
Abstract
The recalcitrance of lignocellulosic biomass is a major constraint to its high-value use at industrial scale. In nature, microbes play a crucial role in biomass degradation, nutrient recycling and ecosystem functioning. Therefore, the use of microbes is an attractive way to transform biomass to produce clean energy and high-value compounds. The microbial degradation of lignocelluloses is a complex process which is dependent upon multiple secreted enzymes and their synergistic activities. The availability of the cutting edge proteomics and highly sensitive mass spectrometry tools make possible for researchers to probe the secretome of microbes and microbial consortia grown on different lignocelluloses for the identification of hydrolytic enzymes of industrial interest and their substrate-dependent expression. This review summarizes the role of secretomics in identifying enzymes involved in lignocelluloses deconstruction, the development of enzyme cocktails and the construction of synthetic microbial consortia for biomass valorization, providing our perspectives to address the current challenges.
Similar content being viewed by others

Background
Lignocellulosic biomass is a gift of nature and its seen as an inexhaustible source of organic carbon for the green synthesis of diverse and biofuels and other bio-based compounds with very low (or null) carbon and sulphur emissions. The emergence of biomass-based industries in developed and developing countries has provided opportunities in diverse types of employment, as the use of biomass involves a series of logistics processes such as bulk collection, transportation and pretreatment [1].
Nearly 94% of the global lignocellulosic production comes from agricultural sources and represents ~ 140 gigatons annually [2], and is expected to further increase in response due to population growth, food demand and intensive agriculture. In addition, lignocellulosic biomass provides more environmental benefits than fossil-derived fuels and other processed compounds due to its renewable nature, high abundance and its capacity to degrade by microorganisms. Hence lignocellulosic biomass is recognized as an abundant and economic feedstock for the synthesis of valuable chemicals. However, most of the valuable biomass has being disposed by burning and landfilling in agricultural/domestic environments, or burned for electric power generation in several industries. In recent years, physical, thermochemical and biochemical approaches have been used as biomass pretreatment methods to produce syngas, fuel gas, bio-oil, biochar and bioethanol and other compounds. Though chemical and thermochemical processing methods are robust and efficient, biochemical methods such as bacterial, fungal and lignocellulolytic bacterial/fungal secretome-based pretreatment are becoming more relevant in recent years because of their eco-friendly nature and controlled depolymerization of biomass.
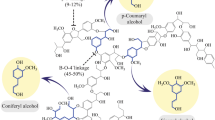
According to the global energy assessment (GEA), the energy derived from renewable resources is expected to cover 10–40% of the global energy requirements by 2050 [3, 4]. Typically, lignocellulosic biomass comprised cellulose (40–50%), hemicellulose (25–30%), lignin (3–30%), proteins, resins and others depending on the plant species (pectin, fatty acids, amino acids, pigments and secondary metabolites) [5, 6]. Cellulose is a linear polysaccharide consisting of glucose repeats linked by β − 1 → 4 glycosidic bonds. Whereas hemicellulose/polyose is a heteropolysaccharide made up of several different monosaccharides and its depolymerization is far easier than cellulose. Lignin is an aromatic heteropolymer composed of main three monolignols [coumaryl (H), coniferyl (G), and sinapyl alcohol (S)] and are cross-linked especially through β-O-4, 5-5, β-5, β-1, α-O-4, 4-O-5 and β–β bonds. In addition, O–H, C–H, C–O, C=C, and C–C bonds are also present in lignin [7, 8]. Since lignin is considered as the main source of aromatic compounds from renewable resources, it has attracted the attention of many researchers to produce a wide variety of value-added chemicals.
The massive use of lignocellulosic biomass still faces a series of challenges, some of them are the structural complexity of lignocellulosic biomass, the high lignin content, cellulose coverage by hemicellulose and lignin, cellulose’s crystalline nature, and the propensity of lignin to repolymerize. In the particular case of cellulose valorization, physical, chemical and biological pretreatment methods are being used to remove lignin and hemicellulose for its effective processing by microorganisms [9]. After lignocelluloses fractionation and cellulose conversion, in order to achieve a successful approach for lignin bioconversion to produce aromatic compounds at commercial scale, research has become more involved in the development of microbial tools. If the available biomass is used efficiently to produce biofuels and other several bio-based compounds, it would be possible to reduce fossil fuel consumption, to promote the circular economy and to decrease the global environmental burdens in the future.
Under natural circumstances, bacteria and fungi are actively involved in biomass deconstruction by hydrolysis, as these microorganisms secrete a series of hydrolytic enzymes, such as cellulases, hemicellulases, laccases, peroxidases, etc. [10] (Fig. 1). As a result, bacteria and fungi are used to transform lignocellulosic biomass into fermentable sugars and aliphatic/aromatic monomers. Microorganisms employ various mechanisms and secretory enzymes for deconstructing lignocellulosic biomass. However, the identification and purification of ligninolytic enzymes is a time-consuming process. Secretion of enzymes by lignocellulose degrading microbes depends on the nature and macromolecular composition of the biomass. In this regard, profiling of the bacterial/fungal secretomes using proteomic analyses is a befitting way to study the expression kinetics and identification of substrate-specific enzymes [11]. Numerous secretome analyses have catalogued the presence of active ligninolytic enzymes and their substrate-dependent expression pattern. Consequently, substrate-specific synthetic enzyme cocktails are developed and successfully used at laboratory scale as well as at industrial scale for biomass conversion.

An overview of biomass degradation
To date, a number of synthetic enzyme cocktails and cellulase preparations are commercially available, for example, Accellerase 1000, Accellerase® 1500, Novozyme 188, Celluclast 1.5 L, Cellic Ctec2, GC220, Depol ™ 692L, Spezyme-CP, etc. [11]. Microbial consortia are more efficient biomass degraders than single strains, due to the secretion of a repertoire of hydrolytic enzymes. However, sugars produced from biomass fermentation by microbial consortia treatment are rapidly utilized by some of the bacterial species present in the same microbial consortia, hampering their separation and use. Therefore, several studies demonstrated a significant increase in biofuel production from secretome/metasecretome pretreated biomass. Given the application of secretomes, metasecretomes and synthetic enzyme cocktails in biomass deconstruction/pretreatment for an environmentally friendly production of high-value products, this review focuses on the evaluation of the application of secretomic and metasecretomic technologies in the mining of enzymes associated with lignocelluloses fractionation and valorization.
Glycoside hydrolases (GH)
Lignocellulosic degradation is a complex and multi-enzymatic process carried out predominantly by bacterial and fungal communities under natural conditions. Though fungi have been studied extensively for their application in lignocellulosic deconstruction, bacteria have attracted more attention owing to their genome size and suitability for genetic engineering and heterologous expression of desired enzymes/proteins and the expression of synthetic metabolic pathways [12]. Carbohydrate-active enzymes (CAZymes) are classified into four classes, namely glycoside hydrolases/glycosidases (GHs [EC 3.2.1.]), glycosyltransferases (GTs [EC 2.4.x.y]), polysaccharide lyases (PLs [EC 4.2.2.-]), carbohydrate esterases (CEs) and auxiliary activities (AAs). Nearly all life forms on Earth contain glycoside hydrolases (GHs) and can hydrolyse O-, N- and S-linked glycosidic linkages. Numerous investigations have reported the presence of GHs in bacteria, fungi, plants and animals. In particular, terrestrial fungi produce a plethora of extracellular and intracellular GHs to acquire nutrients from biomass [13,14,15]. GHs acts on glycosidic bonds in crystalline polysaccharides, amino polysaccharides and other complex polysaccharides present in the biomass and transform them into fermentable sugars [16]. Moreover, GHs are also associated with developmental processes, such as the defence against pathogens, basic cellular functions in higher eukaryotes [17, 18], parasitism in nematodes [19] and virulence in pathogenic fungi and bacteria [20]. It is interesting to note that GHs have been shown to participate in the inhibition of biofilm formation and the disruption of preformed biofilms by clinically identified bacterial pathogens [21,22,23]. Beta-galactosidase, amylases, cellulases and hemicellulases are the well-studied GHs and are used in a number of important biotechnological processes.
GHs are classified based on the location (intracellular and extracellular), the stereochemistry of hydrolysis (retaining and inverting) and the cleavage position (exo and endo) in the polysaccharide chain [24]. Likewise, based on the amino acid sequences and folding similarities, GHs are classified into 171 GH families. Among them, the GH families GH 5, GH13, GH16, GH30 and GH 43 are further classified into 56, 43, 27, 9 and 37 different sub-families, respectively [24, 67]. In addition, some of the GH families were classified under 18 clans (GH-A to GH-R) depending on the conserved folding patterns [25]. Based on the catalytic mechanisms, GHs are classified under the Enzyme Commission (EC) number. So far, terrestrial fungi have been reported to produce copious GHs which can degrade the complex lignocellulosic biomass. A recent study by Chrismas and Cunliffe (2020) have shown the presence of numerous GHs in the open ocean mycoplankton communities and the members of GH7 family was found to be the most abundant, followed by GH17, GH5, GH72, GH16, GH13, GH3, GH47, GH18, etc. [26]. Besides biomass pretreatment, GHs are mainly used in food, pharmaceutical, and paper and pulp industries. Despite the identification of numerous GHs, only a few of them are widely produced on a large scale for industrial applications due to their stability and requirement of stringent reaction conditions.
Role of auxiliary activity (AA) lytic polysaccharide monooxygenases (LPMOs) in biomass hydrolysis
Numerous reports have demonstrated that the copper-containing lytic polysaccharide monooxygenases (LPMOs) are mainly present in fungal and bacterial kingdoms. Most of them have been identified over the past decade and have shown to improve the degradation of lignocellulosic biomass through oxidative cleavage of glycosidic linkages [27, 28]. For the first time, a secreted LMPO called catalytic chitin-binding protein (CBP21) by Serratia marcescens was shown to enhance the chitin degradation by binding with chitin and inducing structural changes, which makes chitin more accessible for chitinases [29].
According to the information available at the Carbohydrate-Active enZymes (CAZy) database, to date, the identified auxiliary activity (AA) enzymes/proteins are classified into 16 families, among them 6 families such as AA9, AA10, AA11, AA13, AA14 and AA15, consisted of LPMOs [30]. The most common AA enzyme in fungi is AA9 which acts on cellulose, while AA13 and AA14 act on chitin, starch [31] and xylan [32], respectively. Chitin-active LPMOs are classified under the AA10, AA11, and AA15 families and are present in bacteria, fungi, viruses and arthropods [33]. A new AA16 family identified in Aspergillus aculeatus was shown to increase the activity of T. reesei cellobiohydrolase I by increasing the substrate availability [34]. The prevalence of LPMOs in the bacterial and fungal population involved in the lignocellulosic biomass degradation highlight the important role of LPMOs in the recycling of organic carbon in nature [35]. However, LPMOs are also associated with the virulence in phytopathogenic bacteria/fungi as well as in some human pathogenic Vibrio cholerae and Listeria monocytogenes [36, 37]. LPMOs are used as a key component in the widely used cellulase and hemicellulase cocktail called Cellic CTec enzyme products (Novozymes A/S) at industrial scale for ethanol production using lignocellulosic biomass [35].
The presence of β-sandwich folds in the core structure and a flat substrate binding site in LPMOs provides the much-needed flexibility to act on the crystalline cellulose and other polymers [38, 39]. Fungal LPMOs contain histidine brace with a copper ion and N-methylated N-terminal histidine (His1) [40, 41], whereas bacterial LPMOs contains only histidine brace with copper in their active sites [39, 42]. The catalytic activity of LPMOs depends on the reduction of copper followed by oxidative cleavage of β−(1 → 4) glycosidic linkage and the oxidation of the C1 or C4 position of the substrate in the presence of O2 as a co-substrate [27, 43,44,45]. Studies by Müller et al., [46] and Bissaro et al. [47] suggested that H2O2 can be used as a co-substrate by LPMOs and the supplementation of H2O2 was found to enhance the saccharification of cellulose by the Cellulase cocktail Cellic® CTec2 containing LPMOs.
Occurrence of AA LPMOs in bacteria and fungi
According to the CAZy database, it is apparent that the AA10 family (6938 entries-accessed on 15th June 2020) is very common in bacteria. However, only a small fraction of AA10 LPMOs has been experimentally well studied and validated. For example, AA10 LPMOs from Bacillus amyloliquefaciens DSM7, Bacillus licheniformis, Bacillus thuringensis, Caldibacillus cellulovorans, Cellvibrio japonicus Ueda107, Enterococcus faecalis V583, Hahella chejuensis KCTC 2396, Jonesia denitrificans DSM20603, Listeria monocytogenes EGD-e, Photorhabdus laumondii subsp. laumondii TTO1, Serratia marcescens BJL200, Streptomyces ambofaciens ATCC 23877, Streptomyces coelicolor A3(2), Streptomyces griseus, Streptomyces lividans 1326, Streptomyces pratensis ATCC 33331, Teredinibacter turnerae T7901, Thermobifida fusca YX and Vibrio cholerae O1 biovar El were characterized. Recently identified LPMO from Photorhabdus luminescen called Pl AA10 can act on both cellulose and chitin [48].
Furthermore, members of Ascomycetes such as Neurospora crassa [49, 50], Pestalotiopsis sp. [51], Pestalotiopsis sp NCi6 [52], Trichoderma asperellum [53], Fusarium graminearum [54, 55], Fusarium verticillioides [56], Penicillium oxalicum [57], Aspergillus niger [58], Aspergillus nidulans [59, 60], Aspergillus fumigatus [31], Myceliophthora thermophila [61], Penicillium echinulatum [62], Malbranchea Cinnamomea [63] and Podospora anserina [64], and members of Basidiomycetes such as Gloeophyllum trabeum [65, 66], Laetisaria arvalis [67], Phanerochaete chrysosporium [68, 69], Pycnoporus coccineus [70], Pycnoporus sanguineus, Pycnoporus cinnabarinus [71] and Schizophyllum communae [72] were reported to have AA enzyme genes and their expression is dependent on biomass nature. In addition, Myceliophthora thermophila [73, 74], Thielavia terrestris [75], Geotrichum candidum [76], Heterobasidion irregulare [77], Thermoascus aurantiacus [41, 78] were also shown to have LMPOs of biotechnological importance. However, experimental validation is required for the addition of one or more appropriate AAs into commercially available enzyme cocktails to expand their application in the degradation of various biomass resources.
Ligninolytic enzymes
Lignin removal is an important and difficult process in biomass conversion as it acts as a protective barrier to cellulose. Chemical degradation of lignin is an energy-intensive process which requires extreme reaction conditions (high temperature, high pressure and acidic/alkaline pH). Microbes break down lignin aerobically and anaerobically and are considered as the best choice for lignin degradation. Furthermore, ligninolytic microbes are useful in the synthesis of aromatic compounds of interest of the food flavouring and pharmaceutical industries [79, 80]. The ligninolytic system of bacteria actinomycetes and fungi is mainly comprised laccases (LACs [EC 1.10.3.2]), versatile peroxidases (VP [EC 1.11.1.16]), lignin peroxidases (LiPs [EC 1.11.1.14]) and manganese peroxidases (MnPs [EC 1.11.1.13]) [79,80,81]. Although ligninolytic enzymes were first identified from fungus, it was later found that the ligninolytic enzymes from bacterial sources are more effective in the deconstruction of various types of lignin such as kraft, alkali, organosolv, soda and sulfolignins. Among the ligninolytic enzymes, LACs find a place in the alcohol and food industries in the improvement of wine, beer, fruit juice, bread and sugar beet pectin’s quality [82,83,84]. LACs are muliti-copper oxidoreductases (MCOs), ubiquitously present in bacteria and fungi, and have been shown to deconstruct lignin and polyaromatic hydrocarbons, dye discolouration as well [85,86,87]. The availability of the microbial whole genome and whole metagenomic data paved the way for exploring the presence of lignin deconstructing enzymes using bioinformatic tools.
Ausec et al., reported more than 1200 putative MCOs in 2,200 whole and draft bacterial genomes, and four metagenomic sequence sets. Among the identified putative MCOs encoding genes, 76% of the genes are predicted to have a signal peptide indicating that the majority of MCOs are secreted into the environment to degrade lignin [88]. Genomic reconstruction of lignin degradation pathways in Cupriavidus basilensis B-8 revealed the presence of three functional pathways [89]. Briefly, β-ketoadipate pathway with the catechol branch and the protocatechuate branch, phenol degradation pathway and gentisate pathway for the catabolism of lignin and lignin-derived aromatics. Further analysis showed that manganese peroxidase (MnP) is required for the growth of C. basilensis in the presence of lignin and in a subsequent stage of growth laccase has been found to degrade lignin [89]. In a recent functional genomic study, 13 bacterial genomes were examined to identify pathways involved in lignin deconstruction. The results showed that lignin degradation is a complex process involving DyP-type peroxidases and MCOs, aromatic compounds degrading gene clusters and the β-ketoadipate pathway [90].
What is the need for proteomic analysis of lignocellulolytic microorganisms?
Under different growth conditions (pH, temperature and media composition/biomass) lignocellulose-hydrolysing bacteria and fungi secret different types of GHs (Table 1). Moreover, studies have shown the difference in the secretion and proportion of GHs by lignocellulolytic microbes when they have grown in solid state, static and in submerged fermentation modes (Table 1). Thus, it is important to analyse the secretome of lignocellulolytic strains grown on specific biomass to study the substrate-dependent secretion pattern of GHs, identification of novel and potent GHs for biomass hydrolysis. Genomic analysis of T. reesei has revealed the presence of genes encoding extracellular cellobiohydrolases (N = 2), extracellular endoglucanases (N = 8), xylanases (extracellular, N = 4 and intracellular, N = 1), intracellular xylanase (N = 1), glycosidases (extracellular, N = 5, intracellular, N = 6 and mitochondrion, N = 1) and xylosidases (extracellular, N = 1 and intracellular, N = 1) [91]. Nevertheless, proteomic analysis of secretomes of T. reesei QM6a and Rut C30 have grown on different lignocellulosic biomass revealed that the expression of lignocellulolytic enzymes are modulated by the nature and complex composition of the biomass to be degraded [92,93,94]. For example, enzymes such as endoglucanases (N = 18), glucosidases (N = 13), xylanases, arabinofuranosidases, β-xylosidases, α- and β-glucuronidases, CBM1 acetylxylan esterase, CBM1 acetyl esterase, cellobiohydrolase, β-mannase, α- and β-mannosidases, polysaccharide lyases, exo-rhamnogalacturonase, endopolygalacturonase, 3-phytase, and glucuronan lyase A were secreted by T. reesei (QM6a and Rut C30) when cultured on fibrous cellulose, corn stover, and sawdust (1% W/V) [92]. In addition, proteomic analysis highlighted the expression of substrate binding proteins such as cellulose-binding domain (CBM1) Cip2, carbohydrate-binding module family 13 and hydrophobin-1 and 2 [92]. Furthermore, enzymes involved in lignin degradation such as laccase, superoxide dismutase, glyoxal oxidase, peroxidases and oxidoreductases were identified and quantified from the secretome of T. reesei QM6a and Rut C30. Importantly, proteomic analysis leads to a better understanding of the importance of auxiliary proteins in degrading lignocellulosic biomass [92]. Global proteomic analysis of T. reesei QM9414 cultured on glucose, cellulose and sophorose as carbon sources revealed the differential regulation of several genes encoding accessory proteins, transcriptional regulators, transporters, major facilitator superfamily (MFS) permeases and CAZymes. Hierarchical clustering and gene regulatory network analyses demonstrated that 75 and 107 genes were specific to sophorose and cellulose utilization, respectively [95].
In addition to the identification of novel enzymes and regulatory mechanisms, proteomic studies are useful in selecting substrate-specific lignocellulolytic bacterial and fungal strains (Table 1). For example, through a proteomic study, Gong et al., have validated the suitability of A. niger ATCC1015 secretome composed of glucanases, esterases, cellulases, mannases, xyloglucanases, xylanases and β-pectinases to breakdown of dicotyledon biomass [63]. While the secretomes of T. reesei QM9414, and Penicillium oxalicum 114-2 were found to be suitable for the degradation of monocotyledon biomass [57]. Considering the recalcitrance nature of biomass and the abundance of lignocellulolytic enzymes, secretome from different fungi can be combined to improve the biomass degradation. For example, combining T. reesei and A. niger secretome has been reported to increase the rate of biomass saccharification [58]. Thus, it is important to analyse the secretome of lignocellulolytic strains grown on specific biomass to study the substrate-dependent secretion pattern of GHs, and the identification of novel and powerful GHs for biomass hydrolysis.
Gel-based and gel-free proteomics approaches
As proteins regulate and perform numerous biochemical functions, among the omics approaches, the proteomic approach is very important for accessing the functional status of different life forms. The availability of whole-genome sequences and advances in bioinformatic analysis have made proteomics as one of the best ways to unravel the biological mechanisms and pathways. Furthermore, proteomic analyses are very useful in bridging the knowledge gap between the genetic and functional status of an organism grown under specific conditions [96, 97]. A typical proteomic analysis workflow involves sample preparation, protein separation, mass spectrometric analysis, data acquisition, protein identification and quantification, and the validation of the protein of interest. Sample preparation is an important step in proteomic analysis to obtain accurate and reproducible results. Sample preparation consists on the protein extraction from the samples through physical and enzymatic treatment, solubilization, denaturation, reduction, alkylation, protein labelling, removal of highly abundant proteins to study the low-abundant proteins and tryptic digestion (Fig. 2) [98]. Prefractionation of proteins using SDS-PAGE based on the molecular weight or two-dimensional gel electrophoresis based on isoelectric pH and molecular weight, and prefractionation of tryptic peptides or using reverse-phase high-performance nano liquid chromatography based on the hydrophobicity (Fig. 2). After the separation phase, peptides are analysed by mass spectrometry in which the peptide mixture is ionized and separated to determine the mass-to-charge ratio (m/z) [99], followed by protein identification using database search tools such as Mascot [100], SEQUEST [101], OLAV [102], MS-FIT (http://prospector.ucsf.edu/), Open mass spectrometry search algorithm (OMSSA) and X! Tandem [103].

Steps and tools involved in secretomic analysis
In the modern proteomics era, the combination of gel-based and gel-free techniques is successfully used to analyse the complex proteomes using advanced mass spectrometry tools (Fig. 2). Mass spectrometry contains three important components, such as ionization, mass analysis, and detection. Matrix-assisted laser desorption/ionization (MALDI), surface-enhanced laser desorption/ionization (SELDI) liquid chromatography coupled electrospray ionization (ESI) are the major ionization techniques used for peptide analyses [104]. Regarding mass analysers, time-of-flight (TOF and TOF–TOF), ion trap (linear trap, quadrupole and orbitrap trap), Fourier transform ion cyclotron resonance (FT-ICR) and quadrupole-TOF (hybrid analyser) are widely used in proteomic analyses [105, 106]. In recent years, mass spectrometry-based modern proteomics approaches such as top-down [107], bottom-up/shotgun proteomics [108], multidimensional protein identification technology (MudPIT) [109] and gel-enhanced liquid chromatography–mass spectrometry (GeLCMS) [110] have been used to analyse complex proteomes.
The availability of the hybrid mass spectrometer with high sensitivity and mass accuracies such as hybrid linear trap orbitrap mass spectrometer, triple quadrupole mass spectrometer, quadrupole-orbitrap mass spectrometer, triple quadrupole/linear ion trap mass spectrometer, and orbitrap fusion tribrid mass spectrometer, has driven researchers to carry out label -based and label-free relative and absolute quantification of protein expression [111,112,113]. For label-based mass spectrometric approaches, stable isotope labelling by amino acids in cell culture (SILAC), isotope-coded affinity tag (ICAT), tandem mass tag (TMT) and isobaric tags for relative and absolute quantitation (iTRAQ) are being used to spike the proteins from various biological samples to analyse and quantify the proteins through LC–MS/MS [114].
Label-free mass proteomic analysis is straightforward, less expensive and less time-consuming as compared to other proteomics approaches. This type of proteomic analysis relies on the tryptic digestion of several thousand proteins from the complex biological samples in solution, LC separation of peptides and quantification of proteins based on peak intensity and/or spectral counting [115]. Given that the label-free proteomic approach simultaneously identifies and quantifies thousands of proteins from the biological samples in a single run, thus it is widely used in the field of such as biomedicine, microbiology, bioenergy, biotransformation, etc., to decode several complex biochemical pathways and its regulation, post-translational modifications, the response of an organism to physical, chemical and environmental factors.
Proteomic approaches are successfully used to identify the carbohydrate-active enzymes (CAZymes) from the secretome of biomass deconstructing microbes [116]. Additionally, proteomic analyses provide the quantitative expression of enzymes in response to the available substrates. Secretomic analysis of lignocellulolytic microbes are advantageous in designing synthetic enzyme cocktails and synthetic microbial consortia for the robust and efficient biomass pretreatment and deconstruction. However, the extraction of a whole set of proteins secreted by bacteria/fungi grow in lignocellulosic biomass remains as a difficult task; this is due to difficulties associated with the elimination of contaminants affecting trypsin digestion and the minimization of contaminating cellular proteins, masking the absolute quantification of secreted proteins. The development of activity-based protein profiling methods is necessary to identify substrate-specific and highly active lignocellulose degrading enzymes to facilitate the formation of effective enzyme cocktails.
Proteomics in profiling of glycoside hydrolases in bacterial and fungal secretomes
Secretome of lignocellulolytic bacteria/fungi composed of proteins anchored on the cell surface, secreted enzymes and proteins involved in biomass hydrolysis. Thus proteomic analysis of the secretome of lignocellulosic biomass-degrading microorganisms helps to formulate enzyme cocktails for better biomass degradation [10, 117,118,119]. Comparative analysis of the commercially available enzyme cocktails Celluclast 1.5 L and an enzyme cocktail comprising T. reesei secretome that comprised cellobiohydrolases, endoglucanase and β-glucosidase on the sugarcane bagasse deconstruction showed higher glucose yield from T. reesei secretome treated sugarcane bagasse than Celluclast 1.5 L treated [120]. The use of fungal secretome would be helpful in reducing the cost and increasing lignocellulosic biomass utilization efficiency. Studies have reported increased cellulolytic activity of commercially available enzyme cocktails when completed with fungal secretomes [121].
Especially secretome/enzymes from fungi have received considerable attention due to their wide range of applications in the food, pulp and paper, textile, biofuel and bioenergy industries [122]. In addition, fungal secretomes can be used in combination with lignin-depolymerizing bacteria to prevent the re-polymerization of low molar mass lignin fractions. For example, Salvachúa et al. have reported that the complementation of Pleurotus eryngii secretome rich in laccases with Pseudomonas putida KT2440 is useful for lignin depolymerization in solid biomass [123].
The secretome of any organism can be predicted through bioinformatics tools using the available whole-genome data [124, 125]. However, experimental identification of enzymes/proteins present in the secretome is more preferential than bioinformatics prediction as it represents the functional status of a genome [126, 127]. Hence, the secretome of industrially important microorganisms has been studied extensively using traditional proteomic approaches and contemporary cutting-edge proteomic approaches (Table 1). Microorganisms can be stimulated to produce the desired composition of enzymatic cocktails by supplementing different sugars in the growth medium [128,129,130].
Secretome of industrially important T. reesei strains: an arsenal of lignocellulolytic enzymes
T. reesei is one of the well-studied lignocellulolytic fungus at the genome, transcriptome and proteome level as it contains robust cellulolytic and hemicellulolytic systems [131]. Several researchers have reported the potential of T. reesei hypercellulolytic mutant Rut C30 to degrade lignocellulosic biomass and analysed its secretome using proteomic approaches. When T. reesei Rut C30 is cultured on lignocellulosic biomass, it secretes a variety of hydrolytic enzymes targeting cellulose, hemicellulose and lignin [92]. Complementarily, T. reesei Rut C30 can be stimulated to secrete the desired enzyme/enzymes by supplementing the appropriate carbon source in the growth medium and hence becoming a promising candidate for economically feasible lignocellulolytic enzymes production [93]. Replacement of transcriptional factors in T. reesei Rut C30 with minimal/artificial transcriptional factors [132] and expression of artificial zinc finger proteins [133] are the promising approaches for increasing cellulase secretion. For example, substituting natural transcriptional factors with minimal transcriptional activators such as ACEII and CREI in T. reesei Rut C30 increased the cellulase production [134]. Similarly, the introduction of artificial transcriptional factors such as XYR1VP, ACE2VP, and ACE1VP into T. reesei Rut C30 has improved the degradation of pretreated corn stover with increased glucose yield [135]. As a result, T. reesei is being successfully used for biofuel production at industrial scale.
Proteotranscriptomic analysis of T. reesei and genetic engineering has paved a way to understand and overcome factors limiting lignocelluloses degradation. For example, when T. reesei has grown on corn steep liquor as a nitrogen source, in the later stages of the fermentation process proteases were found to have a negative effect on the cellulolytic activity [136]. Since T. reesei contains nearly 200 protease encoding genes [137], the knocking out and deletion of all the genes encoding proteases become impossible [138], and this can have an adverse effect on strain’s growth. However, Qian et al., have identified the major proteases in the secretome of T. reesei grown on corn steep liquor using biochemical assays, LC–MS/MS analysis and successfully constructed a triple protease mutant strain called ΔP70 with reduced protease activity and enhanced the cellulolytic activity on a six-fold basis [136]. Moreover, CRISPR/Cas9 mediated deletion of the cellulase repressor gene (ACE1) and secreted protease genes (SLP1 and PEP1), constitutive expression of cellulase master regulator gene (XYR1) and heterologous expression of β-glucosidase CEL3A and invertase SUC1 from Talaromyces emersonii and A. niger, respectively, in T. reesei RUT-C30 has remarkably increase the cellulase, hemicellulase, β-glucosidase and xylanase production [135]. These results have further strengthened the prospects for combining genetic engineering and multi-omics technologies to develop of industrially important strains for biomass conversion.
It has been shown that T. reesei Rut C30 cultured on spent hydrolysate model medium as a carbon source produces more lignocellulose degrading enzymes such as cellobiohydrolases and endoglucanases compared to control [139]. Genetic engineering of industrially relevant strains for heterologous expression and secretion of desired enzymes has been proved to be one of the best ways to accelerate the utilization of lignocellulosic biomass. Ayrinhac et al., have genetically modified T. reesei to secrete a β-glucosidases (evolved through L-Shuffling of putative glycosidase genes from Chaetomium globosum, T. reesei and Neurospora crassa) with 242-fold, increased cellulolytic activity [140]. The recombinant T. reesei secretome that comprised the above mentioned β-glucosidases can enable to perform wheat straw saccharification in combination with a fourfold reduced level of commercial cellulase preparation, when compared to the wild-type strain [140]. In a recent investigation, a two-step hydrolyzation of lignocellulosic biomass was conducted to produce glucose. Briefly, in the first step T. reesei secretome was used and in the second step the mixture of Laetisaria arvalis, Artolenzites elegans and Trametes ljubarskyi secretomes were deployed for the maximum biomass degradation [11]. Proteomic analysis of the secretome of Ganoderma lucidum grown on sugar cane bagasse revealed the presence of lignin-degrading enzymes, cellulases, hemicellulases, glycoside hydrolases, proteases and phosphatases. Complementarily, the authors have also demonstrated the hydrolytic activity of the identified enzymes using biochemical assays [141].
Secretomic analysis of phytopathogenic fungi for hunting lignocellulolytic enzymes
Since the strategy of using enzyme cocktails has proven to be more effective than the use of single enzymes in lignocelluloses degradation, the identification and characterization of enzymes from bacterial and fungal secretomes would be useful in formulating enzyme cocktails to accelerate lignocellulosic biomass degradation. In addition to the identification of novel enzymes/proteins from lignocellulolytic microbes (Table 1), secretome profiling of phytopathogenic microbes certainly offers the best opportunity to harness very active lignocellulolytic enzymes (Table 1) [142]. For example, LC–MS/MS-based identification of proteins present in the secretome of a phytopathogenic fungus Ustilago maydis showed the presence of carbohydrate-active enzymes (CAZymes), hydrolases and oxido-reductases. Similarly, profiling of Aspergillus niger, Aspergillus nidulans secretome has demonstrated their ability to produce several CAZymes, oxidoreductases, proteases, lipases and esterases of biotechnological importance [143]. Additionally, the authors have demonstrated a significant increase in the yield of total sugars and glucose, when T. reesei CL847 enzymes were supplemented with U. maydis secretome for the saccharification of micronised wheat straw [143]. Secretome analysis of T. reesei Rut C3 cultured using a mixture of glucose, disaccharide and alkali-pretreated corn stover as inducers revealed the depletion in the production of β-glucosidase. Hence, a β-glucosidase gene from Aspergillus aculeatus was heterologously expressed in T. reesei Rut C3 under the pdc1 promoter. As a result, β-glucosidase production has increased 71-fold compared to the parent strain. Subsequent application of the secretome for corn stover degradation led to significant increase in ethanol production (54.2 g L−1) [144]
Secretomic analysis of other important lignocellulosic-degrading microbes
Members of the Aspergillus family such as Aspergillus nidulans, Aspergillus oryzae, Aspergillus niger and Aspergillus fumigatus are receiving more attention, due to the presence of the arsenal of biomass-deconstruction enzymes. In several studies, secretomes of various Aspergillus species have been successfully used to degrade biomass for biofuels production. A. fumigatus is known for its thermostable enzymes such as endoglucanases [145, 146], β-xylosidase [147, 148], β-glucosidases [149] and xylanses [150, 151], thus its secretome is considered as one of the promising candidates for the biomass pretreatment in combination with commercially available enzyme cocktails. As a result, secretomes of several A. fumigatus strains were studied (Table 1) [152, 153]. For example, the proteomic analysis of the secretome of A. fumigatus AF293 cultured on steam-exploded bagasse revealed the presence of endoglucanases, endo-1,4-beta-xylanases, acetyl xylan esterase, feruloyl esterase, feruloyl esterase C, extracellular GDSL-like lipase/acylhydrolase, beta-glucosidase, glycosyl hydrolases, arabinanases, glucanases, endoglucanases, cellobiohydrolases, arabinofuranosidases, 1,4-beta-d-glucancellobiohydrolyase, swollenin and BNR/Asp-box repeat domain protein [154]. These identified enzymes and proteins are associated with the degradation of cellulose, pectin, xylan, arabinoxylan, mannan, galactomannan, glucomannan, β-1,6-glucan and arabinogalactan.
The LC–MS/MS analysis of the secretome of a thermophilic ascomycete Malbranchea cinnamomea grown on sorghum straw showed the presence of a wide range of catalytically active and Mn2+/Cu2+ dependent cellulases (N = 8), hemicellulases (N = 9), polysaccharide lyases (N = 12), proteases (N = 9), glyoxal oxidase (N = 1) and other enzymes [63]. In a recent study on the secretome of Talaromyces emersonii cultured on cellulose and wheat grain flour highlighted the presence of 93, 13, 14 and 8, glycosyl hydrolases (covering 40 different glycosyl hydrolase families), cellulose-binding modules, carbohydrate esterases and auxiliary activity proteins, respectively [155]. However, a genome analysis of T. emersonii has speculated the presence of 221 glycosyl hydrolase genes covering 55 different glycosyl hydrolase families, 57 auxiliary activity protein genes and 34 carbohydrate esterase genes [155]. These studies have further confirmed the substrate-dependent secretion of enzymes, and accessory proteins, and highlighted the need of secretome analysis to formulate the suitable and efficient enzyme cocktails to obtain the best results. More interestingly, Claes et al., succeeded in developing a Saccharomyces cerevisiae to secrete seven ligninolytic enzymes such as β-glucosidase, β-xylosidase, xylanase, endoglucanase, cellobiohydrolase I, cellobiohydrolase II and acetylxylan esterase from different fungi, and the engineered S. cerevisiae was shown to produce bioethanol from various substrates [156]. Co-culturing of lignocellulolytic bacteria and fungi is an emerging area of research in lignocellulosic biomass degradation. Recent investigation on co-cultivation of Thermomyces lanuginosus and Thermobifida fusca on corn stalks revealed the rapid degradation of xylan by T. lanuginosus that enables the breakdown of cellulose by T. fusca through secretion of multiple glycoside hydrolases [89]. However, detailed studies on the secretome of co-culturing microbes on different lignocellulosic biomass are required to harness numerous synergistic mechanisms.
Proteomics in the identification of AALPMOs from the secretome of biomass deconstructing bacteria and fungi
The presence of numerous genes encoding AA9 LPMO in most of the biomass-degrading Ascomycetes and Basidiomycetes, substrate-dependent expression patterns confirming their role in the deconstruction of biomass along with other major hydrolytic enzymes. Accumulating scientific data highlighting the role of auxiliary proteins in lignocellulose deconstruction, for example, LPMOs [50, 65, 157] enhance the lignocellulose deconstruction through oxidative cleavage, which in turn eases the activity of other major enzymes [65, 157, 158]. AA9 LPMOs disrupt the recalcitrant ultrastructure of cellulose by introducing nicks and weakening its mechanical strength through oxidation, thereby allowing rapid deconstruction of cellulose by cellulases [159]. A recent study has demonstrated that the heterologous expression of a thermostable LPMO of Talaromyces cellulolyticus in T. reesei significantly enhanced the breakdown of delignified corncob and cellulose [160]. In addition, a LMPO called PoLPMO9A from Pleurotus ostreatus has improved the degradation of natural and kraft lignin by a versatile peroxidase through H2O2 production and introducing structural modifications [161]. In a recent investigation, two LPMOs such as AtAA9A and AtAA9B from Aspergillus tamarii were identified to cleave the β-(1 → 4)-glucan linkage in xyloglucan [162]. Previously, it was believed that LPMOs were present in bacteria, fungi and certain viruses, but researchers identified LPMOs from the digestive tract of insects that thrives on lignocellulose biomass. Sabbadin et al., have identified more than 20 AA15 LPMOs accounting for 20.2% of the gut proteome of Thermobia domestica (firebrat). Furthermore, in T. domestica’s gut several GHs belong to GH1, GH2, GH5, GH9, GH13, GH16, GH18, GH20, GH27, GH30, GH31, GH35, GH38 and GH65 were identified [163]. Similarly, proteomic analysis of the tracheal and mid gut region of Drosophila melanogaster showed the presence of two AA15 LPMOs such as DmAA15A and DmAA15B [163]. Further evaluation of these novel LPMOs and their heterologous expression in a suitable lignocellulose degrading fungi/bacteria is expected to built-up a new experimental platform to enhance biomass utilization.
Substrate-dependent expression of AALPMOs in lignocellulolyitc microbes
Secretome profiling of Gloeophyllum trabeum and Pleurotus ostreatus cultured on sugarcane bagasse showed the involvement of secreted oxalate decarboxylase, intracellular AA6 quinone oxidoreductases along with cellulases, hemicellulases and auxiliary proteins in deconstructing lignocellulosic biomass [164]. Similarly, proteomic analysis of T. reesei Rut C-30 grown on sugar cane bagasse and various cellulosic substrates revealed the secretion of AA9 family enzymes (EGIV, EGVII), whereas AA9 family enzyme ECIV was secreted when T. reesei Rut C-30 was grown on xylose [165]. Genomic analysis of ascomycete Podospora anserina showed the presence of 39 genes encoding auxiliary activity (AA) enzymes (AA9, N = 33; AA11, N = 5 and AA13, N = 1). However, when it grew on sugar beet pulp, avicel cellulose and soybean hulls, 7, 14 and 20 AA9 enzymes were secreted, respectively [166, 167]. It is worth mentioning that P. anserina strain S mat + (CBS143333) harbours 146 genes encoding different AA enzymes (AA1 = 16, AA2 = 4, AA3 = 30, AA4 = 4, 2, AA6 = 1, AA7 = 32, AA8 = 9, AA9 = 33, AA11 = 6, AA12 = 4, AA13 = 1 and AA16 = 1). Nonetheless, CBS143333 secretes 60 AA enzymes (AA1 = 4, AA2 = 1, AA3 = 12, AA5 = 1, AA7 = 15, AA8 = 7, AA9 = 17, AA12 = 2 and AA16 = 1) when cultured in medium containing glucuronoarabinoxylan and wheat straw lignin isolate [168]. Substrate-dependent expression of several cellulases, hemicellulases, ligninolytic enzymes and LPMOs acting on cellulose and hemicellulose by P. anserina offers an opportunity for the researchers to prepare custom-designed enzyme cocktails for the maximum utilization of lignocellulose biomass at industrial level [169]. Recently, a novel AA16 from Aspergillus aculeatus was identified through proteomic analysis and was expressed in Pichia pastoris. Further analysis revealed its oxidative cleavage property on cellulose and showed that increased T. reesei cellobiohydrolase I activity synergistically [33]. A recent investigation has showed that LPMO ScAA10C from Streptomyces coelicolor photocatalytically induced cellulose oxidation [47, 137]. In addition, AA10 called SgLPMO10A identified from Streptomyces griseus was reported to enhance cellulose degradation by GHs [170]. Similarly, the AA10 identified from Photorhabdus luminescens contains isoleucine in place of conserved alanine (present in other bacterial LPMOs) in its active site with the ability to act on α‐ and β‐chitin and C1‐oxidation [48]. These findings highlight and reinforce the role of functional proteomics in the identification of appropriate enzyme cocktails with necessary LPMOs for efficient biomass utilization.
Probing microbial secretomes for lignin-degrading enzymes
Secretomic analysis of Fusarium solani MYA 4552 grown on medium containing solid wood rich in lignin revealed the presence of several proteins such as laccases, oxidoreductases, dioxygenase, superoxide dismutases and catalase. Among them, laccases and Mn-independent peroxidase were found to be the major enzymes responsible for the lignin depolymerization [171]. Similarly, proteomic studies have revealed that lignin degradation in Sphingobacterium sp. T2 and Pseudomonas putida A514 was carried out by Mn dependent superoxide dismutases (Sod1 and Sod2) [172] and Mn independent B subfamily dye-decolorizing peroxidases (DypBs 1 and 2) [173], respectively. Conversely, Enterobacter lignolyticus SCF1 depolymerizes lignin using catalases, peroxidases, oxidoreductases, ABC transporter proteins, enzymes involved in the glutathione biosynthesis pathway and the 4-hydroxyphenylacetate pathway [174]. Whereas proteomic investigation of co-cultivation of Rhodococcus strains on lignin-containing medium has shown that the depolymerization of lignin through up-regulation of extracellular peroxidases and oxidases, as well as enzymes involved in the β-ketoadipate and the phenylacetic acid pathway for metabolism of lignin degradation products [175].
A recent proteomic investigation on the outer membrane vesicles of Pseudomonas putida KT2440 sheds novel insights on the catabolism of lignin. Following important enzymes such as vanillin dehydrogenase, vanillate O-demethylase, 4-carboxymuconolactone decarboxylase, 3-oxoadipate enol-lactonase, betaketoadipyl-CoA thiolase, reductases (FMN-dependent NADH-azoreductase and glutathionyl-hydroquinone reductase), alcohol dehydrogenases, ABC transporter substrate-binding proteins (phosphonate, methionine and polyamine) and quercetin-2,3-dioxygenase are selectively packed inside the outer membrane vesicles [176]. Interestingly, supplementation of outer membrane vesicles from wild-type P. putida KT2440 cultured in lignin-rich medium enhanced the growth of P. putida KT2440 mutant strains lacking protocatechuate 3,4-dioxygenase and 4-hydroxybenzoate 3-monooxygenase [176]. These encouraging findings certainly offer a new avenue to overcome the hindrance of bacterial growth during lignin depolymerization.
Comparative genomic and proteomic analysis of Pandoraea sp. ISTKB cultured in the presence of kraft lignin and vanillic acid have revealed the presence of functionally active uncommon aerobic ‘-CoA’-mediated, ortho and meta ring-breaking aromatic compound degradation pathways and robust oxidoreductases. In addition, Pandoraea sp. ISTKB harbours an active polyhydroxyalkanoate synthesis pathway that can utilize lignin and lignin-derived aromatic compounds as carbon source [177]. In a recent secretomic investigation, marine Agaricomycete Peniophora sp. CBMAI 1063 has been shown to produce 2 laccases (Pnh_LAC1 [major secretory protein] and Pnh_LAC2) to degrade lignin. Further analysis involving the addition of the Pnh_LAC1-mediator system has shown that effectively removes lignin from sugarcane bagasse and makes the cellulose and hemicellulose more accessible to CAZymes [178]. Similarly, the secretome of Podospora anserina cultured on wheat straw lignin resulted in 20 and a threefold increase in the secretion of laccases and H2O2-producing enzymes, respectively [168]. These findings emphasize the importance of H2O2 in the deconstruction of lignin by laccases. Although numerous bacteria and fungi have been reported to depolymerize lignin, yet, only a few have undergone in-depth proteomic analyses (Table 2). Hence, it is necessary to analyse the secretome of lignin-degrading microorganisms to decode the underlying mechanism(s). In addition, the global proteomic analysis of lignin-degrading microbes should be carried out to understand the metabolism of lignin derived aromatic compounds, associated bottlenecks and selection of target genes/pathways for genetic manipulation to develop high efficiency lignin-degrading strains as well as to produce the desired end products.
Metaproteomics in lignocellulosic biomass degradation
Metaproteomics is a promising approach to identify the structure, function and dynamics of the complete proteome of the microbial community present in any environmental sample [179]. In addition, it offers an opportunity to analyse the proteome of solitary species within the microbial community. In general, the metaproteomic analysis involved seven major steps namely, sample collection, cell lysis, protein extraction, protein separation, trypsin digestion, peptide fractionation, mass spectrometry analysis followed by database search for further protein identification [179]. Lignocellulosic biomass degradation is carried out by numerous bacteria, actinomycetes and fungi together by complementing each other through secretion of different CAZymes, esterases and oxidoreductases [180]. Insect gut microbiomes are considered as an endless reservoir for the construction of synthetic microbial consortia for biomass utilization and identification of enzymes of industrial importance [181,182,183,184]. Hence gut microbiome of insects has been extensively analysed through whole metagenome sequencing to identify novel glycoside hydrolases, lignin-degrading enzymes, transport proteins, etc. [185,186,187]. In addition, several cellulase and hemicellulose-encoding genes derived from the insect gut microbiomes have been heterologously expressed [184, 188]. However, integrated metagenomic, metatranscriptomic and metaproteomic analyses are required to analyse the metabolic pathways, enzymes involved in biomass degradation and their regulation at the community level [189,190,191,192]. Sulfo-NHS-SS-biotin labelling of metasecretome and biomass bound proteome, i.e. meta-surface-proteome of the biomass-degrading microbial community has been shown to increase the depth of identification of enzymes [193, 194].
When compared to other ‘omics’ technologies, metaproteomics provides more valuable precedence for functional analyses. It can also be used as a precision tool to emphasize the investigation of subtle functional genes from large metagenomic data. Metaproteomics has an enormous perspective in the field of biomass utilization, particularly by identifying the functional metasecretome to give a new angle to environmental catalysis. In particular, metaproteomics helps select specific enzymes from the synthetic and naturally occurring microbial communities for heterologous expression in industrially demanded strains, making synthetic enzyme cocktails and augmenting the hydrolysis potential of currently available secretome preparations [195]. For example, metasecretomic analysis of a microbial consortium EMSD5 harbouring members of the phyla Proteobacteria, Firmicutes and Bacteroidetes cultured on corn stover showed the presence of various glycosyl hydrolases especially multiple xylanases followed by cellulases. By breaking down xylan, EMSD5 secretome significantly increased the hydrolysis of pretreated corn stover by commercially available T. reesei secretome preparation [196]. Researchers have identified several CAZymes from the metasecretome of switchgrass thermophilic bacterial consortium and rice straw adapted microbial consortia [197, 198].
Though metaproteomic analysis is considered as a promising tool to evaluate the functional state of the microbiota present in the different environmental samples, proteomic information related to the lignocellulosic biomass-degrading microbial community remains limited. Due to challenges associated with metaproteomic analysis such as complexity of the environmental samples, quality of extracted proteins, peptide extraction, requirement of long nano-LC run time, lack of protein sequence database for protein identification, less coverage of proteomic information from low abundant species, hence limiting the application of metaproteomic analysis [198]. In spite of the above mentioned challenges, the development of specific metaproteomic data analysis tools such as MetaProteomAnalyzer (MPA portable) [199], Unipept 4.0 [200], Calis-p [201], ComPIL 2.0 [202], Unipept Desktop [203] and metaQuantome [204] are encouraging the in-depth metaproteomic research. Furthermore, a newly developed mass spectrometer called Trapped Ion Mobility Mass Spectrometer (TIMS) can achieve the acquisition of more than 60,0000 spectra in 2 h of liquid chromatography runs [205]. In light of the newly developed analysis methods, research combining metagenomic and metasecretomic analyses is expected to shed more light on the identification of novel enzymes, synergistic mechanisms and microbial consortia for biomass utilization.
Conclusion and future perspectives
The replacement of fossil fuels with biofuels produced from renewable sources such as lignocellulosic biomass is an inevitable phenomenon in the coming years due to the necessity for greenhouse gas emissions reduction and the energy crisis. Forestry and agricultural biomass wastes are an abundant and renewable source of carbon for the production of bio-energy and numerous high-value chemicals. At industrial scale, physical, chemical and thermochemical methods are preferably used rather than biochemical methods for biomass utilization. However, a full utilization of biomass is not yet reached, due its chemical recalcitrance.
In addition, reducing the cost of converting biomass into fermentable sugars is a major challenge for biomass-processing industries. Microbes are the major players involved in biomass degradation, nutrient cycling and their application is considered as an environmentally friendly and a promising economically feasible way to obtain fermentable sugars. In recent years proteomic analysis has been used extensively to explore the secretomes of biomass-degrading bacterial/fungal strains and microbial consortia to identify novel enzymes, to decode the substrate-dependent expression of lignocellulases and post-translational modifications.
With the availability of optimized protein extraction protocols, and absolute quantitative proteomic approaches such as ITRAQ and TMT, each protein can be identified and quantified simultaneously in multiple samples. This milestone can enable researchers to design specific enzyme cocktails with the perfect combination and absolute composition of enzymes for an effective biomass pretreatment and utilization. Although currently available proteomic analysis methods are very good for analysing the secretome of microorganisms grown on different biomass, the extraction and purification of secreted proteins from the environmental samples still requires further improvement. Furthermore, several key enzymes identified from the secretome of lignocellulolytic microbes should be heterologously expressed and studied further to validate their involvement in biomass deconstruction.
Lignin provides recalcitrance to lignocellulosic biomass and acts as a barrier against the saccharification of cellulose. Thus, delignification of lignocellulosic biomass is required to achieve a maximum saccharification of cellulose. For a biological deconstruction of lignocellulosic biomass, enzyme cocktails should be complemented by ligninolytic enzymes. Several proteomics studies showed the consecutive secretion of CAZymes (GHs, GTs, PLs, CEs and AAs) in lignocellulolytic bacteria and fungi. Delignification is essential for an effective saccharification and reduction of cellulolytic enzyme consumption. Although enzyme cocktails and microbial consortia have been used for lignocellulosic biomass deconstruction, less attention has been paid to ligninolytic enzymes. Given the advantages of delignification, the engineering of hyper ligninolytic enzymes secreting strains and the development of ligninolytic microbial consortia should be carried out for biomass pretreatment.
In order to develop a lignocellulosic biomass-specific enzyme cocktails, a comprehensive screening of so far identified lignocellulolytic enzymes from the secretomes of various bacteria, fungi and synthetic/natural microbial communities should be done to select potential candidate enzymes, capable of performing at a wide pH and temperature range. Post-translational modifications (PTMs) (for example N- and O-glycosylation) play a key role in the stability and activity of biomass-hydrolyzing enzymes, but most of the secretomic studies lack information about PTMs. Therefore, future studies involving the identification of important PTMs and finding suitable hosts for heterologous production of target enzymes with appropriate PTMs is expected to overcome the low lignocellulolytic efficiency of heterologously produced enzymes.
In addition, the engineering of industrial strains to simultaneously secrete multiple ligninolytic enzymes is required to reduce biofuels and other commercially important chemicals production costs. The combination of the secretome of different organisms is one of the ways to cut down the enzyme production costs and make use of the synergistic effect among the enzymes in biomass hydrolysis. For example, T. reesei secretome is rich in endoglucanases and exoglucanases, whereas members of Aspergillus secrete more β-glucosidases and combining the secretomes of T. reesei and Aspergillus, along with a bacterial/fungal secretome-rich in the auxiliary enzymes could improve the biomass hydrolysis and reduce the enzymes’s production costs. Similarly, further attention should be paid on the development of more hyper lignocellulolytic enzymes-secreting strains. The development of enzymes can withstand under harsh reaction conditions through enzyme engineering, development of simple methods for reusing/immobilizing enzyme cocktails to obtain complete biomass hydrolysis.
Availability of data and materials
Not applicable.
Abbreviations
- MnP:
-
Manganese peroxidase
- LiP:
-
Lignin peroxidase
- VP:
-
Versatile peroxidase
- DyP:
-
Dye-decolorizing peroxidase
- SMF:
-
Submerged fermentation
- SSF:
-
Solid-state fermentation
- SQF:
-
Sequential fermentation
- CCP:
-
Commercial cellulase preparations
- MALDI-TOF:
-
Matrix-assisted laser desorption/ionization
- SDS-PAGE:
-
Sodium dodecyl sulphate–polyacrylamide gel electrophoresis
- LC–MS:
-
Liquid chromatography–mass spectrometry
- iTRAQ:
-
Isobaric tags for relative and absolute quantitation
- CDH:
-
Cellobiose dehydrogenase
- GHs:
-
Glycosyl hydrolases
- LPMOs:
-
Lytic polysaccharide monooxygenases
- AA:
-
Auxiliary activity
- PLs:
-
Polysaccharide lyases
- CEs:
-
Carbohydrate esterases
- GTs:
-
Glycosyltransferases
- CAZymes:
-
Carbohydrate-active enzymes
- CMC:
-
Carboxy methyl cellulose
- MCOs:
-
Mulita-copper oxidoreductases
- SILAC:
-
Stable isotope labelling by amino acids in cell culture
- ICAT:
-
Isotope-coded affinity tag
- TMT:
-
Tandem mass tag
- GeLCMS:
-
Gel-enhanced liquid chromatography–mass spectrometry
- MudPIT:
-
Multidimensional protein identification technology
- FT-ICR:
-
Fourier transform ion cyclotron resonance
- PTMs:
-
Post-translational modifications
References
Bajpai P, Chapter 7: Advantages and disadvantages of biomass utilization. In: Pratima B, editors. Biomass to energy conversion technologies. Elsevier; 2020, pp. 169–173, ISBN 9780128184004. https://doi.org/10.1016/B978-0-12-818400-4.00007-4.
Centore M, Hochman G, Zilberman D (2014) Worldwide survey of biodegradable feedstocks, waste-to-energy technologies, and adoption of technologies. In: Pinto A, Zilberman D. editors, Modeling, dynamics, optimization and bioeconomics I. Springer Proceedings in Mathematics and Statistics, vol. 73. Springer, Cham. https://doi.org/10.1007/978-3-319-04849-9_11.
GEA, 2012. Global energy assessment—toward a sustainable future. Scenario Database Connected to Assessment. Cambridge University Press. Available from: http://www.iiasa.ac.at/web-apps/ene/geadb/. Lauri P, Havlík P, Kindermann G, Forsell N, Böttcher H, Obersteiner M. Woody biomass energy potential in 2050. Energy Policy. 2014;66:19–31.
Lauri P, Havlík P, Kindermann G, Forsell N, Böttcher H, Obersteiner M. Woody biomass energy potential in 2050. Energy Policy. 2014;66:19–31. https://doi.org/10.1016/j.enpol.2013.11.033.
Mood SH, Golfeshan AH, Tabatabaei M, Jouzani GS, Najafi GH, Gholami M, Ardjmand M. Lignocellulosic biomass to bioethanol, a comprehensive review with a focus on pretreatment. Renew Sustain Energy Rev. 2013;27:77–93. https://doi.org/10.1016/j.rser.2013.06.033.
Mori T, Tsuboi Y, Ishida N, Nishikubo N, Demura T, Kikuchi J. Multidimensional high-resolution magic angle spinning and solution-state NMR characterization of 13 C-labeled plant metabolites and lignocellulose. Sci Rep. 2015;5:1–2. https://doi.org/10.1038/srep11848.
Ralph J, Lundquist K, Brunow G, Lu F, Kim H, Schatz PF, et al. Lignins: natural polymers from oxidative coupling of 4-hydroxyphenyl-propanoids. Phytochem Rev. 2004;3(1):29–60. https://doi.org/10.1023/B:PHYT.0000047809.65444.a4.
Patil RA. Cleavage of acetyl groups for acetic acid production in Kraft pulp mills. Thesis, University of Maine; 2012.
Chen H, Liu J, Chang X, Chen D, Xue Y, Liu P, et al. A review on the pretreatment of lignocellulose for high-value chemicals. Fuel Process Technol. 2017;160:196–206. https://doi.org/10.1016/j.fuproc.2016.12.007.
Lopes AM, Ferreira Filho EX, Moreira LR. An update on enzymatic cocktails for lignocellulose breakdown. J Appl Microbiol. 2018;125(3):632–45. https://doi.org/10.1111/jam.13923.
Paës G, Navarro D, Benoit Y, Blanquet S, Chabbert B, Chaussepied B, et al. Tracking of enzymatic biomass deconstruction by fungal secretomes highlights markers of lignocellulose recalcitrance. Biotechnol Biofuels. 2019;12(1):76. https://doi.org/10.1186/s13068-019-1417-8.
Bugg TD, Ahmad M, Hardiman EM, Rahmanpour R. Pathways for degradation of lignin in bacteria and fungi. Nat Prod Rep. 2011;28(12):1883–96. https://doi.org/10.1039/C1NP00042J.
Murphy C, Powlowski J, Wu M, Butler G, Tsang A. Curation of characterized glycoside hydrolases of fungal origin. Database. 2011. https://doi.org/10.1093/database/bar020.
Cragg SM, Beckham GT, Bruce NC, Bugg TD, Distel DL, Dupree P, Etxabe AG, Goodell BS, Jellison J, McGeehan JE, McQueen-Mason SJ. Lignocellulose degradation mechanisms across the Tree of Life. Curr Opin Chem Biol. 2015;29:108–19. https://doi.org/10.1016/j.cbpa.2015.10.018.
Andlar M, Rezić T, Marđetko N, Kracher D, Ludwig R, Šantek B. Lignocellulose degradation: an overview of fungi and fungal enzymes involved in lignocellulose degradation. Eng Life Sci. 2018;18(11):768–78. https://doi.org/10.1002/elsc.201800039.
Coines J, Raich L, Rovira C. Modeling catalytic reaction mechanisms in glycoside hydrolases. Curr Opin Chem Biol. 2019;53:183–91. https://doi.org/10.1016/j.cbpa.2019.09.007.
Szymanski DB, Cosgrove DJ. Dynamic coordination of cytoskeletal and cell wall systems during plant cell morphogenesis. Curr Biol. 2009;19(17):R800–11. https://doi.org/10.1016/j.cub.2009.07.056.
Tyler L, Bragg JN, Wu J, Yang X, Tuskan GA, Vogel JP. Annotation and comparative analysis of the glycoside hydrolase genes in Brachypodium distachyon. BMC Genomics. 2010;11(1):1–21. https://doi.org/10.1186/1471-2164-11-600.
Palomares-Rius JE, Hirooka Y, Tsai IJ, Masuya H, Hino A, Kanzaki N, et al. Distribution and evolution of glycoside hydrolase family 45 cellulases in nematodes and fungi. BMC Evol Biol. 2014;14(1):1–2. https://doi.org/10.1186/1471-2148-14-69.
Shen D, Wang J, Dong Y, Zhang M, Tang Z, Xia Q, Nyawira KT, Jing M, Dou D, Xia A. The glycoside hydrolase 18 family chitinases are associated with development and virulence in the mosquito pathogen Pythium guiyangense. Fungal Genet Biol. 2020;135: 103290. https://doi.org/10.1016/j.fgb.2019.103290.
Baker P, Hill PJ, Snarr BD, Alnabelseya N, Pestrak MJ, Lee MJ, et al. Exopolysaccharide biosynthetic glycoside hydrolases can be utilized to disrupt and prevent Pseudomonas aeruginosa biofilms. Sci Adv. 2016;2(5): e1501632. https://doi.org/10.1126/sciadv.1501632.
Snarr BD, Baker P, Bamford NC, Sato Y, Liu H, Lehoux M, Gravelat FN, Ostapska H, Baistrocchi SR, Cerone RP, Filler EE. Microbial glycoside hydrolases as antibiofilm agents with cross-kingdom activity. Proc Natl Acad Sci USA. 2017;114(27):7124–9. https://doi.org/10.1073/pnas.1702798114.
Su T, He J, Li N, Liu S, Xu S, Gu L. A Rational designed PslG with normal biofilm hydrolysis and enhanced resistance to trypsin-like protease digestion. Front Microbiol. 2020;11:760. https://doi.org/10.3389/fmicb.2020.00760.
Sinnott ML. Catalytic mechanism of enzymic glycosyl transfer. Chem Rev. 1990;90(7):1171–202. https://doi.org/10.1021/cr00105a006.
Cazy. CAZy: Carbohydrate-active enzyme database. Available at www.cazy.org. 2020. Accessed 20 Jul 2020.
Chrismas N, Cunliffe M. Depth-dependent mycoplankton glycoside hydrolase gene activity in the open ocean—evidence from the Tara Oceans eukaryote metatranscriptomes. ISME J. 2020;14(9):2361–5. https://doi.org/10.1038/s41396-020-0687-2.
Vaaje-Kolstad G, Westereng B, Horn SJ, Liu Z, Zhai H, Sørlie M, Eijsink VG. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science. 2010;330(6001):219–22. https://doi.org/10.1126/science.1192231.
Agger JW, Isaksen T, Várnai A, Vidal-Melgosa S, Willats WG, Ludwig R, Horn SJ, Eijsink VG, Westereng B. Discovery of LPMO activity on hemicelluloses shows the importance of oxidative processes in plant cell wall degradation. Proc Natl Acad Sci USA. 2014;111(17):6287–92. https://doi.org/10.1073/pnas.1323629111.
Vaaje-Kolstad G, Horn SJ, van Aalten DM, Synstad B, Eijsink VG. The non-catalytic chitin-binding protein CBP21 from Serratia marcescens is essential for chitin degradation. J Biol Chem. 2005;280(31):28492–7. https://doi.org/10.1074/jbc.M504468200.
Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels. 2013;6(1):1–4. https://doi.org/10.1186/1754-6834-6-41.
Leggio LL, Simmons TJ, Poulsen JC, Frandsen KE, Hemsworth GR, Stringer MA, Von Freiesleben P, Tovborg M, Johansen KS, De Maria L, Harris PV. Structure and boosting activity of a starch-degrading lytic polysaccharide monooxygenase. Nat Commun. 2015;6(1):1–9. https://doi.org/10.1038/ncomms6961.
Couturier M, Ladeveze S, Sulzenbacher G, Ciano L, Fanuel M, Moreau C, et al. Lytic xylan oxidases from wood-decay fungi unlock biomass degradation. Nat Chem Biol. 2018;14(3):306. https://doi.org/10.1038/nchembio.2558.
Courtade G, Aachmann FL. Chitin-active lytic polysaccharide monooxygenases. In: Yang Q, Fukamizo T, editors, Targeting chitin-containing organisms. Advances in Experimental Medicine and Biology, vol. 1142. 2019; Springer, Singapore. https://doi.org/10.1007/978-981-13-7318-3_6.
Filiatrault-Chastel C, Navarro D, Haon M, Grisel S, Herpoël-Gimbert I, Chevret D, Fanuel M, Henrissat B, Heiss-Blanquet S, Margeot A, Berrin JG. AA16, a new lytic polysaccharide monooxygenase family identified in fungal secretomes. Biotechnol Biofuels. 2019;12(1):1–5. https://doi.org/10.1186/s13068-019-1394-y.
Johansen KS. Discovery and industrial applications of lytic polysaccharide mono-oxygenases. Iochem Soc Trans. 2016;44(1):143–9. https://doi.org/10.1042/BST20150204.
Loose JS, Forsberg Z, Fraaije MW, Eijsink VG, Vaaje-Kolstad G. A rapid quantitative activity assay shows that the Vibrio cholerae colonization factor GbpA is an active lytic polysaccharide monooxygenase. FEBS Lett. 2014;588(18):3435–40. https://doi.org/10.1016/j.febslet.2014.07.036.
Paspaliari DK, Loose JS, Larsen MH, Vaaje-Kolstad G. Listeria monocytogenes has a functional chitinolytic system and an active lytic polysaccharide monooxygenase. The FEBS J. 2015;282(5):921–36. https://doi.org/10.1111/febs.13191.
Quinlan RJ, Sweeney MD, Leggio LL, Otten H, Poulsen JC, Johansen KS, et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc Natl Acad Sci USA. 2011;108(37):15079–84. https://doi.org/10.1073/pnas.1105776108.
Hemsworth GR, Johnston EM, Davies GJ, Walton PH. Lytic polysaccharide monooxygenases in biomass conversion. Trends Biotechnol. 2015;33(12):747–61. https://doi.org/10.1016/j.tibtech.2015.09.006.
Harris PV, Welner D, McFarland KC, Re E, Navarro Poulsen JC, Brown K, et al. Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry. 2010;49(15):3305–16. https://doi.org/10.1021/bi100009p.
Petrović DM, Bissaro B, Chylenski P, Skaugen M, Sørlie M, Jensen MS, et al. Methylation of the N-terminal histidine protects a lytic polysaccharide monooxygenase from auto-oxidative inactivation. Protein Sci. 2018;27(9):1636–50. https://doi.org/10.1002/pro.3451.
Vaaje-Kolstad G, Forsberg Z, Loose JS, Bissaro B, Eijsink VG. Structural diversity of lytic polysaccharide monooxygenases. Curr Opin Struct Biol. 2017;44:67–76. https://doi.org/10.1016/j.sbi.2016.12.012.
Beeson WT, Phillips CM, Cate JH, Marletta MA. Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J Am Chem Soc. 2012;134(2):890–2. https://doi.org/10.1021/ja210657t.
Isaksen T, Westereng B, Aachmann FL, Agger JW, Kracher D, Kittl R, Ludwig R, Haltrich D, Eijsink VG, Horn SJ. A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J Biol Chem. 2014;289(5):2632–42. https://doi.org/10.1074/jbc.M113.530196.
Eijsink VG, Petrovic D, Forsberg Z, Mekasha S, Røhr ÅK, Várnai A, Bissaro B, Vaaje-Kolstad G. On the functional characterization of lytic polysaccharide monooxygenases (LPMOs). Biotechnol Biofuels. 2019;12(1):1–6. https://doi.org/10.1186/s13068-019-1392-0.
Müller G, Chylenski P, Bissaro B, Eijsink VG, Horn SJ. The impact of hydrogen peroxide supply on LPMO activity and overall saccharification efficiency of a commercial cellulase cocktail. Biotechnol Biofuels. 2018;11(1):1–7. https://doi.org/10.1186/s13068-018-1199-4.
Bissaro B, Kommedal E, Røhr ÅK, Eijsink VG. Controlled depolymerization of cellulose by light-driven lytic polysaccharide oxygenases. Nat Commun. 2020;11(1):1–2. https://doi.org/10.1038/s41467-020-14744-9.
Munzone A, El Kerdi B, Fanuel M, Rogniaux H, Ropartz D, Réglier M, et al. Characterization of a bacterial copper-dependent lytic polysaccharide monooxygenase with an unusual second coordination sphere. FEBS J. 2020;287(15):3298–314. https://doi.org/10.1111/febs.15203.
Phillips CM, Iavarone AT, Marletta MA. Quantitative proteomic approach for cellulose degradation by Neurospora crassa. J Proteome Res. 2011;10(9):4177–85. https://doi.org/10.1021/pr200329b.
Sun P, Laurent CV, Scheiblbrandner S, Frommhagen M, Kouzounis D, Sanders MG, van Berkel WJ, Ludwig R, Kabel MA. Configuration of active site segments in lytic polysaccharide monooxygenases steers oxidative xyloglucan degradation. Biotechnol Biofuels. 2020;13:1–9. https://doi.org/10.1186/s13068-020-01731-x.
Arfi Y, Chevret D, Henrissat B, Berrin JG, Levasseur A, Record E. Characterization of salt-adapted secreted lignocellulolytic enzymes from the mangrove fungus Pestalotiopsis sp. Nat Commun. 2013;4(1):1–9. https://doi.org/10.1038/ncomms2850.
Patel I, Kracher D, Ma S, Garajova S, Haon M, Faulds CB, Berrin JG, Ludwig R, Record E. Salt-responsive lytic polysaccharide monooxygenases from the mangrove fungus Pestalotiopsis sp NCi6. Biotechnol Biofuels. 2016;9(1):1–2. https://doi.org/10.1186/s13068-016-0520-3.
Marx IJ, Van Wyk N, Smit S, Jacobson D, Viljoen-Bloom M, Volschenk H. Comparative secretome analysis of Trichoderma asperellum S4F8 and Trichoderma reesei Rut C30 during solid-state fermentation on sugarcane bagasse. Biotechnol Biofuels. 2013;6(1):1–3. https://doi.org/10.1186/1754-6834-6-172.
Debeire P, Delalande F, Habrylo O, Jeltsch JM, Van Dorsselaer A, Phalip V. Enzymatic cocktails produced by Fusarium graminearum under submerged fermentation using different lignocellulosic biomasses. FEMS Microbiol. 2014;355(2):116–23. https://doi.org/10.1111/1574-6968.12467.
Nekiunaite L, Petrović DM, Westereng B, Vaaje-Kolstad G, Hachem MA, Várnai A, et al. Fg LPMO 9A from Fusarium graminearum cleaves xyloglucan independently of the backbone substitution pattern. FEBS Lett. 2016;590(19):3346–56. https://doi.org/10.1002/1873-3468.12385.
Vu VV, Beeson WT, Span EA, Farquhar ER, Marletta MA. A family of starch-active polysaccharide monooxygenases. Proc Natl Acad Sci USA. 2014;111(38):13822–7. https://doi.org/10.1073/pnas.1408090111.
Gong W, Zhang H, Liu S, Zhang L, Gao P, Chen G, Wang L. Comparative secretome analysis of Aspergillus niger, Trichoderma reesei, and Penicillium oxalicum during solid-state fermentation. Appl Biochem Biotechnol. 2015;177(6):1252–71. https://doi.org/10.1007/s12010-015-1811-z.
Borin GP, Sanchez CC, de Souza AP, de Santana ES, de Souza AT, Leme AF, Squina FM, Buckeridge M, Goldman GH, de Castro Oliveira JV. Comparative secretome analysis of Trichoderma reesei and Aspergillus niger during growth on sugarcane biomass. PLoS ONE. 2015;10(6): e0129275. https://doi.org/10.1371/journal.pone.0129275.
Nekiunaite L, Arntzen MØ, Svensson B, Vaaje-Kolstad G, Abou HM. Lytic polysaccharide monooxygenases and other oxidative enzymes are abundantly secreted by Aspergillus nidulans grown on different starches. Biotechnol Biofuels. 2016;9(1):1–6. https://doi.org/10.1186/s13068-016-0604-0.
Jagadeeswaran G, Gainey L, Prade R, Mort AJ. A family of AA9 lytic polysaccharide monooxygenases in Aspergillus nidulans is differentially regulated by multiple substrates and at least one is active on cellulose and xyloglucan. Appl Microbiol Biotechnol. 2016;100(10):4535–47. https://doi.org/10.1007/s00253-016-7505-9.
Dos Santos HB, Bezerra TM, Pradella JG, Delabona P, Lima D, Gomes E, et al. Myceliophthora thermophila M77 utilizes hydrolytic and oxidative mechanisms to deconstruct biomass. AMB Express. 2016;6(1):1–2. https://doi.org/10.1186/s13568-016-0276-y.
Schneider WD, Gonçalves TA, Uchima CA, Couger MB, Prade R, Squina FM, et al. Penicillium echinulatum secretome analysis reveals the fungi potential for degradation of lignocellulosic biomass. Biotechnol Biofuels. 2016;9(1):1–26. https://doi.org/10.1186/s13068-016-0476-3.
Mahajan C, Basotra N, Singh S, Di Falco M, Tsang A, Chadha BS. Malbranchea cinnamomea: a thermophilic fungal source of catalytically efficient lignocellulolytic glycosyl hydrolases and metal dependent enzymes. Bioresour Technol. 2016;200:55–63. https://doi.org/10.1016/j.biortech.2015.09.113.
Fanuel M, Garajova S, Ropartz D, McGregor N, Brumer H, Rogniaux H, Berrin JG. The Podospora anserina lytic polysaccharide monooxygenase Pa LPMO9H catalyzes oxidative cleavage of diverse plant cell wall matrix glycans. Biotechnol Biofuels. 2017;10(1):1. https://doi.org/10.1186/s13068-017-0749-5.
Kojima Y, Várnai A, Ishida T, Sunagawa N, Petrovic DM, Igarashi K, et al. A lytic polysaccharide monooxygenase with broad xyloglucan specificity from the brown-rot fungus Gloeophyllum trabeum and its action on cellulose–xyloglucan complexes. Appl Environ Microbiol. 2016;82(22):6557–72. https://doi.org/10.1128/AEM.01768-16.
Hegnar OA, Petrovic DM, Bissaro B, Alfredsen G, Várnai A, Eijsink VG. PH-dependent relationship between catalytic activity and hydrogen peroxide production shown via characterization of a lytic polysaccharide monooxygenase from Gloeophyllum trabeum. Appl Environ Microbiol 2019;85(5). https://doi.org/10.1128/AEM.02612-18.
Navarro D, Rosso MN, Haon M, Olivé C, Bonnin E, Lesage-Meessen L, Chevret D, Coutinho PM, Henrissat B, Berrin JG. Fast solubilization of recalcitrant cellulosic biomass by the basidiomycete fungus Laetisaria arvalis involves successive secretion of oxidative and hydrolytic enzymes. Biotechnol Biofuels. 2014;7(1):1–4. https://doi.org/10.1186/s13068-014-0143-5.
Martinez D, Larrondo LF, Putnam N, Gelpke MD, Huang K, Chapman J, et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat Biotechnol. 2004;22(6):695–700. https://doi.org/10.1038/nbt967.
Liu J, Yang J, Wang R, Liu L, Zhang Y, Bao H, Jang JM, Wang E, Yuan H. Comparative characterization of extracellular enzymes secreted by Phanerochaete chrysosporium during solid-state and submerged fermentation. Int J Biol Macromol. 2020;152:288–94. https://doi.org/10.1016/j.ijbiomac.2020.02.256.
Couturier M, Navarro D, Chevret D, Henrissat B, Piumi F, Ruiz-Dueñas FJ, et al. Enhanced degradation of softwood versus hardwood by the white-rot fungus Pycnoporus coccineus. Biotechnol Biofuels. 2015;8(1):1–6. https://doi.org/10.1186/s13068-015-0407-8.
Miyauchi S, Navarro D, Grigoriev IV, Lipzen A, Riley R, Chevret D, Grisel S, Berrin JG, Henrissat B, Rosso MN. Visual comparative omics of fungi for plant biomass deconstruction. Front Microbiol. 2016;7:1335. https://doi.org/10.3389/fmicb.2016.01335.
Zhu N, Liu J, Yang J, Lin Y, Yang Y, Ji L, Li M, Yuan H. Comparative analysis of the secretomes of Schizophyllum commune and other wood-decay basidiomycetes during solid-state fermentation reveals its unique lignocellulose-degrading enzyme system. Biotechnol Biofuels. 2016;9(1):1–22. https://doi.org/10.1186/s13068-016-0461-x.
Frommhagen M, Koetsier MJ, Westphal AH, Visser J, Hinz SW, Vincken JP, et al. Lytic polysaccharide monooxygenases from Myceliophthora thermophila C1 differ in substrate preference and reducing agent specificity. Biotechnol Biofuels. 2016;9(1):1–7. https://doi.org/10.1186/s13068-016-0594-y.
Kadowaki MA, Varnai A, Jameson JK, Leite TAE, Costa-Filho AJ, Kumagai PS, Prade RA, Polikarpov I, Eijsink VG. Functional characterization of a lytic polysaccharide monooxygenase from the thermophilic fungus Myceliophthora thermophila. PLoS ONE. 2018;13(8):e0202148. https://doi.org/10.1371/journal.pone.0202148.
Cannella D, Möllers KB, Frigaard NU, Jensen PE, Bjerrum MJ, Johansen KS, et al. Light-driven oxidation of polysaccharides by photosynthetic pigments and a metalloenzyme. Nat Commun. 2016;7(1):1–8. https://doi.org/10.1038/ncomms11134.
Ladevèze S, Haon M, Villares A, Cathala B, Grisel S, Herpoël-Gimbert I, Henrissat B, Berrin JG. The yeast Geotrichum candidum encodes functional lytic polysaccharide monooxygenases. Biotechnol Biofuels. 2017;10(1):1–1. https://doi.org/10.1186/s13068-017-0903-0.
Liu B, Kognole AA, Wu M, Westereng B, Crowley MF, Kim S, Dimarogona M, et al. Structural and molecular dynamics studies of a C1-oxidizing lytic polysaccharide monooxygenase from Heterobasidion irregulare reveal amino acids important for substrate recognition. FEBS J. 2018;285(12):2225–42. https://doi.org/10.1111/febs.14472.
Singh RK, Oort BV, Möllers B, Russo DA, Singh R, Weihe H, et al. Detection and characterization of a novel copper-dependent intermediate in a lytic polysaccharide monooxygenase. Chem Eur J. 2020;26(2):454–63. https://doi.org/10.1002/chem.201903562.
Kumar A, Chandra R. Ligninolytic enzymes and its mechanisms for degradation of lignocellulosic waste in environment. Heliyon. 2020;6(2): e03170. https://doi.org/10.1016/j.heliyon.2020.e03170.
Bilal M, Iqbal HM. Ligninolytic enzymes mediated ligninolysis: an untapped biocatalytic potential to deconstruct lignocellulosic molecules in a sustainable manner. Catal Lett. 2020;150(2):524–43. https://doi.org/10.1007/s10562-019-03096-9.
Chaudhary I, Verma SR. Ligninolysis: roles of microbes and their extracellular enzymes. In: Shah M. editors, Microbial bioremediation and biodegradation. 2020; Springer, Singapore. https://doi.org/10.1007/978-981-15-1812-6_14.
Madhavi V, Lele SS. Laccase: properties and applications. BioResources. 2009;4(4):1694–717.
Maciel MJ, Ribeiro HC. Industrial and biotechnological applications of ligninolytic enzymes of the basidiomycota: a review. Electron J Biotechnol. 2010;13(6):14–5. https://doi.org/10.2225/vol13-issue6-fulltext-2.
Chowdhary P, More N, Yadav A, Bharagava RN. Ligninolytic enzymes: an introduction and applications in the food industry. In: Enzymes in food biotechnology. Academic Press, 2019; pp. 181–195. https://doi.org/10.1016/B978-0-12-813280-7.00012-8.
Zhu D, Adebisi WA, Ahmad F, Sethupathy S, Danso B, Sun J. Recent development of extremophilic bacteria and their application in biorefinery. Front Bioeng Biotechnol. 2020;8:483. https://doi.org/10.3389/fbioe.2020.00483.
Zhu D, Liang N, Zhang R, Ahmad F, Zhang W, Yang B, et al. Insight into depolymerization mechanism of bacterial laccase for lignin. ACS Sustain Chem Eng. 2020;8(34):12920–33. https://doi.org/10.1021/acssuschemeng.0c03457.
Zhu D, Si H, Zhang P, Geng A, Zhang W, Yang B, Qian WJ, Gabriel M, Sun J. Genomics and biochemistry investigation on the metabolic pathway of milled wood and alkali lignin-derived aromatic metabolites of Comamonas serinivorans SP-35. Biotechnol Biofuels. 2018;11(1):1–5. https://doi.org/10.1186/s13068-018-1341-3.
Ausec L, Zakrzewski M, Goesmann A, Schlüter A, Mandic-Mulec I. Bioinformatic analysis reveals high diversity of bacterial genes for laccase-like enzymes. PLoS ONE. 2011;6(10): e25724. https://doi.org/10.1371/journal.pone.0025724.
Shi Z, Han C, Zhang X, Tian L, Wang L. Novel synergistic mechanism for lignocellulose degradation by a thermophilic filamentous fungus and a thermophilic actinobacterium based on functional proteomics. Front Microbiol. 2020;11:2265. https://doi.org/10.3389/fmicb.2020.539438.
Díaz-García L, Bugg TD, Jiménez DJ. Exploring the lignin catabolism potential of soil-derived lignocellulolytic microbial consortia by a gene-centric metagenomic approach. Microb Ecol. 2020;80(4):885–96. https://doi.org/10.1007/s00248-020-01546-1.
Ouyang J, Yan M, Kong D, Xu L. A complete protein pattern of cellulase and hemicellulase genes in the filamentous fungus Trichoderma reesei. Biotechnol J Healthcare Nutr Technol. 2006;1(11):1266–74. https://doi.org/10.1002/biot.200600103.
Adav SS, Chao LT, Sze SK. Quantitative secretomic analysis of Trichoderma reesei strains reveals enzymatic composition for lignocellulosic biomass degradation. Mol Cell Proteomics. 2012;11(7):M111-012419. https://doi.org/10.1074/mcp.M111.012419.
Novy V, Nielsen F, Seiboth B, Nidetzky B. The influence of feedstock characteristics on enzyme production in Trichoderma reesei: a review on productivity, gene regulation and secretion profiles. Biotechnol Biofuels. 2019;12(1):1–6. https://doi.org/10.1186/s13068-019-1571-z.
Herpoël-Gimbert I, Margeot A, Dolla A, Jan G, Mollé D, Lignon S, Mathis H, Sigoillot JC, Monot F, Asther M. Comparative secretome analyses of two Trichoderma reesei RUT-C30 and CL847 hypersecretory strains. Biotechnol Biofuels. 2008;1(1):1–2. https://doi.org/10.1186/1754-6834-1-18.
dos Santos CL, Pedersoli WR, Antoniêto AC, Steindorff AS, Silva-Rocha R, Martinez-Rossi NM, et al. Comparative metabolism of cellulose, sophorose and glucose in Trichoderma reesei using high-throughput genomic and proteomic analyses. Biotechnol Biofuels. 2014;7(1):1–8. https://doi.org/10.1186/1754-6834-7-41.
Apaliya MT, Yang Q, Zhang H, Zheng X, Zhao L, Zhang X, Kwaw E, et al. Proteomics profile of Hanseniaspora uvarum enhanced with trehalose involved in the biocontrol efficacy of grape berry. Food chem. 2019;274:907–14. https://doi.org/10.1016/j.foodchem.2018.09.060.
Zhu D, Zhang P, Xie C, Zhang W, Sun J, Qian WJ, Yang B. Biodegradation of alkaline lignin by Bacillus ligniniphilus L1. Biotechnol Biofuels. 2017;10(1):1–4. https://doi.org/10.1186/s13068-017-0735-y.
Bianco L, Perrotta G. Methodologies and perspectives of proteomics applied to filamentous fungi: from sample preparation to secretome analysis. Int J Mol Sci. 2015;16(3):5803–29. https://doi.org/10.3390/ijms16035803.
Noor Z, Ahn SB, Baker MS, Ranganathan S, Mohamedali A. Mass spectrometry-based protein identification in proteomics—a review. Brief Bioinform. 2020;22:1620–38. https://doi.org/10.1093/bib/bbz163.
Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–67.
Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5(11):976–89. https://doi.org/10.1002/(SICI)1522-2683(19991201)20:18%3c3551::AID-ELPS3551%3e3.0.CO;2-2.
Colinge J, Masselot A, Giron M, Dessingy T, Magnin J. OLAV: towards high-throughput tandem mass spectrometry data identification. Proteomics. 2003;3(8):1454–63. https://doi.org/10.1002/pmic.200300485.
Duncan DT, Craig R, Link AJ. Parallel tandem: a program for parallel processing of tandem mass spectra using PVM or MPI and X! Tandem J Proteome Res. 2005;4(5):1842–7. https://doi.org/10.1021/pr050058i.
Mikesh LM, Ueberheide B, Chi A, Coon JJ, Syka JE, Shabanowitz J, Hunt DF. The utility of ETD mass spectrometry in proteomic analysis. Biochim Biophys Acta. 2006;1764(12):1811–22. https://doi.org/10.1016/j.bbapap.2006.10.003.
Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207. https://doi.org/10.1038/nature01511.
March RE. Ion Trap Mass Spectrometers. In: Lindon JC, George E. Tranter, Koppenaal DW, editors, Encyclopedia of spectroscopy and spectrometry. 3rd edn, Academic Press, 2017, pp. 330–337, ISBN 9780128032244, https://doi.org/10.1016/B978-0-12-409547-2.12675-7.
Fornelli L, Toby TK, Schachner LF, Doubleday PF, Srzentić K, DeHart CJ, Kelleher NL. Top-down proteomics: where we are, where we are going? J Proteomics. 2018;175:3. https://doi.org/10.1016/j.jprot.2017.02.002.
Kellie JF, Tran JC, Lee JE, Ahlf DR, Thomas HM, Ntai I, et al. The emerging process of Top Down mass spectrometry for protein analysis: biomarkers, protein-therapeutics, and achieving high throughput. Mol Biosyst. 2010;6(9):1532–9. https://doi.org/10.1039/C000896F.
Schirmer EC, Yates JR 3rd, Gerace L. MudPIT: a powerful proteomics tool for discovery. Discov Med. 2003;3(18):38–9.
Lundby A, Olsen JV. GeLCMS for in-depth protein characterization and advanced analysis of proteomes. In: Gevaert K, Vandekerckhove J. editors, Gel-free proteomics. Methods in molecular biology (methods and protocols), vol 753. Humana Press; 2011. https://doi.org/10.1007/978-1-61779-148-2_10.
Doerr A. Mass spectrometry-based targeted proteomics. Nat Methods. 2013;10(1):23. https://doi.org/10.1038/nmeth.2286.
Mann M. Origins of mass spectrometry-based proteomics. Nat Rev Mol Cell Biol. 2016;17(11):678. https://doi.org/10.1038/nrm.2016.135.
Pino LK, Rose J, O’Broin A, Shah S, Schilling B. Emerging mass spectrometry-based proteomics methodologies for novel biomedical applications. Biochem Soc Trans. 2020;48(5):1953–66. https://doi.org/10.1042/BST20191091.
Anand S, Samuel M, Ang CS, Keerthikumar S, Mathivanan S. Label-based and label-free strategies for protein quantitation. In: Keerthikumar S, Mathivanan S. editors, Proteome bioinformatics. Methods in Molecular Biology, vol 1549. 2017, Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-6740-7_4.
Dupree EJ, Jayathirtha M, Yorkey H, Mihasan M, Petre BA, Darie CC. A critical review of bottom-up proteomics: The good, the bad, and the future of this field. Proteomes. 2020;8(3):14. https://doi.org/10.3390/proteomes8030014.
Guo H, Wang XD, Lee DJ. Proteomic researches for lignocellulose-degrading enzymes: a mini-review. Bioresour Technol. 2018;265:532–41. https://doi.org/10.1016/j.biortech.2018.05.101.
Mohanram S, Amat D, Choudhary J, Arora A, Nain L. Novel perspectives for evolving enzyme cocktails for lignocellulose hydrolysis in biorefineries. Sustain Chem Process. 2013;1(1):1–2. https://doi.org/10.1186/2043-7129-1-15.
Berrin JG, Herpoel-Gimbert I, Ferreira NL, Margeot A, Heiss-Blanquet S. Use of cellulases from Trichoderma reesei in the twenty-first century—part II: optimization of cellulolytic cocktails for saccharification of lignocellulosic feedstocks. In: Biotechnology and biology of Trichoderma. Elsevier. 2014; 263–280.
Adsul M, Sandhu SK, Singhania RR, Gupta R, Puri SK, Mathur A. Designing a cellulolytic enzyme cocktail for the efficient and economical conversion of lignocellulosic biomass to biofuels. Enzyme Microb Technol. 2020;133:109442. https://doi.org/10.1016/j.enzmictec.2019.109442.
Bussamra BC, Freitas S, da Costa AC. Improvement on sugar cane bagasse hydrolysis using enzymatic mixture designed cocktail. Bioresour Technol. 2015;187:173–81. https://doi.org/10.1016/j.biortech.2015.03.117.
Peciulyte A, Pisano M, de Vries RP, Olsson L. Hydrolytic potential of five fungal supernatants to enhance a commercial enzyme cocktail. Biotechnol Lett. 2017;39(9):1403–11. https://doi.org/10.1007/s10529-017-2371-9.
Kango N, Jana UK, Choukade R. Fungal enzymes: sources and biotechnological applications. In: Advancing frontiers in mycology & mycotechnology 2019. Springer, Singapore. pp. 515–538. https://doi.org/10.1007/978-981-13-9349-5_21.
Salvachúa D, Katahira R, Cleveland NS, Khanna P, Resch MG, Black BA, Purvine SO, Zink EM, Prieto A, Martínez MJ, Martínez AT. Lignin depolymerization by fungal secretomes and a microbial sink. Green Chem. 2016;18(22):6046–62. https://doi.org/10.1039/C6GC01531J.
Hathout Y. Approaches to the study of the cell secretome. Expert Rev Proteomics. 2007;4(2):239–48. https://doi.org/10.1586/14789450.4.2.239.
Tsang A, Butler G, Powlowski J, Panisko EA, Baker SE. Analytical and computational approaches to define the Aspergillus niger secretome. Fungal Genetics Biol. 2009;46(1):S153–60. https://doi.org/10.1016/j.fgb.2008.07.014.
Bouws H, Wattenberg A, Zorn H. Fungal secretomes—nature’s toolbox for white biotechnology. Appl Microbiol Biotechnol. 2008;80(3):381–8. https://doi.org/10.1007/s00253-008-1572-5.
Barrett K, Jensen K, Meyer AS, Frisvad JC, Lange L. Fungal secretome profile categorization of CAZymes by function and family corresponds to fungal phylogeny and taxonomy: example Aspergillus and Penicillium. Sci Rep. 2020;10(1):1–2. https://doi.org/10.1038/s41598-020-61907-1.
Olsson L, Christensen TM, Hansen KP, Palmqvist EA. Influence of the carbon source on production of cellulases, hemicellulases and pectinases by Trichoderma reesei Rut C-30. Enzyme Microb Technol. 2003;33(5):612–9. https://doi.org/10.1016/S0141-0229(03)00181-9.
Xiong H, Turunen O, Pastinen O, Leisola M, Von Weymarn N. Improved xylanase production by Trichoderma reesei grown on l-arabinose and lactose or d-glucose mixtures. Appl Microbiol Biotechnol. 2004;64(3):353–8. https://doi.org/10.1007/s00253-003-1548-4.
Xiong H, von Weymarn N, Turunen O, Leisola M, Pastinen O. Xylanase production by Trichoderma reesei Rut C-30 grown on l-arabinose-rich plant hydrolysates. Bioresour Technol. 2005;96(7):753–9. https://doi.org/10.1016/j.biortech.2004.08.007.
Gupta VK, Steindorff AS, de Paula RG, Silva-Rocha R, Mach-Aigner AR, Mach RL, Silva RN. The post-genomic era of Trichoderma reesei: what’s next? Trends Biotechnol. 2016;34(12):970–82. https://doi.org/10.1016/j.tibtech.2016.06.003.
Zhang J, Wu C, Wang W, Wei D. Construction of enhanced transcriptional activators for improving cellulase production in Trichoderma reesei RUT C30. Bioresour Bioprocess. 2018;5(1):1–2. https://doi.org/10.1186/s40643-018-0226-4.
Zhang F, Bai F, Zhao X. Enhanced cellulase production from Trichoderma reesei Rut-C30 by engineering with an artificial zinc finger protein library. Biotechnol J. 2016;11(10):1282–90. https://doi.org/10.1002/biot.201600227.
Zhang J, Zhang G, Wang W, Wei D. Enhanced cellulase production in Trichoderma reesei RUT C30 via constitution of minimal transcriptional activators. Microb Cell Fact. 2018;17(1):1–4. https://doi.org/10.1186/s12934-018-0926-7.
Fonseca LM, Parreiras LS, Murakami MT. Rational engineering of the Trichoderma reesei RUT-C30 strain into an industrially relevant platform for cellulase production. Biotechnol Biofuels. 2020;13:1–5. https://doi.org/10.1186/s13068-020-01732-w.
Qian Y, Zhong L, Sun Y, Sun N, Zhang L, Liu W, Qu Y, Zhong Y. Enhancement of cellulase production in Trichoderma reesei via disruption of multiple protease genes identified by comparative secretomics. Front Microbiol. 2019;10:2784. https://doi.org/10.3389/fmicb.2019.02784.
Martinez D, Berka RM, Henrissat B, Saloheimo M, Arvas M, Baker SE, et al. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotechnol. 2008;26(5):553–60. https://doi.org/10.1038/nbt1403.
Landowski CP, Huuskonen A, Wahl R, Westerholm-Parvinen A, Kanerva A, Hänninen AL, et al. Enabling low cost biopharmaceuticals: a systematic approach to delete proteases from a well-known protein production host Trichoderma reesei. PLoS ONE. 2015;10(8): e0134723. https://doi.org/10.1371/journal.pone.0134723.
Jun H, Kieselbach T, Jönsson LJ. Enzyme production by filamentous fungi: analysis of the secretome of Trichoderma reesei grown on unconventional carbon source. Microb Cell Fact. 2011;10(1):1. https://doi.org/10.1186/1475-2859-10-68.
Ayrinhac C, Margeot A, Ferreira NL, Chaabane FB, Monot F, Ravot G, Sonet JM, Fourage L. Improved saccharification of wheat straw for biofuel production using an engineered secretome of Trichoderma reesei. Org Process Res Dev. 2011;15(1):275–8. https://doi.org/10.1021/op100218a.
Manavalan T, Manavalan A, Thangavelu KP, Heese K. Secretome analysis of Ganoderma lucidum cultivated in sugarcane bagasse. J Proteomics. 2012;77:298–309. https://doi.org/10.1016/j.jprot.2012.09.004.
Tavares MP, Morgan T, Gomes RF, Rodrigues MQ, Castro-Borges W, de Rezende ST, de Oliveira Mendes TA, Guimarães VM. Secretomic insight into the biomass hydrolysis potential of the phytopathogenic fungus Chrysoporthe cubensis. J Proteomics. 2021;236: 104121. https://doi.org/10.1016/j.jprot.2021.104121.
Couturier M, Navarro D, Olivé C, Chevret D, Haon M, Favel A, et al. Post-genomic analyses of fungal lignocellulosic biomass degradation reveal the unexpected potential of the plant pathogen Ustilago maydis. BMC Genomics. 2012;13(1):1–4. https://doi.org/10.1186/1471-2164-13-57.
Li YH, Zhang XY, Zhang F, Peng LC, Zhang DB, Kondo A, et al. Optimization of cellulolytic enzyme components through engineering Trichoderma reesei and on-site fermentation using the soluble inducer for cellulosic ethanol production from corn stover. Biotechnol Biofuels. 2018;11(1):1–4. https://doi.org/10.1186/s13068-018-1048-5.
Saqib AA, Farooq A, Iqbal M, Hassan JU, Hayat U, Baig S. A thermostable crude endoglucanase produced by Aspergillus fumigatus in a novel solid state fermentation process using isolated free water. Enzyme Res. 2012;2012:1–6. https://doi.org/10.1155/2012/196853.
Vianna Bernardi A, Kimie Yonamine D, Akira Uyemura S, Magnani DT. A thermostable Aspergillus fumigatus GH7 endoglucanase over-expressed in Pichia pastoris stimulates lignocellulosic biomass hydrolysis. Int J Mol Sci. 2019;20(9):2261. https://doi.org/10.3390/ijms20092261.
Lenartovicz V, de Souza CG, Moreira FG, Peralta RM. Temperature and carbon source affect the production and secretion of a thermostable β-xylosidase by Aspergillus fumigatus. Process Biochem. 2003;38(12):1775–80. https://doi.org/10.1016/S0032-9592(02)00261-3.
Jin X, Song J, Ma J, Liu GQ. Thermostable β-xylosidase from Aspergillus fumigatus: purification, characterization and potential application in lignocellulose bioethanol production. Renew Energy. 2020;155:1425–31. https://doi.org/10.1016/j.renene.2020.04.054.
Liu D, Zhang R, Yang X, Zhang Z, Song S, Miao Y, Shen Q. Characterization of a thermostable β-glucosidase from Aspergillus fumigatus Z5, and its functional expression in Pichia pastoris X33. Microb Cell Fact. 2012;11(1):1–5. https://doi.org/10.1186/1475-2859-11-25.
Bin Abdul Wahab MK, BinJonet MA, Illias RM. Thermostability enhancement of xylanase Aspergillus fumigatus RT-1. J Mol Catal. 2016;134:154–63. https://doi.org/10.1016/j.molcatb.2016.09.020.
Damis SI, Murad AM, Bakar FD, Rashid SA, Jaafar NR, Illias RM. Protein engineering of GH11 xylanase from Aspergillus fumigatus RT-1 for catalytic efficiency improvement on kenaf biomass hydrolysis. Enzyme Microb Technol. 2019;131: 109383. https://doi.org/10.1016/j.enzmictec.2019.109383.
Liu D, Li J, Zhao S, Zhang R, Wang M, Miao Y, et al. Secretome diversity and quantitative analysis of cellulolytic Aspergillus fumigatus Z5 in the presence of different carbon sources. Biotechnol Biofuels. 2013;6(1):1–6. https://doi.org/10.1186/1754-6834-6-149.
Adav SS, Ravindran A, Sze SK. Quantitative proteomic study of Aspergillus fumigatus secretome revealed deamidation of secretory enzymes. J Proteomics. 2015;24(119):154–68. https://doi.org/10.1016/j.jprot.2015.02.007.
de Gouvêa PF, Bernardi AV, Gerolamo LE, de Souza SE, Riaño-Pachón DM, Uyemura SA, et al. Transcriptome and secretome analysis of Aspergillus fumigatus in the presence of sugarcane bagasse. BMC Genomics. 2018;19(1):1–8. https://doi.org/10.1186/s12864-018-4627-8.
Raheja Y, Kaur B, Falco M, Tsang A, Chadha BS. Secretome analysis of Talaromyces emersonii reveals distinct CAZymes profile and enhanced cellulase production through response surface methodology. Ind Crop Prod. 2020;152: 112554. https://doi.org/10.1016/j.indcrop.2020.112554.
Claes A, Deparis Q, Foulquié-Moreno MR, Thevelein JM. Simultaneous secretion of seven lignocellulolytic enzymes by an industrial second-generation yeast strain enables efficient ethanol production from multiple polymeric substrates. Metab Eng. 2020;59:131–41. https://doi.org/10.1016/j.ymben.2020.02.004.
Zhang R. Functional characterization of cellulose-degrading AA9 lytic polysaccharide monooxygenases and their potential exploitation. Appl Microbiol Biotechnol. 2020;104(8):3229–43. https://doi.org/10.1007/s00253-020-10467-5.
Monclaro AV, Ferreira Filho EX. Fungal lytic polysaccharide monooxygenases from family AA9: recent developments and application in lignocelullose breakdown. Int J Biol Macromol. 2017;102:771–8. https://doi.org/10.1016/j.ijbiomac.2017.04.077.
Villares A, Moreau C, Bennati-Granier C, Garajova S, Foucat L, Falourd X, et al. Lytic polysaccharide monooxygenases disrupt the cellulose fibers structure. Sci Rep. 2017;7(1):1–9. https://doi.org/10.1038/srep40262.
Zhang R, Liu Y, Zhang Y, Feng D, Hou S, Guo W, Niu K, Jiang Y, Han L, Sindhu L, Fang X. Identification of a thermostable fungal lytic polysaccharide monooxygenase and evaluation of its effect on lignocellulosic degradation. Appl Microbiol Biotechnol. 2019;103(14):5739–50. https://doi.org/10.1007/s00253-019-09928-3.
Li F, Ma F, Zhao H, Zhang S, Wang L, Zhang X, Yu H. A lytic polysaccharide monooxygenase from a white-rot fungus drives the degradation of lignin by a versatile peroxidase. Appl Environ Microbiol. 2019;85(9). https://doi.org/10.1128/AEM.02803-18.
Monclaro AV, Petrović DM, Alves GS, Costa MM, Midorikawa GE, Miller RN, Filho EX, Eijsink VG, Várnai A. Characterization of two family AA9 LPMOs from Aspergillus tamarii with distinct activities on xyloglucan reveals structural differences linked to cleavage specificity. PLoS ONE. 2020;15(7): e0235642. https://doi.org/10.1371/journal.pone.0235642.
Sabbadin F, Hemsworth GR, Ciano L, Henrissat B, Dupree P, Tryfona T, et al. An ancient family of lytic polysaccharide monooxygenases with roles in arthropod development and biomass digestion. Nat Com. 2018;9(1):1–2. https://doi.org/10.1038/s41467-018-03142-x.
Valadares F, Gonçalves TA, Damasio A, Milagres AM, Squina FM, Segato F, Ferraz A. The secretome of two representative lignocellulose-decay basidiomycetes growing on sugarcane bagasse solid-state cultures. Enzyme Microb Technol. 2019;130: 109370. https://doi.org/10.1016/j.enzmictec.2019.109370.
Peciulyte A, Anasontzis GE, Karlström K, Larsson PT, Olsson L. Morphology and enzyme production of Trichoderma reesei Rut C-30 are affected by the physical and structural characteristics of cellulosic substrates. Fungal Genet Biol. 2014;72:64–72. https://doi.org/10.1016/j.fgb.2014.07.011.
Poidevin L, Berrin JG, Bennati-Granier C, Levasseur A, Herpoël-Gimbert I, Chevret D, et al. Comparative analyses of Podospora anserina secretomes reveal a large array of lignocellulose-active enzymes. Appl Microbiol Biotechnol. 2014;98(17):7457–69. https://doi.org/10.1007/s00253-014-5698-3.
Mäkelä MR, Bouzid O, Robl D, Post H, Peng M, Heck A, Altelaar M, DeVries RP. Cultivation of Podospora anserina on soybean hulls results in an efficient enzyme cocktail for plant biomass hydrolysis. New Biotechnol. 2017;37:162–71. https://doi.org/10.1016/j.nbt.2017.02.002.
Van Erven G, Kleijn AF, Patyshakuliyeva A, Di Falco M, Tsang A, De Vries RP, Van Berkel WJ, Kabel MA. Evidence for ligninolytic activity of the ascomycete fungus Podospora anserina. Biotechnol Biofuels. 2020;13:1–2. https://doi.org/10.1186/s13068-020-01713-z.
Benocci T, Daly P, Aguilar-Pontes MV, Lail K, Wang M, Lipzen A, et al. Enzymatic adaptation of Podospora anserina to different plant biomass provides leads to optimized commercial enzyme cocktails. Biotechnology J. 2019;14(4):1800185. https://doi.org/10.1002/biot.201800185.
Sato K, Chiba D, Yoshida S, Takahashi M, Totani K, Shida Y, Ogasawara W, Nakagawa YS. Functional analysis of a novel lytic polysaccharide monooxygenase from Streptomyces griseus on cellulose and chitin. Int J Biol Macromol. 2020;164:2085–91. https://doi.org/10.1016/j.ijbiomac.2020.08.015.
Scully ED, Hoover K, Carlson J, Tien M, Geib SM. Proteomic analysis of Fusarium solani isolated from the Asian longhorned beetle, Anoplophora glabripennis. PLoS ONE. 2012;7(4): e32990. https://doi.org/10.1371/journal.pone.0032990.
Rashid GM, Taylor CR, Liu Y, Zhang X, Rea D, Fülöp V, Bugg TD. Identification of manganese superoxide dismutase from Sphingobacterium sp. T2 as a novel bacterial enzyme for lignin oxidation. ACS Chem Biol. 2015;10(10):2286–94. https://doi.org/10.1021/acschembio.5b00298.
Lin L, Wang X, Cao L, Xu M. Lignin catabolic pathways reveal unique characteristics of dye-decolorizing peroxidases in Pseudomonas putida. Environ Microbiol. 2019;21(5):1847–63. https://doi.org/10.1111/1462-2920.14593.
DeAngelis KM, Sharma D, Varney R, Simmons BA, Isern NG, Markillie LM, Nicora CD, Norbeck AD, Taylor RC, Aldrich JT, Robinson EW. Evidence supporting dissimilatory and assimilatory lignin degradation in Enterobacter lignolyticus SCF1. Front Microbiol. 2013;4:280. https://doi.org/10.3389/fmicb.2013.00280.
Li X, He Y, Zhang L, Xu Z, Ben H, Gaffrey MJ, Yang Y, Yang S, Yuan JS, Qian WJ, Yang B. Discovery of potential pathways for biological conversion of poplar wood into lipids by co-fermentation of Rhodococci strains. Biotechnol Biofuels. 2019;12(1):1–6. https://doi.org/10.1186/s13068-019-1395-x.
Salvachúa D, Werner AZ, Pardo I, Michalska M, Black BA, Donohoe BS, Haugen SJ, Katahira R, Notonier S, Ramirez KJ, Amore A. Outer membrane vesicles catabolize lignin-derived aromatic compounds in Pseudomonas putida KT2440. Proc Natl Acad Sci USA. 2020;117(17):9302–10. https://doi.org/10.1073/pnas.1921073117.
Kumar M, Verma S, Gazara RK, Kumar M, Pandey A, Verma PK, et al. Genomic and proteomic analysis of lignin degrading and polyhydroxyalkanoate accumulating β-proteobacterium Pandoraea sp. ISTKB Biotechnol Biofuels. 2018;11(1):1–23. https://doi.org/10.1186/s13068-018-1148-2.
Brenelli LB, Persinoti GF, Cairo JP, Liberato MV, Gonçalves TA, et al. Novel redox-active enzymes for ligninolytic applications revealed from multiomics analyses of Peniophora sp. CBMAI 1063, a laccase hyper-producer strain. Sci Rep. 2019;9(1):1–5. https://doi.org/10.1038/s41598-019-53608-1.
Heyer R, Schallert K, Büdel A, Zoun R, Dorl S, Behne A, Kohrs F, Püttker S, Siewert C, Muth T, Saake G. A Robust and universal metaproteomics workflow for research studies and routine diagnostics within 24 h using phenol extraction, FASP digest, and the MetaProteomeAnalyzer. Front Microbiol. 2019;10:1883. https://doi.org/10.3389/fmicb.2019.01883.
Bai DP, Lin XY, Hu YQ, Chen ZZ, Chen L, Huang YF, et al. Metagenomics approach to identify lignocellulose-degrading enzymes in the gut microbiota of the Chinese bamboo rat cecum. Elect J Biotech. 2021;50:29–36. https://doi.org/10.1016/j.ejbt.2020.12.001.
Lazuka A, Auer L, O’Donohue M, Hernandez-Raquet G. Anaerobic lignocellulolytic microbial consortium derived from termite gut: enrichment, lignocellulose degradation and community dynamics. Biotechnol Biofuels. 2018;11(1):1–4. https://doi.org/10.1186/s13068-018-1282-x.
Thornbury M, Sicheri J, Slaine P, Getz LJ, Finlayson-Trick E, Cook J, Guinard C, et al. Characterization of novel lignocellulose-degrading enzymes from the porcupine microbiome using synthetic metagenomics. PLoS ONE. 2019;14(1): e0209221. https://doi.org/10.1371/journal.pone.0209221.
Marynowska M, Goux X, Sillam-Dussès D, Rouland-Lefèvre C, Halder R, Wilmes P, et al. Compositional and functional characterisation of biomass-degrading microbial communities in guts of plant fibre-and soil-feeding higher termites. Microbiome. 2020;8(1):1–8. https://doi.org/10.1186/s40168-020-00872-3.
Victorica MR, Soria MA, Batista-García RA, Ceja-Navarro JA, Vikram S, Ortiz M, Ontañon O, Ghio S, Martínez-Ávila L, García OJ, Etcheverry C. Neotropical termite microbiomes as sources of novel plant cell wall degrading enzymes. Sci Rep. 2020;10(1):1–4. https://doi.org/10.1038/s41598-020-60850-5.
Scully ED, Geib SM, Hoover K, Tien M, Tringe SG, Barry KW, del Rio TG, Chovatia M, et al. Metagenomic profiling reveals lignocellulose degrading system in a microbial community associated with a wood-feeding beetle. PLoS ONE. 2013;8(9): e73827. https://doi.org/10.1371/journal.pone.0073827.
Malacrinò A. Meta-omics tools in the world of insect-microorganism interactions. Biology. 2018;7(4):50. https://doi.org/10.3390/biology7040050.
Muñoz-Benavent M, Pérez-Cobas AE, García-Ferris C, Moya A, Latorre A. Insects’ potential: understanding the functional role of their gut microbiome. J Pharm Biomed Anal. 2020;2020:113787. https://doi.org/10.1016/j.jpba.2020.113787.
Woon JS, King PJ, Mackeen MM, Mahadi NM, Seman WM, Broughton WJ, Murad AM, Bakar FD. Cloning, production and characterization of a glycoside hydrolase family 7 enzyme from the gut microbiota of the termite Coptotermes curvignathus. Mol Biotechnol. 2017;59(7):271–83. https://doi.org/10.1007/s12033-017-0015-x.
Jiménez DJ, Chaib De Mares M, Salles JF. Temporal expression dynamics of plant biomass-degrading enzymes by a synthetic bacterial consortium growing on sugarcane bagasse. Front Microbiol. 2018;9:299. https://doi.org/10.3389/fmicb.2018.00299.
He B, Jin S, Cao J, Mi L, Wang J. Metatranscriptomics of the Hu sheep rumen microbiome reveals novel cellulases. Biotechnol Biofuels. 2019;12(1):1–5. https://doi.org/10.1186/s13068-019-1498-4.
Levy-Booth DJ, Hashimi A, Roccor R, Liu LY, Renneckar S, Eltis LD, et al. Genomics and metatranscriptomics of biogeochemical cycling and degradation of lignin-derived aromatic compounds in thermal swamp sediment. ISME J. 2021;15(3):879–93. https://doi.org/10.1038/s41396-020-00820-x.
Meyer-Cifuentes IE, Werner J, Jehmlich N, Will SE, Neumann-Schaal M, Öztürk B. Synergistic biodegradation of aromatic-aliphatic copolyester plastic by a marine microbial consortium. Nat Commun. 2020;11(1):1–3. https://doi.org/10.1038/s41467-020-19583-2.
Alessi AM, Bird SM, Bennett JP, Oates NC, Li Y, Dowle AA, et al. Revealing the insoluble metasecretome of lignocellulose-degrading microbial communities. Sci Rep. 2017;7(1):1–10. https://doi.org/10.1038/s41598-017-02506-5.
Alessi AM, Bird SM, Oates NC, Li Y, Dowle AA, Novotny EH, et al. Defining functional diversity for lignocellulose degradation in a microbial community using multi-omics studies. Biotechnol Biofuels. 2018;11(1):1–6. https://doi.org/10.1186/s13068-018-1164-2.
Speda J, Jonsson BH, Carlsson U, Karlsson M. Metaproteomics-guided selection of targeted enzymes for bioprospecting of mixed microbial communities. Biotechnol Biofuels. 2017;10(1):1–17. https://doi.org/10.1186/s13068-017-0815-z.
Zhu N, Yang J, Ji L, Liu J, Yang Y, Yuan H. Metagenomic and metaproteomic analyses of a corn stover-adapted microbial consortium EMSD5 reveal its taxonomic and enzymatic basis for degrading lignocellulose. Biotechnol Biofuels. 2016;9(1):1–23. https://doi.org/10.1186/s13068-016-0658-z.
D’haeseleer P, Gladden JM, Allgaier M, Chain PSG, Tringe SG, , et al. Proteogenomic analysis of a thermophilic bacterial consortium adapted to deconstruct switchgrass. PLoS ONE. 2013;8(7): e68465. https://doi.org/10.1371/journal.pone.0068465.
Singh R, Bennett JP, Gupta M, et al. Mining the biomass deconstructing capabilities of rice yellow stem borer symbionts. Biotechnol Biofuels. 2019;12:265. https://doi.org/10.1186/s13068-019-1603-8.
Muth T, Kohrs F, Heyer R, Benndorf D, Rapp E, Reichl U, Martens L, Renard BY. MPA portable: a stand-alone software package for analyzing metaproteome samples on the go. Anal Chem. 2018;90:685–9. https://doi.org/10.1021/acs.analchem.7b03544.
Gurdeep Singh R, Tanca A, Palomba A, Van der Jeugt F, Verschaffelt P, Uzzau S, Martens L, Dawyndt P, Mesuere B. Unipept 4.0: functional analysis of metaproteome data. J Proteome Res. 2019;18:606–15. https://doi.org/10.1021/acs.jproteome.8b00716.
Kleiner M, Dong X, Hinzke T, Wippler J, Thorson E, Mayer B, Strous M. Metaproteomics method to determine carbon sources and assimilation pathways of species in microbial communities. Proc Natl Acad Sci USA. 2018;115:E5576–84. https://doi.org/10.1073/pnas.1722325115.
Sung K, Robin P, Titus J, Thuy-Boun PS, Wang AY, Yates JR, Dennis III, Wolan W. ComPIL 2.0: an updated comprehensive metaproteomics database. J Proteome Res 2019;18(2):616–622. https://doi.org/10.1021/acs.jproteome.8b00722
Verschaffelt P, Van Den Bossche T, Martens L, Dawyndt P, Mesuere B. Unipept desktop: a faster, more powerful metaproteomics results analysis tool. J Proteome Res. 2021;20(4):2005–9. https://doi.org/10.1021/acs.jproteome.0c00855.
Subina M, Praveen K, Marie C, Johnson JE, Sajulga R, Duy D, Nguyen A, McGowan T, Arntzen MØ, Griffin TJ, Jagtap PD. Updates on metaQuantome Software for Quantitative Metaproteomics. J Proteome Res. 2021;20(4):2130–7. https://doi.org/10.1021/acs.jproteome.0c00960.
Meier F, Brunner AD, Koch S, Koch H, Lubeck M, Krause M, Goedecke N, Decker J, Kosinski T, Park MA, Bache N, Hoerning O, Cox J, Räther O, Mann M. Online parallel accumulation-serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol Cell Proteomics. 2018;17(12):2534–45. https://doi.org/10.1074/mcp.TIR118.000900 (Epub 2018 Nov 1. PMID: 30385480; PMCID: PMC6283298).
Peterson R, Grinyer J, Nevalainen H. Secretome of the coprophilous fungus Doratomyces stemonitis C8, isolated from koala feces. Appl Environ Microbiol. 2011;77(11):3793–801. https://doi.org/10.1128/AEM.00252-11.
Ravalason H, Grisel S, Chevret D, Favel A, Berrin JG, Sigoillot JC, et al. Fusarium verticillioides secretome as a source of auxiliary enzymes to enhance saccharification of wheat straw. Bioresour Technol. 2012;114:589–96. https://doi.org/10.1016/j.biortech.2012.03.009.
Florencio C, Cunha FM, Badino AC, Farinas CS, Ximenes E, Ladisch MR. Secretome data from Trichoderma reesei and Aspergillus niger cultivated in submerged and sequential fermentation methods. Data Brief. 2016;8:588–98. https://doi.org/10.1016/j.dib.2016.05.080.
Wang D, Zhang L, Zou H, Wang L. Secretome profiling reveals temperature-dependent growth of Aspergillus fumigatus. Science China Life Sciences. 2018;61(5):578–92. https://doi.org/10.1007/s11427-017-9168-4.
Manavalan A, Adav SS, Sze SK. iTRAQ-based quantitative secretome analysis of Phanerochaete chrysosporium. J Prot. 2011;75(2):642–54. https://doi.org/10.1016/j.jprot.2011.09.001.
Yang Y, Yang J, Liu J, Wang R, Liu L, Wang F, Yuan H. The composition of accessory enzymes of Penicillium chrysogenum P33 revealed by secretome and synergistic effects with commercial cellulase on lignocellulose hydrolysis. Bioresour Technol. 2018;257:54–61. https://doi.org/10.1016/j.biortech.2018.02.028.
Rai R, Kaur B, Singh S, Di Falco M, Tsang A, Chadha BS. Evaluation of secretome of highly efficient lignocellulolytic Penicillium sp. Dal 5 isolated from rhizosphere of conifers. Bioresour Technol. 2016;216:958–67. https://doi.org/10.1016/j.biortech.2016.06.040.
Song W, Han X, Qian Y, Liu G, Yao G, Zhong Y, Qu Y. Proteomic analysis of the biomass hydrolytic potentials of Penicillium oxalicum lignocellulolytic enzyme system. Biotechnol Biofuels. 2016;9(1):1–5. https://doi.org/10.1186/s13068-016-0477-2.
Ribeiro DA, Cota J, Alvarez TM, Brüchli F, Bragato J, Pereira BM, Pauletti BA, Jackson G, Pimenta MT, Murakami MT, Camassola M. The Penicillium echinulatum secretome on sugar cane bagasse. PLoS ONE. 2012;7(12): e50571. https://doi.org/10.1371/journal.pone.0050571.
Elmahdy MH, Azmy AF, El-Gebaly E, Saafan A, Gaber Y. A comparative proteomic study of Thermobifida cellulosilytica TB100T secretome grown on carboxymethylcellulose and rice straw. Open Biotech. 2020;14(1):42–51. https://doi.org/10.2174/1874070702014010042.
Wakarchuk WW, Brochu D, Foote S, Robotham A, Saxena H, Erak T, et al. Proteomic analysis of the secretome of Cellulomonas fimi ATCC 484 and Cellulomonas flavigena ATCC 482. PLoS ONE. 2016;11(3): e0151186. https://doi.org/10.1371/journal.pone.0151186.
Cai Y, Gong Y, Liu W, Hu Y, Chen L, Yan L, Zhou Y, Bian Y. Comparative secretomic analysis of lignocellulose degradation by Lentinula edodes grown on microcrystalline cellulose, lignosulfonate and glucose. J Prot. 2017;163:92–101. https://doi.org/10.1016/j.jprot.2017.04.023.
Xie C, Gong W, Zhu Z, Zhou Y, Xu C, Yan L, Hu Z, Ai L, Peng Y. Comparative secretome of white-rot fungi reveals co-regulated carbohydrate-active enzymes associated with selective ligninolysis of ramie stalks. Microb Biotechnol. 2020. https://doi.org/10.1111/1751-7915.13647.
Gomes HA, Moreira LR, Júnior AC, Fontes W, Santana RH, Kruger RH, et al. Evaluation of different secretomes produced by Clonostachys byssicola as tools to holocellulose breakdown. Int Biodet Biodeg. 2020;148: 104880. https://doi.org/10.1016/j.ibiod.2019.104880.
Zhang Y, Yang J, Luo L, Wang E, Wang R, Liu L, Liu J, Yuan H. Low-cost cellulase-hemicellulase mixture secreted by Trichoderma harzianum EM0925 with complete saccharification efficacy of lignocellulose. Int J Mol Sci. 2020;21(2):371. https://doi.org/10.3390/ijms21020371.
Rocha ALV, Maeda NR, Pereira N Jr, Kern FM, Simister ELR, et al. Characterization of the cellulolytic secretome of Trichoderma harzianum during growth on sugarcane bagasse and analysis of the activity boosting effects of swollenin. Biotechnol Prog. 2016;32(2):327–36. https://doi.org/10.1002/btpr.2217.
da Silva DS, Dantzger M, Assis MA, Gallardo JC, Teixeira GS, Missawa SK, et al. Lignocellulolytic characterization and comparative secretome analysis of a Trichoderma erinaceum strain isolated from decaying sugarcane straw. Fungal Biol. 2019;123(4):330–40. https://doi.org/10.1016/j.funbio.2019.01.007.
Basotra N, Kaur B, Di Falco M, Tsang A, Chadha BS. Mycothermus thermophilus (Syn. Scytalidium thermophilum): repertoire of a diverse array of efficient cellulases and hemicellulases in the secretome revealed. Bioresour Technol. 2016;222:413–21. https://doi.org/10.1016/j.biortech.2016.10.018.
Kolbusz MA, Di Falco M, Ishmael N, Marqueteau S, Moisan MC, da Silva BC, et al. Transcriptome and exoproteome analysis of utilization of plant-derived biomass by Myceliophthora thermophila. Fungal Genet Biol. 2014;72:10–20. https://doi.org/10.1016/j.fgb.2014.05.006.
Valadares F, Gonçalves TA, Gonçalves DS, Segato F, Romanel E, Milagres AM, et al. Exploring glycoside hydrolases and accessory proteins from wood decay fungi to enhance sugarcane bagasse saccharification. Biotechnol Biofuels. 2016;9(1):1–2. https://doi.org/10.1186/s13068-016-0525-y.
Salvachúa D, Martínez AT, Tien M, López-Lucendo MF, García F, De Los RV, et al. Differential proteomic analysis of the secretome of Irpex lacteus and other white-rot fungi during wheat straw pretreatment. Biotechnol Biofuels. 2013;6(1):1–4. https://doi.org/10.1186/1754-6834-6-115.
Hori C, Song R, Matsumoto K, Matsumoto R, Minkoff BB, Oita S, Hara H, Takasuka TE. Proteomic characterization of lignocellulolytic enzymes secreted by the insect-associated fungus Daldinia decipiens oita, isolated from a forest in northern Japan. Appl. Environ. Microbiol. 2020;86(8). https://doi.org/10.1128/AEM.02350-19.
Meng X, Ma L, Li T, Zhu H, Guo K, Liu D, et al. The functioning of a novel protein, Swollenin, in promoting the lignocellulose degradation capacity of Trichoderma guizhouense NJAU4742 from a proteomic perspective. Bioresour Technol. 2020;317: 123992. https://doi.org/10.1016/j.biortech.2020.123992.
Presley GN, Panisko E, Purvine SO, Schilling JS. Coupling secretomics with enzyme activities to compare the temporal processes of wood metabolism among white and brown rot fungi. Appl Environ Microbiol. 2018;84(16):e00159-e218. https://doi.org/10.1128/AEM.00159-18.
Machado AS, Valadares F, Silva TF, Milagres AM, Segato F, Ferraz A. The secretome of Phanerochaete chrysosporium and Trametes versicolor grown in microcrystalline cellulose and use of the enzymes for hydrolysis of lignocellulosic materials. Front Bioeng Biotechnol. 2020;8:826. https://doi.org/10.3389/fbioe.2020.00826.
de Lucas RC, de Oliveira TB, Lima MS, Pasin TM, de Almeida Scarcella AS, Ribeiro LF, et al. The profile secretion of Aspergillus clavatus: Different pre-treatments of sugarcane bagasse distinctly induces holocellulases for the lignocellulosic biomass conversion into sugar. Renew Energy. 2021;165:748–57. https://doi.org/10.1016/j.renene.2020.11.072.
Gaskell J, Blanchette RA, Stewart PE, BonDurant SS, Adams M, Sabat G, et al. Transcriptome and secretome analyses of the wood decay fungus Wolfiporia cocos support alternative mechanisms of lignocellulose conversion. Appl Environ Microbiol. 2016;82(13):3979–87. https://doi.org/10.1128/AEM.00639-16.
Hori C, Gaskell J, Igarashi K, Kersten P, Mozuch M, Samejima M, Cullen D. Temporal alterations in the secretome of the selective ligninolytic fungus Ceriporiopsis subvermispora during growth on aspen wood reveal this organism’s strategy for degrading lignocellulose. Appl Environ Microbiol. 2014;80(7):2062. https://doi.org/10.1128/AEM.03652-13.
Ravalason H, Jan G, Mollé D, Pasco M, Coutinho PM, Lapierre C, Pollet B, Bertaud F, Petit-Conil M, Grisel S, Sigoillot JC. Secretome analysis of Phanerochaete chrysosporium strain CIRM-BRFM41 grown on softwood. Appl Microbiol Biotechnol. 2008;80(4):719–33. https://doi.org/10.1007/s00253-008-1596-x.
Ji XL, Zhang WT, Gai YP, Lu BY, Yuan CZ, Liu QX, et al. Patterns of lignocellulose degradation and secretome analysis of Trametes trogii MT. Int Biodet Biodegr. 2012;75:55–62. https://doi.org/10.1016/j.ibiod.2012.09.001.
Fernández-Fueyo E, Ruiz-Dueñas FJ, López-Lucendo MF, Pérez-Boada M, Rencoret J, Gutiérrez A, et al. A secretomic view of woody and nonwoody lignocellulose degradation by Pleurotus ostreatus. Biotechnol Biofuels. 2016;9(1):1–8. https://doi.org/10.1186/s13068-016-0462-9.
Vasina DV, Moiseenko KV, Fedorova TV, Tyazhelova TV. Lignin-degrading peroxidases in white-rot fungus Trametes hirsuta 072. Absolute expression quantification of full multigene family. PLoS ONE. 2017;12(3):e0173813. https://doi.org/10.1371/journal.pone.0173813.
Acknowledgements
We thank Dr. Laura C. Seithers from the College of Education and Human Development, University of Minnesota, United States of America for the English grammar editing of this manuscript.
Funding
This work was not supported by any funding.
Author information
Authors and Affiliations
Contributions
SS and DZ conceptualized the review. SS wrote the manuscript. SS, GMM and YL prepared the figures and tables. SS, GMM, YL, DZ, WY, JJ and JS reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sethupathy, S., Morales, G.M., Li, Y. et al. Harnessing microbial wealth for lignocellulose biomass valorization through secretomics: a review. Biotechnol Biofuels 14, 154 (2021). https://doi.org/10.1186/s13068-021-02006-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-021-02006-9