
Abstract
Background
Spinal fusion with pedicle screw fixation represents the gold standard for lumbar degenerative disc disease with instability. Although it is an established technique, it is nevertheless an invasive intervention with high complication rates. Therefore, minimally invasive approaches have been developed, the medialized bilateral screw pedicel fixation (mPACT) being one of them. The study objective is to evaluate prospectively the efficacy and safety of the mPACT technique compared with the traditional trajectory for degenerative lumbar spondylolisthesis.
Methods/design
This is a single-center, randomized, controlled, parallel group, superiority trial. A total of 154 adult patients are allocated in a ratio of 1:1. Sample size and power calculation were performed to detect the minimal clinically important difference of 10%, with an expected standard deviation of 20% in the primary outcome parameter, the Oswestry Disability Index, with power of 80%, based on an assumed maximal dropout rate of 20%. Secondary outcome parameters include the EuroQoL 5-Dimension questionnaire, the Beck Depression Inventory, the painDETECT questionnaire and the “timed up and go” test. Furthermore, radiological and health economic outcomes will be evaluated. Follow up is performed until 5 years after surgery. Major inclusion criteria are lumbar degenerative spondylolisthesis with Meyerding grade I or II, which qualifies for decompression and fusion by medialised posterior screw placement with cortical trajectory (mPACT) or by a traditional trajectory for lumbar pedicle screw placement.
Discussion
This trial will contribute to the understanding of the short-term and long-term clinical and radiological postoperative course in patients with lumbar degenerative disc disease, in which the mPACT technique is used.
Trial registration
ISRCTN registry, ISRCTN99263604. Registered on 3 November 2016.
Similar content being viewed by others

Background
Spinal fusion with pedicle screw fixation represents the gold standard of surgical treatment of lumbar degenerative disc disease (DDD) with instability of the spinal segment in recent years. Fusion is achieved by creating a firm osseous bridge between the affected levels. Thus, a cage that is inserted via the transforaminal route (transforaminal lumbar interbody fusion (TLIF)) is commonly used to restore the intervertebral disc and foraminal height [1]. Although pedicle screw fixation with transforaminal lumbar interbody fusion is an established technique, it is nevertheless an invasive intervention with high complication rates [2].
Regardless of the popularization of tube-based unilateral minimally invasive (MIS) approaches for TLIF, bilateral posterior exposure was not initially considered possible to be performed in a less invasive fashion. The introduction of the cortical bone trajectory for the placement of medialized bilateral pedicle screw and rod fixation (mPACT) has changed the requirement for wider exposure in posterior fusion and has led to the development of a further MIS posterior approach [3,4,5]. Although various reports on mPACT have been published recently, there are no randomized, controlled, prospective clinical trials with long-term follow up on this issue. Its application is determined predominantly by the individual surgeon’s preference and skill [3, 6, 7].
The study objective is to prospectively evaluate the efficacy and safety of medialized cortical bone trajectory for lumbar pedicle screw placement compared with the traditional trajectory for degenerative lumbar spondylolisthesis. Both surgical approaches are well-established techniques in clinical practice.
Methods/design
Study design
The mPACT study is a single-center, randomized, controlled, parallel group, superiority trial with 154 patients allocated to the groups in a 1:1 ratio. Any trial involving a surgical procedure precludes surgical investigators from being blinded. Patients, however, will be blinded to the randomized treatment and further evaluations, such as radiologic analyses, will be conducted in a blinded fashion whenever possible. The expected enrollment time is 2 years. The conclusion of the study is estimated 5 years thereafter. The primary study endpoint is the difference in Oswestry Disability Index (ODI) between treatment groups at 2 years after surgery. The ODI is a widely used tool for the assessment of therapeutic effect. It consists of ten sections, with six questions in each section. A lower score indicates a higher level of function. An overall score of all 10 sections of the ODI will be computed and used as the ODI score. The standardized version of the ODI can be computed by re-scaling the score to the range 0–100 [8]. Moreover, the generic health status is assessed with the EuroQoL 5-Dimension questionnaire (EQ-5D) [9]. The Beck Depression Inventory (BDI) is used as a multiple choice self-reported inventory for measuring the severity of depression and responsiveness to treatment [10]. To identify neuropathic pain components the painDETECT questionnaire (PD-Q) will be used [11]. Furthermore, with the “timed up and go” (TUG) test the physical ability of walking is objectively documented and quantified [12]. Preoperative and postoperative magnetic resonance imaging (MRI) of the lumbar spine is performed. The image protocols include sagittal and axial sequences, both T1 and T2 weighted, in all patients [13,14,15,16,17]. A computed tomography (CT) scan is routinely performed one year after the intervention to assess the overall fusion rate [18]. Pre-operative American Society of Anesthesiologists (ASA) grade staging might allow the identification of risk factors. Neurological status and the quality and quantity of current pain medication are documented. Operation time and time of hospitalization is recorded. Pain medication usage, including epidural injections and nerve block injections are documented. Quantitative sensory testing (QST) is performed to assess and quantify low back pain non-invasively [19]. Blood samples will be tested preoperatively and postoperatively for TNF-α, IL-6, C-reactive protein (CRP) and leukocytes [20]. Direct costs of hospital care and the indirect costs of follow-up treatment outside the hospital will be evaluated. Costs of surgery and hospitalization including duration of inpatient stay, costs of nursing, costs of medication and physiotherapy are assessed after discharge from the hospital, according to the hospital’s internal cost-estimate lists. Additionally, the indirect costs after hospital discharge including physiotherapy, rehabilitation centers, pain medications, medical consulting etc. are documented on a routing sheet. Postoperative care is not standardized in the study protocol and will be performed according to the standard of care for degenerative lumbar spondylolisthesis at the study site. Primary and secondary outcome parameters are outlined in more detail in Table 1. Ethics approval was attained at the local ethics committee. This study complies with the World Medical Association Declaration of Helsinki Ethical Principles for Medical Research Involving Human Subjects, 2008. The final report will follow the CONSORT 2010 guidelines and its extension to non-pharmacological interventions. The CONSORT checklist is attached as Additional file 1.
Study population
The mPACT trial will include patients with lumbar degenerative spondylolisthesis of Meyerding grade I or II, who qualify for decompression and fusion of ≤ 3 lumbar spinal levels by a posterior medialized cortical bone trajectory or by a traditional trajectory for lumbar pedicle screw placement. These participants need to have failed adequate conservative or interventional therapy for a minimum of 3 months. Additionally, radiologically determined pathologic change at the treatment level needs to correlate with the primary symptoms. Patients with significant morbidities need to be excluded as this may lead to differences in treatment efficacy. Inclusion and exclusion criteria are described in detail in Table 2. Informed consent is obtained from each patient.
Timetable
The timetable and visit plan is outlined in Fig. 1.

Visit plan. Patients will be followed for 5 years after the intervention. AE, adverse event; ASA, American Society of Anesthesiologists; CT, computed tomography; d, day; intra-OP, intraoperative; m, month; MR imaging, magnetic resonance imaging; QST, quantitative sensory testing; pre-OP, preoperative; SAE, severe adverse event; V, visit; VAS, visual analogue scale
Investigational groups
Population 1 - titanium-based medialized cortical bone trajectory (mPACT)
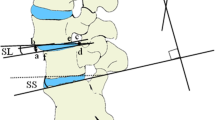
Cortical bone trajectory, medial-to-lateral, pedicle screws are placed under fluororadiographic guidance. It is performed by identifying the uppermost lateral edge of the pars and moving 3 to 5 mm medial to identify the screw entry point. For the cephalad screw, it is important to avoid the adjacent facet capsule and set the entry point slightly inferior. Typically, the screw is placed with an approximately 15° medial-to-lateral trajectory and an approximately 30° interior-to-superior trajectory for the cephalad screw with a less steep trajectory for the caudal screw, limiting the surgical exposure required [21, 22] (Fig. 2). Decompression is added if necessary. The standard approach to the disc space can be attained via a posterolateral transforaminal (TLIF) route. Then poly-ether-ether-ketone (PEEK) or titan cages filled with bone of the decompression side are used [23, 24].

The medialized cortical bone trajectory (mPACT) in comparison to the conventional pedicle srew instrumentation
Population 2 - conventional titanium-based instrumentation
The control group needs to represent the gold standard of care and is represented by rigid interbody fusion with titanium-based pedicle screw instrumentation (Fig. 2) [25].
Randomization
Randomization should avoid selection bias for each group. The allocation ratio is 1:1. Allocation of treatments will be performed using a computer-generated list. Statistical Software Stata 10.0 module Ralloc version 3.5.2 (Statacorp College Station, TX, USA) will be used to generate the random code (CODE: ralloc block size treat, ratio(1) osiz(2) nsubj(77) trtlab(mPACT konventionell) strata(2) seed(20161012) sav(File-Path) idvar(study_ID)). An independent statistician at the Department of Medical Statistics, Informatics and Health Economics, Innsbruck Medical University will administer the randomization code (Fig. 3).

Randomization
Selection bias
Selection bias should be avoided by the randomization of the allocated intervention. All patients accomplishing the inclusion criteria are considered for participation and pre-screening lists are kept to justify non-included patients. Performance bias should be avoided by a standardized protocol of postoperative care at our department. Both groups receive the same preopeative and postoperative treatment until discharge. Attrition bias should be avoided by the “intention-to-treat” or “per-protocol” analysis, respectively. Every patient included in the trial will be considered in the overall evaluation.
Data management and confidentiality
Study data generation at the study sites is clearly separated from data storage, processing and statistical analysis in the Department of Medical Statistics, Informatics and Health Economics. This requires a validated database system programmed in a customized software package, which is provided at the same place. The system includes an audit trial facility and will be used for defining the database structure, data entry, for handling data cleaning processes, and for final storing of the data. The evaluation of the data takes place by double-entry of the data and manual/visual evaluation of plausibility. The database will be closed at the completion of the study after entry of all collected data and clarification of all queries. All study findings and documents will be regarded as confidential. The investigator and members of the research team must not disclose any information without prior written approval from the Sponsor. The pseudonymity of patients participating must be maintained. Throughout documentation and evaluation, the patients will be recognized on case report forms (CRFs) and other documents by age and identification number. Documents that identify the patient personally (e.g., the signed informed consent form) must be maintained in confidence by the investigator. The patients will be told that all study findings will be stored on computer and handled in strictest confidence.
Sample size and power calculation
The sample size calculation is based on the primary endpoint of the study, a difference in the ODI 2 years post intervention [8]. The sample size was calculated based on the two-group t test (GraphPad StatMate Software, version 2.0). Based on a minimally clinical important change of 10% on the ODI, standard deviation for the ODI of 20%, significance level of p ≤ 0.05, and power of 80%, an estimated 64 patients are required in each of the intervention groups, for the primary endpoint. The current sample size of 154 patients allows for a 20% loss to follow up while maintaining > 80% statistical power [23].
Statistical analysis
Primary and secondary variables and their change from baseline will be summarized by treatment group. Endpoints will be analyzed as appropriate in dependence on the data distribution at a two-sided 0.05 level of significance. Detailed descriptive statistics will be provided for the data collected and 95% confidence intervals will be calculated for all relevant estimates. Measurements on the course of follow up will be analyzed by analysis of covariance (ANCOVA) or generalized model alternatives for categorical or semi-quantitative data. Additional covariates, such as baseline measurements, will be included in the model as appropriate. MRI parameters and variables to assess functional neurological status will be summarized descriptively. Imputation of missing data has not been planned. The primary analysis will follow as randomized. All values will be expressed as the mean and standard deviation.
Data are analyzed for normality by the Kolmogorov-Smirnov test. The Wilcoxon signed-rank test or the paired t test is used to analyze continuous paired data, while McNemar’s test is used for categorical data. The Mann-Whitney U test and unpaired t test, are used to perform group comparisons of continuous clinical outcome variables while the chi-square and Fisher’s exact test are used to analyze intergroup differences in categorized outcome variables and demographic characteristics (age, sex ratio, duration of symptoms, etc.), respectively. Statistical analysis will be performed after recruitment, one year postoperatively and after completion of the last visit of the study population at the specified time points. An interim analysis will be performed 6 and 12 months after recruitment and after completion of the last visit of the study population at the specified time points.
Discussion
The high prevalence of spinal disease, especially the increasing number of patients with degenerative disc disease, extensively increases the demand for spinal fusion surgery [26]. The standard surgical technique used is pedicle screw fixation with transforaminal lumbar interbody fusion [1]. Although it is an established technique, it is nevertheless an invasive intervention with high complication rates that appear to increase with age, blood loss, operative time, and the number of levels treated [2]. Therefore, minimally invasive techniques have been developed to minimize tissue damage, reduce blood loss, decrease narcotic requirement, and shorten the length of hospital stay [27]. The mPACT technique was established as one of these techniques. A posterior lumbar interbody fusion (PLIF) is then additionally used to perform an intervertebral fusion [6]. However, the PLIF is associated with higher rates of nerve tissue damage and extends the duration of operative procedure in comparison with TLIF [28]. In our prospective, clinical, randomized, controlled trial we will thus use TLIF only.
Implant loosening, especially in patients with poor bone nutrition, is another mentionable drawback that may be associated with spinal instrumentation [29]. The mPACT technique offers greater cortical bone contact [3] and therefore shows several promising advantages in biomechanical testing in comparison to traditional pedicle screw fixation. The cortical bone trajectory increases the pullout strength and improves the rigidity of spinal instrumentation [4].
Most clinical data on this subject are based on retrospective investigations or small prospective cohort studies without long-term results [3, 6, 7]. At this time, there are no prospective, clinical, randomized, controlled trials available. With current standards, well-validated outcome instruments are mandatory to provide efficient surgical treatment recommendations for patients with lumbar degenerative disc disease. This trial will contribute to the understanding of the short-term and long-term clinical and radiological postoperative course in patients with lumbar degenerative disc disease in which the mPACT technique is used.
Trial status
The trial will start in January 2017 at the Department of Neurosurgery, Medical University of Innsbruck.
Abbreviations
- AE:
-
Adverse event
- BDI:
-
Beck Depression Inventory
- BMI:
-
Body mass index
- CRP:
-
C-reactive protein
- CT:
-
Computed tomography
- EQ-5D:
-
EuroQoL-5 Dimension
- IL:
-
Interleukin
- MIS:
-
Minimally invasive surgery
- mPACT:
-
Medialised posterior screw placement with cortical trajectory
- MRI:
-
Magnetic resonance imaging
- ODI:
-
Oswestry Disability Index
- PD-Q:
-
painDETECT questionnaire
- PEEK:
-
Poly-ether-ether-ketone
- PLIF:
-
Posterior lumbar interbody fusion
- QST:
-
Quantitative sensory testing
- SAE:
-
Severe adverse event
- TLIF:
-
Transforaminal lumbar interbody fusion
- TNF:
-
Tumor necrosis factor
- TUG:
-
Timed up and go test
- VAS:
-
Visual analog scale
References
Mobbs RJ, Phan K, Malham G, Seex K, Rao PJ. Lumbar interbody fusion: techniques, indications and comparison of interbody fusion options including PLIF, TLIF, MI-TLIF, OLIF/ATP, LLIF and ALIF. J Spine Surg. 2015;1:2–18.
Carreon LY, Puno RM, Dimar JR, Glassman SD, Johnson JR. Perioperative complications of posterior lumbar decompression and arthrodesis in older adults. J Bone Joint Surg Am. 2003;85-A:2089–92.
Kojima K, Asamoto S, Kobayashi Y, Ishikawa M, Fukui Y. Cortical bone trajectory and traditional trajectory–a radiological evaluation of screw-bone contact. Acta Neurochir. 2015;157:1173–8.
Phan K, Hogan J, Maharaj M, Mobbs RJ. Cortical bone trajectory for lumbar pedicle screw placement: a review of published reports. Orthop Surg. 2015;7:213–21.
Mobbs RJ. The "medio-latero-superior trajectory technique": an alternative cortical trajectory for pedicle fixation. Orthop Surg. 2013;5:56–9.
Sakaura H, Miwa T, Yamashita T, Kuroda Y, Ohwada T. Posterior lumbar interbody fusion with cortical bone trajectory screw fixation versus posterior lumbar interbody fusion using traditional pedicle screw fixation for degenerative lumbar spondylolisthesis: a comparative study. J Neurosurg Spine. 2016;25:591–5.
Chen YR, Deb S, Pham L, Singh H. Minimally invasive lumbar pedicle screw fixation using cortical bone trajectory - a prospective cohort study on postoperative pain outcomes. Cureus. 2016;8:e714.
Fairbank JC, Pynsent PB. The Oswestry Disability Index. Spine (Phila Pa 1976). 2000;25:2940–52. discussion 2952
Brazier J, Roberts J, Tsuchiya A, Busschbach J. A comparison of the EQ-5D and SF-6D across seven patient groups. Health Econ. 2004;13:873–84.
Smarr KL, Keefer AL. Measures of depression and depressive symptoms: Beck Depression Inventory-II (BDI-II), Center for Epidemiologic Studies Depression Scale (CES-D), Geriatric Depression Scale (GDS), Hospital Anxiety and Depression Scale (HADS), and Patient Health Questionnaire-9 (PHQ-9). Arthritis Care Res (Hoboken). 2011;63(Suppl 11):S454–66.
Freynhagen R, Baron R, Gockel U, Tölle TR. painDETECT: a new screening questionnaire to identify neuropathic components in patients with back pain. Curr Med Res Opin. 2006;22:1911–20.
Hartmann S, Hegewald AA, Tschugg A, Neururer S, Abenhardt M, Thomé C. Analysis of a performance-based functional test in comparison with the visual analog scale for postoperative outcome assessment after lumbar spondylodesis. Eur Spine J. 2016;25(5):1620–6.
Pfirrmann CW, Dora C, Schmid MR, Zanetti M, Hodler J, Boos N. MR image-based grading of lumbar nerve root compromise due to disk herniation: reliability study with surgical correlation. Radiology. 2004;230:583–8.
Pfirrmann CW, Metzdorf A, Zanetti M, Hodler J, Boos N. Magnetic resonance classification of lumbar intervertebral disc degeneration. Spine (Phila Pa 1976). 2001;26:1873–8.
Modic MT, Steinberg PM, Ross JS, Masaryk TJ, Carter JR. Degenerative disk disease: assessment of changes in vertebral body marrow with MR imaging. Radiology. 1988;166:193–9.
Modic MT, Masaryk TJ, Ross JS, Carter JR. Imaging of degenerative disk disease. Radiology. 1988;168:177–86.
Modic MT, Pavlicek W, Weinstein MA, Boumphrey F, Ngo F, Hardy R, et al. Magnetic resonance imaging of intervertebral disk disease. Clinical and pulse sequence considerations. Radiology. 1984;152:103–11.
Schimmel JJ, Poeschmann MS, Horsting PP, Schönfeld DH, van Limbeek J, Pavlov PW. PEEK cages in lumbar fusion: mid-term clinical outcome and radiological fusion. Clin Spine Surg. 2016;9(5):E252–8J.
Rolke R, Baron R, Maier C, Tölle TR, Treede RD, Beyer A, et al. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): standardized protocol and reference values. Pain. 2006;123:231–43.
Pedersen LM, Schistad E, Jacobsen LM, Røe C, Gjerstad J. Serum levels of the pro-inflammatory interleukins 6 (IL-6) and −8 (IL-8) in patients with lumbar radicular pain due to disc herniation: a 12-month prospective study. Brain Behav Immun. 2015;46:132–6.
Khanna N, Deol G, Poulter G, Ahuja A. Medialized, Muscle-Splitting Approach for Posterior Lumbar Interbody Fusion: Technique and Multicenter Perioperative Results. Spine (Phila Pa 1976). 2016;41(Suppl 8):S90–6.
Matsukawa K, Yato Y, Kato T, Imabayashi H, Asazuma T, Nemoto K. In vivo analysis of insertional torque during pedicle screwing using cortical bone trajectory technique. Spine (Phila Pa 1976). 2014;39:E240–5.
Hägg O, Fritzell P, Nordwall A, Swedish Lumbar Spine Study Group. The clinical importance of changes in outcome scores after treatment for chronic low back pain. Eur Spine J. 2003;12:12–20.
Fritzell P, Hägg O, Wessberg P, Nordwall A, Swedish Lumbar Spine Study Group. 2001 Volvo Award Winner in Clinical Studies. Lumbar fusion versus nonsurgical treatment for chronic low back pain: a multicenter randomized controlled trial from the Swedish Lumbar Spine Study Group. Spine (Phila Pa 1976). 2001;26:2521–32. discussion 2532-4
Resnick DK, Watters WC, Sharan A, Mummaneni PV, Dailey AT, Wang JC, et al. Guideline update for the performance of fusion procedures for degenerative disease of the lumbar spine. Part 9: lumbar fusion for stenosis with spondylolisthesis. J Neurosurg Spine. 2014;21:54–61.
Deyo RA, Nachemson A, Mirza SK. Spinal-fusion surgery — the case for restraint. N Engl J Med. 2004;350:722–6.
Khan NR, Clark AJ, Lee SL, Venable GT, Rossi NB, Foley KT. Surgical outcomes for minimally invasive vs open transforaminal lumbar interbody fusion: an updated systematic review and meta-analysis. Neurosurgery. 2015;77:847–74. discussion 874
Zhang Q, Yuan Z, Zhou M, Liu H, Xu Y, Ren Y. A comparison of posterior lumbar interbody fusion and transforaminal lumbar interbody fusion: a literature review and meta-analysis. BMC Musculoskelet Disord. 2014;15:367.
Wittenberg RH, Shea M, Swartz DE, Lee KS, White AA, Hayes WC. Importance of bone mineral density in instrumented spine fusions. Spine (Phila Pa 1976). 1991;16:647–52.
Mannion AF, Porchet F, Kleinstück FS, Lattig F, Jeszenszky D, Bartanusz V, et al. The quality of spine surgery from the patient's perspective. Part 1: the Core Outcome Measures Index in clinical practice. Eur Spine J. 2009;18(Suppl 3):367–73.
Mannion AF, Porchet F, Kleinstück FS, Lattig F, Jeszenszky D, Bartanusz V, et al. The quality of spine surgery from the patient's perspective: part 2. Minimal clinically important difference for improvement and deterioration as measured with the Core Outcome Measures Index. Eur Spine J. 2009;18(Suppl 3):374–9.
Le Cara EC, Marcus RL, Dempsey AR, Hoffman MD, Hebert JJ. Morphology versus function: the relationship between lumbar multifidus intramuscular adipose tissue and muscle function among patients with low back pain. Arch Phys Med Rehabil. 2014;95:1846–52.
Acknowledgements
We would like to thank DePuy Synthes (Johnson & Johnson Medial Products GmbH, Vorgartenstrasse 206B, 1020 Wien) who will enable full clinical PhD education. Furthermore, thanks Prof. Dr. Elke Gizewski (Department of Neuroradiology, Medical University of Innsbruck) for the imaging support.
Funding
There is no funding available.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
AT conceived the study, participated in its design and coordination, and drafted the protocol. PK contributed his extensive spinal surgical expertise to plan the trial. SH participated in the design of the trial. SL and CW substantially coordinated the trial. WNL gave substantial contribution to conception of the trial. MW supervised the imaging support. SN consulted in drafting the statistical part of the protocol and revised it critically, participated in designing the study, and was involved in drafting and revising the protocol. CT participated in designing the study and gave final approval as Sponsor and Principal Investigator of the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval was attained in Austria from the committee of the Medical University Innsbruck (AN2016–0168 365/4.1). The study complies with the World Medical Association Declaration of Helsinki Ethical Principles for Medical Research Involving Human Subjects and Good Clinical Practice (GCP). Registration was required at Current Controlled Trials. (http://www.isrctn.com). Informed consent will be obtained from all study participants. All authors have read the manuscript and have made approval for publication of the manuscript, the figures and tables.
Consent for publication
Not applicable.
Competing interests
CT and PK are consultants for DePuy Synthes. None of the other authors has any conflict of interest in connection with the study.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
SPIRIT 2013 Checklist: recommended items to address in a clinical trial protocol and related documents. (DOC 122 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tschugg, A., Kavakebi, P., Hartmann, S. et al. Clinical and radiological effect of medialized cortical bone trajectory for lumbar pedicle screw fixation in patients with degenerative lumbar spondylolisthesis: study protocol for a randomized controlled trial (mPACT). Trials 19, 129 (2018). https://doi.org/10.1186/s13063-018-2504-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-018-2504-z