
Abstract
Background
Stable transfection systems have been described in many protozoan parasites, including Plasmodium falciparum, Cryptosporidium parvum, Babesia bovis, Babesia ovata, and Babesia gibsoni. For Babesia sp. Xinjiang (Bxj), which is the causative pathogen of ovine babesiosis and mainly prevails across China, the platform of those techniques remains absent. Genetic manipulation techniques are powerful tools to enhance our knowledge on parasite biology, which may provide potential drug targets and diagnostic markers.
Methods
We evaluated the inhibition efficiency of blasticidin (BSD) and WR99210 to Bxj. Then, a plasmid was constructed bearing selectable marker BSD, green fluorescent protein (GFP) gene, and rhoptry-associated protein-1 3′ terminator region (rap 3′ TR). The plasmid was integrated into the elongation factor-1 alpha (ef-1α) site of Bxj genome by cross-over homologous recombination technique. Twenty μg of plasmid was transfected into Bxj merozoites. Subsequently, drug selection was performed 24 h after transfection to generate transfected parasites.
Results
Transfected parasite lines, Bxj-c1, Bxj-c2, and Bxj-c3, were successfully obtained after transfection, drug selection, and colonization. Exogenous genes were integrated into the Bxj genome, which were confirmed by PCR amplification and sequencing. In addition, results of western blot (WB) and indirect immunofluorescence assay (IFA) revealed that GFP-BSD had expressed for 11 months.
Conclusions
In our present study, stable transfection system for Bxj was successfully developed. We anticipate that this platform will greatly facilitate basic research of Bxj.
Graphical abstract

Similar content being viewed by others


Background
Babesiosis, caused by protozoan pathogens of the genus Babesia (phylum Apicomplexa, order Piroplasmida) infective to humans and domestic and wild animals, is one of the emerging and re-emerging tick-borne disease in tropical and subtropical regions of the world [1, 2]. A wide spectrum of clinical manifestation ranges from mild fever to serve anemia hemoglobinuria and even death [3]. Early in the nineteenth century, the first case of babesiosis was reported in Rumania and correlated with bovine hemoglobinuria or red water fever. Shortly after, similar organisms were also determined in sheep red blood cells [4]. Till now, more than 100 Babesia species have been described throughout the world. However, only a few Babesia species have been identified in sheep and cause ovine babesiosis, namely B. ovis, B. motasi, and B. crassa.
In China, ovine babesiosis was firstly reported as early as 1982 in Sichuan Province and 1986 in Heilongjiang Province [5, 6]. Since then, this disease was sporadically reported in Xinjiang Uygur Autonomous region, Henan, Shannxi, Yunnan, and Hebei Province. During the past decades of investigations of ovine babesiosis, great attention had been paid to a novel Babesia species, Babesia sp. Xinjiang, which presented distinct morphologies from B. motasi, B. ovis, and B. crassa in a thin blood smear [7, 8]. This novel Babesia species was initially isolated from a splenectomized sheep infested with partially engorged Rhipicephalus sanguineus and Hyalomma anatolicum anatolicum ticks [9]. In the years since then, systematic studies of this Babesia species have investigated morphological characteristics, transmission patterns, culture features, epidemiology, pathogenicity, and whole-genome sequencing and annotation [10,11,12,13,14]. Available results indicated that this pathogen is widely distributed across China and has caused significantly economic losses to the sheep industry [15].
During recent decades, much attention has been focused on developing diagnostic assays and performing epidemiological investigations. However, available approaches to evaluate virulence factors and vaccine candidate antigens, and even to explore invasion and transmission mechanisms for these parasites, are limited. Ovine babesiosis control suffers from a lack of effective vaccines and limited choices of therapeutic drugs. Development of these relies on a better understanding of the basic biology of Babesia species [16]. Genetic manipulation technologies have been described to discover virulence factors and to investigate the interaction of parasite and host cells in apicomplexan parasites such as Cryptosporidium parvum, Babesia bovis, B. gibsoni, B. ovis, B. ovata, Theileria annulata, and T. parva [17,18,19,20,21,22,23,24,25]. In this study, we developed a stable transfection system of Bxj merozoites to investigate tick–Babesia and host–Babesia interactions in the future.
Methods
In vitro culture system
Bxj was cultured in 24-well plates (Corning, MA, USA) at 37 °C under an atmosphere of 5% CO2 as reported previously [14, 15, 26]. Briefly, the parasite was cultured in 7.5% fresh sheep erythrocytes supplemented with 20% fetal bovine serum (FBS) (Gibco, Carlsbad, CA, USA) in Roswell Park Memorial Institute 1640 medium (Lonza Biologics, Portsmouth, NH, USA).
Evaluation the inhibition efficiency of BSD and WR99210 to Bxj
Bxj was cultured in complete medium with various concentrations of BSD (1 μg/ml, 2 μg/ml, 5 μg/ml, 8 μg/ml, and 10 μg/ml) and WR99210 (10 μg, 25 μg/ml, 50 μg/ml, 100 μg/ml, and 200 μg/ml), and the medium was replaced each day. The proportion of infected red blood cells (PI) was calculated by examining 2000 red blood cells (RBCs) stained with Giemsa in thin blood smear at 48 h [22]. The formula of Bxj growth inhibition was as follows:
Plasmid constructs
The plasmid construct used in this study is listed as Fig. 1 (PBS-bpgb-rap-orf). Initially, the ef1α-B 5′ non-coding region and enhanced green fluorescent protein (eGFP) gene and blasticidin-S (BSD) deaminase gene were amplified from Bxj genomic DNA and plasmid pgfp-bsd-ef (kindly donated by Carlos E. Suarez) using the primer sets listed in Table 1. Then, these two sequences were cloned into the NotI site of plasmid pBluescript SK(+) using a ClonExpress MultiS One Step Cloning Kit (Vzayme, Nanjing, China) according to the manufacturer’s instructions. This plasmid was designated as PBS-bpgb. Meanwhile, Bxj rap3′ terminal region (Bxj rap3′ TR) and ef1α–B open reading frame (ef1α–B-orf) sequences were amplified from Bxj genomic DNA and cloned into the NotI site of plasmid pBluescript SK(+) as mentioned above, and the generated plasmid was designated as PBS-TR-orf. Subsequently, the first large fragment of the ef1a-B non-coding region and gfp-bsd was amplified from plasmid PBS-bpgb, and the second fragment of the Bxj rap3' TR and ef1α–B open reading frame sequences was derived from plasmid PBS-TR-ORF. Finally, these two large fragments were ligated into the NotI site of plasmid pBluescript SK(+), designated as PBS-bpgb-rap-orf. Furthermore, cloned plasmids were validated by polymerase chain reaction (PCR) amplification using two sets of primers (set 1: ef1a-B-F and ef1a-B-R; set 2: ef1a-orf-F and ef1a-orf-R) and confirmed by sequencing and extracted using a QIAGEN Plasmid Maxi Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.

Babesia sp. Xinjiang sensitivity to BSD. All data are presented as means ± SD of triplicate cultures
Electroporation of Bxj merozoites and drug selection
When infected RBCs reached 10–20%, the cultures in a 75 cm2 flask were centrifuged at 800×g for 10 min. Then, the supernatant was removed, and cell pellets of Bxj-infected RBCs were washed twice in cold cytomix buffer (120 mM KCl, 0.15 mM CaCl2, 10 mM K2HPO4/KH2PO4, pH 7.6, 25 mM HEPES. pH 7.6, 2 mM EGTA, 5 mM MgCl2, final pH 7.6) [17, 18, 22, 27,28,29]. Mixture, containing 1 × 108 infected RBCs and 20 μg of circular PBS-bpgb-rap-orf plasmid in a final volume of 100 μl was transfected with parameters of 1200 V and 25 μF using a Gene Pulser Xcell™ Electroporation System (Bio-Rad Laboratories, Hercules, CA, USA). After transfection, the mixtures were transferred into wells of 24-well culture plates containing 7.5% fresh sheep RBCs and 20% FBS. After transfection for 24 h, 2 μg/ml of blasticidin-S (BSD, Gibco, R21001) was added to the cultures, and the concentration of BSD was gradually increased to 10 μg. Parasites, maintained in medium with BSD for 2–3 weeks, could be observed in thin blood smear stained with Giemsa under microscopy. To obtain a clonal strain, the population of parasites were diluted to 2.5 infected RBCs/ml with completed medium containing 7.5% of fresh sheep RBCs. Then, 100 μl of the diluted culture was added to each well of a 96-well culture plate, maintained at 37 °C in an atmosphere of 5% CO2 for 14–17 days. During this period, the medium was completely replaced every 3 days. Three monoclonal strains were able to grow in a high concentration of BSD (10 μg/ml) and were designated as Bxj-c1, Bxj-c2, and Bxj-c3.
Identification of monoclonal parasite Bxj stably expressing eGFP-BSD
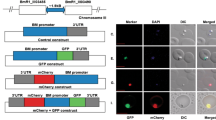
Three pairs of primer (Table 1) were designed to confirm whether the egfp-bsd-rap fragments were correctly integrated into the Bxj ef1α locus. The first set of primers (efbs-F and efbs-R) was designed to amplify the F1 fragment with the size of 1924 bp to determine 5′ recombination, while the second set of primers (gfor-F and gfor-R) were generated to amplify the F2 fragment of 3394 bp to confirm 3′ recombination. Additionally, a large-size fragment (F3, 5960 bp) was amplified using the third primer pair (efbs-F and orf-R) targeting the ef1α locus (Fig. 2).

Plasmid structures and ef1α locus in Bxj genome. PBS-bpgd-rap-orf shows the structure of plasmid used for stable transfection in this study. ef1α locus illustrates the organization of ef1α in the Bxj genome. Transfected Bxj parasite lines, stably expressing eGFP-BSD fusion protein, should be like the Bxj transfected diagram
To identify the expression of eGFP-BSD fusion protein, WB and indirect immunofluorescence assay (IFA) were performed. Briefly, when infected blood cells reached 5–10%, cultures of Bxj-c1, Bxj-c2, Bxj-c3, and wild-type Bxj were added to three times the volume of red blood cell lysis buffer (Solarbio, Beijing, China) to lyse red blood cells. Merozoites of Bxj-c1, Bxj-c2, and Bxj-c3 and wild-type Bxj (WT) were collected using centrifugation at 5000×g for 10 min. The merozoite pellet was mixed with a twofold volume of 1x sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer and heated at 100 °C for 5 min. Soluble proteins were subjected to SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane. Then, the membrane was incubated with monoclonal anti-eGFP antibody (Sigma, SAB2702211, dilution: 1:2000) as the primary antibody followed with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Proteintech, USA, dilution: 1:5000), diluted with 0.1 M Tris-buffered saline (pH 7.6) with 0.1% Tween 20 (TBST). After the membranes were washed three times using TBST, bands were detected using a SuperSignal West Femto Kit (Thermo, USA) in a ChemiDoc MP imaging system (Bio-Rad, USA). IFA was performed using anti-GFP antibody (Sigma, SAB2702211, dilution: 1:200) as the primary antibody and Alexa 488-conjugated goat anti-mouse IgG (Thermo Fisher, USA, dilution: 1:500) as the secondary antibody [30]. The parasite nucleus was stained with Hoechst 33342 (Invitrogen, dilution: 1:2000) and observed with confocal laser scanning microscopy (SP8, Leica, Germany).
Results
An inhibitory effect of BSD on Bxj merozoites in in vitro culture
The inhibitory efficiency of BSD on Bxj merozoites in in vitro culture shows an upward trend with gradual increase of drug concentration, ranging from 1 to 10 μg/ml (Fig. 1). After 48 h of drug selection, the 50% inhibitory concentration of BSD (IC50) was 2.26 μg/ml. When the concentration reached 8 μg/ml, the growth of over 90% of Bxj merozoites was inhibited. To inhibit the growth of WT, 2–10 μg/ml BSD was employed in our following experiments.
Regarding the inhibition assays with WR99210, no efficient growth inhibition was observed. When the concentration of WR99210 increased to 150 μg/ml, the growth inhibition of Bxj was as low as ~ 30%. Thus, the BSD gene was selected to be included in the following constructs as a selectable marker.
Stable transfected Bxj
After 2 weeks of drug selection with BSD, transfected Bxj was cloned by limiting dilution. Three clonal parasite lines, designated as Bxj-c1, Bxj-c2, and Bxj-c3, were obtained. At this point, no fluorescence was observed in the three lines. To determine whether the fragment of egfp-bsd-rap was correctly integrated into the target locus, PCR amplifications were performed using three primer pairs. As show in Figs. 2 and 3a, three fragments (1924, 3394, and 5960 bp) were successfully obtained from each of the parasite lines (Bxj-c1, Bxj-c2, and Bxj-c3) and validated by gene sequencing. On the contrary, only one fragment (approximately 3500 bp), corresponding to the third primer pair, was amplified from WT (Fig. 3a).

Identification of monoclonal parasite Bxj stably expressing eGFP-BSD. a PCR amplification of F1, F2, and F3 fragments. b Western blot analysis of stable expression of eGFP-BSD in Bxj-c1, Bxj-c2, and Bxj-c3. c IFA confirms stable eGFP expression of Bxj. The parasite nucleus was stained with Hoechst 33342
Expression analysis of eGFP-BSD fusion protein in Bxj merozoites
Merozoites collected from Bxj-c1, Bxj-c2, Bxj-c3, and WT were subjected to Western blot analysis to determine eGFP-BSD fusion protein (around 38 kDa) using monoclonal anti-eGFP. Available results showed that antibodies specifically bound to a protein of ~ 38 kDa; however, they did not react with merozoite proteins of WT (Fig. 3b). Similarly, the evidence of IFA also demonstrated expression of eGFP-BSD in Bxj merozoites (Fig. 3c). These three lines stably expressed eGFP for 11 months.
Discussion
The first description of a stable transfection system in Babesia genus is reported in B. bovis in 2009; subsequently, this technique has been developed in B. gibsoni and B. ovata [17, 22, 29]. For most of public health/economically important Babesia spp., including B. microti, B. divergens, B. duncani, B. ovis, and Theileria spp., such as T. annulata and T. parva, genetic manipulation platforms have not been described. In addition, Bxj was firstly isolated from sheep infested with R. sanguineus and H. anatolicum anatolicum collected from the Xinjiang Uygur Autonomous Region in China in 2001 [31]. Prevalence of Bxj has been systematically investigated across China using molecular and sera techniques, such as multiplex PCR, loop-mediated isothermal amplification method, reverse line blot assay, and enzyme-linked immunosorbent assays [11, 12, 15]. Those data indicated that this pathogen was widely prevalent in sheep and goat in almost all investigated regions across China; however, genetic manipulation of Bxj has not been documented. In our previous study, we systematically evaluated the transient transfection parameters from Bxj, including transfection solution, programs, amount of plasmid DNA, and promoter activities. Eventually, a Bxj transient transfection system was developed with the most favorable transfection conditions (human T cell nucleoporation solution, program V-024, 20 μg of plasmid DNA, and ef1α promoter). However, an alternative transfection parameter (cytomix transfection solution, 1200 V and 25 μF, 20 μg of plasmid DNA, and ef1α promoter) also achieved preferable results.
A drug selection marker is required to develop a stable transfection system for Bxj. In this study, we firstly evaluated the sensitivity of Bxj to WR99210, which had been reported to provide suitable transfection of Plasmodium, B. bovis, and B. gibsoni. However, Bxj showed resistance to this drug. Similar situations have been described in B. bovis and Plasmodium spp. [17, 32]. This can be explained by the existence of an associated gene in the Bxj genome. Inhibition assays with BSD resulted in a strong Bxj merozoites growth inhibition with an IC50 of 2.26 μg/ml, validating this drug as preferable to WR99210, the selection maker. Thus, this was the selection maker used for developing Bxj stable transfection. Nevertheless, it is worth mentioning that the value of IC50 is around five times of that B. bovis (~ 0.4 μg/ml).
With regard to the target locus for genome integration, the ef1a locus was selected as the ideal target site for several Babesia spp., for instance B. bovis, B. ovata, and B. gibsoni [17, 22, 29]. Sequence analysis reveals that the Bxj ef1α locus consists of two identical genes, arranged head to head and separated by a 1454-bp regulator sequence. Disruption one of these two genes had no significantly negative effect on survival and growth of parasites. It is suggested that this gene locus also serves as a suitable target site to introduce foreign genes for Bxj. Transfected parasites could be observed under microscopy after 2 weeks of selection with BSD. PCR identification revealed that the fragment of gfp-bsd-rap 3' UTR was successfully introduced into the ef1α locus of the cloned Bxj-c1, Bxj-c2, and Bxj-c3. Stably expressed eGFP was confirmed by WB and IFA assays; however, green fluorescence could not be observed with fluorescence microscopy. We attempted to obtain transfected Bxj which could directly detect fluorescence under fluorescence microscopy, by replacing the ef-1α promoter with an actin promoter (approximately 2000 bp)/ef1α-IG A and replacing eGFP with a red fluorescent protein gene (data not shown). However, this goal was not achieved. A similar situation was previously reported in C. parvum [19]. The firefly luciferase and fluorescent proteins were not detected in transfected parasites, whereas nanoluciferase showed significant reporter activity at 48 h after transfection [19].
Although there are still some drawbacks in this system, including lack of fluorescence and relatively low transfect efficiency compared with the CRISPR/Cas9-based genome editing strategy, this transfection system of Bxj provides a powerful tool to determine gene function and discover critical gene families of invasion, egress, immune evasion, and even virulence factors. A more convenient, facile, and highly effective technique needs to be developed in the near future.
Conclusions
To conclude, we provide a stable transfection system for Bxj and obtain transfected parasites, which have stably expressed eGFP-BSD for 11 months. This study is the first effort to create a platform for genetic manipulation of Bxj to further illustrate the invasive mechanism of this parasite, together with the parasite–vector and parasite–host interactions.
Availability of data and materials
All data are available upon request.
Abbreviations
- Bxj :
-
Babesia sp. Xinjiang
- FBS:
-
Fetal bovine serum
- RBCs:
-
Red blood cells
- PCR:
-
Polymerase chain reaction
- eGFP:
-
Enhanced green fluorescent protein
- BSD:
-
Blasticidin
- WB:
-
Western blot
- IFA:
-
Indirect immunofluorescence assay
References
Uilenberg G. International collaborative research: significance of tick-borne hemoparasitic diseases to world animal health. Vet Parasitol. 1995;57:19–41.
Uilenberg G. Babesia—a historical overview. Vet Parasitol. 2006;138:3–10.
Yin H, Lu W, Luo J. Babesiosis in China. Trop Anim Health Prod. 1997;29(Suppl):11S-S15.
Babes V. Sur l’ hémoglobinurie bactérienne du boeuf. C R Acad Sci. 1888;107:692–4.
Chen D. Investigation of ovine piroplasmosis. Chin J Vet SciTechnol. 1982;12:31–2.
Zhao X, Li C, Ming Y, Bai S, Chi S, Su G, et al. Investigation of ovine babesiosis. Chin J Vet Sci Technol. 1986;01:26–7.
Bai Q, Liu QY, Liu DK, Ren JX, Li X. Isolation and preliminary characterization of a large Babesia sp. from sheep and goats in the eastern part of Gansu Province, China. Parasitol Res. 2002;88:S16–21.
Yin H, Lu WS, Luo JX. Babesiosis in China. Trop Anim Health Prod. 1997;29:11s-s15.
Guan G, Ma M, Moreau E, Liu J, Lu B, Bai Q, et al. A new ovine Babesia species transmitted by Hyalomma anatolicum anatolicum. Exp Parasitol. 2009;122(4):261–7.
Guan G, Korhonen PK, Young ND, Koehler AV, Wang T, Li Y, et al. Genomic resources for a unique, low-virulence Babesia taxon from China. Parasites Vectors. 2016;9:564.
Guan G, Chauvin A, Luo J, Inoue N, Moreau E, Liu Z, et al. The development and evaluation of a loop-mediated isothermal amplification (LAMP) method for detection of Babesia spp. infective to sheep and goats in China. Exp Parasitol. 2008;120:39–44.
Niu QL, Luo JX, Guan GQ, Liu ZJ, Ma ML, Liu AH, et al. Differentiation of two ovine Babesia based on the ribosomal DNA internal transcribed spacer (ITS) sequences. Exp Parasitol. 2009;121:64–8.
Wang X, Wang J, Liu J, Liu A, He X, Xiang Q, et al. Insights into the phylogenetic relationships and drug targets of Babesia isolates infective to small ruminants from the mitochondrial genomes. Parasites Vectors. 2020;13:378.
Guan G, Ma M, Liu A, Du P, Ren Q, Li Y, et al. Continuous in vitro cultivation of a recently identified Babesia that infects small ruminants in China. Vet Parasitol. 2012;187:3–4.
Guan G, Ma M, Liu A, Ren Q, Wang J, Yang J, et al. A recently identified ovine Babesia in China: serology and sero-epidemiology. Parasitol Int. 2012;61:532–7.
Antunes S, Rosa C, Couto J, Ferrolho J, Domingos A. Deciphering Babesia-vector interactions. Front Cell Infect Microbiol. 2017;7:429.
Suarez CE, McElwain TF. Stable expression of a GFP-BSD fusion protein in Babesia bovis merozoites. Int J Parasitol. 2009;39:289–97.
Asada M, Tanaka M, Goto Y, Yokoyama N, Inoue N, Kawazu S. Stable expression of green fluorescent protein and targeted disruption of thioredoxin peroxidase-1 gene in Babesia bovis with the WR99210/dhfr selection system. Mol Biochem Parasitol. 2012;181:162–70.
Vinayak S, Pawlowic MC, Sateriale A, Brooks CF, Studstill CJ, Bar-Peled Y, et al. Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature. 2015;523:477–80.
Adamson R, Lyons K, Sharrard M, Kinnaird J, Swan D, Graham S, et al. Transient transfection of Theileria annulata. Mol Biochem Parasitol. 2001;114:53–61.
De Goeyse I, Jansen F, Madder M, Hayashida K, Berkvens D, Dobbelaere D, et al. Transfection of live, tick derived sporozoites of the protozoan Apicomplexan parasite Theileria parva. Vet Parasitol. 2015;208:238–41.
Hakimi H, Yamagishi J, Kegawa Y, Kaneko O, Kawazu S, Asada M. Establishment of transient and stable transfection systems for Babesia ovata. Parasites Vectors. 2016;9:171.
Silva MG, Knowles DP, Suarez CE. Identification of interchangeable cross-species function of elongation factor-1 alpha promoters in Babesia bigemina and Babesia bovis. Parasites Vectors. 2016;9:576.
Liu M, Asada M, Cao S, Adjou Moumouni PF, Vudriko P, Efstratiou A, et al. Transient transfection of intraerythrocytic Babesia gibsoni using elongation factor-1 alpha promoter. Mol Biochem Parasitol. 2017;216:56–9.
Rosa C, Asada M, Hakimi H, Domingos A, Pimentel M, Antunes S. Transient transfection of Babesia ovis using heterologous promoters. Ticks Tick Borne Dis. 2019;10: 101279.
Guan GQ, Ma ML, Liu AH, Du PF, Ren QY, Li YQ, et al. Continuous in vitro cultivation of a recently identified Babesia that infects small ruminants in China. Vet Parasitol. 2012;187:371–8.
Suarez CE, McElwain TF. Transient transfection of purified Babesia bovis merozoites. Exp Parasitol. 2008;118:498–504.
Jaijyan DK, Govindasamy K, Singh J, Bhattacharya S, Singh AP. Establishment of a stable transfection method in Babesia microti and identification of a novel bidirectional promoter of Babesia microti. Sci Rep. 2020;10:15614.
Liu M, Adjou Moumouni PF, Asada M, Hakimi H, Masatani T, Vudriko P, et al. Establishment of a stable transfection system for genetic manipulation of Babesia gibsoni. Parasites Vectors. 2018;11:260.
Qin M, Tang X, Yin G, Liu X, Suo J, Tao G, et al. Chicken IgY Fc expressed by Eimeria mitis enhances the immunogenicity of E. mitis. Parasites Vectors. 2016;9:164.
Guiquan G, Hong Y, Jianxun L, Wenshun L, Qicai Z, Milin M. Isolation of a large ovine Babesia sp. in Xinjiang, China. China J Vet Sci Technol. 2001;31:35–6.
Wang P, Wang Q, Sims PF, Hyde JE. Rapid positive selection of stable integrants following transfection of Plasmodium falciparum. Mol Biochem Parasitol. 2002;123:1–10.
Acknowledgements
We acknowledged Carlos E. Suarez from the Department of Veterinary Microbiology and Pathology, Washington State University for technical supports and providing pgfp-bsd-ef and pBluescript SK( +) plasmid. The authors are grateful to Shin-ichiro Kawazu from the National Research Center for Protozoan Diseases, Obihiro University of Agriculture and Veterinary Medicine, Obihiro, Hokkaido 080-8555, Japan, for providing DHFR-gfp plasmid.
Funding
This study was financially supported by the National Science Foundation of China (Grant No. 31972701), National Key Research and Development Program of China (Grant No. 2017YFD0501200), the 973 Program (Grant No. 2015CB150300), ASTIP (Grant No. CAAS-ASTIP-2016-LVRI), NBCIS (Grant No. CARS-37), and the Jiangsu Co-innovation Center Program for the Prevention and Control of Important Animal Infectious Disease and Zoonoses.
Author information
Authors and Affiliations
Contributions
JW and XW carried out the experiments. JW wrote the draft of the manuscript. JY, JL, AL, and YL participated in plasmid construction. GG corrected the manuscript. JLu and HY supervised all parts of the study. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Animal Ethics Committee of the Lanzhou Veterinary Research Institute, CAAS (Permit No. LVRIAEC-2018-001). All the procedures were conducted according to the Animal Ethics Procedures and Guidelines of the People’s Republic of China.
Consent for publication
All authors consent to be published.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, J., Wang, X., Guan, G. et al. Stable transfection system for Babesia sp. Xinjiang. Parasites Vectors 14, 463 (2021). https://doi.org/10.1186/s13071-021-04940-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-021-04940-x