
Abstract
Background
Bovine babesiosis is a tick-borne disease caused by several species of Babesia which produce acute and fatal disease in cattle and affect livestock industry worldwide. Babesia ovata is a benign species widespread in east Asian countries and causes anemia, particularly in cattle which are co-infected with Theileria orientalis. The development of genetic manipulation methods is necessary to improve our understanding of the basic biology of protozoan pathogens toward a better control of disease. Such tools have not been developed for B. ovata, and are the aim of this study.
Methods
In this study we transfected constructs that were designed to evaluate the ability of several B. ovata promoter candidates to drive expression of a reporter luciferase. We found that the elongation factor-1 alpha intergenic region (ef-1α IG) and the actin 5’ non-coding region (NR) had highest promoter activities. To establish a stable transfection system, we generated a plasmid construct in which the ef-1α IG promoter drives gfp expression, and the actin 5’ NR mediates expression of the selectable marker hdhfr. The plasmid was designed for episomal transfection, as well as to integrate by double cross-over homologous recombination into the ef-1α locus. Circular or linearized plasmid was transfected by electroporation into in vitro cultured B. ovata and retention of the plasmid was facilitated by drug selection with 5 nM WR99210 initiated 48 h after transfection.
Results
After one-week cultivation with WR99210, GFP-expressing parasites were observed by fluorescence microscopy. Integration of the plasmid construct into the ef-1α locus was confirmed by PCR, Southern blot analysis, and sequencing of recombination sites. These results confirm successful development of a stable transfection system for B. ovata.
Conclusion
The current study provides a fundamental molecular tool to aid in molecular and cellular studies of B. ovata.
Similar content being viewed by others


Background
Bovine babesiosis remains an economic burden in livestock industry worldwide and is caused by intraerythrocytic apicomplexan parasites of the genus Babesia. The most common and pathogenic species of Babesia infecting cattle are Babesia bovis, B. bigemina and B. divergens, whose geographical distributions are defined by the prevalence of their respective tick vectors. B. ovata is a benign bovine Babesia sp. widespread in several east Asian countries [1] and causes anemia particularly in animals co-infected with Theileria orientalis, since both parasites are transmitted by the ixodid tick, Haemaphysalis longicornis [2].
Obstacles to control bovine babesiosis include the lack of effective and safe vaccines, the emergence of acaricide-resistant ticks [3], limited choices for anti-Babesia drugs in the field, and the emergence of drug-resistant Babesia strains [4]. Better understanding of the basic biology of Babesia spp. is crucial to design and develop new strategies for controlling the disease. In the post-genomic era in which the genome sequence information for several Babesia spp. is available, genetic manipulation using reverse genetic techniques can improve our understanding of the basic biology of these parasites. Transfection systems have been established for several apicomplexan parasites such as Plasmodium falciparum [5], Toxoplasma gondii [6], Babesia bovis [7, 8] and recently Theileria parva [9]. Regarding Babesia spp., only two reports describe stable transfection systems, for B. bovis [7, 8]; one used blasticidin-S/blasticidin deaminase and the other employed WR99210/human dihydrofolate reductase (hdhfr) as selective markers.
Several Babesia spp. are responsible for babesiosis in cattle causing a range of clinical symptoms from acute severe anemia and occasionally death by B. bigemina and B. bovis to a mild anemia or subclinical signs by B. ovata. Our group has recently derived whole genome nucleotide sequence for B. ovata (unpublished data). This advancement, together with the availability of genome sequences from other pathogenic bovine Babesia spp. [10–12], paves the way for comparative functional genomics studies and to find genes responsible for the virulence and pathogenesis of these parasites. To aid in these molecular and cellular studies, we describe herein the development of a transfection system for B. ovata. Since B. ovata infection is benign and does not cause severe clinical symptoms in cattle, stable integration of expression vectors could be used to transfer an immunogenic antigen of pathogenic Babesia spp. and induce protective antibody against virulent bovine Babesia spp. In addition, H. longicornis, the tick vector for B. ovata, is easily maintained in vitro and has been used to study the biology of ixodid ticks [13]. Therefore, B. ovata could be employed as a model organism to study the tick stage of Babesia, the genes responsible for tick transmission as well as parasite and tick interactions.
Since the elongation factor-1 alpha intergenic region (ef-1α IG) and the actin 5’ non-coding region (NR) contain promoter activities which conferred expression of a reporter protein in B. bovis [8, 14], we initially attempted to use these B. bovis promoters for transfection of B. ovata. However, these trials were not successful (unpublished data), suggesting that B. bovis promoters might not work in B. ovata. Therefore, in this study, we initially evaluated several B. ovata promoter candidates and then established a stable transfection system for this parasite using selected strongest promoters.
Methods
Parasite culture
The B. ovata Miyake strain was cultured in vitro using purified bovine red blood cells (RBCs) purchased from Nippon Bio-Test Laboratories (Tokyo, Japan) and GIT medium (Wako Pure Chemical Industries, Osaka, Japan) in a microaerophilous stationary phase system as described [15].
Evaluation of sensitivity of B. ovata to WR99210
B. ovata was cultured in 1 ml culture medium containing 10 % bovine RBC in a 24-well plate with or without WR99210 (0, 0.5, 1, 5, 10 and 50 nM). The initial parasitemia was 0.5 % and the final parasitemia was measured on day 3. For each drug concentration, parasites were cultured in triplicate and culture medium was replaced daily. Parasitemia was calculated by examining 5000 RBCs of a prepared thin blood smear stained with Giemsa's solution.
Plasmid constructs
For evaluation of promoter activity, the firefly luciferase gene was amplified by PCR from the plasmid pENT12luc using specific primers (Additional file 1: Table S1) and inserted into the EcoRV site of pBluescript plasmid vector using an In-Fusion HD Cloning Kit (Takara Bio Inc., Otsu, Japan) (Fig. 1a). The B. ovata rhoptry associated protein-1 intergenic region (rap-1 IG) was amplified by PCR from B. ovata genomic DNA using specific primers and inserted into the BamHI site of pBluescript. The B. ovata ef-1α IG, 5’ NR of actin, heat shock protein 70 (hsp70), calmodulin (cam), thioredoxin peroxidase 1(tpx-1) and B. bovis ef-1α IG were amplified by PCR and cloned into the HindIII site of pBluescript to be evaluated for their promoter activities. To construct an internal control plasmid to normalize the promoter activity, a DNA fragment encoding Renilla reniformis luciferase was amplified from pHRH (a gift from Dr. Kirk Deitsch, Weill Cornell Medical College, USA) using specific primers (Additional file 1: Table S1) and inserted into the EcoRV site of pBluescript plasmid vector (Fig. 1a). Renilla luciferase was driven by ef-1α IG2 and rap-1 IG used as terminator. The sequences from all novel candidate promoters were deposited in DDBJ/EMBL/GenBank nucleotide database with the accession numbers LC101765-69.

Schematic diagram of the plasmids used for transient transfection and evaluation of promoter activity. a Plasmid construct to evaluate the promoter activity and a Renilla luciferase-expressing plasmid for normalization. b Evaluation of the promoter activity of actin 5’NR driving luciferase expression, over a time course of 24–96 h post transfection. c Comparison of luciferase activity in lysates of B. ovata transfected with different constructs at 48 h post transfection. A promoter-less plasmid was used as a negative control. Values from 3 independent transfections are shown. Asterisks indicate statistical significance between promoter-less plasmid (No Promoter) and other promoter candidates by Dunnett’s multiple comparison test (p < 0.001). RLU: Relative luciferase units
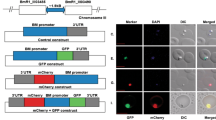
A schematic diagram of GFP-expressing plasmid (pBS-EGRADE) is shown in Fig. 2a. The plasmid was designed for episomal transfection as well as integration into the ef-1α locus of B. ovata. The drug selection and reporter gene cassettes were separated in order to drive hdhfr and gfp with different promoters. All the PCR primers used for plasmid construction are listed in Table 1 and the restriction sites are underlined. To construct the reporter cassette, gfp was amplified from a B. bovis GFP expressing plasmid [8] and cloned into the HindIII site of pBluescript using an In-Fusion HD Cloning Kit. To produce a strongly expressing GFP parasite, the ef-1α IG2 which showed strong promoter activity by luciferase assay was cloned into a ClaI site at the 5’ flanking region of gfp. B. ovata rap-1 IG was cloned into an EcoRV site at the 3’ flanking region of gfp. For construction of the drug selection cassette, hdhfr was amplified from a B. bovis GFP-expressing plasmid [8] and cloned into the BamHI site of pBluescript. The hdhfr gene was flanked by actin 5’ NR and ef-1α 3’ NR at the 5’ and 3’ flanking regions, respectively. The resulting hdhfr expression cassette was amplified with a primer pair, BoADE-F-EcoRI-IF and BoADE-R-EcoRI-IF (Table 1), and cloned into the EcoRI site of the reporter cassette to produce the pBS-EGRADE plasmid. All plasmids were purified using a Qiagen Plasmid Maxi kit (Qiagen, MD, USA) following the manufacturer’s instructions and the inserted DNA sequences were confirmed by sequencing. The sequence of pBS-EGRADE was deposited into DDBJ/EMBL/GenBank nucleotide database with the accession number LC101772.

Schematic diagram of GFP-expressing plasmid construct, growth inhibition of B. ovata by WR99210 and fluorescence microscopy images of stably expressing GFP B. ovata. a Plasmid construct for stable GFP expression (pBS-EGRADE) showing the recombination sites for integration into the ef-1α locus by homologous double cross-over recombination. The restriction sites for linearization (SalI and SacII) are shown. b Growth inhibition rate of B. ovata in the absence or presence of different concentrations of WR99210. All data are expressed as mean + SEM of triplicate culture. c Live fluorescence microscopy images of GFP-expressing parasites. The pictures are taken from the pBS-EGRADE genome integrated isolate 1. The parasite nucleus was stained with Hoechst33342. Scale-bar: 10 μm
Transfection of parasites
To prepare B. ovata-infected RBCs (iRBCs) for transfection, cell pellets were washed twice with PBS and one time with cytomix buffer (120 mM KCl, 0.15 mM CaCl2, 10 mM K2HPO4, 10 mM KH2PO4, 25 mM HEPES, pH 7.6, 2 mM EGTA, 5 mM MgCl2, 100 μg/ml bovine serum albumin, and 1 mM hypoxanthine). One hundred microgram of the circular plasmid in 50 μl of cytomix was mixed with 50 μl of Amaxa nucleofector human T-cell solution, and then combined with 100 μl of washed iRBCs. To promote genome integration of the plasmid in order to produce B. ovata stably expressing GFP, the pBS-EGRADE plasmid was linearized by incubating overnight with SalI and SacII. The resulting linear plasmid would favor integration into the ef-1α locus of B. ovata. Parasite-iRBCs were transfected with the circular or linearized plasmids by electroporation using a nucleofector device with a program v-024 (Amaxa Biosystems, Cologne, Germany) [8] and were immediately transferred to 1 ml culture containing 10 % of bovine RBCs. To select GFP-expressing transgenic parasites, WR99210 was added two days after the transfection. The parasite population transfected with the linearized plasmid was cloned by limiting dilution.
Luciferase assay
A luciferase assay was performed to evaluate activities of candidate promoters. Transient transfections were conducted by introducing 50 μg of each promoter plasmid expressing firefly luciferase together with 50 μg of renilla luciferase-expressing plasmid into B. ovata-iRBCs. The luciferase assay was performed 48 h after transfection. A parasite pellet was prepared for the luciferase assay as described [16] and resuspended in 230 μl of freshly-prepared Promega’s 1X cell culture lysis reagent (Promega, Madison, WI, USA). The pellet was incubated for 15 min at room temperature (RT) for complete lysis and briefly centrifuged to remove the cell debris. Seventy-five microliters of cell lysate were mixed with 75 μl of the Dual-Glo luciferase substrate. The mixture was kept for 10 min at RT and the luminescence was measured for 10 s using a microplate reader (Wallac 1420; PerkinElmer, Turku, Finland). Immediately after measurement of firefly luciferase activity, 75 μl of Dual-Glo Stop & Glo reagent was added to each well of the plate. Renilla luciferase activity was measured after 10 min incubation at RT. Readings from mock transfected parasites were subtracted from the firefly and renilla luciferase readings and the resulting values of firefly luciferase activity were normalized using the renilla luciferase activity for each sample. To evaluate promoter activities, three independent transfections were done for each promoter and each luciferase assay was performed in triplicate.
Biostatistical analysis of promoter activities
The normalized luciferase activities were plotted using GraphPad Prism6. Activity of each promoter candidates was examined by Dunnett’s multiple comparison test with a promoter-less (control) plasmid. The promoter activity was considered to be significantly different from the control if P-value was below 0.05.
PCR to confirm the integration of pBS-EGRADE into ef-1α locus
Three sets of primers were designed to confirm the integration of pBS-EGRADE into the ef-1α locus by specific amplification of DNA fragments surrounding the 5’ recombination site, the 3’ recombination site and the ef-1α locus (Additional file 1: Table S1). To examine the 5’ recombination event of the plasmid construct, a primer pair Boef1α-integ-F and eGFP-R-HindIII-IF was used to amplify a 2.2 kb DNA fragment. To confirm the 3’ recombination event of the plasmid construct, a primer pair hDHFR-F-BamHI-IF and Boef1α-integ-R was used to amplify a 1.8 kb DNA fragment. To examine if the ef-1α gene was replaced with the plasmid construct, a primer pair Boef1α-F2 and Boef1α-integ-R was used, by which a 7.7 kb DNA fragment would be amplified for the pBS-EGRADE-integrated parasites or a 3.6 kb DNA fragment for the wild type and episomally transfected parasites. The nucleotide sequences of the amplified DNA fragments containing the 5’ or 3’ recombination sites were confirmed by sequencing.
Southern blot analysis
Genomic DNA was extracted from wild type and 2 parasite isolates transfected with linearized pBS-EGRADE. Five micrograms of DNA were digested overnight with 100 units of NotI and PacI, separated by agarose gel electrophoresis and transferred onto HyBond N+ (GE Healthcare, Buckinghamshire, UK). Two probes were used, one corresponding to the complete open reading frame (ORF) of gfp and the other 1 kb length of ef-1α 3’NR, and were labeled and hybridized with the AlkPhos Direct kit (GE Healthcare) according to the manufacturer’s instructions. The signal was developed with CDP-star detection reagent (GE Healthcare) and detected with a multipurpose CCD camera system (LAS-4000 mini EPUV; Fujifilm, Japan).
Results
Evaluation of the B. ovata promoter activities
In order to evaluate promoter activity and to select promoters to be used for stable transfections, candidate promoters were chosen which were previously used for B. bovis or Plasmodium. The evaluated candidate promoters were (i) ef-1α IG, which has a bidirectional promoter activity in B. bovis [17]; (ii) actin 5’NR, which showed strong promoter activity in B. bovis [8]; (iii) tpx-1 5’NR, which was shown to constitutively express in Plasmodium spp. [18, 19]; and (iv) cam 5’NR and hsp70 5’NR, which were used for P. falciparum [5, 20]. Similar to the B. bovis ef-1α gene locus [17], two ef-1α gene copies are oriented head to head in the B. ovata genome, and thus the IG region was expected to have a bidirectional promoter activity. To independently evaluate the two promoter activities in the ef-1α IG region, two plasmids termed IG1 and IG2 were constructed (Fig. 1c). Our initial analysis of rap-1 region in B. ovata revealed at least three copies of rap-1a and rap-1b in the genome. This architecture is similar to what was observed for the B. bigemina genome in which five copies of rap-1a and rap-1b are tandemly arranged and an additional copy of rap-1b and a copy of rap-1c are located at 3’ of these gene loci [21]. The intergenic region between rap-1a and rap-1b which contains the 3’ NR of rap-1a was selected as a terminator.
In the initial trials for transient transfection, we introduced 50 μg of plasmid constructs with transfection conditions of low voltage/high capacitance (0.31 kV, 1 mF) or high voltage/low capacitance (1.25 kV, 25 μF) using a Gene Pulser Xcell apparatus (Bio-Rad, Richmond, CA) [14]. Parasite lysates did not show any luciferase activity indicating the failure in introducing plasmid into the parasite using these conditions, possibly due to harmful effects of electroporation to the parasites. Because P. falciparum is able to uptake plasmid DNA and efficiently express exogenous proteins following infection of erythrocytes which are preloaded by electroporation into the RBC cytosol [22], we preloaded 50 μg of plasmid DNA to the bovine RBCs for the transfection experiment. Again, however, parasite lysates did not show any luciferase activity after this procedure. The erythrocytic cycle of B. ovata (7–10 h) [23] is shorter than that of P. falciparum (48 h), which might be the reason for the failure of the transfection with plasmid-preloaded RBC. In a third attempt nucleofection using an Amaxa nucleofector device with 50 μg of plasmid DNA was successful for transient expression experiments (data not shown). Increasing plasmid DNA to 100 μg showed more efficient transformation and was used for the evaluation of promoter activity and stable transfection.
We first determined the best time for evaluation of the promoter activity for B. ovata using a plasmid expressing luciferase driven by the B. ovata actin 5’NR. Parasite lysates were prepared at 24, 48, 72 and 96 h post transfection for the luciferase assay. We found that while parasites transfected with a promoter-less construct (negative control) did not show luciferase activity, the peak of luciferase activity for actin 5’NR was at 48 h post transfection; thus we selected this time point to evaluate luciferase activities (Fig. 1b). Simultaneous expression of firefly luciferase under different promoters and renilla luciferase under the identical promoter in Plasmodium spp. enables to normalize the firefly luciferase value using renilla luciferase value as a transfection efficiency control [20, 24]. Therefore, firefly luciferase luminescence values were normalized with the renilla luciferase values from the same samples. By three independent transfection experiments, ef-1α IG2 and actin 5’NR showed significantly higher values than promoter-less (control) plasmid (P < 0.001), whereas the luciferase values from hsp70 5’NR, tpx-1 5’NR, cam 5’NR and ef-1α IG1 were not significantly different when compared to the control (P > 0.05) (Fig. 1c). The B. bovis ef-1α IG construct, for which a promoter activity was validated in B. bovis [8], was not able to express firefly luciferase in B. ovata, indicating that B. bovis ef-1α IG was not recognized by the B. ovata transcriptional or translational machinery.
Growth of B. ovata is inhibited by WR99210 at nanomolar concentration
WR99210 has been successfully used to select for stable transfection of B. bovis [8]. To examine whether WR99210 could inhibit the growth of B. ovata at low concentration, and to determine a suitable concentration to select the transgenic parasites, B. ovata was cultured in the presence of different concentrations of WR99210 from 0 to 50 nM. The experiment was done in triplicate wells and parasitemia was calculated on day 3 after adding the drug into the culture (Fig. 2b). The calculated IC50 was 0.56 ± 0.01 nM and 5 nM WR99210 completely inhibited the growth of B. ovata; thus we employed 5 nM thereafter for the selection of stable GFP-expressing parasites.
Establishment of the B. ovata line stably expressing GFP
Approximately one week after addition of WR99210, GFP-expressing parasites appeared in cultures which were transfected with either circular or linearized plasmids (Fig. 2c). The integration of pBS-EGRADE into the ef-1α locus was initially evaluated by PCR. The diagnostic PCR-1 and −2 primer pairs successfully amplified 2.2 and 1.8 kb DNA fragments, respectively, using DNA template from genome integrated (GI) isolates; and the amplified DNA fragments were validated by sequencing (Fig. 3a). These DNA fragments sequences were deposited into DDBJ/EMBL/GenBank nucleotide database with the accession numbers LC101770 and LC101771. No amplified products were detected using DNA template from episomal transfectants or wild type parasites. In addition, PCR-3 primer pair amplified a 7.7 kb DNA fragment, a size expected after the insertion of the pBS-EGRADE construct into the ef-1α locus. In contrast, a 3.6 kb DNA fragment was amplified for wild type and episomal transfectants, indicating that their ef-1α locus was intact. Overall, the PCR results confirmed the integration of the plasmid construct into the ef-1α locus in the GI1 and GI2 parasite lines. We further confirmed the structure of the integration site by Southern blot analysis (Fig. 3b). The gfp and ef-1α 3’NR probes both detected a single 8.9 kb band for GI isolates 1 and 2, while only the ef-1α 3’NR probe detected a single 4.8 kb band and the gfp probe did not detect any band for wild type parasites. These results confirmed the correct integration of pBS-EGRADE into the ef-1α locus. In addition, the growth of GI isolates was comparable with wild type parasites, indicating that disruption of one copy of the ef-1α gene did not affect the growth of parasites in the in vitro culture condition (Fig. 4).

Confirmation of the integration of pBS-EGRADE into the ef-1α locus. a Schematic diagram and PCRs to confirm the integration of pBS-EGRADE into ef-1α locus. PCR-1, −2 and −3 are done with primer sets Boef1α-integ-F/eGFP-R-HindIII-IF, hDHFR-F-BamHI-IF/Boef1α-integ-R and Boef1α-F2/Boef1α-integ-R, respectively. b Schematic diagram and Southern blot analysis to confirm the integration of pBS-EGRADE into ef-1α locus. Five microgram of samples genomic DNA were digested with PacI and NotI, and hybridized with gfp and ef-1α 3’NR probes. GI: genome integrated; Episomal: two independent transfectants with circular plasmids; WT: wild type

Growth curve of different lines of B. ovata. Wild type (WT) and genome integrants (GI) were cultured continuously by sub-culturing every 3 days and parasitemia monitored daily
Discussion
In this study we transiently introduced plasmid DNA into B. ovata in order to evaluate promoter activity. With this transient expression we evaluated the strength of promoter activity for a panel of B. ovata promoters, as well as for the B. bovis ef-1α IG promoter region. Evaluation of a range of promoter types and activities increases the options of promoters to be chosen to express transgenes in accordance the purposes of a given study. Herein, we aimed to establish a transfection system using GFP as a reporter protein, and therefore the ef-1α IG2 was chosen to drive gfp and the actin 5’NR to drive hdhfr based upon their relatively strong promoter activities. Unlike Plasmodium spp., for which ef-1α promoter region from one species works in other species, the B. bovis ef-1α IG promoter region was not able to express luciferase in B. ovata, and this indicates that B. bovis ef-1α IG was not recognized by B. ovata’s transcription or translation system.
The first report of stable transfection of B. bovis targeted the ef-1α locus for genome integration of a plasmid construct [7]. Preliminary investigation of the ef-1α locus sequence in B. ovata showed that two identical genes are located head to head and are separated by a 1.4 kb IG region (unpublished data), similar to the pattern reported for B. bovis and Plasmodium [17, 25]. Replacing one of two ef-1α in Plasmodium or B. bovis by homologous recombination did not significantly affect the growth of the parasites [7, 26, 27] and thus we considered that one of the ef-1α gene loci could be targeted as an integration site of exogenous genes into the genome. As expected, we were able to use this gene locus for the genome integration in B. ovata, and destruction of one of the ef-1α gene loci did not affect the growth of GI isolates, suggesting the feasibility of this locus as a site for the introduction of foreign genes.
For the selection of GFP-expressing parasites we employed hdhfr as a selectable marker which confers resistance to WR99210 and pyrimethamine [28]. It was reported that in most cases the selection of B. bovis using hdhfr/pyrimethamine resulted in the selection of naturally resistant parasites, which challenged the use of this selection system for B. bovis [14]. However, the hdhfr/WR99210 system was successfully employed for B. bovis in our former study, in which natural resistant parasites were not selected [8]. Thus we used WR99210 instead of pyrimethamine. The IC50 of WR99210 against B. ovata was 0.56 ± 0.01 nM, which is comparable to that of B. bovis (1 nM) [8]. The transfectants selected with hdhfr/WR99210 in this study emerged as early as 7 days after adding the drug and continued expressing GFP for more than 7 months after transfection (data not shown), indicating the feasibility of using this selection system for B. ovata.
In a recent report two selection systems, blasticidin-S/blasticidin deaminase and WR99210/hdhfr, were successfully used sequentially in a double transfection system for complementation of tpx-1 gene knockout and subsequent recovery of the phenotype for B. bovis [29]. This method could be used not only for gene knockout complementation studies but also for the development of double gene knockouts to investigate biological aspects of this parasite, such as invasion and egress, where multiple genes may be involved. In addition, the available gene knockout technologies for T. gondii and P. falciparum were further advanced by the CRISPR/Cas9-based genome editing strategy [30, 31]. The CRISPR/Cas9 system has also been successfully employed for genetic modification of the diarrheal protozoan parasite, Cryptosporidium parvum [32], indicating the feasibility of this genome editing system in various organisms. These developments together with conditional systems for regulation of gene expression at the genomic, transcriptional or protein level could be applied for Babesia spp. to investigate genes responsible for survival, virulence, transmission and immunity of bovine babesiosis.
Conclusion
Here, we established a transient and stable transfection systems for B. ovata and successfully inserted a foreign gene via integration and disruption of one copy of the ef-1α gene. This advancement in methodology enables us to investigate gene functions in B. ovata by targeted gene disruption, complementation and tagging of target proteins.
Abbreviations
- ef-1α :
-
Elongation factor-1 alpha
- IG:
-
Intergenic region
- rap-1 :
-
Rhoptry associated protein-1
- hdhfr :
-
Human dihydrofolate reductase
- RBC:
-
Red blood cell
- hsp70 :
-
Heat shock protein 70
- tpx-1 :
-
Thioredoxin peroxidase-1
- cam :
-
Calmodulin
- iRBC:
-
Infected RBC
- RT:
-
Room temperature
- GI:
-
Genome integrated
- ORF:
-
Open reading frame
References
Yoshinari T, Sivakumar T, Asada M, Battsetseg B, Huang X, Lan DT, et al. A PCR based survey of Babesia ovata in cattle from various Asian, African, and South American countries. J Vet Med Sci. 2013;75:211–4.
Sivakumar T, Tagawa M, Yoshinari T, Ybañez AP, Igarashi I, Ikehara Y, et al. PCR Detection of Babesia ovata from cattle reared in Japan and clinical significance of coinfection with Theileria orientalis. J Clin Microbiol. 2012;50:2111–3.
Abbas RZ, Zaman MA, Colwell DD, Gilleard J, Iqbal Z. Acaricide resistance in cattle ticks and approaches to its management: the state of play. Vet Parasitol. 2014;203(1–2):6–20.
Zintl A, Mulcahy G, Skerrett HE, Taylor SM, Gray JS. Babesia divergens: a bovine blood parasite of veterinary and zoonotic importance. Clin Microbiol Rev. 2003;16:622–36.
Crabb BS, Cowman AF. Characterization of promoters and stable transfection by homologous and nonhomologous recombination in Plasmodium falciparum. Proc Natl Acad Sci USA. 1996;93:7289–94.
Donald RGK, Roos DS. Stable molecular transformation of Toxoplasma gondii: a selectable dihydrofolate reductase-thymidylate synthase marker based on drug-resistance mutations in malaria. Proc Natl Acad Sci USA. 1993;90:11703–7.
Suarez CE, McElwain TF. Stable expression of a GFP-BSD fusion protein in Babesia bovis merozoites. Int J Parasitol. 2009;39(3):289–97.
Asada M, Tanaka M, Goto Y, Yokoyama N, Inoue N, Kawazu SI. Stable expression of green fluorescent protein and targeted disruption of thioredoxin peroxidase-1 gene in Babesia bovis with the WR99210/dhfr selection system. Mol Biochem Parasitol. 2012;181:162–70.
De Goeyse I, Jansen F, Madder M, Hayashida K, Berkvens D, Dobbelaere D, et al. Transfection of live, tick derived sporozoites of the protozoan apicomplexan parasite Theileria parva. Vet Parasitol. 2015;208(3–4):238–41.
Brayton KA, Lau AO, Herndon DR, Hannick L, Kappmeyer LS, Berens SJ, et al. Genome sequence of Babesia bovis and comparative analysis of apicomplexan hemoprotozoa. PLoS Pathog. 2007;3:1401–13.
Jackson AP, Otto TD, Darby A, Ramaprasad A, Xia D, Echaide IE, et al. The evolutionary dynamics of variant antigen genes in Babesia reveal a history of genomic innovation underlying host-parasite interaction. Nucleic Acids Res. 2014;42:7113–31.
Cuesta I, González LM, Estrada K, Grande R, Zaballos Á, Lobo CA, et al. High-quality draft genome sequence of Babesia divergens, the etiological agent of cattle and human babesiosis. Genome Announc. 2014;2(6):e01194–14.
Umemiya-Shirafuji R, Matsuo T, Liao M, Boldbaatar D, Battur B, Suzuki H, et al. Increased expression of ATG genes during nonfeeding periods in the tick Haemaphysalis longicornis. Autophagy. 2010;6(4):473–81.
Suarez CE, McElwain TF. Transfection systems for Babesia bovis: a review of methods for the transient and stable expression of exogenous genes. Vet Parasitol. 2010;167:205–15.
Igarashi I, Avarzed A, Tanaka T, Inoue N, Ito M, Omata Y, et al. Continuous in vitro cultivation of Babesia ovata. J Protozool Res. 1994;4:111–293.
Suarez CE, Palmer GH, LeRoith T, Florin-Christensen M, Crabb B, McElwain TF. Intergenic regions in the rhoptry associated protein-1 (rap-1) locus promote exogenous gene expression in Babesia bovis. Int J Parasitol. 2004;34:1177–84.
Suarez CE, Norimine J, Lacy P, McElwain TF. Characterization and gene expression of Babesia bovis elongation factor-1alpha. Int J Parasitol. 2006;36:965–73.
Kawazu S, Komaki-Yasuda K, Oku H, Kano S. Peroxiredoxins in malaria parasites: parasitologic aspects. Parasitol Int. 2008;57(1):1–7.
Hakimi H, Asada M, Angeles JMM, Inoue N, Kawazu S. Cloning and characterization of Plasmodium vivax thioredoxin peroxidase-1. Parasitol Res. 2012;111:525–9.
Militello KT, Dodge M, Bethke L, Wirth DF. Identification of regulatory elements in the Plasmodium falciparum genome. Mol Biochem Parasitol. 2004;134(1):75–88.
Suarez CE, Palmer GH, Florin-Christensen M, Hines SA, Hötzel I, McElwain TF. Organization, transcription, and expression of rhoptry associated protein genes in the Babesia bigemina rap-1 locus. Mol Biochem Parasitol. 2003;127:101–12.
Deitsch K, Driskill C, Wellems T. Transformation of malaria parasites by the spontaneous uptake and expression of DNA from human erythrocytes. Nucleic Acids Res. 2001;29:850–3.
Schuster FL. Cultivation of Babesia and Babesia-like blood parasites: agents of an emerging zoonotic disease. Clin Microbiol Rev. 2002;15:365–73.
Militello KT, Wirth DF. A new reporter gene for transient transfection of Plasmodium falciparum. Parasitol Res. 2003;89:154–7.
Vinkenoog R, Speranca MA, van Breemen O, Ramesar J, Williamson DH, Ross-MacDonald PB, et al. Malaria parasites contain two identical copies of an elongation factor 1 alpha gene. Mol Biochem Parasitol. 1998;94:1–12.
Janse CJ, Haghparast A, Speranca MA, Ramesar J, Kroeze H, del Portillo HA, et al. Malaria parasites lacking eef1a have a normal S/M phase yet grow more slowly due to a longer G1 phase. Mol Microbiol. 2003;50:1539–51.
Suarez CE, Johnson WC, Herndon DR, Laughery J, Davis WC. Integration of a transfected gene into the genome of Babesia bovis occurs by legitimate homologous recombination mechanisms. Mol Biochem Parasitol. 2015;202:23–8.
de Koning-Ward TF, Fidock DA, Thathy V, Menard R, van Spaendonk RM, Waters AP, et al. The selectable marker human dihydrofolate reductase enables sequential genetic manipulation of the Plasmodium berghei genome. Mol Biochem Parasitol. 2000;106:199–212.
Asada M, Yahata K, Hakimi H, Yokoyama N, Igarashi I, Kaneko O, et al. Transfection of Babesia bovis by double selection with WR99210 and blasticidin-S and its application for functional analysis of thioredoxin peroxidase-1. PLoS ONE. 2015;10(5), e0125993.
Jimenez-Ruiz E, Wong EH, Pall GS, Meissner M. Advantages and disadvantages of conditional systems for characterization of essential genes in Toxoplasma gondii. Parasitology. 2014;141(11):1390–8.
de Koning-Ward TF, Gilson PR, Crabb BS. Advances in molecular genetic systems in malaria. Nat Rev Microbiol. 2015;13(6):373–87.
Vinayak S, Pawlowic MC, Sateriale A, Brooks CF, Studstill CJ, Bar-Peled Y, et al. Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature. 2015;523:477–80.
Acknowledgements
The authors are grateful to Takeshi Q Tanaka from the National Research Center for Protozoan Diseases, Obihiro University and Thomas J. Templeton from our institute for critical reading of the manuscript. pHRH is a kind gift from Kirk W. Deitsch, Weill Cornell Medical College, USA. This research was supported by a grant from Science and Technology Research Promotion Program for Agriculture, Forestry, Fisheries and Food Industry to SK, MA, and JY (25018A). This work was also supported in part by Grants-in-Aids for Scientific Research on Innovative Areas, MEXT, Japan, to OK (23117008). This study was conducted at the Joint Usage/Research Center on Tropical Disease, Institute of Tropical Medicine, Nagasaki University, Japan with a support of Cooperative Research Grant of NEKKEN to SK (26-A-1, 27-A-2), and the National Research Center for Protozoan Diseases, Obihiro University of Agriculture and Veterinary Medicine, Japan with a support of Cooperative Research Grant of NRCPD to MA (27-joint-3). YK is a recipient of the scholarship from the Program for Nurturing Global Leaders in Tropical and Emerging Communicable Diseases, Graduate School of Biomedical Sciences, Nagasaki University.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
HH, MA and SK designed the study. HH, MA and YK carried out the experiments. JY performed the bioinformatics analysis. HH, MA, SK and OK performed data analysis. HH wrote the paper and all the authors read and approved the content of the paper.
Additional file
Additional file 1: Table S1.
List of primers used for promoter evaluation constructs and PCRs to confirm the integration of pBS-EGRADE into ef-1α locus. (PDF 162 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hakimi, H., Yamagishi, J., Kegawa, Y. et al. Establishment of transient and stable transfection systems for Babesia ovata . Parasites Vectors 9, 171 (2016). https://doi.org/10.1186/s13071-016-1439-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-016-1439-z