
Abstract
Background
Rumen flukes parasitize the rumen and reticulum of ruminants, causing paramphistomiasis. Over the years, there has been considerable debate as to whether Paramphistomum leydeni and Paramphistomum cervi are the same or distant species.
Methods
In the present study, the complete mitochondrial (mt) genome of P. leydeni was amplified using PCR-based sequencing and compared with that of P. cervi. The second internal transcribed spacer (ITS-2) of nuclear ribosomal DNA (rDNA) of P. leydeni specimens (n = 6) and P. cervi specimens (n = 8) was amplified and then sequenced. Phylogenetic relationship of the concatenated amino acid sequence data for 12 protein-coding genes of the two rumen flukes and selected members of Trematoda was evaluated using Bayesian inference (BI).
Results
The complete mt genome of P. leydeni was 14,050 bp in size. Significant nucleotide difference between the P. leydeni mt genome and that of P. cervi (14.7%) was observed. For genetic divergence in ITS-2, sequence difference between P. leydeni and P. cervi was 3.1%, while no sequence variation was detected within each of them. Phylogenetic analysis indicated that P. leydeni and P. cervi are closely-related but distinct rumen flukes.
Conclusions
Results of the present study support the proposal that P. leydeni and P. cervi represent two distinct valid species. The mt genome sequences of P. leydeni provide plentiful resources of mitochondrial markers, which can be combined with nuclear markers, for further comparative studies of the biology of P. leydeni and its congeners from China and other countries.
Similar content being viewed by others


Background
Species of Paramphistomum (Trematoda: Digenea), known as the ‘rumen flukes’ or ‘amphistomes’, are the pathogens of paramphistomiasis of ruminants, such as cattle, buffalo, sheep, goat and deer [1-5]. Although rumen flukes are considered neglected parasites, they are widely distributed in many continents of the world, (e.g., Asia, the Americas, Europe, Africa and Oceania) [1,2,4,6-12]. Rumen flukes require aquatic snails as intermediate hosts and the pre-parasitic stages of miracidia and stages in snails (sporocyst, redia and cercaria) are similar to those of liver flukes, such as Fasciola hepatica [13]. Cercaria escape from snails and attach to aquatic plants forming infectious metacercaria. Ruminants acquire infection through ingestion of infectious metacercaria attached to plants. Infection with adult Paramphistomum can cause chronic clinical signs, such as emaciation, anemia, diarrhea and edema [8]. The immature paramphistomes might migrate through intestine towards rumen, reticulum, abomasums, bile duct and gallbladder. The migration could lead to significant morbidity in ruminants, even death.
Paramphistomum leydeni and Paramphistomum cervi are common rumen flukes in many countries [1,2], particularly in Argentina [3]. Various host animals are often infected concurrently with P. leydeni, P. cervi and other paramphistomums globally, and the host or geographical preference of the two rumen flukes has not been documented. In spite of the economic loss and morbidity of paramphistomiasis, over the years, there has been a significant controversy as to whether P. leydeni and P. cervi represent the same or distinct fluke species. The taxonomy of P. leydeni and P. cervi is still unclear [1]. Although the amphistome species are morphologically very similar [2], reports have documented that P. leydeni and P. cervi are morphologically distinct species based on morphological features of the adult (e.g., genital opening type, pharynx type, ventral pouch and tegumental papillae absent or present) [13,14]. Furthermore, some studies have shown that Cotylophoron cotylophorum was re-classified as P. leydeni [1,2,5]. P. leydeni, as well as Paramphistomum hiberniae, Paramphistomum scotiae and Cotylophoron skriabini, was regarded as established synonym of P. cervi [5,14-17].
Molecular tools, using genetic markers in mitochondrial (mt) DNA and in the internal transcribed spacer (ITS) regions of nuclear ribosomal DNA (rDNA), have been used effectively to identify trematode species [18-21]. For rumen flukes, Yan et al. (2013) reported that mtDNA might be an useful molecular marker for studies of inter- and intra-specific differentiation of the Paramphistomidae [21]. Additionally, the ITS-2 rDNA has also proved to be a valuable marker for identification of amphistomes [1,2]. Advancements in long PCR-coupled sequencing and bioinformatic methods are providing effective approaches to probe into the biology of these parasites [22,23]. Therefore, in the present study, the complete mt genome of P. leydeni, and ITS-2 rDNA sequences of P. leydeni and P. cervi were sequenced, analyzed and compared to test the hypothesis that P. leydeni and P. cervi are two genetically distinct species.
Methods
Ethics statement
This study was approved by the Animal Ethics Committee of Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences. Adult specimens of Paramphistomum were collected from bovids and caprids, in accordance with the Animal Ethics Procedures and Guidelines of the People’s Republic of China.
Parasites, total genomic DNA extraction and the ascertainment of specimen identity
Adult specimens of Paramphistomum were collected, post-mortem, from the rumens of naturally infected goats in Nimu County, Tibet Autonomous Region; from livers and rumens of naturally infected yaks in Tianzhu and Maqu counties, Gansu Province; Ruoergai County, Sichuan Province; and Shaoyang City, Hunan Province, China. Samples were washed in physiological saline extensively, fixed in 70% (v/v) ethanol and preserved at −20°C until use.
Because the specimens were kept in 70% ethyl alcohol, it was difficult to acquire the accurate morphological data of the paramphistomums, thus molecular identification was performed to ascertain the identities of the two paramphistomums. Total genomic DNA of each sample was extracted separately by sodium dodecyl sulfate (SDS)/proteinase K digestion system [24] and mini-column purification (Wizard-SV Genomic DNA Purification System, Promega) according to the existing instructions.
ITS-2 rDNA of individual Paramphistomum specimens was amplified by PCR and sequenced according to established methods [25-27], and the identity of individual Paramphistomum specimens was ascertained by comparison with corresponding sequences available in GenBank [2].
Long-range PCR-based sequencing of mt genome
The primers (Table 1) were designed to relatively conserved regions of mtDNA nucleotide sequences from P. cervi and other closely-related taxa. The mt DNA was amplified from one specimen of P. leydeni collected from a goat in Nimu County, Tibet Autonomous Region, China. The full mt genome of P. leydeni was amplified in 4 overlapping long fragments between cox3 and atp6 (approximately 3.5 kb), between atp6 to cox1 (approximately 4 kb), between cox1 to rrnS (approximately 2.6 kb) and between rrnL to cox3 (approximately 5.5 kb) (Table 1). PCR reactions were conducted in a total volume of 50 μl using 4 mM MgCl2, 0.4 mM each of dNTPs, 5 μl 10× LATaq buffer, 5 mM of each primer, 0.5 μl LA Taq DNA polymerase (Takara, Dalian, China) and 2 μl DNA templates in a thermocycler (Biometra, Göttingen, Germany). The PCR cycling conditions began with an initial denaturation at 92°C for 2 min, then 12 cycles of denaturation at 92°C for 20 s, annealing at 55–62°C for 30 s and extension at 60°C for 3–5 min, followed by 92°C denaturation for 2 min, plus 28 cycles of 92°C for 20 s (denaturation), 55–62°C for 30 s (annealing) and 66°C for 3–5 min, with 10 min of the final extension at 66°C. A cycle elongation of 10 s was added for each cycle. A negative control containing nuclease-free water was included in every amplification run. Each amplicon (4 μl) was evidenced by electrophoresis in a 1.2% agarose gel, stained with Gold View I (Solarbio, Beijing, China) and photographed by GelDoc-It TSTM Imaging System (UVP, USA). Amplified products were sent to Genewiz Company (Beijing, China) for sequencing using ABI3730 sequencer from both directions using the primer walking strategy [28]. Sequencing results were tested by Seq Scanner 2 and artificial secondary interpretation was performed by professional technical personnel to ensure that the fragment of 50–800 bp of each sequencing result was read accurately. The walking primers were designed for approximately 600 to 700 bp of each sequence to assure the accuracy of two adjacent sequencing reactions by the sequencing company. The sequences were assembled manually to avoid errors by visualization of the chromatograms.
Amplification and sequencing of ITS-2 rDNA
The ITS rDNA region, spanning partial 18S, complete ITS-1, complete 5.8S, complete ITS-2 and partial 28S rDNA sequences, was amplified from the extracted DNA of each specimens using primers 18SF (forward; 5’-CACCGCCCGTCGCTACTACC-3’) and 28SR (reverse; 5’-ACTTTTCAACTTTCCCTC-3’) described previously [27]. The amplicons were approximately 2582 bp in length.
Assembling, annotation and bioinformatic analysis
P. leydeni mtDNA sequences were assembled manually and aligned against the whole mt DNA sequences of P. cervi (KF_475773) [21] and Paragonimus westermani (AF_219379) using MAFFT 7.122 to define specific gene boundaries. Twelve protein-coding genes were translated into amino acid sequences using MEGA 6.06 selecting the trematode mt genetic code option. The tRNA genes were identified using the program tRNAscan-SE [29] and ARWEN (http://130.235.46.10/ARWEN/) or by visual inspection [30]. The two rRNA genes were annotated by comparison with those of P. cervi and P. westermani.
Sliding window analysis of nucleotide variability
Pairwise alignment of the complete mt genomes of P. leydeni and P. cervi, including tRNAs and all intergenic spacers, was conducted by MAFFT 7.122 to locate variable nucleotide sites between the two rumen flukes. A sliding window analysis (window length =300 bp, overlapping step size =10 bp) was performed using DnaSP v. 5 [31] to estimate nucleotide diversity Pi (π) for each mt genes in the alignment. Nucleotide diversity was plotted against mid-point positions of each window, and gene boundaries were identified.
Phylogenetic analysis
For comparative purposes, the concatenated amino acid sequences conceptually translated from individual genes of the mt genomes of the two rumen fluke were aligned with published mt genomes from selected Digenea, including Clonorchis sinensis (FJ_381664) [32], Opisthorchis felineus (EU_921260) [32] and Opisthorchis viverrini (JF_739555) [33] [family Opisthorchiidae]; Haplorchis taichui (KF_214770) [34] [Heterophyidae]; P. westermani (AF_219379) [Paragonimidae]; Fasciola hepatica (NC_002546) [35], Fasciola gigantica (NC_024025) [19] and Fasciola sp. (KF_543343) [19] [Fasciolidae]; Dicrocoelium chinensis (NC_025279) [20] and Dicrocoelium dendriticum (NC_025280) [20] [Dicrocoeliidae] and P. cervi (KF_475773) [21] [Paramphistomidae]. The sequence of Schistosoma turkestanicum (HQ_283100) [36] [Schistosomatidae] was included as an outgroup.
All amino acid sequences were aligned using MAFFT 7.122 and excluding ambiguously aligned regions using Gblocks v. 0.91b selecting the defaults choosing options for less strict flanking positions. Then the alignment was modified into nex format and subjected to phylogenetic analysis using Bayesian inference (BI) applying the General Time Reversible (GTR) model as described previously [37]. Four Monte Carlo Markov Chain (MCMC) were run and two independent runs for 10000 metropolis-coupled MCMC generations were used, sampling a tree every 10 generation in MrBayes 3.1.2. Phylograms were viewed using FigTree v. 1.42 [38].
Results and discussion
Identity of P. leydeni and P. cervi
The ITS-2 sequences of P. leydeni specimens (n = 6) (GenBank accession nos. KP341666 to KP341671) were 100% homologous to previously published sequences of P. leydeni from sheep and cattle in Buenos Aires and Entre Ríos provinces, Argentina (HM_209064 and HM_209067), deer in Ireland (AB_973398) and ruminants in northern Uruguay (KJ_995524 to KJ_995529). The ITS-2 sequences of P. cervi specimens (n = 8) (GenBank accession nos. KP341658 to KP341665) were 100% identical to those of P. cervi from cattle in Heilongjiang Province, China (KJ_459934, KJ_459935).
Content and organization of mt genome of P. leydeni
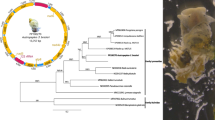
The complete mt genome sequence of P. leydeni (GenBank accession no. KP341657) is 14,050 bp in size, 38 bp larger than that of P. cervi. The circular genome of P. leydeni contains 36 genes that transcribing in the same direction, covering 12 protein-coding genes (nad1–6, nad4L, cox1–3, cytb and atp6), 22 tRNA genes and two rRNA genes (rrnL and rrnS) (Table 2) which is consistent with those of all the trematode species available to date (Figure 1) [18-21,32,33,36,39,40]. A comparison of nucleotide sequences of each protein coding gene, the amino acid sequences, two ribosomal DNA genes and two NCRs is given in Tables 2 and 3.

Organization of the mitochondrial genome of Paramphistomum leydeni. The scale is accurate. All genes are transcribed in the clockwise direction, and use standard nomenclature including 22 tRNA genes. “LNCR” and “SNCR” refer to a large non-coding region and small non-coding region. The A + T content also showed in each gene or region and represented by color.
The gene arrangement of the mt genome of P. leydeni is identical to that of P. cervi, but is obviously different from some species of Schistosoma, such as Schistosoma mansoni, Schistosoma spindale and Schistosoma haematobium [36,39-42]. The two rumen flukes, together with Opisthorchis spp. [32,33], Fasciola spp. [19,35], Dicrocoelium spp. [20], C. sinensis [32,33] and S. turkestanicum [36], share the same protein-coding gene and rRNA gene arrangement, which are interrupted by different tRNA genes or tRNA gene combinations, indicating important phylogenetic signal for Paramphistomatidae from the switched position of tRNA genes [39].
The nucleotide compositions of the whole mt genomes of two flukes reveal high T content and low C content, with T content being 44.53% in P. leydeni and 44.95% in P. cervi and C content being 9.44% in P. leydeni and 9.10% in P. cervi. The nucleotide composition of these two entire mt genomes is biased toward A and T, with an overall A + T content of 63.77% for P. leydeni and 63.40% for P. cervi respectively, which is within the range of magnitude of the trematode mt genomes (51.68% in P. westermani to 72.71% in S. spindale) [36,39-42].
The A + T content for the mt genomes of the two rumen flukes is shown in Additional file 1: Table S1. The A + T content of each gene and region range from 53.23% to 74.19% for P. leydeni and 52.24% to 69.84% for P. cervi. Both the highest and the lowest A + T content of two mt genomes exist in tRNA genes of P. leydeni and P. cervi, while the other genes and regions occupy more steady A + T content of 60.94% to 67.29% and 60.88% to 66.78%, respectively. The A + T content of 12 protein-coding genes of P. leydeni are generally higher than that of P. cervi, except for atp6, nad2, nad6 and nad5. Other than high A + T content of NCRs in Schistosomatidae (>72% in S. spindale and >97% in S. haematobium) [39], the A + T content of NCRs of Paramphistomatidae are at around 62%, with 60.94% to 63.90% in P. leydeni, and 62.07% to 64.33% in P. cervi, as shown in Additional file 1: Table S1.
Annotation of mt genome of P. leydeni
In the P. leydeni mt genome, the open reading-frames of 12 protein-coding genes have ATG or GTG or ATA as initiation codons, TAG or TAA as termination codons. It is noticable that P. leydeni is the only trematode found initiating nad2 with ATA so far. None of the 12 genes in the mt genome of P. cervi uses ATA as initial codons, nor TAA as termination codons (Table 2). No incomplete terminal codons were observed in either of genomes of the two Paramphistomum. In the mt genomes of P. leydeni, 22 tRNA genes, ranging from 59 to 73 bp in size, have similar predicted secondary structures to the corresponding genes from P. cervi [21]. In both mt genomes, the rrnL gene is situated between tRNA-Thr and tRNA-Cys, and rrnS locates between tRNA-Cys and cox2 (Table 2). The length of the rrnL gene is 995 bp for P. leydeni, 9 nt longer than that in P. cervi. The length of the rrnS gene is 749 bp for both P. leydeni and P. cervi. For these two mt genomes, the long non-coding regions (LNCR) and short non-coding regions (SNCR) are situated between the tRNA-Glu and cox3, and cytb and nad4L, respectively (Table 2). Though the NCRs reveal no remarkable features, it is speculated that the AT-rich domain could be connected with the replication and transcription initiation [43,44].
Comparative analyses of mt genomes of P. leydeni and P. cervi
The magnitude of sequence difference across the entire mt genome between the two paramphistomums is 14.7% (2088 nucleotide substitutions in all), slightly larger than that between F. hepatica and F. gigantica (11.8%) [19] and D. chinensis and D. dentiticum (11.81%) [20].
For the 12 protein genes of P. leydeni and P. cervi, comparisons also reveal sequence differences at both nucleotide (13.3%, a total of 1336 nucleotide substitutions) and amino acid level (9.05%, a total of 304 amino acid substitutions), which are larger than those between F. hepatica and F. gigantica (11.6% and 9.83%, respectively) [19], and between D. chinensis and D. dentriticum (11.7% and 11.36%, respectively) [20].
A comparison of the nucleotide and amino acid sequences inferred from individual mt protein-coding genes of P. leydeni and P. cervi is shown in Table 3. The nucleotide sequence differences of 12 protein coding-genes range from 9.45% to 16.10%, with cox2 and nad5 being the most and the least conserved genes, respectively. It is notable that the nad5 gene is regarded as the most conserved protein-coding gene in Dicrocoelium, based on nucleotide sequences comparison between D. dendriticum and D. chinensis [20]. The amino acid sequence differences of P. leydeni and P. cervi range from 5.25% to 14.14%. Based on the inferred amino acid sequence differences, cox1 and nad2 are the most and the least conserved protein-coding genes respectively. It is noteworthy that the nad6 gene possesses the highest level of sequence difference in Fasciolidae and Dicrocoeliidae [19,20].
Nucleotide differences also exist in ribosomal RNA genes [rrnL (10.53%) and rrnS (11.67%)], tRNA genes (13.20%) and non-coding regions [LNCR (38.33%) and SNCR (35.94%)] (Table 3). Through the comparison of entire mt genomes of P. leydeni and P. cervi, cox2 is the most conserved gene (Table 3). It is worth noting that the most conserved gene in Dicrocoelium is rrnS [20]. Results of these comparative analyses indicate that P. leydeni and P. cervi represent distinct fluke species.
Sliding window analysis of nucleotide variability
By computing the number of variable positions per unit length of gene, the sliding window indicated that the highest and lowest levels of sequence variability were within the genes nad5 and cox2, respectively. In this study, protein-coding genes of cox2, nad3 and nad1 are the most conserved protein-coding genes, while nad5, nad6 and nad2 are the least conserved (Figure 2). These results are slightly different from those among Fasciola spp. that cytb and nad1 were the most conserved genes, while nad6, nad5 and nad4 were the least conserved [19].

Sliding window of nucleotide variation in complete mt genome sequences of Paramphistomum leydeni and P. cervi . The folding line indicates nucleotide variation in a window of 300 bp (steps in 10 bp). Regions and boundaries of 12 protein-coding genes are indicated by color.
Phylogenetic analysis
Phylogenetic analysis of the concatenated amino acid sequence datasets for all 12 mt proteins (Figure 3) reflected the clear genetic distinctiveness between P. leydeni and P. cervi and also the grouping of these two members of Paramphistomum with other members of families Opisthorchiidae, Heterophyidae, Paragonimidae, Fasciolidae, Dicrocoeliidae and Schitosomatidae, with strong nodal support (posterior probability = 1.00). The difference between the two Paramphistomum spp. is similar to that between F. hepatica and F. gigantica [19], D. chinensis and D. dentriticum [20], and C. sinensis and O. felineus [33] by observing the lengths of the branches. The phylogenetic analysis further confirmed that P. leydeni and P. cervi are different Paramphistomum species.

Phylogenetic relationships of Paramphistomum leydeni and P. cervi , and other trematodes. Phylogenetic analysis of the concatenated amino acid sequence datasets representing 12 protein-coding genes was performed by Bayesian inference (BI), using Schistosoma turkestanicum (HQ_283100) as an outgroup.
Nucleotide differences in ITS-2 rDNA between P. leydeni and P. cervi
The rDNA region sequenced from individual P. leydeni samples was approximately 2582 bp in length, including partial 18S rDNA, complete ITS-1, complete 5.8 rDNA, complete ITS-2, and partial 28S rDNA. ITS-2 was 286 bp in length. Sequence difference in ITS-2 rDNA was 3.1% between the P. leydeni and P. cervi, which is slightly lower than that between D. chinensis and D. dentriticum (3.8-6.3%), but higher than that between F. hepatica and F. gigantica (1.7%) [19], while no sequence variation was observed within P. leydeni and P. cervi. These results provided additional strong support that P. leydeni and P. cervi are different trematode taxa.
In spite of the evidence of genetic difference between two Paramphistomum species, elaborate population genetic investigations still need to be conducted. Further studies could (i) explore nucleotide variation in mtDNAs among Paramphistomum populations in various hosts of numerous countries from different continents, (ii) establish accurate molecular tools and rapid detection methods, (iii) decipher the genomes of Paramphistomum using next generation sequencing (NGS) technologies. It is believed that elucidating the transcriptomes, proteomes and genomes of Paramphistomum would assist in future efforts in deciphering biology and taxonomy of more trematode parasites including the important family Paramphistomatidae.
Conclusions
The present study determined the complete mt genome sequences and ITS-2 rDNA sequences of P. leydeni, and provided reliable genetic evidence that P. leydeni and P. cervi are closely-related but distinct paramphistome species based on mt and nuclear ribosomal DNA dataset. The accurate identification of the two rumen flukes will contribute to the diagnosis and control of paramphistomiasis. The availability of the complete mt genome sequences and nuclear rDNA sequences of P. leydeni could provide additional genetic markers for studies of the epidemiology, population genetics and phylogenetic systematics of trematodes.
References
O’Toole A, Browne JA, Hogan S, Bassière T, DeWaal T, Mulcahy G, et al. Identity of rumen fluke in deer. Parasitol Res. 2014;113:4097–103.
Sanabria R, Moré G, Romero J. Molecular characterization of the ITS-2 fragment of Paramphistomum leydeni (Trematoda: Paramphistomidae). Vet Parasitol. 2011;177:182–5.
Sanabria R, Martorelli S, Romero J. First report of Paramphistomum leydeni Näsmark, 1937 (Trematoda: Paramphistomidae) in Argentina, and re-examination of Cotylophoron cotylophorum sensu Racioppi et al. (1994). Helminthologia. 2009;46:225–9.
Sey O. The amphistomes of Hungarian vertebrates. Parasitol Hung. 1991;24:59–68.
Eduardo S. The taxonomy of the family Paramphistomidae Fischoeder, 1901 with special reference to the morphology of species occurring in ruminants. III. Revision of the genus Calicophoron Näsmark, 1937. Syst Parasitol. 1982;4:7–57.
Graber M, Euzeby J, Gevrey J. Presence in France of Paramphistomum leydeni Näsmark, 1937 (Trematoda: Paramphistomata) (author’s transl). Ann Parasitol Hum Comp. 1979;55:565–9.
Samnaliev P, Pino L, Bayssade Dufour C, Albaret J. Superficial argentophilic structures of miracidia and cercaria of Paramphistomum leydeni Näsmark 1937. Ann Parasitol Hum Comp. 1983;59:151–9.
Dorny P, Stoliaroff V, Charlier J, Meas S, Sorn S, Chea B, et al. Infections with gastrointestinal nematodes, Fasciola and Paramphistomum in cattle in Cambodia and their association with morbidity parameters. Vet Parasitol. 2011;175:293–9.
Ozdal N, Gul A, Ilhan F, Deger S. Prevalence of Paramphistomum infection in cattle and sheep in Van Province, Turkey. Helminthologia. 2010;47:20–4.
Melaku S, Addis M. Prevalence and intensity of Paramphistomum in ruminants slaughtered at Debre Zeit industrial abattoir, Ethiopia. Global Vet. 2012;8:315–9.
Range Ruiz L, Albores Brahms S, Gamboa Aguilar J. Seasonal trends of Paramphistomum cervi in Tabasco, Mexico. Vet Parasitol. 2003;116:217–22.
Roberts F. The occurrence and prevalence of gastro-intestinal helminths in apparently healthy cattle in Queensland, Australia. J Comp Pathol Therap. 1939;52:160–5.
Nikander S, Saari S. Notable seasonal variation observed in the morphology of the reindeer rumen fluke (Paramphistomum leydeni) in Finland. Rangifer. 2009;27:47–57.
Sey O. Revision of the amphistomes of European ruminants. Parasitol Hung. 1980;13:13–25.
Kotrlá B, Kotrlý A. The incidence of flukes of the genus Paramphistomum in Czechoslovakia. Vet Med (Prague, Czech Repub). 1982;27:483–90.
Odening K. What is Paramphistomum cervi? Angew Parasitol. 1983;24:205–16.
Albaret J, Bayssade Dufour C, Ngendahayo L, Postal J, Picot H. Chaetotaxy of the cercaria of Paramphistomum sp., a parasite of cattle in Vendee. Ann Parasitol Hum Comp. 1986;62:271–5.
Biswal DK, Chatterjee A, Bhattacharya A, Tandon V. The mitochondrial genome of Paragonimus westermani (Kerbert, 1878), the Indian isolate of the lung fluke representative of the family Paragonimidae (Trematoda). Peer J. 2014;2:e484.
Liu GH, Gasser RB, Young ND, Song HQ, Ai L, Zhu XQ. Complete mitochondrial genomes of the ‘intermediate form’ of Fasciola and Fasciola gigantica, and their comparison with F. hepatica. Parasit Vectors. 2014;7:150.
Liu GH, Yan HB, Otranto D, Wang XY, Zhao GH, Jia WZ, et al. Dicrocoelium chinensis and Dicrocoelium dendriticum (Trematoda: Digenea) are distinct lancet fluke species based on mitochondrial and nuclear ribosomal DNA sequences. Mol Phylogenet Evol. 2014;79:325–31.
Yan HB, Wang XY, Lou ZZ, Li L, Blair D, Yin H, et al. The mitochondrial genome of Paramphistomum cervi (Digenea), the first representative for the family Paramphistomidae. PLoS One. 2013;8:e71300.
Hu M, Chilton N, Gasser R. Long PCR-based amplification of the entire mitochondrial genome from single parasitic nematodes. Mol Cell Probes. 2002;16:261–7.
Jex AR, Hu M, Littlewood DTJ, Waeschenbach A, Gasser RB. Using 454 technology for long-PCR based sequencing of the complete mitochondrial genome from single Haemonchus contortus (Nematoda). BMC Genomics. 2008;9:11.
Gasser RB, Hu M, Chilton NB, Campbell BE, Jex AJ, Otranto D, et al. Single-strand conformation polymorphism (SSCP) for the analysis of genetic variation. Nat Protoc. 2007;1:3121–8.
Otranto D, Rehbein S, Weigl S, Cantacessi C, Parisi A, Lia RP, et al. Morphological and molecular differentiation between Dicrocoelium dendriticum (Rudolphi, 1819) and Dicrocoelium chinensis (Sudarikov and Ryjikov, 1951) Tang and Tang, 1978 (Platyhelminthes: Digenea). Acta Trop. 2007;104:91–8.
Zhu X, D’Amelio S, Gasser R, Yang T, Paggi L, He F, et al. Practical PCR tools for the delineation of Contracaecum rudolphii A and Contracaecum rudolphii B (Ascaridoidea: Anisakidae) using genetic markers in nuclear ribosomal DNA. Mol Cell Probes. 2007;21:97–102.
Zheng X, Chang QC, Zhang Y, Tian SQ, Lou Y, Duan H, et al. Characterization of the complete nuclear ribosomal DNA sequences of Paramphistomum cervi. Sci World J. 2014;2014:751907.
Hu M, Jex AR, Campbell BE, Gasser RB. Long PCR amplification of the entire mitochondrial genome from individual helminths for direct sequencing. Nat Protoc. 2007;2:2339–44.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:0955–64.
Hu M, Chilton NB, Gasser RB. The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea). Int J Parasitol. 2002;32:145–58.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Shekhovtsov SV, Katokhin AV, Kolchanov NA, Mordvinov VA. The complete mitochondrial genomes of the liver flukes Opisthorchis felineus and Clonorchis sinensis (Trematoda). Parasitol Int. 2010;59:100–3.
Cai X, Liu G, Song H, Wu C, Zou F, Yan H, et al. Sequences and gene organization of the mitochondrial genomes of the liver flukes Opisthorchis viverrini and Clonorchis sinensis (Trematoda). Parasitol Res. 2012;110:235–43.
Lee D, Choe S, Park H, Jeon HK, Chai JY, Sohn WM, et al. Complete mitochondrial genome of Haplorchis taichui and comparative analysis with other trematodes. Korean J Parasitol. 2013;51:719–26.
Le T, Blair D, McManus D. Complete DNA sequence and gene organization of the mitochondrial genome of the liverfluke, Fasciola hepatica L. (Platyhelminthes; Trematoda). Parasitology. 2001;123:609–21.
Wang Y, Wang CR, Zhao GH, Gao JF, Li MW, Zhu XQ. The complete mitochondrial genome of Orientobilharzia turkestanicum supports its affinity with African Schistosoma spp. Infect Genet Evol. 2011;11:1964–70.
Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4.
Chen Y, Chen S, Kang J, Fang H, Dao H, Guo W, et al. Evolving molecular epidemiological profile of Human Immunodeficiency Virus 1 in the southwest border of China. PLoS One. 2014;9:e107578.
Littlewood DTJ, Lockyer AE, Webster BL, Johnston DA, Le TH. The complete mitochondrial genomes of Schistosoma haematobium and Schistosoma spindale and the evolutionary history of mitochondrial genome changes among parasitic flatworms. Mol Phylogenet Evol. 2006;39:452–67.
Webster BL, Rudolfová J, Horák P, Littlewood DTJ. The complete mitochondrial genome of the bird schistosome Trichobilharzia regenti (Platyhelminthes: Digenea), causative agent of cercarial dermatitis. J Parasitol. 2007;93:553–61.
Le TH, Blair D, Agatsuma T, Humair PF, Campbell NJ, Iwagami M, et al. Phylogenies inferred from mitochondrial gene orders-a cautionary tale from the parasitic flatworms. Mol Biol Evol. 2000;17:1123–5.
Zhao GH, Li J, Song HQ, Li XY, Chen F, Lin RQ, et al. A specific PCR assay for the identification and differentiation of Schistosoma japonicum geographical isolates in mainland China based on analysis of mitochondrial genome sequences. Infect Genet Evol. 2012;12:1027–36.
Cantatore P, Attardi G. Mapping of nascent light and heavy strand transcripts on the physical map of HeLa cell mitochondrial DNA. Nucleic Acids Res. 1980;8:2605–26.
Goddard JM, Wolstenholme DR. Origin and direction of replication in mitochondrial DNA molecules from the genus Drosophila. Nucleic Acids Res. 1980;8:741.
Acknowledgements
Project support was provided in part by the International Science & Technology Cooperation Program of China (Grant No. 2013DFA31840), the “Special Fund for Agro-scientific Research in the Public Interest” (Grant No. 201303037) and the Science Fund for Creative Research Groups of Gansu Province (Grant No. 1210RJIA006).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XQZ and GHL conceived and designed the study, and critically revised the manuscript. JM and JJH performed the experiments, analysed the data and drafted the manuscript. DHZ, JZL and YL helped in study design, study implementation and manuscript revision. All authors read and approved the final manuscript.
Additional file
Additional file 1: Table S1.
Comparison of A + T content of mitochondrial genomes of Paramphistomum leydeni (PL) and Paramphistomum cervi (PC).
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Ma, J., He, JJ., Liu, GH. et al. Mitochondrial and nuclear ribosomal DNA dataset supports that Paramphistomum leydeni (Trematoda: Digenea) is a distinct rumen fluke species. Parasites Vectors 8, 201 (2015). https://doi.org/10.1186/s13071-015-0823-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-015-0823-4