
Abstract
Background
Epigenetic alterations are known contributors to cancer development and aggressiveness. Additional to alterations in cancer cells, aberrant epigenetic marks are present in cells of the tumor microenvironment, including lymphocytes and tumor-associated macrophages, which are often overlooked but known to be a contributing factor to a favorable environment for tumor growth. Therefore, the main aim of this review is to give an overview of the epigenetic alterations affecting immune cells in the tumor microenvironment to provoke an immunosuppressive function and contribute to cancer development. Moreover, immunotherapy is briefly discussed in the context of epigenetics, describing both its combination with epigenetic drugs and the need for epigenetic biomarkers to predict response to immune checkpoint blockage.
Main body
Combining both topics, epigenetic machinery plays a central role in generating an immunosuppressive environment for cancer growth, which creates a barrier for immunotherapy to be successful. Furthermore, epigenetic-directed compounds may not only affect cancer cells but also immune cells in the tumor microenvironment, which could be beneficial for the clinical response to immunotherapy.
Conclusion
Thus, modulating epigenetics in combination with immunotherapy might be a promising therapeutic option to improve the success of this therapy. Further studies are necessary to (1) understand in depth the impact of the epigenetic machinery in the tumor microenvironment; (2) how the epigenetic machinery can be modulated according to tumor type to increase response to immunotherapy and (3) find reliable biomarkers for a better selection of patients eligible to immunotherapy.
Similar content being viewed by others
Background
The epigenome is defined by heritable alterations in gene expression, either activation or suppression, without altering the DNA nucleotide sequence. The mechanisms responsible for these changes can be broadly divided into altered gene accessibility for the transcriptional machinery, disrupted chromatin organization or modulation of gene expression at the post-transcriptional level through altered mRNA translation mainly by non-coding RNAs, including miRNAs. Fundamental mechanisms for epigenetic regulation include DNA methylation, histone modifications, chromatin remodeling and non-coding RNA interference [1]. Nevertheless, these modifications of the RNA sequence and their associated regulatory factors represent functionally relevant changes to the transcriptome without altering the RNA ribonucleotide sequence, recently encompassed by the term ‘epitranscriptomics’ [2]. Since epitranscriptomics is recognized as a different area of study that goes beyond the scope of this review, we will only focus on DNA methylation, histone modifications and chromatin remodeling as epigenetic regulatory mechanisms. Those mechanisms are briefly discussed below.
Epigenome functions are essential for normal gene expression, and their modifications affect primary cellular processes like proliferation, differentiation, and apoptosis. Even though its effect on human carcinogenesis is not entirely acknowledged, epigenetic dysfunction is a rising hallmark of malignancy. As epigenetic modifications are essential in the regulation of normal gene expression, epigenetic deregulation results in aberrant gene expression patterns which have been found to favor tumorigenesis, among others [1].
Whereas the role of epigenetic modifications in cancer research has been mainly focused on cancer cells, rising evidence indicates their contribution to the development of a favorable tumor microenvironment (TME), including their effect on surrounding cell phenotypes like fibroblasts, immune cells, endothelial and inflammatory cells, blood and lymphatic vascular networks, and the extracellular matrix (Table 1) [3]. Nevertheless, due to the extreme complexity of the variety of cells and their potential epigenetic modifications affecting tumorigenesis, this review will be focused on the epigenetic regulation of different immune cell types in the TME and their involvement in the generation of a cancer-prone TME.
We further discuss the inhibition of epigenetic modulators as therapeutic option to modify the immunosuppressive TME, and we provide an overview on immunotherapy and the potential of epigenetic biomarkers of response to this therapy. Finally, the application and success of immunotherapy as well as the inhibition of epigenetic processes involved in immune activation will be briefly discussed in the context of bladder cancer (BC).
Epigenetic regulatory mechanisms
DNA methylation
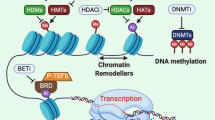
DNA methylation represents a process by which methyl groups are transferred onto the 5′ position of a cytosine molecule without altering the DNA sequence. Methylation commonly occurs on the cytosine of CpG sites, meaning that the cytosine molecule precedes a guanine. DNA regions with a higher density of these CpG sites (so-called CpG islands) have been found throughout the genome, mostly coinciding with gene regulatory regions. This way, methylation of CpG islands plays an important role in the regulation of normal gene expression (Fig. 1) [62, 63].

DNA methylation, histone modifications and chromatin remodeling as regulatory mechanisms of epigenetic gene regulation. DNA methylation represents a process by which methyl groups are transferred onto the 5′ position of a cytosine molecule, commonly in the context of CpG sites, without altering the DNA sequence. Histone modifications include post-translational modifications at the histone N-terminal tails, such as acetylation, methylation and phosphorylation, causing chromatin structure alterations. Changes in nucleosome position are also responsible for rearrangement of chromatin structure, a process known as chromatin remodeling. Nucleosomes can be affected in several ways, including nucleosome sliding, nucleosome ejection and histone eviction. Histone variants add further complexity to epigenetic regulation of the genome. Various histone variants are characterized for H2A, H2B and H3. All these mechanisms are highly interrelated and play an important role in the regulation of gene accessibility to the transcriptional machinery
DNA methyltransferases (DNMTs) are the enzymes responsible for the control of DNA methylation patterns through maintenance DNA methylation and de novo DNA methylation. In general, DNMT1 is the enzyme responsible for maintenance of inherited DNA methylation, whereas DNMT3a and DNMT3b provide de novo DNA methylation. Nevertheless, DNMT3a and DNMT3b methyltransferases have been described to perform maintenance methylation as well, and DNMT1 has also been found to carry out de novo DNA methylation [63].
Alterations in DNA methylation status have been described in various diseases, including cancer. For example, gene silencing of tumor suppressor genes (TSGs) is the result of the hypermethylation of CpG islands in the promoter regions of those genes. TSGs are mainly involved in biological pathways like cell cycle control, DNA repair and apoptosis, and its silencing has been frequently found in tumorigenesis [64].
Histone modifications
Chromatin structure changes as a result of dynamic processes involving post-translational modifications (PTMs) at the histone N-terminal tails. Various PTMs can be distinguished, including histone acetylation, methylation and phosphorylation as well as less known ubiquitylation, deamination and sumoylation, which affect chromatin packaging and availability to the gene transcription machinery (Fig. 1) [65]. Currently known histone PTMs have been extensively reviewed. Here, we briefly mention the three most common histone PTM activities.
Histone acetylation consists of the reversible addition of acetyl groups to the histone tail by histone acetyltransferases (HATs), which weakens the DNA-histone bonds and allows binding of transcription factors. Contrarily, histone deacetylases (HDACs) remove those acetyl groups, allowing for compact wrapping of the DNA around histones, disabling the access of other enzymes. Regarding histone methylation, the transfer of methyl groups is a reversible process regulated by histone methyltransferases and demethylases. The attachment of a methyl group to the histone tail may differentially affect gene expression depending on the specific residue modified. Likewise, the interaction between the DNA and histone tails is regulated by histone (de)phosphorylation processes [66].
Chromatin remodeling
Changes in nucleosome position have also been found responsible for rearrangement of chromatin structure, a process known as chromatin remodeling. Nucleosomes, consisting of a histone core (H2A, H2B, H3 and H4) wrapped by an approximately 150-bp DNA sequence, can be affected in several ways, including nucleosome sliding, nucleosome ejection and histone eviction. Nucleosome sliding represents the movement of the histone octamer across the DNA sequence, whereas nucleosome ejection implies the complete segregation of the histone core from the DNA. Histone eviction includes the disintegration of the core histone octamer trough removal or replacement of H2A–H2B dimers (Fig. 1) [67].
Since nucleosome sliding and ejection as well as removal of the H2A–H2B dimers result in DNA exposition and nucleosome destabilization, these processes play an important role in the regulation of gene accessibility to the transcriptional machinery.
Histone variants
Histone variants add further complexity to epigenetic regulation of the genome. They represent a unique protein sequence compared to core histones and can be identified by a combination of variant-specific proteins and chromatin remodeling complexes which control their localization within the genome. Various histone variants are characterized for H2A (such as macroH2A, H2A.B, H2A.J, H2A.X and H2A.Z.1/2), H2B (including TSH2B) and H3 (like H3.1 till H3.8, H3.X, H3.Y and CENP-A), whereas no variants have been described for H4. Additionally, histone variants are subjected to post-translational modifications which elaborates the epigenetic control of gene expression (Fig. 1) [68, 69].
Epigenetic regulation of immune cell function in TME
A favorable TME is characterized by immune tolerance. Cancer cells employ a variety of epigenetic regulated-immune escape mechanisms, including downregulation of tumor-associated antigens (TAAs), loss of antigen processing and presentation machinery (APM) as well as expression of a tumor-promoting balance in co-stimulatory and co-inhibitory molecules (also known as immune checkpoint receptors). Whereas these processes have been extensively studied and reviewed, epigenetic alterations affecting immune cell function in the TME represent a growing area of investigation. The epigenetic-induced immunosuppressive function of dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), tumor-infiltrating lymphocytes (TILs), regulatory T cells (Tregs) and natural killer cells (NK cells) in the TME will be discussed below.
Dendritic cells
DCs represent important antigen-presenting cells (APCs) that mediate antigen-specific anti-tumor immune responses mainly through the activation of T cells. Whereas these cells are normally able to capture TAAs expressed on cancer cells through accurate MHC expression and cross-present them to cytotoxic T cells (CTLs) by the expression of co-stimulatory molecules, resulting in tumor elimination, tumor-infiltrating DCs show an immune-tolerant phenotype favoring tumor growth [18]. Next to low MHC expression and repression of various co-stimulatory molecules in tumor-infiltrating DCs, epigenetic alterations affecting DC polarization and activity are suggested to impair an effective anti-tumor immune response.
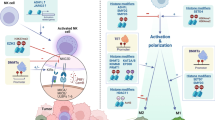
For example, dynamic changes in the levels of chromatin regulator ‘special AT-rich sequence binding 1 (SATB1)’ are essential for the generation of inflammatory DCs and their anti-tumorigenic activity. SATB1 recruits chromatin remodeling complexes to anchored DNA regions, consisting of a characteristic ‘ATC-sequence context’ (well-mixed A’s, T’s and C’s but not G’s on a single strand), thereby controlling gene transcription over long-distance DNA sequences through the regulation of nucleosomal positioning and histone modification [70]. Additionally, the recruitment of HATs and HDACs directly to gene promoter regions has been reported [71]. A continuous increased expression of SATB1 has been described to convert inflammatory anti-tumor DCs into pro-tumor DCs by enhanced secretion of pro-tumorigenic cytokine IL-6 and immunosuppressive factor Galectin-1, activating immune-evasive pathways in these cells [7]. Accordingly, SATB1 has been found to be overexpressed in a wide range of tumors, including breast, lung, pancreas, colorectal, liver, bladder, prostate and ovarian cancer, and has been associated with tumor progression and poor prognosis [19]. Additionally, next to its role in the direct activation of IL-6 transcription, Kruppel-like factor 4 (KLF4) modulates IL-6 production at the post-translational level through histone acetylation. Decreased expression of KLF4 has been described in many tumors, including esophageal, lung, gastric, intestinal, colon and prostate cancer, leading to altered production of cytokine IL-6 in DCs (Fig. 2, Table 1) [20, 21].

Epigenetic regulation of immune cells in the tumor microenvironment. Decreased KLF4 and increased SATB1 expression affect IL-6 (upregulation) and Galectin (downregulation) expression, remodeling anti-tumor DCs into pro-tumor DCs. MDSCs expansion, accumulation and recruitment are favored by STAT3-induced expression of immunosuppressive factors S100A8, Arg1 and S100A9. In this cell population, STAT3 expression is controlled by DNMTT3a/b, HDAC6 and HDAC11. Macrophages can convert into TAMs under the influence of multiple epigenetic factors, including DNMT3b, PRMT1, HDAC3/4, HDAC9 and SIRT2, favoring acquisition of the M2 phenotype through various pathways, such as increased PPARγ and Arg1 expression as well as downregulation of inflammatory factors TNF-α and IL-1β. SMYD3 activates M2 marker ALOX15. Impaired NK-cell anti-tumor cytotoxicity can be the result of increased EZH2 expression, which downregulates activating NK-cell receptor NKG2D through enhanced H3K27me3 levels. The same way, EZH2 also regulates inhibition of regulatory T-cell pro-inflammatory activities. Naïve CD8 + T-cells differentiate into TILs or exhausted CD8 + cells dependent on epigenetic profile. Whereas specific DNA methylation patterns of CTLA4, PDCD1 and LAG3 are identified in exhausted CD8 + T-cells, DNMT1 and EZH2 inhibit CD8 + TILs infiltration through downregulation of CXCL9 and CXCL10 chemokines. TGF-β and SATB1 affect TILs infiltration by controlling PD-1 expression. DCs, dendritic cells; MDSCs, myeloid-derived suppressor cells; TAMs, tumor-associated macrophages; NK, natural killer; Tregs, regulatory T-cells; TILs, tumor-infiltrating lymphocytes
Taken together, increasing our knowledge on tumor-induced epigenetic modifications affecting DC polarization and activity might help modifying the TME to become more “receptive” to the development of an effective anti-tumor response.
Myeloid-derived suppressor cells
MDSCs represent immature myeloid cells and are mainly characterized by their immunosuppressive function providing tumor immune evasion [27]. These cells are known to have a major impact on cancer progression as the TME has been found to support this cell population, promoting MDSC persistence, proliferation and function. Indeed, the presence of MDSCs has been associated with poor prognosis and reduced patients’ survival in many cancer types, including head and neck, breast, lung, kidney and prostate [26, 27]. Several studies have suggested the role of epigenetic mechanisms in MDSC accumulation and functions.
For example, an elevated expression of signal transducer and activator of transcription 3 (STAT3) has been defined in several tumor types including lung, pancreas and renal cancer [28,29,30,31]. Overexpression of STAT3 can be the result of promoter silencing of DNMT3a and DNMT3b through hypermethylation, followed by promoter hypomethylation of the STAT3 gene. Besides, Villagra et al. proposed HDAC11 as a transcriptional repressor of IL-10 (a STAT3-activating cytokine) through interaction with the IL-10 promoter at the chromatin level and indicated that elevated levels of STAT3 in APCs might be associated with the absence of HDAC11 [72]. More recently, HDAC11 has been described as an essential regulator of IL-10 levels in myeloid cells and its role in the MDSC expansion was demonstrated [73]. Moreover, Cheng et al. showed that HDAC6 has a regulatory function in STAT3 activation in the MDSC population. Surprisingly, HDAC6 seems to act as a transcriptional activator of IL-10 expression [74]. Next to their possible individual implications, HDAC6 and HDAC11 have been reported to interact and be recruited together toward the IL-10 promoter site where they control IL-10 transcription and subsequent STAT3 expression [74]. Increased STAT3 expression leads to augmented expression of immunosuppressive factors S100A8 and Arginase 1 (Arg1) in MDSCs. Additionally, the induction of these proteins, together with the STAT3-mediated induction of S100A9 expression, has been shown to provide expansion, accumulation and recruitment of immunosuppressive MDSCs in TME (Fig. 2, Table 1) [29, 75].
As abovementioned, this MDSC population represents immature myeloid cells which fail to differentiate into macrophages and DCs. Several studies already described the accumulation of immature myeloid cells as a result of retinoblastoma gene (Rb) transcriptional silencing [76, 77]. Concordantly, Young et al. proposed that HDAC2 might be the epigenetic regulator provoking Rb transcriptional silencing in this cell population through its direct interaction with the Rb1 gene promoter [78].
Taken together, the MDSC population represents a major barrier for immunotherapy. Accordingly, further research is needed to increase our knowledge on MDSCs in the TME and be able to improve the ability to revert their immunosuppressive function.
Tumor-associated macrophages
TAMs represent the main component of the immune infiltrates in TME of solid tumors and have frequently been associated with worse prognosis [79]. By continuously sensing their surrounding environment, this cell population has refined regulatory epigenetic mechanisms to manage their polarization state. Depending on their polarization into classically activated (M1) or alternatively activated (M2) macrophages, they inhibit or promote tumor growth, respectively. Epigenetic modifications have been widely shown to be involved in macrophage differentiation, activation and survival [80].
Yang et al. indicated a significant role for DNMT3b in macrophage polarization. They showed that DNMT3b knockdown induces elevated expression of M2 macrophage markers, such as Arg1, as well as increased Arg1 function. In concordance with these results, Arg1 activity has been reported to define immunosuppressive subsets of TAMs. Additionally, DNMT3 knockdown resulted in significantly decreased expression of inflammatory genes, such as TNF-α and IL-1β, emphasizing the importance of DNMT3b in the regulation of both macrophage differentiation and inflammation [81, 82]. Moreover, DNMT3b has been reported to methylate the peroxisome proliferator-activated receptor γ (PPARγ) promoter region.
Protein arginine methyltransferase 1 (PRMT1) has been reported as a positive regulator of PPARγ-dependent M2 polarization through methylation of the arginine located at residue 3 on the tail of histone 4 (H4R3me2a). Furthermore, Ishii et al. demonstrated that expression of M2 macrophage markers seems to be epigenetically controlled by convertible changes in H3K4 and H3K27 methylation [83]. Accordingly, H3K4 methyltransferase SET and MYND Domain 3 (SMYD3) has been shown to play a role in M2 differentiation. Kittan et al. [84] showed that the increased expression of SMYD3 is associated with the methylation and activation of the M2 marker arachidonate 15-lipoxygenase (ALOX15). The only histone demethylase recognized as a crucial regulator of M2 polarization is Jumonji domain-containing protein D3 (JMJD3), a H3K27 demethylase. IL-4-induced STAT6 activation leads to STAT6-mediated increased expression of JMJD3, provoking H3K27me2/3 demethylation and subsequent transcriptional activation of several M2 marker genes, including Arg1 [83, 85]. IL-4 increased expression has been found in various tumor types, including breast, lung, pancreatic, colon, bladder and ovarian carcinomas [42].
Whereas the role of HATs in macrophage polarization remains unclear, the function of HDACs as epigenetic modifiers in the regulation of M2 differentiation and phenotypic control has been explored by various studies. Mullican et al. indicated that HDAC3 activity leads to suppressed IL-4 activity through deacetylation of histone tails at regulatory sequences. Together with the finding that HDAC3 knockdown resulted in decreased inflammatory gene expression, HDAC3 has been proposed to negatively regulate M2 polarization [86, 87]. HDAC9 has also been described as a negative regulator of M2 phenotype as HDAC9 deficiency results in PPARγ promoter acetylation and increased PPARγ expression levels, promoting M2 polarization and downregulating M1 phenotype inflammatory genes [88]. Contrarily, HDAC4 positively regulates the M2 phenotype through IL-4-activated HDAC4-induced STAT6 signaling and Arg1 expression [89]. Another HDAC, SIRT2, positively controls M2 polarization through its function in the expression of M2 macrophage markers, such as Arg1, and downregulation of M1 polarization by NFkB acetylation, provoking decreased NFkB signaling and suppression of IL-1β expression (Fig. 2, Table 1) [90].
Accordingly, targeting these epigenetic enzymes responsible for polarization of TAMs into M2 macrophages would prevent their tumor-supporting function. Nevertheless, it should be considered that these regulators disclose secondary functions and that histone-modifying enzymes also affect proteins other than histones.
Tumor-infiltrating lymphocytes
TILs represent the major component of the adaptive immune system in the TME and can be classified into two main categories: 1) CD4-expressing T cells (CD4 + T cells), which can differentiate into the T helper1 (Th1) or T helper2 (Th2) phenotype, and 2) CD8-expressing T cells (CD8 + T cells), which are able to eliminate tumor cells after differentiation into cytotoxic effector T lymphocytes.
Whereas the important role of CD8 + CTLs in anti-tumor immune response has been known for many years, the potential importance of CD4 + Th cells in the generation and maintenance of anti-tumor activity has only recently been reported [91]. Even though further research is needed to find out whether and, if so, how epigenetic mechanisms affect CD4 + cells in an immunosuppressive TME, DNA demethylation has been reported to play an important role in differentiation of CD4 + T cells toward Th1/Th2 lymphocytes [92].
Additionally, epigenetic modulation has been found to control rapid activation and differentiation of naïve CD8 + into CTLs upon antigen stimulation. For example, Peng et al. associated DNMT1-mediated DNA methylation and enhancer of zeste homolog 2 (EZH2)-mediated H3K27 trimethylation with impaired T-cell infiltration in the TME through downregulation of CXCL9 and CXCL10 chemokine expression [93]. Furthermore, Yang et al. reported that whole-genome methylation profiling showed a distinct methylome pattern for tumor-reactive CD8 + T cells compared to the naïve subtype. Moreover, specific DNA methylation patterns have been discovered in exhausted CD8 + T cells. PDCD1 and CTLA4 expression in exhausted CD8 + T cells has been found to be epigenetically controlled by DNA demethylation, and the LAG3 gene has been found methylated in naïve cells but demethylated during the activation of naïve CD8 + T cells [94]. Ghoneim et al. [95] found that high programmed death 1 (PD-1)-expressing tumor-infiltrating CD8 + T cells in prostate cancer display exhaustion-associated DNA methylation patterns. Stephen et al. further showed that chromatin organizer Satb1 recruits the nucleosome remodeling deacetylase complex to regulatory regions of the Pdcd1 gene, reducing PD-1 expression levels upon T-cell activation. Nevertheless, Satb1 is known to be downregulated by Smad proteins under the influence of TGF-β, an immunosuppressive cytokine found to play a relevant role in cancer, resulting in elevated PD-1 levels. Accordingly, Satb1 prevents premature T-cell exhaustion by controlling PD-1 expression, a pathway that is altered in cancer, causing reduced anti-tumor activity [96, 97]. Another mechanism underlying tumor-specific T-cell dysfunction in tumor progression is represented by chromatin state dynamics. Philip et al. reported that naïve T cells differentiate into a dysfunctional but reprogrammable chromatin state upon tumor antigen recognition in premalignant lesions, which converts into a fixed non-reprogrammable dysfunctional state during tumor progression. The presence of surface markers CD101 and CD38 has been associated with reduced reprogrammability of high PD-1-expressing tumor-infiltrating CD8 + T cells, a finding with important clinical relevance as these markers can be used to discriminate reprogrammable from non-reprogrammable PD-1 high T cells within the heterogeneous TIL populations [98]. This mechanism might explain why certain patients do not respond to therapies based on immune-checkpoint blockade as well as it provides new insights in possible strategies to revert non-reprogrammable PD-1 high T cells into tumor-reactive CD8 + T cells (Fig. 2, Table 1).
Taken together, impaired CD8 + T cell functions seem to play a major role in the generation of an immunosuppressive TME. Importantly, the prevention of T-cell exhaustion might represent a potential strategy to reverse a TIL-mediated immunosuppressive TME.
Regulatory T cells
Tregs represent a functionally different T cell population which is essential for the maintenance of homeostasis and immune tolerance. Accordingly, mature Tregs provide a tumor-supportive microenvironment [47]. Various studies have reported a key role for Foxp3 in the development of these cells as well as their function, and epigenetic regulation of Tregs through Foxp3 has been emphasized by recent studies [99, 100].
Moreover, epigenetic modifications controlling Treg development and function have been found to play an important role in the establishment of an immunosuppressive TME. Ohkura et al. reported that Treg maturation involves the generation of genome-wide CpG DNA hypomethylation pattern, needed for Treg-specific gene expression and immunosuppressive activity [101]. Besides, Foxp3 seems to exert an EZH2-mediated repressive role upon CD28-mediated Treg activation as target genes show elevated H2K27me3 levels. As CD28 not only provides a key role in the stimulation of Tregs, but also in effector T-cell (CD4+/CD8+) activation, this suppressive role of Foxp3 might be essential to preserve the Treg-specific gene expression profile upon T cell stimulation through downregulation of genes involved in the effector T-cell activation [102]. Indeed, Wang et al. showed that inhibition of EZH2 resulted in Treg-mediated pro-inflammatory activities in the TME, supporting the generation of an effector T-cell-mediated anti-tumor immune response (Fig. 2, Table 1) [103].
Taken together, targeting the maturation of functional Tregs might be a potential strategy to convert an immunosuppressive TME into a microenvironment able to provide anti-tumor activity.
Natural killer cells
The NK cell population forms part of the innate immune system and is able to control tumor growth by their ability to recognize and eliminate tumor cells. Epigenetic modification has been reported to play an key role in the NK cell maturation, differentiation and activation [104]. Regulation of the effector function of this cell population mainly depends on the balance between inhibiting and activating receptors present on NK cell surface, the activation status of which seems to be epigenetically modulated, as well. Accordingly, rising evidence indicates the involvement of epigenetic processes in impairing NK-cell mediated anti-tumor immune response.
An impaired NK-cell-mediated anti-tumor immune response is highly associated with NK-cell exhaustion because of diminished expression of activating receptors and increased expression of inhibitory receptors, among others. Specific activating NK-cell receptors include NKG2D, NKp30, NKp44, NKp46 and DNAM-1/CD226, whereas inhibitory receptors are represented by PD-1, TIM-3, TIGIT and CD94-NKG2A. Fernandez-Sanchez et al. reported the involvement of DNA methylation and histone acetylation in the regulation of NKG2D levels, with NKG2D gene demethylation and H3K9 acetylation providing NKG2D expression [105]. Nevertheless, whether reduced expression of this receptor is due to NKG2D hypermethylation remains unknown. Additionally, Ogbomo et al. proposed that the regulation of NKp30 and NKp46 expression levels is controlled by histone acetylation [106]. Using an HDAC inhibitor (HDACi), they showed that the suppression of NK-cell activity is caused by decreased expression of NKp30 and NKp46, but is independent of activating NKG2D, NKp44 and DNAM-1 expression levels as well as inhibitory NKG2A expression. Finally, Yin et al. revealed that enhanced levels of the activating NKG2D receptor are associated with elevated NK cell expansion and cytotoxicity against the tumor. Inhibition of EZH2 activity has been associated with decreased H3K27me3 levels, providing increased expression of the NKG2D receptor (Fig. 2, Table 1) [107, 108].
Although epigenetic modifications affecting NK cell development and function are widely examined, further studies are needed to increase our knowledge on the epigenetic regulation of NK cells and the potential of epigenetic enzymes/markers as therapeutic targets.
Inhibition of epigenetic modulators as therapeutic option to modify the immunosuppressive TME
A favorable TME, created by tumor cells affecting different immune cell populations, forms a major barrier for cancer therapy. Nevertheless, the study into epigenetic mechanisms underlying the generation of this immunosuppressive TME currently represents subject of utmost interest. As previously stated, a variety of epigenetic modifications affects the phenotypes of diverse immune cell populations in the TME to become immunosuppressive. Accordingly, modifying the TME to become more “receptive” to the development of an effective anti-tumor response could be achieved by molecular re-wiring using pharmacologic modulators of epigenetic enzymes. In this regard, when considering the use of compounds targeting the epigenetic machinery, it is worth considering that these compounds not only affect tumor cells, but also TME cells. Accordingly, a proper selection of inhibitors could become a two-edge sword to tackle tumors.
For example, SATB1 represents an attractive therapeutic target as it modifies different immune cell populations under tumor's influence, including DCs and CTLs. The statins fluvastatin and simvastatin have been found to suppress SATB1 expression, probably acting at the post-translational level [109, 110]. Additionally, in vivo silencing of Satb1 expression in tumor-associated DCs was found to diminish immunosuppression in the TME, boost T-cell mediated anti-tumor activity and delay tumor progression [7]. In the context of immunotherapy, Satb1 has been described as a possible TAA that can be recognized by CD8 + T cells. Accordingly, Wang et al. proposed that Satb1-derived epitope might be used as immune target for cancer vaccine generation [111].
Furthermore, the importance of HDAC activation and IL-6 signaling in controlling the immunosuppressive function of MDSCs as well as its recruitment to the TME has been reported by Nair et al. [26]. Various studies demonstrated the potential of entinostat, a class I HDACi, as therapeutic compound to modulate the immunosuppressive TME through inhibition of MDSC activity. Besides, the application of entinostat not only resulted in decreased MDSC function, but also augmented the effect of checkpoint inhibitor treatment [112,113,114,115]. Kim et al. reported that entinostat reduces the MDSC population and the combination of entinostat with PD-1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) antibodies resulted in an elimination of approximately 80% of the tumor, whereas the use of immune checkpoint inhibitors against PD-1 and CTLA-4 alone did not provide an anti-tumor immune response [113]. The potential role of HDACi in priming the TME for enhanced response to immunotherapy has been further emphasized by Briere et al., who obtained similar results using a class I/IV HDACi (mocetinostat) in combination with anti-programmed death-ligand 1 (PD-L1) antibody [114]. Additionally, Orillion et al. observed reduced macrophage population in the TME after entinostat treatment [112]. Other potential therapeutic targets to suppress MDSCs function would be HDAC2, HDAC6 and HDAC11. Nevertheless, further studies are needed to investigate the therapeutic potential of their corresponding inhibitors [78, 115, 116].
Regarding the polarization of TAMs into M2 macrophages, targeting the enzymes responsible for the acquisition of M2 phenotype would attenuate their tumor-promoting function. Accordingly, Tikhanovich et al. reported the therapeutic potential of AMI-1, a PRMT1 inhibitor, inhibiting M2 processes [117]. Additionally, GSK-J4 might diminish the immunosuppressive, tumor-supporting function of M2 macrophages through KDM6B (a lysine-specific demethylase that demethylates H3K27me2 or H3K27me3) inhibition. Nevertheless, in concordance with the essential role of KDM6B in both M1 and M2 polarization, GSK-J4 has also been found to inhibit the expression of TNF and other M1 inflammatory cytokines [118, 119]. Noteworthy is the effect of histone-modifying enzymes on proteins other than histones and the importance of non-histone protein modifications in macrophage-polarizing process. Moreover, macrophage polarization is a complex dynamic process in which most epigenetic enzymes are neither involved exclusively in the polarization toward M1 or M2 phenotype nor do they all have opposing roles in M1 versus M2 phenotype acquisition [120]. Accordingly, the discovery of therapeutic epigenetic targets in this cell population is very challenging.
Another attractive therapeutic target is EZH2, responsible for the immunosuppressive phenotype of several immune cell populations in the TME, including TILs, Tregs and NK cells. This epigenetic regulator has already been extensively studied for its potential as therapeutic target to convert the immunosuppressive TME into an immune-promoting microenvironment. Various studies described an enhanced effector-T cell infiltration and cytotoxic activation in the TME upon EZH2 inhibition as well as functional alterations of the Treg population resulting in Treg-mediated pro-inflammatory activities [93, 103, 121, 122]. Additionally, targeting EZH2 has been reported as an attractive strategy to combine with immunotherapy, as it might overcome resistance to immune checkpoint therapies, including CTLA-4, PD-1 and PD-L1 [93, 121,122,123]. Among the EZH2-inhibiting compounds, small molecule inhibitors of EZH2, GSK-126, PF-06821497, MAK683, CPI-0209, CPI-1205 and DS-3201 have entered into clinical trials, although none of these has been approved for cancer treatment, yet [122]. Tazemetostat, a selective inhibitor of EZH2, has been approved by the Food and Drug Administration (FDA) in 2020.
Another relevant aspect is the profound interaction between the different immune cell populations in immune response regulation. Therefore, it is important to take into account that epigenetic reprogramming of a certain immunosuppressive immune cell population might positively co-opt other immune cell populations to provoke an effective anti-tumor immune response. One example is the Treg-mediated pro-inflammatory function upon EZH2 inhibition causing increased effector-T cell infiltration and activity. Furthermore, MDSCs play a central role in the immunosuppressive, tumor-promoting TME and have been found to interact with many of the other immune cell populations. Accordingly, epigenetic targeting of this cell type might be sufficient to modify the TME to become more “receptive” to the development of an effective anti-tumor response.
Immunotherapy—an overview
The immune system plays a critical role in cancer development and progression by both eliminating cancer cells and determining tumor immunogenicity [124]. Thus, cancer immunoediting helps to understand how tumors escape the immune system by dividing the process in three distinct phases: “elimination,” “equilibrium” and “escape.” At first, when cancer cells are present, the immune system can recognize these and eliminate them. However, when not all cancer cells are eradicated in this process and an equilibrium is reached, the adaptive immune system impedes tumor’s growth associated with a dormancy state and high genomic instability. T cells, IL-12 and interferon (IFN)-γ are needed to maintain tumor dormancy [125, 126]. Subsequently, cancer cells escape from the immune system by expressing suppressive effects and losing target antigen expression. At this stage, tumor immune escape occurs, since the adaptive immune system fails to recognize cancer cells, which became resistant to immune effector mechanisms and induced an immunosuppressive state [127].
Immune responses are regulated by an interplay of costimulatory and inhibitory signals that balance the immune response and self-tolerance [128]. Immune checkpoint inhibitors are essential as negative signals to stop immune response and impede autoimmunity [129]. PD-1 is expressed in T lymphocytes and prevents the activation of these cells by binding to its ligands PD-L1 and PD-L2 [130]. Additionally, cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) leads to suppression of T-cells activation by competing with the costimulatory signal CD28 for binding to B7-1 and B7-2, attenuating the activation signals of CD28 [131, 132]. Interestingly, PD-L1 is overexpressed in cancer cells, facilitating cancer cells to escape immune surveillance by T cells [133].
Checkpoint inhibitor immunotherapies consist of monoclonal antibodies that target CTLA-4 or the programmed cell death protein 1 pathway (PD-L1, PD-1) [133]. When the antibodies bind to PD-L1/PD-1 or CTLA-4, the inhibitory effect is canceled and an immunological response against cancer cells starts by activation of tumor-reactive T cells [134]. Of note, several clinical trials have demonstrated increased efficacy of combining anti PD-L1/PD1 and anti-CTLA4, although with increased risk of adverse reactions. The use of immune checkpoint inhibitors as cancer therapy was firstly approved for treatment of metastatic melanoma. Since then, several antibodies have been approved for treatment of non-small cell lung cancer, head and neck squamous cell carcinoma, hepatocellular carcinoma and BC [134].
A significant subset of cancer patients does not respond or respond poorly to immune checkpoint blockage treatments [135]. This can be a consequence of primary resistance that occurs prior to treatment, associated with a reduction of antigen expression and changes in metabolic pathways or through acquired resistance during the course of the treatment [136]. The one and foremost biomarker used for prediction of response to PD-L1/PD-1 blockade is PD-L1 expression [137]. It seems to be a biomarker of aggressive disease and it might also be considered a prognostic biomarker. However, the evaluation of PD-L1 as a single biomarker across clinical trials was shown to be heterogeneous [138]. Several reasons can be appointed for the presence of this heterogeneity: (1) clinical trials used different PD-L1 immunohistochemistry scoring assays [137]; (2) the scoring compartment differs for each specific therapy, namely pembrolizumab and nivolumab use PD-L1 tumor cell expression, whereas atezolizumab uses PD-L1 immune cell expression; (3) intratumoral heterogeneity; and (4) the sample available may not represent the full intratumoral heterogeneity [139]. All these data indicate that PD-L1 expression as a single biomarker is probably not adequate to accurately predict immunotherapy response and more reliable biomarkers could help to better predict and improve patient selection for these therapies [139].
Other approaches have been used to try to predict immunotherapy response including tumor mutational burden, tumor mismatch-repair deficiency, grade of TILs [140,141,142], among others, depending on tumor type. For example, the TCGA-based molecular subtypes in BC have been associated with response to immune checkpoint blockage. Specifically, the neuronal subtype seems to display a better response to immunotherapy [143]. However, the criteria to define BC subtypes have to be uniformed before it may be considered a possible biomarker for immunotherapy [144]. Moreover, tumor mutational burden has been proposed to predict response to immunotherapy [145]. With a high rate of mutations, novel antigens emerge regularly so it could potentiate the use of immunotherapy. Nonetheless, some patients with low mutational burden endure response to immunotherapy, which demonstrates that criteria to define the tumor mutational burden have to be clarified [145]. Interestingly, pembrolizumab was approved for patients with microsatellite instability and mismatch repair-deficient malignancies showing progression after failure of other approved treatments [142]. Finally, immune expression profiling has the potential to correctly identify “hot” or “cold” tumors by assessing levels of chemokines, cytokines and cell surface proteins reflecting the inflammatory status [146]. Also, it can take into account the several cell types present in the TME, which can be useful in defining immunotherapy response [147]. Examples include IFN-γ, CXCL9 and CXLC10 whose expression correlates with response to immunotherapy [146, 148].
Epigenetic mechanisms are known to regulate several aspects related to immune regulation and actions [149]. 5-Azacytidine (5-aza), a demethylating agent, was shown to upregulate innate and adaptative immune-related genes, specifically to immune invasion, such as PD-L1 at both transcript and protein levels without altering CD80 and CD86. Furthermore, genes related to antigen presentation including HLA class I, B2M, CD58, TAP1, PMSB9 and PSMB8 were upregulated after 5-aza treatment [150]. Treatment of leukemia cells with decitabine (DAC) translated into upregulation of PD-L1, PD-L2, PD-1 and CTLA-4 in these cells [151]. This topic is thoroughly discussed in a recently published review [152]. A DNA methylation-based profile—EPIMMUNE signature—of stage IV non-small cell lung cancer patients treated with anti-PD-1 therapies associated with improved progression-free and overall survival. EPIMMUNE-negative tumors disclosed a TME enriched in TAMs, cancer-associated fibroblasts and neutrophils. Moreover, unmethylated FOXP1 associated with better progression-free and overall survival [153]. Promoter methylation of RAD51B seems to associate with PD-L1 expression in lung cancer patients, with high levels of RAD51B methylation associating with lower risk of disease progression. Remarkably, combining RAD51B methylation and PD-L1 improved sensitivity to predict response to anti-PD-1 blockade and associated with a lower risk of death [154].
Although several studies showed that modulating epigenetic marks can improve therapeutic response to immune checkpoint inhibitors, the search for biomarkers is ongoing and needs further exploitation (Additional file 1: Table 1).
Epidrugs in cancer
Epigenetic mechanisms including DNA methylation and histone post-translational modifications represent an alluring target for cancer therapy since they are reversible alterations important for tumor cells’ development [155, 156]. Therefore, a class of compounds that came to be known as “epidrugs” were developed targeting these alterations (Table 2) [157]. Currently, DNMT and HDAC inhibitors are the most used epidrugs for cancer treatment in both clinic and clinical trials, namely in combination with chemo- and immunotherapy (Table 2).
DNMT inhibitors are classified into two main groups: nucleoside and non-nucleoside analogues (Table 2) [158]. 5-aza was the first compound developed and approved for clinical usage for DNMT inhibition. Indeed, 5-aza and decitabine (5-aza-2′-deoxycytidine) are already approved for treatment of hematological malignancies including acute myeloid leukemia (AML), myelodysplastic syndrome and chronic myelomonocytic leukemia (CMML) by European Medicines Agency (EMA) and FDA (Table 3) [157, 159, 160]. Nucleoside analogues include analogues of cytosine that are integrated in DNA and lead to the formation of a covalent bond with the DNMT, which results in its degradation and DNA methylation inhibition [157, 161]. Both are nucleoside analogues characterized by replacing cytosine during DNA replication, which translates in the formation of a covalent bond when DNMTs exert their function, resulting in DNMTs’ inhibition and further degradation [161,162,163]. Despite both showing to have anti-tumoral effects, namely by inducing apoptosis, cell and re-expression of tumor suppressor genes [164, 165], 5-aza and DAC display low bioavailability and a limited half-life, which hinders their wide implementation in clinical practice [161, 166]. More stable compounds with the same effect and mechanism of action have been described in the past years including zebularine, 5′-fluoro-2′-deoxycytidine (FdCyd), guadecitabine and RX-3117. For example, FdCyd, a fluoropyrimidine analogue, is less toxic and more stable when compared to 5-aza and DAC (77). Furthermore, FdCyd showed to stop cell arrest at G2/M in HCT116 cells though the DNA damage response pathway [167]. RX-3117 (fluorocyclopentenylcytosine) is a recent cytidine analogue with a modified ribose molecule being activated specifically by uridine-cytidine kinase 2 (UCK2), leading to DNA damage and lower DNA methylation levels though DNMT1 inhibition [168]. Another compound that shows great stability, including being possible to administer orally, is zebularine [169], which showed to have an specific effect on cancer cells and not fibroblasts [170, 171]. However, since it showed great toxicity in primates, it was not continued for clinical trials (Table 3) [172, 173]. Guadecitabine is a CpG dinucleotide analogue hypomethylating agent [174] with proven anti-tumoral activity, such as in combination with cisplatin in platinum-refractory germ cell tumors [175].
Another heterogeneous group of compounds with DNMT inhibition activity is the non-nucleoside analogs, which inhibit DNMTs independently from DNA incorporation (Table 2) [176]. These include natural compounds, DNA binders, SAM competitors and repurposed drugs [176, 177]. Regarding natural compounds, molecules such as genistein, the natural polyphenol epigallocatechin-3-gallate (EGCG) and curcumin that displays anti-inflammatory and anti-oxidant properties showed to inhibit DNA methylation, translating in anti-tumoral activity [158, 178,179,180]. 3-Halo-3-nitroflavanones, a novel class of DNMT inhibitors described recently, showed anti-tumoral activity associated with higher stability and low cytotoxicity than the most used DNMT inhibitors [181]. Interestingly, a compound from this family MLo1302 caused a decrease in cell viability, namely in cisplatin-resistant cell lines, pluripotency markers and an activation of apoptosis and cell cycle arrest in germ cell tumors cell lines [182]. RG108 is a SAM competitor that binds to the catalytic pockets of DNMTs, forming covalent bonds and inhibiting the enzyme action [183]. Treatment with RG108 caused radiosensitivity of esophageal cancer cells, enhancing apoptosis and G2/M phase arrest by radiation [184]. SGI-1027, a quinolone-based molecule which binds to both DNMT3a and DNMT1 [185], combined with AH057 (JAK inhibitor) increased cervical cancer cells apoptotic cell death and cell-cycle arrest [186]. Furthermore, MG98 is a second-generation 20-nucleotide antisense oligonucleotide that binds specifically to DNMT1 mRNA, decreasing DNMT1 levels [187].
Selected repurposed drugs, designed for a specific treatment that were found to have other therapeutic targets, also showed effect against DNMTs [176, 177]. For example, procaine and procainamide, approved as anesthetic and anti-arrhythmic drugs, respectively, bind to the catalytic center of DNMTs, which translated in a decrease in DNA methylation levels [188, 189]. The arterial vasodilator hydralazine also leads to the loss of promoter hypermethylation of TSGs in cancer cell lines and primary tumors [190, 191]. In this line, the antibiotic nanaomycin A also causes the same effects, being selective for DNMT3b [192].
HDAC inhibitors can be classified according to chemical group as hydroxamic acids, short-chain and aromatic fatty acids, benzamides and cyclic peptides (Table 2). The hydroxamic acids and cycle peptides constitute the most potent inhibitors, with IC50 in the low micro- or nanomolar range, while short-chain fatty acids require doses in the millimolar range [193].
Hydroxamic acids include belinostat (approved for peripheral T-cell lymphoma), givinostat, panobinostat, trichostatin A and vorinostat, all pan-HDAC inhibitors. Vorinostat or suberanilohydroxamic acid (SAHA) was the first HDAC inhibitor to be approved for the treatment of advanced cutaneous T-cell lymphoma, and it shows several anti-tumor effects in several hematological and solid tumors in vitro and in vivo (Table 3) [194]. It inhibits class I and II HDACs by binding to the catalytic domain of the enzymes [195]. Similar to vorinostat, trichostatin A (TSA), also a class I and II inhibitor, acts as a non-competitive inhibitor of HDAC by mimicking the lysin substrate as a chelating agent to the zinc atom in the active site [195]. Interestingly, it showed to reverse chemoresistance in lung cancer cell lines with high expression of IGFBP2, with IGFBP2 being a biomarker of chemoresistance poor outcome in lung cancer patients [196]. Panobinostat is approved for patients with recurrent multiple myeloma who have received at least two prior treatment regiments (Table 3) [193]. Indeed, the progression-free survival of patients treated with panobinostat was 10.6 months in comparison with 5.8 months in the control arm [197]. Belinostat also inhibits HDACs by binding to the zinc finger in the enzymes’ active catalytic site and, recently, showed to be active in testicular germ cell tumor cell lines resistant to cisplatin, with IC50 in the low nanomolar range for all cell lines [198].
Entinostat belongs to the benzamide group of HDAC inhibitors and inhibits class I HDACs [193]. Remarkably, entinostat boosted the effects of PD-1 inhibition in in vivo models of lung and renal cancers by impairing the immunosuppressive function of polymorphonuclear and monocytic-myeloid derived suppressor cell populations [112]. Another inhibitor of class I HDACs is romidepsin (FK228) belonging to the cyclic peptide family [193]. Romidepsin has been approved by the FDA for the treatment of advanced cutaneous T-cell lymphoma and peripheral T-cell lymphoma (Table 3) [199]. The triple combination of romidepsin, gemcitabine and cisplatin acted synergically and induced death by creating reactive oxygen species in triple-negative breast cancer cell lines [200].
The short-chain and aromatic fatty acids sodium butyrate and valproic acid also were shown to be pan-HDACs inhibitors [193]. Sodium butyrate is a short-chain fatty acid produced by fermentation by anaerobic bacterial fermentation that inhibits growth, induces apoptosis, migration and EMT of colorectal cancer cells [201]. Valproic acid, used for the treatment of epilepsy and bipolar disorder, in combination with cisplatin and cetuximab, showed antiproliferative and pro-apoptotic effects in 3D-self-assembled spheroid models of ad and neck squamous cell carcinoma cells [202].
Bladder cancer
BC is estimated to be the tenth most frequent cancer worldwide and the ninth cause of death by cancer [203]. About 70% of the patients are diagnosed as a non-muscle invasive BC (NMIBC), while 30% are diagnosed with muscle invasive BC (MIBC) [204]. NMIBC is mostly comprised of urothelial papillary neoplasms with varying propensity for recurrence and progression, which may be predicted based on grading [205]. According to the 2016 WHO classification, the spectrum of papillary neoplasms includes urothelial papilloma, papillary urothelial neoplasm of low malignant potential, low-grade and high-grade papillary urothelial carcinoma, in ascending order of biological and clinical aggressiveness [206]. On the other hand, urothelial carcinoma in situ (CIS) represents a high-grade form of non-papillary NMIBC, with substantial risk of progression to invasive disease [206]. Although NMIBC mostly contributes to the overall BC 5-year survival rate of 77.1%, 80% of high-grade papillary carcinomas and CIS recur and 20–50% progress to MIBC [207]. In addition to grade, evaluation of disease stage [by means of clinical examination, cystoscopy, radiographic evaluation and/or pathological examination using tissue collected by transurethral resection of the bladder tumor (TURBT)] is mandatory to define the best therapeutic strategy [208, 209]. For NMIBC, treatment mostly consists of TURBT eventually complemented with mitomycin or Bacillus Calmette-Guérin (BCG) instillation, whereas radical cystectomy with lymphadenectomy remains the gold standard for MIBC, complemented with neo-adjuvant or adjuvant cisplatin-based chemotherapy, which is also the main option for metastatic BC [209]. Recently, immunotherapies targeting PD-1/PD-L1 immune checkpoint were approved for BC patients that are refractory or ineligible to cisplatin-based chemotherapy [210]. Although chemotherapy and immunotherapy have improved the outcome of locally advanced and metastatic disease, 5-year survival remains poor (36% and 5%, respectively) [207].
BCG, which is a weakened strain of Mycobacterium bovis, was the first form of immunotherapy approved for cancer treatment and, specifically, for BC. Currently, it is administrated by intravesical instillation after TURBT in NMIBC patients with high risk of recurrence [211]. Although the mechanism is not fully known, BCG leads to localized innate and adaptative immune responses, including CD4 and CD8 lymphocytes, NK cells, macrophages, granulocytes and DCs [212]. About 55–75% of the high-risk patients suffering from papillary tumors to CIS respond to this therapy. However, 25 to 45% of these eventually relapse and progress to invasive disease. Hypermethylation of CDKN2B and of MUS81a and MSH6 involved in DNA repair and THBS1, important for cell adhesion, have been associated with response to BCG therapy [210, 213]. Likewise, low methylation levels of PMF1 have been associated with disease recurrence, poor outcome and lack of response to BCG in BC patients (Fig. 3) [214].

Modalities of immunotherapy in bladder cancer. BCG is a weakened strain of Mycobacterium bovis and was the first of immunotherapy approved for BC. NMIBC patients with high risk of recurrence are subjected to BCG therapy. The administration of BCG leads to a stimulation of both adaptative and innate immune response by recruiting lymphocytes, macrophages, NK cells and neutrophils, leading to the elimination of the remaining tumor cells. On the other hand, BC patients with MIBC are candidates for immune checkpoint blockage. Tumor cells express repression signals that lead to the inhibition of the immune response, namely by expressing PD-L1/PD-L2 and B7-1/B7-2, that will bind to PD-1 and CTLA-4 present in T lymphocytes, respectively. Nevertheless, with the administration of antibodies against PD-1, PD-L1 or CTL4-A, this process is reverted, leading to the activation of T cells and the start of an immune response against tumor cells, leading ultimately to their death
The immune landscape of BC is composed by different immune populations, including CD8 + T lymphocytes and Th1 CD4 + T lymphocytes. Interestingly, tumor-infiltrating CD4 + lymphocytes were found to be hypomethylated in four lineage loci compared to CD4 + lymphocytes in lymph nodes and blood. Patients with complete response to neoadjuvant chemotherapy (NACT) showed hypomethylation in CD4 + T cells, namely in IFN- γ. Furthermore, shifts in methylation patterns of Th1 CD4 + T cells after NACT show a relocation of cells from blood to the tumor (Fig. 3) [215]. Tissue-resident memory T cells showed low PRF1 DNA methylation levels concomitantly with increased perforin expression [216]. The analysis of DNA methylation in neutrophils and lymphocytes predicted the outcome of BC patients, i.e., high levels of DNA methylation-derived neutrophil-to-lymphocyte ratio associated with poor outcome [217].
Demethylating agents lead to reactivation of TSGs, inhibition of cancer cells’ proliferation and migration, increased apoptosis and activation of IFN pathway in BC [218]. Ramakrishnan et al. showed that low concentrations of DAC lead to the activation of NOTCH1, which may prevent epithelial-mesenchymal transition of tumor cells, thus impairing cancer cell dissemination [219, 220]. Moreover, increased IL-6 levels were observed in DAC-treated cells, and reduction of cytokeratin 5 expression associated with cell differentiation and impaired BC progression [221]. Another epigenetic inhibitor for G9a, CM-272, in combination with cisplatin caused an increase in expression of genes associated with immune response, such as TNF-α, IFN-α and IFN-γ, which correlated with an endogenous retrovirus response. Furthermore, an extensive infiltration of CD8 + T cells and NK cells was observed in tumors and metastases in an in vivo immunocompetent model of MIBC. This was also observed with CM-272 in combination with an anti-PD-L1 antibody, with immune infiltration by CD3 + , CD8 + and NK cells, and the absence of CD4 + and CD163 + cells [222].
Conclusions
Epigenetic alterations in cells of the TME play a major role in creating an immunosuppressive environment ideal for tumor development, which translates in a lack of effectiveness of immune checkpoint blockage therapies. The inhibition of epigenetic modulators might be an interesting therapeutic option to modify the immunosuppressive TME, and their potential in combination with immunotherapy has already been discussed. Additionally, refining patient selection for immunotherapy by exploring new biomarkers with higher sensitivity and specificity might improve the success rate of this therapy. Combining these findings, exploring aberrant epigenetic marks in both cancer cells and in cells of the TME might provide potential biomarkers for this purpose.
Thus, further studies are needed to increase our knowledge on the epigenetic mechanisms underlying the acquisition of immunosuppressive immune cell phenotypes and how these affect immunotherapy response. Additionally, a promising strategy to generate an immune-promoting TME might be the combination of epigenetic modulator targeting and immunotherapy.
Availability of data and materials
Not applicable.
Abbreviations
- 5-aza:
-
5-Azacytidine
- FdCyd:
-
5′-fluoro-2′-deoxycytidine
- ALOX15:
-
Arachidonate 15-lipoxygenase
- AML:
-
Acute myeloid leukemia
- APCs:
-
Antigen-presenting cells
- APM:
-
Antigen processing and presentation machinery
- Arg1:
-
Arginase 1
- BC:
-
Bladder cancer
- BCG:
-
Bacillus Calmette-Guérin
- CAFs:
-
Cancer-associated fibroblasts
- CIS:
-
Carcinoma in situ
- CMML:
-
Chronic myelomonocytic leukemia
- CTLs:
-
Cytotoxic T cells
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- DAC:
-
Decitabine
- DCs:
-
Dendritic cells
- DNMTs:
-
DNA methyltransferases
- ECM:
-
Extracellular matrix
- EGCG:
-
Epigallocatechin-3-gallate
- EMA:
-
European Medicines Agency
- EZH2:
-
Enhancer of zeste homolog 2
- FDA:
-
Food and Drug Administration
- HATs:
-
Histone acetyltransferases
- HDACs:
-
Histone deacetylases
- IFN:
-
Interferon
- JMJD3:
-
Jumonji domain-containing protein D3
- KLF4:
-
Kruppel-like factor 4
- MDSCs:
-
Myeloid-derived suppressor cells
- MIBC:
-
Muscle invasive BC
- NACT:
-
Neoadjuvant chemotherapy
- NK:
-
Natural killer
- NMIBC:
-
Non-muscle invasive BC
- PD-1:
-
Programmed death 1
- PD-L1:
-
Programmed death-ligand 1
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- PRMT1:
-
Protein arginine methyltransferase 1
- PTMs:
-
Post-translational modifications
- Rb:
-
Retinoblastoma gene
- SATB1:
-
Special AT-rich sequence binding 1
- SAHA:
-
Suberanilohydroxamic acid
- SMYD3:
-
SET and MYND Domain 3
- STAT3:
-
Signal transducer and activator of transcription 3
- TAAs:
-
Tumor-associated antigens
- TAMs:
-
Tumor-associated macrophages
- TECs:
-
Tumor endothelial cells
- Th:
-
T helper
- TILs:
-
Tumor-infiltrating lymphocytes
- TME:
-
Tumor microenvironment
- Tregs:
-
Regulatory T cells
- TSA:
-
Trichostatin A
- TSGs:
-
Tumor suppressor genes
- TURBT:
-
Transurethral resection of the bladder
- UCK2:
-
Uridine-cytidine kinase 2
References
Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. 2016;8:a019505.
Song H, Liu D, Dong S, Zeng L, Wu Z, Zhao P, et al. Epitranscriptomics and epiproteomics in cancer drug resistance: therapeutic implications. Signal Transduct Target Ther. 2020;5:193.
Rinaldi G, Rossi M, Fendt SM. Metabolic interactions in cancer: cellular metabolism at the interface between the microenvironment, the cancer cell phenotype and the epigenetic landscape. Wiley Interdiscip Rev Syst Biol Med. 2018;10:e1397.
Mao L, Yang C, Wang J, Li W, Wen R, Chen J, et al. SATB1 is overexpressed in metastatic prostate cancer and promotes prostate cancer cell growth and invasion. J Transl Med. 2013;11:111.
Guo L, Zheng J, Yu T, Liu Y, Duo L. Elevated expression of SATB1 is involved in pancreatic tumorigenesis and is associated with poor patient survival. Mol Med Rep. 2017;16:8842–8.
Nodin B, Hedner C, Uhlén M, Jirström K. Expression of the global regulator SATB1 is an independent factor of poor prognosis in high grade epithelial ovarian cancer. J Ovarian Res. 2012;5:24.
Tesone AJ, Rutkowski MR, Brencicova E, Svoronos N, Perales-Puchalt A, Stephen TL, et al. Satb1 overexpression drives tumor-promoting activities in cancer-associated dendritic cells. Cell Rep. 2016;14:1774–86.
Zhang H, Qu S, Li S, Wang Y, Li Y, Wang Y, et al. Silencing SATB1 inhibits proliferation of human osteosarcoma U2OS cells. Mol Cell Biochem. 2013;378:39–45.
Selinger CI, Cooper WA, Al-Sohaily S, Mladenova DN, Pangon L, Kennedy CW, et al. Loss of special at-rich binding protein 1 expression is a marker of poor survival in lung cancer. J Thorac Oncol. 2011;6:1179–89.
Tu W, Luo M, Wang Z, Yan W, Xia Y, Deng H, et al. Upregulation of SATB1 promotes tumor growth and metastasis in liver cancer. Liver Int Liver Int. 2012;32:1064–78.
Chu SH, Ma YB, Feng DF, Zhang H, Zhu ZA, Li ZQ, et al. Upregulation of SATB1 is associated with the development and progression of glioma. J Transl Med. 2012;10:149.
Yuan CL, Li L, Zhou X, Liz H, Han L. Expression of SATB1 and HER2 in gastric cancer and its clinical significance. Eur Rev Med Pharmacol Sci. 2016;20:2256–64.
Nodin B, Johannesson H, Wangefjord S, O’Connor DP, Lindquist KE, Uhlén M, et al. Molecular correlates and prognostic significance of SATB1 expression in colorectal cancer. Diagn Pathol. 2012;7:1–9.
Fromberg A, Rabe M, Aigner A. Multiple effects of the special AT-rich binding protein 1 (SATB1) in colon carcinoma. Int J Cancer. 2014;135:2537–46.
Palucka K, Coussens LM, O’Shaughnessy J. Dendritic cells, inflammation, and breast cancer. Cancer J. (United States). 2013; 511–6.
Crispen PL, Kusmartsev S. Mechanisms of immune evasion in bladder cancer. Cancer Immunol Immunother. 2020;69:3–14.
Han B, Luan L, Xu Z, Wu B. Expression and biological roles of SATB1 in human bladder cancer. Tumor Biol. 2013;34:2943–9.
Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL. Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol. 2015;194:2985–91.
Glatzel-Plucí Nska N, Piotrowska A, Dzięgiel P, Podhorska-Okołów M. The role of SATB1 in tumour progression and metastasis. Int J Mol Sci. 2019;20:4156.
Ding B, Liu P, Liu W, Sun P, Wang CL. Emerging roles of krüppel-like factor 4 in cancer and cancer stem cells. Asian Pac J Cancer Prev. 2015;16:3629–33.
Rosenzweig JM, Glenn JD, Calabresi PA, Whartenby KA. KLF4 modulates expression of IL-6 in dendritic cells via both promoter activation and epigenetic modification. J Biol Chem. 2013;288:23868–74.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74.
Sanaei M, Salimzadeh L, Bagheri N. Crosstalk between myeloid-derived suppressor cells and the immune system in prostate cancer. J Leukoc Biol. 2020;107:43–56.
Medina-Echeverz J, Eggert T, Han M, Greten TF. Hepatic myeloid-derived suppressor cells in cancer. Cancer Immunol Immunother. 2015;64:931–40.
Ma J, Xu H, Wang S. Immunosuppressive role of myeloid-derived suppressor cells and therapeutic targeting in lung cancer. J Immunol Res. 2018;6319649.
Sasidharan Nair V, Saleh R, Toor SM, Taha RZ, Ahmed AA, Kurer MA, et al. Transcriptomic profiling disclosed the role of DNA methylation and histone modifications in tumor-infiltrating myeloid-derived suppressor cell subsets in colorectal cancer. Clin Epigenet. 2020;12:13.
Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells [1]. Cancer Res. 2007;67:425.
Wu L, Du H, Li Y, Qu P, Yan C. Signal transducer and activator of transcription 3 (Stat3C) promotes myeloid-derived suppressor cell expansion and immune suppression during lung tumorigenesis. Am J Pathol. 2011;179:2131–41.
Trikha P, Carson WE. Signaling pathways involved in MDSC regulation. Biochim Biophys Acta Rev Cancer. 2014;1846:55–65.
Mace TA, Bloomston M, Lesinski GB. Pancreatic cancer-associated stellate cells: a viable target for reducing immunosuppression in the tumor microenvironment. Oncoimmunology. 2013;2:e24891.
Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69:2506–13.
Lissbrant IF, Stattin P, Wikstrom P, Damber JE, Egevad L, Bergh A. Tumor associated macrophages in human prostate cancer: relation to clinicopathological variables and survival. Int J Oncol. 2000;17:445–51.
Mielgo A, Schmid MC. Impact of tumour associated macrophages in pancreatic cancer. BMB Rep. 2013;46:131–8.
Yin M, Shen J, Yu S, Fei J, Zhu X, Zhao J, et al. Tumor-associated macrophages (Tams): a critical activator in ovarian cancer metastasis. OncoTargets Ther. 2019;12:8687–99.
Malfitano AM, Pisanti S, Napolitano F, Di Somma S, Martinelli R, Portella G. Tumor-associated macrophage status in cancer treatment. Cancers (Basel). 2020;12:1987.
Sumitomo R, Hirai T, Fujita M, Murakami H, Otake Y, Huang C. M2 tumor-associated macrophages promote tumor progression in non-small-cell lung cancer. Exp Ther Med. 2019;18:4490–8.
Vidyarthi A, Agnihotri T, Khan N, Singh S, Tewari MK, Radotra BD, et al. Predominance of M2 macrophages in gliomas leads to the suppression of local and systemic immunity. Cancer Immunol Immunother. 2019;68:1995–2004.
Gambardella V, Castillo J, Tarazona N, Gimeno-Valiente F, Martínez-Ciarpaglini C, Cabeza-Segura M, et al. The role of tumor-associated macrophages in gastric cancer development and their potential as a therapeutic target. Cancer Treat Rev. 2020;86:102015.
Liu Y, Li L, Li Y, Zhao X. Research Progress on Tumor-Associated Macrophages and Inflammation in Cervical Cancer. Biomed Res Int. 2020;6842963.
Zhong X, Chen B, Yang Z. The role of tumor-associated macrophages in colorectal carcinoma progression. Cell Physiol Biochem. 2018;45:356–65.
Choi J, Gyamfi J, Jang H, Koo JS. The role of tumor-associated macrophage in breast cancer biology. Histol Histopathol. 2018;33:133–45.
Setrerrahmane S, Xu H. Tumor-related interleukins: old validated targets for new anti-cancer drug development. Mol Cancer. 2017;16:1–17.
Hendry S, Salgado R, Gevaert T, Russell PA, John T, Thapa B, et al. Assessing tumor-infiltrating lymphocytes in solid tumors: a practical review for pathologists and proposal for a standardized method from the international immuno-oncology biomarkers working group: part 2: TILs in melanoma, gastrointestinal tract carcinomas, non-small cell lung carcinoma and mesothelioma, endometrial and ovarian carcinomas, squamous cell carcinoma of the head and neck, genitourinary carcinomas, and primary brain tumors. Adv Anat Pathol. 2017;24:311–35.
Hendry S, Salgado R, Gevaert T, Russell PA, John T, Thapa B, et al. Assessing tumor-infiltrating lymphocytes in solid tumors: a practical review for pathologists and proposal for a standardized method from the international immunooncology biomarkers working group: part 1: assessing the host immune response, TILs in invasive breast carcinoma and ductal carcinoma in situ, metastatic tumor deposits and areas for further research. Adv Anat Pathol. 2017;24:235–51.
Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Harrison Farber S, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res. 2018;24:4175–86.
Baitsch L, Baumgaertner P, Devêvre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–60.
Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer. 2020;19:116.
Halim L, Romano M, McGregor R, Correa I, Pavlidis P, Grageda N, et al. An atlas of human regulatory T helper-like cells reveals features of Th2-like tregs that support a tumorigenic environment. Cell Rep. 2017;20:757–70.
Tao H, Mimura Y, Aoe K, Kobayashi S, Yamamoto H, Matsuda E, et al. Prognostic potential of FOXP3 expression in non-small cell lung cancer cells combined with tumor-infiltrating regulatory T cells. Lung Cancer. 2012;75:95–101.
Sayour EJ, McLendon P, McLendon R, De Leon G, Reynolds R, Kresak J, et al. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol Immunother. 2015;64:419–27.
Judge SJ, Murphy WJ, Canter RJ. Characterizing the dysfunctional NK Cell: assessing the clinical relevance of exhaustion, anergy, and senescence. Front Cell Infect Microbiol. 2020;10:49.
Peng YP, Zhu Y, Zhang JJ, Xu ZK, Qian ZY, Dai CC, et al. Comprehensive analysis of the percentage of surface receptors and cytotoxic granules positive natural killer cells in patients with pancreatic cancer, gastric cancer, and colorectal cancer. J Transl Med. 2013;11:262.
Bi J, Tian Z. NK cell exhaustion. Front Immunol. 2017;8:760.
Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest. 2011;121:3609–22.
Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582–98.
Tao L, Huang G, Song H, Chen Y, Chen L. Cancer associated fibroblasts: An essential role in the tumor microenvironment (review). Oncol Lett. 2017;14:2611–20.
Ziani L, Chouaib S, Thiery J. Alteration of the antitumor immune response by cancer-associated fibroblasts. Front Immunol. 2018;9:1.
Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156:1363–80.
Hida K, Hida Y, Amin DN, Flint AF, Panigrahy D, Morton CC, et al. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004;64:8249–55.
Farnsworth RH, Lackmann M, Achen MG, Stacker SA. Vascular remodeling in cancer. Oncogene. 2014;33:3496–505.
Henke E, Nandigama R, Ergün S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci. 2020;6:160.
Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19:81–92.
Chen Z, Chen Z, Chen Z, Zhang Y, Zhang Y, Zhang Y, et al. Role of mammalian DNA methyltransferases in development. Annu Rev Biochem. 2020;89:135–58.
Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007;16:50–9.
Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta Gene Regul Mech. 2014;1839:627–43.
Zhao Z, Shilatifard A. Epigenetic modifications of histones in cancer. Genome Biol. 2019;20:1–16.
Cairns BR. Chromatin remodeling: insights and intrigue from single-molecule studies. Nat Struct Mol Biol. 2007;14:989–96.
Martire S, Banaszynski LA. The roles of histone variants in fine-tuning chromatin organization and function. Nat Rev Mol Cell Biol. 2020;21:522–41.
Kurumizaka H, Kujirai T, Takizawa Y. Contributions of histone variants in nucleosome structure and function. J Mol Biol. 2020;166678.
Yasui D, Miyano M, Cai S, Varga-Weisz P, Kohwi-Shigematsu T. SATB1 targets chromatin remodelling to regulate genes over long distances. Nature. 2002;419:641–5.
Pavan Kumar P, Purbey PK, Sinha CK, Notani D, Limaye A, Jayani RS, et al. Phosphorylation of SATB1, a global gene regulator, acts as a molecular switch regulating its transcriptional activity in vivo. Mol Cell. 2006;22:231–43.
Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol. 2009;10:92–100.
Sahakian E, Powers JJ, Chen J, Deng SL, Cheng F, Distler A, et al. Histone deacetylase 11: a novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol Immunol. 2015;63:579–85.
Cheng F, Lienlaf M, Perez-Villarroel P, Wang HW, Lee C, Woan K, et al. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol Immunol. 2014;60:44–53.
Sido JM, Yang X, Nagarkatti PS, Nagarkatti M. Δ 9 -Tetrahydrocannabinol-mediated epigenetic modifications elicit myeloid-derived suppressor cell activation via STAT3/S100A8. J Leukoc Biol. 2015;97:677–88.
Viatour P, Somervaille TC, Venkatasubrahmanyam S, Kogan S, McLaughlin ME, Weissman IL, et al. Hematopoietic stem cell quiescence is maintained by compound contributions of the retinoblastoma gene family. Cell Stem Cell. 2008;3:416–28.
Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007;129:1081–95.
Youn JI, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol. 2013;14:211–20.
Ojalvo LS, Whittaker CA, Condeelis JS, Pollard JW. Gene expression analysis of macrophages that facilitate tumor invasion supports a role for Wnt-signaling in mediating their activity in primary mammary tumors. J Immunol. 2010;184:702–12.
Rubio C, Munera-Maravilla E, Lodewijk I, Suarez-Cabrera C, Karaivanova V, Ruiz-Palomares R, et al. Macrophage polarization as a novel weapon in conditioning tumor microenvironment for bladder cancer: can we turn demons into gods? Clin Transl Oncol. 2019;21:391–403.
Yang X, Wang X, Liu D, Yu L, Xue B, Shi H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol Endocrinol. 2014;28:565–74.
Arlauckas SP, Garren SB, Garris CS, Kohler RH, Oh J, Pittet MJ, et al. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics. 2018;8:5842–54.
Ishii M, Wen H, Corsa CAS, Liu T, Coelho AL, Allen RM, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood. 2009;114:3244–54.
Kittan NA, Allen RM, Dhaliwal A, Cavassani KA, Schaller M, Gallagher KA, et al. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. Zissel G, editor. PLoS ONE. 2013;8:e78045.
Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11:936–44.
Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, et al. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci U S A. 2012;109:E2865–74.
Mullican SE, Gaddis CA, Alenghat T, Nair MG, Giacomin PR, Everett LJ, et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev Genes Dev. 2011;25:2480–8.
Cao Q, Rong S, Repa JJ, Clair RS, Parks JS, Mishra N. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler Thromb Vasc Biol. 2014;34:1871–9.
Yang Q, Wei J, Zhong L, Shi M, Zhou P, Zuo S, et al. Cross talk between histone deacetylase 4 and STAT6 in the transcriptional regulation of arginase 1 during mouse dendritic cell differentiation. Mol Cell Biol. 2015;35:63–75.
Lo Sasso G, Joe Menzies K, Mottis A, Piersigilli A, Perino A, Yamamoto H, et al. SIRT2 deficiency modulates macrophage polarization and susceptibility to experimental colitis. PLoS ONE. 2014;9:e103573.
Tay RE, Richardson EK, Toh HC. Revisiting the role of CD4+ T cells in cancer immunotherapy—new insights into old paradigms. Cancer Gene Ther. 2020;28:5–17.
Janson PCJ, Marits P, Thörn M, Ohlsson R, Winqvist O. CpG methylation of the IFNG gene as a mechanism to induce immunosupression in tumor-infiltrating lymphocytes. J Immunol. 2008;181:2878–86.
Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249–53.
Yang R, Cheng S, Luo N, Gao R, Yu K, Kang B, et al. Distinct epigenetic features of tumor-reactive CD8+ T cells in colorectal cancer patients revealed by genome-wide DNA methylation analysis. Genome Biol. 2019;21:2.
Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De Novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell. 2017;170:142–57.
Massagué J. TGFβ in Cancer. Cell. 2008; 215–30.
Stephen TL, Payne KK, Chaurio RA, Allegrezza MJ, Zhu H, Perez-Sanz J, et al. SATB1 expression governs epigenetic repression of PD-1 in tumor-reactive T cells. Immunity. 2017;46:51–64.
Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017;545:452–6.
Waight JD, Takai S, Marelli B, Qin G, Hance KW, Zhang D, et al. Cutting edge: epigenetic regulation of Foxp3 defines a stable population of CD4 + regulatory T cells in tumors from mice and humans. J Immunol. 2015;194:878–82.
Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity. 2013;38:414–23.
Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T Cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for treg cell development. Immunity. 2012;37:785–99.
DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity. 2015;42:227–38.
Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A, et al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep. 2018;23:3262–74.
Schenk A, Bloch W, Zimmer P. Natural killer cells—an epigenetic perspective of development and regulation. Int J Mol Sci. 2016;17:326.
Fernández-Sánchez A, Raneros AB, Palao RC, Sanz AB, Ortiz A, Ortega F, et al. DNA demethylation and histone H3K9 acetylation determine the active transcription of the NKG2D gene in human CD8+ T and NK cells. Epigenetics. 2013;8:66–78.
Ogbomo H, Michaelis M, Kreuter J, Doerr HW, Cinatl J. Histone deacetylase inhibitors suppress natural killer cell cytolytic activity. FEBS Lett. 2007;581:1317–22.
Yin J, Leavenworth JW, Li Y, Luo Q, Xie H, Liu X, et al. Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity. Proc Natl Acad Sci U S A. 2015;112:15988–93.
Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7:299–313.
Gao XY, Xue XH, Ma YN, Zhang SQ. Effect of baicalein on the expression of SATB1 in human breast cancer cells. Exp Ther Med. 2015;9:1665–9.
Lakshminarayana Reddy CN, Vyjayanti VN, Notani D, Galande S, Kotamraju S. Down-regulation of the global regulator SATB1 by statins in COLO205 colon cancer cells. Mol Med Rep. 2010;3:857–61.
Wang M, Yin B, Matsueda S, Deng L, Li Y, Zhao W, et al. Identification of special AT-rich sequence binding protein 1 as a novel tumor antigen recognized by CD8+ T cells: implication for cancer immunotherapy. PLoS ONE. 2013;8:e56730.
Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res. 2017;23:5187–201.
Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014;111:11774–9.
Briere D, Sudhakar N, Woods DM, Hallin J, Engstrom LD, Aranda R, et al. The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy. Cancer Immunol Immunother. 2018;67:381–92.
Hashimoto A, Fukumoto T, Zhang R, Gabrilovich D. Selective targeting of different populations of myeloid-derived suppressor cells by histone deacetylase inhibitors. Cancer Immunol Immunother. 2020;69:1929–36.
Liu SS, Wu F, Jin YM, Chang WQ, Xu TM. HDAC11: a rising star in epigenetics. Biomed Pharmacother. 2020;131:110607.
Tikhanovich I, Zhao J, Olson J, Adams A, Taylor R, Bridges B, et al. Protein arginine methyltransferase 1 modulates innate immune responses through regulation of peroxisome proliferator-activated receptor γ-dependent macrophage differentiation. J Biol Chem. 2017;292:6882–94.
Yildirim-Buharalioğlu G, Bond M, Sala-Newby GB, Hindmarch CCT, Newby AC. Regulation of epigenetic modifiers, including KDM6B, by interferon-γ and interleukin-4 in human macrophages. Front Immunol. 2017;8:1.
Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–8.
de Groot AE, Pienta KJ. Epigenetic control of macrophage polarization: Implications for targeting tumor-associated macrophages. Oncotarget. 2018;9:20908–27.
Goswami S, Apostolou I, Zhang J, Skepner J, Anandhan S, Zhang X, et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J Clin Invest. 2018;128:3813–8.
Qiu J, Sharma S, Rollins RA, Paul TA. The complex role of EZH2 in the tumor microenvironment: opportunities and challenges for immunotherapy combinations. Future Med Chem. 2020;12:1415–30.
Zingg D, Arenas-Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J, et al. The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. 2017;20:854–67.
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70.
Aqbi HF, Wallace M, Sappal S, Payne KK, Manjili MH. IFN-γ orchestrates tumor elimination, tumor dormancy, tumor escape, and progression. J Leukoc Biol. 2018;103:1219–23.
Jahanban-Esfahlan R, Seidi K, Manjili MH, Jahanban-Esfahlan A, Javaheri T, Zare P. Tumor cell dormancy: threat or opportunity in the fight against cancer. Cancers (Basel). 2019;11:1207.
O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16:151–67.
Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–42.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol. 2007;19:813–24.
Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAAS, Andrews MC, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell. 2017;170:1120–33.
Van Der Merwe PA, Bodian DL, Daenke S, Linsley P, Davis SJ. CD80 (B7–1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J Exp Med. 1997;185:393–403.
Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y, Zang X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol Med. 2015;21:24–33.
Lee HT, Lee SH, Heo YS. Molecular interactions of antibody drugs targeting PD-1, PD-L1, and CTLA-4 in immuno-oncology. Molecules. 2019;24:1190.
Borcoman E, Nandikolla A, Long G, Goel S, Le Tourneau C. Patterns of Response and Progression to Immunotherapy. Am Soc Clin Oncol Educ B. 2018;38:169–78.
Rieth J, Subramanian S. Mechanisms of intrinsic tumor resistance to immunotherapy. Int J Mol Sci. 2018;19:1340.
Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 2015;847–56.
Haragan A, Field JK, Davies MPA, Escriu C, Gruver A, Gosney JR. Heterogeneity of PD-L1 expression in non-small cell lung cancer: Implications for specimen sampling in predicting treatment response. Lung Cancer. 2019;134:79–84.
Aggen DH, Drake CG. Biomarkers for immunotherapy in bladder cancer: a moving target. J Immunother Cancer. 2017;5:94.
Lee N, Zakka LR, Mihm MC, Schatton T. Tumour-infiltrating lymphocytes in melanoma prognosis and cancer immunotherapy. Pathology. 2016;48:177–87.
Fumet JD, Truntzer C, Yarchoan M, Ghiringhelli F. Tumour mutational burden as a biomarker for immunotherapy: current data and emerging concepts. Eur J Cancer. 2020; 40–50.
Zhao P, Li L, Jiang X, Li Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J Hematol Oncol. 2019;12:54.
Kim J, Kwiatkowski D, McConkey DJ, Meeks JJ, Freeman SS, Bellmunt J, et al. The cancer genome atlas expression subtypes stratify response to checkpoint inhibition in advanced urothelial cancer and identify a subset of patients with high survival probability. Eur Urol. 2019;75:961–4.
Kamoun A, de Reyniès A, Allory Y, Sjödahl G, Robertson AG, Seiler R, et al. A consensus molecular classification of muscle-invasive bladder cancer[Formula presented]. Eur Urol. 2020;77:420–33.
Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598–608.
Qin Y, Bollin K, De MacEdo MP, Carapeto F, Kim KB, Roszik J, et al. Immune profiling of uveal melanoma identifies a potential signature associated with response to immunotherapy. J Immunother Cancer. 2020;8.
Wargo JA, Reddy SM, Reuben A, Sharma P. Monitoring immune responses in the tumor microenvironment. Curr Opin Immunol 2016; p. 23–31.
House IG, Savas P, Lai J, Chen AXY, Oliver AJ, Teo ZL, et al. Macrophage-derived CXCL9 and CXCL10 are required for antitumor immune responses following immune checkpoint blockade. Clin Cancer Res. 2020;26:487–504.
Ali MA, Matboli M, Tarek M, Reda M, Kamal KM, Nouh M, et al. Epigenetic regulation of immune checkpoints: Another target for cancer immunotherapy?. Immunotherapy. 2017; 99–108.
Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, et al. Alterations of immune response of non-small cell lung cancer with Azacytidine. Oncotarget. 2013;4:2067–79.
Yang H, Bueso-Ramos C, Dinardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28:1280–8.
Chen X, Pan X, Zhang W, Guo H, Cheng S, He Q, et al. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm Sin B. 2020;10:723–33.
Duruisseaux M, Martínez-Cardús A, Calleja-Cervantes ME, Moran S, Castro de Moura M, Davalos V, et al. Epigenetic prediction of response to anti-PD-1 treatment in non-small-cell lung cancer: a multicentre, retrospective analysis. Lancet Respir Med. 2018;6:771–81.
Guerreiro IM, Barros-Silva D, Lopes P, Cantante M, Cunha AL, Lobo J, et al. RAD51Bme levels as a potential predictive biomarker for PD-1 blockade response in non-small cell lung cancer. J Clin Med. 2020;9:1000.
Kandimalla R, van Tilborg AA, Zwarthoff EC. DNA methylation-based biomarkers in bladder cancer. Nat Rev Urol. 2013;10:327–35.
Wang X, Chen E, Yang X, Wang Y, Quan Z, Wu X, et al. 5-azacytidine inhibits the proliferation of bladder cancer cells via reversal of the aberrant hypermethylation of the hepaCAM gene. Oncol Rep. 2016;35:1375–84.
Erdmann A, Halby L, Fahy J, Arimondo PB. Targeting DNA methylation with small molecules: what’s next? J Med Chem. 2015;58:2569–83.
Marques-Magalhães Â, Graça I, Henrique R, Jerónimo C. Targeting DNA methyltranferases in urological tumors. Front Pharmacol. 2018;9:366.
Pasculli B, Barbano R, Parrella P. Epigenetics of breast cancer: Biology and clinical implication in the era of precision medicine. Semin Cancer Biol. 2018;51:22–35.
Bohl SR, Bullinger L, Rücker FG. Epigenetic therapy: azacytidine and decitabine in acute myeloid leukemia. Expert Rev Hematol. 2018;11:361–71.
Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123:8–13.
Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc Natl Acad Sci U S A. 1984;81:6993–7.
Chen L, MacMillan AM, Chang W, Ezaz-Nikpay K, Lane WS, Verdine GL. Direct identification of the active-site nucleophile in a DNA (cytosine-5)-methyltransferase. Biochemistry. 1991;30:11018–25.
Liu X, Dai X, Wu B. Study of 5-Aza-CdR on transcription regulation of RASSF1A gene in the BIU87 cell line. Urol Int. 2009;82:108–12.
Nguyen AN, Hollenbach PW, Richard N, Luna-Moran A, Brady H, Heise C, et al. Azacitidine and decitabine have different mechanisms of action in non-small cell lung cancer cell lines. Lung Cancer. 2010;1:119–40.
Gros C, Fahy J, Halby L, Dufau I, Erdmann A, Gregoire JM, et al. DNA methylation inhibitors in cancer: recent and future approaches. Biochimie. 2012;94:2280–96.
Zhao Q, Fan J, Hong W, Li L, Wu M. Inhibition of cancer cell proliferation by 5-fluoro-2’-deoxycytidine, a DNA methylation inhibitor, through activation of DNA damage response pathway. Springerplus. 2012;1:65.
Balboni B, El Hassouni B, Honeywell RJ, Sarkisjan D, Giovannetti E, Poore J, et al. RX-3117 (fluorocyclopentenyl cytosine): a novel specific antimetabolite for selective cancer treatment. Expert Opin Investig Drugs. 2019;28:311–22.
Yoo CB, Cheng JC, Jones PA. Zebularine: a new drug for epigenetic therapy. Biochem Soc Trans. 2004;32:910–2.
Takemura Y, Satoh M, Hatanaka K, Kubota S. Zebularine exerts its antiproliferative activity through S phase delay and cell death in human malignant mesothelioma cells. Biosci Biotechnol Biochem. 2018;82:1159–64.
Cheng JC, Yoo CB, Weisenberger DJ, Chuang J, Wozniak C, Liang G, et al. Preferential response of cancer cells to zebularine. Cancer Cell. 2004;6:151–8.
Raynal NJM, Issa JPJ. DNA methyltransferase inhibitors. Drug Discov Cancer Epigenet. 2015; 169–90.
Johnson WD, Harder JB, Naylor J, McCormick DL, Detrisac CJ, Glaze ER, et al. A pharmacokinetic/pharmacodynamic approach to evaluating the safety of zebularine in non-human primates. Cancer Res. 2006;66.
Ganesan A, Arimondo PB, Rots MG, Jeronimo C, Berdasco M. The timeline of epigenetic drug discovery: from reality to dreams. Clin Epigenet. 2019;11:174.
Albany C, Fazal Z, Singh R, Bikorimana E, Adra N, Hanna NH, et al. A phase 1 study of combined guadecitabine and cisplatin in platinum refractory germ cell cancer. Cancer Med. 2021;10:156–63.
Castillo-Aguilera O, Depreux P, Halby L, Arimondo PB, Goossens L. DNA methylation targeting: the DNMT/HMT crosstalk challenge. Biomolecules. 2017;7.
Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18:41–58.
Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, et al. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003;63:7563–70.
Nandakumar V, Vaid M, Katiyar SK. (-)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis. 2011;32:537–44.
Link A, Balaguer F, Shen Y, Lozano JJ, Leung HC, Boland CR, et al. Curcumin modulates DNA methylation in colorectal cancer cells. PLoS ONE. 2013;8:e57709.
Pechalrieu D, Dauzonne D, Arimondo PB, Lopez M. Synthesis of novel 3-halo-3-nitroflavanones and their activities as DNA methyltransferase inhibitors in cancer cells. Eur J Med Chem. 2020;186.
Lobo J, Cardoso AR, Miranda-Gonçalves V, Looijenga LHJ, Lopez M, Arimondo PB, et al. Targeting germ cell tumors with the newly synthesized flavanone-derived compound mlo1302 efficiently reduces tumor cell viability and induces apoptosis and cell cycle arrest. Pharmaceutics. 2021;13:1–18.
Rondelet G, Fleury L, Faux C, Masson V, Dubois J, Arimondo PB, et al. Inhibition studies of DNA methyltransferases by maleimide derivatives of RG108 as non-nucleoside inhibitors. Futur Med Chem. 2017;9:1465–81.
Ou Y, Zhang Q, Tang Y, Lu Z, Lu X, Zhou X, et al. DNA methylation enzyme inhibitor RG108 suppresses the radioresistance of esophageal cancer. Oncol Rep. 2018;39:993–1002.
Yoo J, Choi S, Medina-Franco JL. Molecular modeling studies of the novel inhibitors of DNA methyltransferases SGI-1027 and CBC12: implications for the mechanism of inhibition of DNMTs. PLoS ONE. 2013;8:e62152.
She S, Zhao Y, Kang B, Chen C, Chen X, Zhang X, et al. Combined inhibition of JAK1/2 and DNMT1 by newly identified small-molecule compounds synergistically suppresses the survival and proliferation of cervical cancer cells. Cell Death Dis. 2020;11.
Amato RJ. Inhibition of DNA methylation by antisense oligonucleotide MG98 as cancer therapy. Clin Genitourin Cancer. 2007;5:422–6.
Villar-Garea A, Fraga MF, Espada J, Esteller M. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res. 2003;63:4984–9.
Li YC, Wang Y, Li DD, Zhang Y, Zhao TC, Li CF. Procaine is a specific DNA methylation inhibitor with anti-tumor effect for human gastric cancer. J Cell Biochem. 2018;119:2440–9.
Dueñas-Gonzalez A, Coronel J, Cetina L, González-Fierro A, Chavez-Blanco A, Taja-Chayeb L. Hydralazine-valproate: a repositioned drug combination for the epigenetic therapy of cancer. Expert Opin Drug Metab Toxicol. 2014;10:1433–44.
Segura-Pacheco B, Trejo-Becerril C, Perez-Cardenas E, Taja-Chayeb L, Mariscal I, Chavez A, et al. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin Cancer Res. 2003;9:1596–603.
Kuck D, Caulfield T, Lyko F, Medina-Franco JL. Nanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cells. Mol Cancer Ther. 2010;9:3015–23.
Giannopoulou AF, Velentzas AD, Konstantakou EG, Avgeris M, Katarachia SA, Papandreou NC, et al. Revisiting histone deacetylases in human tumorigenesis: The paradigm of urothelial bladder cancer. Int J Mol Sci. 2019;20:1291.
Siegel D, Hussein M, Belani C, Robert F, Galanis E, Richon VM, et al. Vorinostat in solid and hematologic malignancies. J Hematol Oncol. 2009;2.
Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–93.
Tang D, Yao R, Zhao D, Zhou L, Wu Y, Yang Y, et al. Trichostatin A reverses the chemoresistance of lung cancer with high IGFBP2 expression through enhancing autophagy. Sci Rep. 2018;8.
Yee AJ, Raje NS. Panobinostat and multiple myeloma in 2018. Oncologist. 2018;23:516–7.
Lobo J, Guimarães-Teixeira C, Barros-Silva D, Miranda-Gonçalves V, Camilo V, Guimarães R, et al. Efficacy of HDAC inhibitors belinostat and panobinostat against cisplatin-sensitive and cisplatin-resistant testicular germ cell tumors. Cancers (Basel). 2020;12:2903.
Vandermolen KM, McCulloch W, Pearce CJ, Oberlies NH. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma. J Antibiot (Tokyo). 2011; 525–531.
Pattarawat P, Wallace S, Pfisterer B, Odoi A, Wang HCR. Formulation of a triple combination gemcitabine plus romidepsin + cisplatin regimen to efficaciously and safely control triple-negative breast cancer tumor development. Cancer Chemother Pharmacol. 2020;85:141–52.
Wang W, Fang D, Zhang H, Xue J, Wangchuk D, Du J, et al. Sodium butyrate selectively kills cancer cells and inhibits migration in colorectal cancer by targeting thioredoxin-1. Onco Targets Ther. 2020;13:4691–704.
Iannelli F, Zotti AI, Roca MS, Grumetti L, Lombardi R, Moccia T, et al. Valproic Acid synergizes with cisplatin and cetuximab in vitro and in vivo in head and neck cancer by targeting the mechanisms of resistance. Front Cell Dev Biol. 2020;8.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Kamat AM, Hahn NM, Efstathiou JA, Lerner SP, Malmström PU, Choi W, et al. Bladder cancer. Lancet. 2016;388:2796–810.
Chen Z, Ding W, Xu K, Tan J, Sun C, Gou Y, et al. The 1973 WHO Classification is more suitable than the 2004 WHO Classification for predicting prognosis in non-muscle-invasive bladder cancer. PLoS ONE. 2012;7:e47199.
Humphrey PA, Moch H, Cubilla AL, Ulbright TM, Reuter VE. The 2016 WHO classification of tumours of the urinary system and male genital organs—part B: prostate and bladder tumours. Eur Urol. 2016;70:106–19.
Woldu SL, Şanli Ö, Lotan Y. Tackling non-muscle invasive bladder cancer in the clinic. Expert Rev Anticancer Ther. 2017;17:467–80.
Amin MB, Edge S, Greene F, Byrd DR, Brookland RK, Washington MK, et al. AJCC Cancer Staging Manual. Eighth edn. Springer. 2017.
Bellmunt J, Orsola A, Leow JJ, Wiegel T, De Santis M, Horwich A. Bladder cancer: ESMO practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(Suppl 3):40–8.
Lobo J, Jerónimo C, Henrique R. Targeting the immune system and epigenetic landscape of urological tumors. Int J Mol Sci. 2020;21:829.
Herr HW, Morales A. History of bacillus Calmette-Guerin and bladder cancer: an immunotherapy success story. J Urol. 2008;179:53–6.
Zhang C, Berndt-Paetz M, Neuhaus J. Identification of key biomarkers in bladder cancer: evidence from a bioinformatics analysis. Diagnostics. 2020;10.
Agundez M, Grau L, Palou J, Algaba F, Villavicencio H, Sanchez-Carbayo M. Evaluation of the methylation status of tumour suppressor genes for predicting bacillus Calmette-Guérin response in patients with T1G3 high-risk bladder tumours. Eur Urol. 2011;60:131–40.
Alvarez-Múgica M, Fernández-Gómez JM, Cebrian V, Fresno F, Escaf S, Sánchez-Carbayo M. Polyamine-modulated factor-1 methylation predicts bacillus Calmette-Guérin response in patients with high-grade non-muscle-invasive bladder carcinoma. Eur Urol. 2013;63:364–70.
Ahlén Bergman E, Hartana CA, Johansson M, Linton LB, Berglund S, Hyllienmark M, et al. Increased CD4 + T cell lineage commitment determined by CpG methylation correlates with better prognosis in urinary bladder cancer patients. Clin Epigenet. 2018;10.
Hartana CA, Ahlén Bergman E, Broomé A, Berglund S, Johansson M, Alamdari F, et al. Tissue-resident memory T cells are epigenetically cytotoxic with signs of exhaustion in human urinary bladder cancer. Clin Exp Immunol. 2018;194:39–53.
Koestler DC, Usset J, Christensen BC, Marsit CJ, Karagas MR, Kelsey KT, et al. DNA methylation-derived neutrophil-tolymphocyte ratio: An epigenetic tool to explore cancer inflammation and outcomes. Cancer Epidemiol Biomark. 2017;26:328–38.
Nunes SP, Henrique R, Jerónimo C, Paramio JM. DNA Methylation as a Therapeutic Target for Bladder Cancer. Cells. 2020;9:1850.
Maraver A, Fernandez-Marcos PJ, Cash TP, Mendez-Pertuz M, Dueñas M, Maietta P, et al. NOTCH pathway inactivation promotes bladder cancer progression. J Clin Invest. 2015;125:824–30.
Rampias T, Vgenopoulou P, Avgeris M, Polyzos A, Stravodimos K, Valavanis C, et al. A new tumor suppressor role for the Notch pathway in bladder cancer. Nat Med. 2014;20:1199–205.
Ramakrishnan S, Hu Q, Krishnan N, Wang D, Smit E, Granger V, et al. Decitabine, a DNA-demethylating agent, promotes differentiation via NOTCH1 signaling and alters immune-related pathways in muscle-invasive bladder cancer. Cell Death Dis. 2017;8.
Segovia C, San José-Enériz E, Munera-Maravilla E, Martínez-Fernández M, Garate L, Miranda E, et al. Inhibition of a G9a/DNMT network triggers immune-mediated bladder cancer regression. Nat Med. 2019;25:1073–81.
Funding
This study was cofunded by European Regional Development Fund (FEDER) Grants from Science and Innovation (SAF2015-66015-R, and PID2019-110758RB-I00 to JMP), Instituto de Salud Carlos III (CIBERONC no. CB16/12/00228 to JMP; PI20/00813 and DTS20/00043 to MD) and Programa Operacional Competitividade e Internacionalização (POCI), in the component FEDER, and by national funds (OE) through FCT/MCTES, in the scope of the project HyTherCaP- PO-CI-01-0145-FEDER-29030 to CJ). IL is supported by a predoctoral fellowship from AECC (Spanish Ass. against Cancer), Predoctoral AECC 2019 grant number PRDMA19024LODE. SPN is supported by an FCT-Fundação para a Ciência e Tecnologia Grant (SFRH/BD/ 144241/2019).
Author information
Authors and Affiliations
Contributions
IL, SPN and JMP conceptualized the study; IL and SPN wrote the manuscript; RH, CJ, MD and JMP reviewed and edited the manuscript; RH, CJ, MD and JMP supervised the study; CJ, MD and JMP acquired the funding. All authors have read and agreed to the published version of the manuscript. All figures are original and were created with BioRender.com.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table 1.
Main studies comprising epigenetic drugs and biomarkers associated with immunotherapy.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lodewijk, I., Nunes, S.P., Henrique, R. et al. Tackling tumor microenvironment through epigenetic tools to improve cancer immunotherapy. Clin Epigenet 13, 63 (2021). https://doi.org/10.1186/s13148-021-01046-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-021-01046-0