
Abstract
Cellular senescence is a major hurdle for primary cell-based tissue engineering and regenerative medicine. Telomere erosion, oxidative stress, the expression of oncogenes and the loss of tumor suppressor genes all may account for the cellular senescence process with the involvement of various signaling pathways. To establish immortalized cell lines for research and clinical use, strategies have been applied including internal genomic or external matrix microenvironment modification. Considering the potential risks of malignant transformation and tumorigenesis of genetic manipulation, environmental modification methods, especially the decellularized cell-deposited extracellular matrix (dECM)-based preconditioning strategy, appear to be promising for tissue engineering-aimed cell immortalization. Due to few review articles focusing on this topic, this review provides a summary of cell senescence and immortalization and discusses advantages and limitations of tissue engineering and regeneration with the use of immortalized cells as well as a potential rejuvenation strategy through combination with the dECM approach.
Similar content being viewed by others

Background
Tissue and organ failure is a prominent health issue that cannot be ignored. Surgical intervention, organ transplantation, artificial substitutes and mechanical devices are methods to address this issue but all have undesirable short- and long-term consequences [1]. Tissue engineering is an attractive method that enables fabrication of functional tissue for tissue regeneration as well as the establishment of physiological and pathological models for mechanistic studies [2]. This technique can harness the intrinsic regenerative potential of primary cells and expand them in a controlled environment before reintroduction into the patient’s body. These natural, synthetic or semisynthetic tissue and organ mimics are expected to function normally in a tissue-specific pattern as required [1, 3]. However, primary cells derived from non-cancerous tissues have a finite lifespan and decreased proliferation ability when cultured in vitro. After a limited number of divisions, cells enter a viable state of permanent quiescence, termed cellular senescence [4]. Cellular senescence, regulated by both intrinsic and extrinsic factors, is characterized as two key phenotypes, a stable proliferation arrest and altered secretory pathway, the senescence-associated secretory phenotype (SASP) [5].
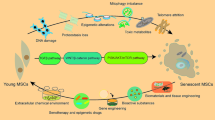
In order to acquire an abundant number of cells for functional tissue engineering, cellular senescence is the major obstacle that needs to be overcome. Numerous attempts have been made in past decades to deal with cellular senescence in order to achieve successful immortalization of primary cells. To establish immortalized cell lines for research and clinical use, strategies have been applied including internal genomic or external matrix microenvironment modification. Considering the potential risks of malignant transformation and tumorigenesis of genetic manipulation, environmental modification methods, especially the decellularized cell-deposited extracellular matrix (dECM)-based preconditioning strategy, appear to be promising for tissue engineering-aimed cell immortalization. Due to few review articles focusing on this topic, this review provides a summary of cell senescence and immortalization and discusses advantages and limitations of tissue engineering and regeneration with the use of immortalized cells as well as a potential rejuvenation strategy when combined with the dECM approach (Fig. 1).

A schematic diagram of immortalization strategy combined with the decellularized cell-deposited extracellular matrix approach to overcome cell senescence and promote tissue regeneration
Cellular senescence
Cellular senescence is a process that imposes irreversible proliferative arrest on cells in response to internal and external environmental changes. Various stressors, including progressive telomere erosion, oxidative stress, the expression of oncogenes and the loss of tumor suppressors, contribute to the occurrence of cellular senescence.
Replicative senescence
As vital structures that cap and protect the ends of linear chromosomes [6], shortening of telomeres happens at every fission which eventually causes cells to reach their “Hayflick limit” which halts growth after approximately 60 population doublings [4, 7]. When telomeres are too short to function normally for capping, replicative senescence (M1 stage, a cellular growth arrest) occurs [8,9,10]. The critically short telomeres are detected by cells as double-strand breaks and trigger a deoxyribonucleic acid (DNA) damage response (DDR) that consists of a series of signaling events centered on two anti-proliferative mechanisms, the p53/p21 and p16/tumor suppressor retinoblastoma protein (Rb) pathways. This cessation allows cells to repair the DNA damage, but if the damage continues to exceed a certain limit, apoptosis or senescence may occur [11]. Regulated by its upstream partner p16, one of the cyclin-dependent kinase inhibitors (CKIs), Rb controls cell cycle progression from G1 into S phase by binding to and suppressing the activity of E2F transcription factor 1 (E2F1) [10, 12, 13].
Many researchers have verified the importance of both pathways in directing senescence as the suppression of either p53 or Rb alone failed to achieve cell immortalization [14,15,16,17]. However, there were still reports showing that, in human mammary epithelial cells and mesenchymal stem cells (MSCs), inactivation of p16 alone allows human cells to avoid senescence [18, 19]. Meanwhile, in human diploid fibroblasts, the p53 mutant alone is able to suppress cellular senescence [20]. These findings raise the possibility that these two pathways may function differently among different cell strains.
However, abrogation of the p53/p21 and p16/Rb pathways will only lead to a “pre-immortal” state instead of an “immortal” status for cells. Terminal telomere shortening still exists and will eventually lead to the M2 stage, characterized as massive cell death [10, 15, 21]. In most cases, the stabilization of telomeres is achieved through the introduction of telomerase, an enzyme that synthesizes telomeric repeats and adds them to the ends of chromosomes for the compensation of inevitable loss with each round of DNA replication [22].
Premature senescence
Senescence also happens in conditions that are not dependent on telomere erosion or dysfunction. This process is often referred to as “premature” since it can arrest growth prior to reaching the “Hayflick limit” [23]. Various conditions have been identified that may result in premature cellular senescence.
Stress-induced senescence
During a long-term in vitro cell expansion, laboratory culture conditions, generally defined as a lack of surrounding cell types and support from extracellular matrix (ECM), abnormal growth factors and oxygen (O2) level, expose the cell to excessive oxidative stress and induce oxidant production [24,25,26,27]. The excessive levels of reactive oxygen species (ROS), including hydrogen peroxide (H2O2), hydroxyl radical (OH−) and superoxide anion (O2−), are detectable during long-term culturing of MSCs, accounting for stress-induced senescence [24,25,26]. H2O2 could directly affect cellular DNA, trigger DDR and subsequent p16/Rb and p53 pathways, leading to cell cycle arrest [28,29,30,31,32].
Oncogene-induced senescence
There is accumulating evidence showing both in vitro [33,34,35] and in vivo [36, 37] oncogene activation, including Ras, Raf, BRAF (human gene that encodes a protein called B-Raf) and E2F1, can cause an irreversible cell growth arrest, termed oncogene-induced senescence. In normal primary cells, Ras activation leads to compulsory replication, triggering DDR and the subsequent senescence-based pathways [33, 38, 39]. Raf encodes proteins that function as a downstream effector of the Ras family and activate the extracellular signal-regulated kinase (MAPK) kinase (MEK) in cascade, which in turn, activates extracellular signal regulated kinase 1/2 (ERK1/2) [40]. Interestingly, Raf itself is able to elicit senescence in IMR-90 cells [34]. The p16/Rb and p53 pathways are crucial mediators of oncogene-induced senescence; however, the p16/Rb pathway in oncogene-induced senescence acts differently than in replicative senescence [33, 41, 42]. The BRAF gene, a downstream effector of Ras, is an intracellular effector of the MAPK signaling cascade that facilitates transmembrane signal transduction [43]. In primary cells, the expression of BRAFV600E is known to induce transient stimulation of proliferation and subsequently trigger cellular senescence as demonstrated in normal cells including melanocytes [44], fibroblasts [45] and stem cells [46]. E2F1 is the founding member of the E2F family, a regulatory protein that drives cell cycle progression through interaction with Rb [47]. When a cell prepares to enter the S phase of the cell cycle, E2F1 is released from the Rb-E2F1 complex, activating the downstream target genes regulating normal entry into S phase [48]. Interestingly, in normal human fibroblasts, E2F1 and its target gene p14 (ARF) are responsible for the induction of cellular senescence [35]. Meanwhile, E2F1 knock-out mouse embryonic fibroblasts demonstrated attenuated senescence and ROS levels [49].
Tumor suppressor loss-induced senescence
Tumor suppressors are the counterpart of oncogenes, and their loss can elicit cellular senescence. Depletion of NF1 (Neurofibromatosis 1), a tumor suppressor gene, induces senescence in human fibroblasts [50]. Similarly, loss of BTG3 (B-cell translocation gene 3), a member of the anti-proliferative BTG gene family and a downstream target of p53, triggers cellular senescence as well [51]. Inactivation of VHL (von Hippel-Lindau tumor suppressor) induces an efficient senescence in mouse fibroblasts and primary renal epithelial cells under atmospheric conditions (21% O2); however, loss of VHL only causes a decreased cell proliferation instead of cell arrest in human renal epithelial cells [52, 53]. Similarly, acute loss of tumor suppressor gene PTEN (phosphatase and tensin homolog) induces growth arrest through the p53-dependent cellular senescence pathway in mouse prostate both in vitro and in vivo whereas, in systemic lupus erythematosus patients, the complete loss is significantly related to advanced cancer and poor outcomes [54,55,56]. These findings raise the possibility that tumor suppressors may function differently according to different species and cell types.
Signaling pathways involved in cellular senescence
Despite the abovementioned p53/p21 and p16/Rb pathways, other signaling pathways are also involved in cellular senescence, including, but not limited to, transforming growth factor β (TGFβ)/bone morphogenetic protein (BMP), Wingless/Int (Wnt)/β-catenin, MAPK, phosphatidylinostitide 3 kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR), Hippo, NOTCH, fibroblast growth factor (FGF) and insulin-like growth factor (IGF) and hypoxia inducible factor (HIF) (Fig. 2).

Signaling pathways mediating the cellular senescence process. In response to telomere erosion, ROS production, the expression of oncogenes and the loss of tumor suppressors, various signaling pathways including TGFβ, BMP, Wnt, MAPK, FGF, IGF, HIF and Hippo pathways are all actively involved in cell cycle regulation, which eventually influences the cellular senescence process of primary cells
TGFβ/BMP signaling pathways
TGFβ is a classic regulator for chondrogenic differentiation but its role in cell expansion remains controversial [57, 58]. TGFβ activation is positively involved in the induction of cellular senescence of all kinds of species [59,60,61]. In human breast cancer cells, TGFβ negatively mediates telomerase activity through its downstream effector, Smad3 [62, 63]. For stress-induced senescence, TGFβ contributes to ROS production and activation of DDR during the senescence of human fibroblasts and bone marrow-derived MSCs (BMSCs) [64, 65]. The kinase ataxia-telangiectasia mutated (ATM) is a key player in nuclear DDR [66]. Meanwhile, TGFβ is required for oncogene-induced senescence that is independent of the p16/Rb and p53 pathways; attenuation of TGFβ inhibits premature senescence in human mammary epithelial cells [67, 68].
BMPs are secreted signal factors belonging to the TGFβ superfamily and are involved in embryonic development and cellular processes [69]. Similar to the function of TGFβ, BMP receptor II/Smad3 contributes to telomerase inhibition and telomere shortening in human breast cancer cells, leading to replicative senescence [70]. Similar results were observed in primary cells as the BMP signaling axis plays an important role in oncogene-induced senescence of mouse fibroblasts [71].
Wnt/β-catenin pathway
Wnts are highly conservative proteins that participate in embryonic development and homeostatic mechanisms in adult tissues [72]. Wnt signals appear to be an important regulator of both premature senescence and replicative senescence. On one hand, the Wnt/β-catenin signaling pathway interacts with the p53/p21 pathway for ROS production to induce MSC senescence [73,74,75,76]. On the other hand, Wnt3a/β-catenin also plays a critical role in hedging replicative senescence of MSCs, probably through regulation of a telomerase subunit—telomerase reverse transcriptase (TERT) [72, 77]. Meanwhile, Wnt/β-catenin signaling enhances rat nucleus pulposus cell senescence as well as induces the expression of TGFβ, another strong promoter of cellular senescence [78].
MAPK pathway
The MAPK signaling cascade, mainly including ERK, c-Jun N-terminal kinase (JNK) and p38, regulates several physiological and pathological processes [40]. p38 is well-recognized to be involved in premature cellular senescence [79, 80]. The major role of the p38 pathway in oncogene-induced senescence is induced by the oncogene Ras or its downstream effector, Raf-1 [81, 82]. Ras provokes premature senescence through activation of the MEK/ERK pathway, followed by p38 activation [81]. Shin and colleagues found that ERK2 is responsible for Ras-induced senescence in mouse embryonic fibroblasts [83]. Despite premature senescence, ERK is also actively involved in replicative senescence and suppression of ERK signaling rescues cardiac progenitor cells from replicative senescence when expanded in vitro [84]. The JNK signal was reported to be active in responding to a wide range of DNA-damaging agents from both endogenous and exogenous causes and JNK phosphorylation is involved in senescence-associated matrix metalloproteinase-1 production in response to ROS in IMPR-90 cells [85,86,87,88,89]. However, the senescence-promoting role of JNK was challenged as it was also revealed to antagonize p38-induced p16 activation [90]. Moreover, JNK acts as a negative regulator of p53 tumor suppressor to suppress p53-dependent senescence in mouse embryonic fibroblasts [91]. An increase of intracellular ROS levels can suppress the growth of cancer cells and induce cellular apoptosis by mediating MAPK signaling components [92].
PI3K/AKT/mTOR pathways
PI3Ks and their downstream mediators AKT and mTOR constitute the core component of the PI3K/AKT/mTOR signaling pathway which is precisely controlled under normal physiological conditions and is a frequently hyperactivated pathway in cancer [93]. Similar to the MEK-ERK pathway, PI3K is one of the main downstream effectors of Ras dependent signaling and its activation plays dual roles in cell cycle regulation as it can promote cell cycle progression as well as cause cell cycle arrest [94]. Recent studies reveal the involvement of PI3K/AKT/mTOR in the regulation of replicative senescence in human vascular smooth muscle cells [95]. Moreover, a constitutively active, myristoylated form of AKT leads to oncogene-induced senescence in primary cultured human endothelial cells and murine fibroblasts; the loss of PTEN triggers senescence through activation of the PI3K/AKT pathway in mouse prostate [56, 96]. However, a recent report subverted the positive role of the PI3K/AKT/mTOR pathway in senescence-induction by introducing the fact that activation of this pathway abolished BRAFV600E-induced senescence in both primary human fibroblasts and primary human melanocytes [97]. Moreover, targets of the PI3K/AKT signaling pathway have been found to promote cell survival [98] and the activation of the PI3K/AKT pathway can be induced by TGFβ, leading to a pro-survival/anti-apoptotic effect in both human nasopharyngeal carcinoma cells [99] and mesenchymal cells/fibroblasts [100].
Hippo pathway
The Hippo pathway is a tumor suppressor pathway; dysregulation of this pathway can lead to uncontrolled cell proliferation and tumorigenesis [101]. Yes-associated protein 1 (YAP), a major downstream effector of the Hippo pathway, is phosphorylated and inactivated by the serine/threonine kinases large tumor suppressor 1 (LATS1) and LATS2 [102, 103]. YAP dephosphorylation is associated with the senescence of rat nucleus pulposus cells and overexpression of YAP in primary human keratinocytes blocks clonal evolution and induces cell immortalization [104, 105]. Coordination of the Hippo pathway and p53 occurs in response to various types of stress signals including replication and oncogenic Ras. LATS2 cooperates with p53 to induce p21 expression, resulting in cellular senescence [106]. LATS2 also plays an important role in the oncogenic H-Ras induced stress checkpoint in a p53-dependent pathway [107].
Notch pathway
The Notch pathway is an evolutionarily highly conserved signaling pathway that is associated with a variety of cellular processes including cell-fate determination, proliferation and death. In mammals, the Notch family has five ligands and four receptors [108, 109]. There is accumulating evidence that abnormal Notch signaling has been implicated in multiple facets of cancer biology, and Notch can behave as either an oncogene or a tumor suppressor depending on cell context [110, 111]. The oncogenic function of Notch has been demonstrated in several types of cancer including melanoma [112], breast cancer [113] and brain tumors [114]. Activated Notch 1 significantly enhances the rate of glycolysis, which prevents cellular senescence of human adipose-derived stromal cells (ADSCs) through HIF1 activation and p53 inactivation [115]. On the other hand, the Notch pathway is found to serve as a tumor suppressor in the progression of carcinoma including bladder cancer [116], medullary thyroid carcinoma [117] and pancreatic cancer [118]. Enforced Notch activation in human endothelial cells is associated with cellular senescence with the involvement of p16 [119]. Down-regulation of Notch 3 in human fibroblasts and mammary epithelial cells delays the onset of senescence and extends cell lifespan [120, 121].
FGF and IGF pathways
FGFs are well-recognized for their critical roles in embryonic development [122]. The mitogenic effect of FGF has been demonstrated by promoting proliferation while maintaining stemness of MSCs in vitro [123,124,125]. FGF2 treatment led to an early increase in telomere size in MSCs, probably due to its ability to increase TERT mRNA expression [126, 127]. FGF signals negatively regulated MSC senescence through interaction with PI3K/AKT/MDM2 (mouse double minute-2 homolog) in the mouse and through down-regulation of TGFβ expression in human MSCs [128, 129]. Surprisingly, FGF23 can also induce premature senescence in human MSCs from skeletal muscle via the p53/p21 oxidative-stress pathway [130].
IGFs are considered detrimental to cell survival due to their role in diminishing tissue resistance to oxidative stress and shortening lifespan [131, 132]. In mouse, rat and human primary vascular smooth muscle cells, IGFI induces cellular senescence dependent on the upregulation of p53 [133]. Additional evidence has revealed that IGF binding protein-5 is upregulated in the regulation of premature senescence of umbilical vein endothelial cells through a p53-controlled mechanism [134, 135]. These findings may be due to the mechanism whereby IGFI is capable of inducing telomere shortening [136]. However, opposite results were found in human annulus fibrosus cells as IGFI alleviates cellular senescence [137]. In this scenario, the regulatory roles of both FGFs and IGFs relating to cellular senescence are context-dependent.
HIF pathway
HIFs are composed of two different basic-helix-loop-helix-PAS transcription factors, HIF1α and HIF1β [138]. It has been proposed that a classic cellular response to hypoxia is cell cycle arrest at the G1/S interface through the regulation of p27 expression in which HIF1α is a major mediator [139, 140]. On the other hand, HIFs are involved in the promotion of cancer growth and the loss of HIFs induces the production of ROS and the activation of proteins p53 and p16 [141, 142]. HIF1α is involved in the suppression of senescence through regulation of p53 and p21 in human diploid fibroblasts [143]. When expanded in hypoxic conditions, human MSCs and the old human endothelial progenitor cells escape senescence through regulation of HIF1α-TWIST-p21 axis [144, 145]. Moreover, hypoxia led to PI3K/AKT pathway activation and elevated expression of Wnt coreceptor low-density lipoprotein receptor-related protein 5 (LRP5), contributing to the promotion of self-renewal and decreased cellular senescence of marrow-isolated adult multilineage inducible cells [146].
Immortalization of cells through genetic modification
To achieve cell lifespan extension, biotechnological methods are often used for direct manipulation of a cell’s genome. However, concerns still exist regarding genomic stability and tumorigenicity after genetic modification. For MSCs and progenitor cells, the potential loss of differentiation ability after genetic modification is a problem that cannot be overlooked.
Genetic modification
The introduction of viral oncogenes/oncoproteins and TERT are two typical methods for this type of genetic modification (Table 1).
Viral oncogenes/oncoproteins
Viral oncogenes that are able to inactivate both pRb and p53 can overcome M1 (a barrier in which normal cells senesce and cease replication) and significantly prolong cell lifespan. For several decades, simian virus 40 (SV40) early region genes have been commonly used for cell immortalization and cell line establishment [147, 148]. SV40 is limited to two proteins as the large T (LT) and small t antigen (ST). LT is mainly responsible for the SV40-extended lifespan based on its ability to interact with growth suppressors—pRb and p53. LT binds to the pRb-E2F complex via its pocket binding site including AA101–118 and the J domain that acts as a chaperone, leading to the dissociation of E2F from the LT-pRb complex [149]. Meanwhile, LT binds to p53 therefore suppressing the p53 pathway [150]. More interestingly, SV40 was reported to induce telomerase activity in primary human mesothelial cells, but not in primary fibroblasts [151].
Human papillomavirus (HPV) is a small, double-stranded DNA virus that infects mucosal and cutaneous epithelial tissue [152]. The high-risk strains including HPV-8, -16, -18 and -31 cause malignant progression of lesions, whereas the low-risk strains including HPV-6 and -11 cause benign warts and lesions [153]. The E6 and E7 proteins encoded by “high-risk” strains including HPV-16 and -18 are oncoproteins that have been shown to have transformation properties [154]. When used in immortalization, E6 causes telomerase activation as well as accelerating the degradation of p53 by the 26S proteasome, whereas E7 inactivates Rb by preventing the binding of pRb to the E2F transcription factor [155, 156].
Human T-lymphotropic virus type 1 (HTLV-1) is the etiologic agent of adult T cell leukemia. Although HTLV-2 is less pathogenic than HTLV-1, both of the HTLV-1 and -2 Tax proteins, p40tax (Tax1) and p37tax (Tax2), share the capacity to immortalize lymphocytes in vitro [157]. HTLV-2 protein Tax2 demonstrates much stronger efficacy than that of Tax1 in immortalizing human T cells [158].
The myc oncogene family consists of several different members including c-myc, N-myc, L-myc and B- myc. c-myc expression is restricted to proliferating cells while N-myc and L-myc expression is associated with cellular differentiation [159, 160]. Moreover, the oncogene myc fulfils many of the expectations for a gene involved in immortalization of primary cells alone [160, 161] or cooperates with oncoproteins [162] or TERT [163].
B cell-specific Moloney murine leukemia virus integration site 1 (BMI1) which was identified as a c-myc-cooperating oncogene, is a critical transcriptional repressor for maintenance of proper gene expression during development [164,165,166]. INK4a locus, which encodes p16 and p19Arf, is an important target of BMI1 and overexpression of BMI1 extends replicative lifespan of human fibroblasts, probably through suppressing the p16-mediated senescence pathway [167, 168].
TERT
Telomerase is composed of two core components: the small nuclear ribonucleic acid (RNA) human telomerase RNA, which serves as an internal template for the synthesis of telomeric repeats, and the protein TERT (or hTERT in humans), which serves as a catalytic subunit that synthesizes the new telomeric DNA from the RNA template [22]. In most human primary cells, telomerase is either absent or present at an insufficient level for telomere maintenance [169]. TERT is the determinant for the presence of active telomerase [170, 171]. The introduction of ectopic expression of TERT is necessary for telomere-dependent senescence as it is able to significantly extend the lifespan of a variety of cell types, but it alone is not sufficient to immortalize them [172,173,174]. Theoretically, the abrogation of the Rb and p53 pathways with oncogenes or at a minimum, low p16 expression, is indispensable for cell immortalization [175, 176]. However, there are still investigations showing that TERT bypassed Rb and p53 pathway-dependent barriers to immortalize cells alone [176,177,178,179,180].
Carcinogenic limitations and strategies
Despite the increasingly sophisticated strategies to immortalize human cells, there is still some debate over the risks upon integration of oncogenes into chromosomes. The primary safety concern with the use of a cell line is the transmission of an oncogenic factor to the host cells. Indeed, cells transduced with these oncogenes underwent additional changes including full carcinogenesis-associated changes (Table 2). The persistent infection by a subset of HPVs, especially HPV-16 and HPV-18, is etiologically linked to cervical cancer in women [181]. Deregulated overexpression of HPV E6 and E7 led to several alterations in cellular pathways and functions, which is associated with malignant transformation of cells and tumorigenesis [182]. In addition, HPV E6 oncoprotein can interact with hTERT to promote carcinogenesis in keratinocytes [183, 184].
SV40 or SV40 sequences were found in several types of human cancers located in bone, brain, chest, etc. [185,186,187,188]. Evidence has shown that SV40 can successfully transform cell lines in vitro and induce tumors in neonatal hamsters in vivo [189,190,191,192]. The injection of SV40-transformed cells into terminally ill human patients caused subcutaneous tumor nodules [193]. Moreover, SV40-transduced cells contained integrated SV40 DNA, which was integrated at random positions on the cellular chromosomes of host cells [194, 195], leading to the controversial question of whether the virus poses a threat for further in vivo use.
Although introduction of hTERT is associated with fewer phenotypic and karyotypic changes of cells compared with SV40, the tumorigenicity of hTERT-transfected human cells remains controversial as well [176]. Previous studies have claimed that the hTERT-transduced primary human fetal lung fibroblasts, ameloblastoma cells and bovine mammary epithelial cells showed no malignant transformation [180, 196, 197]. Similarly, the hTERT-immortalized human MSCs past 290 population doublings showed no sign of malignant transformation or tumorigenesis in vitro and in vivo as the cells maintain contact inhibition and a stable protein expression profile as well as no tumor-like activity in immune-deficient mice [198,199,200]. Meanwhile, after subcutaneous injection with HPV-16 E6/E7 immortalized BMSCs into Nonobese Diabetic/Severe Combined Immunodeficiency (NOD/SCID) mice for 3 days, no tumor mass was observed compared to those injected with Hela cells in which tumor mass was observable [201]. Even the introduction of TERT and SV40 or HPV-16 E6/E7 was sufficient to immortalize ovarian surface epithelial cells and dermal papilla cells but not enough for tumor formation [179, 202,203,204,205].
However, other groups argue about the increased potential for tumor development in TERT-immortalized cells. On monolayer cultures, human MSCs and fibroblasts avoid cell-to-cell contact inhibition, anchor to culture dishes and tend to proliferate limitlessly [206, 207]. In clinical practice, elevated hTERT expression is a diagnostic marker for tumor and the overexpression of hTERT is claimed to be associated with an advanced invasive stage of tumor progression and poor prognosis [208,209,210]. Moreover, there are still concerns about genetic instability after TERT transfection or transduction. Spontaneous changes in c-myc proto-oncogene expression and other genetic alterations have been observed during in vitro culture of hTERT-immortalized human cells [211, 212].
Despite safety concerns for the immortalized cells, there are still some cases that successfully applied these cells for in vivo organ and tissue restoration. For liver impairment, Guo et al. [213] found that SV40-immortalized marmoset hepatic progenitor cells (MHPCs) injected into the injured liver of fumarylacetoacetate hydrolase-deficient mice repopulated with hepatocyte-like cells and MHPCs were also implanted as cholangiocytes into bile ducts of 3.5-diethoxycarbonyl-1,4-dihydrocollodine-induced bile ductular injured mice. Meanwhile, SV40-immortalized human fetal liver cells differentiated into mature hepatocytes after being transplanted into liver injured mice [214]. For brain damage, hTERT-immortalized cord blood MSCs were injected into the traumatically injured brain of a rat model and proliferated efficiently at the injury site for 2 weeks and showed no tumor formation in SCID mice after a 6-month observation [215].
To avoid persistent oncogene expression, conditional immortalization technology was developed. Conditional immortalization includes inserting a reagent mediate, operator controllable gene to create a cell line that can be expanded in a consistent fashion when the transgene is active. When desired clinical quantities of cell material are achieved, the transgene can be deactivated by the operator and the cells will return to a normal, post-mitotic state. The conditional immortalization technology c-MycERTAM uses a combination of growth factor and 4-hydroxytamoxifen (4-HT) to activate the c-MycER transgene. In the absence of 4-HT, c-MycER is inactivated and the cells return to a normal phenotype [216]. Inactivation of SV40 LT was achieved using a temperature-sensitive mutant of the LT (SV40 tsA58) that is biologically active at permissive temperature (33.5 °C) but inactive at a non-permissive temperature (39 °C) [217]. Different vectors can have influence on the expression of transgenes. Unlike lentivirus, adenovirus does not integrate transgenes into the host genome and thereby can only provide a transient expression of the transgenes [218]. However, this kind of expression time is not controllable.
To acquire more accurate excision of oncogenes, site-specific recombination systems were developed. Cre/LoxP technology involves engineering a transgene flanked by LoxP sites. The transgene is activated until Cre recombinase is added. However, the cre-lox system is not 100% efficient and cells that have not deleted the transgene might require elimination [219, 220]. In addition, a Tet on/off system uses tetracycline responsive elements (TRE) that consist of a Tet operator and minimal promoter. The activation of the transgene and the subsequent cell division is related to tetracycline or doxycycline, which acts as a cue for activation (Tet-On) or inactivation (Tet-Off) [221, 222]. However, this technology still has the evident limitation termed “leakiness”, where the transgene continues to express at a low level even when the system is off [223]. Moreover, the transient activation of β-catenin was used to efficiently induce hTERT activation while silencing β-catenin suppresses the expression of hTERT [224]. Meanwhile, therapeutic strategies concerning transient activation of telomerase with small molecules, including the administration of 1% N-acetylcarnosine lubricant eye drops for prevention and treatment of cataracts, have been proven beneficial for dogs and other animals [225]. Huang et al. have identified that anthraquinone derivatives might be able to activate hTERT expression without causing genetic alterations in cells, whereas these cells fail to possess potent proliferative ability [226]. Interestingly, the introduction of some adenovirus derived genes, including the early 4 region (E4) of the adenoviral vector (AdE4), augments survival of human endothelial cells [227, 228]. However, investigations into AdE4 gene products were largely overshadowed by the fact that these proteins not only orchestrated many viral processes, but also overlapped with oncogenic transformation of primary cells [229, 230]. Although creation of conditionally immortalized cell lines has the potential for therapeutic application, complete silence of the transgene before introduction into the patient’s body is still a concern that needs to be addressed.
Potential loss of differentiation capacity
Differentiation capacity of MSCs and progenitor cells after immortalization is another concern that deserves more attention. A variety of reports has claimed that immortalization of progenitor cells will retain proliferative activity without compromising multipotent or specific differentiation potential of primary cells from species including human, mouse and porcine [231,232,233,234,235,236,237]. A similar phenomenon has been mentioned in human MSCs immortalized with SV40 [238, 239]. After serial transduction with hTERT, SV40 and H-Ras, human MSCs still retain their multilineage differentiation potential even during tumorigenesis [240, 241]. Moreover, Yang et al. [242] showed that hTERT-transduced human BMSCs seeded on porous polylactic glycolic acid (PLGA) scaffold have better osteogenic differentiation ability than primary human BMSCs seeded on scaffold. Similarly, human BMSCs immortalized with hTERT and HPV16 E6/E7 displayed greater differentiation potential far beyond the primary human BMSCs or even when human BMSCs expressed HPV-16 E6/E7 alone [243]. Interestingly, Okamoto et al. [244] reported that human BMSCs immortalized with hTERT and HPV-16 E6/E7 demonstrated significant clonal heterogeneity in differentiation potential. A similar phenomenon was found in mouse melanocyte progenitors that displayed distinct melanogenic differentiation potential [232]. More interestingly, there were opposite results as human primary dental pulp stem cells (DPSCs) are found to be approximately 60% more effective than hTERT-immortalized DPSCs in osteogenic differentiation [245]. For human placenta-derived MSCs immortalized with hTERT and BMI1, the differentiation potential was lost [246]. This discrepancy may partially be due to different immortalization strategies as the lost differentiation potential in ADSCs due to “SV40+hTERT” introduction can be preserved by “hTERT+BMI1” [247]. Moreover, cellular senescence counteracts the induction and reprogramming of induced pluripotent stem cells and senescence related INK4A/ARF and p53/p21 pathways are considered to be involved in these processes [248,249,250,251].
Preconditioning of cells through matrix microenvironment optimization
Although genetic manipulation is a popular strategy for functional tissue engineering, it has limited clinical benefit due to its inherent risks [189, 193, 194]. For human cells that are sensitive to external changes, matrix microenvironmental alterations may modify intercellular communication, leading to enhanced proliferation ability without carcinogenic mutation [252, 253].
Cells in the body reside in a niche, a dynamic and complex environment, where extracellular cues provided allow cells to survive and maintain their balance between quiescence, self-renewal and differentiation [254, 255]. ECM, a versatile component that plays a key role in the stem cell niche, interacts with the resident cells by modulating cell behavior through its physical, biochemical and biomechanical properties [256, 257]. There is an increasing number of reports indicating that dECM is a promising substrate to maintain the stemness of expanded cells by mimicking the in vivo niche [258, 259]. dECM was found to improve the expansion capacity of human BMSCs [260], human and porcine SDSCs [261, 262], human umbilical cord MSCs [263] and porcine adipose stem cells derived from the infrapatellar fat pad [264]. Interestingly, dECM deposited by SDSCs could also rejuvenate somatic cells such as porcine nucleus pulposus cells [265, 266] and replicatively senescent porcine chondrocytes [267]. These dECM-expanded cells were smaller in size compared to those grown on plastic flasks. These results are in accordance with the finding from Whitefield and coworkers in which, under time-lapse video microscopy, the smaller cells were observed to continue proliferation, while the larger cells became senescent and exited the cell proliferation cycle [268].
Furthermore, current data indicate that dECM deposited by MSCs yields human adult SDSCs [269, 270] and porcine adipose stem cells derived from the infrapatellar fat pad [264] with better chondrogenic potential in vitro and with better repair capacity for cartilage defects in vivo [271]. Interestingly, BMSCs, a tissue-specific stem cell for endochondral bone formation, could be greatly recharged toward chondrogenic differentiation by expansion on dECM deposited by nonchondrogenic human urine stem cells [272] or human BMSCs themselves [260]. More interestingly, a recent report showed that, despite dECMs deposited by BMSCs and ADSCs enhancing the proliferation ability of MSCs, they failed to yield expansion of cancer cells (HeLa, MCF7 and MDA-MB-231) in terms of inhibiting the expansion ability of these cancer cells [273]. This finding indicates that normal cell derived dECM is not favorable for the growth of cancer cells. Given the undesirable potential of carcinogenesis after genetic modification from cell immortalization, dECM tends to be a better alternative.
In addition to rescuing replicative and differentiation capacities, dECM could reduce intracellular generation of ROS in aged murine BMSCs [274] and in human BMSCs [260] and umbilical cord MSCs [263]. Meanwhile, dECM could enhance the anti-oxidative capacity of human adult SDSCs [269] and protect umbilical cord MSCs from oxidative stress-induced premature senescence [275] to finally achieve better chondrogenic differentiation. Moreover, dECM could repress osteoclastogenesis in bone marrow monocytes through the attenuation of intracellular ROS [276]. All the above mentioned studies confirmed the anti-senescence and anti-oxidative effect of dECM as a culture substrate.
Mechanical cues, including stiffness and elasticity from the surrounding matrix microenvironment, are important cellular inputs that sustain cell proliferation and oppose cell senescence. Integrin-based focal adhesions are the main adhesion complex dominating mechanosensing [277]. In our previous study, dECM-expanded human BMSCs demonstrated increased expression of integrin α2 and β5 [260], which are potentially involved in the process of cell proliferation [278, 279]. As a powerful regulator of cell proliferation and survival, YAP/YAZ act as mechanotransducer that is regulated by F-actin cytoskeleton [280, 281]. Interestingly, dECM expansion was found to induce sustained activation of ERK1/2 as well as phosphorylated cyclin D1 human BMSCs [260] but decreased phosphorylated ERK in human adult source SDSCs [261]. A similar phenomenon was found in phosphorylated p38 expression in human SDSCs [261] and human umbilical cord MSCs [263] after dECM preconditioning. These discrepancies might be explained by the dual role of ERK [282] and p38 [283] signals that play upon cell senescence. The expression of Wnt5a and Wnt11a were also found to be upregulated following dECM expansion [261].
Conclusions and perspective
Primary cells display a stable and long-term loss of proliferative capacity upon in vitro expansion despite continued viability and metabolic activity. This inability to proliferate is due to progressive shortening of telomeres during each replication which ultimately makes cells reach their “Hayflick limit”, termed telomere-dependent or replicative senescence. Meanwhile, there is another kind of senescence referred to as premature senescence since it can arrest cell growth long before reaching the “Hayflick limit”. One type is stress-induced senescence caused by the failure to simulate the in vivo supportive environment, which puts pressure on cell proliferation through the generation of ROS. In addition, both oncogenes and their counteracting tumor suppressors are proven to provoke premature senescence. A variety of signaling pathways are involved in all of these types of senescence, in which the p53/p21 and p16/Rb pathways are the two major signals involved. Oncogenes including SV40 and HPV-16 E6/E7 inhibit the p53/p21 and p16/Rb pathways, but are not able to immortalize primary cells unless followed by the introduction of TERT, which elongates telomeres, thereby abrogating the effect of the end replication problem. However, all of these genetic modification methods have the risk of virus introduction and potential oncogenesis, which must be addressed before its application into tissue engineering.
Optimization of the laboratory culture environment [259], including modulation of oxygen level and cell density and the introduction of growth factors, and recently discovered dECM preconditioning, is also an effective strategy to fight against senescence. Despite the fact that dECM deposited by MSCs from fetal [261] or young donors [272] offers a better rejuvenation effect in promoting aged MSCs in both expansion and differentiation capacities compared to adult donors, the source of these young cells was either allogenic or xenogeneic, which might pose a potential risk of compromising the donor [284] or immune rejection [285]. In this scenario, cells donated by the patients themselves are considered the best candidate. However, the elderly primarily suffer from degenerative disease and most of their autologous cells may suffer from senescence, which was identified as an influential factor in the quality of cells [286]. Given the demand for a younger cell population and the situation of carcinogenic transformation after genetic modification for immortalization purposes, it raises the possibility of combining genetic modification and environmental optimization strategies. In other words, we can immortalize these senescent cells and utilize their deposited dECM instead of the cells themselves to achieve a reduced senescent status and enhanced proliferation potential of expanded cells. The combination strategy might also overcome the potential loss of differentiation capacity of stem cells with the use of immortalization strategy alone. Further investigation into this matrix microenvironmental preconditioning-based rejuvenation strategy may offer important insights into possible means of providing robust primary cells as therapeutic agents.
Abbreviations
- 4-HT:
-
4-hydroxytamoxifen
- ADSCs:
-
adipose-derived stromal cells
- AKT:
-
protein kinase B
- ATM:
-
ataxia-telangiectasia mutated
- BMI1:
-
B cell-specific Moloney murine leukemia virus integration site 1
- BMP:
-
bone morphogenetic protein
- BMSCs:
-
bone marrow-derived MSCs
- BTG3:
-
B cell translocation gene 3
- CKIs:
-
cyclin-dependent kinase inhibitors
- DDR:
-
DNA damage response
- dECM:
-
decellularized cell-deposited extracellular matrix
- DNA:
-
deoxyribonucleic acid
- DPSCs:
-
dental pulp stem cells
- ECM:
-
extracellular matrix
- ERK1/2:
-
extracellular signal-regulated kinase 1/2
- FGF:
-
fibroblast growth factor
- H2O2 :
-
hydrogen peroxide
- HIF:
-
hypoxia inducible factor
- HPV:
-
human papillomavirus
- HTLV-1:
-
human T-lymphotropic virus type 1
- IGF:
-
insulin-like growth factor
- JNK:
-
c-Jun N-terminal kinase
- LATS1:
-
large tumor suppressor 1
- LRP5:
-
low-density lipoprotein receptor-related protein 5
- MAPK:
-
extracellular signal-regulated kinase
- MEK:
-
extracellular signal-regulated kinase kinase
- MSCs:
-
mesenchymal stem cells
- mTOR:
-
mammalian target of rapamycin
- NF1:
-
neurofibromatosis 1
- OH− :
-
hydroxyl radical
- O2 − :
-
superoxide anion
- PI3Ks:
-
phosphatidylinostitide 3 kinases
- PLGA:
-
polylactic glycolic acid
- PTEN:
-
phosphatase and tensin homolog
- RNA:
-
nuclear ribonucleic acid
- Rb:
-
retinoblastoma protein
- ROS:
-
reactive oxygen species
- SASP:
-
senescence-associated secretory phenotype
- SDSCs:
-
synovium-derived stem cell
- SV40:
-
simian virus 40
- SV40 LT:
-
SV40 large T
- SV40 ST:
-
SV40 small t antigen
- TERT:
-
telomerase reverse transcriptase
- TGFβ:
-
transforming growth factor β
- TRE:
-
tetracycline responsive elements
- VHL:
-
von Hippel-Lindau tumor suppressor
- Wnt:
-
Wingless/IntMDM2: mouse double minute-2 homolog
- YAP:
-
yes-associated protein 1
References
Persidis A. Tissue engineering. Nat Biotechnol. 1999;17(5):508–10.
Sengupta D, Waldman SD, Li S. From in vitro to in situ tissue engineering. Ann Biomed Eng. 2014;42(7):1537–45.
Shay JW, Wright WE. The use of telomerized cells for tissue engineering. Nat Biotechnol. 2000;18(1):22–3.
Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621.
Perez-Mancera PA, Young AR, Narita M. Inside and out: the activities of senescence in cancer. Nat Rev Cancer. 2014;14(8):547–58.
Blackburn EH. Telomeres—no end in sight. Cell. 1994;77(5):621–3.
Allsopp RC, Chang E, Kashefiaazam M, Rogaev EI, Piatyszek MA, Shay JW, et al. Telomere shortening is associated with cell-division in-vitro and in-vivo. Exp Cell Res. 1995;220(1):194–200.
Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345(6274):458–60.
Allsopp RC, Harley CB. Evidence for a critical telomere length in senescent human fibroblasts. Exp Cell Res. 1995;219(1):130–6.
Shay JW, Wright WE. Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis. 2005;26(5):867–74.
di Fagagna FD. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8(7):512–22.
Li Y, Nichols MA, Shay JW, Xiong Y. Transcriptional repression of the D-type cyclin-dependent kinase inhibitor p16 by the retinoblastoma susceptibility gene product pRb. Cancer Res. 1994;54(23):6078–82.
Campisi J, di Fagagna FD. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–40.
Shay JW, Pereirasmith OM, Wright WE. A role for both Rb and P53 in the regulation of human cellular senescence. Exp Cell Res. 1991;196(1):33–9.
Ozer HL, Banga SS, Dasgupta T, Houghton J, Hubbard K, Jha KK, et al. SV40-mediated immortalization of human fibroblasts. Exp Gerontol. 1996;31(1–2):303–10.
Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37(5):961–76.
Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120(4):513–22.
Brenner AJ, Stampfer MR, Aldaz CM. Increased p16 expression with first senescence arrest in human mammary epithelial cells and extended growth capacity with p16 inactivation. Oncogene. 1998;17(2):199–205.
Shibata KR, Aoyama T, Shima Y, Fukiage K, Otsuka S, Furu M, et al. Expression of the p161NK4A gene is associated closely with senescence of human mesenchymal stem cells and is potentially silenced by DNA methylation during in vitro expansion. Stem Cells. 2007;25(9):2371–82.
Bond JA, Wyllie FS, Wynford-Thomas D. Escape from senescence in human diploid fibroblasts induced directly by mutant p53. Oncogene. 1994;9(7):1885–9.
Shay JW, Wright WE, Werbin H. Defining the molecular mechanisms of human cell immortalization. Biochim Biophys Acta. 1991;1072(1):1–7.
Vaziri H, Benchimol S. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol. 1998;8(5):279–82.
Toussaint O, Medrano EE, von Zglinicki T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp Gerontol. 2000;35(8):927–45.
Scott JE. Oxygen and the connective tissues. Trends Biochem Sci. 1992;17(9):340–3.
Moussavi-Harami F, Duwayri Y, Martin JA, Moussavi-Harami F, Buckwalter JA. Oxygen effects on senescence in chondrocytes and mesenchymal stem cells: consequences for tissue engineering. Iowa Orthop J. 2004;24:15–20.
Prasad KN, Wu MX, Bondy SC. Telomere shortening during aging: attenuation by antioxidants and anti-inflammatory agents. Mech Ageing Dev. 2017;164:61–6.
Ramirez RD, Morales CP, Herbert BS, Rohde JM, Passons C, Shay JW, et al. Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev. 2001;15(4):398–403.
Ho PJ, Yen ML, Tang BC, Chen CT, Yen BL. H2O2 accumulation mediates differentiation capacity alteration, but not proliferative decline, in senescent human fetal mesenchymal stem cells. Antioxid Redox Signal. 2013;18(15):1895–905.
Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol. 2007;8(9):722–8.
Johnson TM, Yu ZX, Ferrans VJ, Lowenstein RA, Finkel T. Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc Natl Acad Sci U S A. 1996;93(21):11848–52.
Dohi Y, Ikura T, Hoshikawa Y, Katoh Y, Ota K, Nakanome A, et al. Bach1 inhibits oxidative stress-induced cellular senescence by impeding p53 function on chromatin. Nat Struct Mol Biol. 2008;15(12):1246–54.
Yin Y, Solomon G, Deng C, Barrett JC. Differential regulation of p21 by p53 and Rb in cellular response to oxidative stress. Mol Carcinog. 1999;24(1):15–24.
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602.
Zhu J, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998;12(19):2997–3007.
Dimri GP, Itahana K, Acosta M, Campisi J. Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Mol Cell Biol. 2000;20(1):273–85.
Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436(7051):720–4.
Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24(22):2463–79.
Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least 2 cooperating oncogenes. Nature. 1983;304(5927):596–602.
Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444(7119):638–42.
Marshall CJ. MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr Opin Genet Dev. 1994;4(1):82–9.
Nelson DM, McBryan T, Jeyapalan JC, Sedivy JM, Adams PD. A comparison of oncogene-induced senescence and replicative senescence: implications for tumor suppression and aging. Age (Dordr). 2014;36(3):9637.
Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007;21(1):43–8.
Nikiforov YE. Thyroid carcinoma: molecular pathways and therapeutic targets. Mod Pathol. 2008;21(Suppl 2):S37–43.
Ren G, Feng J, Datar I, Yeung AH, Saladi SV, Feng Y, et al. A micro-RNA connection in BRaf(V600E)-mediated premature senescence of human melanocytes. Int J Cell Biol. 2012;2012:913242.
Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132(3):363–74.
Raabe EH, Lim KS, Kim JM, Meeker A, Mao XG, Nikkhah G, et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res. 2011;17(11):3590–9.
Dimova DK, Stevaux O, Frolov MV, Dyson NJ. Cell cycle-dependent and cell cycle-independent control of transcription by the Drosophila E2F/RB pathway. Genes Dev. 2003;17(18):2308–20.
DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995;15(8):4215–24.
Xie Q, Peng S, Tao L, Ruan H, Yang Y, Li TM, et al. E2F transcription factor 1 regulates cellular and organismal senescence by inhibiting Forkhead box O transcription factors. J Biol Chem. 2014;289(49):34205–13.
Larribere L, Wu H, Novak D, Galach M, Bernhardt M, Orouji E, et al. NF1 loss induces senescence during human melanocyte differentiation in an iPSC-based model. Pigment Cell Melanoma Res. 2015;28(4):407–16.
Lin TY, Cheng YC, Yang HC, Lin WC, Wang CC, Lai PL, et al. Loss of the candidate tumor suppressor BTG3 triggers acute cellular senescence via the ERK-JMJD3-p16(INK4a) signaling axis. Oncogene. 2012;31(27):3287–97.
Young AP, Kaelin WG Jr. Senescence triggered by the loss of the VHL tumor suppressor. Cell Cycle. 2008;7(12):1709–12.
Welford SM, Dorie MJ, Li X, Haase VH, Giaccia AJ. Renal oxygenation suppresses VHL loss-induced senescence that is caused by increased sensitivity to oxidative stress. Mol Cell Biol. 2010;30(19):4595–603.
Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–27.
Tan W, Gu Z, Shen B, Jiang J, Meng Y, Da Z, et al. PTEN/Akt-p27(kip1) signaling promote the BM-MSCs senescence and apoptosis in SLE patients. J Cell Biochem. 2015;116(8):1583–94.
Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–30.
Kim YI, Ryu JS, Yeo JE, Choi YJ, Kim YS, Ko K, et al. Overexpression of TGF-beta1 enhances chondrogenic differentiation and proliferation of human synovium-derived stem cells. Biochem Biophys Res Commun. 2014;450(4):1593–9.
Millena AC, Vo BT, Khan SA. JunD is required for proliferation of prostate cancer cells and plays a role in transforming growth factor-beta (TGF-beta)-induced inhibition of cell proliferation. J Biol Chem. 2016;291(34):17964–76.
Kawamura H, Nakatsuka R, Matsuoka Y, Sumide K, Fujioka T, Asano H, et al. TGF-beta signaling accelerates senescence of human bone-derived CD271 and SSEA-4 double-positive mesenchymal stromal cells. Stem Cell Reports. 2018;10(3):920–32.
Rapisarda V, Borghesan M, Miguela V, Encheva V, Snijders AP, Lujambio A, et al. Integrin beta 3 regulates cellular senescence by activating the TGF-beta pathway. Cell Rep. 2017;18(10):2480–93.
Hu C, Zhang Y, Tang K, Luo Y, Liu Y, Chen W. Downregulation of CITED2 contributes to TGFbeta-mediated senescence of tendon-derived stem cells. Cell Tissue Res. 2017;368(1):93–104.
Li H, Xu D, Li J, Berndt MC, Liu JP. Transforming growth factor beta suppresses human telomerase reverse transcriptase (hTERT) by Smad3 interactions with c-Myc and the hTERT gene. J Biol Chem. 2006;281(35):25588–600.
Hu B, Tack DC, Liu T, Wu Z, Ullenbruch MR, Phan SH. Role of Smad3 in the regulation of rat telomerase reverse transcriptase by TGFbeta. Oncogene. 2006;25(7):1030–41.
Kretova M, Sabova L, Hodny Z, Bartek J, Kollarovic G, Nelson BD, et al. TGF-beta/NF1/Smad4-mediated suppression of ANT2 contributes to oxidative stress in cellular senescence. Cell Signal. 2014;26(12):2903–11.
Wu J, Niu J, Li X, Wang X, Guo Z, Zhang F. TGF-beta1 induces senescence of bone marrow mesenchymal stem cells via increase of mitochondrial ROS production. BMC Dev Biol. 2014;14:21.
Zhang Y, Lee JH, Paull TT, Gehrke S, D’Alessandro A, Dou Q, et al. Mitochondrial redox sensing by the kinase ATM maintains cellular antioxidant capacity. Sci Signal. 2018;11(538):eaaq0702.
Cipriano R, Kan CE, Graham J, Danielpour D, Stampfer M, Jackson MW. TGF-beta signaling engages an ATM-CHK2-p53-independent RAS-induced senescence and prevents malignant transformation in human mammary epithelial cells. Proc Natl Acad Sci U S A. 2011;108(21):8668–73.
Lin S, Yang J, Elkahloun AG, Bandyopadhyay A, Wang L, Cornell JE, et al. Attenuation of TGF-beta signaling suppresses premature senescence in a p21-dependent manner and promotes oncogenic Ras-mediated metastatic transformation in human mammary epithelial cells. Mol Biol Cell. 2012;23(8):1569–81.
Walsh DW, Godson C, Brazil DP, Martin F. Extracellular BMP-antagonist regulation in development and disease: tied up in knots. Trends Cell Biol. 2010;20(5):244–56.
Cassar L, Nicholls C, Pinto AR, Chen R, Wang L, Li H, et al. TGF-beta receptor mediated telomerase inhibition, telomere shortening and breast cancer cell senescence. Protein Cell. 2017;8(1):39–54.
Kaneda A, Fujita T, Anai M, Yamamoto S, Nagae G, Morikawa M, et al. Activation of Bmp2-Smad1 signal and its regulation by coordinated alteration of H3K27 trimethylation in Ras-induced senescence. PLoS Genet. 2011;7(11):e1002359.
Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–205.
Gu Z, Tan W, Feng G, Meng Y, Shen B, Liu H, et al. Wnt/beta-catenin signaling mediates the senescence of bone marrow-mesenchymal stem cells from systemic lupus erythematosus patients through the p53/p21 pathway. Mol Cell Biochem. 2014;387(1–2):27–37.
Zhang DY, Wang HJ, Tan YZ. Wnt/beta-catenin signaling induces the aging of mesenchymal stem cells through the DNA damage response and the p53/p21 pathway. PLoS ONE. 2011;6(6):e21397.
Zhang DY, Pan Y, Zhang C, Yan BX, Yu SS, Wu DL, et al. Wnt/beta-catenin signaling induces the aging of mesenchymal stem cells through promoting the ROS production. Mol Cell Biochem. 2013;374(1–2):13–20.
Gajjar M, Candeias MM, Malbert-Colas L, Mazars A, Fujita J, Olivares-Illana V, et al. The p53 mRNA-Mdm2 interaction controls Mdm2 nuclear trafficking and is required for p53 activation following DNA damage. Cancer Cell. 2012;21(1):25–35.
Jeoung JY, Nam HY, Kwak J, Jin HJ, Lee HJ, Lee BW, et al. A decline in Wnt3a signaling is necessary for mesenchymal stem cells to proceed to replicative senescence. Stem Cells Dev. 2015;24(8):973–82.
Hiyama A, Sakai D, Risbud MV, Tanaka M, Arai F, Abe K, et al. Enhancement of intervertebral disc cell senescence by WNT/beta-catenin signaling-induced matrix metalloproteinase expression. Arthritis Rheum. 2010;62(10):3036–47.
Tivey HS, Brook AJ, Rokicki MJ, Kipling D, Davis T. p38 (MAPK) stress signalling in replicative senescence in fibroblasts from progeroid and genomic instability syndromes. Biogerontology. 2013;14(1):47–62.
Borodkina A, Shatrova A, Abushik P, Nikolsky N, Burova E. Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging (Albany NY). 2014;6(6):481–95.
Wang W, Chen JX, Liao R, Deng Q, Zhou JJ, Huang S, et al. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol Cell Biol. 2002;22(10):3389–403.
Iwasa H, Han J, Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells. 2003;8(2):131–44.
Shin J, Yang J, Lee JC, Baek KH. Depletion of ERK2 but not ERK1 abrogates oncogenic Ras-induced senescence. Cell Signal. 2013;25(12):2540–7.
Choi SH, Jung SY, Yoo SY, Yoo SM, Kim DY, Kang S, et al. Regulation of ROS-independent ERK signaling rescues replicative cellular senescence in ex vivo expanded human c-kit-positive cardiac progenitor cells. Int J Cardiol. 2013;169(1):73–82.
Asur R, Balasubramaniam M, Marples B, Thomas RA, Tucker JD. Involvement of MAPK proteins in bystander effects induced by chemicals and ionizing radiation. Mutat Res. 2010;686(1–2):15–29.
Liu ZG, Baskaran R, Lea-Chou ET, Wood LD, Chen Y, Karin M, et al. Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature. 1996;384(6606):273–6.
Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng TL, et al. Jnk1—a protein-kinase stimulated by Uv-light and Ha-Ras that binds and phosphorylates the C-Jun activation domain. Cell. 1994;76(6):1025–37.
Hibi M, Lin AN, Smeal T, Minden A, Karin M. Identification of an oncoprotein-responsive and Uv-responsive protein-kinase that binds and potentiates the C-Jun activation domain. Gene Dev. 1993;7(11):2135–48.
Dasgupta J, Kar S, Liu R, Joseph J, Kalyanaraman B, Remington SJ, et al. Reactive oxygen species control senescence-associated matrix metalloproteinase-1 through c-Jun-N-terminal kinase. J Cell Physiol. 2010;225(1):52–62.
Spallarossa P, Altieri P, Barisione C, Passalacqua M, Aloi C, Fugazza G, et al. p38 MAPK and JNK antagonistically control senescence and cytoplasmic p16INK4A expression in doxorubicin-treated endothelial progenitor cells. PLoS ONE. 2010;5(12):e15583.
Das M, Jiang F, Sluss HK, Zhang C, Shokat KM, Flavell RA, et al. Suppression of p53-dependent senescence by the JNK signal transduction pathway. Proc Natl Acad Sci U S A. 2007;104(40):15759–64.
Jia D, Lu W, Zhang X, Cai G, Teng L, Wang X, et al. Calf Spleen Extractive Injection (CSEI), a small peptides enriched extraction, induces human hepatocellular carcinoma cell apoptosis via ROS/MAPKs dependent mitochondrial pathway. J Pharmacol Sci. 2016;132(2):122–30.
Khan KH, Yap TA, Yan L, Cunningham D. Targeting the PI3K-AKT-mTOR signaling network in cancer. Chin J Cancer. 2013;32(5):253–65.
Stout MC, Asiimwe E, Birkenstamm JR, Kim SY, Campbell PM. Analyzing Ras-associated cell proliferation signaling. Methods Mol Biol. 2014;1170:393–409.
Tan P, Wang YJ, Li S, Wang Y, He JY, Chen YY, et al. The PI3K/Akt/mTOR pathway regulates the replicative senescence of human VSMCs. Mol Cell Biochem. 2016;422(1–2):1–10.
Miyauchi H, Minamino T, Tateno K, Kunieda T, Toko H, Komuro I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. EMBO J. 2004;23(1):212–20.
Vredeveld LC, Possik PA, Smit MA, Meissl K, Michaloglou C, Horlings HM, et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev. 2012;26(10):1055–69.
Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13(22):2905–27.
Li BS, Huang JY, Guan J, Chen LH. Camptothecin inhibits the progression of NPC by regulating TGF-beta-induced activation of the PI3K/AKT signaling pathway. Oncol Lett. 2018;16(1):552–8.
Horowitz JC, Lee DY, Waghray M, Keshamouni VG, Thomas PE, Zhang H, et al. Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem. 2004;279(2):1359–67.
Fernandez LA, Kenney AM. The Hippo in the room: a new look at a key pathway in cell growth and transformation. Cell Cycle. 2010;9(12):2292–9.
Varelas X. The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development. 2014;141(8):1614–26.
Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747–61.
D’Addario I, Abbruzzese C, Lo Iacono M, Teson M, Golisano O, Barone V. Overexpression of YAP1 induces immortalization of normal human keratinocytes by blocking clonal evolution. Histochem Cell Biol. 2010;134(3):265–76.
Zhang C, Wang F, Xie Z, Chen L, Sinkemani A, Yu H, et al. Dysregulation of YAP by the Hippo pathway is involved in intervertebral disc degeneration, cell contact inhibition, and cell senescence. Oncotarget. 2018;9(2):2175–92.
Vigneron AM, Vousden KH. An indirect role for ASPP1 in limiting p53-dependent p21 expression and cellular senescence. EMBO J. 2012;31(2):471–80.
Aylon Y, Yabuta N, Besserglick H, Buganim Y, Rotter V, Nojima H, et al. Silencing of the Lats2 tumor suppressor overrides a p53-dependent oncogenic stress checkpoint and enables mutant H-Ras-driven cell transformation. Oncogene. 2009;28(50):4469–79.
Blaumueller CM, Qi H, Zagouras P, Artavanis-Tsakonas S. Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell. 1997;90(2):281–91.
Yin L, Velazquez OC, Liu ZJ. Notch signaling: emerging molecular targets for cancer therapy. Biochem Pharmacol. 2010;80(5):690–701.
Wilson A, Radtke F. Multiple functions of Notch signaling in self-renewing organs and cancer. FEBS Lett. 2006;580(12):2860–8.
Dotto GP. Notch tumor suppressor function. Oncogene. 2008;27(38):5115–23.
Liu ZJ, Xiao M, Balint K, Smalley KS, Brafford P, Qiu R, et al. Notch1 signaling promotes primary melanoma progression by activating mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt pathways and up-regulating N-cadherin expression. Cancer Res. 2006;66(8):4182–90.
Mittal S, Subramanyam D, Dey D, Kumar RV, Rangarajan A. Cooperation of Notch and Ras/MAPK signaling pathways in human breast carcinogenesis. Mol Cancer. 2009;8:128.
Purow BW, Haque RM, Noel MW, Su Q, Burdick MJ, Lee J, et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005;65(6):2353–63.
Moriyama H, Moriyama M, Ozawa T, Tsuruta D, Iguchi T, Tamada S, et al. Notch signaling enhances stemness by regulating metabolic pathways through modifying p53, NF-kappaB, and HIF-1alpha. Stem Cells Dev. 2018. https://doi.org/10.1089/scd.2017.0260.
Rampias T, Vgenopoulou P, Avgeris M, Polyzos A, Stravodimos K, Valavanis C, et al. A new tumor suppressor role for the Notch pathway in bladder cancer. Nat Med. 2014;20(10):1199–205.
Jaskula-Sztul R, Eide J, Tesfazghi S, Dammalapati A, Harrison AD, Yu XM, et al. Tumor-suppressor role of Notch3 in medullary thyroid carcinoma revealed by genetic and pharmacological induction. Mol Cancer Ther. 2015;14(2):499–512.
Avila JL, Kissil JL. Notch signaling in pancreatic cancer: oncogene or tumor suppressor? Trends Mol Med. 2013;19(5):320–7.
Yoshida Y, Hayashi Y, Suda M, Tateno K, Okada S, Moriya J, et al. Notch signaling regulates the lifespan of vascular endothelial cells via a p16-dependent pathway. PLoS ONE. 2014;9(6):e100359.
Cui H, Kong Y, Xu M, Zhang H. Notch3 functions as a tumor suppressor by controlling cellular senescence. Cancer Res. 2013;73(11):3451–9.
Liu ZJ, Tan Y, Beecham GW, Seo DM, Tian R, Li Y, et al. Notch activation induces endothelial cell senescence and pro-inflammatory response: implication of Notch signaling in atherosclerosis. Atherosclerosis. 2012;225(2):296–303.
Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7(3):165–97.
Oliver LJ, Rifkin DB, Gabrilove J, Hannocks MJ, Wilson EL. Long-term culture of human bone marrow stromal cells in the presence of basic fibroblast growth factor. Growth Factors. 1990;3(3):231–6.
Solchaga LA, Penick K, Porter JD, Goldberg VM, Caplan AI, Welter JF. FGF-2 enhances the mitotic and chondrogenic potentials of human adult bone marrow-derived mesenchymal stem cells. J Cell Physiol. 2005;203(2):398–409.
Solchaga LA, Penick K, Goldberg VM, Caplan AI, Welter JF. Fibroblast growth factor-2 enhances proliferation and delays loss of chondrogenic potential in human adult bone-marrow-derived mesenchymal stem cells. Tissue Eng Part A. 2010;16(3):1009–19.
Bianchi G, Banfi A, Mastrogiacomo M, Notaro R, Luzzatto L, Cancedda R, et al. Ex vivo enrichment of mesenchymal cell progenitors by fibroblast growth factor 2. Exp Cell Res. 2003;287(1):98–105.
Salehinejad P, Alitheen NB, Mandegary A, Nematollahi-Mahani SN, Janzamin E. Effect of EGF and FGF on the expansion properties of human umbilical cord mesenchymal cells. Vitro Cell Dev Biol Anim. 2013;49(7):515–23.
Ito T, Sawada R, Fujiwara Y, Seyama Y, Tsuchiya T. FGF-2 suppresses cellular senescence of human mesenchymal stem cells by down-regulation of TGF-beta2. Biochem Biophys Res Commun. 2007;359(1):108–14.
Coutu DL, Francois M, Galipeau J. Inhibition of cellular senescence by developmentally regulated FGF receptors in mesenchymal stem cells. Blood. 2011;117(25):6801–12.
Sato C, Iso Y, Mizukami T, Otabe K, Sasai M, Kurata M, et al. Fibroblast growth factor-23 induces cellular senescence in human mesenchymal stem cells from skeletal muscle. Biochem Biophys Res Commun. 2016;470(3):657–62.
Kenyon C. A conserved regulatory system for aging. Cell. 2001;105(2):165–8.
Elis S, Wu Y, Courtland HW, Sun H, Rosen CJ, Adamo ML, et al. Increased serum IGF-1 levels protect the musculoskeletal system but are associated with elevated oxidative stress markers and increased mortality independent of tissue igf1 gene expression. Aging Cell. 2011;10(3):547–50.
Handayaningsih AE, Takahashi M, Fukuoka H, Iguchi G, Nishizawa H, Yamamoto M, et al. IGF-I enhances cellular senescence via the reactive oxygen species-p53 pathway. Biochem Biophys Res Commun. 2012;425(2):478–84.
Rombouts C, Aerts A, Quintens R, Baselet B, El-Saghire H, Harms-Ringdahl M, et al. Transcriptomic profiling suggests a role for IGFBP5 in premature senescence of endothelial cells after chronic low dose rate irradiation. Int J Radiat Biol. 2014;90(7):560–74.
Kim KS, Seu YB, Baek SH, Kim MJ, Kim KJ, Kim JH, et al. Induction of cellular senescence by insulin-like growth factor binding protein-5 through a p53-dependent mechanism. Mol Biol Cell. 2007;18(11):4543–52.
Matsumoto R, Fukuoka H, Iguchi G, Odake Y, Yoshida K, Bando H, et al. Accelerated telomere shortening in acromegaly; IGF-I induces telomere shortening and cellular senescence. PLoS ONE. 2015;10(10):e0140189.
Gruber HE, Hoelscher GL, Ingram JA, Bethea S, Hanley EN. IGF-1 rescues human intervertebral annulus cells from in vitro stress-induced premature senescence. Growth Factors. 2008;26(4):220–5.
Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A. 1993;90(9):4304–8.
Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol. 2003;23(1):359–69.
Gardner LB, Li Q, Park MS, Flanagan WM, Semenza GL, Dang CV. Hypoxia inhibits G1/S transition through regulation of p27 expression. J Biol Chem. 2001;276(11):7919–26.
Vindrieux D, Devailly G, Augert A, Le Calve B, Ferrand M, Pigny P, et al. Repression of PLA2R1 by c-MYC and HIF-2alpha promotes cancer growth. Oncotarget. 2014;5(4):1004–13.
Saito S, Lin YC, Tsai MH, Lin CS, Murayama Y, Sato R, et al. Emerging roles of hypoxia-inducible factors and reactive oxygen species in cancer and pluripotent stem cells. Kaohsiung J Med Sci. 2015;31(6):279–86.
Kilic Eren M, Tabor V. The role of hypoxia inducible factor-1 alpha in bypassing oncogene-induced senescence. PLoS ONE. 2014;9(7):e101064.
Tsai CC, Chen YJ, Yew TL, Chen LL, Wang JY, Chiu CH, et al. Hypoxia inhibits senescence and maintains mesenchymal stem cell properties through down-regulation of E2A-p21 by HIF-TWIST. Blood. 2011;117(2):459–69.
Lee SH, Lee JH, Yoo SY, Hur J, Kim HS, Kwon SM. Hypoxia inhibits cellular senescence to restore the therapeutic potential of old human endothelial progenitor cells via the hypoxia-inducible factor-1alpha-TWIST-p21 axis. Arterioscler Thromb Vasc Biol. 2013;33(10):2407–14.
Rios C, D’Ippolito G, Curtis KM, Delcroix GJ, Gomez LA, El Hokayem J, et al. Low oxygen modulates multiple signaling pathways, increasing self-renewal, while decreasing differentiation, senescence, and apoptosis in stromal MIAMI cells. Stem Cells Dev. 2016;25(11):848–60.
Soltoff SP, Grubman SA, Jefferson DM. Development of salivary gland cell lines for studies of signaling and physiology. Ann N Y Acad Sci. 1998;842:100–7.
Sakai D, Mochida J, Yamamoto Y, Toh E, Iwashina T, Miyazaki T, et al. Immortalization of human nucleus pulposus cells by a recombinant SV40 adenovirus vector: establishment of a novel cell line for the study of human nucleus pulposus cells. Spine. 2004;29(14):1515–23.
Srinivasan A, McClellan AJ, Vartikar J, Marks I, Cantalupo P, Li Y, et al. The amino-terminal transforming region of Simian Virus 40 large T and small t antigens functions as a J domain. Mol Cell Biol. 1997;17(8):4761–73.
Symonds H, Krall L, Remington L, Saenz-Robles M, Lowe S, Jacks T, et al. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell. 1994;78(4):703–11.
Foddis R, De Rienzo A, Broccoli D, Bocchetta M, Stekala E, Rizzo P, et al. SV40 infection induces telomerase activity in human mesothelial cells. Oncogene. 2002;21(9):1434–42.
Longworth MS, Laimins LA. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev. 2004;68(2):362–72.
Motoyama S, Ladines-Llave CA, Luis Villanueva S, Maruo T. The role of human papilloma virus in the molecular biology of cervical carcinogenesis. Kobe J Med Sci. 2004;50(1–2):9–19.
Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R. The E6-gene and E7-gene of the human papillomavirus type-16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol. 1989;63(10):4417–21.
Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63(6):1129–36.
Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243(4893):934–7.
Sieburg M, Tripp A, Ma JW, Feuer G. Human T-cell leukemia virus type 1 (HTLV-1) and HTLV-2 tax oncoproteins modulate cell cycle progression and apoptosis. J Virol. 2004;78(19):10399–409.
Imai M, Higuchi M, Kawamura H, Yoshita M, Takahashi M, Oie M, et al. Human T cell leukemia virus type 2 (HTLV-2) Tax2 has a dominant activity over HTLV-1 Tax1 to immortalize human CD4+ T cells. Virus Genes. 2013;46(1):39–46.
Kelly K, Siebenlist U. The regulation and expression of c-myc in normal and malignant cells. Annu Rev Immunol. 1986;4:317–38.
Bernard O, Reid HH, Bartlett PF. Role of the c-myc and the N-myc proto-oncogenes in the immortalization of neural precursors. J Neurosci Res. 1989;24(1):9–20.
Gil J, Kerai P, Lleonart M, Bernard D, Cigudosa JC, Peters G, et al. Immortalization of primary human prostate epithelial cells by c-Myc. Cancer Res. 2005;65(6):2179–85.
Liu X, Disbrow GL, Yuan H, Tomaic V, Schlegel R. Myc and human papillomavirus type 16 E7 genes cooperate to immortalize human keratinocytes. J Virol. 2007;81(22):12689–95.
Bi CM, Zhang SQ, Zhang Y, Peng SY, Wang L, An ZX, et al. Immortalization of bovine germ line stem cells by c-myc and hTERT. Anim Reprod Sci. 2007;100(3–4):371–8.
Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423(6937):302–5.
Pirrotta V. Polycombing the genome: PcG, trxG, and chromatin silencing. Cell. 1998;93(3):333–6.
Vanderlugt NMT, Domen J, Linders K, Vanroon M, Robanusmaandag E, Teriele H, et al. Posterior Transformation, Neurological Abnormalities, and Severe Hematopoietic Defects in Mice with a Targeted Deletion of the Bmi-1 Protooncogene. Gene Dev. 1994;8(7):757–69.
Itahana K, Zou Y, Itahana Y, Martinez JL, Beausejour C, Jacobs JJ, et al. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol. 2003;23(1):389–401.
Jacobs JJL, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397(6715):164–8.
Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33(5):787–91.
Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90(4):785–95.
Masutomi K, Yu EY, Khurts S, Ben-Porath I, Currier JL, Metz GB, et al. Telomerase maintains telomere structure in normal human cells. Cell. 2003;114(2):241–53.
Simonsen JL, Rosada C, Serakinci N, Justesen J, Stenderup K, Rattan SI, et al. Telomerase expression extends the proliferative life-span and maintains the osteogenic potential of human bone marrow stromal cells. Nat Biotechnol. 2002;20(6):592–6.
Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279(5349):349–52.
Piera-Velazquez S, Jimenez SA, Stokes D. Increased life span of human osteoarthritic chondrocytes by exogenous expression of telomerase. Arthritis Rheum. 2002;46(3):683–93.
Graham MK, Principessa L, Antony L, Meeker AK, Isaacs JT. Low p16INK4a expression in early passage human prostate basal epithelial cells enables immortalization by telomerase expression alone. Prostate. 2017;77(4):374–84.
Toouli CD, Huschtscha LI, Neumann AA, Noble JR, Colgin LM, Hukku B, et al. Comparison of human mammary epithelial cells immortalized by simian virus 40 T-Antigen or by the telomerase catalytic subunit. Oncogene. 2002;21(1):128–39.
Chapman EJ, Hurst CD, Pitt E, Chambers P, Aveyard JS, Knowles MA. Expression of hTERT immortalises normal human urothelial cells without inactivation of the p16/Rb pathway. Oncogene. 2006;25(36):5037–45.
Roy NS, Nakano T, Keyoung HM, Windrem M, Rashbaum WK, Alonso ML, et al. Telomerase immortalization of neuronally restricted progenitor cells derived from the human fetal spinal cord. Nat Biotechnol. 2004;22(3):297–305.
Morales CP, Holt SE, Ouellette M, Kaur KJ, Yan Y, Wilson KS, et al. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat Genet. 1999;21(1):115–8.
Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, et al. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat Genet. 1999;21(1):111–4.
Lowy DR, Solomon D, Hildesheim A, Schiller JT, Schiffman M. Human papillomavirus infection and the primary and secondary prevention of cervical cancer. Cancer. 2008;113(7 Suppl):1980–93.
Snijders PJ, Steenbergen RD, Heideman DA, Meijer CJ. HPV-mediated cervical carcinogenesis: concepts and clinical implications. J Pathol. 2006;208(2):152–64.
Horikawa I, Barrett JC. Transcriptional regulation of the telomerase hTERT gene as a target for cellular and viral oncogenic mechanisms. Carcinogenesis. 2003;24(7):1167–76.
James MA, Lee JH, Klingelhutz AJ. HPV16-E6 associated hTERT promoter acetylation is E6AP dependent, increased in later passage cells and enhanced by loss of p300. Int J Cancer. 2006;119(8):1878–85.
Wiman KG, Klein G. An old acquaintance resurfaces in human mesothelioma. Nat Med. 1997;3(8):839–40.
Carbone M, Pass HI, Rizzo P, Marinetti M, Di Muzio M, Mew DJ, et al. Simian virus 40-like DNA sequences in human pleural mesothelioma. Oncogene. 1994;9(6):1781–90.
Huang H, Reis R, Yonekawa Y, Lopes JM, Kleihues P, Ohgaki H. Identification in human brain tumors of DNA sequences specific for SV40 large T antigen. Brain Pathol. 1999;9(1):33–42.
Martini F, Lazzarin L, Iaccheri L, Vignocchi B, Finocchiaro G, Magnani I, et al. Different simian virus 40 genomic regions and sequences homologous with SV40 large T antigen in DNA of human brain and bone tumors and of leukocytes from blood donors. Cancer. 2002;94(4):1037–48.
Kirschstein RL, Gerber P. Ependymomas produced after intracerebral inoculation of SV40 into new-born hamsters. Nature. 1962;195:299–300.
Brown KW, Gallimore PH. Malignant progression of an SV40-transformed human epidermal keratinocyte cell line. Br J Cancer. 1987;56(5):545–54.
Bravard A, Beaumatin J, Luccioni C, Fritsch P, Lefrancois D, Thenet S, et al. Chromosomal, mitochondrial and metabolic alterations in SV40-transformed rabbit chondrocytes. Carcinogenesis. 1992;13(5):767–72.
Cicala C, Pompetti F, Carbone M. SV40 induces mesotheliomas in hamsters. Am J Pathol. 1993;142(5):1524–33.
Jensen F, Pagano JS, Ponten J, Ravdin RG, Koprowski H. Autologous + homologous implantation of human cells transformed in vitro by simian virus 40. J Natl Cancer I. 1964;32(4):917.
Botchan M, Topp W, Sambrook J. The arrangement of simian virus 40 sequences in the DNA of transformed cells. Cell. 1976;9(2):269–87.
Ketner G, Kelly TJ. Integrated simian-virus 40 sequences in transformed-cell DNA—analysis using restriction endonucleases. Proc Natl Acad Sci U S A. 1976;73(4):1102–6.
MacKenzie KL, Franco S, May C, Sadelain M, Moore MA. Mass cultured human fibroblasts overexpressing hTERT encounter a growth crisis following an extended period of proliferation. Exp Cell Res. 2000;259(2):336–50.
Zhao CF, Hu HY, Meng L, Li QQ, Lin AX. Immortalization of bovine mammary epithelial cells alone by human telomerase reverse transcriptase. Cell Biol Int. 2010;34(6):579–86.
Huang G, Zheng Q, Sun J, Guo C, Yang J, Chen R, et al. Stabilization of cellular properties and differentiation mutilpotential of human mesenchymal stem cells transduced with hTERT gene in a long-term culture. J Cell Biochem. 2008;103(4):1256–69.
Huang GP, Pan ZJ, Huang JP, Yang JF, Guo CJ, Wang YG, et al. Proteomic analysis of human bone marrow mesenchymal stem cells transduced with human telomerase reverse transcriptase gene during proliferation. Cell Proliferat. 2008;41(4):625–44.
Bocker W, Yin Z, Drosse I, Haasters F, Rossmann O, Wierer M, et al. Introducing a single-cell-derived human mesenchymal stem cell line expressing hTERT after lentiviral gene transfer. J Cell Mol Med. 2008;12(4):1347–59.
Peng BY, Chiou CS, Dubey NK, Yu SH, Deng YH, Tsai FC, et al. Correction: non-invasive in vivo molecular imaging of intra-articularly transplanted immortalized bone marrow stem cells for osteoarthritis treatment. Oncotarget. 2018;9(38):25383.
Miyanishi M, Mandai M, Matsumura N, Yamaguchi K, Hamanishi J, Higuchi T, et al. Immortalized ovarian surface epithelial cells acquire tumorigenicity by Acrogranin gene overexpression. Oncol Rep. 2007;17(2):329–33.
Thomas M, Yang L, Hornsby PJ. Formation of functional tissue from transplanted adrenocortical cells expressing telomerase reverse transcriptase. Nat Biotechnol. 2000;18(1):39–42.
Lu X, Arbiser JL, West J, Hoedt-Miller M, Sheridan A, Govindarajan B, et al. Tumor necrosis factor-related apoptosis-inducing ligand can induce apoptosis in subsets of premalignant cells. Am J Pathol. 2004;165(5):1613–20.
Shin SH, Park SY, Kim MK, Kim JC, Sung YK. Establishment and characterization of an immortalized human dermal papilla cell line. BMB Rep. 2011;44(8):512–6.
Serakinci N, Guldberg P, Burns JS, Abdallah B, Schrodder H, Jensen T, et al. Adult human mesenchymal stem cell as a target for neoplastic transformation. Oncogene. 2004;23(29):5095–8.
Burns JS, Abdallah BM, Guldberg P, Rygaard J, Schroder HD, Kassem M. Tumorigenic heterogeneity in cancer stem cells evolved from long-term cultures of telomerase-immortalized human mesenchymal stem cells. Cancer Res. 2005;65(8):3126–35.
Hashimoto Y, Murakami Y, Uemura K, Hayashidani Y, Sudo T, Ohge H, et al. Detection of human telomerase reverse transcriptase (hTERT) expression in tissue and pancreatic juice from pancreatic cancer. Surgery. 2008;143(1):113–25.
Nissim S, Idos GE, Wu B. Genetic markers of malignant transformation in intraductal papillary mucinous neoplasm of the pancreas: a meta-analysis. Pancreas. 2012;41(8):1195–205.
Bertorelle R, Briarava M, Rampazzo E, Biasini L, Agostini M, Maretto I, et al. Telomerase is an independent prognostic marker of overall survival in patients with colorectal cancer. Br J Cancer. 2013;108(2):278–84.
Noble JR, Zhong ZH, Neumann AA, Melki JR, Clark SJ, Reddel RR. Alterations in the p16(INK4a) and p53 tumor suppressor genes of hTERT-immortalized human fibroblasts. Oncogene. 2004;23(17):3116–21.
Wang J, Hannon GJ, Beach DH. Risky immortalization by telomerase. Nature. 2000;405(6788):755–6.
Guo Z, Jing R, Rao Q, Zhang L, Gao Y, Liu F, et al. Immortalized common marmoset (Callithrix jacchus) hepatic progenitor cells possess bipotentiality in vitro and in vivo. Cell Discov. 2018;4:23.
Patil PB, Begum S, Joshi M, Kleman MI, Olausson M, Sumitran-Holgersson S. Phenotypic and in vivo functional characterization of immortalized human fetal liver cells. Scand J Gastroenterol. 2014;49(6):705–14.
Hung CJ, Yao CL, Cheng FC, Wu ML, Wang TH, Hwang SM. Establishment of immortalized mesenchymal stromal cells with red fluorescence protein expression for in vivo transplantation and tracing in the rat model with traumatic brain injury. Cytotherapy. 2010;12(4):455–65.
Danielian PS, White R, Hoare SA, Fawell SE, Parker MG. Identification of residues in the estrogen receptor that confer differential sensitivity to estrogen and hydroxytamoxifen. Mol Endocrinol. 1993;7(2):232–40.
Tegtmeyer P. Function of simian virus 40 gene A in transforming infection. J Virol. 1975;15(3):613–8.
Kawabata K, Sakurai F, Koizumi N, Hayakawa T, Mizuguchi H. Adenovirus vector-mediated gene transfer into stem cells. Mol Pharmaceut. 2006;3(2):95–103.
Paillard F. Reversible cell immortalization with the Cre-lox system. Hum Gene Ther. 1999;10(10):1597–8.
Sauer B, Henderson N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc Natl Acad Sci U S A. 1988;85(14):5166–70.
Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992;89(12):5547–51.
Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268(5218):1766–9.
Sipo I, Hurtado Pico A, Wang X, Eberle J, Petersen I, Weger S, et al. An improved Tet-On regulatable FasL-adenovirus vector system for lung cancer therapy. J Mol Med (Berl). 2006;84(3):215–25.
Zhang Y, Toh L, Lau P, Wang XY. Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/beta-catenin pathway in human cancer. J Biol Chem. 2012;287(39):32494–511.
Babizhayev MA, Yegorov YE. Biomarkers of oxidative stress and cataract. Novel drug delivery therapeutic strategies targeting telomere reduction and the expression of telomerase activity in the lens epithelial cells with N-acetylcarnosine lubricant eye drops: anti-cataract which helps to prevent and treat cataracts in the eyes of dogs and other animals. Curr Drug Deliv. 2014;11(1):24–61.
Huang HS, Chiou JF, Fong Y, Hou CC, Lu YC, Wang JY, et al. Activation of human telomerase reverse transcriptase expression by some new symmetrical bis-substituted derivatives of the anthraquinone. J Med Chem. 2003;46(15):3300–7.
Seandel M, Butler JM, Kobayashi H, Hooper AT, White IA, Zhang F, et al. Generation of a functional and durable vascular niche by the adenoviral E4ORF1 gene. Proc Natl Acad Sci U S A. 2008;105(49):19288–93.
Jerebtsova M, Kumari N, Obuhkov Y, Nekhai S. Adenoviral E4 gene stimulates secretion of pigmental epithelium derived factor (PEDF) that maintains long-term survival of human glomerulus-derived endothelial cells. Mol Cell Proteomics. 2012;11(11):1378–88.
Nevels M, Tauber B, Spruss T, Wolf H, Dobner T. “Hit-and-run” transformation by adenovirus oncogenes. J Virol. 2001;75(7):3089–94.
Tauber B, Dobner T. Adenovirus early E4 genes in viral oncogenesis. Oncogene. 2001;20(54):7847–54.
Alexander D, Biller R, Rieger M, Ardjomandi N, Reinert S. Phenotypic characterization of a human immortalized cranial periosteal cell line. Cell Physiol Biochem. 2015;35(6):2244–54.
Yang K, Chen J, Jiang W, Huang E, Cui J, Kim SH, et al. Conditional immortalization establishes a repertoire of mouse melanocyte progenitors with distinct melanogenic differentiation potential. J Invest Dermatol. 2012;132(10):2479–83.
Song D, Zhang F, Reid RR, Ye J, Wei Q, Liao J, et al. BMP9 induces osteogenesis and adipogenesis in the immortalized human cranial suture progenitors from the patent sutures of craniosynostosis patients. J Cell Mol Med. 2017. https://doi.org/10.1111/jcmm.13193.
Gong M, Bi Y, Jiang W, Zhang Y, Chen L, Hou N, et al. Immortalized mesenchymal stem cells: an alternative to primary mesenchymal stem cells in neuronal differentiation and neuroregeneration associated studies. J Biomed Sci. 2011;18:87.
Yang GB, Li XY, Yuan GH, Liu H, Fan MW. Immortalization and characterization of human dental papilla cells with odontoblastic differentiation. Int Endod J. 2013;46(6):565–72.
Moscoso I, Rodriguez-Barbosa JI, Barallobre-Barreiro J, Anon P, Domenech N. Immortalization of bone marrow-derived porcine mesenchymal stem cells and their differentiation into cells expressing cardiac phenotypic markers. J Tissue Eng Regen Med. 2012;6(8):655–65.
Wei P, Li L, Qi H, Zhou HX, Deng CY, Li FR. Reversible immortalization of Nestin-positive precursor cells from pancreas and differentiation into insulin-secreting cells. Biochem Biophys Res Commun. 2012;418(2):330–5.
Houghton A, Oyajobi BO, Foster GA, Russell RG, Stringer BM. Immortalization of human marrow stromal cells by retroviral transduction with a temperature sensitive oncogene: identification of bipotential precursor cells capable of directed differentiation to either an osteoblast or adipocyte phenotype. Bone. 1998;22(1):7–16.
Stolzel K, Schulze-Tanzil G, Olze H, Schwarz S, Feldmann EM, Rotter N. Immortalised human mesenchymal stem cells undergo chondrogenic differentiation in alginate and PGA/PLLA scaffolds. Cell Tissue Bank. 2015;16(1):159–70.
Li N, Yang R, Zhang W, Dorfman H, Rao P, Gorlick R. Genetically transforming human mesenchymal stem cells to sarcomas: changes in cellular phenotype and multilineage differentiation potential. Cancer. 2009;115(20):4795–806.
Yang Y, Yang R, Roth M, Piperdi S, Zhang W, Dorfman H, et al. Genetically transforming human osteoblasts to sarcoma: development of an osteosarcoma model. Genes Cancer. 2017;8(1–2):484–94.
Yang J, Cao C, Wang W, Tong X, Shi D, Wu F, et al. Proliferation and osteogenesis of immortalized bone marrow-derived mesenchymal stem cells in porous polylactic glycolic acid scaffolds under perfusion culture. J Biomed Mater Res A. 2010;92(3):817–29.
Tsai CC, Chen CL, Liu HC, Lee YT, Wang HW, Hou LT, et al. Overexpression of hTERT increases stem-like properties and decreases spontaneous differentiation in human mesenchymal stem cell lines. J Biomed Sci. 2010;17:64.
Okamoto T, Aoyama T, Nakayama T, Nakamata T, Hosaka T, Nishijo K, et al. Clonal heterogeneity in differentiation potential of immortalized human mesenchymal stem cells. Biochem Biophys Res Commun. 2002;295(2):354–61.
el Ikbale A, Goorha S, Reiter LT, Miranda-Carboni GA. Effects of hTERT immortalization on osteogenic and adipogenic differentiation of dental pulp stem cells. Data Brief. 2016;6:696–9.
Zhang X, Soda Y, Takahashi K, Bai Y, Mitsuru A, Igura K, et al. Successful immortalization of mesenchymal progenitor cells derived from human placenta and the differentiation abilities of immortalized cells. Biochem Biophys Res Commun. 2006;351(4):853–9.
Tatrai P, Szepesi A, Matula Z, Szigeti A, Buchan G, Madi A, et al. Combined introduction of Bmi-1 and hTERT immortalizes human adipose tissue-derived stromal cells with low risk of transformation. Biochem Biophys Res Commun. 2012;422(1):28–35.
Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, Geti I, et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009;23(18):2134–9.
Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460(7259):1132–5.
Li H, Collado M, Villasante A, Strati K, Ortega S, Canamero M, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460(7259):1136-U101.
Marion RM, Strati K, Li H, Murga M, Blanco R, Ortega S, et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460(7259):1149–53.
Agur Z, Kogan Y, Levi L, Harrison H, Lamb R, Kirnasovsky OU, et al. Disruption of a Quorum Sensing mechanism triggers tumorigenesis: a simple discrete model corroborated by experiments in mammary cancer stem cells. Biol Direct. 2010;5:20.
Sotiropoulou PA, Perez SA, Salagianni M, Baxevanis CN, Papamichail M. Characterization of the optimal culture conditions for clinical scale production of human mesenchymal stem cells. Stem Cells. 2006;24(2):462–71.
Fuchs E, Tumbar T, Guasch G. Socializing with the neighbors: stem cells and their niche. Cell. 2004;116(6):769–78.
Jones DL, Wagers AJ. No place like home: anatomy and function of the stem cell niche. Nat Rev Mol Cell Biol. 2008;9(1):11–21.
Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochem Biophys Acta. 2014;1840(8):2506–19.
Zhang Y, Chen S, Pei M. Biomechanical signals guiding stem cell cartilage engineering: from molecular adaption to tissue functionality. Eur Cell Mater. 2016;31:59–78.
Pei M, Li JT, Shoukry M, Zhang Y. A review of decellularized stem cell matrix: a novel cell expansion system for cartilage tissue engineering. Eur Cell Mater. 2011;22:333–43 (discussion 43).
Pei M. Environmental preconditioning rejuvenates adult stem cells’ proliferation and chondrogenic potential. Biomaterials. 2017;117:10–23.
Pei M, He F, Kish VL. Expansion on extracellular matrix deposited by human bone marrow stromal cells facilitates stem cell proliferation and tissue-specific lineage potential. Tissue Eng Part A. 2011;17(23–24):3067–76.
Li J, Hansen KC, Zhang Y, Dong C, Dinu CZ, Dzieciatkowska M, et al. Rejuvenation of chondrogenic potential in a young stem cell microenvironment. Biomaterials. 2014;35(2):642–53.
He F, Chen X, Pei M. Reconstruction of an in vitro tissue-specific microenvironment to rejuvenate synovium-derived stem cells for cartilage tissue engineering. Tissue Eng Part A. 2009;15(12):3809–21.
Liu X, Zhou L, Chen X, Liu T, Pan G, Cui W, et al. Culturing on decellularized extracellular matrix enhances antioxidant properties of human umbilical cord-derived mesenchymal stem cells. Mater Sci Eng C Mater Biol Appl. 2016;61:437–48.
He F, Pei M. Extracellular matrix enhances differentiation of adipose stem cells from infrapatellar fat pad toward chondrogenesis. J Tissue Eng Regen Med. 2013;7(1):73–84.
He F, Pei M. Rejuvenation of nucleus pulposus cells using extracellular matrix deposited by synovium-derived stem cells. Spine. 2012;37(6):459–69.
Pei M, Shoukry M, Li J, Daffner SD, France JC, Emery SE. Modulation of in vitro microenvironment facilitates synovium-derived stem cell-based nucleus pulposus tissue regeneration. Spine. 2012;37(18):1538–47.
Pei M, He F. Extracellular matrix deposited by synovium-derived stem cells delays replicative senescent chondrocyte dedifferentiation and enhances redifferentiation. J Cell Physiol. 2012;227(5):2163–74.
Whitfield MJ, Lee WC, Van Vliet KJ. Onset of heterogeneity in culture-expanded bone marrow stromal cells. Stem Cell Res. 2013;11(3):1365–77.
Pei M, Zhang Y, Li J, Chen D. Antioxidation of decellularized stem cell matrix promotes human synovium-derived stem cell-based chondrogenesis. Stem Cells Dev. 2013;22(6):889–900.
Zhang Y, Li J, Davis ME, Pei M. Delineation of in vitro chondrogenesis of human synovial stem cells following preconditioning using decellularized matrix. Acta Biomater. 2015;20:39–50.
Pei M, He F, Li JT, Tidwell JE, Jones AC, McDonough EB. Repair of large animal partial-thickness cartilage defects through intraarticular injection of matrix-rejuvenated synovium-derived stem cells. Tissue Eng Pt A. 2013;19(9–10):1144–54.
Pei M, Li J, Zhang Y, Liu G, Wei L, Zhang Y. Expansion on a matrix deposited by nonchondrogenic urine stem cells strengthens the chondrogenic capacity of repeated-passage bone marrow stromal cells. Cell Tissue Res. 2014;356(2):391–403.
Marinkovic M, Block TJ, Rakian R, Li Q, Wang E, Reilly MA, et al. One size does not fit all: developing a cell-specific niche for in vitro study of cell behavior. Matrix Biol. 2016;52–54:426–41.
Sun Y, Li W, Lu Z, Chen R, Ling J, Ran Q, et al. Rescuing replication and osteogenesis of aged mesenchymal stem cells by exposure to a young extracellular matrix. FASEB J. 2011;25(5):1474–85.
Zhou L, Chen X, Liu T, Zhu C, Si M, Jargstorf J, et al. SIRT1-dependent anti-senescence effects of cell-deposited matrix on human umbilical cord mesenchymal stem cells. J Tissue Eng Regen Med. 2018;12(2):e1008–21.
Li M, Chen X, Yan J, Zhou L, Wang Y, He F, et al. Inhibition of osteoclastogenesis by stem cell-derived extracellular matrix through modulation of intracellular reactive oxygen species. Acta Biomater. 2018;71:118–31.
Takada Y, Ye X, Simon S. The integrins. Genome Biol. 2007;8(5):215.
Kozlova NI, Morozevich GE, Ushakova NA, Berman AE. Implication of integrin alpha2beta1 in proliferation and invasion of human breast carcinoma and melanoma cells: noncanonical function of Akt protein kinase. Biochemistry (Mosc). 2018;83(6):738–45.
Liaw L, Almeida M, Hart CE, Schwartz SM, Giachelli CM. Osteopontin promotes vascular cell adhesion and spreading and is chemotactic for smooth muscle cells in vitro. Circ Res. 1994;74(2):214–24.
Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179–83.
Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol. 2012;13(9):591–600.
Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS J. 2010;277(1):2–21.
Tormos AM, Talens-Visconti R, Nebreda AR, Sastre J. p38 MAPK: a dual role in hepatocyte proliferation through reactive oxygen species. Free Radic Res. 2013;47(11):905–16.
Iffy L, Varadi V, Portuondo N, Ende N. Collection of fetal blood for stem cell research and therapy. Med Law. 2006;25(3):553–61.
Zhang Y, Pizzute T, Li J, He F, Pei M. sb203580 preconditioning recharges matrix-expanded human adult stem cells for chondrogenesis in an inflammatory environment—a feasible approach for autologous stem cell based osteoarthritic cartilage repair. Biomaterials. 2015;64:88–97.
Barbero A, Grogan S, Schafer D, Heberer M, Mainil-Varlet P, Martin I. Age related changes in human articular chondrocyte yield, proliferation and post-expansion chondrogenic capacity. Osteoarthritis Cartilage. 2004;12(6):476–84.
Li J, Li LJ, Cao HC, Sheng GP, Yu HY, Xu W, et al. Establishment of highly differentiated immortalized human hepatocyte line with simian virus 40 large tumor antigen for liver based cell therapy. ASAIO J. 2005;51(3):262–8.
Kobayashi N, Fujiwara T, Westerman KA, Inoue Y, Sakaguchi M, Noguchi H, et al. Prevention of acute liver failure in rats with reversibly immortalized human hepatocytes. Science. 2000;287(5456):1258–62.
Band V, Zajchowski D, Kulesa V, Sager R. Human papilloma-virus DNAs immortalize normal human mammary epithelial-cells and reduce their growth-factor requirements. Proc Natl Acad Sci USA. 1990;87(1):463–7.
Band V, De Caprio JA, Delmolino L, Kulesa V, Sager R. Loss of p53 protein in human papillomavirus type 16 E6-immortalized human mammary epithelial cells. J Virol. 1991;65(12):6671–6.
Rood PM, Calafat J, von dem Borne AE, Gerritsen WR, van der Schoot CE. Immortalisation of human bone marrow endothelial cells: characterisation of new cell lines. Eur J Clin Invest. 2000;30(7):618–29.
Rhim JS, Tsai WP, Chen ZQ, Chen Z, Van Waes C, Burger AM, et al. A human vascular endothelial cell model to study angiogenesis and tumorigenesis. Carcinogenesis. 1998;19(4):673–81.
Sobhan PK, Seervi M, Joseph J, Varghese S, Pillai PR, Sivaraman DM, et al. Immortalized functional endothelial progenitor cell lines from umbilical cord blood for vascular tissue engineering. Tissue Eng Part C Methods. 2012;18(11):890–902.
Hudson JB, Bedell MA, McCance DJ, Laiminis LA. Immortalization and altered differentiation of human keratinocytes in vitro by the E6 and E7 open reading frames of human papillomavirus type 18. J Virol. 1990;64(2):519–26.
Halbert CL, Demers GW, Galloway DA. The E7 gene of human papillomavirus type-16 is sufficient for immortalization of human epithelial-cells. J Virol. 1991;65(1):473–8.
Saleem MA, O’Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol. 2002;13(3):630–8.
Tsutsumi K, Belaguli N, Qi S, Michalak TI, Gulliver WP, Pater A, et al. Human papillomavirus-16 DNA immortalizes 2 types of normal human epithelial-cells of the uterine cervix. Am J Pathol. 1992;140(2):255–61.
Liu MC, Chen WH, Wu LC, Hsu WC, Lo WC, Yeh SD, et al. Establishment of a promising human nucleus pulposus cell line for intervertebral disc tissue engineering. Tissue Eng Part C Methods. 2014;20(1):1–10.
Hawley-Nelson P, Vousden KH, Hubbert NL, Lowy DR, Schiller JT. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 1989;8(12):3905–10.
Noguchi H, Kobayashi N, Westerman KA, Sakaguchi M, Okitsu T, Totsugawa T, et al. Controlled expansion of human endothelial cell populations by Cre-loxP-based reversible immortalization. Hum Gene Ther. 2002;13(2):321–34.
Robbins JR, Thomas B, Tan L, Choy B, Arbiser JL, Berenbaum F, et al. Immortalized human adult articular chondrocytes maintain cartilage-specific phenotype and responses to interleukin-1beta. Arthritis Rheum. 2000;43(10):2189–201.
Grigolo B, Roseti L, Neri S, Gobbi P, Jensen P, Major EO, et al. Human articular chondrocytes immortalized by HPV-16 E6 and E7 genes: maintenance of differentiated phenotype under defined culture conditions. Osteoarthritis Cartilage. 2002;10(11):879–89.
Hering S, Griffin BE, Strauss M. Immortalization of human fetal sinusoidal liver cells by polyoma virus large T antigen. Exp Cell Res. 1991;195(1):1–7.
Lamplot JD, Liu B, Yin L, Zhang W, Wang Z, Luther G, et al. Reversibly immortalized mouse articular chondrocytes acquire long-term proliferative capability while retaining chondrogenic phenotype. Cell Transplant. 2015;24(6):1053–66.
Chen TS, Arslan F, Yin Y, Tan SS, Lai RC, Choo ABH, et al. Enabling a robust scalable manufacturing process for therapeutic exosomes through oncogenic immortalization of human ESC-derived MSCs. J Transl Med. 2011;9:47.
Kim HS, Shin JY, Yun JY, Ahn DK, Le JY. Immortalization of human embryonic fibroblasts by overexpression of c-myc and simian virus 40 large T antigen. Exp Mol Med. 2001;33(4):293–8.
Xu C, Jiang J, Sottile V, McWhir J, Lebkowski J, Carpenter MK. Immortalized fibroblast-like cells derived from human embryonic stem cells support undifferentiated cell growth. Stem Cells. 2004;22(6):972–80.
Wolbank S, Stadler G, Peterbauer A, Gillich A, Karbiener M, Streubel B, et al. Telomerase immortalized human amnion- and adipose-derived mesenchymal stem cells: maintenance of differentiation and immunomodulatory characteristics. Tissue Eng Part A. 2009;15(7):1843–54.
Piper SL, Wang M, Yamamoto A, Malek F, Luu A, Kuo AC, et al. Inducible immortality in hTERT-human mesenchymal stem cells. J Orthop Res. 2012;30(12):1879–85.
Wang L, Song K, Qu X, Wang H, Zhu H, Xu X, et al. hTERT gene immortalized human adipose-derived stem cells and its multiple differentiations: a preliminary investigation. Appl Biochem Biotechnol. 2013;169(5):1546–56.
Kitagawa M, Tahara H, Kitagawa S, Oka H, Kudo Y, Sato S, et al. Characterization of established cementoblast-like cell lines from human cementum-lining cells in vitro and in vivo. Bone. 2006;39(5):1035–42.
Xiaoxue Y, Zhongqiang C, Zhaoqing G, Gengting D, Qingjun M, Shenwu W. Immortalization of human osteoblasts by transferring human telomerase reverse transcriptase gene. Biochem Biophys Res Commun. 2004;315(3):643–51.
Yang JW, Nagavarapu U, Relloma K, Sjaastad MD, Moss WC, Passaniti A, et al. Telomerized human microvasculature is functional in vivo. Nat Biotechnol. 2001;19(3):219–24.
Chen X, Thibeault SL. Novel isolation and biochemical characterization of immortalized fibroblasts for tissue engineering vocal fold lamina propria. Tissue Eng Part C Methods. 2009;15(2):201–12.
Docheva D, Padula D, Popov C, Weishaupt P, Pragert M, Miosge N, et al. Establishment of immortalized periodontal ligament progenitor cell line and its behavioural analysis on smooth and rough titanium surface. Eur Cell Mater. 2010;19:228–41.
Wieser M, Stadler G, Jennings P, Streubel B, Pfaller W, Ambros P, et al. hTERT alone immortalizes epithelial cells of renal proximal tubules without changing their functional characteristics. Am J Physiol Renal. 2008;295(5):F1365–75.
Wege H, Le HT, Chui MS, Liu L, Wu J, Giri R, et al. Telomerase reconstitution immortalizes human fetal hepatocytes without disrupting their differentiation potential. Gastroenterology. 2003;124(2):432–44.
Watanabe T, Shibata N, Westerman KA, Okitsu T, Allain JE, Sakaguchi M, et al. Establishment of immortalized human hepatic stellate scavenger cells to develop bioartificial livers. Transplantation. 2003;75(11):1873–80.
Park Y, Hosomichi J, Ge C, Xu J, Franceschi R, Kapila S. Immortalization and characterization of mouse temporomandibular joint disc cell clones with capacity for multi-lineage differentiation. Osteoarthritis Cartilage. 2015;23(9):1532–42.
Balducci L, Blasi A, Saldarelli M, Soleti A, Pessina A, Bonomi A, et al. Immortalization of human adipose-derived stromal cells: production of cell lines with high growth rate, mesenchymal marker expression and capability to secrete high levels of angiogenic factors. Stem Cell Res Ther. 2014;5(3):63.
Miceli M, Franci G, Dell’Aversana C, Ricciardiello F, Petraglia F, Carissimo A, et al. MePR: a novel human mesenchymal progenitor model with characteristics of pluripotency. Stem Cells Dev. 2013;22(17):2368–83.
Ravassard P, Hazhouz Y, Pechberty S, Bricout-Neveu E, Armanet M, Czernichow P, et al. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J Clin Invest. 2011;121(9):3589–97.
Narushima M, Kobayashi N, Okitsu T, Tanaka Y, Li SA, Chen Y, et al. A human beta-cell line for transplantation therapy to control type 1 diabetes. Nat Biotechnol. 2005;23(10):1274–82.
Krump-Konvalinkova V, Bittinger F, Unger RE, Peters K, Lehr HA, Kirkpatrick CJ. Generation of human pulmonary microvascular endothelial cell lines. Lab Invest. 2001;81(12):1717–27.
Fujii S, Maeda H, Wada N, Kano Y, Akamine A. Establishing and characterizing human periodontal ligament fibroblasts immortalized by SV40T-antigen and hTERT gene transfer. Cell Tissue Res. 2006;324(1):117–25.
Davies BR, Steele IA, Edmondson RJ, Zwolinski SA, Saretzki G, von Zglinicki T, et al. Immortalisation of human ovarian surface epithelium with telomerase and temperature-sensitive SV40 large T antigen. Exp Cell Res. 2003;288(2):390–402.
Wilmer MJ, Saleem MA, Masereeuw R, Ni L, van der Velden TJ, Russel FG, et al. Novel conditionally immortalized human proximal tubule cell line expressing functional influx and efflux transporters. Cell Tissue Res. 2010;339(2):449–57.
Kowolik CM, Liang S, Yu Y, Yee JK. Cre-mediated reversible immortalization of human renal proximal tubular epithelial cells. Oncogene. 2004;23(35):5950–7.
Zhang Y, Nuglozeh E, Toure F, Schmidt AM, Vunjak-Novakovic G. Controllable expansion of primary cardiomyocytes by reversible immortalization. Hum Gene Ther. 2009;20(12):1687–96.
Wang S, Beattie GM, Mally MI, Cirulli V, Itkin-Ansari P, Lopez AD, et al. Isolation and characterization of a cell line from the epithelial cells of the human fetal pancreas. Cell Transplant. 1997;6(1):59–67.
Halvorsen TL, Leibowitz G, Levine F. Telomerase activity is sufficient to allow transformed cells to escape from crisis. Mol Cell Biol. 1999;19(3):1864–70.
Cocce V, Balducci L, Falchetti ML, Pascucci L, Ciusani E, Brini AT, et al. Fluorescent immortalized human adipose derived stromal cells (hASCs-TS/GFP+) for studying cell drug delivery mediated by microvesicles. Anticancer Agents Med Chem. 2017. https://doi.org/10.2174/1871520617666170327113932.
Hung SC, Yang DM, Chang CF, Lin RJ, Wang JS, Low-Tone Ho L, et al. Immortalization without neoplastic transformation of human mesenchymal stem cells by transduction with HPV16 E6/E7 genes. Int J Cancer. 2004;110(3):313–9.
Yang G, Rosen DG, Mercado-Uribe I, Colacino JA, Mills GB, Bast RC Jr, et al. Knockdown of p53 combined with expression of the catalytic subunit of telomerase is sufficient to immortalize primary human ovarian surface epithelial cells. Carcinogenesis. 2007;28(1):174–82.
Yang G, Rosen DG, Colacino JA, Mercado-Uribe I, Liu J. Disruption of the retinoblastoma pathway by small interfering RNA and ectopic expression of the catalytic subunit of telomerase lead to immortalization of human ovarian surface epithelial cells. Oncogene. 2007;26(10):1492–8.
Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64(24):9027–34.
Cao Y, Gao Q, Wazer DE, Band V. Abrogation of wild-type p53-mediated transactivation is insufficient for mutant p53-induced immortalization of normal human mammary epithelial cells. Cancer Res. 1997;57(24):5584–9.
Gao Q, Hauser SH, Liu XL, Wazer DE, Madoc-Jones H, Band V. Mutant p53-induced immortalization of primary human mammary epithelial cells. Cancer Res. 1996;56(13):3129–33.
Wazer DE, Chu Q, Liu XL, Gao Q, Safaii H, Band V. Loss of p53 protein during radiation transformation of primary human mammary epithelial cells. Mol Cell Biol. 1994;14(4):2468–78.
Ko E, Lee KY, Hwang DS. Human umbilical cord blood-derived mesenchymal stem cells undergo cellular senescence in response to oxidative stress. Stem Cells Dev. 2012;21(11):1877–86.
Stampfer MR, Bartley JC. Induction of transformation and continuous cell lines from normal human mammary epithelial cells after exposure to benzo[a]pyrene. Proc Natl Acad Sci U S A. 1985;82(8):2394–8.
Yasaei H, Gilham E, Pickles JC, Roberts TP, O’Donovan M, Newbold RF. Carcinogen-specific mutational and epigenetic alterations in INK4A, INK4B and p53 tumour-suppressor genes drive induced senescence bypass in normal diploid mammalian cells. Oncogene. 2013;32(2):171–9.
Tsutsui T, Taguchi S, Hasegawa K, Ide T, Kojima K, Matsumura M, et al. Reserpine-induced cell transformation without detectable genetic effects in Syrian hamster embryo cells in culture. Carcinogenesis. 1994;15(1):11–4.
Martens U, Lennartsson P, Hogberg J, Stenius U. Low expression of the WAF1/CIP1 gene product, p21, in enzyme-altered foci induced in rat liver by diethylnitrosamine or phenobarbital. Cancer Lett. 1996;104(1):21–6.
Dai F, Yang S, Zhang F, Shi D, Zhang Z, Wu J, et al. hTERT- and hCTLA4Ig-expressing human bone marrow-derived mesenchymal stem cells: in vitro and in vivo characterization and osteogenic differentiation. J Tissue Eng Regen Med. 2017;11(2):400–11.
Bravard A, Hoffschir F, Ricoul M, Cassingena R, Estrade S, Luccioni C, et al. Alterations of the glutathione cycle enzymes during and after SV40-transformation of human fibroblasts. Carcinogenesis. 1993;14(1):21–4.
Li ZH, Tang QB, Wang J, Zhou L, Huang WL, Liu RY, et al. Hepatitis C virus core protein induces malignant transformation of biliary epithelial cells by activating nuclear factor-kappaB pathway. J Gastroenterol Hepatol. 2010;25(7):1315–20.
Shen ZY, Xu LY, Li EM, Cai WJ, Shen J, Chen MH, et al. The multistage process of carcinogenesis in human esophageal epithelial cells induced by human papillomavirus. Oncol Rep. 2004;11(3):647–54.
Uren A, Fallen S, Yuan H, Usubutun A, Kucukali T, Schlegel R, et al. Activation of the canonical Wnt pathway during genital keratinocyte transformation: a model for cervical cancer progression. Cancer Res. 2005;65(14):6199–206.
Lehman JM, Trevor K. Karyology and tumorigenicity of a simian virus 40-transformed Chinese hamster cell clone. J Cell Physiol. 1979;98(3):443–50.
Liang S, Kahlenberg MS, Rousseau DL Jr, Hornsby PJ. Neoplastic conversion of human colon smooth muscle cells: no requirement for telomerase. Mol Carcinog. 2008;47(6):478–84.
Sheibani N, Rhim JS, Allen-Hoffmann BL. Malignant human papillomavirus type 16-transformed human keratinocytes exhibit altered expression of extracellular matrix glycoproteins. Cancer Res. 1991;51(21):5967–75.
Woods Ignatoski KM, Dziubinski ML, Ammerman C, Ethier SP. Cooperative interactions of HER-2 and HPV-16 oncoproteins in the malignant transformation of human mammary epithelial cells. Neoplasia. 2005;7(8):788–98.
Shen ZY, Xu LY, Chen MH, Cai WJ, Shen J, Chen JY, et al. Cytogenetic and molecular genetic changes in malignant transformation of immortalized esophageal epithelial cells. Int J Mol Med. 2003;12(2):219–24.
Sun B, Chen M, Hawks CL, Pereira-Smith OM, Hornsby PJ. The minimal set of genetic alterations required for conversion of primary human fibroblasts to cancer cells in the subrenal capsule assay. Neoplasia. 2005;7(6):585–93.
Rhim JS, Kawakami T, Pierce J, Sanford K, Arnstein P. Cooperation of V-oncogenes in human epithelial cell transformation. Leukemia. 1988;2(12 Suppl):151S–9S.
Sun B, Chen M, Hawks C, Hornsby PJ, Wang X. Tumorigenic study on hepatocytes coexpressing SV40 with Ras. Mol Carcinog. 2006;45(4):213–9.
Rhim JS, Webber MM, Bello D, Lee MS, Arnstein P, Chen LS, et al. Stepwise immortalization and transformation of adult human prostate epithelial cells by a combination of HPV-18 and v-Ki-ras. Proc Natl Acad Sci U S A. 1994;91(25):11874–8.
Sonoda Y, Ozawa T, Hirose Y, Aldape KD, McMahon M, Berger MS, et al. Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res. 2001;61(13):4956–60.
Sato M, Larsen JE, Lee W, Sun H, Shames DS, Dalvi MP, et al. Human lung epithelial cells progressed to malignancy through specific oncogenic manipulations. Mol Cancer Res. 2013;11(6):638–50.
Lundberg AS, Randell SH, Stewart SA, Elenbaas B, Hartwell KA, Brooks MW, et al. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene. 2002;21(29):4577–86.
Akimov SS, Ramezani A, Hawley TS, Hawley RG. Bypass of senescence, immortalization, and transformation of human hematopoietic progenitor cells. Stem Cells. 2005;23(9):1423–33.
Kusakari T, Kariya M, Mandai M, Tsuruta Y, Hamid AA, Fukuhara K, et al. C-erbB-2 or mutant Ha-ras induced malignant transformation of immortalized human ovarian surface epithelial cells in vitro. Br J Cancer. 2003;89(12):2293–8.
Liu J, Yang G, Thompson-Lanza JA, Glassman A, Hayes K, Patterson A, et al. A genetically defined model for human ovarian cancer. Cancer Res. 2004;64(5):1655–63.
MacKenzie KL, Franco S, Naiyer AJ, May C, Sadelain M, Rafii S, et al. Multiple stages of malignant transformation of human endothelial cells modelled by co-expression of telomerase reverse transcriptase, SV40 T antigen and oncogenic N-ras. Oncogene. 2002;21(27):4200–11.
Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400(6743):464–8.
Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15(1):50–65.
Zhang H, Jin Y, Chen X, Jin C, Law S, Tsao SW, et al. Papillomavirus type 16 E6/E7 and human telomerase reverse transcriptase in esophageal cell immortalization and early transformation. Cancer Lett. 2007;245(1–2):184–94.
Thomas M, Suwa T, Yang L, Zhao L, Hawks CL, Hornsby PJ. Cooperation of hTERT, SV40 T antigen and oncogenic Ras in tumorigenesis: a cell transplantation model using bovine adrenocortical cells. Neoplasia. 2002;4(6):493–500.
Rao K, Alper O, Opheim KE, Bonnet G, Wolfe K, Bryant E, et al. Cytogenetic characterization and H-ras associated transformation of immortalized human mammary epithelial cells. Cancer Cell Int. 2006;6:15.
Shima Y, Okamoto T, Aoyama T, Yasura K, Ishibe T, Nishijo K, et al. In vitro transformation of mesenchymal stem cells by oncogenic H-rasVal12. Biochem Biophys Res Commun. 2007;353(1):60–6.
Min B, Woo K, Baek J, Lee G, Park N. Malignant transformation of hpv-immortalized human oral keratinocytes by chemical carcinogens. Int J Oncol. 1995;7(2):249–56.
Nakao Y, Yang X, Yokoyama M, Pater MM, Pater A. Malignant transformation of human ectocervical cells immortalized by HPV 18: in vitro model of carcinogenesis by cigarette smoke. Carcinogenesis. 1996;17(3):577–83.
Yang X, Jin G, Nakao Y, Rahimtula M, Pater MM, Pater A. Malignant transformation of HPV 16-immortalized human endocervical cells by cigarette smoke condensate and characterization of multistage carcinogenesis. Int J Cancer. 1996;65(3):338–44.
Shin KH, Min BM, Cherrick HM, Park NH. Combined effects of human papillomavirus-18 and N-methyl-N’-nitro-N-nitrosoguanidine on the transformation of normal human oral keratinocytes. Mol Carcinog. 1994;9(2):76–86.
Kim MS, Shin KH, Baek JH, Cherrick HM, Park NH. HPV-16, tobacco-specific N-nitrosamine, and N-methyl-N’-nitro-N-nitrosoguanidine in oral carcinogenesis. Cancer Res. 1993;53(20):4811–6.
Cleaver AL, Bhamidipaty K, Wylie B, Connor T, Robinson C, Robinson BW, et al. Long-term exposure of mesothelial cells to SV40 and asbestos leads to malignant transformation and chemotherapy resistance. Carcinogenesis. 2014;35(2):407–14.
Perkins DJ, Barber GN. Defects in translational regulation mediated by the alpha subunit of eukaryotic initiation factor 2 inhibit antiviral activity and facilitate the malignant transformation of human fibroblasts. Mol Cell Biol. 2004;24(5):2025–40.
Authors’ contributions
YW and MP conceived the original idea; YW and SC drafted the manuscript; YW, SC, ZY and MP revised. All authors read and approved the final manuscript.
Acknowledgements
We thank Suzanne Danley for editing the manuscript.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number (AR067747-01A1) and the Musculoskeletal Transplant Foundation to Ming Pei, the National Natural Science Foundation of China (81672157) to Zuoqin Yan and Natural Science Foundation of China (81601889) to Song Chen.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, Y., Chen, S., Yan, Z. et al. A prospect of cell immortalization combined with matrix microenvironmental optimization strategy for tissue engineering and regeneration. Cell Biosci 9, 7 (2019). https://doi.org/10.1186/s13578-018-0264-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-018-0264-9