
Abstract
Cuproptosis is a newly identified form of cell death induced by excessive copper (Cu) accumulation within cells. Mechanistically, cuproptosis results from Cu-induced aggregation of dihydrolipoamide S-acetyltransferase, correlated with the mitochondrial tricarboxylic acid cycle and the loss of iron–sulfur cluster proteins, ultimately resulting in proteotoxic stress and triggering cell death. Recently, cuproptosis has garnered significant interest in tumor research due to its potential as a crucial therapeutic strategy against cancer. In this review, we summarized the cellular and molecular mechanisms of cuproptosis and its relationship with other types of cell death. Additionally, we reviewed the current drugs or strategies available to induce cuproptosis in tumor cells, including Cu ionophores, small compounds, and nanomedicine. Furthermore, we targeted cell metabolism and specific regulatory genes in cancer therapy to enhance tumor sensitivity to cuproptosis. Finally, we discussed the feasibility of targeting cuproptosis to overcome tumor chemotherapy and immunotherapy resistance and suggested future research directions. This study suggested that targeting cuproptosis could open new avenues for developing tumor therapy.
Similar content being viewed by others
Introduction
Copper (Cu) is an essential trace metal element for normal physiological functions primarily obtained from dietary supplements. In biological systems, Cu exists predominantly in two oxidative states: divalent copper ions (Cu2+) and monovalent copper ions (Cu+). Cu+ is the principal oxidative form and plays a significant role in physiological and pathological regulation within cells [1,2,3]. Disruptions in Cu homeostasis can induce disease onset; for instance, Cu overload may lead to Wilson’s disease [4], while Cu deficiency can cause Menkes disease [5]. Furthermore, previous studies have demonstrated that Cu could promote tumor cell proliferation, angiogenesis, and metastasis and reduces the efficacy of tumor treatments [6].
However, excess Cu in the cells can also cause cellular damage. Researchers observed that using Cu ionophores to elevate Cu levels within tumor cells could cause cell death [7, 8]. Although subsequent studies have extensively investigated the molecular mechanisms underlying Cu-induced cell death, such as the association of this type of cell death with reactive oxygen species (ROS), apoptosis, and ferroptosis-related signaling pathways, the key mechanisms remain unclear. Tsvetkov et al. termed the Cu-induced cell death cuproptosis in 2022 based on their findings that this form of cell death depends on the aggregation of lipoylated dihydrolipoamide S-acetyltransferase (DLAT) and the reduction of iron–sulfur cluster (Fe–S) proteins, triggered by Cu accumulation in mitochondria, leading to proteotoxic stress, and cell death [9].
With the concept and mechanism of cuproptosis established, researchers have increasingly focused on cuproptosis in cancer therapy and demonstrated that Cu-based treatments play a pivotal role in inhibiting tumor growth. In this review, we summarized the core molecular mechanisms of cuproptosis and discussed the relationship between it and other forms of cell death. Moreover, we systematically summarized the current understanding of targeting cuproptosis for tumor therapy, including using Cu ionophores, small compounds, and nanomedicine to induce cuproptosis and targeting cell metabolism or certain genes to sensitize cuproptosis. Additionally, we discussed the potential of targeting cuproptosis to overcome tumor drug resistance in chemotherapy, targeted therapy, and immunotherapy. We also discussed the opportunities and challenges in targeting cuproptosis-associated cancer therapy.
Cu metabolism
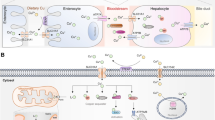
Cu homeostasis is essential for the normal physiological functioning of human life. In humans, Cu uptake, distribution, transport, and elimination are meticulously regulated (Fig. 1A), which is crucial to prevent deficiency or excessive Cu accumulation in various tissues and cells, thereby averting disease onset [10]. Humans predominantly acquire Cu through their diet, with an adult daily requirement ranging from 0.8 to 2.4 mg [3]. Following digestion in the stomach and duodenum, Cu is primarily absorbed in the small intestine, where Cu2+ is reduced to Cu+ by metalloreductases, such as six-transmembrane epithelial antigen of the prostate (STEAP) [11] and duodenal cytochrome b (DCYTB) [12], before being transported into enterocytes by Cu transport protein 1 (CTR1), also known as the solute carrier family 31 member 1 (SLC31A1) [13], located at the apical membrane of enterocytes. Subsequently, Cu is exported to the interstitial fluid or bloodstream by the protein ATPase copper transporting alpha (ATP7A) [14]. Cu in the bloodstream usually binds to plasma proteins, such as ceruloplasmin and human serum albumin, and is transported to the liver, the primary Cu storage organ, via the portal system [15]. Metallothionein1/2 (MT1/2), a thiol-rich protein, binds to and stores Cu in hepatocytes [16]. Hepatocytes can excrete excess Cu into the bile via ATP7B. Beyond the liver, Cu in the bloodstream can also be absorbed by other tissues and organs, including the heart and the brain [15].

Cu metabolism. A. Cu homeostasis in physiological systems. Dietary Cu2+ is reduced to Cu+ by the STEAP family members and subsequently transported into enterocytes via SLC31A1. Within enterocytes, Cu+ is released into the bloodstream by ATP7A, where it typically binds to soluble chaperones such as albumin and ceruloplasmin. Hepatocytes uptake Cu+ from the bloodstream through SLC31A1 on their cell membrane. In hepatocytes, Cu+ can be delivered to MT or GSH for storage, or to ATP7B for re-entry into the bloodstream for uptake by other tissues and organs. Additionally, ATP7B in hepatocytes can excrete excess Cu+ into the bile duct. B. Cu metabolism at the cellular levels. Extracellular Cu+ is taken up by SLC31A1 on the cell membrane, while Cu2+ can be transported into the cell by SLC11A2. Intracellular Cu+ can be sequestered by MT1/2 and GSH or bound to other Cu chaperones such as CCS, COX17, and ATOX1 for further trafficking to proteins. CCS delivers Cu+ to cytosolic SOD1, COX17 transports Cu+ to mitochondrial COX11 or SCO1, and ATOX1 delivers Cu+ to ATP7A/B located on the Golgi membrane in the TGN, ultimately secreting it outside the cell. Cu+ can also form a non-proteinaceous low-molecular-weight complex, CuL, which transports Cu+ into mitochondrion. Cu+ can be transported into the mitochondrial matrix via solute carrier family 25 member 3 (SLC25A3) located in inner mitochondrial membrane. Cu in the matrix is transported back across the IMM to intermembrane space (IMS) by an unknown transporter. Histones H3/H4 in the nucleus can reduce Cu2+ to Cu+. STEAP, six-transmembrane epithelial antigen of the prostate; SLC31A1, the solute carrier family 31 member 1; ATP7A, ATPase copper transporting alpha; MT1/2, Metallothionein1/2; GSH, glutathione; CCS, Cu chaperone for superoxide dismutase; COX, cytochrome oxidase; SOD1, superoxide dismutase 1; ATOX1, antioxidant 1; TGN, trans-Golgi network; SLC25A3, solute carrier family 25 member 3
After being transported into cells by transmembrane proteins, such as CTR1 and divalent metal transporter 1 (DMT1), Cu can be stored by binding to intracellular Cu chaperones, such as MT1/2 and glutathione (GSH), thereby preventing cellular damage [1, 17]. Concurrently, Cu can be delivered to other cellular structures or proteins via Cu chaperones to maintain normal cellular function (Fig. 1B). For example, the Cu chaperone for superoxide dismutase (CCS) binds to Cu and delivers it to superoxide dismutase 1 (SOD1), catalyzing the conversion of superoxide radicals to H2O2, thus maintaining cellular ROS homeostasis [18]. Abnormal SOD1 expression is considered to be linked to tumor development [19]. Additionally, the Cu chaperone antioxidant 1 (ATOX1), which binds Cu+ via two cysteine residues, can transport cytosolic Cu+ to the ATP7A/ATP7B, located in the Golgi network [20, 21]. Excess Cu+ enters the trans-Golgi network (TGN) and can be expelled from the cell via the vesicular system [20, 21]. The absence of ATOX1 leads to perinatal lethality induced by Cu dyshomeostasis [22]. Moreover, increased intracellular Cu levels can promote the distribution of ATP7A and ATP7B within the TGN, thereby facilitating Cu efflux [23]. These studies highlight significant regulatory roles of ATOX1 and ATP7A/B in intracellular Cu homeostasis.
The mitochondria are the primary targets of intracellular Cu (Fig. 1B). Cu contributes to ATP production within the mitochondria by ensuring the catalytic function of cytochrome oxidase (COX) during oxidative phosphorylation (OXPHOS). Cytosolic Cu+ can be transported to the mitochondrial intermembrane space by a Cu ligand (CuL), a non-proteinaceous low-molecular-weight complex, where it can enter the mitochondrial matrix via solute carrier family 25 member 3 (SLC25A3) located in the inner mitochondrial membrane (IMM) [24, 25]. Cu in the matrix is transported back across the IMM to intermembrane space (IMS) by an unknown transporter, where it is delivered to COX and SOD1 [26]. Additionally, COX17 can transfer cytosolic Cu+ to the mitochondrial intermembrane space and deliver Cu+ to other Cu chaperone molecules, such as COX11 and synthesis of cytochrome c oxidase 1/2 (SCO1/2), which then deliver Cu+ to COX1 and COX2, respectively [27, 28].
Cu and cancer
The role of Cu in tumorigenesis and tumor therapy has been of great concern. Compared to the healthy population, elevated Cu levels have been observed in the tumors or serum of patients with various types of cancers, including breast [29, 30], prostate [31], lung [32], cervical [33], and bladder [34], thyroid [35], and oral cancers [36, 37]. Cu can promote the onset and development of tumors, termed cuproplasia, by activating oncogenic signaling pathways [6]. For instance, Cu can directly bind mitogen-activated extracellular signal-regulated kinase (MEK) and activate its downstream mitogen-activated protein kinase (MAPK) pathway to promote tumor cell growth [38, 39]. In melanoma driven by BRAFV600E, chelating Cu can inhibit MAPK signal and reduce tumor cell resistance to BRAFV600E and MEK1/2 inhibitors [40]. Moreover, Cu is considered to contribute to tumor metastasis. Cu can act on metalloenzymes in the extracellular matrix, such as lysyl oxidase (LOX) [41] and secreted protein acidic and rich in cysteine (SPARC) [42], altering cell–matrix and cell–cell interactions, thereby promoting tumor cell migration and metastasis. In breast cancer cells, Cu can bind to the mediator of ErbB2-driven cell motility 1 (MEMO1) and activate its oxidase activity promoting O2− production and enhancing tumor cell migration and invasion [43, 44]. Additionally, Cu can promote angiogenesis, essential for tumor growth and progression, by activating pro-angiogenic factors, such as fibroblast growth factors (FGFs), vascular endothelial growth factor (VEGF), tumor necrosis factor alpha (TNF-α), interleukin (IL)-1, IL-6, and IL-8 [45,46,47,48]. Taken together, Cu is a critical trace metal element in tumor development. Elucidating the molecular mechanisms by which Cu drives tumorigenesis will aid the discovery of new therapeutic targets for cancer.
Cuproptosis
Although Cu-induced cell death has been investigated for decades, the concept of ‘cuproptosis’ was not proposed until 2022 by Tsvetkov and colleagues [9]. A significant feature of Cu ionophores and Cu-treated cells is a sharp increase in ROS levels, which has long been considered the main cause of cell death. ROS scavengers, such as N-acetylcysteine (NAC), can mitigate the extent of Cu-induced cell death in some cells [49,50,51,52,53]. However, eliminating ROS does not always inhibit cell death induced by Cu [9, 54], suggesting that ROS are not primary mediators of Cu-induced cell death. Tsvetkov et al. also observed that antioxidants, such as NAC, JP4-039, ebselen, and α-tocopherol, were unable to rescue cells from elesclomol (a Cu ionophore)–Cu-induced cellular damage, whereas agents that chelate Cu, such as GSH and ammonium tetrathiomolybdate (TTM), could prevent the lethality of elesclomol–Cu to cells [9]. This type of cellular damage could not be rescued by inhibitors of other types of cell death, such as apoptosis, ferroptosis, and necrosis. Tsvetkov et al. found that elesclomol could transport Cu2+ across membranes into the mitochondria, where Cu2+ was reduced to Cu+ by ferredoxin 1 (FDX1) [9], a mitochondrial matrix reductase. However, excess mitochondrial Cu+ can directly bind to lipoylated DLAT, an essential component of mitochondrial TCA cycle, causing DLAT aggregation and cytotoxicity [9]. Additionally, excess mitochondrial Cu+ reduces the stability of the Fe–S cluster proteins [9], which play an important role in OXPHOS-related electron transport. These induce proteotoxic stress, ultimately leading to cell death (Fig. 2). Pharmacological inhibition of the electron transport chain (ETC) and pyruvate uptake could reverse elesclomol–Cu-induced cell death [9], indicating that cuproptosis depends on mitochondrial respiration. Additionally, whole-genome CRISPR-Cas9 screening, combined with single-gene knockout validation experiments, showed that proteins in the lipoic acid (LA) pathway, such as FDX1, LA synthase (LIAS), lipoyl transferase 1 (LIPT1), and dihydrolipoamide dehydrogenase (DLD), and genes in the pyruvate dehydrogenase complex, such as DLAT, pyruvate dehydrogenase E1 subunit alpha 1 (PDHA1), pyruvate dehydrogenase E1 subunit beta (PDHB), metal-regulatory transcription factor-1 (MTF1), glutaminase (GLS), and cyclin-dependent kinase inhibitor 2A (CDKN2A), are important regulators of cuproptosis (Fig. 2) [9]. These results also ascertain the significance of FDX1 and its regulation of mitochondrial protein lipoylation in cuproptosis.

Mechanisms of cuproptosis. The transportation of Cu2+ by Cu ionophores and the uptake of Cu+ by SLC31A1 lead to the excessive accumulation of Cu within cells. Cu ionophores, such as elesclomol, can transport Cu2+ into mitochondria, where Cu2+ is reduced to Cu+ by FDX1. FDX1 is a crucial regulatory protein for the lipoylation of mitochondrial TCA cycle enzymes, particularly DLAT. Accumulated Cu+ in mitochondria induces DLAT aggregation by directly binding to lipoylated DLAT and destabilizes Fe–S cluster proteins, ultimately triggering mitochondrial proteotoxic stress and resulting in cuproptosis. Key positive regulators of cuproptosis include LIAS, DLD, LIPT1, and FDX1 from the LA pathway, as well as DLAT, PDHA1, and PDHB from the PDH complex. Important inhibitors of cuproptosis include MTF1, GLS, and CDKN2A. FDX1 promotes G6PD degradation by binding to it, resulting in GSH reduction and intensified cuproptosis. METTL16 enhances cuproptosis by promoting FDX1 accumulation via m6A modification on FDX1 mRNA, a process inhibited by SIRT2 through delactylating METTL16 at K229. MELK increases DLAT expression through the PI3K/mTOR signaling pathway, enhancing mitochondrial function and cuproptosis. AMPK activated by elesclomol–Cu facilitates cuproptosis. Elesclomol–Cu upregulates PPP1R15A to promote proteotoxic stress by enhancing EIF2S1 and 4E-BP1-associated translation initiation, thereby enhancing cuproptosis. p32 enhances elesclomol–Cu-induced cuproptosis by promoting lipo-DLAT oligomerization. MUC20 induces cuproptosis by inhibiting CDKN2A expression. GAPDH and ARID1A inhibit cuproptosis by promoting cellular glycolysis, while SLC7A11 inhibits cuproptosis by upregulating intracellular GSH. SLC31A1, the solute carrier family 31 member 1; FDX1, ferredoxin 1; ES, elesclomol; Disulfiram, disulfiram; DLAT, dihydrolipoamide S-acetyltransferase; TCA, tricarboxylic acid; LIAS, LA synthase; LIPT1, lipoyl transferase 1; DLD, dihydrolipoamide dehydrogenase; PDHA1, pyruvate dehydrogenase E1 subunit alpha 1; PDHB, pyruvate dehydrogenase E1 subunit beta; MTF1, metal-regulatory transcription factor-1; GLS, glutaminase; CDKN2A, cyclin-dependent kinase inhibitor 2A;G6PD, glucose-6-phosphate dehydrogenease; GSH, glutathione; SIRT2, Sirtuin 2; SLC7A11, solute carrier family 7 membrane 11; MUC20, Mucin 20; MELK, maternal embryonic leucine zipper kinase; ARID1A, AT-rich interactive domain 1A; AMPK, adenosine 5 ‘-monophosphate (AMP)-activated protein kinase; TIGD1, trigger transposable element-derived 1; WASF2, Wiskott-Aldrich syndrome protein family member 2
Cuproptosis and regulated cell death
Although Cu ionophores combined with Cu can induce cuproptosis, previous studies have also found that excessive Cu in cells can cause other ways of regulated cell death (RCD) (Fig. 3), suggesting a molecular mechanism crosstalk between cuproptosis and other forms of cell death. Understanding the role of Cu in other cell death modes can help develop more reasonable tumor treatment strategies targeting cuproptosis.

Cuproptosis and regulated cell death. A Excess Cu triggers ferroptosis. Accumulation of Cu in mitochondria generates ROS, which promotes lipid peroxidation and induces ferroptosis. Cu enhances the ubiquitination of GPX4, a protein that blocks ferroptosis by eliminating phospholipid hydroperoxides, facilitating its autophagic degradation and exacerbating ferroptotic cell death. Ferroptosis inducers, such as sorafenib and erastin, can induce cuproptosis by upregulating FDX1 protein levels, promoting lipoylated protein aggregation, and downregulating GSH. The reduction of Fe–S cluster proteins mediated by excessive Cu in mitochondrion further promotes ferroptosis. B Excess Cu triggers apoptosis. Intracellular accumulation of Cu generates ROS via the Fenton reaction, which induces apoptosis. Mitochondrial Cu accumulation causes mitochondrial stress, leading to the localization of pro-apoptotic proteins (such as BAX and BAK) to the outer mitochondrial membrane, resulting in the release of cytochrome c from mitochondria. Cytosolic cytochrome c induces the formation of the apoptosome, which activates the caspases signaling axis, mediating apoptosis. Cu can inhibit proteasome activity either by directly binding to the proteasome or by causing NPL4/p97 aggregation, inducing ER stress and ultimately leading to apoptosis. Additionally, Cu can activate the MAPK-JNK signaling pathway to trigger cell apoptosis. C Cu regulates autophagy. Cu activates ULK1/2 by directly binding to them, promoting phagophore assembly and subsequently autophagosome formation. Cu can also inhibit mTOR by activating AMPK, facilitating phagophore formation. Excess Cu in cells upregulates the expression of autophagy-related genes, such as MAP1LC3 and ATG5. Cu inhibits the cysteine protease activity of ATG4B by directly binding to it, thus preventing the delipidation of MAP1LC3 and consequently blocking cellular autophagy. Cu promotes the fusion of lysosomes and autophagosomes, enhancing cellular autophagic flux. D. Excess Cu induces pyroptosis. Cu induces ROS production and ER stress, promoting the formation of the NLRP3 inflammasome, which activates caspase 1. Caspase 1 cleaves GSDMD to generate the N-terminal domain that creates membrane pores, promoting pyroptosis. ROS, reactive oxygen species; GPX4, glutathione Peroxidase 4; FDX1, ferredoxin 1; GSH, glutathione; ER, endoplasmic reticulum; BAX, BCL2 Associated X; BAK, BCL2 antagonist/killer 1; cyto.c, cytochrome c; MAPK, mitogen-activated protein kinase; JNK, Jun N-terminal kinase; ULK1/2, Unc-51-like autophagy activating kinase 1/2; mTOR, mechanistic target of rapamycin kinase; AMPK, adenosine 5 ‘-monophosphate (AMP)-activated protein kinase; MAP1LC3, microtubule-associated protein 1 light chain 3; ATG5, autophagy related 5; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; GSDMD, gasdermin D
Cuproptosis and ferroptosis
Ferroptosis is a form of programmed cell death induced by disrupting iron homeostasis and accumulating ROS in lipids. Although Tsvetkov and colleagues found that the ferroptosis inhibitor ferrostatin-1 did not rescue cells from growth inhibition induced by elesclomol–Cu, Gao et al. observed that ferrostatin-1 and liproxstatin-1, another ferroptosis inhibitor, could inhibit elesclomol–Cu-induced cell damage in colorectal cancer (CRC) cells [55]. Gao et al. demonstrated that elesclomol alone treatment could reduce the ATP7A expression levels in CRC cells, potentially mediating the degradation of solute carrier family 7 membrane 11 (SLC7A11), a cysteine-glutamate transporter, resulting in lipid peroxidation and ferroptosis, suggesting that it is unclear whether elesclomol–Cu-induced ferroptosis depends on Cu [55]. Furthermore, previous studies have reported that reducing the Fe–S cluster proteins, which are the main features of cuproptosis, can induce ferroptosis [56]. Cu can exacerbate erastin-induced ferroptotic cell death in pancreatic ductal adenocarcinoma (PDAC) cells through increasing ubiquitination and aggregation of glutathione peroxidase 4, a protein blocking ferroptosis by eliminating phospholipid hydroperoxides, promoting its macroautophagic degradation [57].
These studies indicate a close interplay between cuproptosis and ferroptosis, with a key intersection being mitochondrial metabolism. As essential energy sources for cells, mitochondria play critical roles in regulating ferroptosis [58]. Cellular energy metabolic pathways, such as the TCA cycle and glycolysis, are involved in this regulation. For instance, blocking the TCA cycle or loss of glutamine can attenuate cystine-deprivation or erastin-induced ferroptosis [59], suggesting that the TCA cycle and glutaminolysis are required for ferroptosis. Similarly, in the process of cuproptosis, the TCA cycle in mitochondria is crucial [9]. The aggregation of lipoylated DLAT in the TCA cycle is a major inducer of elesclomol–Cu-mediated cell death [9]. Furthermore, cysteine-deprivation-induced ferroptosis is affected by α-ketoglutarate and other intermediates of the TCA cycle, such as succinate and fumarate [58]. Inhibition of ETC complexes can attenuate cystine-deprivation and erastin-induced lipid peroxidation and cell death [59]. Interestingly, inhibition of ETC complexes can also mitigate elesclomol–Cu-induced cuproptosis [9], suggesting that cuproptosis and ferroptosis share the same mitochondrial energy dependency.
Another important hub for cuproptosis and ferroptosis is GSH, a crucial antioxidant. GSH acts as an inhibitor of both ferroptosis and cuproptosis, indicating that it serves as a significant co-regulator in these processes. Both Cu and iron (Fe) promote the oxidation and subsequent consumption of GSH by binding to it [60]. Additionally, GSH chelates Fe and Cu to reduce metal ion toxicity [60]. Since Fe can consume GSH, over accumulation of Fe in cells may trigger cuproptosis by inhibiting GSH. GSH is also a necessary cofactor for GPX4, which reduces cytotoxic lipid peroxides (L-OOH) to corresponding alcohols (L-OH) while converting reduced GSH to oxidized glutathione (GSSG), thus reducing lipid peroxidation and inhibiting ferroptosis [61]. The accumulation of Cu in cells promotes the consumption of GSH, creating favorable conditions for ferroptosis. SLC7A11 transports glutamate outside and cystine inside cells. Cystine is then converted to cysteine, a component of GSH. SLC7A11 inhibitors, such as sorafenib and erastin, are commonly used to induce ferroptosis by reducing intracellular cysteine levels and GSH synthesis, which also makes cuproptosis more likely. Indeed, Wang et al. recently observed that sorafenib and erastin can induce cuproptosis in primary liver cancer cells by primarily reducing intracellular GSH synthesis and increasing Cu-dependent lipoylated protein aggregation [62]. Additionally, BSO, an inhibitor of GSH synthesis known to induce ferroptosis, has also been found to induce cuproptosis [9]. These studies indicate that GSH is a critical molecule mediating the crosstalk between ferroptosis and cuproptosis. Targeting GSH could be a potential strategy to simultaneously induce ferroptosis and cuproptosis in tumor cells.
ROS are also critical factors in understanding the crosstalk between ferroptosis and cuproptosis. Both Fe and Cu can produce ROS through the Fenton reaction, while rapid GSH depletion mediated by these metals can further exacerbate cellular ROS accumulation. Excessive ROS promotes lipid peroxidation and ferroptosis [63]. Although in elesclomol–Cu-induced cuproptosis, Cu toxicity primarily results from the aggregation of lipoylated proteins in the mitochondria rather than ROS production [9], excessive Cu-mediated ROS generation and GSH depletion can also contribute to ferroptosis onset. In summary, while cuproptosis and ferroptosis have distinct initiation mechanisms and molecular characteristics, they mutually influence each other, creating favorable conditions for both processes. This interplay adds complexity to their regulatory mechanisms but also offers potential advantages for cancer therapy.
Cuproptosis and apoptosis
Excessive Cu accumulation in cells has also been linked to apoptosis. For instance, treatment with CuSO4 can upregulate the C/EBP homologous protein (CHOP), Jun N-terminal kinase (JNK), and caspase-12 expression levels in mouse liver cells, thereby enhancing cell apoptosis-related signaling pathways, such as endoplasmic reticulum (ER) stress [64]. Liu et al. also found that treatment with CuSO4 can increase the ROS levels and protein carbonyl compounds in cells and decrease GSH levels, thereby activating the mitochondrial pathway of apoptosis signaling, such as cytochrome c release into the cytosol and cleavage of caspase-9 and caspase-3 [65]. In myeloma cells and osteosarcoma, disulfiram, another Cu ionophore, combined with Cu, can induce apoptosis by activating ROS and JNK signaling pathways [66, 67]. Furthermore, Cu can induce apoptosis by binding to and inhibiting 20S proteasome subunits and activating the cytochrome c-caspase cascade signaling axis [68, 69].
Before the identification of cuproptosis, the damage to tumor cells treated with Cu ionophores combined with Cu was primarily considered to result from ROS production. Excessive ROS within cells can trigger apoptosis through various pathways, including ER stress, mitochondrial damage, and activation of death receptors. In certain tumor cells, such as lung cancer cells [49, 51], gastric cancer [50], melanoma [52], and osteosarcoma [53], ROS scavengers can mitigate the cell damage induced by Cu ionophores and Cu. However, in breast cancer and glioblastoma cells [9, 54], ROS scavengers do not exhibit this protective effect. Additionally, some studies have demonstrated that Cu indeed induces apoptosis in specific tumor cells [52, 67, 70]. These studies suggest that ROS may serve as a crucial link between cuproptosis and apoptosis. Additionally, different tumor cells exhibit varying tolerance and responses to ROS, which may explain the diverse roles of apoptosis in Cu-induced cell death.
Cuproptosis and autophagy
Cu is also considered to regulate autophagy. Studies have revealed that Cu can upregulate the autophagy-related gene expression in cells, such as LC3b/LC3a, BECN1, Atg3, and Atg5 [71, 72]. Besides, Cu can directly bind to the Unc-51-like autophagy activating kinase 1/2 (ULK1/2), crucial protein kinases regulating autophagy initiation, activating ULK1/2 and its downstream autophagy pathway [73]. In KRASG12D-driven lung cancer, deletion of the Cu transporter SLC31A1 diminished the Cu-mediated activation of ULK1/2, resulting in a blockage in the autophagic flux and tumor growth suppression [73]. However, whether Cu-induced autophagy contributes to Cu toxicity remains uncertain. For instance, Tang et al. found that in ATP7B R778L mutant hepatocytes, Cu can activate autophagy, which is beneficial for inhibiting cell necrosis and reducing Cu toxicity [74]. However, in some tumor cells Cu was found to inhibit autophagy. For instance, inhibiting SLC31A1-dependent copper absorption could enhance autophagic flux of pancreatic cancer cells, leading to the suppression of tumor cell death [75]. Besides, it was found that Cu could directly bind to autophagy-related gene 4B, a crucial regulator in the autophagy process responsible for priming and delipidation of LC3, and suppress its cysteine protease activity, consequently blocking cellular autophagy [76]. These results indicate that the regulation of the autophagy process by Cu is bidirectional.
Cuproptosis and pyroptosis
Moreover, Cu can affect pyroptosis in cells. In jejunal epithelial cells, Cu can upregulate the pyroptosis-related gene expression, such as CASP1, GSDMD, and IL-1β, which is believed to be mainly mediated by the ER stress-triggered IRE1α-XBP1 pathway [77]. In hepatocytes, Cu can similarly upregulate pyroptosis-related gene expression, such as CASP1, NLRP3, IL-1β, and IL-18, and NAC and a caspase inhibitor can reverse this behavior, suggesting that ROS generated by Cu induction may be the main mediators of pyroptosis [78]. Excessive ROS can induce various types of cell death, such as apoptosis, ferroptosis, pyroptosis. Cu can induce ROS generation in numerous tumor cells, which may be a crucial factor in the cross-talk between different types of Cu-triggered RCD. This indicates that in some tumor cells highly sensitive to ROS, cuproptosis may not be irreplaceable in Cu-induced cell damage. In summary, Cu overload can cause cellular damage from multiple angles. Although this complicates the mechanism of Cu-induced cell damage, it provides more possibilities for Cu-based tumor therapy.
Targeting cuproptosis for cancer therapy
Although Cu promotes tumorigenesis to a certain extent, excessive Cu accumulation in tumor cells disrupts cellular homeostasis and induces cuproptosis. Therefore, targeting cuproptosis may be a potential tumor treatment strategy.
Induction of tumor cell cuproptosis
Cu ionophores
Cu ionophores, defined as compounds or chemicals, can bind to Cu and carry it into cells, increasing the intracellular Cu levels. As previously mentioned, the most extensively studied Cu ionophores are elesclomol and disulfiram (Table 1). Elesclomol, a highly lipophilic Cu-binding molecule, can chelate extracellular Cu2+ to form an elesclomol–Cu2+ complex, facilitating the transport of Cu into cells [145, 146]. The anti-tumor activity of elesclomol has been recognized for decades and is believed to be Cu-dependent (Table 1). Although early studies reported that cell death induced by elesclomol–Cu was linked to apoptosis and ferroptosis, Tsvetkov et al. observed that elesclomol–Cu induced cellular damage through cuproptosis [9]. Elesclomol–Cu can increase ROS levels in tumor cells [49, 54, 83], exacerbating cellular damage and suggesting that elesclomol–Cu may kill tumor cells via multiple pathways. Additionally, recent studies have revealed FDX1-independent mechanism(s) of elesclomol–associated Cu release, achieving Cu delivery to non-mitochondrial cuproproteins [147]. Whether these non-mitochondrial Cu participates in cell death other than cuproptosis induced by elesclomol–Cu requires further investigation. The crosstalk mechanisms between cuproptosis and other forms of cell death are poorly understood and necessitate more research to unveil, possibly facilitating the development of effective anticancer strategies based on elesclomol–Cu. Elesclomol has not yet shown effective therapeutic outcomes in clinical trials [148]. A possible reason is that single-agent elesclomol treatment may not elevate Cu levels in tumor cells to those required to trigger cuproptosis. A subsequent phase III trial results revealed that although the combination of elesclomol with paclitaxel did not improve progression-free survival in melanoma patients, elesclomol exhibited better anti-tumor effects in patients with low lactate dehydrogenase (LDH) levels [149]. Low LDH represents diminished glycolysis [150], reflecting enhanced mitochondrial metabolism. This aligns with Tsvetkov’s finding that cuproptosis relies on mitochondrial metabolism.
Disulfiram is an aldehyde dehydrogenase (ALDH) inhibitor FDA-approved for treating alcoholism. Disulfiram has also been deeply studied for an extended period in anti-tumor research (Table 1). Disulfiram interacts with Cu as a Cu ionophore to form the metabolite bis-diethyldithiocarbamate-Cu (CuET), which transports Cu across the cell membrane [151]. Disulfiram–Cu–induced cellular damage is also linked to apoptosis, ferroptosis, and cuproptosis (Table 1). Multiple targets or signaling pathways have been reported to be associated with the anti-tumor activity of disulfiram–Cu, such as ROS levels [94, 108, 109, 121, 122, 132, 134, 137], the ubiquitin–proteasome system [112, 124, 125, 133], the JNK and p38 pathways [66, 67, 114, 122], the NF-kB pathway [90, 92, 93, 109, 111, 122], and NPL4 [107, 127, 133]. In addition, disulfiram–Cu has been reported to overcome tumor drug resistance to cisplatin [91, 110], paclitaxel [108, 110], gemcitabine [109, 111], 5-fluorouracil (5-FU) [90, 118], temozolomide [98, 124], and sunitinib [96]. Table 1 summarizes the anti-tumor function of disulfiram in preclinical studies. Although, like elesclomol, disulfiram has presented significant anti-tumor effects in preclinical experiments, exciting results have yet to emerge from clinical trials [152, 153]. One limiting factor may be the inability to maintain high Cu levels in patient tumor cells. However, given the good clinical safety profile, conducting more clinical trials that combine elesclomol or disulfiram with clinical drugs could facilitate the translation of cuproptosis-associated anti-tumor therapies from the laboratory to clinical practice.
Besides elesclomol and disulfiram, other compounds, such as diacetyl-bis (N4-methylthiosemicarbazone) (ATSM) and glyoxal-bis (N4-methylthiosemicarbazone) (GTSM), have been identified as Cu ionophores (Table 1). Cu complexes with ATSM or GTSM induced cytotoxicity in human prostate hyperplastic and carcinoma cell lines without affecting the primary prostate epithelial cells [140]. This selective cytotoxicity may be associated with differential Cu levels in tumor cells, as Cu concentrations are elevated in prostate cancer cells compared to normal prostate epithelial cells [154]. Additionally, 7-iodo-5-chloro-8-hydroxyquinoline (CQ) has been reported to mediate Cu accumulation in cells [141]. In cancerous prostate cells, rather than normal prostate cells, CQ-Cu complexes promote apoptosis by facilitating the degradation of XIAP, a protein that inhibits caspases [141]. Notably, CQ can induce cellular damage via multiple pathways, including proteasome and lysosome dysfunction, conferring severe toxic side effects that limit its clinical application in cancer therapy [155]. Recent studies have also revealed that curcumin, a natural compound derived from Curcuma longa, can act as a Cu ionophore and promote cuproptosis in CRC cells [142, 143]. Curcumin is a potential anticancer natural product that can inhibit the cell cycle, induce apoptosis, and activate tumor suppressors. Additionally, several clinical studies have shown that curcumin has good efficacy and safety [156]. These characteristics make curcumin a promising cuproptosis inducer for clinical application. Furthermore, salicylaldehyde isonicotinoyl hydrazone (SIH), a lipophilic tridentate iron chelator, can facilitate the transportation and intracellular release of Cu2+ in HepG2 cells, thereby triggering mitochondria-mediated apoptosis, suggesting that SIH is also a Cu ionophore [144]. However, the cell death induced by these Cu ionophores, beyond elesclomol and disulfiram, whether related to the aggregation of lipoylated proteins and the reduction of Fe–S proteins, requires further investigation.
During cuproptosis, the primary function of Cu ionophores is to transport Cu across the cell membrane and release it into the cell. Notably, most metal-ion ionophores are not specific to a single metal element. For instance, elesclomol can directly bind Fe2+ [157]. Besides, elesclomol and disulfiram can increase cellular iron content during transporting Cu into cells [158, 159]. This non-specificity complicates the mechanism of cell damage caused by Cu ionophores combined with Cu. However, treatment with such agents can cause metal dyshomeostasis, exacerbating the side effects of the therapy. Consequently, developing Cu- or tumor-specific ionophores represents a potential breakthrough in accelerating the clinical application of Cu ionophores for cancer treatment.
Small compounds
Cu is a dual-faceted player in tumorigenesis. A high Cu level promotes tumor cell proliferation and growth, suggesting some resistance mechanisms to cuproptosis in tumor cells. As a result, small-molecule compounds that disrupt Cu homeostasis may also induce or increase the sensitivity of tumor cells to cuproptosis. Recent studies have unveiled several small compounds capable of inducing cuproptosis (Table 2). For instance, Yang et al. discovered that zinc pyrithione can induce cuproptosis in triple negative breast cancer (TNBC) cells by disrupting intracellular Cu homeostasis and DLAT oligomerization, potentially contributing to the chemosensitivity of TNBC [160]. In CRC cells, 4-Octyl itaconate (4-OI) inhibits glycolysis by targeting the glyceraldehyde-3-phosphate dehydrogenase (GAPDH), promoting elesclomol–Cu-mediated cuproptosis [161]. Besides, anisomycin, a well-known inhibitor of protein synthesis that binds to the 60S ribosomal subunits, has been found to bind and inhibit Yinyang 1 (YY1), inactivating the transcriptional activity of core genes of the LA pathway (FDX1, DLD, DLAT, and PDHB), potentially leading to cuproptosis in ovarian cancer stem cells [162]. Additionally, as mentioned above, sorafenib, the first multi-tyrosine kinase inhibitor approved for treating many cancers and capable of inducing ferroptosis, and erastin, a commonly used ferroptosis inducer, can aggravate cuproptosis in liver cancer cells [62]. Compared to Cu ionophores, small molecular compounds that disrupt tumor cell Cu homeostasis can induce tumor cell cuproptosis without Cu supplementation, thereby avoiding the imbalance of metal ions in the body and reducing metal-induced side effects during treatment. The cuproptosis research is currently in its infancy. The development and discovery of more cuproptosis inducers, especially those based on drugs approved for clinical use, could significantly promote the clinical application of cuproptosis-targeted cancer treatment strategies.
Nanomedicine
Dissolving, adsorption, encapsulation, or attachment to nanomatrices can transform drugs into nanomedicines. These nanomedicines use the characteristics of tumor tissues or cells, such as acidic environments, elevated GSH and ROS levels, and tumor cell-specific surface markers, to accumulate or release within tumor sites. This strategic approach can increase the precision of drug delivery and minimize the side effects of cancer therapy. Given the relatively low selectivity of Cu ionophores toward tumor cells, using a nanoparticle-based delivery system for the precise delivery of Cu to tumor cells can effectively enhance cuproptosis in tumor tissues while reducing damage to other normal tissues. Since cuproptosis was identified, more studies have focused on this area (Table 3). For instance, DSF@PEG/copper-HMSNs can precisely release Cu2+ and disulfiram in tumor tissues to induce cuproptosis and inhibit tumor growth [166]. Au NCs-Cu2+@SA-HA NHGs can enhance cuproptosis-mediated tumor therapy by depleting GSH and H2O2 in the tumor tissues [180].
Researchers have aimed to precisely deliver Cu, Cu ionophores, and other anticancer agents, such as chemotherapeutic drugs and siRNA (Table 3), to explore tumor therapy strategies based on nanomedicine-induced cuproptosis. This approach enhances cell damage through other mechanisms or sensitization to cuproptosis, thereby synergistically combating tumors. For example, TP-M–Cu–MOF/siATP7a efficiently silences the ATP7A gene and increases Cu intake, thus inducing cuproptosis and enhancing anti-tumor efficacy [178]. OMP contains siRNA targeting PDK1, which, during releasing Cu2+, can reduce cellular glycolysis by decreasing PDK1 expression, thereby sensitizing cells to cuproptosis [186]. LDH/HA/5-FU nanosheets can release 5-FU while delivering Cu2+ to tumor tissues, thus inducing apoptosis and cuproptosis in tumor cells [207].
In addition, unlike drug treatment of vitro cultured cells, nanomedicine delivered to tumor tissues impacts the tumor microenvironment (TME), an important factor influencing tumor therapy outcomes, especially in tumor immunotherapy. Nanomedicine containing Cu relies on the TME for precise delivery to tumor tissues and has the potential to modify the TME, making it more conducive to tumor therapy (Table 3). For instance, PDA-DTC/Cu NPs trigger cuproptosis in tumor cells and facilitate the repolarization of tumor-associated macrophages to mitigate the tumor immunosuppressive microenvironment (TIME) [188]. Similarly, ES@CuO promoted cuproptosis-driven immune responses and remodeled the TIME by enhancing lymphocyte infiltration and increasing the release of inflammatory cytokines within tumors. The synergistic application of ES@CuO with programmed cell death-1 (PD-1) immunotherapy markedly enhanced anti-tumor efficacy in murine melanoma models [196]. Besides, CQG NPs induce cuproptosis and pyroptosis by disrupting antioxidant defense mechanisms within tumor cells [201]. This dual action facilitates the transformation of the TIME, augments the infiltration of immune cells into the tumor, and triggers a robust systemic immune response.
Furthermore, the integration of nanomedicine with dynamic therapies, such as photothermal therapy (PTT), photodynamic therapy (PDT), and chemodynamic therapy (CDT) significantly enhances the precision and efficacy of tumor treatments. This approach represents a promising research direction for future studies on cuproptosis-associated therapeutic strategies (Table 3). For instance, PTT can augment E. coli@Cu2O-induced ferroptosis and cuproptosis, reversing the immunosuppression of colon tumors by initiating dendritic cell maturation and T-cell activation [205]. Through PTT and CDT, PEG@Cu2O-ES can generate ROS to target the ATP-Cu pump, reducing the efflux of Cu ions and exacerbating cuproptosis [208]. In nanomedicine, multiple anti-tumor components can be incorporated, significantly enhancing the medication's plasticity and multifunctionality. For instance, the CCNAs constructed by Wen et al., in addition to Cu2+, contain zinc phthalocyanine (ZnPc), 1-methyl tryptophan (1-MT), and doxorubicin (DOX) [209]. Upon near-infrared laser irradiation, ZnPc released into tumor tissues exhibited a photodynamic effect that generated ROS, effectively promoting the release of DOX and inducing apoptosis while intensifying cuproptosis [209]. Moreover, the release of 1-MT from CCNAs can reverse TIME by inhibiting IDO-1-mediated Trp degradation, triggering an immunogenic cell death (ICD) response [209].
Although nanomedicine offers broad prospects for exploring tumor treatments, numerous issues must be addressed before clinical application, warranting attention in future studies on tumor treatments based on cuproptosis. First, current experimental studies are primarily conducted in animal tumor models, which differ from primary or metastatic tumors in humans, especially regarding the tumor microenvironment. Second, the efficacy of laboratory nanomedicine in delivering human tumor tissues remains unknown. Third, research on cuproptosis-related nanomedicine has mainly focused on a limited array of cancer types in animal models, such as breast and colorectal cancers. Expanding studies on other types of tumors, particularly those that are difficult to treat, is necessary to broaden the scope of targeting cuproptosis for tumor treatment. Lastly, biosafety is a critical concern, representing a significant factor in the transition of experimental drugs to clinical applications.
Sensitization of tumor cell cuproptosis
Targeting cell metabolism sensitizes tumor cell cuproptosis
Given the close relationship between cuproptosis and cellular metabolism, targeting cellular metabolism presents a strategy to sensitize tumor cells to cuproptosis. Cuproptosis is strongly associated with mitochondrial metabolism [9]. Elevated mitochondrial metabolism can sensitize tumor cells to Cu-induced cell death. Inhibition of the mitochondrial ETC or pyruvate uptake diminishes tumor cell responsiveness to Cu ionophores [9]. This suggests that inducing cuproptosis could effectively inhibit tumor growth in cells with high aerobic respiration levels, such as melanoma [217] and leukemia [218]. Additionally, a high mitochondrial metabolic state is characteristic of tumor cell resistance to certain drugs such as proteasome inhibitors [9, 219], cisplatin [220], and 5-FU [221]. Thus, tumor cells that exhibit resistance to these drugs may be more sensitive to Cu ionophores, and inducing cuproptosis may improve treatment outcomes in patients with acquired drug resistance.
Furthermore, high glycolysis levels are considered unfavorable for cuproptosis [9], indicating that targeting glycolysis in tumor cells could also be a method to induce or sensitize cuproptosis. For instance, 4-OI attenuates aerobic glycolysis in CRC cells by targeting GAPDH, thereby sensitizing them to cuproptosis induced by elesclomol–Cu [161]. Aerobic glycolysis is the primary energy source for most tumor cells, and inhibiting glycolysis can suppress their growth [222]. This suggests that inducing cuproptosis may enhance the inhibitory effect on tumor growth during aerobic glycolysis-targeted tumor therapy. Besides, recent studies have linked cuproptosis to protein synthesis, although the mechanism remains unclear. Liu et al. found that in elesclomol–Cu-treated cancer cells, PPP1R15A could promote protein synthesis by downregulating eIF2α phosphorylation and upregulating 4EBP1 phosphorylation, thereby exacerbating proteotoxic stress [223], suggesting tumor cells with the high rate of protein synthesis might be more sensitive to cuproptosis inducers. Thus, although the relationship between cuproptosis and cell metabolism remains relatively vague and requires more basic and clinical research, the differential response of cuproptosis under various cellular metabolic states will help develop effective clinical application strategies.
Targeting cuproptosis regulatory proteins and pathways sensitizes tumor cell cuproptosis
Tsvetkov et al. identified several genes regulating cuproptosis. Among the proteins encoded by these genes, FDX1, LIAS, LIPT1, DLD, DLAT, PDHA1, and PDHB positively regulated cuproptosis, whereas MTF1, GLS, and CDKN2A negatively regulated it [9]. This suggests that these gene expression levels could serve as biomarkers of the sensitivity of tumor cells to cuproptosis. For instance, since MTF1, GLS, and CDKN2A knockout promoted cuproptosis in tumor cells, targeting these proteins or related signal pathways could represent a therapeutic sensitization strategy. Fan et al. found that plicamycin can inhibit head and neck squamous cell carcinoma (HNSCC) cell growth by targeting CDKN2A, implying that plicamycin may be a potential sensitizing agent for cuproptosis [224].
The occurrence of cuproptosis is dependent on Cu over-accumulation within cells. As previously mentioned, various factors, such as SLC31A1 and ATP7A/B, regulate cellular Cu levels [225, 226]. Since ATP7A/B can release Cu into the extracellular environment via the vesicle system [225], targeting ATP7A/B may be a means of inducing or sensitizing tumor cells to cuproptosis. For instance, Tsvetkov et al. found that in a Wilson disease mouse model, deletion of ATP7B resulted in excess Cu accumulation and cuproptosis in aging livers [9]. Additionally, Zhang et al. used nanoparticles to simultaneously deliver Cu and ATP7A-specific siRNA to small-cell lung cancer brain metastasis tumors, enhancing cuproptosis and tumor growth suppression [178]. Although an increase in intracellular Cu can promote ATP7A/B-dependent Cu efflux, Cu ionophores have been found to reduce ATP7A/B expression levels. For example, elesclomol and disulfiram could reduce the ATP7A expression level in CRC and prostate cancer cells, respectively [55, 139]. This suggests that Cu ionophores can transport Cu into cells while reducing Cu efflux, providing favorable conditions for inducing cuproptosis.
Although research into the mechanisms of cuproptosis remains in its initial stages, recent studies have uncovered several key regulatory factors of cuproptosis. For instance, in hepatocellular carcinoma (HCC) cells, maternal embryonic leucine zipper kinase (MELK) can enhance DLAT expression by activating the PI3K/mTOR signaling pathway, thereby augmenting mitochondrial function [227]. MELK overexpression exacerbates elesclomol–induced cuproptosis and enhances its anti-tumor effects [227]. Mucin 20 (MUC20) overexpression in proteasome inhibitor-resistant multiple myeloma cells can induce cuproptosis, which is associated with the reduced cuproptosis inhibitor CDKN2A expression level [228]. Elesclomol–Cu activates the adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) signaling pathway in non-small cell lung cancer (NSCLC) cells, attenuating the cuproptosis [229]. Knocking down or inhibiting AMPK can aggravate elesclomol–Cu-induced cuproptosis [229]. Besides, in gastric cancer cells, METTL16 can promote cuproptosis by facilitating FDX1 accumulation via m6A modification of FDX1 mRNA [230]. Further studies revealed that Sirtuin 2 (SIRT2) can inhibit METTL16 activity via deacetylation, and inhibiting SIRT2 can significantly enhance the anti-tumor effect of elesclomol–Cu in gastric cancer [230]. Additionally, some cuproptosis inhibitory factors have been identified in different tumor cells, such as SLC7A11 [231] and AT-rich interactive domain 1A (ARID1A) [232] in liver cancer cells, trigger transposable element-derived 1 (TIGD1) in CRC [233], and Wiskott-Aldrich syndrome protein family member 2 (WASF2) in ovarian cancer cells [234]. Targeting these factors may sensitize tumor cell cuproptosis and enhance the efficacy of tumor treatment.
Recently, extracellular signaling molecules have also been found to be involved in regulating cuproptosis. For example, in diabetic mice, an increase in blood advanced glycosylation end products (AGEs) and Cu upregulates SLC31A1 expression level in cardiomyocytes, thereby disturbing Cu homeostasis and promoting cuproptosis [235], suggesting that AGEs may be sensitizers for cuproptosis. Moreover, adrenomedullin (ADM), a member of the amylin/calcitonin gene-related peptide superfamily, has been found to promote the phosphorylation and nuclear translocation of Forkhead box O3 (FOXO3) via the p38/MAPK signaling pathway, thereby inhibiting FDX1 transcription and suppressing cuproptosis in renal cell carcinoma (RCC), promoting chemoresistance [236]. Consequently, targeting cuproptosis-related cytokines in the blood is also a strategy to sensitize tumor cells to cuproptosis. Table 4 lists the regulatory proteins and pathways of cuproptosis, which are potential targets for sensitizing Cu-induced tumor cell death.
Targeting cuproptosis to overcome tumor drug resistance
As the duration of drug usage extends, tumor cells may develop resistance to therapeutic agents, diminishing drug efficacy and leading to tumor relapse or progression. Therefore, effectively overcoming cancer drug resistance has always been a significant theme in cancer treatment research. Since cuproptosis has been established as a novel mode of cell death, targeting tumor cell cuproptosis holds potential as a new strategy to overcome tumor drug resistance.
The use of Cu ionophores to overcome tumor chemotherapeutic drug resistance has a long history of research. On one hand, Cu ionophores can transport Cu into the cells to induce Cu-triggered cell damage, thereby exacerbating the death of resistant cells (Table 1). For instance, in prostate cancer, elesclomol–Cu can enhance sensitivity to docetaxel by inducing DLAT/mTOR pathway-dependent cuproptosis in vitro and in vivo [87]. Disulfiram–Cu has been found to sensitize breast cancer cells to paclitaxel by simultaneous induction of ROS and inhibition of NF-κB, suggesting its potential to overcome clinical resistance to paclitaxel [108]. Disulfiram–Cu can enhance the cytotoxicity of gemcitabine by reversing NF-κB activity in gemcitabine-resistant colon cancer cells [111]. Moreover, disulfiram–Cu was found to overcome bortezomib and cytarabine resistance in cell lines from patients with Down syndrome-associated acute myeloid leukemia, which is thought to be associated with the induction of apoptosis and re-inhibition of proteasome activity [138]. In addition to Cu ionophores, small molecule compounds capable of inducing cuproptosis are potential candidates for overcoming tumor treatment resistance. These compounds usually enhance anti-tumor effects by inducing cell damage through multiple mechanisms. For example, zinc pyrithione induces necrosis in prostate cancer cells by activating the PKC and ERK pathways and enhancing ROS production [239]; 4-OI induces ferritinophagy-dependent ferroptosis in multi-drug resistant retinoblastoma cells [240]. Among these compounds, the ferroptosis inducer erastin has garnered significant attention for its ability to reverse the resistance of various tumor cells to chemotherapeutic drugs, including ovarian cancer [241, 242], AML cells [243], NSCLC cells [244], and prostate cancer cells [245]. However, whether cuproptosis contributes to the sensitizing effects of these drugs requires further investigation. Future research should explore combining these compounds with chemotherapeutic drugs to treat drug-resistant tumor cells that are sensitive to cuproptosis. Additionally, nanomedicine-based targeting of cuproptosis can be examined to overcome chemotherapeutic drug resistance. For instance, CuO2/DDP@SiO2, which releases Cu2+ and cisplatin, can induce cuproptosis and block the entire cisplatin efflux pathway by downregulating multidrug resistance-associated protein 2 (MRP2), enhancing the anti-tumor effect of cisplatin [193]. E-C@DOX NPs can inhibit tumor cell stemness and cell survival-related pathways while working with Cu ions to damage mitochondria and induce cuproptosis, suppressing the ATP-dependent drug efflux pathway and reversing DOX resistance [204].
Notably, Cu ionophores also chelate certain drugs, especially platinum-based drugs, which are widely used as first-line clinical treatments for cancer. Disulfiram has been found to form a new platinum (Pt) chelate, Pt(DDTC)3+, which has a stronger anti-NSCLC effect than cisplatin alone [105]. Furthermore, Pt drugs and Cu share molecular mechanisms for intracellular transport and extracellular efflux, such as the Cu transporter CTR1, which can transport Pt into the cell [246, 247], and the Cu chaperone protein ATOX1, which can deliver Pt to ATP7A/B located on TGN, thereby promoting the efflux of Pt and leading to drug resistance during treatment [248,249,250]. Thus, targeting Cu homeostasis can also alter the intracellular Pt drug concentration, a potential mechanism for overcoming resistance. Yuki et al. found that in bladder cancer cells treated with disulfiram and cisplatin, disulfiram could reduce ATP7A expression level and its localization in the TGN, accumulating intracellular cisplatin and enhancing tumor cell death [251]. However, increased Cu levels reduce the expression or activity of Cu uptake proteins, such as CTR1, and enhance the translocation of ATP7A/B from the Golgi to post-Golgi sites or lysosomes to promote Cu efflux, which may be unfavorable for accumulating Pt drugs within cells [252,253,254,255]. Therefore, targeting Cu homeostasis to overcome tumor cell resistance to Pt drugs requires comprehensive consideration of the dosage of Pt drugs and the action of cuproptosis.
Moreover, targeted cuproptosis could be used to solve the problem of drug resistance in targeted therapies. For example, disulfiram–Cu kills and sensitizes BRAF-mutant thyroid cancer to BRAF kinase inhibitor by relieving feedback activation of the MAPK/ERK and PI3K/AKT pathways in a ROS-dependent manner [134]. In HCC cells, researchers observed that disulfiram–Cu could strengthen the cytotoxicity of sorafenib by simultaneously inhibiting the NRF2 and MAPK kinase signaling pathways and arrest tumor growth in vitro and in vivo [136]. Current research on cuproptosis overcoming drug resistance in tumor-targeted therapy remains relatively limited. One of the main reasons is that the molecular regulation mechanism underlying cuproptosis remains unknown. However, the relationship between tumor drug resistance and cuproptosis remains unclear, hindering the progress of targeting cuproptosis to overcome drug resistance in tumor-targeted therapy.
Targeting cuproptosis to enhance tumor immunotherapy
Immunotherapy has become an important clinical strategy for cancer treatment due to its significant efficacy in tumor therapy. Since Cu metabolism and cuproptosis play crucial regulatory roles in tumor immunity, targeting cuproptosis may represent a vital sensitization strategy for tumor immunotherapy. PD-1/ PD-L1 (Programmed death-ligand 1) serves as a crucial immune checkpoint, and inhibiting or eliminating PD-1/PD-L1 can lead to favorable clinical outcomes in patients with cancer [256]. Cu may exert a positive regulatory effect on PD-L1 expression in tumors. For instance, disulfiram–Cu can upregulate PD-L1 expression in HCC cells by inhibiting Poly (ADP-ribose) polymerase 1 (PARP1) activity and promoting glycogen synthase kinase 3β (GSK-3β) phosphorylation, thereby suppressing T-cell infiltration [135]. Hence, simultaneous targeting of cuproptosis and co-administration of PD-1/PD-L1 inhibitors may enhance therapeutic effects. This notion is supported by preclinical studies demonstrating superior tumor growth inhibition when Cu ionophores were combined with Cu and anti-PD-L1 agents in PDAC [102], lung cancer [106], and HCC cells [135].
The TME is a pivotal determinant of the efficacy of tumor immunotherapy. Reshaping the immunosuppressive TME, such as dendritic cell maturation and activation of CD8+ T cells, is poised to enhance tumor suppression [257]. The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) signaling pathway is a critical component of innate immunity, capable of sensing aberrant DNA and triggering the release of type I interferons, thereby promoting dendritic cell maturation and migration, as well as augmenting the cytotoxic effects of T lymphocytes or natural killer cells [258]. Jiang et al. demonstrated that elesclomol–Cu-induced cuproptosis in clear cell RCC can activate the cGAS-STING pathway within dendritic cells, thereby promoting the release of inflammatory mediators, including IFN-γ, TNF-α, IL2, C-X-C motif chemokine ligand 10 (CXCL10), and CXCL11, ultimately enhancing tumor immunotherapy [259]. Additionally, CS/MTO-Cu@AMI, established by Huang et al., can activate anti-tumor immunity by inducing dsDNA damage and activating the cGAS-STING pathway [173]. Similarly, PCM nanoinducers constructed by Dai et al. can trigger the release of mitochondrial DNA during inducing tumor cell cuproptosis, activating the cGAS-STING pathway and stimulating innate and adaptive immune responses, thereby enhancing tumor suppression [184].
As discussed in this review, nanomedicine-based cuproptosis induction systems mediate tumor cell cuproptosis and impact the TME by depleting GSH, aggravating oxidative stress, and inducing other types of cell damage, such as pyroptosis and apoptosis. These alterations result in the remodeling of the TME and initiation of ICD responses, which are conducive to enhanced immunotherapy. Consequently, inducing cuproptosis may represent an effective strategy for sensitizing tumors to immunotherapy. Table 3 summarizes various nanomedicines, such as NP@ESCu [171], BCMD [176], OMP [186], PCD@CM [195], ES@CuO [196], CBS [203], E. coli@Cu2O [205], PEG@Cu2O-ES [208], and CLDCu [213], in combination with anti-PD1 or anti-PD-L1 antibodies, effectively inhibited tumor growth. However, several issues must be addressed before these strategies are applied clinically. For instance, the mechanisms underlying cuproptosis-mediated TME remodeling remain unclear. Additionally, are the induction conditions and regulatory mechanisms of cuproptosis in tumors and immune cells similar or different? How can we target tumor cell cuproptosis more precisely to initiate ICD responses? Addressing these problems holds promise for improving the effectiveness of tumor immunotherapy.
Conclusions and future perspectives
Cuproptosis, characterized by its unique features, represents a novel mode of cell death that has infused new optimism into cancer treatment. Since its conceptualization, cuproptosis has received significant attention in oncology. Conversely, it has emerged as a promising therapeutic target, with ongoing research poised to unveil additional cuproptosis inducers, including small-molecule compounds and nanomedicines. This expanding repertoire of therapeutic options holds the potential to diversify tumor treatment strategies. Targeting cuproptosis offers a novel approach to combatting tumor drug resistance. Exploiting therapeutic-induced metabolic changes in tumor cells, such as heightened mitochondrial metabolism and glycolysis, rendering them susceptible to cuproptosis, provides a pathway to sensitize tumor cells to drug interventions or overcome drug resistance. Furthermore, inducing cuproptosis in tumor tissues can remodel the tumor microenvironment, fostering dendritic cell maturation and immune cell infiltration. Consequently, targeting cuproptosis promises to enhance the response rates and overcome resistance to immunotherapy.
However, understanding and research regarding cuproptosis are still in their infancy. Before practical application, numerous issues must be addressed. For instance, Cu can play a dual role in tumor initiation and progression, promoting tumorigenesis and inducing cell death [260, 261]. Cu chelators reduce Cu bioavailability and exert anticancer effects [262]. Therefore, comprehending how tumor cells balance the dual effects of Cu is crucial. Furthermore, whether cuproptosis or its related signaling pathways have pro-tumor effects during tumor initiation, development, and treatment remains unclear. In addition, reliable biomarkers, initiation mechanisms, and links with other cell death forms for cuproptosis remain lacking, impeding the progress of cuproptosis-associated research in diseases and targeted clinical applications. Moreover, distinguishing between the regulatory mechanisms of cuproptosis in normal and tumor cells is crucial for improving the precision of cuproptosis-targeted therapy and reducing the side effects of treatment. Additionally, currently widely used cuproptosis inducers, such as Cu ionophores elesclomol and disulfiram, have not revealed promising therapeutic effects in clinical trials.
Based on the challenges outlined above, the following recommendations may help promote the translation of cuproptosis-associated anti-tumor therapies from the laboratory to clinical practice in future research. First, unraveling the molecular mechanisms underlying tumor cell tolerance and exploiting high Cu levels may offer insights into inducing cuproptosis by disrupting endogenous Cu metabolism within tumor cells. For example, hepatocytes are the primary storage cells for Cu, and abnormal Cu accumulation in liver cells, as observed in patients with Wilson's disease, promotes the development of HCC [263]. Accordingly, driving accumulated Cu in HCC cells to induce cuproptosis might reverse the pro-carcinogenic effects of Cu to anticarcinogenic effects. Additionally, future research and development efforts should focus on novel cuproptosis inducers or induction strategies, including natural products, small-molecule compounds, and nanomedicine. Furthermore, improving the precision of drug delivery to tumor cells and the stability of drugs in plasma should be a priority in the development of cuproptosis inducers. Moreover, conducting more clinical trials is crucial for promoting the clinical application of cuproptosis-targeted therapies. For example, combining Cu ionophores with frontline clinical drugs that can enhance sensitivity to cuproptosis might address issues of drug resistance during treatment. Finally, similar to other types of cell death, inducing cuproptosis to treat tumors will also face the drug resistance issues. Therefore, uncovering the mechanisms of cuproptosis-related drug resistance, particularly in different tumor cell types, should be a key focus of future research.
In summary, cuproptosis is a new target in cancer treatment. As the regulatory mechanisms of cuproptosis continue to be elucidated and the efficiency of cuproptosis induction methods improves, targeting cuproptosis presents a promising new approach to combat chemotherapy and immunotherapy resistance in cancer treatment, leading to improved therapeutic outcomes.
Availability of data and materials
No datasets were generated or analysed during the current study.
Abbreviations
- ROS:
-
Reactive oxygen species
- DLAT:
-
Dihydrolipoamide S-acetyltransferase
- TCA:
-
Tricarboxylic acid
- STEAP:
-
Six-transmembrane epithelial antigen of the prostate
- DCYTB:
-
Duodenal cytochrome b
- CTR1:
-
Cu transport protein 1
- SLC31A1:
-
The solute carrier family 31 member 1
- ATP7A:
-
ATPase copper transporting alpha
- MT1/2:
-
Metallothionein1/2
- GSH:
-
Glutathione
- SOD1:
-
Superoxide dismutase 1
- ATOX1:
-
Antioxidant 1
- TGN:
-
Trans-Golgi network
- COX:
-
Cytochrome oxidase
- OXPHOS:
-
Oxidative phosphorylation
- MEK:
-
Mitogen-activated extracellular signal-regulated kinase
- MAPK:
-
Mitogen-activated protein kinase
- FGFs:
-
Fibroblast growth factors
- VEGF:
-
Vascular endothelial growth factor
- TNF-α:
-
Tumor necrosis factor alpha
- NAC:
-
N-Acetylcysteine
- TNBC:
-
Triple negative breast cancer
- AMPK:
-
Adenosine 5′-monophosphate (AMP)-activated protein kinase
- ES:
-
Elesclomol
- FDX1:
-
Ferredoxin 1
- ETC:
-
Electron transport chain
- LA:
-
Lipoic acid
- LIAS:
-
LA synthase
- LIPT1:
-
Lipoyl transferase 1
- DLD:
-
Dihydrolipoamide dehydrogenase
- PDHA1:
-
Pyruvate dehydrogenase E1 subunit alpha 1
- PDHB:
-
Pyruvate dehydrogenase E1 subunit beta
- MTF1:
-
Metal-regulatory transcription factor-1
- GLS:
-
Glutaminase
- CDKN2A:
-
Cyclin-dependent kinase inhibitor 2A
- RCD:
-
Regulated cell death
- CRC:
-
Colorectal cancer
- SLC7A11:
-
Solute carrier family 7 membrane 11
- PDAC:
-
Pancreatic ductal adenocarcinoma
- ER stress:
-
Endoplasmic reticulum stress
- DSF:
-
Disulfiram
- ULK:
-
Unc-51-like autophagy activating kinase
- 5-FU:
-
5-Fluorouracil
- 4-OI:
-
4-Octyl itaconate
- HCC:
-
Hepatocellular carcinoma
- TME:
-
Microenvironment
- TIME:
-
Tumor immunosuppressive microenvironment
- PD-1:
-
Programmed cell death-1
- PD-L1:
-
Programmed death-ligand 1
- PTT:
-
Photothermal therapy
- PDT:
-
Photodynamic therapy
- CDT:
-
Chemodynamic therapy
- DOX:
-
Doxorubicin
- ZnPc:
-
Zinc phthalocyanine
- 1-MT:
-
1-Methyl tryptophan
- ICD:
-
Immunogenic cell death
- HNSCC:
-
Head and neck squamous cell carcinoma
- NSCLC:
-
Non-small cell lung cancer
- MELK:
-
Maternal embryonic leucine zipper kinase
- cGAS:
-
Cyclic GMP-AMP synthase
- STING:
-
Stimulator of interferon genes
- CXCL:
-
C-X-C motif chemokine ligand
References
Tsang T, Davis CI, Brady DC. Copper biology. Curr Biol. 2021;31(9):R421–7.
Lutsenko S. Human copper homeostasis: a network of interconnected pathways. Curr Opin Chem Biol. 2010;14(2):211–7.
Bost M, Houdart S, Oberli M, Kalonji E, Huneau JF, Margaritis I. Dietary copper and human health: current evidence and unresolved issues. J Trace Elem Med Biol. 2016;35:107–15.
Wungjiranirun M, Sharzehi K. Wilson’s disease. Semin Neurol. 2023;43(4):626–33.
Tümer Z, Horn N. Menkes disease: recent advances and new insights into copper metabolism. Ann Med. 1996;28(2):121–9.
Ge EJ, Bush AI, Casini A, Cobine PA, Cross JR, DeNicola GM, et al. Connecting copper and cancer: from transition metal signalling to metalloplasia. Nat Rev Cancer. 2022;22:102–13.
Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219:1–14.
Oliveri V. Selective targeting of cancer cells by copper ionophores: an overview. Front Mol Biosci. 2022;9: 841814.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254–61.
Scheiber I, Dringen R, Mercer JF. Copper: effects of deficiency and overload. Met Ions Life Sci. 2013;13:359–87.
Knutson MD. Steap proteins: implications for iron and copper metabolism. Nutr Rev. 2007;65:335–40.
Wyman S, Simpson RJ, McKie AT, Sharp PA. Dcytb (Cybrd1) functions as both a ferric and a cupric reductase in vitro. FEBS Lett. 2008;582:1901–6.
Lutsenko S. Dynamic and cell-specific transport networks for intracellular copper ions. J Cell Sci. 2021;134(21):jcs240523.
Lönnerdal B. Intestinal regulation of copper homeostasis: a developmental perspective. Am J Clin Nutr. 2008;88(3):846S-S850.
Linder MC, Wooten L, Cerveza P, Cotton S, Shulze R, Lomeli N. Copper transport. Am J Clin Nutr. 1998;67(5 Suppl):965S-971S.
Doguer C, Ha JH, Collins JF. Intersection of iron and copper metabolism in the mammalian intestine and liver. Compr Physiol. 2018;8(4):1433–61.
Freedman JH, Ciriolo MR, Peisach J. The role of glutathione in copper metabolism and toxicity. J Biol Chem. 1989;264:5598–605.
Wong PC, Waggoner D, Subramaniam JR, et al. Copper chaperone for superoxide dismutase is essential to activate mammalian Cu/Zn superoxide dismutase. Proc Natl Acad Sci USA. 2000;97(6):2886–91.
Gomez ML, Shah N, Kenny TC, Jenkins EC Jr, Germain D. SOD1 is essential for oncogene-driven mammary tumor formation but dispensable for normal development and proliferation. Oncogene. 2019;38(29):5751–65.
Hamza I, Prohaska J, Gitlin JD. Essential role for Atox1 in the copper-mediated intracellular trafficking of the menkes ATPase. Proc Natl Acad Sci USA. 2003;100:1215–20.
Robinson NJ, Winge DR. Copper metallochaperones. Annu Rev Biochem. 2010;79:537–62.
Hamza I, Faisst A, Prohaska J, Chen J, Gruss P, Gitlin JD. The metallochaperone Atox1 plays a critical role in perinatal copper homeostasis. Proc Natl Acad Sci USA. 2001;98(12):6848–52.
La Fontaine S, Mercer JF. Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis. Arch Biochem Biophys. 2007;463(2):149–67.
Cobine PA, Pierrel F, Bestwick ML, Winge DR. Mitochondrial matrix copper complex used in metallation of cytochrome oxidase and superoxide dismutase. J Biol Chem. 2006;281(48):36552–9.
Boulet A, Vest KE, Maynard MK, Gammon MG, Russell AC, Mathews AT, et al. The mammalian phosphate carrier SLC25A3 is a mitochondrial copper transporter required for cytochrome c oxidase biogenesis. J Biol Chem. 2018;293:1887–96.
Baker ZN, Cobine PA, Leary SC. The mitochondrion: a central architect of copper homeostasis. Metallomics. 2017;9(11):1501–12.
Garza NM, Swaminathan AB, Maremanda KP, Zulkifli M, Gohil VM. Mitochondrial copper in human genetic disorders. Trends Endocrinol Metab. 2023;34(1):21–33.
Horn D, Barrientos A. Mitochondrial copper metabolism and delivery to cytochrome c oxidase. IUBMB Life. 2008;60(7):421–9.
Feng Y, Zeng JW, Ma Q, Zhang S, Tang J, Feng JF. Serum copper and zinc levels in breast cancer: a meta-analysis. J Trace Elem Med Biol. 2020;62: 126629.
Duan F, Li J, Huang J, Hua X, Song C, Wang L, et al. Establishment and validation of prognostic nomograms based on serum copper level for patients with early-stage triple-negative breast cancer. Front Cell Dev Biol. 2021;9: 770115.
Saleh SAK, Adly HM, Abdelkhaliq AA, Nassir AM. Serum levels of selenium, zinc, copper, manganese, and iron in prostate cancer patients. Curr Urol. 2020;14(1):44–9.
Zhang L, Shao J, Tan SW, Ye HP, Shan XY. Association between serum copper/zinc ratio and lung cancer: a systematic review with meta-analysis. J Trace Elem Med Biol. 2022;74: 127061.
Zhang M, Shi M, Zhao Y. Association between serum copper levels and cervical cancer risk: a meta-analysis. Biosci Rep. 2018;38:BSR20180161.
Mazdak H, Yazdekhasti F, Movahedian A, Mirkheshti N, Shafieian M. The comparative study of serum iron, copper, and zinc levels between bladder cancer patients and a control group. Int Urol Nephrol. 2010;42:89–93.
Baltaci AK, Dundar TK, Aksoy F, Mogulkoc R. Changes in the serum levels of trace elements before and after the operation in thyroid cancer patients. Biol Trace Elem Res. 2017;175(1):57–64.
Baharvand M, Manifar S, Akkafan R, Mortazavi H, Sabour S. Serum levels of ferritin, copper, and zinc in patients with oral cancer. Biomed J. 2014;37:331–6.
Khanna SS, Karjodkar FR. Circulating immune complexes and trace elements (copper, iron and selenium) as markers in oral precancer and cancer: a randomised, controlled clinical trial. Head Face Med. 2006;2:33.
Turski ML, Brady DC, Kim HJ, et al. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol Cell Biol. 2012;32(7):1284–95.
Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature. 2014;509:492–6.
Brady DC, Crowe MS, Greenberg DN, Counter CM. Copper chelation inhibits BRAFV600E-driven melanomagenesis and counters resistance to BRAFV600E and MEK1/2 inhibitors. Cancer Res. 2017;77(22):6240–52.
Shanbhag V, Jasmer-McDonald K, Zhu S, Martin AL, Gudekar N, Khan A, et al. ATP7A delivers copper to the lysyl oxidase family of enzymes and promotes tumorigenesis and metastasis. Proc Natl Acad Sci USA. 2019;116:6836–41.
Nagaraju GP, Dontula R, El-Rayes BF, Lakka SS. Molecular mechanisms underlying the divergent roles of SPARC in human carcinogenesis. Carcinogenesis. 2014;35(5):967–73.
MacDonald G, Nalvarte I, Smirnova T, et al. Memo is a copper-dependent redox protein with an essential role in migration and metastasis. Sci Signal. 2014;7(329):ra56.
Zhang X, Walke G, Wittung-Stafshede P. Memo1 reduces copper-mediated reactive oxygen species in breast cancer cells. J Inorg Biochem. 2023;247: 112335.
Sivaraja V, Kumar TK, Rajalingam D, Graziani I, Prudovsky I, Yu C. Copper binding affinity of S100A13, a key component of the FGF-1 nonclassical copper-dependent release complex. Biophys J. 2006;91:1832–43.
Rigiracciolo DC, Scarpelli A, Lappano R, Pisano A, Santolla MF, De Marco P, et al. Copper activates HIF-1α/GPER/VEGF signalling in cancer cells. Oncotarget. 2015;6:34158–77.
Pan Q, Kleer CG, Golen KL, Irani J, Bottema KM, Bias C, et al. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002;62:4854–9.
Mandinov L, Mandinova A, Kyurkchiev S, Kyurkchiev D, Kehayov I, Kolev V, et al. Copper chelation represses the vascular response to injury. Proc Natl Acad Sci USA. 2003;100:6700–5.
Wangpaichitr M, Wu C, You M, Maher JC, Dinh V, Feun LG, et al. N’, N’-Dimethyl-N′, N′-bis(phenylcarbonothioyl) propanedihydrazide (elesclomol) selectively kills cisplatin resistant lung cancer cells through reactive oxygen species (ROS). Cancers. 2009;1:23–38.
Liu Y, Guan X, Wang M, Wang N, Chen Y, Li B, et al. Disulfiram/copper induces antitumor activity against gastric cancer via the ROS/MAPK and NPL4 pathways. Bioengineered. 2022;13:6579–89.
Lee JH, Cho YS, Jung KH, Park JW, Lee KH. Genipin enhances the antitumor effect of elesclomol in A549 lung cancer cells by blocking uncoupling protein-2 and stimulating reactive oxygen species production. Oncol Lett. 2020;20:374.
Morrison BW, Doudican NA, Patel KR, Orlow SJ. Disulfiram induces copper-dependent stimulation of reactive oxygen species and activation of the extrinsic apoptotic pathway in melanoma. Melanoma Res. 2010;20:11–20.
Ren Y, Lin Y, Chen J, Jin Y. Disulfiram chelated with copper promotes apoptosis in osteosarcoma via ROS/mitochondria pathway. Biol Pharm Bull. 2021;44:1557–64.
Buccarelli M, D’Alessandris QG, Matarrese P, Mollinari C, Signore M, Cappannini A, et al. Elesclomol-induced increase of mitochondrial reactive oxygen species impairs glioblastoma stem-like cell survival and tumor growth. J Exp Clin Cancer Res. 2021;40:228.
Gao W, Huang Z, Duan J, Nice EC, Lin J, Huang C. Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol Oncol. 2021;15(12):3527–44.
Alvarez SW, Sviderskiy VO, Terzi EM, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551(7682):639–43.
Xue Q, Yan D, Chen X, et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy. 2023;19(7):1982–96.
Javadov S. Mitochondria and ferroptosis. Curr Opin Physiol. 2022;25: 100483.
Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, Jiang X. Role of mitochondria in ferroptosis. Mol Cell. 2019;73:354-363.e3.
Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283:65–87.
Wang G, Qin S, Chen L, Geng H, Zheng Y, Xia C, et al. Butyrate dictates ferroptosis sensitivity through FFAR2-mTOR signaling. Cell Death Dis. 2023;14:292.
Wang W, Lu K, Jiang X, et al. Ferroptosis inducers enhanced cuproptosis induced by copper ionophores in primary liver cancer. J Exp Clin Cancer Res. 2023;42(1):142.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–82.
Wu H, Guo H, Liu H, et al. Copper sulfate-induced endoplasmic reticulum stress promotes hepatic apoptosis by activating CHOP, JNK and caspase-12 signaling pathways. Ecotoxicol Environ Saf. 2020;191: 110236.
Liu H, Guo H, Jian Z, et al. Copper induces oxidative stress and apoptosis in the mouse liver. Oxid Med Cell Longev. 2020;2020:1359164.
Xu Y, Zhou Q, Feng X, et al. Disulfiram/copper markedly induced myeloma cell apoptosis through activation of JNK and intrinsic and extrinsic apoptosis pathways. Biomed Pharmacother. 2020;126: 110048.
Guo W, Zhang X, Lin L, et al. The disulfiram/copper complex induces apoptosis and inhibits tumour growth in human osteosarcoma by activating the ROS/JNK signalling pathway. J Biochem. 2021;170(2):275–87.
Santoro AM, Monaco I, Attanasio F, et al. Copper(II) ions affect the gating dynamics of the 20S proteasome: a molecular and in cell study. Sci Rep. 2016;6:33444.
Xiao Y, Chen DI, Zhang X, et al. Molecular study on copper-mediated tumor proteasome inhibition and cell death. Int J Oncol. 2010;37(1):81–7.
Cen D, Brayton D, Shahandeh B, Meyskens FL Jr, Farmer PJ. Disulfiram facilitates intracellular Cu uptake and induces apoptosis in human melanoma cells. J Med Chem. 2004;47:6914–20.
Wan F, Zhong G, Ning Z, et al. Long-term exposure to copper induces autophagy and apoptosis through oxidative stress in rat kidneys. Ecotoxicol Environ Saf. 2020;190: 110158.
Wang X, Yang F, Tian X, et al. Toxic effects of copper on duck cerebrum: a crucial role of oxidative stress and endoplasmic reticulum quality control. Environ Sci Pollut Res Int. 2023;30(43):98127–38.
Tsang T, Posimo JM, Gudiel AA, Cicchini M, Feldser DM, Brady DC. Copper is an essential regulator of the autophagic kinases ULK1/2 to drive lung adenocarcinoma. Nat Cell Biol. 2020;22:412–24.
Tang S, Liang C, Hou W, et al. ATP7B R778L mutant hepatocytes resist copper toxicity by activating autophagy and inhibiting necroptosis. Cell Death Discov. 2023;9(1):344.
Yu Z, Zhou R, Zhao Y, Pan Y, Liang H, Zhang JS, et al. Blockage of SLC31A1-dependent copper absorption increases pancreatic cancer cell autophagy to resist cell death. Cell Prolif. 2019;52(2): e12568.
Xia F, Fu Y, Xie H, Chen Y, Fang D, Zhang W, et al. Suppression of ATG4B by copper inhibits autophagy and involves in Mallory body formation. Redox Biol. 2022;52: 102284.
Liao J, Hu Z, Li Q, et al. Endoplasmic reticulum stress contributes to copper-induced pyroptosis via regulating the IRE1α-XBP1 pathway in pig jejunal epithelial cells. J Agric Food Chem. 2022;70(4):1293–303.
Liao J, Yang F, Tang Z, et al. Inhibition of Caspase-1-dependent pyroptosis attenuates copper-induced apoptosis in chicken hepatocytes. Ecotoxicol Environ Saf. 2019;174:110–9.
Ren YJ, Wang XH, Ji C, et al. Silencing of NAC1 expression induces cancer cells oxidative stress in hypoxia and potentiates the therapeutic activity of elesclomol. Front Pharmacol. 2017;8:804.
Qu Y, Wang J, Sim MS, et al. Elesclomol, counteracted by Akt survival signaling, enhances the apoptotic effect of chemotherapy drugs in breast cancer cells. Breast Cancer Res Treat. 2010;121(2):311–21.
Kirshner JR, He S, Balasubramanyam V, et al. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol Cancer Ther. 2008;7(8):2319–27.
Kwan SY, Cheng X, Tsang YT, et al. Loss of ARID1A expression leads to sensitivity to ROS-inducing agent elesclomol in gynecologic cancer cells. Oncotarget. 2016;7(35):56933–43.
Li Y, Yang J, Zhang Q, et al. Copper ionophore elesclomol selectively targets GNAQ/11-mutant uveal melanoma. Oncogene. 2022;41(27):3539–53.
Alli E, Ford JM. Breast cancers with compromised DNA repair exhibit selective sensitivity to elesclomol. DNA Repair (Amst). 2012;11(5):522–4.
Albayrak G, Korkmaz FD, Tozcu D, Dogan TI. The outcomes of an impaired powerhouse in KRAS mutant lung adenocarcinoma cells by Elesclomol. J Cell Biochem. 2019;120(6):10564–71.
Nagai M, Vo NH, Shin Ogawa L, et al. The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic Biol Med. 2012;52(10):2142–50.
Wen H, Qu C, Wang Z, et al. Cuproptosis enhances docetaxel chemosensitivity by inhibiting autophagy via the DLAT/mTOR pathway in prostate cancer. FASEB J. 2023;37(9): e23145.
Huang N, Feng Y, Liu Y, et al. Disulfiram mediated anti-tumour effect in pituitary neuroendocrine tumours by inducing cuproptosis. Int Immunopharmacol. 2024;134: 112159.
Lin J, Haffner MC, Zhang Y, et al. Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. Prostate. 2011;71(4):333–43.
Wang W, McLeod HL, Cassidy J. Disulfiram-mediated inhibition of NF-kappaB activity enhances cytotoxicity of 5-fluorouracil in human colorectal cancer cell lines. Int J Cancer. 2003;104(4):504–11.
Yang Z, Guo F, Albers AE, Sehouli J, Kaufmann AM. Disulfiram modulates ROS accumulation and overcomes synergistically cisplatin resistance in breast cancer cell lines. Biomed Pharmacother. 2019;113: 108727.
Han D, Wu G, Chang C, et al. Disulfiram inhibits TGF-β-induced epithelial-mesenchymal transition and stem-like features in breast cancer via ERK/NF-κB/Snail pathway. Oncotarget. 2015;6(38):40907–19.
Zhang J, Pu K, Bai S, et al. The anti-alcohol dependency drug disulfiram inhibits the viability and progression of gastric cancer cells by regulating the Wnt and NF-κB pathways. J Int Med Res. 2020;48(6):300060520925996.
Cen D, Gonzalez RI, Buckmeier JA, Kahlon RS, Tohidian NB, Meyskens FL Jr. Disulfiram induces apoptosis in human melanoma cells: a redox-related process. Mol Cancer Ther. 2002;1(3):197–204.
Read E, Milford J, Zhu J, Wu L, Bilodeau M, Yang G. The interaction of disulfiram and H2S metabolism in inhibition of aldehyde dehydrogenase activity and liver cancer cell growth. Toxicol Appl Pharmacol. 2021;426: 115642.
Ketola K, Kallioniemi O, Iljin K. Chemical biology drug sensitivity screen identifies sunitinib as synergistic agent with disulfiram in prostate cancer cells. PLoS ONE. 2012;7(12): e51470.
Terashima Y, Toda E, Itakura M, et al. Targeting FROUNT with disulfiram suppresses macrophage accumulation and its tumor-promoting properties. Nat Commun. 2020;11(1):609.
Triscott J, Lee C, Hu K, et al. Disulfiram, a drug widely used to control alcoholism, suppresses the self-renewal of glioblastoma and over-rides resistance to temozolomide. Oncotarget. 2012;3(10):1112–23.
Zheng X, Liu Z, Mi M, Wen Q, Wu G, Zhang L. Disulfiram improves the anti-PD-1 therapy efficacy by regulating PD-L1 expression via epigenetically reactivation of IRF7 in triple negative breast cancer. Front Oncol. 2021;11: 734853.
Tang B, Wu M, Zhang L, et al. Combined treatment of disulfiram with PARP inhibitors suppresses ovarian cancer. Front Oncol. 2023;13:1154073.
Park HJ, Kim MS, Cho K, Yun JH, Choi YJ, Cho CH. Disulfiram deregulates HIF-α subunits and blunts tumor adaptation to hypoxia in hepatoma cells. Acta Pharmacol Sin. 2013;34(9):1208–16.
Huang S, Xie P, Huang X, et al. Disulfiram combined with chemoimmunotherapy potentiates pancreatic cancer treatment efficacy through the activation of cGAS-STING signaling pathway via suppressing PARP1 expression. Am J Cancer Res. 2023;13(5):2055–65.
Zheng P, Wu Y, Wang Y, Hu F. Disulfiram suppresses epithelial-mesenchymal transition (EMT), migration and invasion in cervical cancer through the HSP90A/NDRG1 pathway. Cell Signal. 2023;109: 110771.
Lafi Z, Alshaer W, Gharaibeh L, et al. Synergistic combination of doxorubicin with hydralazine, and disulfiram against MCF-7 breast cancer cell line. PLoS ONE. 2023;18(9): e0291981.
Yuan XX, Duan YF, Luo C, et al. Disulfiram enhances cisplatin cytotoxicity by forming a novel platinum chelate Pt(DDTC)3. Biochem Pharmacol. 2023;211: 115498.
Li P, Sun Q, Bai S, Wang H, Zhao L. Combination of the cuproptosis inducer disulfiram and anti PD L1 abolishes NSCLC resistance by ATP7B to regulate the HIF 1 signaling pathway. Int J Mol Med. 2024;53(2):19.
Skrott Z, Mistrik M, Andersen KK, et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature. 2017;552(7684):194–9.
Yip NC, Fombon IS, Liu P, et al. Disulfiram modulated ROS-MAPK and NFκB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br J Cancer. 2011;104(10):1564–74.
Liu P, Brown S, Goktug T, et al. Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br J Cancer. 2012;107(9):1488–97.
Liu P, Kumar IS, Brown S, et al. Disulfiram targets cancer stem-like cells and reverses resistance and cross-resistance in acquired paclitaxel-resistant triple-negative breast cancer cells. Br J Cancer. 2013;109(7):1876–85.
Guo X, Xu B, Pandey S, et al. Disulfiram/copper complex inhibiting NFkappaB activity and potentiating cytotoxic effect of gemcitabine on colon and breast cancer cell lines. Cancer Lett. 2010;290(1):104–13.
Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006;66(21):10425–33.
Zhang H, Chen D, Ringler J, et al. Disulfiram treatment facilitates phosphoinositide 3-kinase inhibition in human breast cancer cells in vitro and in vivo. Cancer Res. 2010;70(10):3996–4004.
Park YM, Go YY, Shin SH, Cho JG, Woo JS, Song JJ. Anti-cancer effects of disulfiram in head and neck squamous cell carcinoma via autophagic cell death. PLoS ONE. 2018;13(9): e0203069.
Kim YJ, Kim JY, Lee N, et al. Disulfiram suppresses cancer stem-like properties and STAT3 signaling in triple-negative breast cancer cells. Biochem Biophys Res Commun. 2017;486(4):1069–76.
Kim JY, Cho Y, Oh E, et al. Disulfiram targets cancer stem-like properties and the HER2/Akt signaling pathway in HER2-positive breast cancer. Cancer Lett. 2016;379(1):39–48.
Hu Y, Qian Y, Wei J, et al. The disulfiram/copper complex induces autophagic cell death in colorectal cancer by targeting ULK1. Front Pharmacol. 2021;12: 752825.
Cong J, Wang Y, Zhang X, et al. A novel chemoradiation targeting stem and nonstem pancreatic cancer cells by repurposing disulfiram. Cancer Lett. 2017;409:9–19.
Wu X, Xue X, Wang L, et al. Suppressing autophagy enhances disulfiram/copper-induced apoptosis in non-small cell lung cancer. Eur J Pharmacol. 2018;827:1–12.
You SY, Rui W, Chen ST, et al. Process of immunogenic cell death caused by disulfiram as the anti-colorectal cancer candidate. Biochem Biophys Res Commun. 2019;513(4):891–7.
Conticello C, Martinetti D, Adamo L, et al. Disulfiram, an old drug with new potential therapeutic uses for human hematological malignancies. Int J Cancer. 2012;131(9):2197–203.
Zha J, Chen F, Dong H, et al. Disulfiram targeting lymphoid malignant cell lines via ROS-JNK activation as well as Nrf2 and NF-kB pathway inhibition. J Transl Med. 2014;12:163.
Zuhra K, Panagaki T, Randi EB, et al. Mechanism of cystathionine-β-synthase inhibition by disulfiram: the role of bis(N, N-diethyldithiocarbamate)-copper(II). Biochem Pharmacol. 2020;182: 114267.
Lun X, Wells JC, Grinshtein N, et al. Disulfiram when combined with copper enhances the therapeutic effects of temozolomide for the treatment of glioblastoma. Clin Cancer Res. 2016;22(15):3860–75.
Li L, Yang H, Chen D, Cui C, Dou QP. Disulfiram promotes the conversion of carcinogenic cadmium to a proteasome inhibitor with pro-apoptotic activity in human cancer cells. Toxicol Appl Pharmacol. 2008;229(2):206–14.
Kim JY, Lee N, Kim YJ, et al. Disulfiram induces anoikis and suppresses lung colonization in triple-negative breast cancer via calpain activation. Cancer Lett. 2017;386:151–60.
Serra R, Zhao T, Huq S, et al. Disulfiram and copper combination therapy targets NPL4, cancer stem cells and extends survival in a medulloblastoma model. PLoS ONE. 2021;16(11): e0251957.
Wang K, Michelakos T, Wang B, et al. Targeting cancer stem cells by disulfiram and copper sensitizes radioresistant chondrosarcoma to radiation. Cancer Lett. 2021;505:37–48.
Cao HZ, Yang WT, Zheng PS. Cytotoxic effect of disulfiram/copper on human cervical cancer cell lines and LGR5-positive cancer stem-like cells. BMC Cancer. 2022;22(1):521.
Ni YL, Chien PJ, Hsieh HC, et al. Disulfiram/copper suppresses cancer stem cell activity in differentiated thyroid cancer cells by inhibiting BMI1 expression. Int J Mol Sci. 2022;23(21):13276.
Papaioannou M, Mylonas I, Kast RE, Brüning A. Disulfiram/copper causes redox-related proteotoxicity and concomitant heat shock response in ovarian cancer cells that is augmented by auranofin-mediated thioredoxin inhibition. Oncoscience. 2013;1(1):21–9.
Navrátilová J, Hankeová T, Beneš P, Šmarda J. Acidic pH of tumor microenvironment enhances cytotoxicity of the disulfiram/Cu2+ complex to breast and colon cancer cells. Chemotherapy. 2013;59(2):112–20.
Gao X, Huang H, Pan C, et al. Disulfiram/copper induces immunogenic cell death and enhances CD47 blockade in hepatocellular carcinoma. Cancers (Basel). 2022;14(19):4715.
Xie J, Liu J, Zhao M, et al. Disulfiram/Cu kills and sensitizes BRAF-mutant thyroid cancer cells to BRAF kinase inhibitor by ROS-dependently relieving feedback activation of MAPK/ERK and PI3K/AKT pathways. Int J Mol Sci. 2023;24(4):3418.
Zhou B, Guo L, Zhang B, et al. Disulfiram combined with copper induces immunosuppression via PD-L1 stabilization in hepatocellular carcinoma. Am J Cancer Res. 2019;9(11):2442–55.
Ren X, Li Y, Zhou Y, et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol. 2021;46: 102122.
Bagherpoor AJ, Shameem M, Luo X, Seelig D, Kassie F. Inhibition of lung adenocarcinoma by combinations of sulfasalazine (SAS) and disulfiram–copper (DSF–Cu) in cell line models and mice. Carcinogenesis. 2023;44(4):291–303.
Bista R, Lee DW, Pepper OB, Azorsa DO, Arceci RJ, Aleem E. Disulfiram overcomes bortezomib and cytarabine resistance in down-syndrome-associated acute myeloid leukemia cells. J Exp Clin Cancer Res. 2017;36(1):22.
Song L, Nguyen V, Xie J, et al. ATPase copper transporting beta (ATP7B) Is a novel target for improving the therapeutic efficacy of docetaxel by disulfiram/copper in human prostate cancer. Mol Cancer Ther. 2024;23(6):854–63.
Cater MA, Pearson HB, Wolyniec K, et al. Increasing intracellular bioavailable copper selectively targets prostate cancer cells. ACS Chem Biol. 2013;8(7):1621–31.
Cater MA, Haupt Y. Clioquinol induces cytoplasmic clearance of the X-linked inhibitor of apoptosis protein (XIAP): therapeutic indication for prostate cancer. Biochem J. 2011;436(2):481–91.
Yang Y, Liang S, Geng H, et al. Proteomics revealed the crosstalk between copper stress and cuproptosis, and explored the feasibility of curcumin as anticancer copper ionophore. Free Radic Biol Med. 2022;193(Pt 2):638–47.
Zhang W, Chen C, Shi H, et al. Curcumin is a biologically active copper chelator with antitumor activity. Phytomedicine. 2016;23(1):1–8.
Ji Y, Dai F, Zhou B. Designing salicylaldehyde isonicotinoyl hydrazones as Cu(II) ionophores with tunable chelation and release of copper for hitting redox Achilles heel of cancer cells. Free Radic Biol Med. 2018;129:215–26.
Gohil VM. Repurposing elesclomol, an investigational drug for the treatment of copper metabolism disorders. Expert Opin Investig Drugs. 2021;30:1–4.
Guthrie LM, Soma S, Yuan S, Silva A, Zulkifli M, Snavely TC, et al. Elesclomol alleviates Menkes pathology and mortality by escorting Cu to cuproenzymes in mice. Science. 2020;368:620–5.
Zulkifli M, Spelbring AN, Zhang Y, et al. FDX1-dependent and independent mechanisms of elesclomol-mediated intracellular copper delivery. Proc Natl Acad Sci USA. 2023;120: e2216722120.
Zheng P, Zhou C, Lu L, Liu B, Ding Y. Elesclomol: a copper ionophore targeting mitochondrial metabolism for cancer therapy. J Exp Clin Cancer Res. 2022;41:271.
O’Day SJ, Eggermont AM, Chiarion-Sileni V, Kefford R, Grob JJ, Mortier L, et al. Final results of phase III SYMMETRY study: randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J Clin Oncol. 2013;31:1211–8.
Sharma D, Singh M, Rani R. Role of LDH in tumor glycolysis: regulation of LDHA by small molecules for cancer therapeutics. Semin Cancer Biol. 2022;87:184–95.
Lu C, Li X, Ren Y, Zhang X. Disulfiram: a novel repurposed drug for cancer therapy. Cancer Chemother Pharmacol. 2021;87:159–72.
Wang X, Zhou M, Liu Y, Si Z. Cope with copper: from copper linked mechanisms to copper-based clinical cancer therapies. Cancer Lett. 2023;561: 216157.
Werlenius K, Kinhult S, Solheim TS, et al. Effect of disulfiram and copper plus chemotherapy vs chemotherapy alone on survival in patients with recurrent glioblastoma: a randomized clinical trial. JAMA Netw Open. 2023;6(3): e234149.
Denoyer D, Clatworthy SA, Masaldan S, Meggyesy PM, Cater MA. Heterogeneous copper concentrations in cancerous human prostate tissues. Prostate. 2015;75(14):1510–7.
Mao X, Schimmer AD. The toxicology of clioquinol. Toxicol Lett. 2008;182:1–6.
Zoi V, Galani V, Lianos GD, Voulgaris S, Kyritsis AP, Alexiou GA. The role of curcumin in cancer treatment. Biomedicines. 2021;9:1086.
Wu L, Zhou L, Liu DQ, Vogt FG, Kord AS. LC-MS/MS and density functional theory study of copper(II) and nickel(II) chelating complexes of elesclomol (a novel anticancer agent). J Pharm Biomed Anal. 2011;54(2):331–6.
Garza NM, Zulkifli M, Gohil VM. Elesclomol elevates cellular and mitochondrial iron levels by delivering copper to the iron import machinery. J Biol Chem. 2022;298(7): 102139.
Chu M, An X, Fu C, et al. Disulfiram/copper induce ferroptosis in triple-negative breast cancer cell line MDA-MB-231. Front Biosci (Landmark Ed). 2023;28(8):186.
Yang X, Deng L, Diao X, et al. Targeting cuproptosis by zinc pyrithione in triple-negative breast cancer. iScience. 2023;26(11): 108218.
Yang W, Wang Y, Huang Y, et al. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to promote cuproptosis in colorectal cancer. Biomed Pharmacother. 2023;159: 114301.
Nie X, Chen H, Xiong Y, Chen J, Liu T. Anisomycin has a potential toxicity of promoting cuproptosis in human ovarian cancer stem cells by attenuating YY1/lipoic acid pathway activation. J Cancer. 2022;13(14):3503–14.
Gao Y, Jin F, Zhang P, et al. Elesclomol-copper synergizes with imidazole ketone erastin by promoting cuproptosis and ferroptosis in myelodysplastic syndromes. Biomed Pharmacother. 2024;175: 116727.
Xu Y, Liu SY, Zeng L, et al. An enzyme-engineered nonporous copper(I) coordination polymer nanoplatform for cuproptosis-based synergistic cancer therapy. Adv Mater. 2022;34(43): e2204733.
Zhao F, Liang L, Wang H, et al. H2S-activated ion-interference therapy: a novel tumor targeted therapy based on copper-overload-mediated cuproptosis and pyroptosis. Adv Funct Mater. 2023;33:2300941.
Wu W, Yu L, Jiang Q, Huo M, Lin H, Wang L, et al. Enhanced tumor-specific disulfiram chemotherapy by in situ Cu2+ chelation-initiated nontoxicity-to-toxicity transition. J Am Chem Soc. 2019;141:11531–9.
Jia W, Tian H, Jiang J, et al. Brain-targeted HFn-Cu-REGO nanoplatform for site-specific delivery and manipulation of autophagy and cuproptosis in glioblastoma. Small. 2023;19: e2205354.
Zhao F, Yu H, Liang L, et al. Redox homeostasis disruptors based on metal-phenolic network nanoparticles for chemo/chemodynamic synergistic tumor therapy through activating apoptosis and cuproptosis. Adv Healthc Mater. 2023;12: e2301346.
Zhou J, Yu Q, Song J, et al. Photothermally triggered copper payload release for cuproptosis-promoted cancer synergistic therapy. Angew Chem Int Ed Engl. 2023;62: e202213922.
Chan L, Liu Y, Chen M, et al. Cuproptosis-driven enhancement of thermotherapy by sequentially response Cu2−xSe via copper chemical transition. Adv Funct Mater. 2023;33:2302054.
Guo B, Yang F, Zhang L, et al. Cuproptosis induced by ROS responsive nanoparticles with elesclomol and copper combined with αPD-L1 for enhanced cancer immunotherapy. Adv Mater. 2023;35: e2212267.
Xu W, Wang Y, Hou G, et al. Tumor microenvironment responsive hollow nanoplatform for triple amplification of oxidative stress to enhance cuproptosis-based synergistic cancer therapy. Adv Healthc Mater. 2023;12: e2202949.
Tian H, Duan J, Li B, et al. Clinical chemotherapeutic agent coordinated copper-based nanoadjuvants for efficiently sensitizing cancer chemo-immunotherapy by cuproptosis-mediated mitochondrial metabolic reprogramming. Adv Funct Mater. 2023;33: 2306584. https://doi.org/10.1002/adfm.202306584.
Jin XK, Liang JL, Zhang SM, et al. Orchestrated copper-based nanoreactor for remodeling tumor microenvironment to amplify cuproptosis-mediated anti-tumor immunity in colorectal cancer. Mater Today. 2023;68:108–24.
Lu Y, Pan Q, Gao W, Pu Y, He B. Reversal of cisplatin chemotherapy resistance by glutathione-resistant copper-based nanomedicine via cuproptosis. J Mater Chem B. 2022;10:6296–306.
Huang QX, Liang JL, Chen QW, et al. Metal-organic framework nanoagent induces cuproptosis for effective immunotherapy of malignant glioblastoma. Nano Today. 2023;51: 101911.
Zhang J, Peng L, Hao Y, Yang H, Zhao W, Mao C. Biodegradable CuMoO4 nanodots with multienzyme activities for multimodal treatment of tumor. Adv Healthc Mater. 2023;12: e2300167.
Zhang J, Han M, Zhang J, et al. Syphilis mimetic nanoparticles for cuproptosis-based synergistic cancer therapy via reprogramming copper metabolism. Int J Pharm. 2023;640: 123025.
Chen K, Zhou A, Zhou X, Liu Y, Xu Y, Ning X. An intelligent cell-derived nanorobot bridges synergistic crosstalk between sonodynamic therapy and cuproptosis to promote cancer treatment. Nano Lett. 2023;23:3038–47.
Yang Z, Zhao Z, Cheng H, Shen Y, Xie A, Zhu M. In-situ fabrication of novel Au nanoclusters-Cu2+@sodium alginate/hyaluronic acid nanohybrid gels for cuproptosis enhanced photothermal/photodynamic/chemodynamic therapy via tumor microenvironment regulation. J Colloid Interface Sci. 2023;641:215–28.
Liu T, Zhou Z, Zhang M, et al. Cuproptosis-immunotherapy using PD-1 overexpressing T cell membrane-coated nanosheets efficiently treats tumor. J Control Release. 2023;362:502–12.
Zhong J, Zheng X, Wen Y, Wang SB, Zhan G, Chen AZ. In situ sacrificial growth of metastable copper-enriched nanomedicine for cuproptosis-based synergistic cancer therapy. Chem Eng J. 2023;474: 145795.
Zhu G, Wang M, Qiao L, et al. Lysosomal rupture-mediated “broken window effect” to amplify cuproptosis and pyroptosis for high-efficiency cancer immunotherapy. Adv Funct Mater. 2024;34: 2400496. https://doi.org/10.1002/adfm.202400496.
Yu X, Li B, Yan J, et al. Cuproptotic nanoinducer-driven proteotoxic stress potentiates cancer immunotherapy by activating the mtDNA-cGAS-STING signaling. Biomaterials. 2024;307: 122512.
Liu Y, Niu R, Zhao H, et al. Single-site nanozymes with a highly conjugated coordination structure for antitumor immunotherapy via cuproptosis and cascade-enhanced T lymphocyte activity. J Am Chem Soc. 2024;146(6):3675–88.
Yan C, Liu Y, Zhao G, et al. Inhalable metal-organic framework-mediated cuproptosis combined with PD-L1 checkpoint blockade for lung metastasis synergistic immunotherapy. Acta Pharm Sin B. 2024;14(5):2281–97.
Zhu J, You Y, Zhang W, et al. Sensitizing cuproptosis by endogenous copper-triggered bioorthogonal nanoremodeler. Nano Today. 2024;55: 102196.
Chang J, Yin W, Zhi H, et al. Copper deposition in polydopamine nanostructure to promote cuproptosis by catalytically inhibiting copper exporters of tumor cells for cancer immunotherapy. Small. 2024. https://doi.org/10.1002/smll.202308565.
Du C, Guo X, Qiu X, et al. Self-reinforced bimetallic mito-jammer for Ca2+ overload-mediated cascade mitochondrial damage for cancer cuproptosis sensitization. Adv Sci (Weinh). 2024;11(15): e2306031.
Ye L, Yu C, Xia J, et al. Multifunctional nanomaterials via cell cuproptosis and oxidative stress for treating osteosarcoma and OS-induced bone destruction. Mater Today Bio. 2024;25: 100996.
Zhang W, Wang M, Liu B, et al. Glutathione induced in situ synthesis of Cu single-atom nanozymes with anaerobic glycolysis metabolism interference for boosting cuproptosis. Angew Chem Int Ed Engl. 2024;63(18): e202402397.
Xiao C, Li J, Hua A, et al. Hyperbaric oxygen boosts antitumor efficacy of copper-diethyldithiocarbamate nanoparticles against pancreatic ductal adenocarcinoma by regulating cancer stem cell metabolism. Research (Wash D C). 2024;7:0335.
He X, Li M, Fan S, et al. Copper peroxide and cisplatin co-loaded silica nanoparticles-based trinity strategy for cooperative cuproptosis/chemo/chemodynamic cancer therapy. Chem Eng J. 2024;481: 148522.
Pei P, Wang Y, Shen W, et al. Oxygen-driven cuproptosis synergizes with radiotherapy to potentiate tumor immunotherapy. Aggregate. 2024;5: e484. https://doi.org/10.1002/agt2.484.
Dai Y, Zhu L, Li X, et al. A biomimetic cuproptosis amplifier for targeted NIR-II fluorescence/photoacoustic imaging-guided synergistic NIR-II photothermal immunotherapy. Biomaterials. 2024;305: 122455.
Lu X, Chen X, Lin C, et al. Elesclomol loaded copper oxide nanoplatform triggers cuproptosis to enhance antitumor immunotherapy. Adv Sci (Weinh). 2024;11(18): e2309984.
Hao C, Huang L, Zhang H, et al. Chiral CuxOS@ Fe-MOFs for enhanced cancer therapy. Adv Func Mater. 2024;34(10):2312795.
Shen W, Pei P, Zhang C, et al. A polymeric hydrogel to eliminate programmed death-ligand 1 for enhanced tumor radio-immunotherapy. ACS Nano. 2023;17(23):23998–4011.
Xia J, Hu C, Ji Y, et al. Copper-loaded nanoheterojunction enables superb orthotopic osteosarcoma therapy via oxidative stress and cell cuproptosis. ACS Nano. 2023;17(21):21134–52.
Zheng J, Ge H, Guo M, et al. Photoinduced cuproptosis with tumor-specific for metastasis-inhibited cancer therapy. Small. 2024;20(10): e2304407.
Qiao L, Zhu G, Jiang T, et al. Self-destructive copper carriers induce pyroptosis and cuproptosis for efficient tumor immunotherapy against dormant and recurrent tumors. Adv Mater. 2024;36(8): e2308241.
Chen W, Xie W, Gao Z, et al. Mild-photothermal effect induced high efficiency ferroptosis-boosted-cuproptosis based on Cu2O@Mn3Cu3O8 nanozyme. Adv Sci (Weinh). 2023;10(33): e2303694.
Hu P, Li Y, Zhang L, et al. Defect-engineered photothermal nanozyme with NIR-II absorption induces cuproptosis–apoptosis for synergized cancer immunotherapy and fast wound healing. Mater Des. 2024;237: 112568.
Lu S, Tian H, Li B, et al. An ellagic acid coordinated copper-based nanoplatform for efficiently overcoming cancer chemoresistance by cuproptosis and synergistic inhibition of cancer cell stemness. Small. 2024;20(17): e2309215.
Ruan Y, Zhuang H, Zeng X, et al. Engineered microbial nanohybrids for tumor-mediated NIR II photothermal enhanced ferroptosis/cuproptosis and immunotherapy. Adv Healthc Mater. 2024;13(4): e2302537.
Ding H, Ren F, Liu P, et al. Cu2+-anchored carbon nano-photocatalysts for visible water splitting to boost hydrogen cuproptosis. Angew Chem Int Ed Engl. 2023;62(44): e202311549.
Xia Y, Gu M, Wang J, et al. Tumor microenvironment-activated, immunomodulatory nanosheets loaded with copper(II) and 5-FU for synergistic chemodynamic therapy and chemotherapy. J Colloid Interface Sci. 2024;653(Pt A):137–47.
Liang W, Han C, Zhang D, et al. Copper-coordinated nanoassemblies based on photosensitizer-chemo prodrugs and checkpoint inhibitors for enhanced apoptosis-cuproptosis and immunotherapy. Acta Biomater. 2024;175:341–52.
Li W, Xiao Y, Guo G, et al. Cuprous oxide nanocomposites with photothermal (PTT) and chemical dynamics (CDT) effects induce cuproptosis in breast cancer using the strategy of increasing inflow and reducing outflow. Nano Today. 2024;56: 102223.
Deng J, Zhuang H, Shao S, et al. Mitochondrial-targeted copper delivery for cuproptosis-based synergistic cancer therapy. Adv Healthc Mater. 2024;13: 2304522. https://doi.org/10.1002/adhm.202304522.
Chen K, Zhou A, Zhou X, He J, Xu Y, Ning X. Cellular Trojan Horse initiates bimetallic Fe–Cu MOF-mediated synergistic cuproptosis and ferroptosis against malignancies. Sci Adv. 2024;10(15): eadk3201.
Wu H, Zhang Z, Cao Y, et al. A self-amplifying ROS-responsive nanoplatform for simultaneous cuproptosis and cancer immunotherapy. Adv Sci (Weinh). 2024;11: 2401047. https://doi.org/10.1002/advs.202401047.
Yan C, Lv H, Feng Y, et al. Inhalable nanoparticles with enhanced cuproptosis and cGAS–STING activation for synergistic lung metastasis immunotherapy. Acta Pharm Sin B. 2024. https://doi.org/10.1016/j.apsb.2024.04.028.
Xie W, Zhang Y, Xu Q, et al. A unique approach: biomimetic graphdiyne-based nanoplatform to treat prostate cancer by combining cuproptosis and enhanced chemodynamic therapy. Int J Nanomed. 2024;19:3957–72.
Zhang X, Zhu J, Wang S, et al. A copper/ferrous-engineering redox homeostasis disruptor for cuproptosis/ferroptosis co-activated nanocatalytic therapy in liver cancer. Adv Funct Mater. 2024. https://doi.org/10.1002/adfm.202402022.
Zhang N, Ping W, Rao K, et al. Biomimetic copper-doped polypyrrole nanoparticles induce glutamine metabolism inhibition to enhance breast cancer cuproptosis and immunotherapy. J Control Release. 2024;371:204–15.
Huang C, Radi RH, Arbiser JL. Mitochondrial metabolism in melanoma. Cells. 2021;10(11):3197.
Stubbins RJ, Maksakova IA, Sanford DS, Rouhi A, Kuchenbauer F. Mitochondrial metabolism: powering new directions in acute myeloid leukemia. Leuk Lymphoma. 2021;62(10):2331–41.
Luengo A, Gui DY, Vander Heiden MG. Targeting metabolism for cancer therapy. Cell Chem Biol. 2017;24(9):1161–80.
Cruz-Bermúdez A, Laza-Briviesca R, Vicente-Blanco RJ, et al. Cisplatin resistance involves a metabolic reprogramming through ROS and PGC-1α in NSCLC which can be overcome by OXPHOS inhibition. Free Radic Biol Med. 2019;135:167–81.
Denise C, Paoli P, Calvani M, et al. 5-fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget. 2015;6(39):41706–21.
Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152.
Liu C, Chen L, Cong Y, et al. Protein phosphatase 1 regulatory subunit 15 A promotes translation initiation and induces G2M phase arrest during cuproptosis in cancers. Cell Death Dis. 2024;15(2):149.
Fan K, Dong Y, Li T, Li Y. Cuproptosis-associated CDKN2A is targeted by plicamycin to regulate the microenvironment in patients with head and neck squamous cell carcinoma. Front Genet. 2023;13:1036408.
Xue Q, Kang R, Klionsky DJ, Tang D, Liu J, Chen X. Copper metabolism in cell death and autophagy. Autophagy. 2023;19(8):2175–95.
Chen J, Jiang Y, Shi H, Peng Y, Fan X, Li C. The molecular mechanisms of copper metabolism and its roles in human diseases. Pflugers Arch. 2020;472(10):1415–29.
Li Z, Zhou H, Zhai X, et al. MELK promotes HCC carcinogenesis through modulating cuproptosis-related gene DLAT-mediated mitochondrial function. Cell Death Dis. 2023;14(11):733.
Wang X, Shi Y, Shi H, et al. MUC20 regulated by extrachromosomal circular DNA attenuates proteasome inhibitor resistance of multiple myeloma by modulating cuproptosis. J Exp Clin Cancer Res. 2024;43(1):68.
Liu J, Liu Y, Wang Y, Kang R, Tang D. HMGB1 is a mediator of cuproptosis-related sterile inflammation. Front Cell Dev Biol. 2022;10: 996307.
Sun L, Zhang Y, Yang B, et al. Lactylation of METTL16 promotes cuproptosis via m6A-modification on FDX1 mRNA in gastric cancer. Nat Commun. 2023;14(1):6523.
Zhang P, Zhou C, Ren X, et al. Inhibiting the compensatory elevation of xCT collaborates with disulfiram/copper-induced GSH consumption for cascade ferroptosis and cuproptosis. Redox Biol. 2024;69: 103007.
Xing T, Li L, Chen Y, et al. Targeting the TCA cycle through cuproptosis confers synthetic lethality on ARID1A-deficient hepatocellular carcinoma. Cell Rep Med. 2023;4(11): 101264.
Wu Z, Lin C, Zhang F, et al. TIGD1 function as a potential cuproptosis regulator following a novel cuproptosis-related gene risk signature in colorectal cancer. Cancers (Basel). 2023;15(8):2286.
Wang K, Zhang Y, Ao M, Luo H, Mao W, Li B. Multi-omics analysis defines a cuproptosis-related prognostic model for ovarian cancer: implication of WASF2 in cuproptosis resistance. Life Sci. 2023;332: 122081.
Huo S, Wang Q, Shi W, et al. ATF3/SPI1/SLC31A1 signaling promotes cuproptosis induced by advanced glycosylation end products in diabetic myocardial injury. Int J Mol Sci. 2023;24(2):1667.
Wang X, Jia JH, Zhang M, et al. Adrenomedullin/FOXO3 enhances sunitinib resistance in clear cell renal cell carcinoma by inhibiting FDX1 expression and cuproptosis. FASEB J. 2023;37(10): e23143.
Lu J, Ling X, Sun Y, et al. FDX1 enhances endometriosis cell cuproptosis via G6PD-mediated redox homeostasis. Apoptosis. 2023;28(7–8):1128–40.
Tian S, Wang R, Wang Y, et al. p32 regulates glycometabolism and TCA cycle to inhibit ccRCC progression via copper-induced DLAT lipoylation oligomerization. Int J Biol Sci. 2024;20(2):516–36.
Carraway RE, Dobner PR. Zinc pyrithione induces ERK- and PKC-dependent necrosis distinct from TPEN-induced apoptosis in prostate cancer cells. Biochim Biophys Acta. 2012;1823:544–57.
Liu K, Huang J, Liu J, Klionsky DJ, Kang R, Tang D. Induction of autophagy-dependent ferroptosis to eliminate drug-tolerant human retinoblastoma cells. Cell Death Dis. 2022;13:521.
Cheng Q, Bao L, Li M, Chang K, Yi X. Erastin synergizes with cisplatin via ferroptosis to inhibit ovarian cancer growth in vitro and in vivo. J Obstet Gynaecol Res. 2021;47:2481–91.
Zhou HH, Chen X, Cai LY, Nan XW, Chen JH, Chen XX, et al. Erastin reverses ABCB1-mediated docetaxel resistance in ovarian cancer. Front Oncol. 2019;9:1398.
Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. 2015;2: e1054549.
Li Y, Yan H, Xu X, Liu H, Wu C, Zhao L. Erastin/sorafenib induces cisplatin-resistant non-small cell lung cancer cell ferroptosis through inhibition of the Nrf2/xCT pathway. Oncol Lett. 2020;19:323–33.
Chen F, Wu S, Kuang N, Zeng Y, Li M, Xu C. ABCB1-mediated docetaxel resistance reversed by erastin in prostate cancer. FEBS J. 2024;291:3249–66.
Holzer AK, Samimi G, Naerdemann, et al. Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Proc Am Assoc Cancer Res. 2005;46:347.
Song IS, Savaraj N, Siddik ZH, et al. Roles of copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and resistant cells. Mol Cancer Ther. 2004;3:1543–9.
Komatsu M, Sumizawa T, Mutoh M, et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000;60(5):1312–6.
Samimi G, Safaei R, Katano K, et al. Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, and oxaliplatin in ovarian cancer cells. Clin Cancer Res. 2004;10(14):4661–9.
Lasorsa A, Nardella MI, Rosato A, et al. Mechanistic and structural basis for inhibition of copper trafficking by platinum anticancer drugs. J Am Chem Soc. 2019;141(30):12109–20.
Kita Y, Hamada A, Saito R, et al. Systematic chemical screening identifies disulfiram as a repurposed drug that enhances sensitivity to cisplatin in bladder cancer: a summary of preclinical studies. Br J Cancer. 2019;121(12):1027–38.
Guo Y, Smith K, Lee J, Thiele DJ, Petris MJ. Identification of methionine-rich clusters that regulate copper-stimulated endocytosis of the human Ctr1 copper transporter. J Biol Chem. 2004;279(17):17428–33.
Wee NK, Weinstein DC, Fraser ST, Assinder SJ. The mammalian copper transporters CTR1 and CTR2 and their roles in development and disease. Int J Biochem Cell Biol. 2013;45(5):960–3.
Horn N, Wittung-Stafshede P. ATP7A-regulated enzyme metalation and trafficking in the menkes disease puzzle. Biomedicines. 2021;9(4):391.
Polishchuk EV, Concilli M, Iacobacci S, et al. Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Dev Cell. 2014;29(6):686–700.
Cha JH, Chan LC, Song MS, Hung MC. New approaches on cancer immunotherapy. Cold Spring Harb Perspect Med. 2020;10(8): a036863.
Tiwari A, Trivedi R, Lin SY. Tumor microenvironment: barrier or opportunity towards effective cancer therapy. J Biomed Sci. 2022;29(1):83.
Samson N, Ablasser A. The cGAS-STING pathway and cancer. Nat Cancer. 2022;3(12):1452–63.
Jiang A, Luo P, Chen M, et al. A new thinking: deciphering the aberrance and clinical implication of copper-death signatures in clear cell renal cell carcinoma. Cell Biosci. 2022;12(1):209.
Tang D, Kroemer G, Kang R. Targeting cuproplasia and cuproptosis in cancer. Nat Rev Clin Oncol. 2024;21(5):370–88.
Li L, Zhou H, Zhang C. Cuproptosis in cancer: biological implications and therapeutic opportunities. Cell Mol Biol Lett. 2024;29:91.
Wang Y, Chen Y, Zhang J, et al. Cuproptosis: a novel therapeutic target for overcoming cancer drug resistance. Drug Resist Update. 2024;72: 101018.
Xu R, Bu-Ghanim M, Fiel MI, Schiano T, Cohen E, Thung SN. Hepatocellular carcinoma associated with an atypical presentation of Wilson’s disease. Semin Liver Dis. 2007;27(1):122–7.
Acknowledgements
We thank Home for Researchers editorial team (www.home-for-researchers.com) for language editing service.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Numbers: 32000533); Sichuan Science and Technology Program (Grant Number: 2021YFS0215).
Author information
Authors and Affiliations
Contributions
C. Z. contributed to conception and design of the manuscript; C. Z. and T. H. drafted the manuscript and conducted image processing. L. L. contributed to reviewing. C. Z. given the final approval of the version to be published.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, C., Huang, T. & Li, L. Targeting cuproptosis for cancer therapy: mechanistic insights and clinical perspectives. J Hematol Oncol 17, 68 (2024). https://doi.org/10.1186/s13045-024-01589-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-024-01589-8