
Abstract
Background
Ovarian carcinoma (OC) is a common cause of death among women with gynecological cancer. MicroRNAs (miRNAs) are believed to have vital roles in tumorigenesis of OC. Although miRNAs are broadly recognized in OC, the role of has-miR-182-5p (miR-182) in OC is still not fully elucidated.
Methods
We evaluated the significance of miR-182 expression in OC by using analysis of a public dataset from the Gene Expression Omnibus (GEO) database and a literature review. Furthermore, we downloaded three mRNA datasets of OC and normal ovarian tissues (NOTs), GSE14407, GSE18520 and GSE36668, from GEO to identify differentially expressed genes (DEGs). Then the targeted genes of hsa-miR-182-5p (TG_miRNA-182-5p) were predicted using miRWALK3.0. Subsequently, we analyzed the gene overlaps integrated between DEGs in OC and predicted target genes of miR-182 by Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. STRING and Cytoscape were used to construct a protein-protein interaction (PPI) network and the prognostic effects of the hub genes were analyzed.
Results
A common pattern of up-regulation for miR-182 in OC was found in our review of the literature. A total of 268 DEGs, both OC-related and miR-182-related, were identified, of which 133 genes were discovered from the PPI network. A number of DEGs were enriched in extracellular matrix organization, pathways in cancer, focal adhesion, and ECM-receptor interaction. Two hub genes, MCM3 and GINS2, were significantly associated with worse overall survival of patients with OC. Furthermore, we identified covert miR-182-related genes that might participate in OC by network analysis, such as DCN, AKT3, and TIMP2. The expressions of these genes were all down-regulated and negatively correlated with miR-182 in OC.
Conclusions
Our study suggests that miR-182 is essential for the biological progression of OC.
Similar content being viewed by others

Background
Ovarian carcinoma (OC) is a common cause of death among women with gynecological cancer [1]. Owing to the lack of specific symptoms and methods for early screening, approximately 75% of women have an advanced stage of the disease at diagnosis, which is consequently associated with poor outcome [2]. Therefore, it is urgent to determine the underlying mechanisms and develop new strategies for OC treatment.
MicroRNAs (miRNAs or miRs), a group of endogenous non-coding RNA molecules, can repress gene expression by mRNA degradation/destabilization or through impaired translation [3,4,5]. The abnormal expression of miRNAs occurs in a variety of tumors and often appears to be associated with altered malignant potential, such as changes in tumor cell development, cell proliferation, and apoptosis [6,7,8].
Increasing studies have revealed that aberrant expression of has-miR-182-5p (miR-182) contributes to the biological processes of various types of cancer. For example, some studies have indicated that over-expression of miR-182 shows increased tumour cell growth and proliferation, highly aggressive features, and tumor progression through repression of a plethora of targets (e.g., CAMK2N1 [9], RAB27A [10] and activation of Wnt/β-catenin signal pathway [11]; Perilli et al. [12] have found that circulating miR-182 can serve as a biomarker for tumor progression. However, some studies have shown different conclusions. For example, one study has shown that over-expression of miR-182 inhibits the epithelial to mesenchymal transition and metastasis via inactivation of Met/AKT/Snail in non-small cell lung cancer cells [13], Sun et al. [14] have reported that the miR-182 expression in cervical cancer is down-regulated and miR-182 induces cervical cancer cell apoptosis by suppressing DNMT3a expression.
MiR-182-related aggressive growth is mainly mediated by the direct regulation of genes associated with tumor invasion and metastasis [15, 16]. Furthermore, it has been found that miR-182 plays oncogenic roles by directly targeting and negatively regulating PDCD4 in ovarian cancer [17]. Previous studies have shown that miR-182 expression levels are significantly up-regulated in ovarian cancer [15, 18], whereas one study has found that miR-182 is down-regulated [19].
Owing to data published on miR-182 expression in OC are partly conflicting and heterogeneous, our study aims to unveil the role of miR-182 in ovarian cancer through investigation of miRNAs expression and identification of putative molecular targets by bioinformatics analysis and analysis based on GEO and literature reviews.
Materials and methods
Selection of GEO dataset
We obtained the microarray profiles of OC from the GEO database (Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/geo/). The following keywords were used in the GEO database: (ovarian) AND (cancer OR carcinoma OR tumor OR neoplasia OR neoplasm OR malignant OR malignancy) AND (microRNA OR miRNA OR noncoding RNA OR ncRNA OR small RNA). The microarray datasets reporting miR-182 expression between OC and normal ovarian tissues (NOTs) were included in our study.
Study selection and data extraction for literature review
A full-scale literature search was performed in PubMed and Embase (up to March 31, 2019) by using the following terms: (microRNA OR miRNA OR noncoding RNA OR ncRNA OR small RNA) AND (182 OR 182-5p) AND (ovarian) AND (cancer OR carcinoma OR tumor OR neoplasia OR neoplasm OR malignant OR malignancy). Publications were considered eligible if they met the following criteria: (1) studies examining the expression of miR-182 in OC; and (2) NOTs were used as control group. The studies were considered ineligible based on the following criteria: (1) reviews, non-clinical studies, case reports, meta-analyses, and conference abstracts; and (2) absence of control groups.
Gene ontology enrichment and target prediction analysis
The gene expression profile of GSE14407, GSE18520, and GSE36668 were obtained from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo) database. The array data of GSE14407, GSE18520, and xGSE36668 consisted of 12, 53, and 4 OC and 12, 10, and 4 NOTs samples, respectively. All data were analyzed on the GPL570 Platform Affymetrix Human Genome U133 Plus 2.0 (Affymetrix; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
The Limma package (version 3.36.5) in R/Bioconductor was used to identify the differentially expressed genes (DEGs) between OC and NOTs [20]. The adjusted P value (adj. P.Value) was applied to correct for the occurrence of false positive results using Benjamini and Hochberg false discovery rate (FDR) method by default [21]. The adj. P. Value < 0.05 and |log2(FC)| > 1 were set as the cut-off criterion. The original probe-level data in Series Matrix Files were converted into gene symbol based on the downloaded platform annotation files. The expression values of multiple probes corresponding to the same gene were selected by the minimum adj. P.Value.
The targeted genes of hsa-miR-182-5p (TG_miRNA-182-5p) were predicted using miRWALK3.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/miRretsys-self.html) [22]. Subsequently, we analyzed the gene overlaps integrated between DEGs in OC and predicted TG_miRNA-182-5p by bioinformatics software. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed for the gene overlaps using DAVID database. FDR < 0.05 was set as the cut-off criterion. Protein-protein interaction (PPI) network was constructed based on the gene overlaps using the Search Tool for the Retrieval of Interacting Genes (STRING, version 11.0, https://string-db.org/) database, which was then visualized by Cytoscape software (version 3.7.1) [23]. And confidence score C ≥ 0.7 was set as the cut-off criterion. Then, the Molecular Complex Detection (MCODE) was performed to screen modules of PPI network with degree cutoff = 2, node score cutoff = 0.2, k-core = 2, and max. Depth = 100 [24].
Survival analysis
Kaplan–Meier plotter (KM plotter, www.kmplot.com) was capable to assess the effect of 54,675 genes on survival using 18,674 cancer samples, include 5143 breast, 1816 ovarian, 2437 lung, 364 liver, and 1065 gastric cancer patients. Based on the median expression level of a particular gene, the patients with OC were divided into two groups (high vs. low). The overall survival of patients with OC was analyzed using a Kaplan–Meier plot. The hazard ratio (HR) with 95% confidence intervals (CI) and log rank P value were calculated and displayed.
Statistical analysis
All data are displayed as mean ± standard deviation (SD) from each group. Student’s t-test was performed to analyze the differences between two groups. Standardized mean difference (SMD) was applied to evaluate the association between miR-182 levels and OC by RevMan 5.3 software. We pooled SMD across GEO datasets using the Mantel–Haenszel formula (fixed-effect model) or the DerSimonian–Laird formula (random effects model). A random-effect model was adopted when the Q statistic was considered significant (p < 0.1 or I2 > 50%), otherwise, a fixed-effect model was used. The relationship of DEGs expression with miR-182 level was analyzed by Spearman’s rank correlation. A two-sided P-value < 0.05 was considered statistically significant.
Results
MiR-182 expression in OC based on GEO
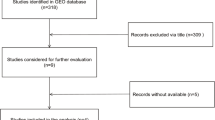
MiR-182 expression was initially assessed in a series of OC and NOTs based on GEO dataset (Fig. 1). A total of four GEO datasets (GSE47841, GSE83693, GSE53829, and GSE23338) were collected in our study. The expression levels of miR-182 in OC tissues were significantly higher than in NOTs in GSE47841 and GSE83693 datasets (p < 0.001 and p = 0.006; respectively), the expression levels of miR-182 in OC tissues were significantly lower than in NOTs in GSE53829 dataset (p < 0.001), whereas no significant difference was found in GSE23338 dataset. Characteristics of studies based on GEO dataset are presented in Table 1 and Fig. 2. However, no significant difference was found between ovarian cancer and normal ovarian tissue groups based on all the included GEO datasets (SMD = 1.42; 95% CI: − 7.62 to 10.46; p = 0.76) with significant heterogeneity by random-effected model (p < 0.0001, I2 = 98%). The results of forest plot are shown in Fig. 3.

Flow chart of study selection for GEO dataset

Expression of miR-182 in ovarian cancer and normal ovarian tissues in GEO datasets. OV: ovarian cancer tissue; Normal: normal ovarian tissue; miR-182: hsa-miR-182-5p

Forest plot (A) and funnel plot (B) of the combined SMD for hsa-miR-182-5p expression between ovarian cancer and normal ovarian tissue by the random effects models
Literature review of miR-182 expression profiles in OC versus NOTs
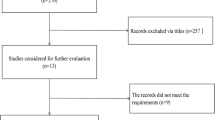
Next, we explored miR-182 expression in OC based on literature data. As shown in Fig. 4, six studies that met the criteria for selection were selected from the literature [15, 17, 19, 25,26,27]. Five of the six studies included showed that the expression level of miR-182 in OC tissues was significantly higher than that in the NOTs, while one study showed the opposite conclusion (Table 2).

Flow chart of study selection for the literature review
miR-182 prediction and bioinformatics analyses
Data preprocessing and DEGs screening
A total of 5751, 5484, and 5115 DEGs were identified from GSE14407, GSE18520, and GSE36668 datasets, respectively; 1213 common DEGs were screened out in these three datasets with Venny 2.1.0(http://bioinfogp.cnb.csic.es/tools/venny/index.html) [28] (Fig. 5, Fig. 6). Following, based on miRWALK3.0, 3240 predicted TG_hsa-miR-182-5p were obtained, of which 268, were validated in 1213 commonly identified DEGs. There were 130 up-regulated and 138 down-regulated hsa-miR-182-5p-related genes in OC tissues compared with NOTs according to data from Gene Expression Omnibus (Fig. 6, Table 3).

Volcano plot of detectable genome-wide mRNA profiles in ovarian cancer tissue and normal ovarian tissue samples from GSE14407, GSE18520, and GSE36668, respectively. Blue and red plots represent aberrantly expressed mRNAs with P<0.05 and |log(FC)|>1. Blue plots indicate up-regulated genes, red plots indicate down-regulated genes and green plots indicate normally expressed mRNAs. The x-axis is the fold-change value between the expression of mRNAs in ovarian cancer tissues and normal ovarian tissues. The y-axis is the -log10 of the adj.P.Value for each mRNA, representing the strength of the association. adj.P.Value, adjusted P value; FC, fold change

Venn plots of hsa-miR-182-5p-related differentially expressed genes from four datasets ( GSE14407, GSE18520, GSE36668, and TG_miR-182-5p), the overlapping area corresponds to the commonly identified DEGs. DEGs: differentially expressed genes; TG_miR-182-5p, target genes of hsa-miRNA-182-5p.
Functional analysis of miR-182-related DEGs in OC
Functional and pathway enrichment analysis was performed using DAVID. The analysis revealed that numerous target genes were involved in the biological processes, such as nucleus, protein binding, and extracellular matrix organization. Moreover, three KEGG pathways were over-represented in these potential target genes, that is, pathways in cancer, focal adhesion, and ECM-receptor interaction (Table 4).
PPI network construction and modules selection
The PPI network of miR-182-related DEGs consisted of 153 nodes and 439 edges, including 73 up-regulated genes and 60 down-regulated genes (Fig. 7). Degrees ≥10 was set as the cutoff criterion [29], a total of 28 genes were selected as hub genes, Moreover, there were close correlations among hub genes (Fig. 8a, Additional file 1: Table S1). A significant module was obtained from PPI network of miR-182-related DEGs using MCODE, including 17 nodes and 122 edges (Fig. 8b).

Protein-protein interaction network of hsa-miR-182-5p-related DEGs. Blue nodes stand for up-regulated genes, while red nodes stand for down-regulated genes. The lines represent interaction relationship between nodes. DEGs, differentially expressed genes

Protein-protein interaction network. (A): Protein-protein interaction network of hube genes of hsa-miR-182-5p-related DEGs. (B): A significant module selected from protein-protein interaction network of hsa-miR-182-5p-related DEGs. Blue nodes stand for up-regulated genes, while red nodes stand for down-regulated genes. The lines represent interaction relationship between nodes. DEGs, differentially expressed genes
Survival analysis
The prognostic value of 28 hub genes in PPI network was assessed in www.kmplot.com. The overall survival of patients with OC was analyzed depending on the high and low expression of each hub gene. It was found that low mRNA expression of MCM3 (HR 0.75 [0.58–0.97], P = 0.027) was associated with worse overall survival for ovarian cancer patients, as well as GINS2 (HR 0.75 [0.58–0.97], P = 0.026) (Fig. 9). In the PPI network, a total of 21 genes (TPM1, COL1A1, PDGFRA, UBE2B, MEF2C, SNAI2, CACNA2D1, RECK, FOXO1, FBN1, ANTXR2, NKX3–1, TIMP2, AKT3, RBPMS, EGLN3, DERL1, PRKD1, SLC2A13, MAF, and DCN) were revealed to exert their potential roles in OC by interactions with miR-182 (Table 5, Additional file 2: Figure S1).

Overall survival analysis of MCM3 and GINS2 expression with prognosis of ovarian cancer patients. The patients with ovarian cancer were divided into two groups (high vs. low), according to the median expression level of MCM3 and GINS2
Discussion
In current study, we identified aberrantly expressed miR-182 associated with OC through the comparison of miRNA expression profiles in OC tissues with that of NOTs based on data from GEO datasets and published studies. In addition, we identified and analyzed novel markers and potential targets for miR-182 that were involved in the regulation of crucial biological processes in OC by GO analysis, KEGG pathway annotation, protein-protein interaction (PPI) network, and Kaplan-Meier plotter.
To date, there have been only a few studies on the characteristics of miR-182 in OC. In 2012, Liu et al. [15] demonstrated that the expression level of miR-182 was higher in high-grade papillary serous carcinoma (HG-PSC) than in fallopian tube tissues. Subsequently, several other studies also supported the conclusion that miR-182 expression was up-regulated in ovarian cancer tissues [17, 25,26,27]. Interestingly, a recent study have suggested that the expression level of miR-182 is down-regulated in ovarian cancer tissues compared with paracancerous and NOTs [19]. Based on the data of included GEO datasets, only two sets of GSE datasets (GSE47841 and GSE83693) showed the expression level of miR-182 was up-regulated in OC. Due to the result of the expression level of miR-182 in OC is still controversial, further investigation is necessary to elucidate the role of miR-182 in OC.
MiR-182 is one of the most frequently deregulated miRNA in OC. Marzec-Kotarska et al. [25] demonstrated that miR-182 expression was significantly increased, higher miR-182 expression was linked with significantly shorter overall survival, and Deletion of the PRDM5 locus may play a supportive role in miR-182 overexpression in OC. Additionally, Wang et al. [17] found that in ovarian cancer, miR-182, as an oncogenic miRNA, promoted cell growth, invasion, and chemoresistance by directly and negatively regulating PDCD4. Liu et al. [15] proposed that the oncogenic properties of miR-182 in ovarian cancer may be partly due to its impaired repair of DNA double-strand breaks, negative regulation of breast cancer 1 (BRCA1) and metastasis suppressor 1 (MTSS1) expressions, and positive regulation of oncogene high-mobility group AT-hook 2 (HMGA2). Interestingly, a recent study has found that miR-182 could induce apoptosis of ovarian cancer by regulating of DNTM3a expression [19]. In view of the current situation, it is necessary to further elucidate the molecular mechanism and clinical value that is associated with abnormal expression of miR-182 in OC.
MCM3 is a member of minichromosome maintenance protein family with a critical role in initiation of DNA replication [30]. It is present during cellular proliferation of normal cells, premalignant and neoplastic cells but absent in cells that are in G0 phase [31]. MCM3, presented in a variety of human tumors, is involved in tumor proliferation [32], diagnosis [33], and prognosis. For example, Hua et al. [34] reported that high mRNA expression levels of MCM2 and MCM3 were correlated with a poor outcome and thus might be clinically useful molecular prognostic markers in glioma. Jankowska-Konsur et al. [35] found that MCM3 was reliable parameters for the correlation with clinical stage of mycosis fungoides and MCM3 expression was of prognostic value in mycosis fungoides. In current study, survival analysis of the hub genes related to target genes of miR-182 revealed that MCM3 was highly associated with poor prognosis of patients with OC. GINS complex subunit 2 (GINS2), is a member of the GINS complex, composed of GINS1, GINS2, GINS3, and GINS4, which is involved in DNA replication [36]. GINS2 is up-regulated in a variety of aggressive tumors. For example, Liu et al. [37] found that lung cancer tissues over-expressed GIN2, which was connected to lung cancer metastasis. Recent years, some studies have found that GINS2 is a novel prognostic biomarker and promoted tumor progression in early-stage cervical cancer [38]; GINS2 is closely related to the occurrence and development of glioma, and may become a prognostic marker for glioma patients [39]; GINS2 is markedly expressed in EOC tissues and cell lines, stable GINS2 knockdown in SKOV-3 cells could significantly inhibit cell proliferation and induce cell cycle arrest and cell apoptosis [40]. In current study, survival analysis of the hub genes related to target genes of miR-182 showed that GINS2 was highly associated with poor prognosis of patients with OC.
In current study, we identified that novel candidate target genes for miR-182 were involved in the regulation of crucial biological processes in OC, such as DCN, AKT3, and TIMP2. DCN is a component of connective tissue, binds to type I collagen fibrils and plays a role in collagen fibrillogenesis when helps to orient fibers. Some previous studies showed that DCN played an important function in cancer development and metastasis. For example, Ehlen et al. [41] found that DCN gene was up-regulated in OC cells and played an important role in SEOC angiogenesis and tumor progression; highly expressed DCN could increase angiogenesis and tumor cell invasiveness in bladder cancer [42]. Our current study revealed that DCN mRNA was decreased in OC compared with NOTs, which was consistent with some previous studies [43,44,45,46], we hypothesized that DCN can act as a tumor suppressor in OC. AKT3 is a homologous gene that belongs to the serine/threonine protein kinase AKT subfamily and is critical in the AKT signal pathway [41, 47]. The expression and activation of AKT3 mediates cancer progression and controls cellular processes such as cell growth, proliferation, apoptosis, and invasion [48]. Some studies found that AKT3 was up-regulated in many types of cancers and knockdown of AKT3 isoforms could abrogate the growth of tumors through inducing cell apoptosis and inhibiting proliferation [49,50,51]. Moreover, lots of evidence suggested that AKT3 was regulated by miRNAs, such as miR-497 [52], miR-338 [53], and miR-16 [54], which could suppress cancer progression. In line with these findings, we hypothesized that AKT3 can be identified as a target gene of miR-182 in OC. TIMP2 is one of four well-known members of the TIMP family: TIMP1, TIMP2, TIMP3, and TIMP4. The function of TIMP2 in carcinogenesis is multifaceted. For example, some previous studies showed that an increasing level of TIMP2 can promote cellular proliferation and invasion in some tumors [55, 56]. On the contrary, some studies found that TIMP2 could inhibit vascular endothelial growth factor A (VEGF-A)-induced endothelial cell proliferation and angiogenesis by binding to α3β1 integrin [57]. Moreover, TIMP2 could prevent the activation of tyrosine kinase receptors in tumor cells, including focal adhesion kinase [58], AKT [59] and epithelial growth factor receptor [60], which played key roles in tumor migration and growth. Our current study revealed that TIMP2 mRNA was decreased in OC compared with NOTs, which was consistent with some previous studies [61,62,63]. To the best of our knowledge, few studies have shown that the down-regulation pattern of TIMP2 occurs in OC tissues. Thus, further investigation is required to explore the ectopic expression of TIMP2 in OC.
Conclusions
In summary, the results presented here suggest that miR-182 plays an important role in the biology of OC. However, further studies in vitro and in vivo are still needed on the pathogenesis to validate the role of miR-182-regulated molecular networks in OC.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
To SKY, Mak ASC, Eva Fung YM, Che CM, Li SS, Deng W, et al. Beta-catenin downregulates dicer to promote ovarian cancer metastasis. Oncogene. 2017;36(43):5927–38. https://doi.org/10.1038/onc.2017.185.
Shapira I, Oswald M, Lovecchio J, Khalili H, Menzin A, Whyte J, et al. Circulating biomarkers for detection of ovarian cancer and predicting cancer outcomes. Br J Cancer. 2014;110(4):976–83. https://doi.org/10.1038/bjc.2013.795.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97.
Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med. 2009;60:167–79. https://doi.org/10.1146/annurev.med.59.053006.104707.
Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610. https://doi.org/10.1038/nrg2843.
An HJ, Kwak SY, Yoo JO, Kim JS, Bae IH, Park MJ, et al. Novel miR-5582-5p functions as a tumor suppressor by inducing apoptosis and cell cycle arrest in cancer cells through direct targeting of GAB1, SHC1, and CDK2. Biochim Biophys Acta. 2016;1862(10):1926–37. https://doi.org/10.1016/j.bbadis.2016.07.017.
Hemmatzadeh M, Mohammadi H, Karimi M, Musavishenas MH, Baradaran B. Differential role of microRNAs in the pathogenesis and treatment of Esophageal cancer. Biomed Pharmacother. 2016;82:509–19. https://doi.org/10.1016/j.biopha.2016.05.009.
Wo L, Lu D, Gu X. Knockdown of miR-182 promotes apoptosis via regulating RIP1 deubiquitination in TNF-alpha-treated triple-negative breast cancer cells. Tumour Biol. 2016;37(10):13733–42. https://doi.org/10.1007/s13277-016-5174-z.
Li N, Nan CC, Zhong XY, Weng JQ, Fan HD, Sun HP, et al. miR-182-5p promotes growth in Oral squamous cell carcinoma by inhibiting CAMK2N1. Cell Physiol Biochem. 2018;49(4):1329–41. https://doi.org/10.1159/000493411.
Li Y, Chen S, Shan Z, Bi L, Yu S, Li Y et al. miR-182-5p improves the viability, mitosis, migration, and invasion ability of human gastric cancer cells by down-regulating RAB27A. Bioscience Reports. 2017;37(3). doi:https://doi.org/10.1042/BSR20170136.
Wang D, Lu G, Shao Y, Xu D. MiR-182 promotes prostate cancer progression through activating Wnt/beta-catenin signal pathway. Biomed Pharmacother. 2018;99:334–9. https://doi.org/10.1016/j.biopha.2018.01.082.
Perilli L, Vicentini C, Agostini M, Pizzini S, Pizzi M, D'Angelo E, et al. Circulating miR-182 is a biomarker of colorectal adenocarcinoma progression. Oncotarget. 2014;5(16):6611–9. https://doi.org/10.18632/oncotarget.2245.
Li Y, Zhang H, Li Y, Zhao C, Fan Y, Liu J, et al. MiR-182 inhibits the epithelial to mesenchymal transition and metastasis of lung cancer cells by targeting the met gene. Mol Carcinog. 2018;57(1):125–36. https://doi.org/10.1002/mc.22741.
Sun J, Ji J, Huo G, Song Q, Zhang X. miR-182 induces cervical cancer cell apoptosis through inhibiting the expression of DNMT3a. Int J Clin Exp Pathol. 2015;8(5):4755–63.
Liu Z, Liu J, Segura MF, Shao C, Lee P, Gong Y, et al. MiR-182 overexpression in tumourigenesis of high-grade serous ovarian carcinoma. J Pathol. 2012;228(2):204–15. https://doi.org/10.1002/path.4000.
Segura MF, Hanniford D, Menendez S, Reavie L, Zou X, Alvarez-Diaz S, et al. Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor. Proc Natl Acad Sci U S A. 2009;106(6):1814–9. https://doi.org/10.1073/pnas.0808263106.
Wang YQ, Guo RD, Guo RM, Sheng W, Yin LR. MicroRNA-182 promotes cell growth, invasion, and chemoresistance by targeting programmed cell death 4 (PDCD4) in human ovarian carcinomas. J Cell Biochem. 2013;114(7):1464–73. https://doi.org/10.1002/jcb.24488.
McMillen BD, Aponte MM, Liu Z, Helenowski IB, Scholtens DM, Buttin BM, et al. Expression analysis of MIR182 and its associated target genes in advanced ovarian carcinoma. Modern Pathol. 2012;25(12):1644–53. https://doi.org/10.1038/modpathol.2012.118.
Lu W, Lu T, Wei X. Downregulation of DNMT3a expression increases miR-182-induced apoptosis of ovarian cancer through caspase-3 and caspase-9-mediated apoptosis and DNA damage response. Oncol Rep. 2016;36(6):3597–604. https://doi.org/10.3892/or.2016.5134.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research. 2015;43(7):e47. https://doi.org/10.1093/nar/gkv007.
Hardcastle TJ. Generalized empirical Bayesian methods for discovery of differential data in high-throughput biology. Bioinformatics. 2016;32(2):195–202. https://doi.org/10.1093/bioinformatics/btv569.
Sticht C, De La Torre C, Parveen A, Gretz N. miRWalk: An online resource for prediction of microRNA binding sites. PloS one. 2018;13(10):e0206239. https://doi.org/10.1371/journal.pone.0206239.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. https://doi.org/10.1101/gr.1239303.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC bioinformatics. 2003;4:2.
Marzec-Kotarska B, Cybulski M, Kotarski JC, Ronowicz A, Tarkowski R, Polak G, et al. Molecular bases of aberrant miR-182 expression in ovarian cancer. Genes, chromosomes & cancer. 2016;55(11):877–89. https://doi.org/10.1002/gcc.22387.
Vilming Elgaaen B, Olstad OK, Haug KB, Brusletto B, Sandvik L, Staff AC, et al. Global miRNA expression analysis of serous and clear cell ovarian carcinomas identifies differentially expressed miRNAs including miR-200c-3p as a prognostic marker. BMC Cancer. 2014;14:80. https://doi.org/10.1186/1471-2407-14-80.
Wang L, Zhu MJ, Ren AM, Wu HF, Han WM, Tan RY, et al. A ten-microRNA signature identified from a genome-wide microRNA expression profiling in human epithelial ovarian cancer. PLoS One. 2014;9(5):e96472. https://doi.org/10.1371/journal.pone.0096472.
Oliveros JC. (2007-2015) Venny. An interactive tool for comparing lists with Venn's diagrams.
Xu Z, Zhou Y, Cao Y, Dinh TL, Wan J, Zhao M. Identification of candidate biomarkers and analysis of prognostic values in ovarian cancer by integrated bioinformatics analysis. Med Oncol. 2016;33(11):130. https://doi.org/10.1007/s12032-016-0840-y.
Scott IS, Odell E, Chatrath P, Morris LS, Davies RJ, Vowler SL, et al. A minimally invasive immunocytochemical approach to early detection of oral squamous cell carcinoma and dysplasia. Br J Cancer. 2006;94(8):1170–5. https://doi.org/10.1038/sj.bjc.6603066.
Gonzalez MA, Tachibana KE, Laskey RA, Coleman N. Control of DNA replication and its potential clinical exploitation. Nat Rev Cancer. 2005;5(2):135–41. https://doi.org/10.1038/nrc1548.
Valverde LF, de Freitas RD, Pereira TA, de Resende MF, Agra IMG, Dos Santos JN, et al. MCM3: a novel proliferation marker in Oral squamous cell carcinoma. Appl Immunohistochem Mol Morphol. 2018;26(2):120–5. https://doi.org/10.1097/PAI.0000000000000397.
Ashkavandi ZJ, Najvani AD, Tadbir AA, Pardis S, Ranjbar MA, Ashraf MJ. MCM3 as a novel diagnostic marker in benign and malignant salivary gland tumors. Asian Pacific J Cancer Prevention. 2013;14(6):3479–82.
Hua C, Zhao G, Li Y, Bie L. Minichromosome Maintenance (MCM) Family as potential diagnostic and prognostic tumor markers for human gliomas. BMC Cancer 2014;14:526. https://doi.org/10.1186/1471-2407-14-526.
Jankowska-Konsur A, Kobierzycki C, Reich A, Grzegrzolka J, Maj J, Dziegiel P. Expression of MCM-3 and MCM-7 in primary cutaneous T-cell lymphomas. Anticancer Res. 2015;35(11):6017–26.
MacNeill SA. Structure and function of the GINS complex, a key component of the eukaryotic replisome. Biochem J. 2010;425(3):489–500. https://doi.org/10.1042/BJ20091531.
Liu M, Pan H, Zhang F, Zhang Y, Zhang Y, Xia H, et al. Identification of TNM stage-specific genes in lung adenocarcinoma by genome-wide expression profiling. Oncol Lett. 2013;6(3):763–8. https://doi.org/10.3892/ol.2013.1469.
Ouyang F, Liu J, Xia M, Lin C, Wu X, Ye L, et al. GINS2 is a novel prognostic biomarker and promotes tumor progression in early-stage cervical cancer. Oncol Rep. 2017;37(5):2652–62. https://doi.org/10.3892/or.2017.5573.
Shen YL, Li HZ, Hu YW, Zheng L, Wang Q. Loss of GINS2 inhibits cell proliferation and tumorigenesis in human gliomas. CNS Neurosci Ther. 2019;25(2):273–87. https://doi.org/10.1111/cns.13064.
Yan T, Liang W, Jiang E, Ye A, Wu Q, Xi M. GINS2 regulates cell proliferation and apoptosis in human epithelial ovarian cancer. Oncol Lett. 2018;16(2):2591–8. https://doi.org/10.3892/ol.2018.8944.
Robey RB, Hay N. Is Akt the "Warburg kinase"?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19(1):25–31. https://doi.org/10.1016/j.semcancer.2008.11.010.
El Behi M, Krumeich S, Lodillinsky C, Kamoun A, Tibaldi L, Sugano G, et al. An essential role for decorin in bladder cancer invasiveness. EMBO molecular medicine. 2013;5(12):1835–51. https://doi.org/10.1002/emmm.201302655.
Bayani J, Brenton JD, Macgregor PF, Beheshti B, Albert M, Nallainathan D, et al. Parallel analysis of sporadic primary ovarian carcinomas by spectral karyotyping, comparative genomic hybridization, and expression microarrays. Cancer Res. 2002;62(12):3466–76.
Grisaru D, Hauspy J, Prasad M, Albert M, Murphy KJ, Covens A, et al. Microarray expression identification of differentially expressed genes in serous epithelial ovarian cancer compared with bulk normal ovarian tissue and ovarian surface scrapings. Oncol Rep. 2007;18(6):1347–56.
Ono K, Tanaka T, Tsunoda T, Kitahara O, Kihara C, Okamoto A, et al. Identification by cDNA microarray of genes involved in ovarian carcinogenesis. Cancer Res. 2000;60(18):5007–11.
Shridhar V, Lee J, Pandita A, Iturria S, Avula R, Staub J, et al. Genetic analysis of early- versus late-stage ovarian tumors. Cancer Res. 2001;61(15):5895–904.
Hers I, Vincent EE, Tavare JM. Akt signalling in health and disease. Cell Signal. 2011;23(10):1515–27. https://doi.org/10.1016/j.cellsig.2011.05.004.
Kim M, Kim YY, Jee HJ, Bae SS, Jeong NY, Um JH, et al. Akt3 knockdown induces mitochondrial dysfunction in human cancer cells. Acta Biochim Biophys Sin. 2016;48(5):447–53. https://doi.org/10.1093/abbs/gmw014.
Madhunapantula SV, Robertson GP. Targeting protein kinase-b3 (akt3) signaling in melanoma. Expert Opin Ther Targets. 2017;21(3):273–90. https://doi.org/10.1080/14728222.2017.1279147.
Paul-Samojedny M, Pudelko A, Kowalczyk M, Fila-Danilow A, Suchanek-Raif R, Borkowska P, et al. Knockdown of AKT3 and PI3KCA by RNA interference changes the expression of the genes that are related to apoptosis and autophagy in T98G glioblastoma multiforme cells. Pharmacol Reports. 2015;67(6):1115–23. https://doi.org/10.1016/j.pharep.2015.04.012.
Paul-Samojedny M, Suchanek R, Borkowska P, Pudelko A, Owczarek A, Kowalczyk M, et al. Knockdown of AKT3 (PKBgamma) and PI3KCA suppresses cell viability and proliferation and induces the apoptosis of glioblastoma multiforme T98G cells. Biomed Res Int. 2014;2014:768181. https://doi.org/10.1155/2014/768181.
Zhuang J, Ye Y, Wang G, Ni J, He S, Hu C, et al. MicroRNA497 inhibits cellular proliferation, migration and invasion of papillary thyroid cancer by directly targeting AKT3. Mol Med Rep. 2017;16(5):5815–22. https://doi.org/10.3892/mmr.2017.7345.
Sui GQ, Fei D, Guo F, Zhen X, Luo Q, Yin S, et al. MicroRNA-338-3p inhibits thyroid cancer progression through targeting AKT3. Am J Cancer Res. 2017;7(5):1177–87.
Wang X, Li GH. MicroRNA-16 functions as a tumor-suppressor gene in oral squamous cell carcinoma by targeting AKT3 and BCL2L2. J Cell Physiol. 2018;233(12):9447–57. https://doi.org/10.1002/jcp.26833.
Hayakawa T, Yamashita K, Tanzawa K, Uchijima E, Iwata K. Growth-promoting activity of tissue inhibitor of metalloproteinases-1 (TIMP-1) for a wide range of cells. A possible new growth factor in serum. FEBS Lett. 1992;298(1):29–32.
Lu KV, Jong KA, Rajasekaran AK, Cloughesy TF, Mischel PS. Upregulation of tissue inhibitor of metalloproteinases (TIMP)-2 promotes matrix metalloproteinase (MMP)-2 activation and cell invasion in a human glioblastoma cell line. Laboratory Investigation. 2004;84(1):8–20. doi:https://doi.org/10.1038/sj.labinvest.3700003.
Seo DW, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, et al. TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell. 2003;114(2):171–80.
Kandalam V, Basu R, Moore L, Fan D, Wang X, Jaworski DM, et al. Lack of tissue inhibitor of metalloproteinases 2 leads to exacerbated left ventricular dysfunction and adverse extracellular matrix remodeling in response to biomechanical stress. Circulation. 2011;124(19):2094–105. https://doi.org/10.1161/CIRCULATIONAHA.111.030338.
Valacca C, Tassone E, Mignatti P. TIMP-2 interaction with MT1-MMP activates the AKT pathway and protects tumor cells from apoptosis. PLoS One. 2015;10(9):e0136797. https://doi.org/10.1371/journal.pone.0136797.
Hoegy SE, Oh HR, Corcoran ML, Stetler-Stevenson WG. Tissue inhibitor of metalloproteinases-2 (TIMP-2) suppresses TKR-growth factor signaling independent of metalloproteinase inhibition. J Biol Chem. 2001;276(5):3203–14. https://doi.org/10.1074/jbc.M008157200.
Hsu CH, Peng KL, Kang ML, Chen YR, Yang YC, Tsai CH, et al. TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2012;2(3):568–79. https://doi.org/10.1016/j.celrep.2012.08.030.
Shin YJ, Kim JH. The role of EZH2 in the regulation of the activity of matrix metalloproteinases in prostate cancer cells. PLoS One. 2012;7(1):e30393. https://doi.org/10.1371/journal.pone.0030393.
Yang SW, Cody JJ, Rivera AA, Waehler R, Wang M, Kimball KJ, et al. Conditionally replicating adenovirus expressing TIMP2 for ovarian cancer therapy. Clin Cancer Res. 2011;17(3):538–49. https://doi.org/10.1158/1078-0432.CCR-10-1628.
Acknowledgements
We thank the data provided by the authors of included studies and GEO.
Funding
This study was funded by the Scientific Research and Technology Development Program of Guangxi (NO.Guike 14140004) and the Natural Science Foundation of Guangxi (NO. 2014jjAA40673).
Author information
Authors and Affiliations
Contributions
YL conceived, designed this study and wrote the manuscript; YL and LL performed data analysis; LL reviewed the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Node-degree analysis of the 28 hub genes (Degree ≥10).
Additional file 2: Figure S1.
The correlated expression of gene and hsa-miR-182-5p (miR-182) in patients with ovarian cancer. a: TPM1, b: COL1A1, c: PDGFRA, d: UBE2B, e: MEF2C, f: SNAI2, g: CACNA2D1, h: RECK, i: FOXO1, j: FBN1, k: ANTXR2, l: NKX3–1, m: TIMP2, n: AKT3, o: RBPMS, p: EGLN3, q: DERL1, r: PRKD1, s: SLC2A13, t: MAF, u: DCN.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, Y., Li, L. Prognostic values and prospective pathway signaling of MicroRNA-182 in ovarian cancer: a study based on gene expression omnibus (GEO) and bioinformatics analysis. J Ovarian Res 12, 106 (2019). https://doi.org/10.1186/s13048-019-0580-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-019-0580-7