
Abstract
Objective
MicroRNAs (miRNAs) play a vital role in the development of ovarian cancer (OC). The aim of this study to investigate the prognostic value and potential signaling pathways of hsa-miR-9-5p (miR-9) in OC through literature review and bioinformatics methods.
Methods
The expression of miR-9 in OC was assessed using the public datasets from the Gene Expression Omnibus (GEO) database. And a literature review was also performed to investigate the correlation between miR-9 expression and the OC prognosis. Two mRNA datasets (GSE18520 and GSE36668) of OC tissues and normal ovarian tissues (NOTs) were downloaded from GEO to identify the differentially expressed genes (DEGs). The target genes of hsa-miR-9-5p (TG-miR-9-5p) were predicted using miRWALK3.0 and TargetScan. Then the gene overlaps between DEGs in OC and the predicted TG-miR-9-5p were confirmed using a Venn diagram. After that, overlapping genes were subjected to Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. Finally, a protein-protein interaction (PPI) network was constructed using STRING and Cytoscape, and the impact of hub genes on OC prognosis was analyzed.
Results
It was found that OC patients with miR-9 low expression had poor prognosis. A total of 107 DEGs related to both OC and miR-9 were identified. Dozens of DEGs were enriched in developmental process, extracellular matrix structural constituent, cell junction, axon guidance. In the PPI network analysis, 5 of the top 10 hub genes was significantly associated with decreased overall survival of OC patients, namely FBN1 (HR = 1.64, P < 0.05), PRRX1 (HR = 1.76, P < 0.05), SMC2 (HR = 1.22, P < 0.05), SMC4 (HR = 1.31, P < 0.05), and VCAN (HR = 1.48, P < 0.05).
Conclusion
Low expression of miR-9 indicates poor prognosis of OC patients. MiR-9 plays a crucial role in the biological process of OC by binding to target genes, thus affecting the prognosis of patients.
Similar content being viewed by others


Introduction
Ovarian cancer (OC) is one of the most common gynecological malignancies, which poses a serious threat to women’s life and health. OC is clinically characterized by insidious- and late-onset, high grade of malignancy, high metastasis rate, and poor prognosis. Its incidence is increasing yearly, and its mortality rate ranks first compared with other female genital tract malignancies, such as endometrial cancer, cervical cancer and vulvar cancer [1, 2]. The American Cancer Society (ACS) has estimated that there will be approximately 22,530 new cases of OC and 13,980 deaths from OC in the United States in 2019 [2]. Because OC is anatomically located deep within the pelvis, the early lesions have no obvious signs and specific diagnostic methods. Therefore, about 59% of patients have progressed to an advanced stage when they seek medical treatment due to abdominal distension, ascites, and abdominal pain [3]. The 5-year survival rate of patients with early OC can be greater than 75%, while that of patients with advanced OC is only 29% [2]. With the continuous improvement of diagnosis and treatment, the 5-year survival rate of OC patients has increased from 36% in 1975 to 47% in 2014 [4]. However, more than 50% of patients with advanced OC relapse within the first 5 years after treatment and develop cross-resistance to standard chemotherapy and other functionally and structurally unrelated chemotherapeutic agents [5].
Because of the low survival rate and high recurrence rate, it is of great significance to find new targets related to the prognosis of OC, and explore the specific mechanism, so as to perform personalized treatment and to improve clinical efficacy and quality of life of OC patients. Besides,, new targets will also have a significant impact on the detection of cancer recurrence and the guide to rehabilitation.MicroRNAs (miRNAs) are a class of highly conserved endogenous non-coding small RNAs composed of 18–22 nucleotides [6, 7], which play a role in gene regulation in eukaryotes. MiRNAs, as small RNAs that control post-transcriptional gene expression, regulate about 30% of genes in the human genome. One miRNA can regulate the expression of various genes, while one gene can also be regulated by various miRNAs [8]. MiRNAs play a crucial role in cell proliferation, migration, differentiation and apoptosis [9, 10]. It has been reported that miRNAs are also involved in the development of a variety of diseases, such as cancers [11] and cardiovascular diseases [12], and are used as targets for clinical diagnosis and treatment. Increasing studies have shown that aberrant expression of hsa-miR-9-5p (miR-9) is involved in biological processes in various cancers. For example, miR-9 can inhibit the proliferation, migration, and invasion of choroidal melanoma by targeting BRAF [13]. Bi et al. [14] have also revealed that lncRNA LINC01116 can promote colorectal cancer development by targeting miR-9-5p/STMN1. MiR-9 can inhibit epithelial OC cell proliferation via targeting the SDF-1/CXCR4 pathway [15]. And the overexpression of miR-9 promotes metastasis of serous OC by targeting E-cadherin [16].
Numerous studies showed that there was a significant correlation between miR-9 expression and the prognosis of various cancers, such as breast cancer [17], osteosarcoma [18], lung cancer [19], OC [20]. Nevertheless, the mechanism of miR-9 affecting OC is still unclear. Here, based on literature review and bioinformatic analysis of GEO datasets, we identified the possible molecular targets of OC and revealed the mechanism of miR-9 in OC.
Materials and methods
Selection of GEO datasets
To obtain gene chip profiles of OC, a search was performed in the Gene Expression Omnibus database (GEO, http://www.ncbi.nlm.nih.gov/geo/). The search strategy was as follows: (“ovarian cancer” OR “ovarian carcinoma” OR “ovarian tumor”) AND (“microRNA” OR “miRNA” OR “non-coding RNA” OR “small RNA”). The datasets related to miR-9 expression in OC tissues and normal ovarian tissues (NOTs) were included in our analysis.
The original data files were downloaded directly from the GEO database, and were preprocessed by R software (version 3.6.3). Based on the groups (OC group and normal ovarian group), the data in the original files were sorted into a Excel table. By using the gene probe file provided by the platform, the gene probes in the sorted Excel document were converted into the gene symbols. Finally, the gene expression data of miR-9 in OC and NOTs were screened.
Literature retrieval
PubMed, Web of Science, Cochrane Library, and Embase databases were comprehensively searched for the literature on the correlation between miR-9 expression and prognosis of OC. The time of literature retrieval was up to November 2020. The search strategy was as follows: (“microRNA-9” OR “miRNA-9” OR “miR-9”) AND (“ovarian cancer” OR “ovarian carcinoma” OR “ovarian tumor”). Literature retrieval was performed independently by two investigators and finally cross-checked.
The studies published in English on the relationship between miR-9 expression and OC prognosis were included. While reviews, nonclinical studies, case reports, abstracts were excluded. The quality evaluation of the included studies was performed according to the Newcastle-Ottawa scale (NOS) [21]. Only those with 6 stars or above, which were considered to be of high quality, were included in this study.
Gene ontology enrichment and target prediction analysis
Gene expression profiles of datasets GSE18520 and GSE36668 were obtained from GEO. The dataset GSE18520 consists of 53 OC samples and 10 NOTs samples, while GSE36668 consists of 4 OC samples and 4 NOTs samples. The data were analyzed on the GPL570 platform Affymetrix Human Genome U133 Plus 2.0 Array (Affymetrix; Thermo Fisher Science, Inc., Waltham, MA, USA).
To identify differentially expressed genes (DEGs) between OC tissues and NOTs, we employed the Limma package (version 3.6.3) in R/BioManager for processing. The adjusted P-value (adj. P.Value) was calculated using Benjamini-Hochberg method for controlling the false discovery rate (FDR), thus correcting false positives. Cut-off criterion was defined as P < 0.05 and |log2 (FC)| > 1. The platform annotation files downloaded from the database were adopted to convert the probe data in the matrix files into gene symbols.
The online websites miRWALK3.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/miRretsys-self.html) and TargetScan (http://www.targetscan.org/vert_72/) were used to predict the target genes of hsa-miR-9-5p (TG-miR-9-5p). The overlapping genes between DEGs in OC and TG-miRNA-9-5p were revealed, and TG-miR-9-5p was also predicted using the online tool Venny 2.1.0 (https://bioinfogp.cnb.csic.es/tools/venny/). The gene overlaps were analyzed by Gene Ontology (GO) and visualized using the Bingo plugin of Cytoscape software (version3.7.2). Kyoto Encyclopedia of Genes and Genomes (KEGG) was analyzed by using DAVID database. FDR < 0.05 was considered as the cutoff criterion. In addition, the protein-protein interaction (PPI) network based on the gene overlaps was constructed by using STRING 11.0 (https://string-db.org/), and the interactions were visualized with CytoHubba plugin for Cytoscape software (version 3.7.2). Then the hub genes of PPI network were obtained. A confidence score of C ≥ 0.7 was set as the cutoff criterion.
Survival analysis
The effect of the top 10 hub genes obtained in the PPI network on OC prognosis was assessed using Kaplan-Meier plotter (www.kmplot.com). The Kaplan-Meier plotter is capable to assess the effect of 54 k genes (mRNA, miRNA, protein) on survival in 21 cancer types, including breast (n = 6234), ovarian (n = 2190), lung (n = 3452), and gastric (n = 1440) cancers. Sources for the databases include GEO, European Genome-phenome Archive (EGA), and The Cancer Genome Atlas (TCGA). OC patients were divided into the high expression group and the low expression group based on the median expression of hub genes. The overall survival risk of OC was evaluated using Kaplan-Meier method, and the hazard ratio (HR) was used as a measure of effect.
Statistical analysis
Stata 15.0 statistical software was used for data processing. MiR-9 expression levels in microarray datasets downloaded from GEO were expressed as mean ± standard deviation (SD). Independent two-sample t-test was used for analysis. Pooled analysis of miR-9 expression levels in different GEO datasets was performed using a random-effects model. If HR and its 95% CI of miR-9 expression and OC survival risk were provided in the included literature, they were directly adopted. If HR was not directly provided in the literature, but with survival curves, the data were then extracted using Engauge Digitizer software and then HR was calculated. Overall survival (OS) and progression-free survival (PFS) were used to evaluate the effect of miR-9 expression on OC prognosis. A fixed-effects model was used for the pooled analysis of these prognostic indicators. P < 0.05 was considered statistically significant.
Results
miR-9 expression in ovarian cancer based on GEO
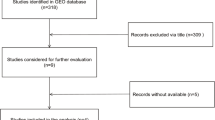
The screening process of GEO datasets is shown in Fig. 1. A total of 4 GEO datasets (GSE47841, GSE83693, GSE53829, and GSE23338) were collected to assess miR-9 expression in OC tissues and NOTs. The miR-9 expression in OC tissues was significantly higher than that in NOTs in the datasets GSE47841 and GSE53829, while it was significantly lower than that in NOTs in the dataset GSE83693 (all P < 0.05). And no significant difference was identified in the GSE23338 dataset. The above comparison results are shown in Table 1 and Fig. 2a. However, the results of the random-effects model showed no significant difference in miR-9 expression between the OC tissues and NOTs (SMD = 0.03, 95% CI: − 0.99-1.05, P = 0.952). The forest plot is shown in Fig. 2b.

Research and screening flow diagram in GEO database. GEO: gene expression omnibus

Expression of miR-9 in ovarian cancer and normal ovarian tissues in GEO. a:miR-9 expression in the datasets of GSE47841, GSE23383, GSE53829, GSE83693; b: Forest plot of differential expression of miR-9-5p in the GEO datasets. GEO: gene expression omnibus
Effect of miR-9 expression on the prognosis of ovarian cancer
The screening process of the literature is shown in Fig. 3. A total of 3 studies with NOS scores of more than 6 stars that met the inclusion criteria were identified [20, 22, 23]. Among them, 2 studies contained OS data, and 3 studies contained PFS data. The results of the fixed-effect model (Fig. 4) showed that high expression of miR-9 was beneficial for the survival of OC patients (OS: HR = 0.60, 95% CI: 0.41–0.88; PFS: HR = 0.58, 95% CI: 0.43–0.77). The differences were statistically significant.

Flow diagram for screening of studies on the relationship between miR-9 expression with the prognosis of patients with ovarian cancer

Forest plot of the correlation between miR-9 expression and prognosis in patients with ovarian cancer. OS:Overall survival; PFS: progression-free survival
miR-9 target gene prediction and DEGs screening
In the GSE18520 and GSE36668 datasets, 3146 and 2051 DEGs were identified, respectively. Volcano plots of gene expression differences in these two datasets are shown in Fig. 5. A total of 935 common DEGs were selected in these two datasets (Fig. 6) by using a Venn diagram. Then, based on miRWALK3.0 and TargetScan, 2072 TG-miR-9-5p were predicted, 101 of which were validated in the 935 common DEGs (Fig. 6). Among these genes associated with hsa-miR-9-5p, 50 were up-regulated and 51 were down-regulated in OC tissues compared with NOTs. The top 10 up-regulated and down-regulated hsa-miR-9-5p-related DEGs in the dataset GSE18520 are listed in Table 2. Among them, genes such as MUM1L1, CALB2, and VGLL3, were important has-miR-9-5p-related target genes of OC.

Volcano plot of Genome-wide mRNA detected by two ovarian cancer-related datasets from GEO. Aberrantly expressed mRNAs with P < 0.05 and |log (FC)| > 1 were represented by blue. up-regulated genes were indicated by blue plots above, down-regulated genes were indicated by blue plots below, and normally expressed mRNAs were indicated by black plots . The y-axis shows the fold-change value between the mRNAs expression in ovarian cancer and normal ovarian tissues. The x-axis represents -log10 of adj. P-Value. adj. P-Value, adjusted P value; FC, fold change; GEO, gene expression omnibus

The Venn map of hsa-miR-9-5p related DEGs in TG_miR-9-5p and other two GEO datasets, and the overlapping region represents the recognized DEGs. DEGs: differentially expressed genes; TG_miR-9-5p, target genes of hsa-miR-9-5p
GO and KEGG analysis of miR-9-related DEGs in ovarian cancer
As shown in Table 3, the partial results of GO and KEGG enrichment analyses revealed that a number of miR-9-related target genes were involved in biological processes such as developmental process, anatomical structure development, extracellular matrix structural constituent, cell junction, and axon guidance. The results of GO enrichment analysis were visualized using the Bingo plugin for Cytoscape software (Fig. 7).

Results of GO enrichment Analysis of the recognized DEGs by using the Bingo plugin of Cytoscape software. The yellow circle represents functional enrichment, and the larger the circle, the darker the color, the more genes are enriched in this pathway. The connecting lines represent the association between gene and gene. GO: Gene Ontology.DEGs: differentially expressed genes
PPI network construction and hub gene selection
The PPI network of miR-9-related DEGs was constructed with 101 nodes and 48 edges, which included 50 up-regulated genes and 51 down-regulated genes (Fig. 8). By using the degree as the ranking criterion, the top 10 genes were selected as hub genes (Fig. 9). Thus, there was a close relationship among hub genes.

Protein-protein interaction network of hsa-miR-9-5p-related DEGs. The lines represent interaction relationship between nodes. DEGs, differentially expressed genes. The line between the circle nodes represents the interaction between the two proteins linked by the line. Colored nodes:query proteins and first shell of interactors;white nodes:second shell of interactors;empty nodes:proteins of unknown 3D structure;filled nodes:some 3D structure is known or predicted..DEGs: differentially expressed genes

Protein-protein interaction network. (A): Protein-protein interaction network of hube genes of hsa-miR-9-5p-related DEGs. The red and yellow represent the top 10 Hub genes. The darker the color, the stronger the association with other genes in the PPI network. The lines represent interaction relationship between nodes. DEGs:differentially expressed genes

Association between the expressions of FBN1, PRRX1, SMC2, SMC4, and VCAN and the prognosis of patients with ovarian cancer. According to the median expression level, patients with ovarian cancer were divided into high expression group and low expression group. HR:hazard ratio
Survival analysis
The prognostic values of the top 10 hub genes selected from the PPI network were evaluated using Kaplan-Meier plotter. The results showed that 5 of the top 10 hub genes were significantly associated with the risk of survival in OC patients. High expression of these 5 hub genes led to a decrease of survival rate in OC patients (Fig. 10), namely FBN1 (HR = 1.64, P < 0.05), PRRX1 (HR = 1.76, P < 0.05), SMC2 (HR = 1.22, P < 0.05), SMC4 (HR = 1.31, P < 0.05), and VCAN (HR = 1.48, P < 0.05). The differences were statistically significant.
Discussion
In this study, the GEO datasets related to expression of miRNAs in OC were systematically searched. Based on the comparison of expression profiles of miRNAs in OC tissues and NOTs, the abnormal expression of miR-9 associated with OC was identified. Based on the comprehensive literature retrieval, it was found that high expression of miR-9 was favorable for OC patients, which could be confirmed by both OS and PFS data. Although the number of included studies is relatively small, this conclusion is still of great significance. In addition, GO, KEGG, PPI network, and survival analysis were adopted to identify and analyze the novel makers and potential targets of miR-9 involved in the regulation of essential biological processes in OC.
To date, few studies have investigated miR-9 characteristics in OC, and there is a lack of data on the miR-9 expression in literature. Therefore, we analyzed the expression of miR-9 in 4 microarray datasets from GEO. The results showed that miR-9 expression was significantly up-regulated in 2 datasets and significantly down-regulated in 1 dataset, while no significant difference in 1 dataset. However, the pooled analysis of miR-9 expression in these 4 datasets revealed no significant difference. Thus it is still inconclusive about the expression of miR-9 in OC. The reason for the above inconsistency may be related to the histological types of OC. The survival analysis pointed out a positive relationship between high expression of miR-9 and OC prognosis. But the specific mechanism is still unknown. In the study of Sun et al. [23], miR-9 mediates the down-regulation of BRCA1 gene, which in turn hinders the repair of DNA damage in OC, suggesting that the therapeutic effect of OC can be improved by increasing the sensitivity of cancer cells to DNA damage. Li et al. [20] have proved that miR-9 may have affected paclitaxel chemosensitivity in OC patients by targeting CCNG1.
MiR-9 is one of the most critical miRNAs in the regulation of OC. Zhang et al. [24] have reported that CircPLEKHM3 exerts tumor-suppressive effects in OC cells by targeting the miR-9/BRCA1/DNAJB6/KLF4/AKT1 axis and can be used as a prognostic indicator and therapeutic target for OC. MiR-9 has been demonstrated to promote the epithelial-to-mesenchymal transition of OC cells by inhibiting E-cadherin [25]. Moreover, epigenetic modification of miR-9 is involved in the pathogenesis and progression of OC, and it has a certain diagnostic value for OC [26].
In the current study, we found that novel candidate target genes of miR-9 are involved in the regulation of vital biological processes in OC, such as Calbindin2 (CALB2), Vestigial like family member3 (VGLL3). CALB2 is a 29-kDa calcium-binding protein of the EF-Hand family, which is a family of proteins containing calcium-binding motifs composed of two helices (E and F) [27]. CALB2 has been shown to regulate colorectal cancer patient sensitivity to 5-fluorouracil by modulating the intrinsic apoptosis pathway [28]. But there are few studies on the role of CALB2 in OC. VGLL3, as a member of vestigial like family of proteins, is associated with epithelial OC [29] and soft tissue sarcoma [30].
Our study also found 5 hub genes with a significant correlation with OC prognosis, such as FBN1, PRRX1, and SMC4. Their high expression contributes to the poor prognosis of OC patients. FBN1 is the coding gene for fibrillin-1 and the main component of extracellular matrix microfibers, which is crucial in maintaining the morphological integrity and normal function of connective tissue [31]. Studies have reported that high expression of FBN1 decreases the OS of serous ovarian cancer [32], and it increases the risk of lymph node metastasis [33]. PRRX1 has been demonstrated as a novel inducer of the epithelial-mesenchymal transition of tumor cells, such as breast cancer [34], colorectal cancer [35], and OC [36]. SMC4 protein is associated with tumor development, and the main biological function of SMC4 protein is the involvement in the dynamic changes of higher-order chromosomal structures, such as chromosome condensation and separation, DNA recombination, and repair of DNA damage [37, 38]. These hub genes are expected to be new predictors of OC prognosis, and become new targets for OC treatment.
The findings of this study have great and far-reaching significance for the clinical targeted therapy of OC patients. It is a long-term and arduous task for medical workers to explore the mechanism of miRNA in OC, to find effective targets, to develop effective targeted drugs for targeted therapy, so as to prolong the survival time of OC patients and improve their quality of life.
Conclusion
In summary, miR-9 is critical in the biological processes of OC. However, further in vitro and in vivo experiments are still required for its pathogenesis, thus validating the role of miR-9-regulated molecular networks in OC.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- miRNAs:
-
MicroRNAs
- OC:
-
Ovarian cancer
- miR-9:
-
hsa-miR-9-5p
- GEO:
-
Gene Expression Omnibus
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PPI:
-
Protein-protein interaction
- ACS:
-
American Cancer Society
- NOTs:
-
Normal ovarian tissues
- FDR:
-
False discovery rate
- SD:
-
Standard deviation
- OS:
-
Overall survival
- PFS:
-
Progression-free survival
- HR:
-
Hazard ratio
References
Kalampokas E, Young H, Bednarek A, Habib M, Parkin DE, Gurumurthy M, et al. Surgical outcomes and morbidity after radical surgery for ovarian cancer in Aberdeen Royal Infirmary, the northeast of Scotland Gynaecologic oncology Centre. Anticancer Res. 2018;38(2):923.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34.
Hirte H. Profile of erlotinib and its potential in the treatment of advanced ovarian carcinoma. Onco Targets Ther. 2013;6:427–35.
Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowl JH, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–89.
Coleman RL, Monk BJ, Sood AK, Herzog TJ. Latest research and treatment of advanced-stage epithelial ovarian cancer. Nat Rev Clin Oncol. 2013;10(4):211–24.
Orellana EA, Li C, Lisevick A, Kasinski AL. Identification and validation of microRNAs that synergize with miR-34a - a basis for combinatorial microRNA therapeutics. Cell Cycle (Georgetown, Tex). 2019;18(15):1798–811.
Zhang YJ, Hu Y, Li J, Chi YJ, Jiang WW, Zhang F, et al. Roles of microRNAs in immunopathogenesis of non-alcoholic fatty liver disease revealed by integrated analysis of microRNA and mRNA expression profiles. Hepatobiliary Pancreatic Dis Int. 2017;16(1):65–79.
Cai Y, Yu X, Hu S, Yu J. A brief review on the mechanisms of miRNA regulation. Genom Proteomic Bioinform. 2009;7(4):147–54.
Feng D, Zhu N, Yu C, Lou D. MicroRNA-34a suppresses human lens epithelial cell proliferation and migration via downregulation of c-met. Clin Chim Acta. 2019;495:326–30.
Sun XX, Zhang SS, Dai CY, Peng J, Pan Q, Xu LF, et al. LukS-PV-regulated MicroRNA-125a-3p promotes THP-1 macrophages differentiation and apoptosis by Down-regulating NF1 and Bcl-2. Cell Physiol Biochem. 2017;44(3):1093–105.
Seo HA, Moeng S, Sim S, Kuh HJ, Choi SY, Park JK. MicroRNA-based combinatorial cancer therapy: effects of MicroRNAs on the efficacy of anti-cancer therapies. CELLS-BASEL. 2019;9:1.
Paseban M, Marjaneh RM, Banach M, Riahi MM, Bo S, Sahebkar A. Modulation of microRNAs by aspirin in cardiovascular disease. Trends Cardiovas Med. 2020;30(5):249–54.
Ying M, Feng H, Zhang X, Liu R, Ning H. MiR-9-5p inhibits the proliferation, migration and invasion of Choroidal melanoma by targeting BRAF. Technol Cancer Res T. 2020;19:1079224635.
Bi C, Cui H, Fan H, Li L. LncRNA LINC01116 promotes the development of colorectal cancer by targeting miR-9-5p/STMN1. Oncotargets Ther. 2020;13:10547–58.
He L, Zhang L, Wang M, Wang W. miR-9 functions as a tumor inhibitor of cell proliferation in epithelial ovarian cancer through targeting the SDF-1/CXCR4 pathway. Exp Ther Med. 2017;13(4):1203–8.
Zhou B, Xu H, Xia M, Sun C, Li N, Guo E, et al. Overexpressed miR-9 promotes tumor metastasis via targeting E-cadherin in serous ovarian cancer. Front Med-Prc. 2017;11(2):214–22.
Gwak JM, Kim HJ, Kim EJ, Chung YR, Yun S, Seo AN, et al. MicroRNA-9 is associated with epithelial-mesenchymal transition, breast cancer stem cell phenotype, and tumor progression in breast cancer. Breast Cancer Res Tr. 2014;147(1):39–49.
Fei D, Li Y, Zhao D, Zhao K, Dai L, Gao Z. Serum miR-9 as a prognostic biomarker in patients with osteosarcoma. J Int Med Res. 2014;42(4):932–7.
Xu T, Liu X, Han L, Shen H, Liu L, Shu Y. Up-regulation of miR-9 expression as a poor prognostic biomarker in patients with non-small cell lung cancer. Clin Transl Oncol. 2014;16(5):469–75.
Li X, Pan Q, Wan X, Mao Y, Lu W, Xie X, et al. Methylation-associated has-miR-9 deregulation in paclitaxel- resistant epithelial ovarian carcinoma. BMC Cancer. 2015;15:509.
Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol. 2010;25(9):603–5.
Li X, Lu Y, Chen Y, Lu W, Xie X. MicroRNA profile of paclitaxel-resistant serous ovarian carcinoma based on formalin-fixed paraffin-embedded samples. BMC Cancer. 2013;13(1):216.
Sun C, Li N, Yang Z, Zhou B, He Y, Weng D, et al. miR-9 regulation of BRCA1 and ovarian cancer sensitivity to cisplatin and PARP inhibition. J Natl Cancer Inst. 2013;105(22):1750–8.
Zhang L, Zhou Q, Qiu Q, Hou L, Wu M, Li J, et al. CircPLEKHM3 acts as a tumor suppressor through regulation of the miR-9/BRCA1/DNAJB6/KLF4/AKT1 axis in ovarian cancer. Mol Cancer. 2019;18(1):144.
Sui X, Jiao YN, Yang LH, Liu J. MiR-9 accelerates epithelial-mesenchymal transition of ovarian cancer cells via inhibiting e-cadherin. Eur Rev Med Pharmaco. 2019;23:209–16.
Braga EA, Loginov VI, Filippova EA, Burdennyi AM, Pronina IV, Kazubskaya TP, et al. Diagnostic value of a group of MicroRNA genes Hypermethylated in ovarian carcinoma. B Exp Biol Med+. 2018;166(2):253–6.
Hack NJ, Wride MC, Charters KM, Kater SB, Parks TN. Developmental changes in the subcellular localization of Calretinin. J Neuro-Oncol. 2000;20(7):C67.
Stevenson L, Allen WL, Proutski I, Stewart G, Johnston L, McCloskey K, et al. Calbindin 2 (CALB2) regulates 5-fluorouracil sensitivity in colorectal cancer by modulating the intrinsic apoptotic pathway. PLoS One. 2011;6(5):e20276.
Gambaro K, Quinn MC, Wojnarowicz PM, Arcand SL, de Ladurantaye M, Barres V, et al. VGLL3 expression is associated with a tumor suppressor phenotype in epithelial ovarian cancer. Mol Oncol. 2013;7(3):513–30.
Cody NA, Shen Z, Ripeau JS, Provencher DM, Mes-Masson AM, Chevrette M, et al. Characterization of the 3p12.3-pcen region associated with tumor suppression in a novel ovarian cancer cell line model genetically modified by chromosome 3 fragment transfer. Mol Carcinog. 2009;48(12):1077–92.
Pfaff M, Reinhardt DP, Sakai LY, Timpl R. Cell adhesion and integrin binding to recombinant human fibrillin-1. FEBS Lett. 1996;384(3):247–50.
Millstein J, Budden T, Goode EL, Anglesio MS, Talhouk A, Intermaggio MP, et al. Prognostic gene expression signature for high-grade serous ovarian cancer. Ann Oncol. 2020;31(9):1240–50.
Yue H, Wang J, Chen R, Hou X, Li J. Lu X. gene signature characteristic of elevated stromal infiltration and activation is associated with increased risk of hematogenous and lymphatic metastasis in serous ovarian cancer. BMC Cancer. 2019;19(1):1266.
Lv ZD, Kong B, Liu XP, Jin LY, Dong Q, Li FN, et al. miR-655 suppresses epithelial-to-mesenchymal transition by targeting Prrx1 in triple-negative breast cancer. J Cell Mol Med. 2016;20(5):864–73.
Takahashi Y, Sawada G, Kurashige J, Uchi R, Matsumura T, Ueo H, et al. Paired related homoeobox 1, a new EMT inducer, is involved in metastasis and poor prognosis in colorectal cancer. Brit J Cancer. 2013;109(2):307–11.
Song IH, Kim KR, Lim S, Kim SH, Sung CO. Expression and prognostic significance of epithelial-mesenchymal transition-related markers and phenotype in serous ovarian cancer. Pathol Res Pract. 2018;214(10):1564–71.
Griese JJ, Hopfner KP. Structure and DNA-binding activity of the Pyrococcus furiosus SMC protein hinge domain. Protns Structure Function Bioinform. 2011;79(2):558–68.
Griese JJ, Gregor W, Karl-Peter H. Structure and DNA binding activity of the mouse condensin hinge domain highlight common and diverse features of SMC proteins. Nucleic Acids Res. 2010;38(10):3454–65.
Acknowledgements
None.
Funding
Our paper is supported by Subject of Science and Technology Commission of Minhang District (Grant NO: 2020MHZ087).
Author information
Authors and Affiliations
Contributions
ZL, LXL, TY, XM: Critical revision of the manuscript; ZL, LXL, XM: Substantial contribution to the conception and design of the work, manuscript drafting; ZL, TY, ZHL: Acquisition, analysis, and interpretation of the data; ZL, LXL, TY, ZHL, XM: Revising the manuscript critically, final approval of the version to be published. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval was not needed because this is a Bioinformatics analysis.
Consent for publication
Not applicable.
Competing interests
All the authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zuo, L., Li, X., Tan, Y. et al. Prospective pathway signaling and prognostic values of MicroRNA-9 in ovarian cancer based on gene expression omnibus (GEO): a bioinformatics analysis. J Ovarian Res 14, 29 (2021). https://doi.org/10.1186/s13048-021-00779-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-021-00779-z