
Abstract
Transcription factor RelB is a member of the nuclear factror-kappa B (NF-κB) family, which plays a crucial role in mediating immune responses. Plenty of studies have demonstrated that RelB actively contributes to lymphoid organ development, dendritic cells maturation and function and T cells differentiation, as well as B cell development and survival. RelB deficiency may cause a variety of immunological disorders in both mice and humans. Multiple sclerosis (MS) is an inflammatory and demyelinating disease of the central nervous system which involves a board of immune cell populations. Thereby, RelB may exert an impact on MS by modulating the functions of dendritic cells and the differentiation of T cells and B cells. Despite intensive research, the role of RelB in MS and its animal model, experimental autoimmune encephalomyelitis, is still unclear. Herein, we give an overview of the biological characters of RelB, summarize the updated knowledge regarding the role of RelB in different cell types that contribute to MS pathogenesis and discuss the potential RelB-targeted therapeutic implications for MS.
Similar content being viewed by others


Introduction
Transcription factors of the nuclear factor-kappa B (NF-κB) family play a critical role in regulating the innate and adaptive immune responses [1]. In mammalian cells, this family includes five members: c-Rel, p65 (RelA), RelB, p105/p50 (NF-κB1) and p100/p52 (NF-κB2) [2]. RelB, first described in 1992 [3], is considered an extraordinary member of the NF-κB family with diverse and unique features [4,5,6,7]. Over the past two decades, a variety of biological characteristics and immunological effects of RelB have been reported [8, 9].
Multiple sclerosis (MS) is a neuro-inflammatory disease that is mainly characterized by multicentric white matter demyelination of the central nervous system (CNS) [10]. It is a disabling disease that primarily affects young adults, particularly young women [11]. MS can be commonly clinically classified into four categories, including relapsing-remitting MS (RRMS), primary progressive MS (PPMS), secondary progressive MS (SPMS) and progressive relapsing MS (PRMS), among which RRMS is the most common and classical form [12]. In the early course of MS, acute attacks of neurological impairment are followed by partial or complete remission. The relapsing-remitting neurological dysfunctions lead to chronic neurological damage and neurodegeneration, which results in disability accumulation and disease progression. The clinical symptoms of MS are complex and diverse, including motor, sensory, visual and autonomic system dysfunctions [13, 14]. Current disease-modifying therapies could reduce the frequency of relapses; however, the progression of MS can be not effectively prevented [15,16,17]. Hence, new therapeutic strategies for MS still need to be proposed.
Given the significant role of NF-κB in immune response [18,19,20,21], a better understanding of the role of RelB in MS is potentially beneficial for exploring the pathogenesis and looking for new immunotherapies for treatment. This review will outline the features of RelB and RelB-associated pathogenic mechanisms in MS, as well as the therapeutic implications of targeting RelB.
Biological characteristics of transcription factor RelB
The gene structure and expression of RelB
The latest data has illustrated that the human RelB gene is located on chromosome 19q13.32, where 12 exons encode a protein with 579 amino acids [22]. The 5′ genomic region of RelB is characterized by a TATA-less promoter containing two κB cis-acting sites. Furthermore, potential vitamin D response elements have been recognized in the RelB promoter region, which are essential for negative transcriptional regulation and mediated by 1,25-Dihydroxyvitamin D3 (1,25-(OH)2D3) and its analogs [23]. Higher expression of the RelB gene is observed in the thymus medulla, the periarterial lymphatic sheaths of the spleen and the deep cortex of the lymph nodes [6, 24]. At the cellular level, RelB expression is mainly restricted to dendritic cells (DCs) [24]. Furthermore, RelB can also be expressed in other immune cells, such as T cells, B cells and monocytes [6, 25,26,27].
Protein structure and functions of RelB
The RelB protein contains three important domains: the C-terminal transcriptional activation domain (TAD), the Rel homology region (RHD) and the N-terminal leucine zipper domain (LZ) [28]. The RHD, highly conserved sequences on all NF-κB family members, consists of 300 amino acids and is responsible for dimerization, nuclear translocation and DNA-binding activity [29]. The TAD is indispensable, but not sufficient to motivate expression of NF-κB-dependent genes [7]. The LZ, recognized for its unique characteristics, differs from other family members and participates in activating transcription of target genes. The structural integrity of both N- and C-terminals domains is necessary for the fully transcriptional activity of RelB [7].
Nevertheless, RelB protein is unstable. In the cytoplasm of unstimulated cells, RelB prevents its degradation by forming a steady heterodimer with p100/p52 or p105/p50 [30]. Differing from the other NF-κB members, a stable RelB homodimer is nonexistent [31]. Accumulating evidence suggests that RelB can act as both an activator and a repressor to regulate NF-κB-responsive gene expression [3, 5]. In addition, RelB plays a dual regulatory role in targeting gene expression by recruiting co-activators or co-repressors, like human epithelial growth factor receptor 2 (HER2) [32], histone H3 lysine methyltransferase G9a [33] and death domain-associated protein (Daxx) [34]. The accurate mechanisms underlying these divergent functions are currently unclear. One widely accepted notion is that RelB post-translational modifications, such as phosphorylation [35,36,37], ubiquitination [38] and SUMOylation [39], have a direct effect on RelB transcriptional activity, which results in functional diversity [40].
The RelB-associated activation pathways
NF-κB family members can be activated by either canonical or non-canonical NF-κB pathways. The canonical pathway can be triggered by various stimuli that bind to immune receptors, like the Toll-like receptors (TLRs), tumor necrosis factor receptor (TNFR), T cell receptor (TCR) and B cell receptor (BCR). Then, the inhibitor of κB kinase (IKK) complex, including two catalytic subunits IKKα and IKKβ, and one regulator IKKγ, can be activated, and in turn, phosphorylates IκBα (a member of κB inhibitors). After that, phosphorylated IκBα undergoes proteasome-dependent degradation and then releases the RelA/p50 complex. The freed RelA/p50 complex translocates into the nucleus and induces the expression of multiple inflammatory genes [41] (Fig. 1). The non-canonical NF-κB pathway is triggered by a series of tumor necrosis factor superfamily receptors (TNFSFRs) members, such as B cell activating factor receptor (BAFFR), lymphotoxin β receptor (LTβR), receptor activator of NF-κB (RANK), CD40, CD30, CD27 and fibroblast growth factor-inducible factor 14 (FN14) [42]. Once TNF superfamily molecules link to their corresponding TNFSFRs, NF-κB inducing kinase (NIK) phosphorylates and activates IKKα. The activated IKKα phosphorylates p100 at the site of C-terminal serine residues, leading to the partial degradation of p100 in the proteasome. Processing of p100 transforms it into p52, which then forms a RelB/p52 heterodimer that in turn migrates from the cytoplasm to the nucleus and promotes the expression of target genes through binding to the promoter or enhancer of target genes (Fig. 1) [43]. While the activation of the canonical pathway depends on the rapid and transient nuclear translocation of RelA/p50 dimers, the non-canonical pathway is activated in a slow and persistent manner via a RelB/p52 complex [2, 42]. Interestingly, the canonical and non-canonical pathways are not completely independent in most cases, but have an impact on each other [44]. The RelB-associated non-canonical pathway plays a critical role in regulating immune homeostasis, and its dysregulation contributes to inflammatory and autoimmune diseases [42, 43, 45,46,47].

Canonical and non-canonical NF-κB pathways. The canonical pathway is triggered by various immune receptors, for example, TLRs, TNFR, BCR and TCR. Various receptors activate the IKK complex, resulting in phosphorylation and proteasome-dependent degradation of IκBα, which in turn frees RelA/p50 and promotes its nuclear import. The non-canonical pathway is induced by the TNFSFRs, such as BAFFR, LTβR, CD40 and RANK. Then, downstream molecules NIK and IKKα are activated, leading to p100 processing and the liberation of RelB/p52 heterodimers. Finally, the uncontrolled dimers translocate into nucleus and bind to target genes, triggering their expression
Abbreviations: TLRs: Toll-like receptors; TNFR: tumor necrosis factor receptor; TCR: T cell receptor; BCR: B cell receptor; TNFSFRs: tumor necrosis factor superfamily receptors; BAFFR: B cell activating factor receptor; LTβR: lymphotoxin β receptor; RANK: receptor activator of NF-κB; FN14: fibroblast growth factor-inducible factor 14; IκB: κB inhibitor; IKK: IκB kinase; NIK: NF-κB-inducing kinase
Immunomodulatory role of RelB
Accumulating evidence suggests that RelB deficiency can lead to a range of immune disorders in both mice and humans (Table 1) [9, 48]. In the following parts, we will discuss the role of RelB in immune organs, immune cells and immune responses.
Lymphoid organ development
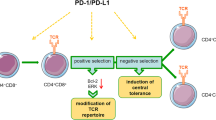
Serving as the primary lymphoid organ, the thymus is a location for the development of T lymphocytes and the formation of central immunologic tolerance [68]. Thymus stromal cell microenvironments, in particular medullary thymic epithelial cells (mTECs), play a key role in these processes [69]. The mTECs are not only involved in the generation of Forkhead box protein 3-expressing regulatory T cells (FoxP3+ Tregs) [70], but can also express autoimmune regulator (Aire; Aire+ mTECs) that can contribute to negative thymocyte selection and suppress the initiation of autoimmune diseases [71,72,73]. The development of mTECs can be regulated by members of the TNFR superfamily, such as LTβR, CD40 and RANK, all of which can play their role through the canonical and non-canonical NF-κB pathways [74, 75]. Interestingly, a recent study revealed that the canonical pathways mediate mTECs differentiation by directly inducing RelB expression [49]. Acting mainly as a downstream signaling molecule of the TNFR superfamily, RelB is closely related to the development and functions of mTECs [50]. In RelB-deficient mice, the thymic medullary architecture is highly disorganized, mTECs and dendritic cells (DCs) are absent, and negative selection is impaired [49, 51,52,53,54]. Along this line, RelB deficiency in humans causes thymic dysplasia and decreased Hassall’s corpuscles [48]. Significantly, RelB is a necessary regulator for the expression of thymic Aire [54], and the development of Aire+ mTECs is primarily mediated by RANK signaling [76,77,78,79].
As secondary lymphoid organs (SLOs), the spleen, lymph nodes and Peyer’s patches provide accommodation for inactivated lymphocytes that can efficiently respond to diverse antigens, thereby making them essential for adaptive immunity [80]. An analysis of RelB-deficient mice suggested that RelB plays an important role in the development of secondary lymphoid organs. RelB-deficient mice lack Peyer’s patches and peripheral lymph nodes [53, 55]. Furthermore, RelB-deficient mice and spleens with severe structural damage, containing impaired follicular dendritic cells (FDCs) networks, a dispersed reticular fibroblast network throughout the white pulp, deficient germinal center (GC) and marginal zone development [56]. The anatomical imperfection in SLOs is closely related to the activation of the non-canonical NF-κB pathway by LTβR signaling via the RelB-related heterodimer [55,56,57, 81]. Once lymphotoxin-α1β2 (LTα1β2) expressed by lymphoid-tissue inducer cells binds to its relative LTβR, which is expressed by stromal organizer cells, non-canonical signaling is activated, inducing the expression of RelB-dependent homeostatic chemokines and cell adhesion molecules, which in turn attract and recruit lymphocytes to developing and mature SLOs [82]. During the expression of these homeostatic chemokines, secondary lymphoid tissue chemokine (SLC) and Epstein-Barr virus-induced molecule 1 ligand chemokine (ELC) are primarily responsible for the migration of T cells into SLOs, while B lymphocyte chemoattractant (BLC) plays a central role in attracting B cells [83, 84]. Furthermore, BCL and SCL generation can be prominently decreased in RelB-deficient mice [56]. Collectively, RelB is required by SLO formation and maintenance.
The maturation and function of DCs
DCs are professional antigen presenting cells (APCs), that are required for initiating adaptive immunity, since they provide signaling to antigen-specific naïve T cells that differentiate into functional mature T cells [85]. RelB plays a key role in DC maturation [24, 52, 58], particularly in myeloid-related DCs [86] that serve as conventional DCs (cDC) [87]. Surface markers associated with myeloid-derived DC maturation, such as major histocompatibility complex (MHC) class-II, CD11c, CD80, CD86 and CD40, were decreased in RelB-deficient mice. Furthermore, these deficiencies were not found in RelB-Venus knock-in mice [58]. RelB deficiency profoundly impaired DCs, both in their maturation and function [59]. In RelB-deficient bone marrow chimera mice, DCs showed a lower capacity of antigen presentation and T cell activation [59]. Aryl hydrocarbon receptor (AhR) signaling promotes RelB expression during DC maturation, and AhR deficiency in DCs may alter the control of RelB in DC maturation and function [60]. However, there are still different opinions on this topic. In 2017, Briseno et al. claimed that the development of most mouse cDC subsets did not rely on cell-intrinsic requirements for RelB [61]. Similarly, another study suggested that RelB/p50 promotes chemokine CCL19 expression instead of facilitating human DC maturation [88]. In summary, RelB plays key roles in the maturation and function of DCs. However, future studies are still needed to thoroughly investigate the association between RelB activation and DC development, making the present paradox clear.
T cell differentiation and T cell-mediated immunity
Responding to diverse antigens, T cells are involved in multiple processes of adaptive immunity [89]. During an immune response, activated naïve T cells can differentiate into effector cells and memory T cells in order to eliminate pathogens and keep long-term immunity [90]. Effector T cells are roughly divided into several subsets, including CD4+ helper T cells (Th1, Th2, Th17), CD8+ T cells and Tregs [91, 92]. Memory T cells are commonly classified into two categories: central memory T cells and effector memory T cells. Human naïve and memory T cells express CD45RA and CD45RO, respectively [93]. Several components of the non-canonical NF-κB pathway, like NIK, NF-κB2 and p52, have been confirmed to participate in T cell activation and T cell-mediated immunity [94,95,96]. Similarly, RelB-deficient mice presented with damaged T cell immunity, a reduction of interferon-γ (IFN-γ) and multiorgan inflammation [53, 62]. Emerging evidence reveals that RelB plays a negative role in Th17 differentiation [63]. Kurosawa et al. observed that effector memory cells from RelB-deficient mice displayed significantly elevated migratory activity than that in the WT mice [64]. Humans with RelB-deficiency present with T cells dysmaturity, reduced output of T cells from thymus and abnormal clonal expansion of T cell subtypes, which results in severe T cell immunodeficiency [48]. Specifically, complex phenotypes were observed, including increased memory cells, weakened T cell responses, significantly reduced IFN-γ and IL-2 generation and decreased expression of signal transducer and activator of transcription 1 (STAT1) and T-bet, which facilitate Th1 differentiation [48]. Taken together, RelB has a pleiotropic effect on T cell differentiation and T cell-mediated immunity.
B cell development, survival, germinal center formation and humoral immunity
B cells are essential for humoral immunity. After T cell-dependent antigenic stimulation, GC B cells undergo somatic hypermutation, negative selection and eventually differentiate into memory B cells and high-affinity plasma cells, which are responsible for immunological memory formation and antigen-specific antibody secretion, respectively [97, 98]. FDCs are limited to GCs and contribute to negative selection of B cells by expressing antigens on their surfaces [98]. In addition, marginal zone B cells play a critical role in T cell-independent humoral immune response [99]. GC and FDC network shortages, follicular B cell reduction and marginal zone B cell absence can be found in the spleens of RelB-deficient mice [56]. Consistently, mice with combined deficiency of RelB/NF-κB2 or RelB/cRel also show B-cell progenitor developmental disorders, the tumble of established GCs and remarkable reduction of peripheral mature B cells [65, 66], which were also observed in BAFF or BAFFR-deficient mice [100,101,102,103,104]. Weih et al. illustrated that RelB activation in stromal cells was responsible for the formation of GCs and FDC networks, whereas RelB expression in hemopoietic cells was required for the generation of marginal zone B cells [56]. RelB mediates GC B cell maturation via CD40 and BAFFR signaling [65, 67] and maintains B-cell survival via BAFFR signaling [65, 66]. BAFFR-mediated survival signaling in mature B cells functions by activating the non-canonical pathway [66, 105, 106]. In humans with RelB deficiency, B cell development is halted and CD27+ memory B cells are absent, leading to shortage of specific antibodies and severe B cell immunodeficiency [8, 48]. Furthermore, B cells from these patients show decreased expression of the surface molecule BAFFR and impaired CD40 signaling [48]. Collectively, RelB plays a crucial role in B cell development, survival, GC formation and humoral immunity.
RelB in the pathogenesis of multiple sclerosis and its animal model
MS is a chronic and progressive autoimmune disease in the CNS, characterized by inflammatory demyelination of the brain and spinal cord [10]. Experimental autoimmune encephalomyelitis (EAE), a conventional animal model of MS, is widely applied to study the pathophysiology and treatment for MS [107]. MS is traditionally thought of as a T cell-mediated autoimmune disorder [108]. Autologous myelin antigen-derived CD4+ T cells migrate from the periphery into the CNS, where they produce cytokines, chemokines and inflammatory molecules to impair the myelin sheath and axons [109]. It is generally accepted that Th1 and Th17 cells are responsible for MS and EAE initiation [109, 110]. However, a growing body of evidence suggests that the occurrence of MS is always accompanied by diverse immune cells infiltration, which mainly contains a variety of activated T cells subtypes, DCs and B cells (Fig. 2) [10]. The invading immune cells mediate MS pathology by secreting a mass of pro-inflammatory or anti-inflammatory cytokines (Fig. 2) [111]. RelB is a powerful molecule that regulates lymphoid organ formation, as well as lymphocyte development and function [9]. While the relationship between RelB and a variety of tumors is widely studied, such as in laryngeal cancer, lung adenocarcinoma and colon cancer [112,113,114,115], the role of RelB in MS is still obscure. This section summarizes the RelB-associated mechanism in MS and EAE, which may provide new insights into the treatment of MS.

Role of different cells in the pathogenesis of MS and EAE. In the thymus, thymocyte precursor cells develop into γδT1, γδT17, nTregs and naïve CD4+ T cells. Upon neuroinflammation, γδT1 and γδT17 cells can cross the endothelial BBB and traffic into the central nervous system CNS, whereas naïve T cells migrate into the peripheral immune tissue. Naïve T cells connected with APCs (DCs and B cells), thereby differentiating into various effector T cells (iTregs, Tr1, Th17 and Th1). Th1, Th17, γδT1 and γδT17 cells secrete pro-inflammatory cytokines that trigger neuroinflammation and impair the myelin sheath and axons. Meanwhile, Tregs (Tr1, iTregs and nTregs) secrete anti-inflammatory cytokines and restrain immune responses mediated by T cells, B cells and DCs, thereby promoting tissue repair. Further, with the help of Tfh cells, naïve B cells differentiate into plasma cells, memory B cells and Bregs. While plasma cells damage the myelin sheath and axons on neurons via secreting antibodies, Bregs play a protective role via producing IL10, IL35 and TGF-β. Memory B cells and several activated B cells can produce a series of pathogenic cytokines
Abbreviations: MS: multiple sclerosis; EAE: experimental autoimmune encephalomyelitis; γδ: gamma delta; BBB: blood-brain barrier; CNS: central nervous system; APC: professional antigen presenting cells; DC: dendritic cell; iTreg: induced regulatory T cell; Tr1: type 1 Treg; Th: T helper; nTreg: natural regulatory T cell; Tfh: follicular helper T; Breg: regulatory B cell; IL: interleukin; IFN-γ: interferon-γ; TGF-β: transforming growth factor-β; TNF-α: tumor necrosis factor α; GM-CSF: granulocyte monocyte-colony stimulating factor
Th17 cells
Th17 cells play a central role in the pathogenesis of MS, and their differentiation depends on the transactivation of the orphan nuclear receptors γt and α (RORγt and RORα) [116, 117]. Autoreactive Th17 cells infiltrate the CNS, where they secrete IL-17A, IL-17F and granulocyte monocyte-colony stimulating factor (GM-CSF). They attract and activate diverse immune cells, eventually resulting in neuroinflammation [118,119,120,121]. IL-17A (also known as IL-17), a hallmark cytokine of Th17 cells, is crucial for the development of EAE [122]. Functional blockage of IL-17 leads to a remissive disease course and improves the outcomes of EAE [123,124,125], whereas its high expression is related to MS severity [126]. GM-CSF−/− mice fail to induce EAE because of the inability of autoreactive lymphocytes to proliferate and the ceasing of immune cell infiltration [120, 127]. The expression of GM-CSF and its receptor is upregulated in brain tissues from acute and chronic MS patients [128]. Interestingly, a recent study puts forwards a new theory that Th1-like Th17 effector memory cells, especially Th17.1 cells, dominantly contribute to MS pathology. Th17.1 cells were affluent and showed increased production of IFN-γ and GM-CSF in patients with MS [129].
There is growing evidence that RelB is a negative factor regarding mediating Th17 cell differentiation and function, which affects the induction and progression of EAE [63, 130, 131]. Park et al. showed that lipopolysaccharides directly stimulated Th17 cell differentiation, enhanced the frequency of IL-17-producing cells and aggravated EAE via modulating phosphorylation of RelB and NF-κB1 [132]. Xiao and his colleagues reported that OX40, a T cell costimulatory molecule in the TNFSFR family, activated downstream molecule RelB, which inhibited IL-17 expression and alleviated EAE through triggering chromatin modification and forming a “closed” chromatin structure at the IL-17 gene [131]. Besides, mucosa-associated lymphoid tissue lymphoma translocation gene 1 (MALT1) is closely related to the level of RelB protein. Using MALT1−/− Th cells and mice, Brustle et al. demonstrated that the deficiency of MALT1 can reduce the degradation of RelB in Th17 cells and decrease the production of IL17 and GM-CSF, thereby preventing mice from EAE induction [130]. In conclusion, RelB regulates Th17 differentiation negatively in EAE, and the treatment that activates RelB in Th17 cells may be a potentially therapy for MS.
Th1 cells
Prior to the discovery of Th17 cells, Th1 cells were thought to be responsible for neuroinflammation in MS and EAE [133]. Th1 cells express the transcription factor T-bet that acts as a positive regulator to promote IFN-γ production [134]. The level of IFN-γ could be increased in EAE and MS patients [135, 136]. Accumulating evidence indicates that both IFN-γ inactivation [137,138,139,140] and T-bet depletion [141] protect mice from developing EAE. Furthermore, Cron and his colleagues observed a noteworthy reduction of IFN-γ and defective Th1 differentiation in RelB-deficient mice. Meanwhile, a remarkable reduction in expression of T-bet occurs in RelB-deficient Th1 cells [142]. Collectively, RelB plays a role in MS and EAE by mediating T-bet expression and Th1 differentiation. Exploring approaches to suppress RelB expression in Th1 cells may be an implicit treatment for MS.
Gamma delta T cells
In the thymus, some of the double negative thymocytes differentiate into gamma delta (γδ) T cells that further develop into IFN-γ-secreting (γδT1) and IL-17-secreting (γδT17) cells under the conditions of the transactivation of T-bet and the orphan nuclear receptors RORγt and RORα4, respectively [143, 144]. γδ T cells play an important role in the pathogenesis of MS and EAE [145, 146]. By using frozen CNS specimens from acute MS patients, Wucherpfennig et al. observed the accumulation and clonal expansion of γδ T cells in lesions [147]. Coincidentally, recent investigations indicated that γδ T cells exert a harmful effect on EAE, evidenced by a significant remission of the clinical symptoms in γδ T cell-deficient animals [148,149,150]. Some evidence demonstrated that γδT17 cells aggravate EAE by enhancing IL-17 production, suppressing Tregs responses and improving antigen-specific T cell responses [151]. Interestingly, another study has illustrated that RelB regulates γδT17 cell differentiation in the thymus and IL-17 production through controlling the expression of RORγt and RORα4, which requires the activation of LTβR signaling [144]. Therefore, RelB may worsen EAE by regulating γδT17 cell differentiation.
Regulatory T cells and steady-state migratory DCs
Tregs are well accepted for their central role in restraining autoreactive immune responses and maintaining peripheral tolerance. There are two widely-studied types of Tregs: FoxP3+ Tregs and IL-10-secreating type 1 Tregs (Tr1) [152]. FoxP3+ Tregs encompass two categories: thymus-derived natural Tregs (nTregs) and periphery-induced Tregs (iTregs), and both of them secrete immunosuppressive cytokines, such as IL-10, IL-35 and transforming growth factor-β (TGF-β) [153]. Tr1 cells are induced in the periphery and primarily produce IL-10 and TGF-β [154]. Tregs can restrain various immune responses mediated by T cells, B cells and DCs [155]. The protective role of FoxP3+ Tregs and Tr1 cells was observed in MS and EAE [156,157,158,159,160].
Mature DCs are essential for the immune response, whereas immature DCs improve immune tolerance by inducing T cell anergy or Tregs generation [161]. Steady-state migratory DCs, known as semi-mature DCs, transport self-antigens from peripheral tissues to the draining lymph nodes [162]. Idoyaga et al. proved that steady state Langerin+ migratory skin DCs exert an unparalleled effect on inducing Foxp3+ Treg generation in vivo, which prominently improved the prognosis of EAE [163, 164]. Moreover, the activation of the non-canonical NF-κB pathway via RelB/p52 is essential for maintaining the frequency of steady-state migratory DCs and inducing Foxp3+ iTreg formation by steady state migratory RelB+ Langerin+ dermal DCs [165]. RelB+ Langerin− dermal DC subset controls the peripheral pool of Foxp3+ nTregs [166]. Further, some researchers found Foxp3+ Tregs markedly expanded in mice with RelB depletion because of increased levels of IL-2, a growth factor for Foxp3+ Tregs that is produced by hyperactive T effector cells [167]. In conclusions, the role of RelB in EAE via regulated Tregs and steady-state migratory DCs are complex. Further studies are still needed to uncover exact mechanisms, and steady-state migratory DCs may be a therapeutic target for MS or other autoimmune diseases.
Dendritic cells
DCs play a critical role in activating immune response. In MS and EAE, DCs present autologous myelin antigen to naïve CD4+ T cells, which then differentiate into myelin-reactive Th1 and Th17 cells that induce neuroinflammation and CNS damage [168,169,170]. Moreover, one of their major effector molecules, cytokine IL-23, is also increased in MS patients [171]. RelB is essential for DC maturation [86], and silencing RelB generates stable tolerogenic properties, creating what is known as tolerogenic DCs [172]. Tolerogenic DCs exhibit an immature phenotype, with lower levels of costimulatory molecules, repressed effector T cell responses and Treg induction [172]. In a previous study, our lab successfully induced tolerogenic DCs by applying 1,25-(OH)2D3, which repressed EAE via the induction of Tregs and the reduction of Th1/Th17 [173]. Coincidentally, monocyte-derived DCs that were treated with 1,25-(OH)2D3 also showed a decreased ability to induce a T cell response and an increase in anti-inflammatory cytokines in MS patients, compared to healthy controls [174]. Moreover, 1,25-(OH)2D3 may function by regulating RelB expression in DCs [23, 175]. In addition, dimethyl fumarate (DMF), an immunotherapeutic drug for MS approved by the United States Food and Drug Administration (FDA), has a therapeutic effect via impairing human myeloid DC maturation. Compared to untreated cells, myeloid DCs from DMF-treated MS patients showed an immature phenotype, decreased expression of RelB, limited capacity to activate T cells and reduced secretion of pro-inflammatory cytokines IFN-γ, IL-17 and GM-CSF [176]. Currently, RelB-silenced tolerogenic DCs are used to study autoimmune diseases, such as systemic lupus erythematosus and myasthenia gravis. Moreover, significant protective effects have been observed in disease conditions [177,178,179]. In a very recent research, Andreas and his team found that mice with RelB-deficient DCs are almost resistant to induction of the EAE model because of the accumulation of FoxP3+ Tregs and the reduction of pathogenic T cells [180]. Therefore, RelB-silenced tolerogenic DCs may be a promising cell therapy for MS.
It should be noted that macrophages are also effective antigen-presenting cells and Recent reports indicate that macrophages play dual roles in the pathogenesis of MS as they contribute to lesion formation and axonal damage, but also present repair mechanisms through the production of neurotrophic factors and anti-inflammatory molecules as well as clearance of myelin debris (Abdul-Majid et al.,2002; Kigerl et al., 2009).
B cells
For decades, MS was generally known as a mainly T cell-mediated disease, and the role of B cells in MS was overlooked; however, a growing amount of evidence shows the significant involvement of B cells in MS pathology [181, 182]. When naïve B cells encounter myelin antigens, they are activated and differentiate into plasma cells, memory B cells and regulatory B cells (Bregs) with the help of follicular helper T (Tfh) cells [108]. In MS patients, a portion of activated B cells act as APCs, presenting myelin antigens to CD4+ T cells and improving Th1 and Th17 responses [183,184,185]. Plasma cells produce myelin specific antibodies that not only cause functional myelin impairment, but also form oligoclonal bands (OCBs) within the CNS and peripheral blood [186]. The detection of OCBs from cerebrospinal fluid are used to diagnose MS with high sensitivity [187, 188]. In untreated RRMS patients, circulating effector memory B cells significantly increase, producing abundant GM-CSF, TNF-α and IL-6 [189]. Additionally, activated B cells from MS patients or EAE mice also secrete pathogenic cytokines IL-6, TNF-α and LT-α [189, 190]. By contrast, Bregs play a protective role in EAE by producing several anti-inflammatory cytokines, such as IL-10, IL-35 and TGF-β [191,192,193,194]. RelB is critical for the maturation and survival of B cells. Mice and humans with RelB deficiency present with developmental disorders of B-cell progenitors and a significant reduction of peripheral mature B cells [48, 65, 66]. Furthermore, RelB/NF-κB2-deficient GC B cells have reduced the expression of inducible T cell co-stimulator ligand (ICOSL), which connect with ICOS expressed on Tfh cells to mediate the selection of high-affinity B cells [195]. Hence, the inhibition of RelB expression in B cells may be beneficial for MS.
Other immune cells
Macrophages and microglia are prominent innate immune cells and play a dual role in the pathogenesis of MS [196]. While macrophages and microglia are induced into the M1 phenotype in the acute phase, which contribute to demyelination and MS lesion formation; macrophages and microglia are activated into M2 phenotype in the later stage, which exhibit neuroprotective effect by clearance of myelin debris and secretion of neuroprotective molecules [196, 197]. Therefore, shifting the phenotype of macrophages and microglia from M1 into M2 may be attractive therapy for MS. Interestingly, overexpression of RelB was observed in LPS-stimulated macrophages, which suppressed the production of TNFα, a pro-inflammatory cytokine [198]. Some studies also observed that the expression of RelB was enhanced in M1 macrophages and RelB deficiency inhibited the differentiation of M1 macrophage [199, 200]. Therefore, strategies to degrade RelB could suppress the polarization of macrophages toward pro-inflammatory phenotype M1 cells, which might be beneficial for the trentment of MS.
Non-immune CNS cells
The role of oligodendrocytes and astrocytes in MS cannot be ignored by researchers. MS lesions are featured by oligodendrocyte death and axon degeneration. Gupta et al. found that the deficiency of RelB in oligodendrocytes decreased the severity of EAE through promoting survival of mature oligodendrocytes [201]. As the most abundant cell type in the CNS, astrocytes are important regulators of inflammation and essential for maintaining CNS homeostasis [202]. Highly expressed RelB in astrocytes may induce immune tolerance in experimental neuroinflammation due to decreased pro-inflammatory cytokines such as IL-1β, IL-6 and IL-8 [203]. Moreover, the severity of EAE with RelB specifically deleted in astrocytes is similar with control mice [201]. Taken together, regulating the expression of RelB in oligodendrocytes and astrocytes may be an option to treat MS in the future.
RelB as a future therapeutic target for MS
As mentioned above, RelB has pleiotropic effects on MS or EAE pathogenesis via a cell type-specific manner. RelB activation or inhibition in specific cell types could be achieved by regulating upstream signaling pathway. While decreased RelB expression in Th1, γδT17, DCs, B cells, macrophages and oligodendrocytes may have a beneficial role in MS or the EAE animal model, suppressive processes in other cell types may also cause greater severity. In this scenario, the use of RelB inhibitors in vivo is still in its infancy, and potential harmful effects must not be ignored by researchers. Therefore, we propose that targeted therapies in more specific cell types, such as RelB-inhibited Th1, γδT17, DCs, B cells, macrophages and oligodendrocytes need to be further investigated. Considering the fact that our research group has successfully alleviated established EAE by adoptive transfer of 1,25-(OH)2D3-induced tolerogenic DCs [173], in the future, the adoptive transfer of RelB-silenced tolerogenic DCs may be a promising strategy for the precise treatment of MS. However, to our knowledge, limited data about RelB in patients with MS has been reported and most work has been mainly focused on the animal models of MS. Only when we are sufficiently knowledgeable should we consider targeting RelB as a clinical approach to treat patients with MS.
Conclusion
Transcription factor RelB, a member of NF-κB family, is essential for lymphoid organ formation and lymphocyte development and function. In MS and its animal model EAE, RelB exerts an impact on Th17, Th1, γδT17, steady-state migratory DCs, DCs, B cells, macrophages, microglia, oligodendrocytes and astrocytes, which provide the theoretic foundation for possible therapies that target RelB. Further studies are still needed to better understand RelB-associated mechanisms and applications.
Availability of data and materials
All work cited is in the public domain.
References
Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25(51):6758–80.
Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4):a000034.
Ryseck RP, Bull P, Takamiya M, Bours V, Siebenlist U, Dobrzanski P, et al. ReiB, a new Rel family transcription activator that can interact with p5O-NF-KB. Mol Cell Biol. 1992;12(2):674–84.
Bours V, Azarenko V, Dejardin E, Siebenlist U. Human RelB (I-Rel) functions as a kappa B site-dependent transactivating member of the family of Rel-related proteins. Oncogene. 1994;9(6):1699–702.
Ruben SM, Klement JF, Coleman TA, Maher M, Chen CH, Rosen CA. I-Rel_ a novel rei-related protein that inhibits NF-KB transcriptional activity. Genes Dev. 1992;6(5):745–60.
Carrasco D, Ryseck RP, Bravo R. Expression of relB transcripts during lymphoid organ development: specific expression in dendritic antigen-presenting cells. Development. 1993;118(4):1221–31.
Dobrzanski P, Ryseck RP, Bravo R. Both N- and C-terminals domains of Relb are required for full transactivation: role of the N-terminal leucine zipper-like motif. Mol Cell Biol. 1993;13(3):1572–82.
Merico D, Sharfe N, Hu P, Herbrick J-A, Roifman CM. RelB deficiency causes combined immunodeficiency. LymphoSign J. 2015;2(3):147–55.
Millet P, McCall C, Yoza B. RelB: an outlier in leukocyte biology. J Leukoc Biol. 2013;94(5):941–51.
Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, et al. Multiple sclerosis. Nat Rev Dis Prim. 2018;4(1):43.
Magyari M, Koch-Henriksen N, Pfleger CC, Sorensen PS. Gender and autoimmune comorbidity in multiple sclerosis. Mult Scler. 2014;20(9):1244–51.
Milo R, Miller A. Revised diagnostic criteria of multiple sclerosis. Autoimmun Rev. 2014;13(4–5):518–24.
Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–17.
Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162–73.
Katrych O, Simone TM, Azad S, Mousa SA. Disease-modifying agents in the treatment of multiple sclerosis: a review of long-term outcomes. CNS Neurol Disord Drug Targets. 2009;8(6):512–9.
Ulzheimer JC, Meuth SG, Bittner S, Kleinschnitz C, Kieseier BC, Wiendl H. Therapeutic approaches to multiple sclerosis: an update on failed, interrupted, or inconclusive trials of immunomodulatory treatment strategies. BioDrugs. 2010;24(4):249–74.
Vargas DL, Tyor WR. Update on disease-modifying therapies for multiple sclerosis. J Investig Med. 2017;65(5):883–91.
Yue Y, Stone S, Lin W. Role of nuclear factor kappaB in multiple sclerosis and experimental autoimmune encephalomyelitis. Neural Regen Res. 2018;13(9):1507–15.
Z Q, L MJ, B D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell. 2017;168(null):37–57.
S O, R CM. NF-κB pathway and the goldilocks principle: lessons from human disorders of immunity and inflammation. J Allergy Clin Immunol. 2019;143(5):1688–701.
Miraghazadeh B, Cook MC. Nuclear factor-kappaB in autoimmunity: Man and mouse. Front Immunol. 2018;9:613.
Data from: RELB RELB proto-oncogene, NF-kB subunit [ Homo sapiens (human) ] in the Gene database. Gene ID: 5971, updated on 21-Apr-2019. https://www.ncbi.nlm.nih.gov/gene/5971.
Dong X, Craig T, Xing N, Bachman LA, Paya CV, Weih F, et al. Direct transcriptional regulation of RelB by 1alpha,25-dihydroxyvitamin D3 and its analogs: physiologic and therapeutic implications for dendritic cell function. J Biol Chem. 2003;278(49):49387–5.
Carrasco D, Ryseck RP, Bravo R. Expression of relB transcripts during lymphoid organ development: specific expression in dendritic antigen-presenting cells. Development (Cambridge, England). 1993;118(4):1221–31.
Lernbecher T, Muller U, Wirth T. Distinct NF-kappa B/Rel transcription factors are responsible for tissue-specific and inducible gene activation. Nature. 1993;365(6448):767–70.
Ammon C, Mondal K, Andreesen R, Krause SW. Differential expression of the transcription factor NF-kappaB during human mononuclear phagocyte differentiation to macrophages and dendritic cells. Biochem Biophys Res Commun. 2000;268(1):99–105.
Lernbecher T, Kistler B, Wirth T. Two distinct mechanisms contribute to the constitutive activation of RelB in lymphoid cells. EMBO J. 1994;13(17):4060–9.
Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344–62.
Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60.
Fusco AJ, Savinova OV, Talwar R, Kearns JD, Hoffmann A, Ghosh G. Stabilization of RelB requires multidomain interactions with p100/p52. J Biol Chem. 2008;283(18):12324–32.
Dobrzanski P, Ryseck RP, Bravo R. Differential interactions of Rel-NF-kappa B complexes with I kappa B alpha determine pools of constitutive and inducible NF-kappa B activity. Embo J. 1994;13(19):4608–16.
Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, et al. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol Cell. 2011;43(5):798–810.
Chen X, El Gazzar M, Yoza BK, McCall CE. The NF-kappaB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J Biol Chem. 2009;284(41):27857–65.
Croxton R, Puto LA, de Belle I, Thomas M, Torii S, Hanaii F, et al. Daxx represses expression of a subset of antiapoptotic genes regulated by nuclear factor-kappaB. Cancer Res. 2006;66(18):9026–35.
Marienfeld R, Berberich-Siebelt F, Berberich I, Denk A, Serfling E, Neumann M. Signal-specific and phosphorylation-dependent RelB degradation: a potential mechanism of NF-kappaB control. Oncogene. 2001;20(56):8142–7.
Authier H, Billot K, Derudder E, Bordereaux D, Riviere P, Rodrigues-Ferreira S, et al. IKK phosphorylates RelB to modulate its promoter specificity and promote fibroblast migration downstream of TNF receptors. Proc Natl Acad Sci U S A. 2014;111(41):14794–9.
Maier HJ, Marienfeld R, Wirth T, Baumann B. Critical role of RelB serine 368 for dimerization and p100 stabilization. J Biol Chem. 2003;278(40):39242–50.
Leidner J, Palkowitsch L, Marienfeld U, Fischer D, Marienfeld R. Identification of lysine residues critical for the transcriptional activity and polyubiquitination of the NF-kappaB family member RelB. Biochem J. 2008;416(1):117–27.
Leidner J, Voogdt C, Niedenthal R, Moller P, Marienfeld U, Marienfeld RB. SUMOylation attenuates the transcriptional activity of the NF-kappaB subunit RelB. J Cell Biochem. 2014;115(8):1430–40.
Baud V, Collares D. Post-Translational Modifications of RelB NF-kappaB Subunit and Associated Functions. Cells. 2016;5(2):22.
Sun SC. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011;21(1):71–85.
Sun SC. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17(9):545–58.
Razani B, Reichardt AD, Cheng G. Non-canonical NF-kappaB signaling activation and regulation: principles and perspectives. Immunol Rev. 2011;244(1):44–54.
Mukherjee T, Chatterjee B, Dhar A, Bais SS, Chawla M, Roy P, et al. A TNF-p100 pathway subverts noncanonical NF-kappaB signaling in inflamed secondary lymphoid organs. EMBO J. 2017;36(23):3501–16.
Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25(6):280–8.
Sun SC. The noncanonical NF-kappaB pathway. Immunol Rev. 2012;246(1):125–40.
Cildir G, Low KC, Tergaonkar V. Noncanonical NF-kappaB signaling in health and disease. Trends Mol Med. 2016;22(5):414–29.
Sharfe N, Merico D, Karanxha A, Macdonald C, Dadi H, Ngan B, et al. The effects of RelB deficiency on lymphocyte development and function. J Autoimmun. 2015;65:90–100.
Riemann M, Andreas N, Fedoseeva M, Meier E, Weih D, Freytag H, et al. Central immune tolerance depends on crosstalk between the classical and alternative NF-kappaB pathways in medullary thymic epithelial cells. J Autoimmun. 2017;81:56–67.
Akiyama T, Shinzawa M, Akiyama N. TNF receptor family signaling in the development and functions of medullary thymic epithelial cells. Front Immunol. 2012;3:278.
Naspetti M, Aurrand-Lions M, DeKoning J, Malissen M, Galland F, Lo D, et al. Thymocytes and RelB-dependent medullary epithelial cells provide growth-promoting and organization signals, respectively, to thymic medullary stromal cells. Eur J Immunol. 1997;27(6):1392–7.
Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, et al. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373(6514):531–6.
Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, et al. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. 1995;80(2):331–40.
Zuklys S, Balciunaite G, Agarwal A, Fasler-Kan E, Palmer E, Hollander GA. Normal thymic architecture and negative selection are associated with Aire expression, the gene defective in the autoimmune-polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). J Immunol. 2000;165(4):1976–83.
Yilmaz ZB, Weih DS, Sivakumar V. RelB is required for Peyer’s patch development_ differential regulation of p52–RelB by lymphotoxin and TNF. EMBO J. 2003;22(1):121–30.
Weih DS, Yilmaz ZB, Weih F. Essential role of RelB in germinal center and marginal zone formation and proper expression of homing chemokines. J Immunol. 2001;167(4):1909–19.
Lo JC, Basak S, James ES, Quiambo RS, Kinsella MC, Alegre ML, et al. Coordination between NF-kappaB family members p50 and p52 is essential for mediating LTbetaR signals in the development and organization of secondary lymphoid tissues. Blood. 2006;107(3):1048–55.
Seki T, Yamamoto M, Taguchi Y, Miyauchi M, Akiyama N, Yamaguchi N, et al. Visualization of RelB expression and activation at the single-cell level during dendritic cell maturation in Relb-Venus knock-in mice. J Biochem. 2015;158(6):485–95.
Zanetti M, Castiglioni P, Schoenberger S, Gerloni M. The role of relB in regulating the adaptive immune response. Ann N Y Acad Sci. 2003;987:249–57.
Vogel CF, Wu D, Goth SR, Baek J, Lollies A, Domhardt R, et al. Aryl hydrocarbon receptor signaling regulates NF-kappaB RelB activation during dendritic-cell differentiation. Immunol Cell Biol. 2013;91(9):568–75.
Briseno CG, Gargaro M, Durai V, Davidson JT, Theisen DJ, Anderson DA 3rd, et al. Deficiency of transcription factor RelB perturbs myeloid and DC development by hematopoietic-extrinsic mechanisms. Proc Natl Acad Sci U S A. 2017;114(15):3957–62.
Caamano J, Alexander J, Craig L, Bravo R, Hunter CA. The NF-kappa B family member RelB is required for innate and adaptive immunity to toxoplasma gondii. J Immunol. 1999;163(8):4453–61.
Koliesnik IO, Andreas N, Romanov VS, Sreekantapuram S, Krljanac B, Haenold R, et al. RelB regulates Th17 differentiation in a cell-intrinsic manner. Immunobiology. 2018;223(2):191–9.
Kurosawa M, Arakaki R, Yamada A, Tsunematsu T, Kudo Y, Sprent J, et al. NF-kappaB2 Controls the Migratory Activity of Memory T Cells by Regulating Expression of CXCR4 in a Mouse Model of Sjogren's Syndrome. Arthritis Rheumatol. 2017;69(11):2193–202.
Almaden JV, Liu YC. B-cel survival and development controlled by the coordination of NF-kappaB family members RelB and cRel. Blood. 2016;127(10):1276–86.
De Silva NS, Silva K, Anderson MM, Bhagat G, Klein U. Impairment of mature B cell maintenance upon combined deletion of the alternative NF-kappaB transcription factors RELB and NF-kappaB2 in B cells. J Immunol. 2016;196(6):2591–601.
Zhang TT, Gonzalez DG, Cote CM, Kerfoot SM, Deng S, Cheng Y, et al. Germinal center B cell development has distinctly regulated stages completed by disengagement from T cell help. eLife. 2017;6:e19552.
Takahama Y. Journey through the thymus: stromal guides for T-cell development and selection. Nat Rev Immunol. 2006;6(2):127–35.
Anderson G, Takahama Y. Thymic epithelial cells: working class heroes for T cell development and repertoire selection. Trends Immunol. 2012;33(6):256–63.
Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–64.
Proekt I, Miller CN, Lionakis MS, Anderson MS. Insights into immune tolerance from AIRE deficiency. Curr Opin Immunol. 2017;49:71–8.
Kappler JW, Roehm N, Marrack P. T cell tolerance by clonal elimination in the thymus. Cell. 1987;49(2):273–80.
Jenkinson WE, McCarthy NI, Dutton EE, Cowan JE, Parnell SM, White AJ, et al. Natural Th17 cells are critically regulated by functional medullary thymic microenvironments. J Autoimmun. 2015;63:13–22.
Abramson J, Anderson G. Thymic epithelial cells. Annu Rev Immunol. 2017;35:85–118.
Rodrigues PM, Peterson P, Alves NL. Setting up the perimeter of tolerance: insights into mTEC physiology. Trends Immunol. 2018;39(1):2–5.
Baik S, Sekai M, Hamazaki Y, Jenkinson WE, Anderson G. Relb acts downstream of medullary thymic epithelial stem cells and is essential for the emergence of RANK(+) medullary epithelial progenitors. Eur J Immunol. 2016;46(4):857–62.
Akiyama T, Shimo Y, Yanai H, Qin J, Ohshima D, Maruyama Y, et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity. 2008;29(3):423–37.
Hikosaka Y, Nitta T, Ohigashi I, Yano K, Ishimaru N, Hayashi Y, et al. The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity. 2008;29(3):438–50.
Roberts NA, White AJ, Jenkinson WE, Turchinovich G, Nakamura K, Withers DR, et al. Rank signaling links the development of invariant gammadelta T cell progenitors and Aire(+) medullary epithelium. Immunity. 2012;36(3):427–37.
Ruddle NH, Akirav EM. Secondary lymphoid organs: responding to genetic and environmental cues in ontogeny and the immune response. J Immunol. 2009;183(4):2205–12.
Weih F, Caamano J. Regulation of secondary lymphoid organ development by the nuclear factor-kappaB signal transduction pathway. Immunol Rev. 2003;195:91–105.
van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10(9):664–74.
Cyster JG. Chemokines and cell migration in secondary lymphoid organs. Science (New York, NY). 1999;286(5447):2098–102.
Gunn MD, Ngo VN, Ansel KM, Ekland EH, Cyster JG, Williams LT. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt's lymphoma receptor-1. Nature. 1998;391(6669):799–803.
Reis e Sousa C. Activation of dendritic cells: translating innate into adaptive immunity. Curr Opin Immunol. 2004;16(1):21–5.
Wu L, D'Amico A, Winkel KD, Suter M, Lo D, Shortman K. RelB is essential for the development of myeloid-related CD8alpha-dendritic cells but not of lymphoid-related CD8alpha+ dendritic cells. Immunity. 1998;9(6):839–47.
Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604.
Gasparini C, Foxwell BM, Feldmann M. RelB/p50 regulates CCL19 production, but fails to promote human DC maturation. Eur J Immunol. 2009;39(8):2215–23.
Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity. 2018;48(2):202–13.
Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. 2014;14(1):24–35.
Gajewski TF, Schell SR, Nau G, Fitch FW. Regulation of T-cell activation: differences among T-cell subsets. Immunol Rev. 1989;111:79–110.
Sakaguchi S, Wing K, Miyara M. Regulatory T cells - a brief history and perspective. Eur J Immunol. 2007;37(Suppl 1):S116–23.
Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Pillars article: two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999. 401: 708-712. J Immunol. 2014;192(3):840–4.
Matsumoto M, Yamada T, Yoshinaga SK, Boone T, Horan T, Fujita S, et al. Essential role of NF-kappa B-inducing kinase in T cell activation through the TCR/CD3 pathway. J Immunol. 2002;169(3):1151–8.
Ishimaru N, Kishimoto H, Hayashi Y, Sprent J. Regulation of naive T cell function by the NF-kappaB2 pathway. Nat Immunol. 2006;7(7):763–72.
Yu J, Zhou X, Nakaya M, Jin W, Cheng X, Sun SC. T cell-intrinsic function of the noncanonical NF-kappaB pathway in the regulation of GM-CSF expression and experimental autoimmune encephalomyelitis pathogenesis. J Immunol. 2014;193(1):422–30.
MacLennan IC. Germinal centers. Annu Rev Immunol. 1994;12:117–39.
Kosco-Vilbois MH, Bonnefoy JY, Chvatchko Y. The physiology of murine germinal center reactions. Immunol Rev. 1997;156:127–36.
Fagarasan S, Honjo T. T-Independent immune response: new aspects of B cell biology. Science. 2000;290(5489):89–92.
Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, et al. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science. 2001;293(5537):2108–11.
Shulga-Morskaya S, Dobles M, Walsh ME, Ng LG, MacKay F, Rao SP, et al. B cell-activating factor belonging to the TNF family acts through separate receptors to support B cell survival and T cell-independent antibody formation. J Immunol. 2004;173(4):2331–41.
Yan M, Brady JR, Chan B, Lee WP, Hsu B, Harless S, et al. Identification of a novel receptor for B lymphocyte stimulator that is mutated in a mouse strain with severe B cell deficiency. Curr Biol. 2001;11(19):1547–52.
Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, et al. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. 2001;293(5537):2111–4.
Sasaki Y, Casola S, Kutok JL, Rajewsky K, Schmidt-Supprian M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J Immunol. 2004;173(4):2245–52.
Morrison MD, Reiley W, Zhang M, Sun SC. An atypical tumor necrosis factor (TNF) receptor-associated factor-binding motif of B cell-activating factor belonging to the TNF family (BAFF) receptor mediates induction of the noncanonical NF-kappaB signaling pathway. J Biol Chem. 2005;280(11):10018–24.
Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol. 2002;3(10):958–65.
Croxford AL, Kurschus FC, Waisman A. Mouse models for multiple sclerosis: historical facts and future implications. Biochim Biophys Acta. 2011;1812(2):177–83.
Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: mechanisms and immunotherapy. Neuron. 2018;97(4):742–68.
Segal BM. The Diversity of Encephalitogenic CD4+ T Cells in Multiple Sclerosis and Its Animal Models. J Clin Med. 2019;8(1):120.
Murphy AC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24(4):641–51.
Palle P, Monaghan KL, Milne SM, ECK W. Cytokine Signaling in Multiple Sclerosis and Its Therapeutic Applications. Medical sciences. 2017;5(4):23.
Tegowski M, Baldwin A. Noncanonical NF-kappaB in Cancer. Biomedicines. 2018;6(2):66.
Giopanou I, Lilis I, Papadaki H, Papadas T, Stathopoulos GT. A link between RelB expression and tumor progression in laryngeal cancer. Oncotarget. 2017;8(69):114019–30.
Qin H, Zhou J, Xu J, Cheng L, Tang Z, Ma H, et al. The nuclear transcription factor RelB functions as an oncogene in human lung adenocarcinoma SPC-A1 cells. Cancer Cell Int. 2018;18:88.
Zhou X, Shan Z, Yang H, Xu J, Li W, Guo F. RelB plays an oncogenic role and conveys chemo-resistance to DLD-1 colon cancer cells. Cancer Cell Int. 2018;18:181.
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126(6):1121–33.
Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28(1):29–39.
Rostami A, Ciric B. Role of Th17 cells in the pathogenesis of CNS inflammatory demyelination. J Neurol Sci. 2013;333(1–2):76–87.
El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12(6):568–75.
Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12(6):560–7.
Tahmasebinia F, Pourgholaminejad A. The role of Th17 cells in auto-inflammatory neurological disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2017;79(Pt B):408–16.
Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177(1):566–73.
Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205(7):1535–41.
Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116(5):1317–26.
Uyttenhove C, Van Snick J. Development of an anti-IL-17A auto-vaccine that prevents experimental auto-immune encephalomyelitis. Eur J Immunol. 2006;36(11):2868–74.
Matusevicius D, Kivisakk P, He B, Kostulas N, Ozenci V, Fredrikson S, et al. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5(2):101–4.
McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med. 2001;194(7):873–82.
Imitola J, Rasouli J, Watanabe F, Mahajan K, Sharan AD, Ciric B, et al. Elevated expression of granulocyte-macrophage colony-stimulating factor receptor in multiple sclerosis lesions. J Neuroimmunol. 2018;317:45–54.
van Langelaar J, van der Vuurst de Vries RM, Janssen M, Wierenga-Wolf AF, Spilt IM, Siepman TA, et al. T helper 17.1 cells associate with multiple sclerosis disease activity: perspectives for early intervention. Brain. 2018;141(5):1334–49.
Brustle A, Brenner D, Knobbe CB, Lang PA, Virtanen C, Hershenfield BM, et al. The NF-kappaB regulator MALT1 determines the encephalitogenic potential of Th17 cells. J Clin Invest. 2012;122(12):4698–709.
Xiao X, Shi X, Fan Y, Wu C, Zhang X, Minze L, et al. The costimulatory receptor OX40 inhibits Interleukin-17 expression through activation of repressive chromatin remodeling pathways. Immunity. 2016;44(6):1271–83.
Park JH, Jeong SY, Choi AJ, Kim SJ. Lipopolysaccharide directly stimulates Th17 differentiation in vitro modulating phosphorylation of RelB and NF-kappaB1. Immunol Lett. 2015;165(1):10–9.
Jadidi-Niaragh F, Mirshafiey A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand J Immunol. 2011;74(1):1–13.
Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol. 2013;13(11):777–89.
Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev. 2012;248(1):205–15.
Traugott U, Lebon P. Multiple sclerosis: involvement of interferons in lesion pathogenesis. Ann Neurol. 1988;24(2):243–51.
Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157(8):3223–7.
Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol. 1996;156(1):5–7.
Tran EH, Prince EN, Owens T. IFN-gamma shapes immune invasion of the central nervous system via regulation of chemokines. J Immunol. 2000;164(5):2759–68.
Krakowski M, Owens T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol. 1996;26(7):1641–6.
Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200(1):79–87.
Corn RA, Hunter C, Liou HC, Siebenlist U, Boothby MR. Opposing roles for RelB and Bcl-3 in regulation of T-box expressed in T cells, GATA-3, and Th effector differentiation. J Immunol. 2005;175(4):2102–10.
Korn T, Petermann F. Development and function of interleukin 17-producing gammadelta T cells. Ann N Y Acad Sci. 2012;1247:34–45.
Powolny-Budnicka I, Riemann M, Tanzer S, Schmid RM, Hehlgans T, Weih F. RelA and RelB transcription factors in distinct thymocyte populations control lymphotoxin-dependent interleukin-17 production in gammadelta T cells. Immunity. 2011;34(3):364–74.
McGinley AM, Edwards SC, Raverdeau M, Mills KHG. Th17cells, gammadelta T cells and their interplay in EAE and multiple sclerosis. J Autoimmun. 2018;S0896-8411(18):30007–6.
Zarobkiewicz MK, Kowalska W, Rolinski J, Bojarska-Junak AA. Gammadelta T lymphocytes in the pathogenesis of multiple sclerosis and experimental autoimmune encephalomyelitis. J Neuroimmunol. 2019;330:67–73.
Wucherpfennig KW, Newcombe J, Li H, Keddy C, Cuzner ML, Hafler DA. Gamma delta T-cell receptor repertoire in acute multiple sclerosis lesions. Proc Natl Acad Sci U S A. 1992;89(10):4588–92.
Rajan AJ, Gao YL, Raine CS, Brosnan CF. A pathogenic role for gamma delta T cells in relapsing-remitting experimental allergic encephalomyelitis in the SJL mouse. J Immunol. 1996;157(2):941–9.
Odyniec A, Szczepanik M, Mycko MP, Stasiolek M, Raine CS, Selmaj KW. Gammadelta T cells enhance the expression of experimental autoimmune encephalomyelitis by promoting antigen presentation and IL-12 production. J Immunol. 2004;173(1):682–94.
Spahn TW, Issazadah S, Salvin AJ, Weiner HL. Decreased severity of myelin oligodendrocyte glycoprotein peptide 33–35-induced experimental autoimmune encephalomyelitis in mice with a disrupted TCR delta chain gene. Eur J Immunol. 1999;29(12):4060–71.
Petermann F, Rothhammer V, Claussen MC, Haas JD, Blanco LR, Heink S, et al. Gammadelta T cells enhance autoimmunity by restraining regulatory T cell responses via an interleukin-23-dependent mechanism. Immunity. 2010;33(3):351–63.
Takenaka MC, Quintana FJ. Tolerogenic dendritic cells. Semin Immunopathol. 2017;39(2):113–20.
Buc M. Role of regulatory T cells in pathogenesis and biological therapy of multiple sclerosis. Mediat Inflamm. 2013;2013:963748.
Roncarolo MG, Gregori S, Bacchetta R, Battaglia M. Tr1 cells and the counter-regulation of immunity: natural mechanisms and therapeutic applications. Curr Top Microbiol Immunol. 2014;380:39–68.
Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, present, and future of regulatory T cell therapy in transplantation and autoimmunity. Front Immunol. 2019;10:43.
Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162(1):1–11.
Kleinewietfeld M, Hafler DA. Regulatory T cells in autoimmune neuroinflammation. Immunol Rev. 2014;259(1):231–44.
Haas J, Hug A, Viehover A, Fritzsching B, Falk CS, Filser A, et al. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol. 2005;35(11):3343–52.
Mastorodemos V, Ioannou M, Verginis P. Cell-based modulation of autoimmune responses in multiple sclerosis and experimental autoimmmune encephalomyelitis: therapeutic implications. Neuroimmunomodulation. 2015;22(3):181–95.
Astier AL, Hafler DA. Abnormal Tr1 differentiation in multiple sclerosis. J Neuroimmunol. 2007;191(1–2):70–8.
Manicassamy S, Pulendran B. Dendritic cell control of tolerogenic responses. Immunol Rev. 2011;241(1):206–27.
Lutz MB, Schuler G. Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol. 2002;23(9):445–9.
Idoyaga J, Fiorese C, Zbytnuik L, Lubkin A, Miller J, Malissen B, et al. Specialized role of migratory dendritic cells in peripheral tolerance induction. J Clin Invest. 2013;123(2):844–54.
Ali N, Zirak B, Truong HA, Maurano MM, Gratz IK, Abbas AK, et al. Skin-resident T cells drive dermal dendritic cell migration in response to tissue self-antigen. J Immunol. 2018;200(9):3100–8.
Azukizawa H, Dohler A, Kanazawa N, Nayak A, Lipp M, Malissen B, et al. Steady state migratory RelB+ langerin+ dermal dendritic cells mediate peripheral induction of antigen-specific CD4+ CD25+ Foxp3+ regulatory T cells. Eur J Immunol. 2011;41(5):1420–34.
Dohler A, Schneider T, Eckert I, Ribechini E, Andreas N, Riemann M, et al. RelB+ steady-state migratory dendritic cells control the peripheral Pool of the natural Foxp3+ regulatory T cells. Front Immunol. 2017;8:726.
Li J, Chen S, Chen W. Role of the NF-kappaB family member RelB in regulation of Foxp3(+) regulatory T cells. In Vivo. 2018;200(4):1325–34.
Xie ZX, Zhang HL, Wu XJ, Zhu J, Ma DH, Jin T. Role of the immunogenic and tolerogenic subsets of dendritic cells in multiple sclerosis. Mediat Inflamm. 2015;2015:513295.
Karni A, Abraham M, Monsonego A, Cai G, Freeman GJ, Hafler D, et al. Innate immunity in multiple sclerosis: myeloid dendritic cells in secondary progressive multiple sclerosis are activated and drive a proinflammatory immune response. J Immunol. 2006;177(6):4196–202.
Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Capello E, Mancardi GL, et al. Dendritic cells in multiple sclerosis lesions: maturation stage, myelin uptake, and interaction with proliferating T cells. J Neuropathol Exp Neurol. 2006;65(2):124–41.
Vaknin-Dembinsky A, Balashov K, Weiner HL. IL-23 is increased in dendritic cells in multiple sclerosis and down-regulation of IL-23 by antisense oligos increases dendritic cell IL-10 production. J Immunol. 2006;176(12):7768–74.
Qiu T, Zhu HC, Liu XH, Dong WC, Weng XD, Hu CH, et al. Lentiviral-mediated shRNA against RelB induces the generation of tolerogenic dendritic cells. Int Immunopharmacol. 2012;12(3):501–9.
Xie Z, Chen J, Zheng C, Wu J, Cheng Y, Zhu S, et al. 1,25-dihydroxyvitamin D3 -induced dendritic cells suppress experimental autoimmune encephalomyelitis by increasing proportions of the regulatory lymphocytes and reducing T helper type 1 and type 17 cells. Immunology. 2017;152(3):414–24.
Lee WP, Willekens B, Cras P, Goossens H, Martinez-Caceres E, Berneman ZN, et al. Immunomodulatory effects of 1,25-Dihydroxyvitamin D3 on dendritic cells promote induction of T cell Hyporesponsiveness to myelin-derived antigens. J Immunol Res. 2016;2016:5392623.
Dong X, Lutz W, Schroeder TM, Bachman LA, Westendorf JJ, Kumar R, et al. Regulation of relB in dendritic cells by means of modulated association of vitamin D receptor and histone deacetylase 3 with the promoter. Proc Natl Acad Sci U S A. 2005;102(44):16007–12.
Mazzola MA, Raheja R, Regev K, Beynon V, von Glehn F, Paul A, et al. Monomethyl fumarate treatment impairs maturation of human myeloid dendritic cells and their ability to activate T cells. Mult Scler. 2019;25(1):63–71.
Zhu HC, Qiu T, Liu XH, Dong WC, Weng XD, Hu CH, et al. Tolerogenic dendritic cells generated by RelB silencing using shRNA prevent acute rejection. Cell Immunol. 2012;274(1–2):12–8.
Wu H, Lo Y, Chan A, Law KS, Mok MY. Rel B-modified dendritic cells possess tolerogenic phenotype and functions on lupus splenic lymphocytes in vitro. Immunology. 2016;149(1):48–61.
Yang H, Zhang Y, Wu M, Li J, Zhou W, Li G, et al. Suppression of ongoing experimental autoimmune myasthenia gravis by transfer of RelB-silenced bone marrow dentritic cells is associated with a change from a T helper Th17/Th1 to a Th2 and FoxP3+ regulatory T-cell profile. Inflamm Res. 2010;59(3):197–205.
Andreas N, Potthast M, Geiselhoringer AL, Garg G, de Jong R, Riewaldt J, et al. RelB Deficiency in Dendritic Cells Protects from Autoimmune Inflammation Due to Spontaneous Accumulation of Tissue T Regulatory Cells. J Immunol. 2019;203(10):2602–13.
Sospedra M. B cells in multiple sclerosis. Curr Opin Neurol. 2018;31(3):256–62.
Rahmanzadeh R, Weber MS, Bruck W, Navardi S, Sahraian MA. B cells in multiple sclerosis therapy-a comprehensive review. Acta Neurol Scand. 2018;137(6):544–56.
Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8(4):345–50.
Menard LC, Minns LA, Darche S, Mielcarz DW, Foureau DM, Roos D, et al. B cells amplify IFN-gamma production by T cells via a TNF-alpha-mediated mechanism. J Immunol. 2007;179(7):4857–66.
Probstel AK, Hauser SL. Multiple sclerosis: B cells take center stage. J Neuroophthalmol. 2018;38(2):251–8.
Nguyen AL, Gresle M, Marshall T, Butzkueven H, Field J. Monoclonal antibodies in the treatment of multiple sclerosis: emergence of B-cell-targeted therapies. Br J Pharmacol. 2017;174(13):1895–907.
Brandle SM, Obermeier B, Senel M, Bruder J, Mentele R, Khademi M, et al. Distinct oligoclonal band antibodies in multiple sclerosis recognize ubiquitous self-proteins. Proc Natl Acad Sci U S A. 2016;113(28):7864–9.
Bankoti J, Apeltsin L, Hauser SL, Allen S, Albertolle ME, Witkowska HE, et al. In multiple sclerosis, oligoclonal bands connect to peripheral B-cell responses. Ann Neurol. 2014;75(2):266–76.
Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P, et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med. 2015;7(310):310ra166.
Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med. 2012;209(5):1001–10.
Staun-Ram E, Miller A. Effector and regulatory B cells in Multiple Sclerosis. Clin Immunol. 2017;184:11–25.
Bjarnadottir K, Benkhoucha M, Merkler D, Weber MS, Payne NL, Bernard CC, et al. B cell-derived transforming growth factor-beta1 expression limits the induction phase of autoimmune neuroinflammation. Sci Rep. 2016;6:34594.
Han J, Sun L, Fan X, Wang Z, Cheng Y, Zhu J, et al. Role of regulatory b cells in neuroimmunologic disorders. J Neurosci Res. 2016;94(8):693–701.
Ray A, Mann MK, Basu S, Dittel BN. A case for regulatory B cells in controlling the severity of autoimmune-mediated inflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J Neuroimmunol. 2011;230(1–2):1–9.
De Silva NS, Anderson MM, Carette A, Silva K, Heise N, Bhagat G, et al. Transcription factors of the alternative NF-kappaB pathway are required for germinal center B-cell development. Proc Natl Acad Sci U S A. 2016;113(32):9063–8.
Chu F, Shi M, Zheng C, Shen D, Zhu J, Zheng X, et al. The roles of macrophages and microglia in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neuroimmunol. 2018;318:1–7.
Fani Maleki A, Rivest S. Innate Immune Cells: Monocytes, Monocyte-Derived Macrophages and Microglia as Therapeutic Targets for Alzheimer's Disease and Multiple Sclerosis. Front Cell Neurosci. 2019;13:355.
Gasparini C, Foxwell BM, Feldmann M. RelB/p50 regulates TNF production in LPS-stimulated dendritic cells and macrophages. Cytokine. 2013;61(3):736–40.
Tan HY, Wang N, Man K, Tsao SW, Che CM, Feng Y. Autophagy-induced RelB/p52 activation mediates tumour-associated macrophage repolarisation and suppression of hepatocellular carcinoma by natural compound baicalin. Cell Death Dis. 2015;6(10):e1942 e.
Zhao Z, Hou X, Yin X, Li Y, Duan R, Boyce BF, et al. TNF induction of NF-kappaB RelB enhances RANKL-induced Osteoclastogenesis by promoting inflammatory macrophage differentiation but also limits it through suppression of NFATc1 expression. PLoS One. 2015;10(8):e0135728.
Gupta AS, Biswas DD, Brown LSN, Mockenhaupt K, Marone M, Hoskins A, et al. A detrimental role of RelB in mature oligodendrocytes during experimental acute encephalomyelitis. J Neuroinflammation. 2019;16(1):161.
Ya W, S D, W X. Astrocytes in multiple sclerosis and experimental autoimmune encephalomyelitis: Star-shaped cells illuminating the darkness of CNS autoimmunity. Brain Behav Immun. 2019;80(undefined):10–24.
Gupta AS, Waters MR, Biswas DD, Brown LN, Surace MJ, Floros C, et al. RelB controls adaptive responses of astrocytes during sterile inflammation. Glia. 2019;67(8):1449–61.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the General Program of the National Natural Science Foundation of China (No. 81671177), the Natural Science Foundation of Jilin Province Science and Technology Development Plan Project (20190201043JC), the Technology Innovation Program of Jilin Provincial Health and Family Planning Commission of China (No. 2016 J040), as well as grants from the Swedish Research Council (No. 2015–03005) and grants from the First Hospital, Jilin University of China.
Author information
Authors and Affiliations
Contributions
YMG and ZY contributed equally to drafting the main text. HJ, ZC, LH and ZJ reviewed the literature, proofread and revised the manuscript. JT designed and supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
There are no ethical approval requirements for this review.
Consent for publication
All researchers consent to publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yang, Mg., Sun, L., Han, J. et al. Biological characteristics of transcription factor RelB in different immune cell types: implications for the treatment of multiple sclerosis. Mol Brain 12, 115 (2019). https://doi.org/10.1186/s13041-019-0532-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-019-0532-6