
Abstract
Background
Babesiosis, a tick-borne disease caused by protozoans of the genus Babesia, is widespread in subtropical and tropical countries. Mitochondria are essential organelles that are responsible for energy transduction and metabolism, calcium homeostasis and cell signaling. Mitochondrial genomes could provide new insights to help elucidate and investigate the biological features, genetic evolution and classification of the protozoans. Nevertheless, there are limited data on the mitochondrial genomes of ovine Babesia spp. in China.
Methods
Herein, we sequenced, assembled and annotated the mitochondrial genomes of six ovine Babesia isolates; analyzed the genome size, gene content, genome structure and cytochrome b (cytb) amino acid sequences and performed comparative mitochondrial genomics and phylogenomic analyses among apicomplexan parasites.
Results
The mitochondrial genomes range from 5767 to 5946 bp in length with a linear form and contain three protein-encoding genes, cytochrome c oxidase subunit 1 (cox1), cytochrome c oxidase subunit 3 (cox3) and cytb, six large subunit rRNA genes (LSU) and two terminal inverted repeats (TIR) on both ends. The cytb gene sequence analysis indicated the binding site of anti-Babesia drugs that targeted the cytochrome bc1 complex. Babesia microti and Babesia rodhaini have a dual flip-flop inversion of 184–1082 bp, whereas other Babesia spp. and Theileria spp. have one pair of TIRs, 25–1563 bp. Phylogenetic analysis indicated that the six ovine Babesia isolates were divided into two clades, Babesia sp. and Babesia motasi. Babesia motasi isolates were further separated into two small clades (B. motasi Hebei/Ningxian and B. motasi Tianzhu/Lintan).
Conclusions
The data provided new insights into the taxonomic relationships and drug targets of apicomplexan parasites.

Similar content being viewed by others

Background
Babesiae are tick-transmitted hemoprotozoans that cause babesiosis, which is characterized by fever, anemia, jaundice and hemoglobinuria. The main causative agents in small ruminants are Babesia ovis and B. motasi, transmitted by the ticks Rhipicephalus spp. and Haemaphysalis spp. in Asia, South America, Africa, the Far East and Europe [1,2,3]. In China, several Babesia isolates were isolated from sheep during the period of 2000–2010, Babesia sp. Dunhuang (BspDH), Babesia sp. Xinjiang (BspXJ), B. motasi Tianzhu (BmTZ), B. motasi Lintan (BmLT), B. motasi Hebei (BmHB) and B. motasi Ningxian (BmNX). The six ovine Babesia parasites have different characteristics in serology, pathogenicity, vector specificity and virulence. For instance, BspXJ and BspDH have low virulence and are transmitted by Hyalomma spp. ticks, whereas the vector ticks of BmLT, BmHB, BmNX and BmTZ are Haemaphysalis spp. and cause a range of clinical manifestations. BspXJ/DH is serologically distinct from B. motasi, and there are also differences between isolates of B. motasi [4,5,6,7,8,9,10,11,12]. The prevalences of B. motasi and Babesia sp. were between 30.4–31.7 and 36.0–43.5%, respectively, which indicates that Babesia infection is prevalent in sheep and goats in China [11, 13,14,15].
Mitochondria are crucial organelles that are responsible for energy transduction and metabolism, calcium homeostasis and cell signaling [16]. The mitochondrial cytochrome bc1 complex, known as complex III, is a multimeric enzyme that is an indispensable element of the respiratory chain and energy conversion [17]. Complex III comprises three redox active subunits, cytochrome b (encoded by the mitochondrial genome), cytochrome c oxidase subunit 1 and the Rieske iron-sulfur protein. The catalytic cycle of cytochrome bc1 (Q cycle) is in quinone reduction (Qi) and quinol oxidation (Qo) sites that are mainly form by cytochrome b. Inhibitors of the bc1 complex may be divided into two types that act on Qi and Qo sites according to the binding site. Therefore, the bc1 complex has been considered a promising target for the development of antimicrobial compounds. Some studies suggest that the yeast bc1 complex structure could be used as a model for discovering new antimalarial drugs [18, 19]. However, mutations in the cytochrome b (cytb) gene have been shown to be the molecular basis of drug resistance of some microorganisms [19,20,21,22]. It has been demonstrated that several anti-Babesia drugs, such as diminazene aceturate and atovaquone, which possibly inhibit mitochondrial respiratory activity and electron transport, are ineffective owing to drug resistance [23,24,25]. Therefore, it is necessary to understand the mechanism of resistance of these anti-Babesia drugs and develop new drugs.
Studies of mitochondrial genomes could provide new insights into the biological features, genetic evolution and classification of the causative agents, as well as providing data for designing anti-Babesia compounds. To date, sequencing of the mitochondrial genomes has been performed for B. bigemina, Babesia bovis, B. caballi, B. canis, B. conradae, B. duncani, B. gibsoni, B. microti, B. orientalis, B. rodhaini, B. rossi, B. vogeli, Theileria equi and T. orientalis [26,27,28,29,30,31]. However, there are limited data on the mitochondrial genomes of ovine Babesia spp. in China. Although the mitochondrial genomes of BmLT and BspXJ were sequenced by using Illumina sequencing technology, they have not been sequenced using the Sanger dideoxy chain-termination method for verification [32]. In this study, we sequenced six mitochondrial genomes of Babesia isolates that can infect small ruminants, and the assembled and annotated sequences were submitted to GenBank after comparison with those of other piroplasms. These data were used to clarify the phylogenetic relationships and classification of the babesiae and to determine novel molecular markers for identification of Babesia species. The present study provides valuable information for understanding mitogenome evolution among apicomplexan parasites, identifying diagnostic markers and screening drug targets.
Methods
Parasites and isolation of genomic DNA
The purified merozoites of six ovine Babesia isolates were provided by our laboratory [33]. Genomic DNA extraction, concentration measurement and quality evaluation were performed as previously described by Wang et al. [34]. All genomic DNA samples were kept at − 20 °C until use.
Amplification and sequencing
The PCR primers were designed based on the reported genomic sequences of B. bovis and B. bigemina (GenBank: AB499088 and AB499085) (Additional file 1: Table S1). Mitochondrial genome fragments were amplified and cloned into the pGEM®-T Easy Vector (Promega, Beijing, China) for subsequent sequencing by Sangon Biotech Company.
Genome assembly and annotation
Mitochondrial genomic fragments were assembled with CLC Genomics Workbench v.7.5.1. The mitogenome annotation was performed using DOGMA [35] and Artemis [36], followed by application of BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) to identify homologous proteins in apicomplexan parasites in the GenBank database. The tRNA genes were searched using tRNAscan-SE v.2.0 with the default search mode and other mitochondrial sequence sources [37]. The rRNA genes were annotated by searching previously reported rRNA sequences of B. bovis, B. microti, B. orientalis and T. parva. The genome comparisons were aligned using Mauve [38]. The sequences were deposited in the GenBank database under the accession numbers MK962313, MK962314 and MN605889-MN605892.
Sequence alignment and amino acid conservation of the cytb gene
We referred to previous studies [18,19,20,21,22, 39,40,41,42,43] that used the mitochondrial cytochrome bc1 complex from Saccharomyces cerevisiae to determine the binding residues of the inhibitor (acting on the bc1 complex) by X-ray crystallography, spectroscopy and cytb sequence alignment and analysis. In a previous report, sequence alignments of the cytb amino acid residue from S. cerevisiae, Plasmodium falciparum, Toxoplasma gondii, B. microti and Bos taurus indicated that the drug binding residues of cytb are conserved. Therefore, we used MegAlign software to compare the cytb sequences of S. cerevisiae, B. taurus, T. gondii, T. parva, B. microti, B. duncani, P. falciparum, BspXJ/DH, BmLT/TZ and BmNX/HB to identify the main resistance-related mutations and drug-binding residues in the genus Babesia.
Phylogenetic analysis
The concatenated sequences of cytb and cytochrome c oxidase subunit 1 (cox1) amino acid residues of 26 apicomplexan parasites (Table 1) were aligned using Clustal W implemented in MEGA v.6.06 (http://www.megasoftware.net/) software. Subsequently, a phylogenetic tree was constructed using MEGA v.6.06 with maximum likelihood (ML) or neighbor-joining (NJ) analysis based on the JTT with the Freqs model. T. gondii and Plasmodium spp. were used as the outgroup. Furthermore, phylogenetic analysis of the whole mitochondrial nucleotide sequence was conducted by the ML method using the Kimura 2-parameter nucleotide substitution model. Consensus trees were created after bootstrap analyses with 1000 replications.
Results
Sequence analysis
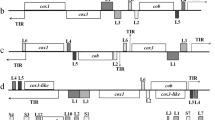
Sequence analysis revealed that the ovine Babesia mitochondrial genomes were linear DNA of 5767 to 5946 bp, with 70.05–70.87% A + T content (Table 1). Mitochondrial genomes of six ovine Babesia isolates contained three protein-encoding genes, cox1, cytochrome c oxidase subunit 3 (cox3), cytb, six large subunit rRNA genes (LSU) and two terminal inverted repeats (TIRs). The transcriptional direction of cox3, LSU3, LSU6, LSU2, cytb and LSU5 was from 3′ to 5′, whereas the direction of cox1, LSU1 and LSU4 was from 5′ to 3′ (Fig. 1, Additional file 2: Figure S1). The start codons of the cox3 and cytb genes of ovine Babesia were ATA and ATG, respectively. The initiation codon of the cox1 gene of BspXJ/DH was ATA, whereas that of B. motasi was ATG. Most of the protein-coding genes had TAA as a termination codon, followed by TGA (Table 2).

Comparison of the mitochondrial genomes of six ovine Babesia isolates and other apicomplexan parasites. The analysis was performed using SnapGene software and Adobe Photoshop. The different shades of gray represent different gene types. In detail, white represents TIR, 40% gray represents protein-encoding genes and 80% gray represents LSU
Comparison of mitochondrial genomes sequenced by Sanger and Illumina technology
The alignment of BspXJ and BmLT mitochondrial genomes sequenced using the Illumina method in a previous study [32] and the Sanger technique in this study was performed. The results showed that two sets of data from Sanger and Illumina have some differences in the size of the mitochondrial genome, the A + T contents, the number of LSUs, and the start and stop codons (Tables 1, 2, Additional file 3: Table S2) [32]. Furthermore, there were base differences at several positions between the two sets of sequences (Table 3).
cytb gene sequence analysis
The results of amino acid sequence alignment indicated that all cytb sequences contain the highly conserved PEWY motif. Leu275 of S. cerevisiae is a key determinant of the efficacy of atovaquone and myxothiazol binding to the bc1 complex. This position is occupied by a Leu in the T. parva, B. microti, B. duncani, BspXJ/DH, BmLT/TZ and BmHB/NX sequences, whereas a Phe is present in the T. gondii, bovine and P. falciparum sequences (Fig. 2). The inhibitors of Qi and Qo sites include atovaquone, stigmatellin, myxothiazol, endochin-like quinolone (ELQ), antimycin A and NQNO. The amino acid changes conferring atovaquone resistance in the yeast numbering system included five mutations (I269M, F278I/A, Y279C/S, L275F and L282V). The drug (target protein is Qo site of bc1 complex) binding residues of cytb of ovine Babesia included Met128, Gly132, Glu259, Leu262, Phe265 and Tyr266. Drug (target protein is Qi site of bc1 complex) binding residues of cytb of ovine Babesia included His187, Ser191 and Asp214 (Table 4).

Amino acid sequence alignment of the conserved PEWY region of cytochrome b of six ovine Babesia isolates, Theileria parva, Toxoplasma gondii, B. microti, Plasmodium falciparum, Saccharomyces cerevisiae, Bos taurus and B. duncani. The alignment was constructed using MegAlign. The number 275 indicates to the residues at position Leu275 of S. cerevisiae. The residue at position 275 is a key determinant of the efficacy of ligand (atovaquone) binding to the bc1 complex. Arrows indicate amino acid positions altered in atovaquone-resistant parasites. Red horizontal lines indicate positions that are completely conserved in all species
Phylogenetic analysis
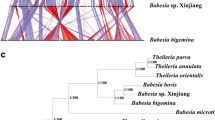
Phylogenetic trees were constructed with the concatenated amino acid sequences of cytb and cox1 using the ML and NJ methods. The two approaches showed no significant changes in the topology. The piroplasms were divided into seven groups: (i) classical Babesia species that could infect ruminants, canines and equines; (ii) classical Theileria species that could infect bovines; (iii) Theileria equi; (iv) Cytauxzoon felis; (v) Babesia duncani; (vi) Babesia conradae; and (vii) Babesia microti/Babesia rodhaini. Babesia infective to ruminants were separated into four clades: Babesia motasi, B. bigemina, B. orientalis/B. bovis and Babesia sp. XJ/DH. Furthermore, B. motasi were further divided into two subclades: B. motasi LT/TZ and B. motasi NX/HB (Fig. 3). The phylogenetic tree using the whole mitochondrial nucleotide sequence was constructed by the ML method based on the Kimura 2-parameter model. The result was similar to that using concatenated amino acid sequences of cytb and cox1, with the exception of T. orientalis, which was located in the B. conradae clade (Additional file 4: Figure S2).

Phylogenetic relationships of Babesia infective to small ruminants in China and other apicomplexan parasites. Phylogeny was inferred with a maximum likelihood analysis of the amino acid sequences of the cox1 and cytb genes based on distances calculated with the JTT with the Freqs model. Bootstrap values > 50% from 1000 replicates are shown on the nodes. Babesia isolates examined in this study are indicated with triangles
Discussion
In the present study, we assembled and annotated the mitochondrial genomes of six ovine Babesia isolates and performed a mitochondrial genomic analysis with published mitochondrial genomes of apicomplexan parasites. The mitochondrial genomes of the six Babesia isolates infective to small ruminants are highly similar to those of most Babesia spp. with respect to genome size, high A + T content, genome form, and gene content. However, they are smaller than those of B. microti, B. rodhaini and T. equi and larger than that of T. gondii [28, 29, 44]. Consistent with the results from other apicomplexan parasites, tRNA genes were not found in the mitochondrial genomes. We speculate that they may be directly encoded by the nuclear genome. The order and transcriptional direction of three protein-encoding genes are the same in B. bovis, B. bigemina and B. gibsoni [28] but different from those in B. microti, T. equi, T. orientalis and P. falciparum [28, 29].
In the reported mitochondrial genomes of piroplasms, B. microti and B. rodhaini have a dual flip-flop inversion system, ranging from 184 to 1082 bp in length [29]. However, one pair of TIRs was found in the mitochondrial genomes of other Babesia spp. and Theileria spp., ranging from 25 to 1563 bp in length [26,27,28, 30, 32]. These findings indicated that the number and size of TIRs are one of the main causes of different mitochondrial genome sizes. We also found that different numbers of LSUs are responsible for the size of the mitochondrial genomes. TIRs are considered to play a crucial role in the replication and stabilization of linear mitochondrial genomes [45]. In the published mitochondrial genomes of apicomplexan parasites, the sequences of the coding genes and LSU are basically the same. Therefore, we speculated that differences in the lengths and sequences of TIRs may be responsible for divergence in the host-specific and in vitro culture characteristics of protozoans.
The difference between the Illumina and Sanger sequencing data is mainly caused by nucleotide substitutions, deletions and insertions, which result from the use of different sequencing techniques assembly and annotation software. The settings of the parameters are different, which is one of the reasons for the difference between the two datasets. The results of the Illumina sequencing method are more prone to errors than those of the Sanger method. The Sanger approach is more accurate and thus more appropriate for further studies of small genome sequencing.
Mitochondria are essential organelles and play an important role in energy metabolism, growth and development of apicomplexan protozoa [46]. Cytochrome bc1 is an integral membrane protein complex that is vital to cellular respiration. The highly conserved binding site of inhibitors in cytochrome bc1 is the molecular basis of the drug effect on yeast, fungi and parasites [43]. The drug binding residues in the cytb sequences of six ovine Babesia isolates are completely consistent. In the conserved PEWY region, Phe278 (yeast number of cytb) is present in most organisms, whereas T. gondii and B. taurus have Tyr and Ala, respectively, at this position. Studies have reported that the L275F mutation in yeast has no effect on enzyme activity, but the IC50 increased ten-fold [22]. Compared with the wild-type yeast, the Y279S mutant had a 40-fold increased IC50 (B1.7 mM) for atovaquone [22], and Y268S in the P. falciparum numbering system resulted in a 3000-fold loss of atovaquone sensitivity. In addition, the mutations M133I, L271V and Y268N of P. berghei confer resistance to atovaquone [43]. Therefore, we conclude that mutations in cytb are largely responsible for the efficacy of drugs (the target protein is the bc1 complex) in apicomplexan parasites.
Currently, atovaquone and ELQs have been reported for the treatment of human babesiosis and malaria by modifying the drug target through disruption of the cytochrome bc1 complex [19, 21, 22, 41, 43]. In 2019, a B. motasi-like parasite was detected in human blood in Korea [47], which suggests that B. motasi may be potentially zoonotic. Therefore, we should investigate the infection of B. motasi in humans in China and evaluate the zoonotic potential of B. motasi and the effect of inhibitors binding to the cytochrome bc1 complex. Our data showed that atovaquone, stigmatellin, myxothiazol, endochin-like quinolone (ELQ), antimycin A and NQNO drugs can be used in the treatment of babesiosis in the future. The molecular mechanism of the resistance of these drugs is the mutation of cytb, which suggests that a combined drug strategy is possible to avoid drug resistance during treatment of babesiosis.
In this study, the taxonomical relationships of Babesia isolates infective to sheep and goats are consistent with the reported phylogenetic analyses based on cytb, cox1, cox3, nuclear small subunit (SSU) and internal transcribed spacer (ITS) [5, 6, 8, 32, 48]. The ovine Babesia isolates are divided into two species: Babesia sp. and B. motasi. Babesia motasi further fell into two small clades, named BmLT/TZ and BmNX/HB. With the exception of B. conradae, piroplasm infective to the same host fell into one clade. These findings are consistent with the phylogenetic position of B. gibsoni, B. duncani and B. orientalis based on the amino acid sequences of cox1 and cytb [26, 27, 30].
Conclusions
In conclusion, we reported the mitochondrial genome of six ovine Babesia isolates that infect small ruminants in China. The phylogeny based on the concatenated amino acid sequences indicated that there are two Babesia species (Babesia sp. and B. motasi) infective to sheep and goats in China and that the B. motasi isolates possibly belong to two subspecies (BmHB/NX and BmTZ/LT). The possible efficacy of the inhibitor (target protein is bc1 complex) should be evaluated on these six ovine Babesia isolates in future work. Further studies are needed to analyze the TIR and protein functions, which could provide new insights into the phylogenetic relationships, biology and therapy of Babesia.
Availability of data and materials
The datasets are included in the article and its additional files. The sequences were submitted to the GenBank database under the accession numbers MK962313, MK962314 and MN605889-MN605892.
Abbreviations
- DNA:
-
Deoxyribonucleic acid
- PCR:
-
Polymerase chain reaction
- cytb:
-
Cytochrome b
- cox1:
-
Cytochrome c oxidase subunit 1
- cox3:
-
Cytochrome c oxidase subunit 3
- LSU :
-
Large subunit ribosomal RNA
- TIR:
-
Terminal inverted repeats
- Qi:
-
Quinone reduction
- Qo:
-
Quinol oxidation
- ELQ:
-
Endochin-like quinolone
- SSU:
-
Small subunit ribosomal RNA
- ITS:
-
Internal transcribed spacer
References
Schnittger L, Rodriguez AE, Florin-Christensen M, Morrison DA. Babesia: a world emerging. Infect Genet Evol. 2012;12:1788–809.
Uilenberg G. Babesia–a historical overview. Vet Parasitol. 2006;138:3–10.
Ozubek S, Aktas M. Molecular evidence of a new Babesia sp. in goats. Vet Parasitol. 2017;233:1–8.
Guan G, Yin H, Luo J, Lu W, Zhang Q, Gao Y, et al. Transmission of Babesia sp. to sheep with field-collected Haemaphysalis qinghaiensis. Parasitol Res. 2002;88(Suppl. 1):22–4.
Tian Z, Liu G, Hong Y, Luo J, Guan G, Xie J, et al. Cytochrome c oxidase subunit III (COX3) gene, an informative marker for phylogenetic analysis and differentiation of Babesia species in China. Infect Genet Evol. 2013;18:13–7.
Tian Z, Luo J, Zheng J, Xie J, Shen H, Yin H, et al. Phylogenetic analysis of Babesia species in China based on cytochrome b (COB) gene. Infect Genet Evol. 2013;13:36–40.
Liu AH, Yin H, Guan GQ, Schnittger L, Liu ZJ, Ma ML, et al. At least two genetically distinct large Babesia species infective to sheep and goats in China. Vet Parasitol. 2007;147:246–51.
Niu Q, Luo J, Guan G, Liu Z, Ma M, Liu A, et al. Differentiation of two ovine Babesia based on the ribosomal DNA internal transcribed spacer (ITS) sequences. Exp Parasitol. 2009;121:64–8.
Guan G, Moreau E, Liu J, Hao X, Ma M, Luo J, et al. Babesia sp. BQ1 (Lintan): molecular evidence of experimental transmission to sheep by Haemaphysalis qinghaiensis and Haemaphysalis longicornis. Parasitol Int. 2010;59:265–7.
Guan G, Ma M, Moreau E, Liu J, Lu B, Bai Q, et al. A new ovine Babesia species transmitted by Hyalomma anatolicum anatolicum. Exp Parasitol. 2009;122:261–7.
Guan G, Ma M, Liu A, Ren Q, Wang J, Yang J, et al. A recently identified ovine Babesia in China: serology and sero-epidemiology. Parasitol Int. 2012;61:532–7.
Bai Q, Liu G, Liu D, Ren J, Li X. Isolation and preliminary characterization of a large Babesia sp. from sheep and goats in the eastern part of Gansu Province. China. Parasitol Res. 2002;88(Suppl. 1):16–21.
Niu Q, Liu Z, Yang J, Yu P, Pan Y, Zhai B, et al. Expression of sheep pathogen Babesia sp. Xinjiang rhoptry-associated protein 1 and evaluation of its diagnostic potential by enzyme-linked immunosorbent assay. Parasitology. 2016;143:1990–9.
Niu Q, Liu Z, Yang J, Yu P, Pan Y, Zhai B, et al. Expression analysis and biological characterization of Babesia sp. BQ1 (Lintan) (Babesia motasi-like) rhoptry-associated protein 1 and its potential use in serodiagnosis via ELISA. Parasit Vectors. 2016;9:313.
Wang JM, Ma ML, Liu AH, Ren QY, Li AY, Liu ZJ, et al. A sero-epidemiological survey of Chinese Babesia motasi for small ruminants in China. Parasitol Res. 2013;112:2387–91.
Frederick RL, Shaw JM. Moving mitochondria: establishing distribution of an essential organelle. Traffic. 2007;8:1668–75.
Hunte C, Solmaz S, Palsdóttir H, Wenz T. A Structural perspective on mechanism and function of the cytochrome bc (1) complex. Results Probl Cell Differ. 2008;45:253–78.
Carrasco MP, Gut J, Rodrigues T, Ribeiro MH, Lopes F, Rosenthal PJ, et al. Exploring the molecular basis of Qo bc1 complex inhibitors activity to find novel antimalarials hits. Mol Inform. 2013;32:659–70.
Birth D, Kao WC, Hunte C. Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat Commun. 2014;5:4029.
Siregar JE, Kurisu G, Kobayashi T, Matsuzaki M, Sakamoto K, Mi-ichi F, et al. Direct evidence for the atovaquone action on the Plasmodium cytochrome bc1 complex. Parasitol Int. 2015;64:295–300.
Gao X, Wen X, Esser L, Quinn B, Yu L, Yu CA, et al. Structural basis for the quinone reduction in the bc1 complex: a comparative analysis of crystal structures of mitochondrial cytochrome bc1 with bound substrate and inhibitors at the Qi site. Biochemistry. 2003;42:9067–80.
Kessl JJ, Lange BB, Merbitz-Zahradnik T, Zwicker K, Hill P, Meunier B, et al. Molecular basis for atovaquone binding to the cytochrome bc1 complex. J Biol Chem. 2003;278:31312–8.
Rożej-Bielicka W, Stypułkowska-Misiurewicz H, Gołąb E. Human babesiosis. Przegl Epidemiol. 2015;69:489–94.
Vial HJ, Gorenflot A. Chemotherapy against babesiosis. Vet Parasitol. 2006;138:147–60.
Baneth G. Antiprotozoal treatment of canine babesiosis. Vet Parasitol. 2018;254:58–63.
Guo J, Miao X, He P, Li M, Wang S, Cui J, et al. Babesia gibsoni endemic to Wuhan, China: mitochondrial genome sequencing, annotation, and comparison with apicomplexan parasites. Parasitol Res. 2019;118:235–43.
He L, Zhang Y, Zhang QL, Zhang WJ, Feng HH, Khan MK, et al. Mitochondrial genome of Babesia orientalis, apicomplexan parasite of water buffalo (Bubalus babalis, Linnaeus, 1758) endemic in China. Parasit Vectors. 2014;7:82.
Hikosaka K, Watanabe Y, Tsuji N, Kita K, Kishine H, Arisue N, et al. Divergence of the mitochondrial genome structure in the apicomplexan parasites, Babesia and Theileria. Mol Biol Evol. 2010;27:1107–16.
Hikosaka K, Tsuji N, Watanabe Y, Kishine H, Horii T, Igarashi I, et al. Novel type of linear mitochondrial genomes with dual flip-flop inversion system in apicomplexan parasites, Babesia microti and Babesia rodhaini. BMC Genomics. 2012;13:622.
Virji AZ, Thekkiniath J, Ma W, Lawres L, Knight J, Swei A, et al. Insights into the evolution and drug susceptibility of Babesia duncani from the sequence of its mitochondrial and apicoplast genomes. Int J Parasitol. 2019;49:105–13.
Schreeg ME, Marr HS, Tarigo JL, Cohn LA, Bird DM, Scholl EH, et al. Mitochondrial genome sequences and structures aid in the resolution of Piroplasmida phylogeny. PLoS ONE. 2016;11:e0165702.
Wang T, Guan G, Korhonen PK, Koehler AV, Young ND, Hall RS, et al. Mitochondrial genomes of two Babesia taxa from sheep in China as a foundation for population genetic and epidemiological investigations. Infect Genet Evol. 2017;47:51–5.
He X, Liu J, Liu A, Wang J, Niu Q, Li Y, et al. The structural and phylogenetic analysis of trap gene in ovine Babesia species in China. Acta Vet et Zoo Sinica. 2017;48:1332–41.
Wang X, Wang J, Liu J, Liu A, He X, Xu J, et al. Comparative analysis of apicoplast genomes of Babesia infective to small ruminants in China. Parasit Vectors. 2019;12:312.
Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20:3252–5.
Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2012;28:464–9.
Lowe TM, Chan PP. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44:W54–7.
Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010;5:e11147.
Srivastava IK, Morrisey JM, Darrouzet E, Daldal F, Vaidya AB. Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol Microbiol. 1999;33:704–11.
Kessl JJ, Ha KH, Merritt AK, Lange BB, Hill P, Meunier B, et al. Cytochrome b mutations that modify the ubiquinol-binding pocket of the cytochrome bc1 complex and confer anti-malarial drug resistance in Saccharomyces cerevisiae. J Biol Chem. 2005;280:17142–8.
Kessl JJ, Meshnick SR, Trumpower BL. Modeling the molecular basis of atovaquone resistance in parasites and pathogenic fungi. Trends Parasitol. 2007;23:494–501.
Barton V, Fisher N, Biagini GA, Ward SA, O’Neill PM. Inhibiting Plasmodium cytochrome bc1: a complex issue. Curr Opin Chem Biol. 2010;14:440–6.
Stickles AM, Almeida MJD, Morrisey JM, Sheridan KA, Forquer IP, Nilsen A, et al. Subtle changes in endochin-like quinolone structure alter the site of inhibition within the cytochrome bc1 complex of Plasmodium falciparum. Antimicrobial Agents Chemother. 2015;59:1977–82.
Gjerde B. Characterisation of full-length mitochondrial copies and partial nuclear copies (numts) of the cytochrome b and cytochrome c oxidase subunit I genes of Toxoplasma gondii, Neospora caninum, Hammondia heydorni and Hammondia triffittae (Apicomplexa: Sarcocystidae). Parasitol Res. 2013;112:1493–511.
Nosek J, Tomaska L. Mitochondrial genome diversity: evolution of the molecular architecture and replication strategy. Curr Genet. 2003;44:73–84.
Kehr S, Sturm N, Rahlfs S, Przyborski JM, Becker K. Compartmentation of redox metabolism in malaria parasites. PLoS Pathog. 2010;6:e1001242.
Hong SH, Kim SY, Song BG, Rho JR, Cho CR, Kim CN, et al. Detection and characterization of an emerging type of Babesia sp. similar to Babesia motasi for the first case of human babesiosis and ticks in Korea. Emerg Microbes Infect. 2019;8:869–78.
Gou H, Guan G, Ma M, Liu A, Liu Z, Ren Q, et al. Phylogenetic analysis based on 28S rRNA of Babesia spp. in ruminants in China. Exp Appl Acarol. 2013;59:463–72.
Acknowledgments
Not applicable.
Funding
This study was financially supported by the National Key Research and Development Programme of China (2017YFD0501200), the 973 Programme (2015CB150300), ASTIP (CAAS-ASTIP-2016-LVRI), NBCIS (CARS-37) and the Jiangsu Co-innovation Center Programme for Prevention and Control of Important Animal Infectious Diseases and Zoonoses.
Author information
Authors and Affiliations
Contributions
GQG and XXW designed the study, wrote the manuscript and performed the experiments. JMW, JLL, QJX, AHL and XH extracted the merozoite genomic DNA of ovine Babesia. HY, JXL and YQL reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Primers used for amplifying the mitochondrial genome of the six ovine Babesia isolates.
Additional file 2: Figure S1.
Mitochondrial genome alignment of six ovine Babesia isolates with B. bovis, B. bigemina, B. orientalis, Babesia sp. Coco, Cytauxzoon felis, T. parva, B. gibsoni, B. microti, B. rodhaini and P. falciparum. The color blocks are linked by lines to similar blocks in genomes. The region of genomes covered by a color block is entirely collinear and homologous in the mitochondrial genomes.
Additional file 3: Table S2.
Comparison of six ovine Babesia mitochondrial genomes.
Additional file 4: Figure S2.
Phylogenetic tree of ovine Babesia isolates and other apicomplexan parasites. Phylogeny was created with a Maximum Likelihood method of mitochondrial nucleotide sequences using the Kimura 2-parameter nucleotide substitution model. The triangles represent the ovine Babesia obtained in our study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Wang, X., Wang, J., Liu, J. et al. Insights into the phylogenetic relationships and drug targets of Babesia isolates infective to small ruminants from the mitochondrial genomes. Parasites Vectors 13, 378 (2020). https://doi.org/10.1186/s13071-020-04250-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-04250-8