Health Science and Technology Project of Zhejiang Province (2018KY089 and 2020KY143).
Author information
Authors and Affiliations
Contributions
YW, YZ and SZ conceived and designed the research; YW, YZ, and YD searched the literature and analyzed the data; YW and YZ wrote the manuscript and created the figures; YD, YH and MZ reviewed and made significant revisions to the manuscript. YW and YZ contributed equally to this work as the first authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Y., Zhang, Y., Du, Y. et al. Emerging roles of N6-methyladenosine (m6A) modification in breast cancer. Cell Biosci 10, 136 (2020). https://doi.org/10.1186/s13578-020-00502-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-020-00502-3