
Abstract
The FGFR signaling pathway is integral to cellular activities, including proliferation, differentiation, and survival. Dysregulation of this pathway is implicated in numerous human cancers, positioning FGFR as a prominent therapeutic target. Here, we conduct a comprehensive review of the function, signaling pathways and abnormal alterations of FGFR, as well as its role in tumorigenesis and development. Additionally, we provide an in-depth analysis of pivotal phase 2 and 3 clinical trials evaluating the performance and safety of FGFR inhibitors in oncology, thereby shedding light on the current state of clinical research in this field. Then, we highlight four drugs that have been approved for marketing by the FDA, offering insights into their molecular mechanisms and clinical achievements. Our discussion encompasses the intricate landscape of FGFR-driven tumorigenesis, current techniques for pinpointing FGFR anomalies, and clinical experiences with FGFR inhibitor regimens. Furthermore, we discuss the inherent challenges of targeting the FGFR pathway, encompassing resistance mechanisms such as activation by gatekeeper mutations, alternative pathways, and potential adverse reactions. By synthesizing the current evidence, we underscore the potential of FGFR-centric therapies to enhance patient prognosis, while emphasizing the imperative need for continued research to surmount resistance and optimize treatment modalities.
Similar content being viewed by others


Background
The Fibroblast Growth Factor Receptor (FGFR) comprises a subset of Receptor Tyrosine Kinases (RTKs), encompassing five members (FGFR 1–5) and exhibiting significant sequence homology [1]. Notably, FGFR 5 (also recognized as FGFRL 1) lacks the tyrosine kinase domain yet assumes a role in modulating excessive activation of the FGF-FGFR signaling pathway [2,3,4].
FGFR signaling predominantly entails Fibroblast Growth Factor (FGF) binding to the receptor, receptor dimerization, and subsequent intracellular kinase auto-phosphorylation cascades [5]. Additionally, FGFR can undergo ligand-independent activation, exemplified by fusion events between the FGFR gene and other gene components initiated by chromosomal translocation [6]. The FGF/FGFR signaling pathway intricately intersects embryonic development, angiogenesis, tissue homeostasis, and wound healing, while concurrently orchestrating pivotal functions in cellular proliferation, differentiation, apoptosis, and migration [7, 8]. Nevertheless, the abnormalities of FGF/FGFR signaling axis can promote a variety of diseases, especially malignant tumors, mainly due to the occurrence of gene amplification, mutation, and gene fusion [9, 10].
In vertebrates, the FGF family reflects substantial diversity, encompassing 22 FGF ligands identified in mice and humans. These FGFs can be classified into six subfamilies [11], comprising paracrine subclasses such as FGF1, FGF2, FGF3, FGF4, FGF5, FGF6, FGF7, FGF8, FGF9, FGF10, FGF16, FGF17, FGF18, FGF16, FGF20, FGF22, as well as an endocrine subclass involving FGF19, FGF21, and FGF23. The paracrine subfamily controls multiple events during embryonic development, while members of the endocrine subfamily play a significant role in regulating metabolism [12,13,14,15,16].
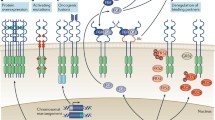
FGFR assumes the architecture of a single-pass transmembrane protein, encompassing extracellular, transmembrane, and intracellular tyrosine kinase domains [17, 18] (Fig. 1). The interaction between FGF and FGFR necessitates the presence of heparan sulfate (HS) as a co-receptor, while heparan sulfate proteoglycans (HSPG) stabilize this binding phenomenon [19, 20]. The canonical downstream signaling routes of FGF/FGFR encompass the Ras/Raf-MEK-MAPK (mitogen-activated protein kinase) pathway, phosphatidylinositol-3 kinase/protein kinase B (PI3K/AKT) pathway, PLCγ pathway, as well as signaling intermediates and transcription activators (STATs) [7, 21]. A unique docking protein, FRS2, exists in the FGFR signaling pathway. FRS2 specifically binds to members of the FGFR family through its phosphorylated tyrosine-binding structural domain, which is rare in other receptor tyrosine kinases [22]. Upon binding to the FGFR, FRS2 is phosphorylated and serves as a hub for the recruitment of downstream signaling molecules (Grb2 and Shp2), which in turn activate pathways such as the MAPK and PI3K-AKT pathways [23]. This signaling interplay is not only shaped by binding specificity, expression levels, and alternative splicing but is also subject to cross-regulation with other pathways such as BMP and Wnt [24,25,26,27,28]. Furthermore, post-translational modifications (including phosphorylation, glycosylation, ubiquitination) and cytoplasmic trafficking contribute substantively to the orchestration of signal specificity and strength [29,30,31,32,33].

FGFR signals and inhibitors. This interaction triggers a downstream signaling network that encompasses the Ras-Raf-MAPK, PI3K-AKT, PLCγ, and STATs pathways, resulting in a range of cellular responses, including proliferation, differentiation, survival, migration, and angiogenesis
In a word, the FGF/FGFR signaling pathway assumes multifarious and pivotal roles within biological systems. A comprehensive exploration of this signaling pathway promises enhanced insights into its functional intricacies and potential therapeutic applications across disease contexts.
Prevalence and diversity of FGFR abnormalities in various malignancies
Multiple sequencing investigations have shown that FGFR abnormalities were detected in 1.9–7.1% of tumor patients. Most of these abnormalities were gene amplifications (53.7–66%), followed by mutations (26–38.8%) and rearrangements/fusions (5.6–8%). The frequencies of aberration for FGFR1, FGFR2, FGFR3, and FGFR4 were 49–56.8%, 14.2–19%, 17.7–26% and 2.8–7%, respectively (Fig. 2). The prevalence of various tumor types was as follows: uroepithelial carcinoma (32–14.8%), colorectal carcinoma (31%), breast carcinoma (12.6–18%), gastric carcinoma (16.8–25.6%), endometrial carcinoma (13%), squamous lung carcinoma (6.8–13%), esophageal carcinoma (12.7%), ovarian carcinoma (9%), and lung adenocarcinoma (1.3%) [34,35,36,37,38,39].

Displays a summary of FGFR alterations in cancers. a FGFR alterations may be categorized as amplification, point mutation, and rearrangement. The graphic illustrates the usage of FGFR3 S249C mutation and FGFR3-TACC3 for a basic mechanism explanation. b Presence of FGFR in various tumors. (NSCLC: non-Small cell lung carcinoma; SqCC: squamous cell cancer; SCLC: small cell lung carcinoma; ESCC: esophageal squamous cell carcinoma; HNSCC: head and neck squamous cell carcinoma; LGG: low-grade glioma; MNGT: mixedneuronal-glialtumors; DMG: diffuse midline gliomas; GBM: glioblastoma; GC: gastric cancer; UCEC: uterine corpus endometrial carcinoma; ICC: intrahepatic cholangiocarcinoma; PAAD: pancreatic cancer; CHOL: cholangiocarcinoma; BLCA: bladder cancer; MM: multiple myeloma; HCC: hepatocellular carcinoma)
FGFR amplification in tumors
The phenomenon of increased copy number of the FGFR gene in cells is called gene amplification. A genetic sequencing of more than 4000 cancer patients showed that FGFR aberrations were found in 7.1% of tumor patients, most of which were gene amplifications (66% of aberrations) [40]. Carrying FGFR amplification is associated with adverse clinical outcomes. FGFR1 amplification was significantly associated with shorter overall survival (OS) (58.6 months vs. 80.0 months) in patients with squamous cell lung cancer (SqCC) [41]. FGFR1 amplification occurs in approximately 10% of breast cancers and functions to drive enhanced ligand-dependent signaling and suppress progesterone receptor expression, associated with poor prognosis [42]. The analysis of circulating tumor DNA (ctDNA) shows that gastric cancer patients with FGFR2 amplification have significantly shorter OS than those without FGFR2 amplification [43]. FGFR autophosphorylation (Tyr653/654 cyclic phosphorylation) is significantly increased in cell lines with high levels of FGFR2 amplification (arbitrarily defined as FISH ratio > 5), leading to FGFR hyperactivation [43]. Patients with high levels of FGFR2 amplification are thought to be likely to respond to FGFR inhibitors, but this occurs in only 5% of GC [43]. Studies have shown that FGFR1 is amplified in 19% of lung squamous cell carcinoma cases (14/73) and is more common in poorly differentiated tumors, suggesting a possible role in tumor aggressiveness [44]. Of these, 50% (7/14) showed high amplification (defined as real-time PCR fold greater than tenfold). And patients with high copy numbers showed better response rates to the FGFR inhibitor AZD4547 [45]. The question was raised as the research progressed: Does FGFR amplification have to reach a certain threshold to benefit from FGFR inhibitors?
FGFR point mutation in tumors
FGFR mutations lead to abnormal activation of the receptor, resulting in continued activation of the FGFR signaling pathway. This sustained signaling pathway activation is a key factor in the development of many cancers, including bladder cancer, breast cancer, and non-small cell lung cancer (NSCLC) [7].
Common FGFR1 point mutation sites are: N546K, K656E and V561M. These mutation sites are mainly located in the kinase domain of FGFR1 and are associated with abnormal activation of FGFR1 and various diseases, including cancer. The presence of FGFR1 point mutations (N546K and K656E) was exclusively observed in H3K27M-mutant diffuse midline gliomas (DMG) (64/304, 21%), a subset that displayed a higher incidence in older individuals with diencephalic tumors [46]. Additionally, analogous findings have been documented for other central nervous system neoplasms, thereby establishing a correlation with heightened malignancy, diminished responsiveness to FGFR inhibitors, and occurrence of spontaneous hemorrhage [47,48,49,50].
However, in tumors with FGFR2 point mutations, the common sites are: S252W, N549K, E565A, K660N/K660E and V565I/V565L. The development of triple-negative breast cancer is facilitated by activating mutations in FGFR2-S252W, which induce epithelial-mesenchymal transition through FGFR2-STAT3 signaling [51]. The pan-cancer analysis demonstrated that uterine endometrial carcinoma (UCEC) exhibits the highest prevalence of FGFR2 mutations, with the most frequent mutations occurring at S252W and N549K. These mutations have been found to have an oncogenic functional impact; however, they show limited responsiveness to targeted therapy [52].
FGFR3 mutation sites are also different, with S249C, Y373C, G370C, R248C and V555M being the most common. The analysis of data obtained from The Cancer Genome Atlas (TCGA) demonstrated that the presence of targetable mutations in the FGFR3 gene was predominantly observed in bladder cancer. Specifically, the mutations S249C, Y373C, G370C, and R248C were identified as hotspot mutations, which can be effectively targeted by the FDA-approved drug erdafitinib [53]. Additionally, the occurrence of FGFR3 S249C mutation was less frequent in upper tract urothelial carcinoma patients compared to bladder cancer patients (37.5% vs. 59.3%) [54].
FGFR4 mutation sites with high incidence are as follows: V550L/V550M/V550E/N535D/N535K and G388R. Genomic analysis of rhabdomyosarcoma reveals high prevalence of FGFR4 overexpression and mutations in tumor tissues as a result of PAX-FOXO1 oncogene transcription [55].
FGFR gene rearrangement and fusion in tumors
Gene rearrangement is the process by which genes are rearranged on chromosomes. This rearrangement can be caused by chromosome breakage, transposition, inversion, or translocation [56]. These changes can result in the loss of gene function, the production of abnormal gene proteins, or the activation of previously dormant oncogenes [57].
Gene fusions involving FGFR1, and various partner genes have been identified in diverse tumor types. The FGFR1-TACC1 fusion is detected in glioblastoma and SqCC. This fusion is associated with the hyperactivation of FGFR1, which promotes cellular proliferation and inhibits apoptosis [58]. Additionally, it might augment FGFR1 signaling either by intensifying its tyrosine kinase activity or by modulating its intracellular positioning [59]. BCR-FGFR1, FGFR1OP-RET, and FGFR1OP-FGFR1 Fusions, which are identified in myeloproliferative disorders, have been associated with the initiation and advancement of the disease [60,61,62]. They might bolster cell survival by facilitating cell cycle progression and suppressing apoptosis.
Gene fusions between FGFR2 and various genes are commonly identified in cholangiocarcinoma (CCA), playing a crucial role in tumorigenesis and tumor progression [63]. The presence of the FGFR2-BICC1 fusion is associated with the aggressive and malignant characteristics of cholangiocarcinoma [63]. The FGFR2 fusion necessitates the involvement of the downstream effector Mek1/2, indicating the potential clinical efficacy of dual blockade targeting FGFR2 and MEK1/2 in patients with intrahepatic cholangiocarcinoma (ICC) [63]. FGFR2- PPHLN1, AHCYL1, and TACC3 fusions might potentially result in continuous activation of FGFR2, which in turn promotes the initiation and progression of CCA [64]. A noteworthy finding is that 90% of patients diagnosed with ICC and having KRAS mutations also showed positive findings for FGFR2 fusions, suggesting a potential collaborative role in promoting the progression of malignant tumors [65]. Consequently, it is hypothesized that the occurrence of FGFR2 fusion in ICC might serve as an initial genetic occurrence that fosters the initiation and progression of tumorigenesis [66].
Gene fusions of FGFR3 with multiple partners are prevalently reported in bladder cancer, gliomas, and multiple myeloma, playing a central role in tumorigenesis and progression [67,68,69]. The FGFR3-TACC3 fusion protein is shown to be localized to mitotic spindle poles, according to research results, possesses inherent kinase activity, elicits abnormalities in mitosis and chromosome segregation, and instigates aneuploidy, ultimately leading to the development of tumorigenicity [70]. The fusion alteration in female bladder cancer patients is regarded as a significant risk factor [71]. The fusion of FGFR3-TACC3 has recently been identified as a relatively uncommon yet potentially significant mechanism of resistance to EGFR inhibitors, specifically osimertinib, in cases of lung cancer [72]. The t(4;14) transforming events lead to activation of FGFR3 in myeloma, and such patients have poor survival and response to chemotherapy [69]. Efficacy is currently being evaluated in a combination trial of erdafitinib + dexamethasone in these patients (NCT02952573). Bladder cancer is associated with FGFR3-BAIAP2L1 fusions, which are known to contribute to the aggressive nature and malignancy of the disease [73]. They might boost FGFR3 signaling by increasing its stability or modifying its intracellular positioning.
Gene fusions of FGFR4 are relatively rare, and one fusion mutation, FGFR4-RAPGEFL1, has been identified in NSCLC. Its function remains unclear and requires further investigation [74].
Detection of FGFR alterations in tumors
Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) employs fluorescently labeled probes to hybridize with target DNA sequences. This allows researchers to directly visualize the location and quantity of chromosomes or genes within cellular or tissue sections. In the realm of FGFR gene studies, FISH has been established as a method of high sensitivity and specificity, adept at identifying amplifications, mutations, and fusions associated with FGFR genes [75]. The excellent spatial resolution of FISH compared to other assays makes FISH particularly effective at mapping heterogeneous changes in the FGFR gene at the single-cell level [76]. As with the Next-Generation Sequencing (NGS) assay, the latest National Comprehensive Cancer Network (NCCN) guidelines recommend it as a test for FGFR2 fusion/other FGFR aberrations in cholangiocarcinoma [77].
Reverse transcription-polymerase chain reaction
Reverse transcription-polymerase chain reaction (RT-PCR) is an experimental method by first converting RNA template into cDNA, and then using PCR technology for DNA amplification. With repeated cycles, RT-PCR can significantly enhance the signal of the target sequence, which allows RT-PCR to detect and quantify specific RNA molecules starting from extremely small amounts of RNA [78, 79]. Both NCCN and European Society for Medical Oncology (ESMO) guidelines recommend the detection of FGFR2/3 mutations and fusions in bladder cancer, and the FDA has also approved the therascreen® FGFR RGQ RT-PCR kit as a PCR-based companion diagnostic kit [80, 81]. The combination of peptide nucleic acid mediated RT-PCR clamping has been shown to be sensitive enough to detect FGFR3 mutations in bladder cancer from urine sediments, clearly distinguishing mutant DNA from wild-type DNA at concentrations greater than 1% [82]. In contrast to alternative methodologies, such as sequencing, RT-PCR can deliver results in a significantly shorter timeframe, often within hours. It has been instrumental in identifying FGFR fusions, particularly evident in malignancies like cholangiocarcinoma [83].
Next-generation sequencing
NGS is a revolutionary DNA sequencing technology. The basic principle is to break a DNA sample into millions of small fragments, and then these fragments are amplified, sequenced, and analyzed through different methods, which can simultaneously sequence millions to billions of DNA fragments [84]. Targeted NGS panels, focusing on specific genes or gene regions, have been developed for comprehensive FGFR profiling [85]. These panels can detect point mutations, copy number variations, and gene fusions with high sensitivity and specificity. Several studies employing NGS have reported a diverse spectrum of FGFR alterations across different tumor types [40, 75]. Based on the advantages of NGS testing and past practical experience, the latest NCCN and ESMO guidelines recommend it as a detection method for FGFR2 fusion/other FGFR aberrations in cholangiocarcinoma [77, 86]. ESMO guidelines emphasize that detection involving FGFR2 is best performed at the RNA level to help identify fusion transcripts with unknown fusion partners [86]. In bladder cancer, ESMO guidelines recommend NGS for FGFR2/3 mutation and fusion detection [80].
Hybridization-capture
Hybridization-capture is a genomic technique that uses specialized probes to capture and enhance target DNA sequences, allowing for rapid and precise sequencing analysis [87]. Hybridization-capture enables a thorough examination of the genetic makeup of tumors, allowing for the detection of hitherto unidentified FGFR fusions [88]. By using this technique in NGS, it becomes possible to analyze a substantial quantity of samples concurrently, therefore offering a comprehensive depiction of FGFR abnormalities among the whole population [89]. Hybridization capture may be used to discover FGFR fusions in patients who do not respond to conventional treatments. These fusions can serve as acquired resistance mechanisms and studying them can assist improve the prognosis of the patients [89].
Liquid biopsy
Liquid biopsy is an innovative diagnostic technique that analyzes biomarkers in body fluids like blood, saliva, or urine to detect and monitor diseases, particularly cancers. This technology captures circulating tumor cells, ctDNA, microRNAs, and other biomolecules, offering a non-invasive alternative to traditional tissue biopsies [90, 91]. Research has underscored the potential of liquid biopsy in identifying FGFR alterations. For instance, FGFR mutations and fusions, once identified in tissue samples, have been detected in ctDNA with a high degree of concordance [92]. NCCN guidelines also recognize that the use of cell free DNA (cfDNA) testing can detect some FGFR2 fusion breakpoints in cholangiocarcinoma, but the sensitivity is lower than that of tumor tissue testing [77]. Moreover, liquid biopsies facilitate continuous monitoring, capturing the evolving profile of FGFR alterations during treatment, which could signal treatment resistance or disease progression [93].
Targeting FGFR in the clinic
The contribution of aberrant FGFR signaling to tumorigenesis has led to the development of multiple therapies targeting the FGFR pathway (Fig. 3), many of which have shown promise in phase II and III clinical studies in various tumor types with FGFR abnormalities (Tables 1 and 2). We focused on selective FGFRs inhibitors with Phase II/III clinical trial results and representative multi-target kinase inhibitors.

Take Futibatinib as an example, showing the functions of FGFR inhibitors. a Cell apoptosis. b Antiangiogenic impact. c Regulation of the immunological microenvironment. d Anti-metastatic impact
Listed drugs
Infigratinib
Infigratinib is a potent and selective inhibitor of FGFR1, 2 and 3 that has demonstrated significant anti-tumor efficacy in preclinical studies [94,95,96,97,98] (Fig. 4a). The results of a phase II study indicate that infigratinib exhibits significant clinical activity in patients with CCA who have previously received gemcitabine treatment containing an FGFR2 fusion. The overall response rate (OR) was found to be 14.8%, and the disease control rate was 75.4% [99]. Following a median follow-up duration of 10.6 months, the ORR was determined to be 23.1% (25 out of 108 patients), with a patient demonstrating a complete response (CR) [100]. In particular, several phase III trials are underway evaluating infigratinib versus standard of care as a first-line treatment in patients with CCA and infigratinib versus placebo in patients with urothelial carcinoma [101, 102]. Another phase II study demonstrated restricted effectiveness of infigratinib monotherapy in patients with recurrent gliomas and various FGFR gene alterations. However, patients with FGFR1 or FGFR3 point mutations or FGFR3-TACC3 fusions exhibited enduring disease control exceeding 1 year [103]. Intriguingly, infigratinib appears to possess therapeutic potential in the treatment of tumour-induced osteochondrosis (TIO), an uncommon paraneoplastic syndrome [104]. However, in October 2022, Helsinn Group announced the withdrawal of infigratinib's application for listing in the United States based on business plan considerations.

a Crystal structures of the compound Infigratinib and the compound Infigratinib in FGFR1 (PDB ID 3TT0). b Crystal structures of the compound Pemigatinib and the compound Pemigatinib in FGFR1 (PDB ID 7WCL). c Crystal structures of the compound Erdafitinib and the compound Erdafitinib in FGFR1 (PDB ID 5EW8). d Crystal structures of the compound Futibatinib and the compound Futibatinib in FGFR1 (PDB ID 6MZW)
Pemigatinib
Pemigatinib, an inhibitor targeting FGFR 1, 2, and 3, effectively inhibits the growth of FGFR-abnormalized xenografts [105,106,107] (Fig. 4b). In a phase II clinical study, it was observed that among patients with CCA who had previously undergone therapy and experienced of FGFR2 fusions or rearrangements, the administration of 13.5 mg of oral pemigatinib once daily resulted in an ORR of 35.5% with 38 out of 107 patients achieving a response, including 3 CR and 35 partial responses [108]. Furthermore, post hoc analyses of the study revealed that the median Progression-Free Survival (PFS) for patients with FGFR2 fusion/rearrangement (n = 65) who received second line pemigatinib was 7.0 months [109]. Another similar study found that 15 of 30 patients with FGFR2 fusions or rearrangements who were evaluated for efficacy achieved a partial response (ORR, 50.0%) [110]. Considering these findings, a randomized controlled phase III study (FIGHT-302) is currently underway to assess and compare the effectiveness and safety of first line pemigatinib in contrast to gemcitabine plus cisplatin in the management of patients diagnosed with advanced CCA exhibiting FGFR2 rearrangements [111]. In the cohort of patients with relapsed or refractory myeloid or lymphoid neoplasms harboring recombinant FGFR1 genes and treated with pemigatinib, the evaluation of efficacy was conducted on a total of 33 patients. The rates of complete cytogenetic response were in rates of 72.7% and 75.8%, respectively. The median duration of CR has not yet been determined [112]. Furthermore, Pemigatinib has demonstrated favorable clinical efficacy and a satisfactory safety profile in the treatment of gliomas, gynecological tumors, and pancreatic cancer [113].
Erdafitinib
Erdafitinib exhibits potent inhibitory activity against FGFR1-4 and shows robust antitumor effects in preclinical studies [114, 115] (Fig. 4c). In a phase II clinical trial, the objective response rate (ORR) of individuals diagnosed with locally advanced, unresectable, metastatic urothelial carcinoma (mUC) and treated with erdafitinib was found to be 40% [116]. The duration of the curative effect was observed for a period of 24.0 months. Out of a total of 101 patients, the researchers determined that the ORR for those who received the erdafitinib regimen was 40% (40%; 95% CI 30–49) [117]. The THOR 3 study showed that in patients with mUC with FGFR alterations previously treated with immunotherapy, erdafitinib significantly improved median PFS (5.6 vs. 2.7 months) and ORR (46% vs. 12%) compared with chemotherapy [118]. Data from the THOR 2 study showed a favorable and durable response to erdafitinib in patients with high-risk, BCG-nonresponsive non-muscle invasive bladder cancer (NMIBC) and FGFR genetic alterations, with 16 patients treated for a median of 6.7 months [119]. Additionally, the cohort revealed that erdafitinib did not outperform pembrolizumab in patients with FGFR-altered, anti-PD-(L)1-naive mUC, achieving median OS times of 10.9 and 11.1 months, respectively [120]. The RAGNAR study confirmed the antitumor effect of erdafitinib in patients with tumors with multiple FGFR mutations. Independent evaluations showed an ORR of 30% and a sustained response period of 6.9 months, with an ORR of 56% for pancreatic cancer (18 enrolled) and 52% for CCA(31 enrolled) [121]. TAR-210, a novel intravesical delivery system for erdafitinib, also demonstrated positive clinical activity in patients with high- and intermediate-risk NMIBC with FGFR alterations, with 13 (87%) of the 315 patients in the cohort with available response evaluators. Achieve complete remission [122]. In addition, PR and SD can be achieved in more than 50% of Asian patients with advanced CCA and pediatric central malignancies with FGFR alterations treated with Erdafitinib [123, 124].
Futibatinib
Futibatinib is an irreversible pan-FGFR inhibitor that has shown potent activity in preclinical studies against tumors that are resistant to FGFR inhibitors [125,126,127] (Fig. 4d). In patients with previously treated FGFR2 fusion- or rearrangement-positiven ICC, the use of futibatinib provided a significant clinical benefit, with a total of 43 of 103 patients (42%; 95% confidence interval 32–52) responding, with a median PFS of 9.0 months and OS of 21.7 months [128]. A Phase III trial is currently underway to assess the effectiveness and safety of futibatinib in comparison to gemcitabine-cisplatin chemotherapy as the initial therapeutic approach for individuals with advanced, metastatic, or recurrent unresectable ICC carrying a FGFR2 gene rearrangement [129]. In another Phase II study, TAS-120 demonstrated comparable anti-tumor activity in patients with advanced or metastatic gastric and gastroesophageal cancer harboring FGFR2 amplification, with median PFS and OS estimates of 2.8 months and 5.7 months [130].
As mentioned above, there are currently four FGFR inhibitors approved for marketing by the FDA, with infigratinib having been removed from the market due to commercial reasons (Table 3). In 04/2019 Erdafitinib was approved by the FDA for the treatment of uroepithelial carcinoma at a daily oral dose of 8–9 mg. Pemigatinib and Futibatinib were also approved by the FDA for the treatment of CCA in 04/2020 and 09/2022 at a daily oral dose of 13.5 mg and 20 mg, respectively. Despite their limitations, the four approved FGFR inhibitors offer relief to FGFR-altered patients who have failed other treatments, prolonged survival and bringing hope to these patients.
Selective inhibitors
AZD4547
AZD4547 is a small-molecule tyrosine inhibitor that selectively targets FGFR 1, 2, and 3 [131]. In the phase II S1400D trial, AZD4547 demonstrated limited efficacy in squamous cell NSCLC patients with FGFR alterations, despite its satisfactory safety profile [132]. Similarly, the NCI-MATCH trial revealed restricted effectiveness of AZD4547 in refractory cancers exhibiting FGFR1-3 aberrations, with responses observed solely in tumors harboring FGFR1-3 point mutations or fusions [133]. The SHINE study findings indicate that AZD4547 does not yield a statistically significant enhancement in PFS when compared to paclitaxel in patients with advanced GC who exhibit FGFR2 amplification/polysomy [134]. Meanwhile, These results underscore the presence of intratumor heterogeneity and a lack of concordance between FGFR2 amplification and mRNA expression. The RADICAL study discovered that the combination of AZD4547 with anastrozole or letrozole exhibited limited efficacy but notable toxicity in patients with estrogen receptor-positive metastatic breast cancer who had developed resistance to aromatase inhibitors [135]. A subsequent study revealed that AZD4547 exhibited no discernible efficacy among patients diagnosed with malignant pleural mesothelioma who experienced disease progression after initial treatment involving platinum-based chemotherapy [136].
HMPL-453
HMPL-453 is a potent inhibitor of FGFR 1, 2, and 3 and exhibits potent antitumor activity in tumor models with FGFR alterations [137]. In previously treated patients with advanced ICC and FGFR fusion, the HMPL-453 (300 mg, QD, 2w on/1w off) dosing arm demonstrated acceptable levels of toxicity and excellent efficacy: an ORR of 50% and a DCR of 90% [138]. Multiple additional studies have been devised to assess the effectiveness, safety, and pharmacokinetics of HMPL-453 in advanced malignant mesothelioma and other advanced solid tumors, as well as to investigate the potential synergistic effects of combining HMPL-453 with chemotherapy or anti-PD-1 antibodies (NCT05173142, NCT04290325, NCT03160833).
RLY-4008
RLY-4008 is a selective FGFR2 inhibitor and remains inhibitory to tumors that develop resistance mutations [139, 140]. In a clinical investigation examining the effects of RLY-4008 on patients diagnosed with bile duct cancer characterized by FGFR2 fusion/rearrangement, a combined phase I/II trial demonstrated favorable efficacy, as evidenced by an ORR of 88% at the phase II dosage level. The administered treatment exhibited a high level of tolerability, primarily manifesting as low-grade side effects, and notably, no instances of severe (grade 4/5) treatment-related adverse events were recorded. Furthermore, the sustained nature of most responses suggests the promising prospect of a prolonged and enduring therapeutic response [141].
Rogaratinib
Rogaratinib, a potent inhibitor of FGFR1-4, exhibited significant inhibition of various tumors characterized by FGFR aberrations [142]. In a clinical trial investigating the efficacy of rogaratinib in patients with a variety of solid tumors, 15 of 100 patients had an objective response to treatment (ORR: 15%) [143]. In a separate phase II/III clinical trial conducted on individuals diagnosed with FGFR mRNA-positive advanced/mUC, rogaratinib demonstrated comparable levels of effectiveness and safety when compared to chemotherapy, exhibiting similar rates of ORR (20.7% vs 19.3%). Nevertheless, an examination of patients exhibiting FGFR3 DNA alterations revealed a substantial disparity in OR (52.4% vs. 26.7%) [144]. Another study demonstrated that despite the elevated expression of FGFR mRNA in approximately 50% of tumors in patients with SqCC, the administration of rogaratinib did not result in a significant improvement in PFS among these individuals [145].
FGF401
FGF401 is a reversible, covalent, small-molecule inhibitor of FGFR4 kinase activity that has shown significant antitumor efficacy in preclinical studies [146]. The results of the NCT02325739 trial showed that 74 patients treated with single-agent FGF401 in phase I and 86 patients treated with FGF401 in phase II experienced a total of 8 patients with objective responses (1 CR, 7 PR; 4 each in phases I and II), demonstrating some clinical efficacy and safety [147].
E7090
E7090 is an FGFR1-3 inhibitor that has shown promise in the treatment of mouse tumor models with FGFR abnormalities [148]. Multiple phase I clinical trials investigating the effects of E7090 in patients with advanced solid tumors revealed a tolerable safety profile, absence of dose-limiting toxicities at doses equal to or exceeding 140 mg, and promising indications of therapeutic efficacy [149, 150]. E7090 has been shown in NCT04238715 Phase II to have strong anti-tumor efficacy (ORR of 30%; DCR of 79%) and to be safe and controlled in the treatment of CCA patients with FGFR2 gene fusion [151].
ICP-192
ICP-192, a newly developed covalent inhibitor targeting pan-FGFR, has shown promising results in Phase I clinical trials [152]. A phase IIa dose extension studied the efficacy of ICP-192 in previously treated patients with FGFR2-altered CCA, resulting in an ORR of 52.9% (9/17), an mPFS of 6.93 months, and a discontinuation rate of 0% due to TRAEs, suggesting that ICP-192 had a favorable response rate and was well tolerated [153]. Simultaneously, several Phase II clinical trials are being conducted for the subject (NCT04565275, NCT04492293, NCT05678270, NCT05372120).
Multi-targeting TKIs
Lenvatinib
Lenvatinib, a selective, multi-target tyrosine kinase inhibitor, exerts its action on the vascular endothelial growth factor receptor (VEGFR), FGFR, and platelet-derived growth factor receptor (PDGFR) family [154, 155]. In vitro experiments have provided evidence that lenvatinib effectively suppresses the proliferation signaling pathway of overexpressed VEGFR and FGFR in cancer cells [156]. Interestingly, lenvatinib offers a promising therapeutic option for FGFR 2-driven CCA, especially in cases involving insurmountable adverse effects to selective Tkis or acquired kinase mutations [157]. A Phase III clinical trial showed that lenvatinib was not inferior to sorafenib in patients with previously untreated advanced HCC (median survival time: 13.6 vs. 12.3 months), in contrast to sorafenib, lenvatinib exhibited substantial enhancements across all secondary endpoints, including a greater ORR, extended PFS, and increased time to progression (TTP) [158]. A separate investigation demonstrated that lenvatinib exhibited a noteworthy enhancement in both PFS and response rates among individuals afflicted with iodine 131-refractory thyroid cancer [159]. In the cohort of patients diagnosed with advanced renal cell carcinoma (RCC) and who had not undergone previous systemic therapy, the combination of lenvatinib and pembrolizumab demonstrated a significantly extended PFS compared to sunitinib (median, 23.9 months versus 9.2 months) [160]. Similarly, in patients with advanced endometrial cancer after failure of prior platinum-based chemotherapy, lenvatinib combined with pembrolizumab significantly prolonged PFS (6.6 months vs 3.8 months) and OS compared with chemotherapy (17.4 months vs 12.0 months) [161]. According to the research findings, numerous extensive phase III clinical trials are currently investigating the effectiveness of the lenvatinib and pembrolizumab combination [162,163,164,165].
Surufatinib
Surufatinib, a multi-targeted tyrosine kinase inhibitor, primarily acts on VEGFR1-3 and FGFR1 [166]. In the cohort of individuals diagnosed with advanced neuroendocrine tumors, the duration of PFS was notably extended in those who received surufatinib treatment (9.2 months compared to 3.8 months). Furthermore, among patients with progressive well-differentiated extra pancreatic NETs, surufatinib demonstrated a favorable benefit-risk ratio [167].
Nintedanib
Nintedanib, a derivative of indolinone, has demonstrated the ability to effectively inhibit the kinase activity of VEGFR, PDGFR, and FGFR in the assay [168]. This compound has been identified as a specific therapeutic agent for the treatment of pulmonary fibrosis and has shown promising results in preclinical studies by effectively suppressing the growth of diverse tumor types [169,170,171]. According to the findings of a phase III clinical trial, the combination of nintedanib and docetaxel has demonstrated efficacy as a second-line treatment for patients with advanced NSCLC, particularly those with adenocarcinoma, who have previously undergone platinum-based chemotherapy. Following a median follow-up period of 31.7 months, the median OS was determined to be 12.6 months for the nintedanib plus docetaxel group, compared to 10.3 months for the placebo plus docetaxel group [172]. Nevertheless, despite its high tolerability, nintedanib did not yield a substantial advantage in individuals with refractory colorectal cancer following the ineffectiveness of conventional treatment [173]. Similarly, nintedanib did not confer a significant benefit as a maintenance therapy in patients with malignant pleural mesothelioma who had previously received pemetrexed plus cisplatin [174].
Anlotinib
Anlotinib is an innovative orally administered tyrosine kinase inhibitor that selectively targets VEGFR, FGFR, PDGFR, and c-kit [175]. In a phase III randomized clinical trial in a Chinese cohort of patients with advanced NSCLC, the administration of anlotinib demonstrated a notable extension in both OS (9.6 months vs. 6.3 months) and PFS (5.4 months vs. 1.4 months) [176]. In another phase II clinical trial, a significant proportion of patients (56.9%) diagnosed with unresectable locally advanced or metastatic medullary thyroid cancer and subjected to anlotinib treatment exhibited a partial response. Furthermore, the PFS rate after 48 weeks of treatment was reported to be 85.5% [177]. A separate study was conducted to assess the effectiveness and safety of anlotinib as a primary treatment for metastatic renal cell carcinoma. The findings indicated that anlotinib exhibited comparable efficacy to sunitinib, as evidenced by similar median PFS durations (17.5 months vs. 16.6 months), median OS durations (30.9 months vs. 30.5 months), and ORR (30.3% vs. 27.9%). Furthermore, anlotinib demonstrated superior safety profiles when compared to sunitinib [178]. In the third-line or subsequent treatment of patients with small cell carcinoma, anlotinib showed better PFS and OS compared with placebo, while maintaining a favorable safety profile [179]. In patients with advanced or metastatic HCC who were previously treated with a tyrosine kinase inhibitor, the 12-week PFS rate and median TTP were 72.5% and 4.6 months, respectively, showing favorable efficacy [180].
Lucitanib
Lucitanib, a small molecule inhibitor targeting VEGFR1-3, PDGFRα/β, and FGFR1-3 tyrosine kinases, exhibited notable suppression of tumor growth across diverse xenograft models owing to its robust angiogenesis inhibition [181]. When used in conjunction with fulvestrant, Lucitanib effectively impeded the proliferation of ER+/FGFR1-amplified cells and patient-derived xenografts (PDX) [182]. The findings of a phase II study revealed that among patients diagnosed with HR+/HER2(−) metastatic breast cancer and who had undergone no more than one prior chemotherapy treatment, the group with FGFR1 amplification (consisting of 32 individuals) exhibited an ORR of 19% when treated with Lucitanib [183].
Derazantinib
Derazantinib, a novel multi-kinase inhibitor, shows potent activity against FGFR-addicted cell lines and tumors [184], and in models of GC driven by FGFR, derazantinib exhibited greater efficacy compared to paclitaxel [185]. In a phase I/II clinical trial, derazantinib exhibited encouraging anti-tumor efficacy and tolerable safety profiles among patients diagnosed with advanced, unresectable ICC harboring FGFR2 fusion, resulting in a notable response rate of 20.7% and a disease control rate of 82.8% [186]. Preliminary data analysis from a comparable study additionally indicates that the administration of derazantinib yields favorable clinical outcomes among patients with advanced ICC harboring FGFR2 aberrations [187]. Regrettably, despite demonstrating certain efficacy in select patients with mUC and FGFR1-3 gene alterations, the observed ORR and PFS did not meet the required benchmarks to substantiate the continuation of derazantinib as a standalone therapeutic approach for this indication [188].
Tinengotinib
Tinengotinib is a multi-kinase inhibitor that strongly inhibits Aurora A/B, FGFR1/2/3 and VEGFRs, as demonstrated in kinase assays. In both in vitro and in vivo studies, exposure to tinengotinib specifically suppressed the proliferation of all subtypes of triple-negative breast cancer while preserving the integrity of luminal breast cancer cells [189]. Qualified patients with advanced/metastatic CCA who had exhausted the standard treatment options received tinengotinib 10 mg once daily (QD). In a cohort with acquired resistance to FGFR inhibitors, 2 out of 6 patients (33%) achieved PR with tumor reduction of 34% and 54%, respectively. The overall disease control rate (DCR, including CR or PR + stable disease (SD)) for FGFR2 fusion/rearrangement patients was 90% (9/10) [190].
Monoclonal antibody
Vofatamab
MFGR1877A is a human IgG1 monoclonal antibody (mAb) that can bind to FGFR3 [191]. In the mUC population with prior 1st-line chemotherapy failure or relapse, the combination strategy of vofatamab combined with docetaxel resulted in a < 40% incidence of disease progression in the patient population; when combined with pembrolizumab, the ORR was 30%, and both treatment strategies were well tolerated [192, 193].
Bemarituzumab
Bemarituzumab, a humanized immunoglobulin G1 mAb, specifically targets FGFR2b overexpression in certain tumors. It exhibits a dual mechanism of action, involving the inhibition of FGFR2b signaling and the enhancement of antibody-dependent cell-mediated cytotoxicity (ADCC) [194]. In the phase I trial (FIGHT), patients with HER2-negative, FGFR2b-selected gastric or gastro-esophageal junction adenocarcinoma demonstrated promising clinical efficacy following combination therapy with Bemarituzumab and mFOLFOX6 [195]. After 24 months of follow-up, patients with FGFR2b-overexpressing gastric or gastro-esophageal junction adenocarcinoma treated with bemarituzumab + mFOLFOX6 continued to exhibit clinically meaningful outcomes compared to those treated with placebo + mFOLFOX6 (Median OS: 19.2 months vs. 13.5 months) [196]. Related phase III clinical studies are currently underway [197].
Drug combination therapies
The utilization of multiple anticancer agents in oncology, known as combination drugs, is a strategy aimed at enhancing therapeutic efficacy. By targeting distinct pathways in cancer cells, these combinations can prevent drug resistance, improve effectiveness, and potentially reduce adverse effects. In the application of FGFR inhibitors, there also seems to be surprising prospects for the use of combination drugs.
Combination with chemotherapy drugs
Targeted agents are frequently administered in conjunction with chemotherapy in clinical settings to optimize therapeutic efficacy by selectively targeting molecular pathways while simultaneously utilizing the broad cytotoxic effects of chemotherapeutic agents. A prime illustration of this approach is the administration of trastuzumab in combination with docetaxel for HER2-positive breast cancer [198], which leverages complementary mechanisms of action to attain superior therapeutic outcomes.
The incorporation of anlotinib into the standard etoposide/platinum chemotherapy regimen demonstrates promising PFS and OS outcomes in individuals with previously untreated extensive-stage small cell lung cancer [199]. Its combination with capecitabine and oxaliplatin also showed considerable ORR, DCR, PFS and DOR in the first-line treatment of metastatic colorectal cancer [200]. Presently, an ongoing clinical trial (KY20192111-F-1) is being conducted to evaluate the efficacy of anlotinib in conjunction with the SOX regimen for the treatment of stage IV GC [201]. Nevertheless, when combined with diverse chemotherapeutic agents, the outcomes yielded synergistic, additive, and antagonistic effects, respectively. This implies that the combination approach may hold promise for the treatment of these tumors, but additional research is necessary [202, 203].
Combination with other targeted therapies
The concomitant use of FGFR inhibitors with other targeted therapies has the potential to augment efficacy by addressing tumor heterogeneity and multiple aberrant signaling pathways concurrently [204]. Such combinations may yield synergistic effects and overcome resistance mechanisms commonly encountered with monotherapy, owing to their ability to target diverse pathways. For example, FGFR inhibitors have been used in combination with VEGF/VEGFR inhibitors to combat tumors by a mechanism that inhibits a key process in tumor growth and metastasis: angiogenesis [205]. Other than that, FGFR inhibitors are also used with poly-ADP ribose polymerase (PARP) inhibitors in patients with refractory solid tumors, and preliminary results show that this regimen has an acceptable safety profile [206]. While these combinations have shown some promise, it is important to note that their effectiveness and safety will vary depending on the individual patient's condition and other factors.
Combination with ICB therapies
Preclinical studies have shown that FGF/FGFR signaling is involved in the regulation of the tumor microenvironment (TME), including immune cells, angiogenesis, and epithelial-mesenchymal transition (EMT) [207]. This makes the application of ICB in combination with FGFR tyrosine kinase inhibitors possible [208,209,210].
FGFR-TKI targets FGFR4 to increase the proteasomal degradation of PD-L1, inhibits STAT5 phosphorylation to prevent Treg development, and restores the sensitivity of HCC cells to T cell death [211]. Of note, in a trial of erdafitinib, patients with previous ICB therapy had a higher response rate compared with the entire cohort (59% vs. 40%) [212]. The efficacy of combination treatments has also been supported in real-world studies, in breast cancer patients, 60% of the combination therapy group achieved complete remission, accompanied by a significant increase in CD4 + and CD8 + T cell infiltration [213]. Sintilimab combined with anlotinib is effective and safe as second-line treatment for patients with advanced cervical cancer and endometrial cancer who failed previous chemotherapy [214, 215]. The combination of Lenvatinib with anti-PD-1 therapy also leads to the formation of long-term immune memory, while acting synergistically to regulate the normalization of TME and tumor vasculature and to enhance the cytotoxic effects of T cells, improving the efficacy against HCC [216]. Many clinical studies underway based on positive results from preclinical studies. Even though conclusive results are yet to be disclosed in most studies, several clinical studies have provided preliminary validation of the safety of this combination therapy [217,218,219,220]. However, data from some clinical studies have shown that FGFR-ICB combined therapy did not achieve significant benefits [221]. FGFR3 has been found in previous studies to be associated with a diminished response to ICBs, however, results from two phase II clinical trials Checkmate 275 and IMVigor 210 showed no statistically significant difference in ICB treatment response rate or OS between patients with or without FGFR3 mutations [222]. The researchers posit that this result could potentially be influenced by a confluence of factors, including modifications to the tumor microenvironment, genetic variations among patients, changes in drug resistance, variations in tumor staging, and specific biomarkers [6, 40, 210, 223].
In summary, while preclinical investigations have demonstrated the potential of FGFR in combination with other therapeutic modalities to elicit antitumor effects, this observation has been validated only in a limited number of malignancies in clinical settings, with no direct outcomes observed in other tumor types.
Other potential treatments
Antibody drug conjugates
Antibody–drug conjugates (ADCs) represent an innovative class of medications that combine monoclonal antibodies, toxin payloads, and linkers to achieve targeted cancer therapy. ADC drugs leverage the high specificity of monoclonal antibodies to target overexpressed antigens on tumor cells. The linker connects the antibody to the toxin, which is then delivered into the tumor cells under the guidance of the antibody. AMB302/GR1017, an ADCC derived from FGFR3-targeting antibody AimedBio conjugated with topoisomerase-1 inhibitor TopoIx, exhibits potent antitumor efficacy in glioblastoma and bladder cancer models with FGFR3 amplification or FGFR3-TACC3 fusion, both in vitro and in vivo [224]. It notably demonstrates significant antitumor activity against glioblastoma patient-derived cells (PDCs) in an FGFR3-TACC3-dependent manner, surpassing the performance of similar antibodies and Deruxtecan as payload for ADC drugs. Moreover, the compound extends the survival time of the FGFR3-TACC3 fusion glioblastoma orthotopic PDX model by 200% and induces complete tumor regression in the FGFR3-TACC3 fusion RT112 BC model. Additionally, AMB302/GR1017 shows excellent tolerability in rodent models, even at therapeutic doses up to 200 mg/kg, reinforcing its potential as a well-tolerated and effective treatment option for these malignancies.
FGF traps
A heterogeneous group of molecules known as FGF traps possess the ability to function as FGFR decoys. These molecules bind to FGFs in the extracellular environment, effectively preventing the growth factors from interacting with target cells. An example is FP-1039, an FGF ligand capture agent incorporating the extracellular domain of the FGFR1-IIIc splice isoform. A recent phase I study involving patients with metastatic or locally advanced solid tumors, treated with FP-1039, revealed the most favorable response to be SD (41.7%) among the 39 patients studied [225]. Notably, no discernible relationship between abnormalities in the FGF pathway and the observed antitumor effects was identified.
Radionuclide-conjugated drugs
Radionuclide-conjugated drugs are a category of therapeutic agents that merge radionuclides with distinct drug molecules. The fundamental concept underlying nuclide-conjugated drugs is to guide radionuclides to specific treatment sites, where radiation is emitted through radioactive decay, inducing a lethal impact on targeted cells. [225Ac]-FPI-1966 represents a therapeutic α-emitting drug with FGFR-targeting properties. This compound merges the FGFR3 monoclonal antibody vofatamab with the α-emitting radionuclide Actinium-225. Currently, it is the subject of investigation within a Phase I/II study focused on evaluating FGFR3 expression (NCT05363605).
FGFR-TKI resistance in tumors
FGFR-TKI resistance in tumors refers to the phenomenon where tumors become unresponsive to treatment with tyrosine kinase inhibitors targeting the FGFR pathway. This resistance can arise through various mechanisms, such as genetic mutations in the FGFR gene, activation of alternative signaling pathways, or adaptive cellular responses [226]. Such resistance complicates treatment and necessitates the development of novel therapeutic strategies to overcome or circumvent it, ensuring continued efficacy of anti-cancer therapies in patients with FGFR-driven tumors.
FGFR gatekeeper mutations
Resistance to FGFR inhibitors mainly originates from mutations in the targeted kinase, especially mutations in the “gatekeeper” residues [6, 227,228,229,230,231]. The access control residues are in the hinge region connected to the C-terminal end of the kinase structural domain and have an important function in modulating the availability of hydrophobic pockets [232, 233]. Any mutation in these residues may stabilize the active kinase conformation by detaching the molecular brake or enhancing the hydrophobic spine or mimicking the action of A-ring tyrosine phosphorylation, thereby enhancing resistance to inhibitors [234]. The occurrence of a gatekeeper mutation has the potential to disrupt the crucial hydrogen bonds necessary for establishing strong affinity interactions or induce three-dimensional clashes that impede the binding of inhibitors [235,236,237,238]. All FGFR kinases have a threonine gatekeeper residue at the pathway control position, and mutations in this residue can lead to resistance to a generation of FGFR-TKIs [230]. Considering this alteration, LY2874455 undergoes a conformational change resembling that of a chair, resulting in the folding of the hydrophobic residue Leu619 in FGFR4V550L/M [239]. This conformational property results in an increased spatial distance between the mutant amino acid residue and the compound, effectively bypassing resistance by avoiding collision with the mutant gatekeeper residue. The covalent inhibitor TAS-120 possesses a flexible connector within its core structure. This feature allows the dimethoxybenzene ring of TAS-120 to exhibit significant rotational flexibility [240]. Consequently, the inhibitor can adapt to the hydrophobic pocket of FGFR, thereby overcoming the gatekeeper mutation. In addition, several novel 3-aminopyrazole compounds take the approach of covalently modifying p -cyclic cysteine residues in response to gatekeeper mutations in FGFR2 and FGFR3 [230, 241].
Alternative activation associated with the FGFR pathway
Cancer cells show remarkable flexibility in controlling their proliferation in terms of RTKs, often exhibiting crosstalk between signals with alternative activation [242, 243]. This convergence can result in the activation of additional downstream signals through the process of cross-phosphorylation of RTKs. Preclinical research suggests that in cases of acquired resistance to infigratinib, the AKT pathway experiences positive regulation [244, 245]. In addition, the interaction between FGFR and IGFR prevents complete inhibition of MAPK signaling by inhibiting FGFR, which is associated with intrinsic and acquired resistance to FGFR inhibitors [246]. In FGFR amplification tumors, co-activated RTKs ligands, including EGFR family proteins and MET, serve as alternative RTKs. These alternative RTKs are associated with FGFR2 amplification and confer resistance to FGFR inhibitors in FGFR3 translocation cells [243, 247]. The activation of AKT and ERK signals by MET was facilitated by GAB1, and the intercommunication of signals mediated by GAB1 resulted in the development of resistance to FGFR inhibitors in the cell line [246]. The concurrent administration of MET inhibitors and FGFR inhibitors has demonstrated a potent ability to effectively reverse this phenomenon [243]. The activation of EGFR serves as the mechanism by which PD173074 treatment is evaded. The co-administration of Gefitinib and PD173074 demonstrates the potential to surmount drug resistance in urothelial cancer cases characterized by FGFR3 molecular alterations [247]. Src, HER2 and EphB3 pathways serve as alternative resistance mechanisms in uroepithelial cancer cells and gastric cancer, respectively, and can be overcome by combining FGFR-TKIs with corresponding inhibitors [226, 248,249,250]. The PROTAC drug LC-MB12 developed by FGFR has the potential to interfere with FGFR by inducing asymmetric dephosphorylation dimerization, thereby disrupting the contact points of downstream signaling molecules, including the FRS2/SHP2/Gab1/Grab2 complex [251, 252]. To mitigate the scaffolding effect of FGFR2 and impede the advancement of the compensation feedback pathway.
Lysosomal isolation
Lysosomes comprise of lipoprotein membranes, serving as digestive vesicles. They have acidic hydrolases that eliminate excess macromolecules like proteins, nucleic acids, lipids and polysaccharides from cells [253, 254]. The lysosomal environment is acidic, hence weakly basic FGFR kinase inhibitors become protonated and cannot recross the lysosomal membrane, thus being trapped in lysosomal vesicles [254]. This process necessitates the cooperation of the ABC transporter and is controlled by affirmative feedback from TFEB (TFEB is a transcription factor involved in the regulation of lysosomal and autophagic systems in cells)-induced lysosome production [255,256,257,258]. The intracellular gathering and distribution of nintedanib and PD173074 are demonstrated through three-dimensional fluorescence spectroscopy and other techniques that affirm their resistance mediated by lysosomes [259, 260]. Additionally, autophagy is closely linked to lysosome mediated TKI resistance [261]. Activation of the mTOR signaling pathway triggers autophagy to support survival against drug treatment in FGFR-TKI-resistant GC cell lines, and TAK1 aggravates this process. A synergistic therapeutic approach involving NG25 (a TAK1 inhibitor) and AZD4547 can reverse this phenomenon. Modifying the structure of the TKI, interfering with the lysosome's typical functioning and structure, and obstructing the ABC transporter and kinase are deemed to be efficacious techniques in conquering lysosome-mediated resistance [262,263,264,265].
The side effects of FGFR-inhibitors and the mechanism
With any therapeutic agent, there must be a trade-off between efficacy and toxicity to optimize clinical benefit. Common targeted toxicities associated with FGFR inhibitors include hyperphosphatemia and nail, skin, and ocular toxicity [128, 212]. However, there is still an unmet need for practical insights into AE management in the real world (Fig. 5).

Common side effects of FGFR inhibitor. The side effects of FGFR inhibitors can be seen in many systems throughout the body, among which hyperphosphatemia is the most common. (CSC: Central plasmacytoid choroidal retinopathy; PPES: palmar-plantar erythrodysesthesia syndrome)
Hyperphosphatemia is one of the most reported AEs in clinical trials of FGFR inhibitors, with hyperphosphatemia reported in no less than 60% of patients in the BLC2001, FIGHT-202, and FOENIX-CCA2 trials [109, 117, 128]. FGFR1 is normally involved in the regulation of phosphorus metabolism in vivo, and when FGFR1 is inhibited, this regulation is disturbed, resulting in elevated serum phosphorus levels, which is thought to be a targeted FGFR inhibitor-like effect [266]. In most cases, hyperphosphatemia can be alleviated with appropriate management. In trials such as FIGHT-202 and FOENIX-CCA2, no more than 2% of patients were discontinued or had a change in dose after being placed on a low-phosphorus diet and given phosphate binders and diuretics [109, 117, 128].
Similarly, a 30–59% incidence of nail toxicity and an 11–21% incidence of palmar-plantar erythrodysesthesia syndrome (PPES) were reported in the BLC2001, FIGHT-202, and FOENIX-CCA2 trials [109, 117, 128]. The pathologic mechanism behind this adverse event has not been elucidated and may be the induction of hair follicle homeostasis dysregulation and epidermal proliferation and/or differentiation by inhibition of FGFR in keratin-forming cells, with concomitant down-regulation of tight junction gene expression [267]. Oral antibiotics against infection, partial or total nail removal, topical urea and cortisol creams or interruption/discontinuation of treatment are considered mandatory in some cases [268].
In the BLC2001 and FIGHT-202 studies, dry eye occurred in 19–25% of patients and central plasma retinopathy in 4–21% [109, 117]. In contrast, ocular side effects occurred less frequently in the FOENIX-CCA2 trial, with dry eye occurring in approximately 17% of patients [92]. Retinopathy is commonly associated with MAPK pathway inhibitors, and it so happens that FGFR inhibitors can interfere with downstream MAPK-mediated signaling, which is thought to be a potential mechanism for FGFR inhibitor-associated retinopathy [269, 270]. Therefore, it is recommended that a comprehensive ophthalmologic examination be performed prior to the initiation of FGFR inhibitor therapy and during the first few months of treatment, and that dose adjustments be made to control the associated targeting toxicity to ensure adherence.
Treatment-related side effects of FGFR inhibitors are not uncommon. Therefore, active monitoring during treatment is possible to minimize dose reductions and discontinuations and may be beneficial to patients' quality of life and outcomes.
Conclusions and prospects
FGFR abnormalities are often seen in several forms of cancer, such as bladder, cholangiocarcinoma, gastric cancer, and lung cancer. Presently, both the NCCN and ESMO recommendations endorse the use of FISH, RT-PCR, and NGS techniques to identify this anomaly in cholangiocarcinoma and bladder cancer. The FDA has granted approval for Infigratinib, Pemigatinib, and Futibatinib in the treatment of cholangiocarcinoma with FGFR2 fusion, as well as Erdafitinib in the treatment of bladder cancer with FGFR3 mutation. These medications have shown good clinical effectiveness. Additionally, clinical studies are being conducted for medications such as AZD4547, HMPL-453, and RLY-4008.
Certainly, there remain some inquiries that merit contemplation and investigation. First, is it worth expanding FGFR-targeted therapy clinical indications? FGFR1 amplification in 12.5% of breast tumors makes ER + breast cancer patients resistant to CDK4/6 inhibitors and endocrine treatments, increasing their likelihood of disease recurrence [42, 271, 272]. Therefore, appropriate clinical trials could be attempted to improve the clinical prognosis of such patients. Furthermore, there is potential for improvement in the techniques used to identify the specific subset of the population that has the most favorable response to FGFR-TKI. In the FORT-1 trial, patients with FGFR1-3 mRNA overexpressing uroepithelial carcinoma treated with rogaratinib had similar benefits as those treated with chemotherapy [273]. However, in the subgroup that also had altered FGFR3 DNA levels, the ORR increased to 52% compared to 27% in the chemotherapy group [273]. Does this disparity imply that FGFR3 DNA changes serve as a more precise biomarker compared to mRNA overexpression? Alternatively, does it imply that doctors should consider both FGFR3 DNA changes and mRNA overexpression when determining whether to provide FGFR-TKI treatment to a patient? With the help of AI tools, it may be possible to identify the presence or absence of FGFR changes on HE slides, and the results showed that the area under the receiver operating curve value was 0.76 [274]. Each of these methods needs to be validated in a larger cohort. At present, it is uncertain whether patients with urothelial carcinoma with FGFR alterations can benefit from ICI, and more relevant exploration is needed. In cohort 1 of the phase III THOR trial, OS and PFS were also significantly longer in urothelial carcinoma patients treated with previous ICI with erdafitinib than in those treated with chemotherapy [275]. However, in cohort 2 of the THOR trial, OS was very similar in patients treated with erdafitinib or pembrolizumab [120]. These differences may reflect increased numbers of immunosuppressive macrophages and regulatory T cells, as well as reduced infiltration of inflammatory CAF and T cells in the tumor microenvironment of cancer patients with FGFR3 alterations [276, 277]. Currently, studies are underway to assess the safety and initial effectiveness of combining ICI with FGFR inhibitors [278]. Nevertheless, the existing combination regimens primarily rely on anti-PD-1 therapy, and the combination with other immune checkpoint inhibitors like CTLA4 lacks substantiating evidence. Finally, the emergence of drug resistance during the treatment of cancer patients is an unavoidable topic. There are many mechanisms for the development of drug resistance, among which the most common is the FGFR gatekeeper mutation [226]. In addition, the activation of some related pathways, such as FGFR2 N550H mutation, which can up-regulate the PI3K/AKT/mTOR signaling pathway, is also associated with drug resistance [279]. Several new mutation-targeting agents have emerged, but they still have some limitations [125, 190]. However, the correlation between the development of resistance to FGFR inhibitors and the microenvironment remains unclear. For patients with FGFR inhibitor resistance, whether the therapeutic effect of combined immunotherapy can be restored or improved is also a topic worth exploring.
Availability of data and materials
Not applicable.
Abbreviations
- BLCA:
-
Bladder Cancer
- BMP:
-
Bone Morphogenetic Protein
- CCA:
-
Cholangiocarcinoma
- CHOL:
-
Cholangiocarcinoma
- CKD:
-
Chronic Kidney Disease
- CR:
-
Complete Response
- CSC:
-
Central Plasmacytoid Choroidal Retinopathy
- ctDNA:
-
Circulating Tumor DNA
- DMG:
-
Diffuse Midline Gliomas
- ESCC:
-
Esophageal Squamous Cell Carcinoma
- FDA:
-
Food and Drug Administration
- FGF:
-
Fibroblast Growth Factor
- FGFR:
-
Fibroblast Growth Factor Receptor
- FGFRL1:
-
Fibroblast Growth Factor Receptor-Like 1
- FISH:
-
Fluorescence In Situ Hybridization
- GBM:
-
Glioblastoma
- GC:
-
Gastric Cancer
- HCC:
-
Hepatocellular Carcinoma
- HNSCC:
-
Head and Neck Squamous Cell Carcinoma
- HS:
-
Heparan Sulfate
- HSPG:
-
Heparan Sulfate Proteoglycans
- ICC:
-
Intrahepatic Cholangiocarcinoma
- LGG:
-
Low-grade Glioma
- MKP3:
-
Mitogen-Activated Protein Kinase Phosphatase 3
- MM:
-
Multiple Myeloma
- MNGT:
-
Mixed Neuronal-Glial Tumors
- mUC:
-
Metastatic Urothelial Carcinoma
- NGS:
-
Next-Generation Sequencing
- NMIBC:
-
Non-Muscle Invasive Bladder Cancer
- NSCLC:
-
Non-Small Cell Lung Cancer
- ORR:
-
Objective Response Rate
- OS:
-
Overall Survival
- PAAD:
-
Pancreatic Cancer
- PFS:
-
Progression-Free Survival
- PI3K/AKT:
-
Phosphoinositide 3-Kinase/Protein Kinase B
- PLCγ:
-
Phospholipase C Gamma
- PPES:
-
Palmar-Plantar Erythrodysesthesia Syndrome
- RTKs:
-
Receptor Tyrosine Kinases
- RT-PCR:
-
Reverse Transcription-Polymerase Chain Reaction
- SCLC:
-
Small Cell Lung Carcinoma
- SEF:
-
Similar Expression to FGF
- Spry:
-
Sprouty
- SqCC:
-
Squamous Cell Lung Cancer
- STATs:
-
Signal Transducers and Activators of Transcription
- TKI:
-
Tyrosine Kinase Inhibitor
- UCEC:
-
Uterine Endometrial Carcinoma
- Wnt:
-
Wingless/Integrated
- XFLRT3:
-
Xylosyl transferase 3
References
Xue WJ, Li MT, Chen L, Sun LP, Li YY. Recent developments and advances of FGFR as a potential target in cancer. Future Med Chem. 2018;10(17):2109–26.
Regeenes R, Silva PN, Chang HH, Arany EJ, Shukalyuk AI, Audet J, et al. Fibroblast growth factor receptor 5 (FGFR5) is a co-receptor for FGFR1 that is up-regulated in beta-cells by cytokine-induced inflammation. J Biol Chem. 2018;293(44):17218–28.
Trueb B. Biology of FGFRL1, the fifth fibroblast growth factor receptor. Cell Mol Life Sci. 2011;68(6):951–64.
Wiedemann M, Trueb B. Characterization of a novel protein (FGFRL1) from human cartilage related to FGF receptors. Genomics. 2000;69(2):275–9.
Mikhaylenko DS, Alekseev BY, Zaletaev DV, Goncharova RI, Nemtsova MV. Structural alterations in human fibroblast growth factor receptors in carcinogenesis. Biochemistry (Mosc). 2018;83(8):930–43.
Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer. 2017;17(5):318–32.
Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–29.
Gnatenko DA, Kopantzev EP, Sverdlov ED. Fibroblast growth factors and their effects in pancreas organogenesis. Biomed Khim. 2017;63(3):211–8.
Katoh M. FGFR inhibitors: effects on cancer cells, tumor microenvironment and whole-body homeostasis. Int J Mol Med. 2016;38(1):3–15.
Tiong KH, Mah LY, Leong CO. Functional roles of fibroblast growth factor receptors (FGFRs) signaling in human cancers. Apoptosis. 2013;18(12):1447–68.
Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem. 2011;149(2):121–30.
Itoh N, Ornitz DM. Evolution of the FGF and FGFR gene families. Trends Genet. 2004;20(11):563–9.
Kliewer SA, Mangelsdorf DJ. A dozen years of discovery: Insights into the physiology and pharmacology of FGF21. Cell Metab. 2019;29(2):246–53.
Razzaque MS. The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5(11):611–9.
Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115(6):1627–35.
Cariello M, Piglionica M, Gadaleta RM, Moschetta A. The enterokine fibroblast growth factor 15/19 in bile acid metabolism. Handb Exp Pharmacol. 2019;256:73–93.
Dieci MV, Arnedos M, Andre F, Soria JC. Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov. 2013;3(3):264–79.
Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353(2):172–87.
Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol. 2011;3(7):a004952.
Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, et al. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. 2000;6(3):743–50.
Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol. 2015;4(3):215–66.
Ong SH, Guy GR, Hadari YR, Laks S, Gotoh N, Schlessinger J, et al. FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol Cell Biol. 2000;20(3):979–89.
Hadari YR, Gotoh N, Kouhara H, Lax I, Schlessinger J. Critical role for the docking-protein FRS2α in FGF receptor-mediated signal transduction pathways. Proc Natl Acad Sci U S A. 2001;98(15):8578–83.
Belov AA, Mohammadi M. Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb Perspect Biol. 2013;5(6):a015958.
Eswarakumar VP, Monsonego OE, Pines M, Antonopoulou I, Morriss-Kay GM, Lonai P. The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development. 2002;129(16):3783–93.
Miraoui H, Marie PJ. Fibroblast growth factor receptor signaling crosstalk in skeletogenesis. Sci Signal. 2010;3(146):re9.
Minina E, Kreschel C, Naski MC, Ornitz DM, Vortkamp A. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev Cell. 2002;3(3):439–49.
Katoh M, Katoh M. Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol Ther. 2006;5(9):1059–64.
Zhu X, Lee K, Asa SL, Ezzat S. Epigenetic silencing through DNA and histone methylation of fibroblast growth factor receptor 2 in neoplastic pituitary cells. Am J Pathol. 2007;170(5):1618–28.
Sarabipour S, Hristova K. Mechanism of FGF receptor dimerization and activation. Nat Commun. 2016;7:10262.
Triantis V, Saeland E, Bijl N, Oude-Elferink RP, Jansen PL. Glycosylation of fibroblast growth factor receptor 4 is a key regulator of fibroblast growth factor 19-mediated down-regulation of cytochrome P450 7A1. Hepatology. 2010;52(2):656–66.
Kucińska M, Porębska N, Lampart A, Latko M, Knapik A, Zakrzewska M, et al. Differential regulation of fibroblast growth factor receptor 1 trafficking and function by extracellular galectins. Cell Commun Signal. 2019;17(1):65.
Porębska N, Latko M, Kucińska M, Zakrzewska M, Otlewski J, Opaliński Ł. Targeting cellular trafficking of fibroblast growth factor receptors as a strategy for selective cancer treatment. J Clin Med. 2018;8(1):7.
Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res. 2016;22(1):259–67.
Souza VC, Soares A, Maluf FC, Monteiro FSM, Gidekel R, Ardila-Salcedo J, et al. Clinicopathological characterization, FGFR alteration prevalence, and outcomes of locally advanced or metastatic urothelial cancer in Latin America (LACOG 1518). J Clin Oncol. 2022;40(16):E16549.
Zhou Z, Liu ZC, Ou QX, Wu X, Wang XN, Shao Y, et al. Targeting FGFR in non-small cell lung cancer: implications from the landscape of clinically actionable aberrations of FGFR kinases. Cancer Biol Med. 2021;18(2):490.
Yasui H, Takeno A, Hara H, Imamura H, Akamatsu H, Fujitani K, et al. Prospective analysis of the expression status of FGFR2 and HER2 in colorectal and gastric cancer populations: DS-Screen Study. Int J Colorectal Dis. 2022;37(6):1393–402.
Yi S, Gao L, Wei Z, He QY, Liu YC, Chen XS, et al. A comprehensive pan-cancer study of fibroblast growth factor receptor aberrations in Chinese cancer patients. Ann Transl Med. 2020;8(20):1290.
Loibl S, Treue D, Budczies J, Weber K, Stenzinger A, Schmitt WD, et al. Mutational diversity and therapy response in breast cancer: a sequencing analysis in the neoadjuvant GeparSepto trial. Clin Cancer Res. 2019;25(13):3986–95.
Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res. 2016;22(1):259–67.
Seo AN, Jin Y, Lee HJ, Sun PL, Kim H, Jheon S, et al. FGFR1 amplification is associated with poor prognosis and smoking in non-small-cell lung cancer. Virchows Arch. 2014;465(5):547–58.
Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70(5):2085–94.
Shoji H, Yamada Y, Okita N, Takashima A, Honma Y, Iwasa S, et al. Amplification of FGFR2 gene in patients with advanced gastric cancer receiving chemotherapy: prevalence and prognostic significance. Anticancer Res. 2015;35(9):5055–61.
Chatziandreou I, Psaraki A, Paschidis K, Lazaris AC, Saetta AA. Evidence for frequent concurrent DCUN1D1, FGFR1, BCL9 gene copy number amplification in squamous cell lung cancer. Pathol Res Pract. 2021;221:153412.
Kilgour E, Ferry D, Saggese M, Arkenau HT, Rooney C, Smith NR, et al. Exploratory biomarker analysis of a phase I study of AZD4547, an inhibitor of fibroblast growth factor receptor (FGFR), in patients with advanced solid tumors. J Clin Oncol. 2014;32(15):11010.
Williams EA, Brastianos PK, Wakimoto H, Zolal A, Filbin MG, Cahill DP, et al. A comprehensive genomic study of 390 H3F3A-mutant pediatric and adult diffuse high-grade gliomas, CNS WHO grade 4. Acta Neuropathol. 2023;146(3):515–25.
Cimmino F, Montella A, Tirelli M, Avitabile M, Lasorsa VA, Visconte F, et al. FGFR1 is a potential therapeutic target in neuroblastoma. Cancer Cell Int. 2022;22(1):174.
Engelhardt S, Behling F, Beschorner R, Eckert F, Kohlhof P, Tatagiba M, et al. Frequent FGFR1 hotspot alterations in driver-unknown low-grade glioma and mixed neuronal-glial tumors. J Cancer Res Clin Oncol. 2022;148(4):857–66.
Ishi Y, Yamaguchi S, Hatanaka KC, Okamoto M, Motegi H, Kobayashi H, et al. Association of the FGFR1 mutation with spontaneous hemorrhage in low-grade gliomas in pediatric and young adult patients. J Neurosurg. 2020;134(3):733–41.
Rivera B, Gayden T, Carrot-Zhang J, Nadaf J, Boshari T, Faury D, et al. Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors. Acta Neuropathol. 2016;131(6):847–63.
Lei JH, Lee MH, Miao K, Huang Z, Yao Z, Zhang A, et al. Activation of FGFR2 signaling suppresses BRCA1 and drives triple-negative mammary tumorigenesis that is sensitive to immunotherapy. Adv Sci (Weinh). 2021;8(21):e2100974.
Li J, Hu K, Huang J, Zhou L, Yan Y, Xu Z. A pancancer analysis of the expression landscape and clinical relevance of fibroblast growth factor receptor 2 in human cancers. Front Oncol. 2021;11:14.
Li J, Hu K, Huang J, Zhou L, Yan Y, Xu Z. Insights of fibroblast growth factor receptor 3 aberrations in pan-cancer and their roles in potential clinical treatment. Aging (Albany NY). 2021;13(12):16541–66.
Pal SK, Bajorin D, Dizman N, Hoffman-Censits J, Quinn DI, Petrylak DP, et al. Infigratinib in upper tract urothelial carcinoma versus urothelial carcinoma of the bladder and its association with comprehensive genomic profiling and/or cell-free DNA results. Cancer. 2020;126(11):2597–606.
Taylor JG, Cheuk AT, Tsang PS, Chung JY, Song YK, Desai K, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest. 2009;119(11):3395–407.
Li Z, Qin F, Li H. Chimeric RNAs and their implications in cancer. Curr Opin Genet Dev. 2018;48:36–43.
Wojtas B, Gielniewski B, Wojnicki K, Maleszewska M, Mondal SS, Nauman P, et al. Gliosarcoma is driven by alterations in PI3K/Akt, RAS/MAPK pathways and characterized by collagen gene expression signature. Cancers (Basel). 2019;11(3):284.
Cai X, Xu C, Wang W, Zhang Q, Chen Y, Fang Y, et al. Incidence of FGFR-TACC gene fusions in Chinese non-small cell lung cancer (NSCLC): a multicenter study. J Clin Oncol. 2019;37(15):e13001.
De Luca A, Abate RE, Rachiglio AM, Maiello MR, Esposito C, Schettino C, et al. FGFR fusions in cancer: From diagnostic approaches to therapeutic intervention. Int J Mol Sci. 2020;21(18):6856.
Popovici C, Zhang B, Grégoire MJ, Jonveaux P, Lafage-Pochitaloff M, Birnbaum D, et al. The t(6;8)(q27;p11) translocation in a stem cell myeloproliferative disorder fuses a novel gene, FOP, to fibroblast growth factor receptor 1. Blood. 1999;93(4):1381–9.
Ballerini P, Struski S, Cresson C, Prade N, Toujani S, Deswarte C, et al. RET fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia. 2012;26(11):2384–9.
Chong Y, Liu Y, Lu S, Cai B, Qin H, Chang CS, et al. Critical individual roles of the BCR and FGFR1 kinase domains in BCR-FGFR1-driven stem cell leukemia/lymphoma syndrome. Int J Cancer. 2020;146(8):2243–54.
Cristinziano G, Porru M, Lamberti D, Buglioni S, Rollo F, Amoreo CA, et al. FGFR2 fusion proteins drive oncogenic transformation of mouse liver organoids towards cholangiocarcinoma. J Hepatol. 2021;75(2):351–62.
Li F, Peiris MN, Donoghue DJ. Functions of FGFR2 corrupted by translocations in intrahepatic cholangiocarcinoma. Cytokine Growth Factor Rev. 2020;52:56–67.
Sia D, Losic B, Moeini A, Cabellos L, Hao K, Revill K, et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun. 2015;6:6087.
Kendre G, Marhenke S, Lorz G, Becker D, Reineke-Plaaß T, Poth T, et al. The co-mutational spectrum determines the therapeutic response in murine FGFR2 fusion-driven cholangiocarcinoma. Hepatology. 2021;74(3):1357–70.
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315–22.
Granberg KJ, Annala M, Lehtinen B, Kesseli J, Haapasalo J, Ruusuvuori P, et al. Strong FGFR3 staining is a marker for FGFR3 fusions in diffuse gliomas. Neuro Oncol. 2017;19(9):1206–16.
Stewart JP, Thompson A, Santra M, Barlogie B, Lappin TR, Shaughnessy J. Correlation of TACC3, FGFR3, MMSET and p21 expression with the t(4;14)(p16.3;q32) in multiple myeloma. Br J Haematol. 2004;126(1):72–6.
Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337(6099):1231–5.
Koutros S, Kiemeney LA, Pal Choudhury P, Milne RL, de Maturana EL, Ye Y, et al. Genome-wide association study of bladder cancer reveals new biological and translational insights. Eur Urol. 2023;84(1):127–37.
Chen J, Facchinetti F, Braye F, Yurchenko AA, Bigot L, Ponce S, et al. Single-cell DNA-seq depicts clonal evolution of multiple driver alterations in osimertinib-resistant patients. Ann Oncol. 2022;33(4):434–44.
Nakanishi Y, Akiyama N, Tsukaguchi T, Fujii T, Satoh Y, Ishii N, et al. Mechanism of oncogenic signal activation by the novel fusion kinase FGFR3-BAIAP2L1. Mol Cancer Ther. 2015;14(3):704–12.
Zhou Z, Liu Z, Ou Q, Wu X, Wang X, Shao Y, et al. Targeting FGFR in non-small cell lung cancer: implications from the landscape of clinically actionable aberrations of FGFR kinases. Cancer Biol Med. 2021;18(2):490.
Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3(6):636–47.
Luo B, Li W, Deng CH, Zheng FF, Sun XZ, Wang DH, et al. Utility of fluorescence in situ hybridization in the diagnosis of upper urinary tract urothelial carcinoma. Cancer Genet Cytogenet. 2009;189(2):93–7.
Network NCC. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®): Biliary Tract Cancers 2023.
Bustin SA, Benes V, Nolan T, Pfaffl MW. Quantitative real-time RT-PCR—a perspective. J Mol Endocrinol. 2005;34(3):597–601.
Tarnuzzer R, Macauley S, Farmerie W, Caballero S, Ghassemifar M, Anderson J, et al. Competitive RNA templates for detection and quantitation of growth factors, cytokines, extracellular matrix components and matrix metalloproteinases by RT-PCR. Biotechniques. 1996;20(4):670–4.
Powles T, Bellmunt J, Comperat E, De Santis M, Huddart R, Loriot Y, et al. Bladder cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up. Ann Oncol. 2022;33(3):244–58.
Network. NCC. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®): Bladder Cancer. 2024.
Miyake M, Sugano K, Kawashima K, Ichikawa H, Hirabayashi K, Kodama T, et al. Sensitive detection of FGFR3 mutations in bladder cancer and urine sediments by peptide nucleic acid-mediated real-time PCR clamping. Biochem Biophys Res Commun. 2007;362(4):865–71.
Arai Y, Totoki Y, Hosoda F, Shirota T, Hama N, Nakamura H, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology. 2014;59(4):1427–34.
Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–31.
Vogel A, Bridgewater J, Edeline J, Kelley R, Klümpen H, Malka D, et al. Biliary tract cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34(2):127–40.
Gasc C, Peyretaillade E, Peyret P. Sequence capture by hybridization to explore modern and ancient genomic diversity in model and nonmodel organisms. Nucleic Acids Res. 2016;44(10):4504–18.
Krook MA, Reeser JW, Ernst G, Barker H, Wilberding M, Li G, et al. Fibroblast growth factor receptors in cancer: genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br J Cancer. 2021;124(5):880–92.
Zhou Z, Liu Z, Ou Q, Wu X, Wang X, Shao Y, et al. Targeting FGFR in non-small cell lung cancer: implications from the landscape of clinically actionable aberrations of FGFR kinases. Cancer Biol Med. 2021;18(2):490.
Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol. 2017;14(9):531–48.
Soda N, Rehm BH, Sonar P, Nguyen N-T, Shiddiky MJ. Advanced liquid biopsy technologies for circulating biomarker detection. J Mater Chem B. 2019;7(43):6670–704.
Goyal L, Meric-Bernstam F, Hollebecque A, Valle JW, Morizane C, Karasic TB, et al. Futibatinib for FGFR2-rearranged intrahepatic cholangiocarcinoma. N Engl J Med. 2023;388(3):228–39.
Varghese AM, Patel J, Janjigian YY, Meng F, Selcuklu SD, Iyer G, et al. Noninvasive detection of polyclonal acquired resistance to FGFR inhibition in patients with cholangiocarcinoma harboring FGFR2 alterations. JCO Precis Oncol. 2021;5:44–50.
Guagnano V, Furet P, Spanka C, Bordas V, Le Douget M, Stamm C, et al. Discovery of 3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-1-{6- 4-(4-ethyl-piperazin-1-yl)-p henylamino -pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), A potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J Med Chem. 2011;54(20):7066–83.
Krook MA, Lenyo A, Wilberding M, Barker H, Dantuono M, Bailey KM, et al. Efficacy of FGFR inhibitors and combination therapies for acquired resistance in FGFR2-Fusion cholangiocarcinoma. Mol Cancer Ther. 2020;19(3):847–57.
Capelletti M, Dodge ME, Ercan D, Hammerman PS, Park SI, Kim J, et al. Identification of recurrent FGFR3-TACC3 fusion oncogenes from lung adenocarcinoma. Clin Cancer Res. 2014;20(24):6551–8.
Zhang KQ, Chu KV, Wu XW, Gao HL, Wang JH, Yuan YC, et al. Amplification of FRS2 and activation of FGFR/FRS2 signaling pathway in high-grade liposarcoma. Cancer Res. 2013;73(4):1298–307.
Konecny GE, Kolarova T, O’Brien NA, Winterhoff B, Yang GR, Qi JW, et al. Activity of the fibroblast growth factor receptor inhibitors dovitinib (TKI258) and NVP-BGJ398 in human endometrial cancer cells. Mol Cancer Ther. 2013;12(5):632–42.
Javle M, Lowery M, Shroff RT, Weiss KH, Springfeld C, Borad MJ, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol. 2018;36(3):276.
Javle M, Roychowdhury S, Kelley RK, Sadeghi S, Macarulla T, Weiss KH, et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol Hepatol. 2021;6(10):803–15.
Abou-Alfa GK, Borbath I, Goyal L, Lamarca A, Macarulla T, Oh DY, et al. PROOF 301: A multicenter, open-label, randomized, phase 3 trial of infigratinib versus gemcitabine plus cisplatin in patients with advanced cholangiocarcinoma with an FGFR2 gene fusion/rearrangement. J Clin Oncol. 2022;40(16):TPS4171.
Pal SK, Somford DM, Grivas P, Sridhar SS, Gupta S, Bellmunt J, et al. Targeting FGFR3 alterations with adjuvant infigratinib in invasive urothelial carcinoma: the phase III PROOF 302 trial. Future Oncol. 2022;18(21):2599–614.
Lassman AB, Sepulveda-Sanchez JM, Cloughesy TF, Gil-Gil MJ, Puduvalli VK, Raizer JJ, et al. Infigratinib in patients with recurrent gliomas and FGFR alterations: a multicenter phase II study. Clin Cancer Res. 2022;28(11):2270–7.
Hartley IR, Roszko KL, Li XB, Pozo K, Streit J, del Rivero J, et al. Infigratinib reduces fibroblast growth factor 23 (FGF23) and increases blood phosphate in tumor-induced osteomalacia. JBMR Plus. 2022;6(8):9.
Liu PCC, Koblish H, Wu LX, Bowman K, Diamond S, DiMatteo D, et al. INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models. PLoS ONE. 2020;15(4):16.
Wu LX, Zhang CL, He CH, Qian DQ, Lu L, Sun YP, et al. Discovery of pemigatinib: a potent and selective fibroblast growth factor receptor (FGFR) inhibitor. J Med Chem. 2021;64(15):10666–79.
Chiodelli P, Coltrini D, Turati M, Cerasuolo M, Maccarinelli F, Rezzola S, et al. FGFR blockade by pemigatinib treats naïve and castration resistant prostate cancer. Cancer Lett. 2022;526:217–24.
Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21(5):671–84.
Bibeau K, Feliz L, Lihou CF, Ren HB, Abou-Alfa GK. Progression-free survival in patients with cholangiocarcinoma with or without FGF/FGFR alterations: a FIGHT-202 post hoc analysis of prior systemic therapy response. JCO Precis Oncol. 2022;6:10.
Shi GM, Huang XY, Wen TF, Song TQ, Kuang M, Mou HB, et al. Pemigatinib in previously treated Chinese patients with locally advanced or metastatic cholangiocarcinoma carrying FGFR2 fusions or rearrangements: a phase II study. Cancer Med. 2023;12(4):4137–46.
Bekaii-Saab TS, Valle JW, Van Cutsem E, Rimassa L, Furuse J, Ioka T, et al. FIGHT-302: first-line pemigatinib vs gemcitabine plus cisplatin for advanced cholangiocarcinoma with FGFR2 rearrangements. Future Oncol. 2020;16(30):2385–99.
Gotlib J, Kiladjian J-J, Vannucchi A, Rambaldi A, Reiter A, Shomali W, et al. A phase 2 study of pemigatinib (FIGHT-203; INCB054828) in patients with myeloid/lymphoid neoplasms (MLNs) with fibroblast growth factor receptor 1 (FGFR1) rearrangement (MLN FGFR1). Blood. 2021;138:385.
Rodon J, Damian S, Furqan M, Garcia-Donas J, Imai H, Italiano A, et al. Abstract CT016: clinical and translational findings of pemigatinib in previously treated solid tumors with activating FGFR1-3 alterations in the FIGHT-207 study. Cancer Res. 2023;83(8):CT016-CT.
Perera TPS, Jovcheva E, Mevellec L, Vialard J, De Lange D, Verhulst T, et al. Discovery and pharmacological characterization of JNJ-42756493 (Erdafitinib), a functionally selective small-molecule FGFR family inhibitor. Mol Cancer Ther. 2017;16(6):1010–20.
Lau DK, Luk IY, Jenkins LJ, Martin A, Williams DS, Schoffer KL, et al. Rapid resistance of FGFR-driven gastric cancers to regorafenib and targeted FGFR inhibitors can be overcome by parallel Inhibition of MEK. Mol Cancer Ther. 2021;20(4):704–15.
Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381(4):338–48.
Siefker-Radtke AO, Necchi A, Park SH, Garcia-Donas J, Huddart RA, Burgess EF, et al. Efficacy and safety of erdafitinib in patients with locally advanced or metastatic urothelial carcinoma: long-term follow-up of a phase 2 study. Lancet Oncol. 2022;23(2):248–58.
Loriot Y, Matsubara N, Park SH, Huddart RA, Burgess EF, Houede N, et al. Phase 3 THOR study: results of erdafitinib (erda) versus chemotherapy (chemo) in patients (pts) with advanced or metastatic urothelial cancer (mUC) with select fibroblast growth factor receptor alterations (FGFRalt). J Clin Oncol. 2023;41(17):1.
Catto JW, Tran B, Master VA, Roupret M, Pignot G, Tubaro A, et al. Phase 2 study of the efficacy and safety of erdafitinib in patients (pts) with bacillus Calmette-Guérin (BCG)-unresponsive, high-risk non–muscle-invasive bladder cancer (HR-NMIBC) with FGFR3/2 alterations (alt) in THOR-2: Cohort 2 interim analysis results. J Clin Oncol. 2023;41(6):503.
Siefker-Radtke AO, Matsubara N, Park SH, Huddart RA, Burgess EF, Özgüroğlu M, et al. Erdafitinib versus pembrolizumab in pretreated patients with advanced or metastatic urothelial cancer with select FGFR alterations: cohort 2 of the randomized phase III THOR trial. Ann Oncol. 2024;35(1):107–17.
Pant S, Schuler M, Iyer G, Witt O, Doi T, Qin S, et al. Erdafitinib in patients with advanced solid tumours with FGFR alterations (RAGNAR): an international, single-arm, phase 2 study. Lancet Oncol. 2023;24(8):925–35.
Vilaseca A, Guerrero F, Zainfeld D, Shore ND, Rodriguez Faba O, Meijer RP, et al. Safety and efficacy of the erdafitinib (erda) intravesical delivery system, TAR-210, in patients (pts) with non-muscle-invasive bladder cancer (NMIBC) or muscle-invasive bladder cancer (MIBC) harboring select FGFR mutations or fusions: Phase 1 first-in-human study. J Clin Oncol. 2023;41(6):TPS583.
Lee A, Chou AJ, Williams PM, Roy-Chowdhuri S, Patton DR, Coffey BD, et al. Erdafitinb in patients with FGFR-altered tumors: Results from the NCI-COG pediatric MATCH trial arm B (APEC1621B). J Clin Oncol. 2023;41(16):10007.
Liow E, Howard N, Jung CH, Pope B, Campbell BK, Nguyen A, et al. Phase 2 Study of neoadjuvant FGFR inhibition and androgen deprivation therapy prior to prostatectomy. Clin Genitourin Cancer. 2022;20(5):452–8.
Goyal L, Shi L, Liu LY, de la Cruz FF, Lennerz JK, Raghavan S, et al. TAS-120 overcomes resistance to ATP-compartitive FGFR inhibitors in patients with FGFR2 fusion-positive intrahepatic cholanjiocarcinoma. Cancer Discov. 2019;9(8):1064–79.
Sootome H, Fujita H, Ito K, Ochiiwa H, Fujioka Y, Ito K, et al. Futibatinib is a novel irreversible FGFR 1–4 inhibitor that shows selective antitumor activity against FGFR-deregulated tumors. Cancer Res. 2020;80(22):4986–97.
Sootome H, Fujioka Y, Miura A, Fujita H, Hirai H, Utsugi T. TAS-120, an irreversible FGFR inhibitor, was effective in tumors harboring FGFR mutations, refractory or resistant to ATP competitive inhibitors. Mol Cancer Ther. 2013;12(11):1.
Goyal L, Meric-Bernstam F, Hollebecque A, Valle JW, Morizane C, Karasic TB, et al. Futibatinib for FGFR2-rearranged intrahepatic cholangiocarcinoma. N Engl J Med. 2023;388(3):228–39.
Borad MJ, Bridgewater JA, Morizane C, Shroff RT, Oh DY, Moehler MH, et al. A phase III study of futibatinib (TAS-120) versus gemcitabine-cisplatin (gem-cis) chemotherapy as first-line (1L) treatment for patients (pts) with advanced (adv) cholangiocarcinoma (CCA) harboring fibroblast growth factor receptor 2 (FGFR2) gene rearrangements (FOENIX-CCA3). J Clin Oncol. 2020;38(4):2.
Satoh T, Barthélémy P, Nogova L, Honda K, Iwasa S, Lee K, et al. Phase 2 study of futibatinib in patients with specific FGFR aberrations: activity in patients with gastric or gastroesophageal junction cancer harboring FGFR2 amplification. Ann Oncol. 2023;34:S165–6.
Cimmino F, Montella A, Tirelli M, Avitabile M, Lasorsa VA, Visconte F, et al. FGFR1 is a potential therapeutic target in neuroblastoma. Cancer Cell Int. 2022;22(1):174.
Aggarwal C, Redman MW, Lara PN, Borghaei H, Hoffman P, Bradley JD, et al. SWOG S1400D (NCT02965378), a Phase II study of the fibroblast growth factor receptor inhibitor AZD4547 in previously treated patients with fibroblast growth factor pathway-activated stage IV squamous cell lung cancer (Lung-MAP Substudy). J Thorac Oncol. 2019;14(10):1847–52.
Chae YK, Hong F, Vaklavas C, Cheng HH, Hammerman P, Mitchell EP, et al. Phase II study of AZD4547 in patients with tumors harboring aberrations in the FGFR pathway: results from the NCI-MATCH Trial (EAY131) subprotocol W. J Clin Oncol. 2020;38(21):2407–17.
Van Cutsem E, Bang YJ, Mansoor W, Petty RD, Chao Y, Cunningham D, et al. A randomized, open-label study of the efficacy and safety of AZD4547 monotherapy versus paclitaxel for the treatment of advanced gastric adenocarcinoma with FGFR2 polysomy or gene amplification. Ann Oncol. 2017;28(6):1316–24.
Coombes RC, Badman PD, Lozano-Kuehne JP, Liu X, Macpherson IR, Zubairi I, et al. Results of the phase IIa RADICAL trial of the FGFR inhibitor AZD4547 in endocrine resistant breast cancer. Nat Commun. 2022;13(1):3246.
Lam WS, Creaney J, Chen FK, Chin WL, Muruganandan S, Arunachalam S, et al. A phase II trial of single oral FGF inhibitor, AZD4547, as second or third line therapy in malignant pleural mesothelioma. Lung Cancer. 2020;140:87–92.
Hu J, Ni J, Jiao L, Zhou J, Fan S, Tang R, et al. HMPL-453, a highly selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays potent activity in FGFR-altered tumor models. Cancer Res. 2023;83(7):6321.
Xu J, Xiong J, Gu S, Niu Z, Yin F, Sun B, et al. A phase 2 study of HMPL-453, a selective FGFR tyrosine kinase inhibitor (TKI), in patients with previously treated advanced cholangiocarcinoma containing FGFR2 fusions. American Society of Clinical Oncology; 2023.
Subbiah V, Sahai V, Maglic D, Bruderek K, Toure BB, Zhao S, et al. RLY-4008, the first highly selective FGFR2 inhibitor with activity across FGFR2 alterations and resistance mutations. Cancer Discov. 2023;13(9):2012–31.
Casaletto J, Maglic D, Toure BB, Taylor A, Schoenherr H, Hudson B, et al. RLY-4008, a novel precision therapy for FGFR2-driven cancers designed to potently and selectively inhibit FGFR2 and FGFR2 resistance mutations. Cancer Res. 2021;81(13):2.
Hollebecque A, Borad M, Goyal L, Schram A, Park J, Cassier P, et al. LBA12 Efficacy of RLY-4008, a highly selective FGFR2 inhibitor in patients (pts) with an FGFR2-fusion or rearrangement (f/r), FGFR inhibitor (FGFRi)-naïve cholangiocarcinoma (CCA): ReFocus trial. Ann Oncol. 2022;33:S1381.
Grunewald S, Politz O, Bender S, Heroult M, Lustig K, Thuss U, et al. Rogaratinib: a potent and selective pan-FGFR inhibitor with broad antitumor activity in FGFR-overexpressing preclinical cancer models. Int J Cancer. 2019;145(5):1346–57.
Schuler M, Cho BC, Sayehli CM, Navarro A, Soo RA, Richly H, et al. Rogaratinib in patients with advanced cancers selected by FGFR mRNA expression: a phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2019;20(10):1454–66.
Sternberg CN, Petrylak DP, Bellmunt J, Nishiyama H, Necchi A, Gurney H, et al. FORT-1: Phase II/III study of rogaratinib versus chemotherapy in patients with locally advanced or metastatic urothelial carcinoma selected based on FGFR1/3 mRNA expression. J Clin Oncol. 2023;41(3):629–39.
Addeo A, Rothschild SI, Holer L, Schneider M, Waibel C, Haefliger S, et al. Fibroblast growth factor receptor (FGFR) inhibitor rogaratinib in patients with advanced pretreated squamous-cell non-small cell lung cancer over-expressing FGFR mRNA: the SAKK 19/18 phase II study. Lung Cancer. 2022;172:154–9.
Weiss A, Adler F, Buhles A, Stamm C, Fairhurst RA, Kiffe M, et al. FGF401, a first-in-class highly selective and potent FGFR4 inhibitor for the treatment of FGF19-driven hepatocellular cancer. Mol Cancer Ther. 2019;18(12):2194–206.
Chan TH, Schuler M, Kang YK, Yen CJ, Edeline J, Choo SP, et al. A first-in-human phase 1/2 study of FGF401 and combination of FGF401 with spartalizumab in patients with hepatocellular carcinoma or biomarker-selected solid tumors. J Exp Clin Cancer Res. 2022;41(1):19.
Miyano SW, Yamamoto Y, Kodama K, Miyajima Y, Mikamoto M, Nakagawa T, et al. E7090, a novel selective inhibitor of fibroblast growth factor receptors, displays potent antitumor activity and prolongs survival in preclinical models. Mol Cancer Ther. 2016;15(11):2630–9.
Koyama T, Shimizu T, Iwasa S, Fujiwara Y, Kondo S, Kitano S, et al. First-in-human phase I study of E7090, a novel selective fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. Cancer Sci. 2020;111(2):571–9.
Morizane C, Ueno M, Ioka T, Tajika M, Ikeda M, Yamaguchi K, et al. Expansion part of phase I study of E7090 in patients with cholangiocarcinoma harboring FGFR2 gene fusion and with gastric cancer harboring FGFR2 gene amplification or FGFR2 protein high expression. J Clin Oncol. 2020;38(4):538.
Furuse J, Jiang B, Kuwahara T, Satoh T, Ma X, Yan S, et al. Pivotal single-arm, phase 2 trial of tasurgratinib for patients with fibroblast growth factor receptor (FGFR)-2 gene fusion-positive cholangiocarcinoma (CCA). J Clin Oncol. 2024;42(3):471.
Guo Y, Yuan CW, Ying JE, Zhu X, Luan GD, Zhang B, et al. Phase I result of ICP-192 (gunagratinib), a highly selective irreversible FGFR inhibitor, in patients with advanced solid tumors harboring FGFR pathway alterations. J Clin Oncol. 2021;39(15):2.
Guo Y, Yuan C, Ding W, Gao Y, Zhu X, Ying J, et al. Gunagratinib, a highly selective irreversible FGFR inhibitor, in patients with previously treated locally advanced or metastatic cholangiocarcinoma harboring FGFR pathway alterations: a phase IIa dose-expansion study. J Clin Oncol. 2023;41(4):572.
Yamamoto Y, Matsui J, Matsushima T, Obaishi H, Miyazaki K, Nakamura K, et al. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc Cell. 2014;6:18.
Kudo M. Lenvatinib may drastically change the treatment landscape of hepatocellular carcinoma. Liver Cancer. 2018;7(1):1–19.
Tohyama O, Matsui J, Kodama K, Hata-Sugi N, Kimura T, Okamoto K, et al. Antitumor activity of lenvatinib (e7080): an angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J Thyroid Res. 2014;2014:638747.
Spahn S, Kleinhenz F, Shevchenko E, Stahl A, Rasen Y, Geisler C, et al. The molecular interaction pattern of lenvatinib enables inhibition of wild-type or kinase-mutated FGFR2-driven cholangiocarcinoma. Nat Commun. 2024;15(1):1287.
Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–73.
Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med. 2015;372(7):621–30.
Motzer R, Alekseev B, Rha SY, Porta C, Eto M, Powles T, et al. Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. N Engl J Med. 2021;384(14):1289–300.
Makker V, Colombo N, Herraez AC, Santin A, Colomba E, Miller DS, et al. Lenvatinib plus pembrolizumab for advanced endometrial cancer. N Engl J Med. 2022;386(5):437–48.
Sun JM, Enzinger PC, Adenis A, Shah MA, Kato K, Bennouna J, et al. LEAP-014: an open-label, randomized, phase 3 study of first-line lenvatinib plus pembrolizumab plus chemotherapy in esophageal squamous cell carcinoma. J Clin Oncol. 2022;40(4):TPS367.
Cohen DJ, Tabernero J, Van Cutsem E, Janjigian YY, Bang YJ, Qin S, et al. A randomized phase 3 study evaluating the efficacy and safety of first-line pembrolizumab plus lenvatinib plus chemotherapy versus chemotherapy in patients with advanced/metastatic gastroesophageal adenocarcinoma: LEAP-015. J Clin Oncol. 2022;40(4):TPS369.
Eggermont A, Carlino M, Hauschild A, Ascierto P, Arance A, Daud A, et al. Pembrolizumab (pembro) plus lenvatinib (len) for first-line treatment of patients (pts) with advanced melanoma: Phase III LEAP-003 study. Ann Oncol. 2019;30: v561.
Siu LL, Burtness B, Cohen EE, Harrington KJ, Licitra LF, Rischin D, et al. Phase III LEAP-010 study: first-line pembrolizumab with or without lenvatinib in recurrent/metastatic (R/M) head and neck squamous cell carcinoma (HNSCC). J Clin Oncol. 2020;38(15):TPS6589.
Xu JM, Wang Y, Chen YL, Jia R, Li J, Gong JF, et al. Sulfatinib, a novel kinase inhibitor, in patients with advanced solid tumors: results from a phase I study. Oncotarget. 2017;8(26):42076–86.
Xu JM, Shen L, Zhou ZW, Li J, Bai CM, Chi Y, et al. Surufatinib in advanced extrapancreatic neuroendocrine tumours (SANET-ep): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020;21(11):1500–12.
Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68(12):4774–82.
Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–82.
Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–87.
Zhao YJ, Adjei AA. Targeting angiogenesis in cancer therapy: moving beyond vascular endothelial growth factor. Oncologist. 2015;20(6):660–73.
Reck M, Kaiser R, Mellemgaard A, Douillard JY, Orlov S, Krzakowski M, et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014;15(2):143–55.
Van Cutsem E, Yoshino T, Lenz HJ, Lonardi S, Falcone A, Limon ML, et al. Nintedanib for the treatment of patients win refractory metastatic colorectal cancer (LUME-Colon 1): a phase III, international, randomized placebo-controlled study. Ann Oncol. 2018;29(9):1955–63.
Scagliotti GV, Gaafar R, Nowak AK, Nakano T, van Meerbeeck J, Popat S, et al. Nintedanib in combination with pemetrexed and cisplatin for chemotherapy-naive patients with advanced malignant pleural mesothelioma (LUME-Meso): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet Resp Med. 2019;7(7):569–80.
Shen G, Zheng F, Ren D, Du F, Dong Q, Wang Z, et al. Anlotinib: a novel multi-targeting tyrosine kinase inhibitor in clinical development. J Hematol Oncol. 2018;11(1):120.
Han BH, Li K, Wang QM, Zhang L, Shi JH, Wang ZH, et al. Effect of anlotinib as a third-line or further treatment on overall survival of patients with advanced non–small cell lung cancer: the ALTER 0303 phase 3 randomized clinical trial. JAMA Oncol. 2018;4(11):1569–75.
Sun YK, Du F, Gao M, Ji QH, Li ZD, Zhang Y, et al. Anlotinib for the treatment of patients with locally advanced or metastatic medullary thyroid cancer. Thyroid. 2018;28(11):1455–61.
Zhou AP, Bai YX, Song Y, Luo H, Ren XB, Wang XW, et al. Anlotinib versus sunitinib as first-line treatment for metastatic renal cell carcinoma: a randomized phase II clinical trial. Oncologist. 2019;24(8):E702–8.
Cheng Y, Wang QM, Li K, Shi JH, Liu Y, Wu L, et al. Anlotinib vs placebo as third- or further-line treatment for patients with small cell lung cancer: a randomised, double-blind, placebo-controlled Phase 2 study. Br J Cancer. 2021;125(3):366–71.
Sun YK, Zhou AP, Zhang W, Jiang ZC, Chen B, Zhao JJ, et al. Anlotinib in the treatment of advanced hepatocellular carcinoma: an open-label phase II study (ALTER-0802 study). Hepatol Int. 2021;15(3):621–9.
Guffanti F, Chilà R, Bello E, Zucchetti M, Zangarini M, Ceriani L, et al. In vitro and in vivo activity of lucitanib in FGFR1/2 amplified or mutated cancer models. Neoplasia. 2017;19(1):35–42.
Formisano L, Stauffer KM, Young CD, Bhola NE, Guerrero-Zotano AL, Jansen VM, et al. Association of FGFR1 with ERα maintains ligand-independent ER transcription and mediates resistance to estrogen deprivation in ER+ breast cancer. Clin Cancer Res. 2017;23(20):6138–50.
Hui RN, Pearson A, Cortes J, Campbell C, Poirot C, Azim HA, et al. Lucitanib for the treatment of HR+/HER2− metastatic breast cancer: results from the multicohort phase II FINESSE study. Clin Cancer Res. 2020;26(2):354–63.
Hall TG, Yu Y, Eathiraj S, Wang YX, Savage RE, Lapierre JM, et al. Preclinical activity of ARQ 087, a novel inhibitor targeting FGFR dysregulation. PLoS ONE. 2016;11(9):19.
McSheehy PMJ, Forster-Gross N, El Shemerly M, Bachmann F, Roceri M, Hermann N, et al. The fibroblast growth factor receptor inhibitor, derazantinib, has strong efficacy in human gastric tumor models and synergizes with paclitaxel in vivo. Anticancer Drugs. 2023;34(4):532–43.
Mazzaferro V, El-Rayes BF, Busset MDD, Cotsoglou C, Harris WP, Damjanov N, et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br J Cancer. 2019;120(2):165–71.
Javle MM, Abou-Alfa GK, Macarulla T, Personeni N, Adeva J, Bergamo F, et al. Efficacy of derazantinib in intrahepatic cholangiocarcinoma patients with FGFR2 mutations or amplifications: interim results from the phase 2 study FIDES-01. J Clin Oncol. 2022;40(4):427.
Necchi A, Todenhöfer T, Deville JL, Häckl M, Marszewska M, McKernan P, et al. Efficacy and safety of derazantinib (DZB) in patients with metastatic urothelial carcinoma (mUC) with activating FGFR1/2/3 genetic aberrations (GA): results from the phase 1b/2 FIDES-02 study. J Clin Oncol. 2023;41(6):501.
Peng P, Qiang XY, Li GY, Li L, Ni SM, Yu Q, et al. Tinengotinib (TT-00420), a novel spectrum-selective small-molecule kinase inhibitor, is highly active against triple-negative breast cancer. Mol Cancer Ther. 2023;22(2):205–14.
Javle MM, Fountzilas C, Li D, Fonkoua LAK, Fan J, Peng P, et al. Phase II study of FGFR1-3 inhibitor tinengotinib as monotherapy in patients with advanced or metastatic cholangiocarcinoma: Interim analysis. J Clin Oncol. 2023;41(4):539.
Kamath AV, Lu D, Gupta P, Jin DS, Xin Y, Brady A, et al. Preclinical pharmacokinetics of MFGR1877A, a human monoclonal antibody to FGFR3, and prediction of its efficacious clinical dose for the treatment of t(4;14)-positive multiple myeloma. Cancer Chemother Pharmacol. 2012;69(4):1071–8.
Siefker-Radtke AO, Currie G, Abella E, Vaena DA, Kalebasty AR, Curigliano G, et al. FIERCE-22: clinical activity of vofatamab (V) a FGFR3 selective inhibitor in combination with pembrolizumab (P) in WT metastatic urothelial carcinoma, preliminary analysis. J Clin Oncol. 2019;37(15):4511.
Mellado B, Castellano DE, Pang S, Urun Y, Park SH, Vaishampayan UN, et al. Interim analysis of the fierce-21 phase 2 (P2) study of vofatamab (B-701), a selective inhibitor of FGFR3, as salvage therapy in metastatic urothelial carcinoma (mUC). J Clin Oncol. 2019;37(15):4547.
Xiang H, Chan AG, Ahene A, Bellovin DI, Deng R, Hsu AW, et al. Preclinical characterization of bemarituzumab, an anti-FGFR2b antibody for the treatment of cancer. MAbs. 2021;13(1):1981202.
Wainberg ZA, Enzinger PC, Kang YK, Qin SK, Yamaguchi K, Kim IH, et al. Bemarituzumab in patients with FGFR2b-selected gastric or gastro-oesophageal junction adenocarcinoma (FIGHT): a randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 2022;23(11):1430–40.
Wainberg Z, Kang Y, Lee K, Qin S, Yamaguchi K, Kim I, et al. SO-11 Bemarituzumab for treatment of previously untreated advanced and/or metastatic gastric and gastroesophageal cancer (GC): Final analysis of a randomized phase 2 trial (FIGHT). Ann Oncol. 2023;34:S166.
Catenacci DVT, Tesfaye A, Tejani M, Cheung E, Eisenberg P, Scott AJ, et al. Bemarituzumab with modified FOLFOX6 for advanced FGFR2-positive gastroesophageal cancer: FIGHT Phase III study design. Future Oncol. 2019;15(18):2073–82.
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–92.
Liu C, Liao JT, Wu XH, Zhao XM, Sun S, Wang HJ, et al. A phase II study of anlotinib combined with etoposide and platinum-based regimens in the first-line treatment of extensive-stage small cell lung cancer. Thoracic Cancer. 2022;13(10):1463–70.
Liu Y, Xiao Q, He JJ, Hu HG, Du JL, Zhu YP, et al. Phase II study of anlotinib in combination with oxaliplatin and capecitabine for patients with RAS/BRAF wild-type metastatic colorectal adenocarcinoma as the first-line therapy. Bmc Med. 2022;20(1):155.
Wang J, Wu DX, Meng L, Ji G. Anlotinib combined with SOX regimen (S1 (tegafur, gimeracil and oteracil porassium capsules)+ oxaliplatin) in treating stage IV gastric cancer: study protocol for a single-armed and single-centred clinical trial. BMJ Open. 2020;10(6):e034685.
Holzhauser S, Lukoseviciute M, Papachristofi C, Vasilopoulou C, Herold N, Wickström M, et al. Effects of PI3K and FGFR inhibitors alone and in combination, and with/without cytostatics in childhood neuroblastoma cell lines. Int J Oncol. 2021;58(2):211–25.
Lukoseviciute M, Maier H, Poulou-Sidiropoulou E, Rosendahl E, Holzhauser S, Dalianis T, et al. Targeting PI3K, FGFR, CDK4/6 signaling pathways together with cytostatics and radiotherapy in two medulloblastoma cell lines. Front Oncol. 2021;11:748657.
Yang Z, Liang SQ, Yang H, Xu D, Bruggmann R, Gao Y, et al. CRISPR-mediated kinome editing prioritizes a synergistic combination therapy for FGFR1-amplified lung cancer. Cancer Res. 2021;81(11):3121–33.
Cao R, Ji H, Feng N, Zhang Y, Yang X, Andersson P, et al. Collaborative interplay between FGF-2 and VEGF-C promotes lymphangiogenesis and metastasis. Proc Natl Acad Sci U S A. 2012;109(39):15894–9.
Dumbrava EE, Shapiro GI, Bendell JC, Yap TA, Jeselsohn RM, Lepley DM, et al. Phase 1b/2 SEASTAR trial: Safety, pharmacokinetics, and preliminary efficacy of the poly(ADP)-ribose polymerase (PARP) inhibitor rucaparib and angiogenesis inhibitor lucitanib in patients with advanced solid tumors. J Clin Oncol. 2021;39(15):3102.
Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol. 2019;16(2):105–22.
Porta R, Borea R, Coelho A, Khan S, Araújo A, Reclusa P, et al. FGFR a promising druggable target in cancer: molecular biology and new drugs. Crit Rev Oncol Hematol. 2017;113:256–67.
Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–5.
Lee HW, Seo HK. Fibroblast growth factor inhibitors for treating locally advanced/metastatic bladder urothelial carcinomas via dual targeting of tumor-specific oncogenic signaling and the tumor immune microenvironment. Int J Mol Sci. 2021;22(17):15.
Yi C, Chen L, Lin Z, Liu L, Shao W, Zhang R, et al. Lenvatinib targets FGF receptor 4 to enhance antitumor immune response of anti–programmed cell death-1 in HCC. Hepatology. 2021;74(5):2544–60.
Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381(4):338–48.
Wu Y, Yi Z, Li J, Wei Y, Feng R, Liu J, et al. FGFR blockade boosts T cell infiltration into triple-negative breast cancer by regulating cancer-associated fibroblasts. Theranostics. 2022;12(10):4564.
Xu Q, Wang JJ, Sun Y, Lin YB, Liu J, Zhuo YH, et al. Efficacy and safety of sintilimab plus anlotinib for PD-L1-positive recurrent or metastatic cervical cancer: a multicenter, single-arm, prospective phase II trial. J Clin Oncol. 2022;40(16):1795–805.
Wei W, Ban XH, Yang F, Li JB, Cheng XQ, Zhang R, et al. Phase II trial of efficacy, safety and biomarker analysis of sintilimab plus anlotinib for patients with recurrent or advanced endometrial cancer. J Immunother Cancer. 2022;10(5):e004338.
Deng H, Kan A, Lyu N, Mu L, Han Y, Liu L, et al. Dual vascular endothelial growth factor receptor and fibroblast growth factor receptor inhibition elicits antitumor immunity and enhances programmed cell death-1 checkpoint blockade in hepatocellular carcinoma. Liver Cancer. 2020;9(3):338–57.
Chan SL, Schuler M, Kang YK, Yen CJ, Edeline J, Choo SP, et al. A first-in-human phase 1/2 study of FGF401 and combination of FGF401 with spartalizumab in patients with hepatocellular carcinoma or biomarker-selected solid tumors. J Exp Clin Cancer Res. 2022;41(1):189.
Gutierrez M, Subbiah V, Nemunaitis JJ, Mettu NB, Papadopoulos KP, Barve MA, et al. Safety and efficacy of pemigatinib plus pembrolizumab combination therapy in patients (pts) with advanced malignancies: Results from FIGHT-101, an open-label phase I/II study. J Clin Oncol. 2020;38(15):2.
Subbiah V, Iannotti NO, Gutierrez M, Smith DC, Féliz L, Lihou CF, et al. FIGHT-101, a first-in-human study of potent and selective FGFR 1–3 inhibitor pemigatinib in pan-cancer patients with FGF/FGFR alterations and advanced malignancies. Ann Oncol. 2022;33(5):522–33.
Koshkin VS, Sonpavde GP, Hwang C, Mellado B, Tomlinson G, Shimura M, et al. Futibatinib plus pembrolizumab in patients (pts) with advanced or metastatic urothelial carcinoma (mUC): Preliminary safety results from a phase 2 study. J Clin Oncol. 2022;40(6):501.
Powles T, Carroll D, Chowdhury S, Gravis G, Joly F, Carles J, et al. An adaptive, biomarker-directed platform study of durvalumab in combination with targeted therapies in advanced urothelial cancer. Nat Med. 2021;27(5):793–801.
Wang L, Gong Y, Saci A, Szabo PM, Martini A, Necchi A, et al. Fibroblast growth factor receptor 3 alterations and response to PD-1/PD-L1 blockade in patients with metastatic urothelial cancer. Eur Urol. 2019;76(5):599–603.
Bai A, Meetze K, Vo NY, Kollipara S, Mazsa EK, Winston WM, et al. GP369, an FGFR2-IIIb-specific antibody, exhibits potent antitumor activity against human cancers driven by activated FGFR2 signaling. Cancer Res. 2010;70(19):7630–9.
Min B, Shi L, Kim HJ, Kim S, Fan B, Lv C, et al. AMB302/GR1017, an antibody-drug conjugate (ADC) with topoisomerase 1 inhibitor shows therapeutic potency in orthotopic glioblastoma PDX and bladder cancer models with FGFR3-TACC3 fusion. Cancer Res. 2023;83(7):2634.
Tolcher AW, Papadopoulos KP, Patnaik A, Wilson K, Thayer S, Zanghi J, et al. A phase I, first in human study of FP-1039 (GSK3052230), a novel FGF ligand trap, in patients with advanced solid tumors. Ann Oncol. 2016;27(3):526–32.
Yue S, Li Y, Chen X, Wang J, Li M, Chen Y, et al. FGFR-TKI resistance in cancer: current status and perspectives. J Hematol Oncol. 2021;14(1):23.
André F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, et al. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res. 2013;19(13):3693–702.
Zhou W, Hur W, McDermott U, Dutt A, Xian W, Ficarro SB, et al. A structure-guided approach to creating covalent FGFR inhibitors. Chem Biol. 2010;17(3):285–95.
Goyal L, Saha SK, Liu LY, Siravegna G, Leshchiner I, Ahronian LG, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion–positive cholangiocarcinoma. Cancer Discov. 2017;7(3):252–63.
Tan L, Wang J, Tanizaki J, Huang Z, Aref AR, Rusan M, et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc Natl Acad Sci U S A. 2014;111(45):E4869–77.
Shukla N, Ameur N, Yilmaz I, Nafa K, Lau CY, Marchetti A, et al. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin Cancer Res. 2012;18(3):748–57.
Chell V, Balmanno K, Little AS, Wilson M, Andrews S, Blockley L, et al. Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in FGFR3 as a mechanism of acquired resistance. Oncogene. 2013;32(25):3059–70.
Sohl CD, Ryan MR, Luo B, Frey KM, Anderson KS. Illuminating the molecular mechanisms of tyrosine kinase inhibitor resistance for the FGFR1 gatekeeper mutation: the Achilles’ heel of targeted therapy. ACS Chem Biol. 2015;10(5):1319–29.
Lin Q, Chen X, Qu L, Guo M, Wei H, Dai S, et al. Characterization of the cholangiocarcinoma drug pemigatinib against FGFR gatekeeper mutants. Commun Chem. 2022;5(1):100.
O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–12.
Liu Y, Shah K, Yang F, Witucki L, Shokat KM. A molecular gate which controls unnatural ATP analogue recognition by the tyrosine kinase v-Src. Bioorg Med Chem. 1998;6(8):1219–26.
Andrews Wright NM, Goss GD. Third-generation epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. Transl Lung Cancer Res. 2019;8(Suppl 3):S247–64.
Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305(5682):399–401.
Wu D, Guo M, Min X, Dai S, Li M, Tan S, et al. LY2874455 potently inhibits FGFR gatekeeper mutants and overcomes mutation-based resistance. Chem Commun (Camb). 2018;54(85):12089–92.
Qu L, Chen X, Wei H, Guo M, Dai S, Jiang L, et al. Structural insights into the potency and selectivity of covalent pan-FGFR inhibitors. Commun Chem. 2022;5(1):5.
Brawn RA, Cook A, Omoto K, Ke J, Karr C, Colombo F, et al. Discovery of aminopyrazole derivatives as potent inhibitors of wild-type and gatekeeper mutant FGFR2 and 3. ACS Med Chem Lett. 2021;12(1):93–8.
Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20(6):810–7.
Harbinski F, Craig VJ, Sanghavi S, Jeffery D, Liu L, Sheppard KA, et al. Rescue screens with secreted proteins reveal compensatory potential of receptor tyrosine kinases in driving cancer growth. Cancer Discov. 2012;2(10):948–59.
Datta J, Damodaran S, Parks H, Ocrainiciuc C, Miya J, Yu L, et al. Akt activation mediates acquired resistance to fibroblast growth factor receptor inhibitor BGJ398. Mol Cancer Ther. 2017;16(4):614–24.
Wang J, Mikse O, Liao RG, Li Y, Tan L, Janne PA, et al. Ligand-associated ERBB2/3 activation confers acquired resistance to FGFR inhibition in FGFR3-dependent cancer cells. Oncogene. 2015;34(17):2167–77.
Adachi Y, Watanabe K, Kita K, Kitai H, Kotani H, Sato Y, et al. Resistance mediated by alternative receptor tyrosine kinases in FGFR1-amplified lung cancer. Carcinogenesis. 2017;38(11):1063–72.
Herrera-Abreu MT, Pearson A, Campbell J, Shnyder SD, Knowles MA, Ashworth A, et al. Parallel RNA interference screens identify EGFR activation as an escape mechanism in FGFR3-mutant cancer. Cancer Discov. 2013;3(9):1058–71.
Terai H, Soejima K, Yasuda H, Nakayama S, Hamamoto J, Arai D, et al. Activation of the FGF2-FGFR1 autocrine pathway: a novel mechanism of acquired resistance to gefitinib in NSCLC. Mol Cancer Res. 2013;11(7):759–67.
Lima NC, Atkinson E, Bunney TD, Katan M, Huang PH. Targeting the Src pathway enhances the efficacy of selective FGFR inhibitors in urothelial cancers with FGFR3 alterations. Int J Mol Sci. 2020;21(9):3214.
Kunii K, Davis L, Gorenstein J, Hatch H, Yashiro M, Di Bacco A, et al. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res. 2008;68(7):2340–8.
Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16(2):139–49.
Ma L, Li Y, Luo R, Wang Y, Cao J, Fu W, et al. Discovery of a selective and orally bioavailable FGFR2 degrader for treating gastric cancer. J Med Chem. 2023;66(11):7438–53.
Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14(5):283–96.
Zhitomirsky B, Assaraf YG. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget. 2015;6(2):1143–56.
Yamagishi T, Sahni S, Sharp DM, Arvind A, Jansson PJ, Richardson DR. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J Biol Chem. 2013;288(44):31761–71.
Chapuy B, Panse M, Radunski U, Koch R, Wenzel D, Inagaki N, et al. ABC transporter A3 facilitates lysosomal sequestration of imatinib and modulates susceptibility of chronic myeloid leukemia cell lines to this drug. Haematologica. 2009;94(11):1528–36.
Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8(6):903–14.
Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17(3):288–99.
Englinger B, Kallus S, Senkiv J, Laemmerer A, Moser P, Gabler L, et al. Lysosomal sequestration impairs the activity of the preclinical FGFR inhibitor PD173074. Cells. 2018;7(12):259.
Englinger B, Kallus S, Senkiv J, Heilos D, Gabler L, van Schoonhoven S, et al. Intrinsic fluorescence of the clinically approved multikinase inhibitor nintedanib reveals lysosomal sequestration as resistance mechanism in FGFR-driven lung cancer. J Exp Clin Cancer Res. 2017;36(1):122.
Chen CH, Hsieh TH, Lin YC, Liu YR, Liou JP, Yen Y. Targeting autophagy by MPT0L145, a highly potent PIK3C3 inhibitor, provides synergistic interaction to targeted or chemotherapeutic agents in cancer cells. Cancers (Basel). 2019;11(9):1345.
Zhitomirsky B, Assaraf YG. Lysosomes as mediators of drug resistance in cancer. Drug Resist Updat. 2016;24:23–33.
de Klerk DJ, Honeywell RJ, Jansen G, Peters GJ. Transporter and lysosomal mediated (multi) drug resistance to tyrosine kinase inhibitors and potential strategies to overcome resistance. Cancers (Basel). 2018;10(12):27.
Krchniakova M, Skoda J, Neradil J, Chlapek P, Veselska R. Repurposing tyrosine kinase inhibitors to overcome multidrug resistance in cancer: a focus on transporters and lysosomal sequestration. Int J Mol Sci. 2020;21(9):3157.
Wu C-P, Hung T-H, Hsiao S-H, Huang Y-H, Hung L-C, Yu Y-J, et al. Erdafitinib resensitizes ABCB1-overexpressing multidrug-resistant cancer cells to cytotoxic anticancer drugs. Cancers. 2020;12(6):1366.
Lyou Y, Grivas P, Rosenberg JE, Hoffman-Censits J, Quinn DI, Petrylak DP, et al. Hyperphosphatemia secondary to the selective fibroblast growth factor receptor 1–3 inhibitor infigratinib (BGJ398) is associated with antitumor efficacy in fibroblast growth factor receptor 3–altered advanced/metastatic urothelial carcinoma. Eur Urol. 2020;78(6):916–24.
Yang J, Meyer M, Müller A-K, Böhm F, Grose R, Dauwalder T, et al. Fibroblast growth factor receptors 1 and 2 in keratinocytes control the epidermal barrier and cutaneous homeostasis. J Cell Biol. 2010;188(6):935–52.
Meric-Bernstam F, Hollebecque A, Furuse J, Oh D-Y, Bridgewater JA, Shimura M, et al. Safety profile and adverse event management for futibatinib, an irreversible FGFR1-4 inhibitor: Pooled safety analysis of 469 patients. Clin Cancer Res. 2024;30(8):1466–77.
Francis JH, Foulsham W, Canestraro J, Harding JJ, Diamond EL, Drilon A, et al. Mitogen-activated pathway kinase inhibitor-associated retinopathy: Do features differ with upstream versus downstream inhibition? Ocul Oncol Pathol. 2023;9(1–2):25–31.
Hoe N, Jin K, Ma Y, Kuy C, Mateling M, Zhou J, et al. FGFR inhibition modulates ErbB receptor tyrosine phosphorylation in FGFR amplified cell lines. Cancer Res. 2013;73(8):4273.
Mouron S, Manso L, Caleiras E, Rodriguez-Peralto JL, Rueda OM, Caldas C, et al. FGFR1 amplification or overexpression and hormonal resistance in luminal breast cancer: rationale for a triple blockade of ER, CDK4/6, and FGFR1. Breast Cancer Res. 2021;23(1):21.
Formisano L, Lu Y, Servetto A, Hanker AB, Jansen VM, Bauer JA, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun. 2019;10(1):1373.
Sternberg CN, Petrylak DP, Bellmunt J, Nishiyama H, Necchi A, Gurney H, et al. FORT-1: Phase II/III study of rogaratinib versus chemotherapy in patients with locally advanced or metastatic urothelial carcinoma selected based on FGFR1/3 mRNA expression. J Clin Oncol. 2023;41(3):629–39.
Velmahos CS, Badgeley M, Lo YC. Using deep learning to identify bladder cancers with FGFR-activating mutations from histology images. Cancer Med. 2021;10(14):4805–13.
Loriot Y, Matsubara N, Park SH, Huddart RA, Burgess EF, Houede N, et al. Erdafitinib or chemotherapy in advanced or metastatic urothelial carcinoma. N Engl J Med. 2023;389(21):1961–71.
Song Y, Peng Y, Qin C, Wang Y, Yang W, Du Y, et al. Fibroblast growth factor receptor 3 mutation attenuates response to immune checkpoint blockade in metastatic urothelial carcinoma by driving immunosuppressive microenvironment. J Immunother Cancer. 2023;11(9):e006643.
Okato A, Utsumi T, Ranieri M, Zheng X, Zhou M, Pereira LD, et al. FGFR inhibition augments anti-PD-1 efficacy in murine FGFR3-mutant bladder cancer by abrogating immunosuppression. J Clin Invest. 2024;134(2):e169241.
Koshkin VS, Sonpavde GP, Hwang C, Mellado B, Tomlinson G, Shimura M, et al. Futibatinib plus pembrolizumab in patients (pts) with advanced or metastatic urothelial carcinoma (mUC): preliminary safety results from a phase 2 study. J Clin Oncol. 2022;40(6):501.
Krook MA, Lenyo A, Wilberding M, Barker H, Dantuono M, Bailey KM, et al. Efficacy of FGFR inhibitors and combination therapies for acquired resistance in FGFR2-fusion cholangiocarcinoma. Mol Cancer Ther. 2020;19(3):847–57.
Acknowledgements
Figures 2 in this article created with BioRender.com. Figures 3 and 5 in this article are drawn by Figdraw (https://www.figdraw.com).
Funding
This study was sponsored by the 1·3·5 project for disciplines of excellence–Clinical Research Incubation Project, West China Hospital, Sichuan University (ZYJC21016).
Author information
Authors and Affiliations
Contributions
DC and THY designed the outline. PZ, QQL and CLG participated in the literature search and drafted the manuscript. PZ and LY designed the figures. DC, CC and THY supervised and edited the manuscript. Final draft read and approved by all authors.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
There are no competing interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, P., Yue, L., Leng, Q. et al. Targeting FGFR for cancer therapy. J Hematol Oncol 17, 39 (2024). https://doi.org/10.1186/s13045-024-01558-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-024-01558-1