
Abstract
Background
Cost-effective production of biofuels from lignocellulose requires the fermentation of d-xylose. Many yeast species within and closely related to the genera Spathaspora and Scheffersomyces (both of the order Serinales) natively assimilate and ferment xylose. Other species consume xylose inefficiently, leading to extracellular accumulation of xylitol. Xylitol excretion is thought to be due to the different cofactor requirements of the first two steps of xylose metabolism. Xylose reductase (XR) generally uses NADPH to reduce xylose to xylitol, while xylitol dehydrogenase (XDH) generally uses NAD+ to oxidize xylitol to xylulose, creating an imbalanced redox pathway. This imbalance is thought to be particularly consequential in hypoxic or anoxic environments.
Results
We screened the growth of xylose-fermenting yeast species in high and moderate aeration and identified both ethanol producers and xylitol producers. Selected species were further characterized for their XR and XDH cofactor preferences by enzyme assays and gene expression patterns by RNA-Seq. Our data revealed that xylose metabolism is more redox balanced in some species, but it is strongly affected by oxygen levels. Under high aeration, most species switched from ethanol production to xylitol accumulation, despite the availability of ample oxygen to accept electrons from NADH. This switch was followed by decreases in enzyme activity and the expression of genes related to xylose metabolism, suggesting that bottlenecks in xylose fermentation are not always due to cofactor preferences. Finally, we expressed XYL genes from multiple Scheffersomyces species in a strain of Saccharomyces cerevisiae. Recombinant S. cerevisiae expressing XYL1 from Scheffersomyces xylosifermentans, which encodes an XR without a cofactor preference, showed improved anaerobic growth on xylose as the primary carbon source compared to S. cerevisiae strain expressing XYL genes from Scheffersomyces stipitis.
Conclusion
Collectively, our data do not support the hypothesis that xylitol accumulation occurs primarily due to differences in cofactor preferences between xylose reductase and xylitol dehydrogenase; instead, gene expression plays a major role in response to oxygen levels. We have also identified the yeast Sc. xylosifermentans as a potential source for genes that can be engineered into S. cerevisiae to improve xylose fermentation and biofuel production.
Similar content being viewed by others

Introduction
Xylose is the most abundant pentose that comprises hemicellulose in plants; therefore, robust microorganisms that can ferment this sugar are required for profitable biofuel production from lignocellulosic materials [1, 2]. The budding yeast Saccharomyces cerevisiae is widely used in biotechnological applications and is one of the most understood model microorganisms [3, 4]. However, S. cerevisiae lacks certain traits that limit its usefulness in lignocellulosic biofuel production, prompting some to investigate other yeast species as alternative biocatalysts. For example, S. cerevisiae does not have the ability to ferment xylose [5, 6], while several non-conventional yeast species do so remarkably well [7].
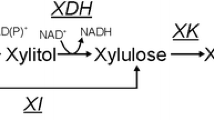
Species belonging to the genera Spathaspora and Scheffersomyces (order Serinales under a recently proposed taxonomy [8], formerly the CUG-Ser1 major clade [9]) are known for their association with insects and their habitats, such as decomposing wood, and by their natural ability to assimilate and/or ferment xylose [10,11,12]. Spathaspora passalidarum, Scheffersomyces stipitis (syn. Pichia stipitis), Scheffersomyces segobiensis, Scheffersomyces shehatae, Scheffersomyces coipomoensis, and Scheffersomyces ergatensis were the first members assigned to their respective clades [13, 14]. Sp. passalidarum and Sc. stipitis have also been used as sources of genes that have been engineered in S. cerevisiae to confer the ability to ferment xylose [15,16,17]. These non-conventional yeasts harbor three genes that encode enzymes for xylose metabolism: XYL1, which encodes xylose reductase (XR) for the reduction of xylose to xylitol; XYL2, which encodes xylitol dehydrogenase (XDH) for the conversion of xylitol to xylulose; and XYL3, which encodes xylulokinase (XK) for the phosphorylation of xylulose to xylulose-5-phosphate [18]. The genomes of most studied xylose-fermenting species contain an XR that preferentially utilizes NADPH as its primary cofactor and has lower affinity for NADH, while the XDH is strictly NAD+ dependent [19,20,21]. A commonly articulated hypothesis is that different cofactor preferences lead to an imbalance during xylose catabolism; specifically, xylitol accumulates in anoxic environments or oxygen-limited conditions because little or no NAD+ can be regenerated without sufficient oxygen to act as a terminal electron acceptor [15, 22].
Some yeast species have adopted genetic mechanisms that manage cofactor imbalances during xylose catabolism. Unlike Sc. stipitis, Sp. passalidarum bears two homologs of XYL1, which are named XYL1.1 and XYL1.2. The first one encodes an XR with NADPH affinity, while the enzyme encoded by the second homolog prefers NADH over NADPH [15]. Interestingly, xylose metabolism in this species is driven toward ethanol production, instead of xylitol accumulation, even at low aeration [23, 24]. Indeed, mutating the XR to prefer NADH over NADPH or inserting Sp. passalidarum XYL1.2 increases the ethanol productivity and alleviates cofactor imbalance in S. cerevisiae [15, 25, 26]. In addition to enzyme specificity, expression of the XYL genes, particularly XYL2, in S. cerevisiae has been observed to impact xylose utilization, as higher expression led to more efficient xylose fermentation [16, 27]. The XR:XDH expression ratios have also been suggested to impact xylitol accumulation and the yield of ethanol produced [28]. Beyond the first steps of xylose metabolism, the overexpression of genes related to the non-oxidative phase of the pentose phosphate pathway (PPP) has also been shown to positively influence the growth rate on xylulose [29]. Although these factors seem to be important for xylose metabolism, genetically modified S. cerevisiae strains still cannot ferment xylose at rates comparable to glucose [5, 30, 31]. The presence of a complete, integrated XYL pathway alone is not sufficient for xylose assimilation and/or fermentation by S. cerevisiae [27, 32], suggesting that other genetic and regulatory mechanisms are required for efficient xylose metabolism. For this reason, investigations of native xylose-fermenting yeast species may provide novel strategies to overcome this problem in S. cerevisiae, which has impeded bioenergy research for decades.
The overall aims of this study are to understand how some species belonging to the genera Spathaspora and Scheffersomyces can efficiently ferment xylose into ethanol, why other species incompletely catabolize the pentose and accumulate xylitol, and how oxygenation can impact these outcomes. Although several members of these genera have been described in the taxonomic literature in recent years, little physiological information is available for most of them, and most earlier studies have been confined to Sp. passalidarum and Sc. stipitis. Our data show that xylose metabolism for species from the order Serinales is highly plastic and that oxygenation has a broad effect on the gene expression of xylolytic and interacting pathways.
Results
Scheffersomyces and Spathaspora switch ethanol production to xylitol or glycerol production during respiration
Species belonging to the order Serinales, particularly members of the genera Scheffersomyces and Spathaspora [33], are known to harbor the uncommon ability to ferment xylose. We first determined the quantities of metabolites that these species produced under moderate oxygen-limited growth conditions with xylose as the primary carbon source (see Methods). Ethanol and xylitol were the major metabolites produced by Spathaspora and Scheffersomyces species during xylose fermentation, while glycerol was produced less frequently. We refer to ethanol and xylitol producers for species that primarily excreted these metabolites (based on yields) under moderate aeration in unbaffled shake flasks (SF). The titers, productivity rates, and yields related to the consumption of d-xylose and the production of biomass, ethanol, and xylitol under moderate and high aeration conditions are summarized in Additional file 1.
The species that produced ethanol under oxygen-limited conditions (41.7% of the yeasts tested) were Scheffersomyces xylosifermentans, Sc. parashehatae, Sc. virginianus, Sc. shehatae, Sc. stipitis, Sc. illinoinensis, Sc. cryptocercus, Spathaspora arborariae, Sp. passalidarum, and Sp. gorwiae. Remarkably, Sc. xylosifermentans, Sc. parashehatae, and Sp. passalidarum consumed all the xylose and reached maximum titers and yields of ethanol within 24 h of fermentation. After this time, the species consumed the ethanol. The xylitol producers (45.8%) included Scheffersomyces coipomoensis, Sc. insectosa, Sc. amazonensis, Sc. quercinus, Sp. brasiliensis, Sp. suhii, Sp. roraimanensis, Sp. girioi, Sp. hagerdaliae, Sp. xylofermentans, and Candida (Spathaspora) materiae. Except for the production of ethanol by Sc. amazonensis, Sc. coipomoensis, and C. materiae, the yield and productivity of xylitol surpassed ethanol production, and these species accumulated the highest titers of xylitol compared to other xylitol producers. Out of the 24 species of Spathaspora and Scheffersomyces tested, three were not capable of fermenting xylose and are not visualized in Fig. 1: Sc. spartinae and Sc. gosingicus showed minimal growth and modest consumption of sugar with no production of any metabolite examined, while Sc. ergatensis did not grow.

Yeast species from the order Serinales generate different metabolic end-products based on oxygen levels. Production of ethanol, xylitol, and/or glycerol by species of Scheffersomyces and Spathaspora genera under moderate (shake flask—SF) and high (baffled flask—BF) aeration conditions. Colored squares: metabolite(s) mainly produced by each species based on yields. Gray squares: the species produced low yields or did not produce the metabolite. Complete data are in Additional file 1
To verify the impact of oxygenation on xylose usage by Spathaspora and Scheffersomyces species, we also tested the species in baffled flasks (BF). This condition increases the volumetric mass transfer coefficient of oxygen (kLa), which is a parameter that determines the rate at which a gaseous compound can transfer between the gas and liquid phases (Li et al., 2013). Using an oxygen meter, we determined that the dissolved oxygen level in BF (4.74 mg L−1) was nearly double the oxygen present in SF (2.63 mg L−1). Although respiration dominated fermentation under high aeration, multiple species still fermented, while several species produced different primary metabolites in BF (Fig. 1). Sc. xylosifermentans, which presented high consumption of xylose and ethanol yields in SF, accumulated xylitol under high aeration and it did not consume xylose completely in 72 h. Sp. passalidarum produced glycerol, instead of ethanol, and the consumption of xylose was delayed in BF. For some species, especially xylitol producers, oxygen increased the rate of xylose utilization, and xylose was consumed faster under high aeration than moderate oxygen-limited conditions. Figure 2 shows the distinction between ethanol producers (Sp. passalidarum and Sc. xylosifermentans) and xylitol producers (Sc. coipomoensis and Sc. amazonensis). In contrast to the former two species, Sc. coipomoensis and Sc. amazonensis had slightly enhanced xylose consumption and produced ethanol, instead of xylitol, under high aeration. Even so, Sc. coipomoensis and Sc. amazonensis produced more biomass under these conditions, which limited their ethanol production relative to the ethanol producers in SF.

Scheffersomyces xylosifermentans and Spathaspora passalidarum ferment xylose into ethanol at high yield and titer. Consumption of xylose and production of biomass, ethanol, xylitol, and glycerol by Sc. xylosifermentans, Sp. passalidarum, Scheffersomyces amazonensis, and Scheffersomyces coipomoensis under moderate (shake flask—SF) and high (baffled flask—BF) aeration conditions. Error bars indicate the standard deviation from the three biological replicates
For all the species studied here, the increased oxygen levels in BF favored the production of cell biomass. With greater oxygen availability in BF, we expected that the metabolism would be directed mainly toward cellular respiration. In general, the yeasts produced higher end-product (ethanol/xylitol) titers in SF compared to BF. Despite the xylitol production by the ethanol producers, this result suggests that most of the carbon was converted into carbon dioxide (CO2) in BF.
Most species with multiple XYL1 homologs accumulate xylitol under moderate and high aeration
Recently, a study reported the presence of multiple homologs of XYL1 in Candida intermedia, which harbors three XYL1 genes, one of which (called XYL1.2) encodes an XR with higher affinity for NADH [19]. Like the XYL1.2 from Sp. passalidarum, C. intermedia XYL1.2 displays dual cofactor specificity [15, 19]. A search for homologs across the order Serinales showed that Candida blattae, Meyerozyma carpophila, Me. guilliermondii, and Me. caribbica also have more than one XYL1 homologs. To determine whether these species ferment xylose as efficiently as Sp. passalidarum and to investigate the influence of multiple XYL1 homologs on xylose metabolism, we tested them, as well as C. intermedia, during growth in YPX (1% yeast extract, 2% peptone, 5% xylose) under moderate and high aeration. Xylose was completely consumed by these strains under high aeration, while a significant amount of xylose remained unused under moderate conditions. These results suggested that xylose utilization by these closely related species was highly dependent on oxygen. All the species outside of the Scheffersomyces and Spathaspora clades accumulated xylitol in both conditions (Additional file 2). Although the yield of xylitol surpassed the yield of ethanol produced during moderate aeration by C. intermedia, it was the only species able to achieve a substantial titer of ethanol. Its fermentative capability may be due to the presence of an enzyme that favors the use of NADH. However, the cofactor preferences of the enzymes encoded by the homologs of XYL1 from the other species are not known.
Xylose reductase from Scheffersomyces xylosifermentans has dual cofactor affinity
Xylitol accumulation during metabolic conversion of xylose has been proposed to result from imbalanced cofactor usage by XR and XDH. To determine the cofactor preference of the enzymes in the first steps of xylose metabolism, we measured the specific activity of XR and XDH from ethanol-producing Sp. passalidarum and Sc. xylosifermentans, as well as from xylitol-producing Sc. amazonensis and Sc. coipomoensis, in SF and BF (Fig. 3A). The rationale for selecting Sc. xylosifermentans, Sc. coipomoensis, and Sc. amazonensis was that they harbor a single homolog of the XYL1 gene, and they exhibited distinct phenotypes (ethanol versus xylitol production) under the tested conditions (Fig. 3B). In SF, XR of Sc. coipomoensis and Sc. amazonensis utilized NADH, but we observed greater XR activities with NADPH (P < 0.05) (Fig. 3A). On the contrary, there were no significant differences in Sc. xylosifermentans XR activities between the two cofactors. This result is interesting because, among these species, Sc. xylosifermentans showed the highest consumption of xylose in SF. In BF, the cofactor preferences of the enzymes were not significantly different, which was expected given the presence of a single homolog of XYL1.

Xylose reductase from Scheffersomyces xylosifermentans lacks a cofactor preference. A XR and XDH activities under moderate aeration and high aeration conditions expressed in units (U) per mg protein [U (mg protein)−1]. B Production of ethanol, xylitol, and/or glycerol by the species tested for XR and XDH activities (reproduced from Fig. 1). Colored squares: metabolite(s) mainly produced by each species based on yields. Gray squares: the species produced low yields or did not produce the metabolite. Spa—Spathaspora passalidarum, Sxy—Scheffersomyces xylosifermentans, Scoip—Scheffersomyces coipomoensis, Sama—Scheffersomyces amazonensis, and Scer—Saccharomyces cerevisiae (negative control). Error bars indicate the standard deviation from the three biological replicates. Asterisks denote significant differences between the activities on NADH and NADPH for each species (P < 0.05)
Previous research showed that XR activities from Sp. passalidarum utilized both NADH and NADPH with higher activity for NADH under moderate oxygen-limited conditions and higher activity for NADPH in high aeration [15, 23]. Our results were consistent with those previous observations: Sp. passalidarum XR significantly preferred NADH over NADPH (P < 0.05) in SF and significantly preferred NADPH (P < 0.05) in BF. This switch in cofactor preference could have been enabled by the differential regulation of the two distinct XYL1 homologs. This species also has two homologs of XYL2 [34], but there is no data available in the literature showing the preference of XDH from each homolog. Our results revealed that XDH was strictly NAD+ dependent not only for Sp. passalidarum, but for all the four species tested. Low activities with NADP+ occurred for Sc. xylosifermentans and Sc. coipomoensis, but these were minimal compared to the activities with NAD+ (Fig. 3A). For species that possess NADPH-preferring XRs, the exclusive utilization of NAD+ by XDH could potentially hinder the conversion of xylitol to xylulose under oxygen-limiting conditions, resulting in xylitol accumulation due to the previously proposed redox imbalance hypothesis [35, 36]. Nonetheless, the xylitol bottleneck observed in Sc. xylosifermentans with high aeration seems to require a different explanation because our enzyme assays suggested that its pathway was more redox balanced due to its NADH-utilizing XR.
Expression of genes related to xylose metabolism is highly affected by oxygen
To determine whether oxygen levels (SF compared to BF) affected gene expression in a redox-independent way that could impact xylose-to-ethanol flux, we sequenced mRNA by taking samples in mid-log phase from the same species used in the enzyme assays. After analyzing the data and filtering the results with specific thresholds (see Methods), we identified 2263, 2546, 2111, and 741 differentially expressed genes (DEGs) for Sp. passalidarum, Sc. xylosifermentans, Sc. coipomoensis, and Sc. amazonensis, respectively. This differential expression analysis showed that xylose metabolism was highly affected by oxygenation. The genes encoding the first three steps of the xylose catabolism pathway were upregulated in SF (relative to BF) for all species, except for Sc. coipomoensis XYL2, which was not differentially expressed. For Sp. passalidarum, XYL1.1 and XYL2.2 were downregulated in SF compared to BF, but XYL1.2 (the homolog that encodes the XR with NADH affinity) and XYL2.1 were upregulated. Interestingly, XYL2 was among the 15 most upregulated genes in SF in Sc. xylosifermentans.
Figure 4 shows the DEGs in key pathways from the comparison of SF and BF. Genes related to the non-oxidative shunt of the PPP, such as TKL1 and TAL1, were also upregulated in SF in ethanol producers under moderate aeration, but there were no DEGs for xylitol producers related to those genes. From the oxidative portion of the PPP, which is not essential for xylose catabolism, some genes, such as SOL1, GND1, and RPE1, were downregulated in Sc. coipomoensis. Outside of the PPP, genes that include TPI1, PDC1, PGK1, GPM1, and ADH1 were also upregulated in SF only in ethanol producers. ADH1 and ADH2 are two genes related to ethanol production and consumption in Sc. stipitis [37, 38]. While ADH1 was differentially expressed only in ethanol producers, ADH2 was also upregulated in xylitol producers and was the highest DEG in Sc. xylosifermentans. Although the ethanol producers upregulated key genes related to xylose metabolism in SF, the genes that were upregulated or downregulated in the four species were enriched in biological processes, such as metabolic and carbohydrate metabolic processes (Fig. 5). These biological processes, along with electron transport, likely have important roles that affect the observed phenotypes, suggesting that relevant pathways and genes related to metabolism were highly affected in the conditions tested.

Several genes related to xylose fermentation were upregulated only for ethanol producers at lower oxygenation. Xylose metabolism and related pathways. Dashed lines correspond to the < -1 and > 1 confidence intervals for the log2-fold change from RNA-Seq analysis (moderate aeration/high aeration). Asterisks indicate genes that did not exhibit differential expression. Positive and negative values indicate upregulated and downregulated genes, respectively, and the colors are associated with the species as shown in the right side of the figure. Protein products encoded by each gene: HXK—hexokinase; PGI1—phosphoglucose isomerase; PFK—phosphofructokinase; FBA1—fructose 1,6-bisphosphate aldolase; TPI1—triose phosphate isomerase; TDH—glyceraldehyde-3-phosphate dehydrogenase; PGK1—3-phosphoglycerate kinase; GPM1—phosphoglycerate mutase; ENO—enolase; PYK1—pyruvate kinase; PDC1—pyruvate decarboxylase, ADH1/2—alcohol dehydrogenase; ALD6—aldehyde dehydrogenase; GPD1—glycerol-3-phosphate dehydrogenase; GPP1—glycerol-3-phosphate phosphatase; ZWF1—glucose-6-phosphate dehydrogenase; SOL—6-phosphogluconolactonase; GND1—6-phosphogluconate dehydrogenase; RKI1—ribose-5-phosphate ketol-isomerase; RPE1—d-ribulose-5-phosphate 3-epimerase; TKL1—transketolase; TAL1—transaldolase; XYL1—xylose reductase; XYL2—xylitol dehydrogenase, and XYL3—xylulokinase. Created with BioRender.com [75] with license number EN2651FNZW

Metabolic and carbohydrate metabolic processes are highly affected by oxygenation. Enrichment analysis for Sc. xylosifermentans, Sc. coipomoensis, Sc. amazonensis, and Sp. passalidarum. On the right are the biological processes enriched in the upregulated gene list, while those the left side are enriched in the downregulated gene list (moderate aeration/high aeration)
XYL genes from Scheffersomyces xylosifermentans enhance anaerobic xylose fermentation by Saccharomyces cerevisiae
Our phenotypic results showed that Sc. xylosifermentans rapidly fermented xylose to ethanol, while Sc. coipomoensis accumulated high titers of xylitol. To test the possibility that the XR and XDH enzymes from Sc. xylosifermentans would enable greater xylose fermentation than those of Sc. coipomoensis enzymes, we used CRISPR-Cas9 to re-engineer a strain of S. cerevisiae that expresses Sc. stipitis XYL1, XYL2, and XYL3. SstipitisXYL1 and SstipitisXYL2 were replaced with the corresponding genes from Sc. xylosifermentans (SxylosiXYL1 and SxylosiXYL2) and Sc. coipomoensis (ScoipXYL1 and ScoipXYL2). We hypothesized that the modified S. cerevisiae would grow faster anaerobically with Sc. xylosifermentans XYL1 because this XR enzyme lacked a cofactor preference (Fig. 3). In contrast, this would not be expected for the strain with XYL1 from Sc. coipomoensis or for the original parental strain containing SstipitisXYL1. Indeed, the strain expressing SxylosiXYL1 and SxylosiXYL1/XYL2 grew rapidly under anaerobic conditions compared to the strain SstipitisXYL1/XYL2/XYL3 (P < 0.05), while the strains expressing ScoipXYL1 alone or ScoipXYL1/XYL2 presented an inferior growth profile (P < 0.05) to the strain containing SstipitisXYL1/XYL2/XYL3 (Fig. 6A). Sc. stipitis showed modest anaerobic growth with very limited production of ethanol or accumulation of intermediates (Additional file 3). This result is interesting because the Sc. stipitis XR can use both NADH and NADPH, but it has a preference for the latter [39]. Surprisingly, Sc. xylosifermentans grew anaerobically and produced ethanol, but it did not excrete xylitol (Fig. 6A–D). Along with the other species and engineered strains, it also did not anaerobically produce glycerol, which is produced by S. cerevisiae under anoxic conditions to alleviate the cytosolic redox balance [40]. Even though the replacement of Sc. stipitis XYL genes with SxylosiXYL1/XYL2 improved xylose fermentation to ethanol by S. cerevisiae and enabled anaerobic growth, the mutants still excreted xylitol (Fig. 6D). This result contrasts what we observed natively with Sc. xylosifermentans, which did not excrete xylitol in anoxic conditions.

Saccharomyces cerevisiae with XYL genes from Scheffersomyces xylosifermentans ferments xylose anaerobically. Growth curves of Sc. xylosifermentans, Sc. coipomoensis, and Sc. cerevisiae strains carrying different XYL genes under anoxic conditions A OD, B xylose consumption, C ethanol production, and D xylitol accumulation. Error bars indicate the standard deviation from the three biological replicates. Asterisks denote significant differences related to S. cerevisiae + SstipitisXYL1/XYL2/XYL3). SxylosiXYL1 and SxylosiXYL1/XYL2—XYL genes from Sc. xylosifermentans; ScoipXYL1 and ScoipXYL1/XYL2—XYL genes from Sc. coipomoensis; and SstipitisXYL1/XYL2/XYL3—XYL genes from Sc. stipitis
Despite employing ATP, instead of NAD(P)H, as a cofactor for the conversion of xylulose to xylulose-5-P, the xylulokinase enzyme encoded by XYL3 might contribute to xylitol accumulation due to the reversible nature of the previous reaction that generates xylulose from xylitol [41] (Fig. 4). We hypothesized that XYL3 could also affect xylitol accumulation, so we also engineered a strain of S. cerevisiae that expressed Sc. xylosifermentans XYL3. Although this strain still accumulated xylitol, ethanol production, xylose consumption, and growth by S. cerevisiae with SxylosiXYL1/XYL2/XYL3 were generally slightly improved compared to the strains carrying SxylosiXYL1 and SxylosiXYL1/XYL2 (P < 0.05), which suggests that this gene is also more active than Sc. stipitis XYL3 (Additional file 3).
Finally, we confirmed that the XR and XDH enzymes maintained their predicted co-factor preferences when expressed in S. cerevisiae by performing enzymatic assays using whole-cell lysates from the S. cerevisiae strains engineered with SxylosiXYL1/SxylosiXYL2 or ScoipXYL1/ScoipXYL2. Although the activities were lower compared to the native activities of XR in the donor species, XR encoded by SxylosiXYL1 in strain GLBRCY1847 (NADH: 1.97 ± 0.01 U mg−1; NADPH: 2.15 ± 0.15 U mg−1 P > 0.05) still had fairly equal preferences between cofactors, while the XR encoded by ScoipXYL1 in GLBRCY1850 showed a preference for NADPH (NADH: 0.63 ± 0.05 U mg−1; NADPH: 1.18 ± 0.03 U mg−1, P < 0.05). Xylitol dehydrogenase activity was high for NAD+ in both GLBRCY1847 (6.69 ± 0.29 U mg−1) and GLBRCY1850 (4.24 ± 0.68 U mg−1), and no activity was detected with NADP+.
Discussion
To better understand the abilities of yeasts from the order Serinales to metabolize xylose, we quantified the metabolites produced by several species cultured in rich medium with xylose as the primary carbon source under high and moderate aeration conditions. Ethanol producers and xylitol producers were both identified. Most of the yeasts tested fermented xylose, but fewer than half primarily produced ethanol under moderate aeration. Instead, most yeasts mainly produced xylitol in the conditions tested. Xylitol, an intermediate of xylose catabolism, is excreted by several species when a bottleneck in oxidation to xylulose occurs. Accumulation of xylitol in the extracellular medium was believed to occur due to the different XR and XDH cofactor requirements, which leads to cofactor imbalance under oxygen-limited (including moderate aeration) or anoxic conditions [15, 42,43,44]. Under these conditions, NADH accumulates, and NAD+ levels fall. Here, we unexpectedly observed that several species accumulated xylitol when sufficient oxygen was available, including species that were among the best ethanol producers in moderate oxygen-limiting conditions (see Figs. 1 and 2, Additional file 1). This result strongly suggests that the different cofactor requirements by XR and XDH are not the sole issue limiting xylose fermentation.
An important question raised by our fermentation experiments was why the top xylose-fermenting species, Sc. xylosifermentans, showed similar or better xylose fermentation than that of Sp. passalidarum in moderate aeration, even though the former has a single homolog of XYL1, while the latter has two homologs. Enzymatic assays were conducted to determine the activities and cofactor preferences of XR and XDH in each condition for key species. Assuming that redox imbalance primarily impacted xylose fermentation, our initial hypothesis was that the enzyme XR from species with high metabolic activity might present the same features as the XR encoded by XYL1.2 from Sp. passalidarum. In contrast, our findings revealed that the enzymes of all the four species could use both NADH and NADPH as cofactors (Fig. 3A), which was previously reported for Sc. stipitis [39]. The Sc. coipomoensis XR showed significant activity with NADH under moderate and high aeration conditions, but it still had a preference for NADPH. An NADPH-dependent XR was also present in Sc. amazonensis, a species that also accumulated xylitol. Surprisingly, the XR of Sc. xylosifermentans did not have a cofactor preference. In this species, the lack of cofactor preference seems to naturally enhance flux for the second reaction of the xylose metabolism due to the oxidation of NADH to NAD+, which is the cofactor used in the preceding step. Our results are consistent with a previous observation in which high aeration caused a switch in cofactor usage from NADH to NADPH for XR from Sp. passalidarum NRRL Y-27907 [23], and our transcriptome data showed that this switch likely occurs because the two paralogous XYL1 genes have different levels of expression under moderate and high aeration conditions (Fig. 6). A similar result was reported by Cadete et al. [15], in which XYL1.2 was 15–16-fold more highly expressed than XYL1.1 in more extreme oxygen-limiting conditions compared to moderate aeration condition. Collectively, these results suggest that XYL1.2 expression increases as the availability of oxygen decreases. Thus, Sp. passalidarum may use its second homolog of XYL1, which encodes the NADH-preferring XR, to limit redox imbalance in low oxygen levels. Rapid consumption of xylose and high yields of ethanol were not observed for other species that harbor multiple paralogs, such as C. intermedia, which was reported to have a NADH-dependent XR [19]. Thus, Sp. passalidarum is unique among the species tested.
To elucidate the effect of oxygen on the expression profile of genes involved in the first steps of xylose metabolism and the PPP, we determined their relative gene expression in Sc. amazonensis, Sc. coipomoensis, Sc. xylosifermentans, and Sp. passalidarum under moderate and high aeration conditions. The genes directly involved in xylose metabolism (XYL1, XYL2, and XYL3) were all upregulated with moderate aeration (Fig. 4) for Sc. xylosifermentans, which is consistent with the higher XR and XDH activities observed in in vitro enzyme assays (Fig. 3). Indeed, Sc. xylosifermentans XYL2 was among the 15 most upregulated genes under oxygen limitation, the condition in which this species presented high levels of xylose consumption. Xylitol to xylulose conversion by XDH enzymes has been proposed to be the limiting step for xylose fermentation, and high expression and codon optimization of XYL2 may be necessary for efficient xylose conversion [16, 45, 46]. Even small expression changes may be associated with large phenotypic effects [47] and, in the case of Sc. xylosifermentans, XYL2 expression decreased significantly under high aeration. The xylitol accumulation by this species in this condition is likely in part due to this reduction in XYL2 expression, rather than the cofactor imbalances postulated for some other species.
Based on what is known about S. cerevisiae metabolism, the production of glycerol by Sp. passalidarum in high aeration is difficult to explain, especially considering the large amount it produced under high aeration. Glycerol can maintain redox balance in the absence of oxygen when ethanol flux is overloaded in S. cerevisiae [40]. Strategies to increase glycerol production by yeasts include cutting off or attenuating ethanol production, shifting the NAD+/NADH ratio to increase the amount of NADH available, and overexpression of the glycerol 3-phosphate dehydrogenase encoded by GPD1 [48,49,50]. The downregulation of PDC1 and ADH1, which encode pyruvate decarboxylase and alcohol dehydrogenase, respectively, in Sp. passalidarum and the upregulation of GPD1 and GPP1 (encoding glycerol-3-phosphate phosphatase) under high aeration suggest a mechanism for the elevated glycerol production by this species in this condition.
Genes from the PPP, such as TKL1, TAL1, and RKI1, were also upregulated under moderate oxygen-limited condition for Sc. xylosifermentans (TKL1 and TAL1) and Sp. passalidarum (TKL1 and RKI1). Overexpression of those genes in engineered strains of S. cerevisiae improved the growth rate on xylose and the ethanol yield [51, 52]. ADH1 and ADH2 are responsible for ethanol production and consumption in Sc. stipitis, and when the availability of oxygen becomes limited, the expression of ADH genes, especially ADH2, increases [37, 53]. While ADH1 was upregulated in the ethanol-producing yeasts under moderate aeration, ADH2 was the most upregulated gene for Sc. xylosifermentans; ADH2 was also differentially expressed in Sc. coipomoensis and Sc. amazonensis, but not Sp. passalidarum (Fig. 4). In addition to the genes required for xylose metabolism, the high expression of the PPP and ethanol pathway in Sp. passalidarum and Sc. xylosifermentans might be related to the greater xylose metabolism by these species under moderate aeration.
Next, we genetically modified S. cerevisiae by the chromosomal integration of XYL genes from Sc. xylosifermentans and Sc. coipomoensis. Given the inability of S. cerevisiae to metabolize xylose, any observed growth must be attributed to the heterologously and constitutively expressed enzymes, which allowed us to investigate how redox balance influences anaerobic fermentation of xylose into ethanol in this model system. With the rest of the XYL pathway coming from Sc. stipitis, S. cerevisiae could grow in anaerobic conditions with SxylosiXYL1 and SxylosiXYL1/XYL2, but not with ScoipXYL1, ScoipXYL1/XYL2, or the Sc. stipitis pathway alone (Fig. 6). This result suggests that cofactor preference has important effects in anoxic conditions, at least when the pathway is constitutively expressed (Fig. 6). S. cerevisiae strains containing SxylosiXYL1 and SxylosiXYL1/XYL2 produced ethanol, but they also accumulated xylitol. Swapping SstipitisXYL3 with SxylosiXYL3 further enhanced ethanol production but only slightly relieved xylitol accumulation, so we hypothesize that other downstream and interacting native metabolic pathways in Sc. xylosifermentans may facilitate its xylitol-to-ethanol flux. Nonetheless, this experiment was essential to test the redox balance hypothesis, and it unexpectedly revealed that Sc. xylosifermentans was able to grow under anaerobic conditions, which are used in numerous industrial applications.
Conclusions
We tested 30 species belonging to the order Serinales for xylose fermentation with a focus on the genera Spathaspora and Scheffersomyces and their close relatives. Collectively, our data reveal that xylose metabolism in the Serinales is highly plastic and oxygen-dependent. Under high aeration conditions, several species switched from ethanol production to xylitol accumulation. This switch was generally accompanied by a decrease in enzyme activity and expression of genes related to xylose catabolism. While we find a global effect of oxygen availability on xylose metabolism, our data support the hypothesis that xylitol accumulation results from redox imbalance generated by differential cofactor preferences for XR and XDH in some species, but they also point to a novel role for oxygen-responsive gene regulation in other species that accumulate xylitol under high aeration, especially Sc. xylosifermentans. Among the species tested, Sc. xylosifermentans is also remarkable for its high xylose consumption and ethanol formation under moderate aeration and even in anaerobic conditions, a phenomenon not previously noted for any xylose-fermenting yeast species. This species may be a novel source of potential genes that can be expressed in industrial microbes, such as S. cerevisiae, for biofuel production from lignocellulosic feedstocks. Alternatively, Sc. xylosifermentans could be subjected to adaptive laboratory evolution or genetic modification to enhance its native potential and transform it into an industrial organism.
Methods
Yeast strains and growth experiment conditions
Candida blattae, C. intermedia, Me. caribbica, Me. carpophila, Me. guilliermondii, and 24 strains of Scheffersomyces and Spathaspora species (see Additional file 4) were obtained from the USDA Agricultural Research Service (ARS) NRRL Culture Collection in Peoria, Illinois, USA; Collection of Microorganisms and Cells of Universidade Federal de Minas Gerais, Belo Horizonte, Minas Gerais, Brazil; and CBS Yeast Collection of the Westerdijk Fungal Biodiversity Institute, Utrecht, the Netherlands.
Saccharomyces cerevisiae strain GLBRCY38 was generated from a haploid spore from the diploid GLBRCY2A strain [54] containing an integrated DNA cassette to express constitutively Sc. stipitis XYL1, XYL2, and XYL3 genes from the HO locus [18]. GLBRCY2A was sporulated, and individual tetrads were dissected as previously described [55]. Haploid spores were verified for the XYL cassette by PCR and selection for the kanMX resistance marker. One spore containing the XYL cassette, GLBRCY25A, was selected, and all XYL gene sequences were fully confirmed for accuracy by PCR and Sanger sequencing. The kanMX marker was excised [56] to generate the GLBRCY38 strain. We used CRISPR-Cas9 to swap the genes XYL1, XYL2, and/or XYL3 to the corresponding genes from Sc. xylosifermentans and Sc. coipomoensis in the strain GLBRCY38 with gRNA expression plasmid (pXIPHOS) [57, 58] targeting XYL1 (CAACAGCCAAAACCCACGGC), XYL2 (CTTAACCAAGAAATCTTCGG), and/or XYL3 (TGCCTCCCCACAACCCGAGG). sgRNAs were designed using CRISpy-pop [59]. Multiple swaps were performed sequentially. Transformation of yeast strains was done using the lithium acetate/PEG-4000/carrier DNA method [60]. After the transformation and recovery, colonies were selected by plating them on YPD agar with 100 mg/L nourseothricin (NAT). All strains were confirmed for gene swaps and antibiotic marker excision by PCR with gene-specific primers or flanking primers. Sanger sequencing of purified PCR products was performed by the University of Wisconsin-Madison Biotechnology Center. The engineered strains used in this study are summarized in Additional file 5.
Strains from all yeast species were initially plated from freezer stocks on 10 g/L yeast extract, 20 g/L peptone, and 2% dextrose (YPD) plates and grown for single colonies. Single colonies of each strain were pre-cultured in 10 mL of YPX medium (1% yeast extract, 2% peptone, 2% xylose) overnight at 30 °C under 200 rpm. Cells were recovered by centrifugation at 2600g for 10 min, washed with sterile water, and suspended in the fermentation medium at 0.5 g dry cell weight L−1 of cell concentration. To evaluate the performance of the yeasts in different aeration conditions, we used 125-mL shake flasks (SF) for moderate aeration and 250-mL baffled flasks (BF) for high aeration, both containing 50 mL of YPX (1% yeast extract, 2% peptone, and 5% xylose). The dissolved oxygen available was measured by a dissolved oxygen meter (Mettler Toledo F4-Field FiveGo, USA). The flasks were incubated at 30 °C under 200 rpm for 72 h. Cell growth was determined by collecting 1 mL of the culture. The cells were recovered by centrifugation and dried in a speed vacuum concentrator. The ethanol, xylitol, and biomass yields (Yp/set, Yp/sxy, Yx/s, g g−1), volumetric productivity of ethanol and xylitol (Qpet, Qpxy, g L−1 h−1), and consumption of d-xylose were determined as described previously by Cadete et al. [15].
Growth experiments using engineered strains were carried out with 125 mL baffled flasks in an anaerobic chamber for 142 h. Sc. xylosifermentans, Sc. coipomoensis, and Sc. stipitis were also tested under this condition. The pre-culture was done exactly as described above, but for inoculation, we shifted into flasks containing 30 mL YPX media (2% xylose) at a concentration of optical density at λ = 600 nm (OD600) = 0.3. Seven-hundred microliters of sample were taken during the experiment for measuring the OD600 and for quantifying end-products using high-performance liquid chromatography (HPLC) and refractive index detection (RID) as described previously [61]. Plots were constructed in R v3.6.3 using the RStudio v1.3.1073 platform. We performed Student’s t tests to determine whether there were significant differences (P < 0.05) between the parental strain and engineered strains.
Enzyme activities
Yeast species Sc. xylosifermentans, Sc. coipomoensis, and Sc. amazonensis were grown in YPX medium as described above or in YPD for the negative control, using both SF and BF. Engineered strains Y1847 and Y1850 were also tested, but they were grown in 125-mL SF with 30 mL YPX (2% xylose) in aerobic conditions. Cells were harvested at mid-log growth phase, washed with cold sterile water, and extracted with Y-PER® Yeast Protein Extraction Reagent (Thermo Fisher). Protein concentrations from the crude cell extracts were determined by BCA Protein Assay Kit (Thermo Fisher). XR activities were obtained from 250-µL reactions containing 100 mM triethanolamine buffer pH 8, 0.2 mM NADPH or NADH, 0.2 M d-xylose, crude cell extract, and deionized water; XDH activities were obtained from 250 µL reactions containing 100 mM glycine buffer pH 9, 50 mM MgCl2, 3 mM NADP+ or NADP+, 0.2 M xylitol, crude cell extract, and deionized water [15]. Enzyme activities were determined by oxidation or reduction of NADH/NADPH or NAD+/NADP+, respectively. Reaction mixtures aliquoted into 96-well microtiter plates (Corning® 96 Well Clear Flat Bottom UV-Transparent, Darmstadt, Germany) were placed in Tecan® (Infinite M-1000, Switzerland) at 25ºC for measuring absorbance at 340 nm for 1 h. Standard curves of NADPH and NADH were used to calculate the concentration of the samples. Extracted proteins from the yeast Sp. passalidarum were used as positive control, while S. cerevisiae 288SC was used as a negative control. In addition, blank measurements with samples lacking either cell lysate or xylose substrate were performed for each sample, and the resulting values were subtracted from the test values. Enzyme activities were determined from three independent biological replicates. The specific activity of each enzyme was estimated by the number of enzyme units per mL divided by the concentration of protein in mg/mL. One unit was defined as the generation of 1 μmol NAD(P)H or NAD(P)+ per min. We performed paired Student’s t tests to determine whether there were significant differences (P < 0.05) between NADH and NADPH usage by XR from each species. Data analyses and plots were performed in R v3.6.3 using the RStudio v1.3.1073 platform.
Genome extraction, sequencing, assembly, and annotation
Genomic DNA (gDNA) of the species Sc. xylosifermentans, Sc. amazonensis, and Sc. coipomoensis was isolated using a modified phenol:chloroform method [62]. The sequencing was performed at the DOE Joint Genome Institute Standard. Genome sequencing was performed using Pacific Biosciences (PacBio) Multiplexed > 10 kb with Blue Pippin Size Selection (AMPure Beads for Sc. coipomoensis). Filtered subread data were processed with the JGI quality control pipeline to remove artifacts. The mitochondrial genome was assembled separately with the circular consensus sequencing (CCS) reads and polished with two rounds of RACON version 1.4.13 [63]. The mitochondria-filtered CCS reads were then assembled with Flye version 2.8.1 [64] to generate an assembly and polished with two rounds of RACON version 1.4.13. The Sc. coipomoensis nuclear genome was assembled using Falcon v. 1.8.8 [65], improved with FinisherSC, and polished with Arrow version SMRTLink v5.0.1.9578 [66]. Ribosomal DNA (rDNA) was assembled separately from a subset of CCS reads identified using kmer matching against the UNITE database with BBTools (http://sourceforge.net/projects/bbmap) version 38.79. Matching reads were subsampled to 600,000 bp with BBTools, assembled with Flye version 2.8.1, and polished with two rounds of RACON version 1.4.13. The eukaryotic internal transcribed spacer (ITS) was identified and extracted from the rDNA assembly using ITSx (Bengtsson-Palme et al., 2013). Results were used to orient and trim the rDNA contig to 100 bp SSU–1 Kb LSU. Contigs less than 1000 bp were excluded. Completeness of the euchromatic portion of the genome assembly was assessed by aligning assembled consensus RNA sequence data with BBTools. Genomes were then annotated using the JGI annotation pipeline [67] (Additional file 6).
RNA extraction, sequencing, and data analysis
Cells from Sp. passalidarum, Sc. xylosifermentans, Sc. coipomoensis, and Sc. amazonensis were grown on YPD agar for 48 h to single colonies, which were each inoculated into 10 mL of YPX medium overnight in a 50-mL glass tube at 30 °C. After pre-culture, aliquots were transferred to 125 mL SF and 250 mL BF, both flasks with 50 mL of YPX, at an initial of inoculum of 0.5 g L−1, and incubated at 30 °C under 200 rpm until mid-log phase. RNA was extracted using the acid phenol protocol [68]. Briefly, the total volume of the cultures was centrifuged with 5% phenol and 95% EtOH, and it was flash-frozen in a dry ice-ethanol bath. Cells were resuspended in TES lysis buffer (10 mM Tris, 10 mM EDTA, 0.5% SDS in water) plus PVP40, acid-washed beads (Sigma #G8772), and one volume of saturated acid phenol (IBI Scientific). Lysates were incubated at 65 °C for 1 h with vortexing every 15 min. Then, the lysates were extracted with 1 volume of acid phenol each and once with 1 volume of chloroform. The aqueous phase of the final chloroform extraction was removed and added to a solution consisting of 2.5 volumes of 95%–100% ethanol and 0.1 volumes of 3 M sodium acetate, and the tubes were placed at − 80 °C overnight to precipitate the RNA. RNA pellets were then collected by centrifugation, washed twice in 70% ethanol each, and resuspended in RNase-free water. Purified RNA was then treated with DNase I (NEB #EN0521) to remove any residual DNA prior to treatment with the RNA Clean & Concentrator kit (Qiagen #74134). Total RNA yields were quantified with the Qubit RNA Assay Kit (Thermo Fisher).
mRNA library preparation, quantification, and sequencing were performed at the DOE Joint Genome Institute. Paired-end libraries were sequenced on Illumina NovaSeq S4 (2 × 151). Raw FASTQ file reads were trimmed to remove adapters and artifact sequences using Trimmomatic version 0.30 [69]. The filtered reads of each species were aligned to their respective reference genome with Bowtie2 version 2.4.5 [70] with average mapping rates of 99.7, 98.8, 98.8, and 98.7% for Sp. passalidarum, Sc. xylosifermentans, Sc. coipomoensis, and Sc. amazonensis, respectively. featureCounts [71] was used to generate the raw gene counts. Raw sequencing reads were normalized using the reads per kilobase per million mapped reads (RPKM). DEseq2 version 1.38.1 [72] was used to perform quality control analysis and identify significantly differentially expressed genes (DEGs) from pairwise analyses with the raw counts; we used a Benjamini–Hochberg false discovery rate (FDR) of less than 0.05 as a significance threshold and log2-fold change > 1 or < − 1 for differentiating between upregulated and downregulated genes, respectively. The enrichment analysis was done by mapping the gene ontology (GO) terms with the gene identifiers, followed by Fisher’s exact and Benjamini-Hochberg-corrected tests using a threshold of 0.05 with SciPy version 1.9.0 [73] and Python Statsmodels version 0.10.2 [74]. The code is available at https://github.com/kathbarros/goea.
For genome annotation, Sc. xylosifermentans, Sc. amazonensis, and Sc. coipomoensis filtered RNASeq reads were also assembled de-novo with Trinity v.2.3.2 using –normalize_reads and –jaccard_clip options [71].
Availability of data and materials
Supporting data are available in additional files. Raw RNA-Seq reads of Sc. xylosifermentans CBS 12540T, Sc. coipomoensis NRRL Y-17651T, and Sc. amazonensis UFMG-HMD-26.3T are available on the JGI portal http://genome.jgi.doe.gov and have been deposited to NCBI’s SRA under the BioProject accessions (PRJNA1015965, PRJNA455445, and PRJNA1015964, respectively). The reference genome of Sp. passalidarum NRRL Y-27907T is publicly available (Wohlbach et al., 2011). The genome assemblies and annotations of Sc. xylosifermentans, Sc. amazonensis, and Sc. coipomoensis are available from the JGI fungal genome portal MycoCosm [67] (https:/mycocosm.jgi.doe.gov) and have been deposited at DDBJ/EMBL/GenBank under the BioProject accessions PRJNA1015837, PRJNA1015963, and PRJNA460959, respectively. RNA-Seq enrichment analysis code is available at https://github.com/kathbarros/goea.
References
Liu Y, Cruz-Morales P, Zargar A, Belcher MS, Pang B, Englund E, Dan Q, Yin K, Keasling J. Biofuels for a sustainable future. Cell. 2021;184(6):1636–47.
Zhao Z, Xian M, Liu M, Zhao G. Biochemical routes for uptake and conversion of xylose by microorganisms. Biotechnol Biofuels. 2020;13:1–2.
Parapouli M, Vasileiadis A, Afendra AS, Hatziloukas E. Saccharomyces cerevisiae and its industrial applications. AIMS Microbiology. 2020;6:1–31.
Hittinger CT. Saccharomyces diversity and evolution: a budding model genus. Trends Genet. 2013;29(5):309–17.
Lee SB, Tremaine M, Place M, Liu L, Pier A, Krause DJ, Xie D, Zhang Y, Landick R, Gasch AP, Hittinger CT, Sato TK. Crabtree/Warburg-like aerobic xylose fermentation by engineered Saccharomyces cerevisiae. Metab Eng. 2021;68:1096–7176.
Osiro KO, Borgström C, Brink DP, Fjölnisdóttir BL, Grauslund MFG. Exploring the xylose paradox in Saccharomyces cerevisiae through in vivo sugar signalomics of targeted deletants. Microb Cell Fact. 2019;18:1–19.
Selim KA, Easa SM, El-Diwany AI. The xylose metabolizing yeast Spathaspora passalidarum is a promising genetic treasure for improving bioethanol production. Fermentation. 2020;6(1):33.
Groenewald M, Hittinger CT, Bensch K, Opulente DA, Shen XX, Li Y, Liu C, et al. A genome-informed higher rank classification of the biotechnologically important fungal subphylum Saccharomycotina. Stud Mycol. 2023;105(1):1–22.
Shen XX, Opulente DA, Kominek J, Zhou X, Steenwyk JL, Buh KV, et al. Tempo and mode of genome evolution in the budding yeast subphylum. Cell. 2018;175(6):1533-1545.e20.
Barros KO, Alvarenga FBM, Magni G, Souza GFL, Abegg MA, Palladino F, Silva SS, et al. The Brazilian Amazonian rainforest harbors a high diversity of yeasts associated with rotting wood, including many candidates for new yeast species. Yeast. 2022;40(2):84–101.
Cadete RM, Rosa CA. The yeasts of the genus Spathaspora: potential candidates for second-generation biofuel production. Yeast. 2018;35(2):191–9.
Urbina H, Blackwell M. Multilocus phylogenetic study of the Scheffersomyces yeast clade and characterization of the N-terminal region of xylose reductase gene. PLoS ONE. 2012;7(6): e39128.
Kurtzman CP, Suzuki M. Phylogenetic analysis of ascomycete yeasts that form coenzyme Q-9 and the proposal of the new genera Babjeviella, Meyerozyma, Millerozyma, Priceomyces, and Scheffersomyces. Mycoscience. 2010;51(1):2–14.
Nguyen NH, Suh SO, Marshall CJ, Blackwell M. Morphological and ecological similarities: wood-boring beetles associated with novel xylose-fermenting yeasts, Spathaspora passalidarum gen. sp. Nov. and Candida jeffriesii sp. nov. Mycol Res. 2006;110(10):1232–41.
Cadete RM, de Heras AM, Sandström AG, Ferreira C, Gírio F, Gorwa-Grauslund MF, Rosa CA, Fonseca C. Exploring xylose metabolism in Spathaspora species: XYL12 from Spathaspora passalidarum as the key for efficient anaerobic xylose fermentation in metabolic engineered Saccharomyces cerevisiae. Biotechnol Biofuels. 2016;9(1):1–4.
Jeffries TW, Jin YS. Metabolic engineering for improved fermentation of pentoses by yeasts. Appl Microbiol Biotechnol. 2004;63:495–509.
Jeffries TW, Grigoriev IV, Grimwood J, Laplaza JM, Aerts A, Salamov A, et al. Genome sequence of the lignocellulose-bioconverting and xylose-fermenting yeast Pichia stipitis. Nat Biotechnol. 2007;25(3):319–26.
Wohlbach DJ, Kuo A, Sato TK, Potts KM, Salamov AA, LaButti KM, et al. Comparative genomics of xylose-fermenting fungi for enhanced biofuel production. Proc Natl Acad Sci. 2011;108(32):13212–7.
Geijer C, Faria-Oliveira F, Moreno AD, Stenberg S, Mazurkewich S, Olsson L. Genomic and transcriptomic analysis of Candida intermedia reveals the genetic determinants for its xylose-converting capacity. Biotechnol Biofuels. 2020;13:1–5.
Jeppsson M, Bengtsson O, Franke K, Lee H, Hahn-Hägerdal B, Gorwa-Grauslund MF. The expression of a Pichia stipitis xylose reductase mutant with higher KM for NADPH increases ethanol production from xylose in recombinant Saccharomyces cerevisiae. Biotechnol Bioeng. 2006;93(4):665–73.
Leitgeb S, Petschacher B, Wilson DK, Nidetzky B. Fine tuning of coenzyme specificity in family 2 aldo-keto reductases revealed by crystal structures of the Lys-274 → Arg mutant of Candida tenuis xylose reductase (AKR2B5) bound to NAD+ and NADP+. FEBS Lett. 2005;579(3):763–7.
Kim SR, Kwee NR, Kim H, Jin YS. Feasibility of xylose fermentation by engineered Saccharomyces cerevisiae overexpressing endogenous aldose reductase (GRE3), xylitol dehydrogenase (XYL2), and xylulokinase (XYL3) from Scheffersomyces stipitis. FEMS Yeast Res. 2013;13(3):312–21.
Bonan CIDG, Biazi LE, Dionísio SR, Soares LB, Tramontina R, Sousa AS, et al. Redox potential as a key parameter for monitoring and optimization of xylose fermentation with yeast Spathaspora passalidarum under limited-oxygen conditions. Bioprocess Biosyst Eng. 2020;43(8):1509–19.
Hou X. Anaerobic xylose fermentation by Spathaspora passalidarum. Appl Microbiol Biotechnol. 2012;94:205–14.
Borelli G, Fiamenghi MB, Dos Santos LV, Carazzolle MF, Pereira GAG, José J, et al. Positive selection evidence in xylose-related genes suggests methylglyoxal reductase as a target for the improvement of yeasts’ fermentation in industry. Genome Biol Evol. 2019;11(7):1923–38.
Bengtsson O, Hahn-Hägerdal B, Gorwa-Grauslund MF. Xylose reductase from Pichia stipitis with altered coenzyme preference improves ethanolic xylose fermentation by recombinant Saccharomyces cerevisiae. Biotechnol Biofuels. 2009;2:1.
Kim SR, Park YC, Jin YS, Seo JH. Strain engineering of Saccharomyces cerevisiae for enhanced xylose metabolism. Biotechnol Adv. 2013;31(6):851–61.
Walfridsson M, Anderlund M, Bao X, Hahn-Hägerdal B. Expression of different levels of enzymes from the Pichia stipitis XYL1 and XYL2 genes in Saccharomyces cerevisiae and its effects on product formation during xylose utilisation. Appl Microbiol Biotechnol. 1997;48(2):218–24.
Johansson B, Hahn-Hägerdal B. The non-oxidative pentose phosphate pathway controls the fermentation rate of xylulose but not of xylose in Saccharomyces cerevisiae TMB3001. FEMS Yeast Res. 2002;2(3):277–82.
Nogueira Moysés D, Castelo Branco Reis V, Ricardo Moreira de Almeida J, Maria Pepe de Moraes L, Araripe Gonçalves Torres F. Xylose fermentation by Saccharomyces cerevisiae: challenges and prospects. Int J Mol Sci. 2016;17(3):207.
Cunha JT, Soares PO, Romaní A, Thevelein JM, Domingues L. Xylose fermentation efficiency of industrial Saccharomyces cerevisiae yeast with separate or combined xylose reductase/xylitol dehydrogenase and xylose isomerase pathways. Biotechnol Biofuels. 2019;12(1):1–14.
Brink DP, Borgström C, Persson VC, Osiro KO, Gorwa-Grauslund MF. D-xylose sensing in Saccharomyces cerevisiae: insights from D-glucose signaling and native D-xylose utilizers. Int J Mol Sci. 2021;22(22):12410.
Ndubuisi IA, Amadi CO, Nwagu TN, Murata Y, Ogbonna JC. Non-conventional yeast strains: unexploited resources for effective commercialization of second-generation bioethanol. Biotechnol Adv. 2023;17: 108100.
Guo J, Huang S, Chen Y, Guo X, Xiao D. Heterologous expression of Spathaspora passalidarum xylose reductase and xylitol dehydrogenase genes improved xylose fermentation ability of Aureobasidium pullulans. Microb Cell Fact. 2018;17(1):1–1.
Zhu Y, Zhang J, Zhu L, Jia Z, Li Q, Xiao W, et al. Minimize the xylitol production in Saccharomyces cerevisiae by balancing the xylose redox metabolic pathway. Front Bioeng Biotechnol. 2021;26(9): 639595.
Xie CY, Yang BX, Song QR, Xia ZY, Gou M, Tang YQ. Different transcriptional responses of haploid and diploid S. cerevisiae strains to changes in cofactor preference of XR. Microb Cell Factories. 2020;19(1):1–6.
Papini M, Nookaew I, Uhlén M, Nielsen J. Scheffersomyces stipitis: a comparative systems biology study with the Crabtree positive yeast Saccharomyces cerevisiae. Microb Cell Fact. 2012;11:1–6.
Cho JY, Jeffries TW. Pichia stipitis genes for alcohol dehydrogenase with fermentative and respiratory functions. Appl Environ Microbiol. 1998;64(4):1350–8.
Bruinenberg PM, De But PHM, Van Dijken JP, Scheffers WA. NADH-linked aldose reductase: the key to anaerobic alcoholic fermentation of xylose by yeasts. Appl Microbiol Biotechnol. 1984;19:256–60.
Jain VK, Divol B, Prior BA, Bauer FF. Elimination of glycerol and replacement with alternative products in ethanol fermentation by Saccharomyces cerevisiae. J Ind Microbiol Biotechnol. 2011;38(9):1427–35.
Veiga L. Polyol dehydrogenases in Candida albicans II. Xylitol oxidation to D-xylulose. J General Appl Microbiol. 1968;14(1):79–87.
Trichez D, Steindorff AS, Soares CEVF, Formighieri EF, Almeida JRM. Physiological and comparative genomic analysis of new isolated yeasts Spathaspora sp. JA1 and Meyerozyma caribbica JA9 reveal insights into xylitol production. FEMS Yeast Res. 2019;19(4):1–15.
Veras HCT, Parachin NS, Almeida JRM. Comparative assessment of fermentative capacity of different xylose-consuming yeasts. Microb Cell Fact. 2017;16(1):1–8.
Cadete RM, Melo-Cheab MA, Viana AL, Oliveira ES, Fonseca C, Rosa CA. The yeast Scheffersomyces amazonensis is an efficient xylitol producer. World J Microbiol Biotechnol. 2016;32:1–5.
Nalabothu RL, Fisher KJ, LaBella AL, Meyer TA, Opulente DA, Wolters JF, et al. Codon optimization improves the prediction of xylose metabolism from gene content in budding yeasts. Mol Biol Evol. 2023;40(6):msad111.
Kim SR, Ha SJ, Kong II, Jin YS. High expression of XYL2 coding for xylitol dehydrogenase is necessary for efficient xylose fermentation by engineered Saccharomyces cerevisiae. Metab Eng. 2012;14(4):336–43.
Lopes-Ramos CM, Chen CY, Kuijjer ML, Paulson JN, Sonawane AR, Fagny M, et al. Sex differences in gene expression and regulatory networks across 29 human tissues. Cell Rep. 2020;31:1–12.
Semkiv MV, Ruchala J, Dmytruk KV, Sibirny AA. 100 Years later, what is new in glycerol bioproduction? Trends Biotechnol. 2020;38(8):907–16.
Murashchenko L, Abbas C, Dmytruk K, Sibirny A, Sibirny A. Overexpression of the truncated version of ILV2 enhances glycerol production in Saccharomyces cerevisiae. Yeast. 2016;33(8):463–9.
Michnick S, Roustan JL, Remize F, Barre P, Dequin S. Modulation of glycerol and ethanol yields during alcoholic fermentation in Saccharomyces cerevisiae strains overexpressed or disrupted for GPD1 encoding glycerol 3-phosphate dehydrogenase. Yeast. 1997;13(9):783–93.
Kobayashi Y, Sahara T, Ohgiya S, Kamagata Y, Fujimori KE. Systematic optimization of gene expression of pentose phosphate pathway enhances ethanol production from a glucose/xylose mixed medium in a recombinant Saccharomyces cerevisiae. AMB Express. 2018;8:1–1.
Diao L, Liu Y, Qian F, Yang J, Jiang Y, Yang S. Construction of fast xylose-fermenting yeast based on industrial ethanol-producing diploid Saccharomyces cerevisiae by rational design and adaptive evolution. BMC Biotechnol. 2013;13(1):1–9.
Passoth V, Schäfer B, Liebel B, Weierstall T, Klinner U. Molecular cloning of alcohol dehydrogenase genes of the yeast Pichia stipitis and identification of the fermentative ADH. Yeast. 1998;14(14):1311–25.
Sato TK, Liu T, Parreiras LS, Williams DL, Wohlbach DJ, Bice BD, et al. Harnessing genetic diversity in Saccharomyces cerevisiae for fermentation of xylose in hydrolysates of alkaline hydrogen peroxide-pretreated biomass. Appl Environ Microbiol. 2014;80(2):540–54.
Parreiras LS, Breuer RJ, Narasimhan RA, Higbee AJ, La Reau A, Tremaine M, et al. Engineering and two-stage evolution of a lignocellulosic hydrolysate-tolerant Saccharomyces cerevisiae strain for anaerobic fermentation of xylose from afex pretreated corn stover. PLoS ONE. 2014;9(9): e107499.
Güldener U, Heck S, Fiedler T, Beinhauer J, Hegemann JH. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 1996;24(13):2519–24.
Higgins DA, Young MKM, Tremaine M, Sardi M, Fletcher JM, Agnew M, et al. Natural variation in the multidrug efflux pump SGE1 underlies ionic liquid tolerance in yeast. Genetics. 2018;210(1):219–34.
Kuang MC, Kominek J, Alexander WG, Cheng JF, Wrobel RL, Hittinger CT. Repeated cis-regulatory tuning of a metabolic bottleneck gene during evolution. Mol Biol Evol. 2018;35(8):1968–81.
Stoneman HR, Wrobel RL, Place M, Graham M, Krause DJ, De Chiara M, et al. CRISpy-Pop: a web tool for designing CRISPR/Cas9-driven genetic modifications in diverse populations. G3 Genes, Genomes, Genetics. 2020;10(11):4287–94.
Woods R, Gietz R. High-efficiency transformation of plasmid DNA into yeast. In: MacDonald PN, editor. Two-hybrid systems. Totowa: Humana Press Inc.; 2001. p. 85–98.
Schwalbach MS, Keating DH, Tremaine M, Marner WD, Zhang Y, Bothfeld W, et al. Complex physiology and compound stress responses during fermentation of alkali-pretreated corn stover hydrolysate by an Escherichia coli ethanologen. Appl Environ Microbiol. 2012;78(9):3442–57.
Moolhuijzen P, See PT, Moffat CS. A new PacBio genome sequence of an Australian Pyrenophora tritici-repentis race 1 isolate. BMC Res Notes. 2019;12:1–5.
Vaser R, Sović I, Nagarajan N, Šikić M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27(5):737–46.
Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37(5):540–6.
Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, et al. Nonhybrid, finished microbial genome assemblies from long-read smrt sequencing data. Nat Methods. 2013;10(6):563–9.
Lam KK, Labutti K, Khalak A, Tse D. FinisherSC: a repeat-aware tool for upgrading de novo assembly using long reads. Bioinformatics. 2015;31(19):3207–9.
Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42(D1):D699-704.
Chomczynski P, Sacchi N. Isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162(1):156–9.
Bushnell B. BBMap: a fast, accurate, splice-aware aligner. Berkeley: Lawrence Berkeley National Lab (LBNL); 2014.
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–15.
Liao Y, Smyth GK, Shi W. FeatureCounts: an efficient general-purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):1–21.
Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 10: fundamental algorithms for scientific computing in Python. Nat Methods. 2020;17(3):261–72.
Seabold S, Perktold J. Statsmodels: econometric and statistical modeling with Python. In: Proceedings of the 9th Python in Science Conference 2010 Jun 28 (Vol. 57, No. 61, pp. 10–25080).
Perkel JM. The software that powers scientific illustration. Nature. 2020;582(7810):137–9.
Acknowledgements
We thank Kevin Myers for assistance with bioinformatic analyses, Mick McGee and the GLBRC Metabolomics Facility for metabolite quantification, and John F. Wolters and Hittinger Lab and Sato Lab members for helpful advice. We thank Vivian Ng and Chris Daum and his team from DOE Joint Genome Institute for providing genome and transcriptome sequencing.
Funding
This material is based upon work supported in part by the Great Lakes Bioenergy Research Center, U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under Award Number DE-SC0018409. Research in the Hittinger Lab is further supported by the National Science Foundation under Grant No. DEB-2110403 and the USDA National Institute of Food and Agriculture (Hatch Projects 1020204 and 7005101). C.T.H. is an H. I. Romnes Faculty Fellow, supported by the Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation. The work (proposal: https://doi.org/10.46936/10.25585/60001116) conducted by the U.S. Department of Energy Joint Genome Institute (https://ror.org/04xm1d337), a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy operated under Contract No. DE-AC02-05CH11231. C.A.R. was funded by “INCT Yeasts: Biodiversity, preservation and biotechnological innovation,” funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brazil, grant #406564/2022-1, and CNPq process numbers 0457499/2014-1, 313088/2020-9, and 408733/2021; and Fundação do Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG, process numbers APQ-01525-14, APQ-03071-17, and APQ-02552-15).
Author information
Authors and Affiliations
Contributions
KOB, TKS, and CTH conceived of the project. KOB designed and performed growth experiments; enzyme assays; RNA extraction; and RNA sequencing analysis and wrote, reviewed, and edited the manuscript. KOB and MM genetically modified S. cerevisiae. KOB and DK extracted gDNA for PacBio genome sequencing. JP, BA, AL, SJM, and IVG performed genome and transcriptome sequencing, assemblies, and annotations. CAR contributed to study design by providing yeast species. TKS and CTH reviewed and edited the manuscript, and all the authors approved.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The Wisconsin Alumni Research Foundation has filed a provisional patent application based on the contents of this manuscript with KOB, TKS, and CTH as inventors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Xylitol and ethanol are the major bioproducts of xylose fermentation by Scheffersomyces and Spathaspora species. Rates and yields related to the consumption of d-xylose and production of biomass, ethanol, and xylitol under moderate (shake flask—SF) and high (baffled flask—BF) aeration conditions. Data are summarized in Fig. 1.
Additional file 2.
Species with multiple homologs of XYL1 accumulate xylitol. Xylose fermentation by closely related species under moderate (shake flask—SF) and high aeration (baffled flask—BF). Error bars indicate the standard deviation from the three independent replicates.
Additional file 3.
Growth curves of Sc. stipitis and S. cerevisiae with Sc. xylosifermentans XYL1, XYL2, and XYL3 under anoxic conditions. Error bars indicate the standard deviation from the three biological replicates. Asterisks denote significant differences between S. cerevisiae + SstipitisXYL1/XYL2/XYL3. The letter a indicates significant differences between S. cerevisiae + SxylosiXYL1 and SxylosiXYL1/XYL2/XYL3, and the letter b indicates significant differences between S. cerevisiae + SxylosiXYL1/XYL2 and SxylosiXYL1/XYL2/XYL3.
Additional file 4.
Yeast species used in this study.
Additional file 5.
S. cerevisiae strains used in this study.
Additional file 6.
Assembly and annotation statistics for the genomes sequenced in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Barros, K.O., Mader, M., Krause, D.J. et al. Oxygenation influences xylose fermentation and gene expression in the yeast genera Spathaspora and Scheffersomyces. Biotechnol Biofuels 17, 20 (2024). https://doi.org/10.1186/s13068-024-02467-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-024-02467-8