
Abstract
Endothelial nitric oxide (NO) is a significant signaling molecule that regulates cerebral blood flow (CBF), playing a pivotal role in the prevention and treatment of cerebrovascular diseases. However, achieving the expected therapeutic efficacy is difficult using direct administration of NO donors. Therefore, endothelial nitric oxide synthase (eNOS) becomes a potential therapeutic target for cerebrovascular diseases. This review summarizes the current evidence supporting the importance of CBF to cerebrovascular function, and the roles of NO and eNOS in CBF regulation.
Similar content being viewed by others


Background
Cerebrovascular diseases are various vascular diseases of the cerebral circulation. Disrupted arterial oxygen supply to the brain results in cerebrovascular diseases such as transient ischemic attack, stroke, subarachnoid hemorrhage and vascular dementia. Many mechanisms contribute to the complex pathophysiological process. Cerebrovascular diseases have become the major causes of long-term disability and mortality throughout the world.
Currently, the treatment of cerebrovascular diseases includes thrombolytic therapy, anti-platelet aggregation drugs, anticoagulants and neuroprotective agents. However, these treatments all have major limitations, including a short treatment time window, side effects, complications, long-term medication and heavy expenses. Therefore, cerebrovascular diseases are not only substantial medical burdens, but also heavy economic and social burdens. These burdens have stimulated tireless efforts to prevent and cure cerebrovascular diseases, and in particular to define the role of cerebrovascular endothelium in the pathogenesis of cerebrovascular diseases. Cerebrovascular endothelium, as a vascular barrier contacting blood directly, is injured first by vascular risk factors and subsequently forms various cerebrovascular diseases. Interestingly, brain microvascular endothelial cells (BMECs), known as the body’s largest endocrine, paracrine and metabolic organ, can generate and release many vasoactive substances. These include nitric oxide (NO) and endothelin-1 (ET-1), which maintain cerebrovascular homeostasis [1]. In addition, cerebrovascular endothelium plays a critical role in the regulation of cerebral blood flow (CBF). It does so partially through synthesis of NO, a major vasodilator, from L-arginine, by the catalytic reaction of enzyme endothelial nitric oxide synthase (eNOS). Adequate CBF is one of the basic conditions for normal brain function. Endothelial NO production mainly depends on the eNOS activity; therefore, eNOS activation in cerebrovascular endothelium should not be ignored. A recent study has found that in patients exposed to vascular risk factors, endothelium-dependent relaxation dysfunction was detectable before any morphological changes on the vessel wall, implying vascular endothelium is the first to be damaged. More importantly, different risk factors for cerebrovascular diseases including hypertension, hypercholesterolemia, diabetes, aging, smoking, and obesity could all impair contraction and dilation of cerebrovascular endothelium [2]. Therefore, eNOS as an important regulator of cerebrovascular endothelium functions should be recognized as a potential therapeutic target for the prevention and treatment of cerebrovascular diseases.
Cerebral blood flow and neurological function
The brain consumes 20 % of the body’s energy and nutrients. The basilar artery loop connects to the common carotid artery and is spread over almost all of the brain with numerous branches (the anterior middle and posterior cerebral arteries). The branches extend from soft membrane to the white matter constituting the cerebrovascular system. This system plays an irreplaceable role in supporting the brain by providing it with oxygen and nutrients. Under normal physiological conditions, the brain can maintain adequate CBF through its vascular autoregulatory mechanisms in order to support the energy needs of its cellular constituents. However, many pathological conditions including cerebral ischemia, trauma, stroke, and vasospasm after subarachnoid hemorrhage (SAH) cause abnormal CBF, subsequently inducing diverse brain disorders. Abnormalities in cerebral hemodynamics, such as changes in local perfusion pressure and vascular integrity, are important pathophysiological elements in ischemic stroke. This may further damage neurons or glial cells beyond the initial ischemia injury caused by either hypo- or hyperperfusion [3]. Existing evidence suggests that the cerebrovascular endothelial dysfunction, induced by cerebral ischemia and anoxia-caused vasospasm and/or thrombosis, will further reduce CBF. Continuously decreased CBF can gradually cause learning and memory disorders and cognitive dysfunction, leading to the development of dementia [4–6]. Thus, the regulation of CBF is closely related to neurological function. Dilation of blood vessels is an important way to improve CBF. Vascular endothelial cells regulate the vasodilatation by synthesis and secretion of cytokines, such as PGI2 and NO. Studies have shown that the endothelial NO generated in cerebrovascular endothelium is one of the most important signaling molecules of CBF autoregulatory mechanisms [7, 8].
Endothelial oxide synthase in cerebrovascular regulatory mechanisms
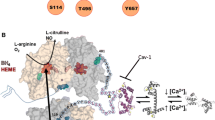
As mentioned above, the brain is endowed with vasoregulatory mechanisms that assure it receives enough blood to support its energy expenditure. Endothelial NO is one of the most pivotal signaling molecules in these mechanisms (Fig. 1) [7, 8]. It is involved in preserving and maintaining the brain’s microcirculation, inhibiting platelet aggregation, leukocyte adhesion and migration, and reducing smooth muscle proliferation. NO plays a pivotal role in regulating cerebrovascular effects by increasing or decreasing oxygen, and elevating carbon dioxide, carbon monoxide and cerebrovascular autoregulation [9, 10]. It is generally acknowledged that the loss of endothelial NO is the central mechanism in the pathogenesis of endothelial dysfunction. In both cerebral and peripheral vasculatures, reduced availability of endothelial NO will result in major detrimental alterations of vascular functions. These include vasoconstriction, an increase in arterial blood pressure, proliferation of vascular smooth muscle cells (VSMCs), platelet aggregation, white blood cell adhesion, and inflammation. Reduced availability of endothelial NO plays an essential role in the initiation and progression of vascular diseases such as atherosclerosis. Therefore, preserving endothelial NO production is an important strategy for the prevention of cerebrovascular diseases [11]. However, diverse vascular risk factors can directly or indirectly reduce endothelial NO production. Oxidative stress caused by excessive production of reactive oxygen species (ROS) and primarily superoxide anions is considered to be the most important mechanism of reducing endothelial NO. On one hand, vascular risk factors promote the up-regulation of NADPH-oxidase activity, generation of superoxide anions, chemical inactivation of NO, and generation of a potent oxidant, peroxynitrite [12]. On the other hand, oxidative stress induced by increased peroxynitrite production may oxidize tetrahydrobiopterin (BH4), an essential co-factor required for eNOS activity. If the concentration of BH4 becomes suboptimal, the eNOS dimer will uncouple from other eNOS monomers, resulting in reducing the production of endothelial NO and increasing the production of superoxide anions and peroxynitrite [13]. It is reported that decreased arterial oxygen supply (hypoxia) increases the CBF baseline in humans [14]. Thus, NO plays a significant role in both the signaling event and the CBF response [15]. Conversely, nonselective NOS inhibition abolishes the CBF increase response to hypoxia in rats [16]. It follows that the NO signaling pathway plays a central role in the CBF response to these alterations, and this post-ischemia pathway dysfunction causes dysregulation of these normal physiological responses. Recent studies on CBF regulation by endothelial NO have provided new insights about the physiological control of cerebrovascular functions and the pathological mechanisms of neurological diseases. They also supply new clues for developing novel therapeutic strategies for cerebral dysfunction [9]. Indeed, eNOS has been paid even more attention than NO because of the instability of NO and the regulatory mechanisms of eNOS on NO production.

Endothelial oxide synthase (eNOS) and its role in cerebral blood flow (CBF) regulatory mechanisms. eNOS is activated by Acetylcholine (ACh), bradykinin, shear stress, etc., and then catalyzes L-arginine to generate NO. It translates into vascular smooth muscle cells, reacts with guanylate cyclase (GC), and promotes the conversion of guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP), resulting in vascular smooth muscle relaxation and the CBF increase
Endothelial nitric oxide synthase and cerebrovascular diseases
eNOS function
Innate eNOS activity determines susceptibility to injury due to its conferring protection against secondary neuronal damage. Impairment of eNOS activity by SAH, traumatic brain injury (TBI) and ischemic stroke has been implicated in many cellular mechanisms of neuronal injury [17].
Delayed cerebral ischemia (DCI) is one of the major causes of morbidity and mortality following SAH. A recent study has demonstrated that the pathogenesis of DCI involves the NO signaling pathway [18]. Meanwhile, genetic variation in the eNOS gene in humans influences the risk of DCI after SAH, because T-786C single nucleotide polymorphism of the eNOS gene promoter can cause lower eNOS activity and result in greater risk [19, 20].
eNOS also plays an important role in maintaining CBF after TBI. Patients with decreased endogenous eNOS levels have poorer outcomes than patients with alleles that do not affect eNOS levels [21]. A previous study has suggested that the immunoreactivity of eNOS is increased in the microvasculature surrounding the area of impact in the first 3 days after experimental trauma [22]. However, eNOS knockout mice have greater reduction in CBF than wildtype variants in the first 2 h after trauma [23]. Genetic variants of eNOS also influence the maintenance of CBF after severe TBI.
Ischemic stroke is most often caused by a thrombotic or embolic blockage of a cerebral artery, which causes blood flow interruption and tissue death. Studies have found that thrombotic cerebral infarctions appear in eNOS+/− mice as early as 3–6 months of age. eNOS knockout mice have bigger infarcts than wildtype variants after ischemic stroke caused by middle cerebral artery occlusion (MCAO) [24, 25]. In contrast, the administration of NO precursor (L-arginine) or donors (sodium nitroprusside (SNP) and 3-morpholino sydnonimine) to rat MCAO models of focal ischemia have improved CBF, prevented cerebral tissue necrosis and decreased brain ATP and glutamate levels. This appears to be time-dependent and limited to the first 30 min after ischemic onset. During this period, NO elicits vasodilatory effects, improves CBF and reduces the infarct size, while it evokes neurovascular toxicity beyond this point [25–27]. Moreover, eNOS not only promotes vasodilation but also increases the proliferation and migration of VSMCs, thereby enhancing arteriogenesis after stroke [28]. Activation of eNOS mediates protection from stroke by preserving CBF and inhibiting inflammation, platelet aggregation, thrombosis, and apoptosis [11]. Continuous voluntary running confers long-term upregulation of eNOS in the vasculature and higher numbers of circulating endothelial progenitor cells (EPCs) in the blood. This nevertheless enhances neovascularization and CBF by eNOS-dependent mechanisms after brain ischemia [29]. eNOS is also essential for mobilization of EPCs. The impaired neovascularization in mice lacking eNOS is related to a defect in progenitor cell mobilization [30].
Uncoupled eNOS in the cerebral arteries has been reported to participate in hypoxic-ischemic brain injury by inducing oxidative stress under ischemia [31]. Uncoupling of eNOS emerges from an absence of either its substrate L-arginine, and/or co-factor BH4, which is required for coupling of the two functional eNOS subunits. This is known to be a common phenomenon in many diseases, such as hypertension, diabetes, and hypercholesterolemia [32, 33]. In an uncoupled state, the electron flow inside the eNOS peptide is diverted to the molecular oxygen, thereby leading to an uncontrolled eNOS uncoupling and enhanced O2 •- production. In summary, normal eNOS function is vital to alleviate cerebrovascular diseases.
Regulatory mechanisms of eNOS activity
eNOS has a homologous dimerized structure consisting of two identical subunits. Each subunit contains a C-terminal reductase domain and an N-terminal oxygenase domain. The former contains binding sites for NADPH, flavin adenine dinucleotide (FAD), and flavin mononucleotide (FMN), in close homology with cytochrome P-450 reductase; the latter binds heme, BH4, and the substrate L-arginine. Between these two regions lies the calmodulin (CaM) binding domain, which plays a key role in both the structure and function of the enzyme. To obtain full catalytic activity under physiological conditions, separate N-terminal oxygenase and C-terminal reductase domains of eNOS must be joined together in the presence of heme, BH4 and Ca2+/CaM complex [7, 34]. Many studies suggest that the enzyme is generally activated by an increase in intracellular Ca2+ via either influx of extracellular Ca2+ or release from intracellular storage sites. However, eNOS activation is also independent of Ca2+, while activated by mechanical forces, including shear stress, cyclic strain, and G protein [35–37].
The regulatory mechanisms of eNOS activity are extremely complex, and can be divided into the genetic and protein level (Fig. 2). At the genetic level, the expression and stability of eNOS genes are related to eNOS activity. The eNOS promoter contains a large number of sites for the binding of transcription factors, including activator protein-1 (AP-1), activator protein-2 (AP-2), endothelin family, nuclear factor-κB (NF-κB), and neurofibromin 1 (NF-1). These transcription factor complexes can regulate eNOS expression. In addition, hypoxia, lipopolysaccharide and several cytokines can induce the expression of two kinds of eNOS cytoplasmic proteins. They bind to the 3′ untranslated region of eNOS mRNA rich in cytosine, subsequently leading to configuration changes and RNA enzyme activation, which cause lower eNOS mRNA stability and shorter half-life. Many factors, such as hypoxia, estrogen, and exercise are now known to affect eNOS expression [38–40]. At the protein level, regulatory mechanisms mainly include eNOS translocation, complex formation and the phosphorylation of the amino acid residues. The association of eNOS with bradykinin B2 receptor and caveolin-1(Cav-1) in the caveolae and microdomains of the endothelial plasma membrane leads to inhibition of eNOS activity. This inhibition is apparently the result of functional interference with CaM binding and electron transfer [41, 42].

The regulatory mechanisms of eNOS activity. The mechanisms work at both the genetic and protein level. At the genetic level, nuclear factor-κB (NF-κB), activator protein-1 (AP-1), activator protein-2 (AP-2), Krűppel-like Factor 2 (KLF2), endothelin family and Forkhead box O1 (Foxo-1) genes regulate eNOS expression; at the protein level, several factors, such as bradykinin, 5-hydroxytryptamine (5-HT), ACh, shear stress, and high density lipoprotein (HDL), promote eNOS phosphorylation by PI3K/Akt, AMPK and MAPK pathways. In addition, binding with calmodulin (CaM) enhances eNOS activation, while caveolin-1(Cav-1) inhibits eNOS activation
Cav-1 on cell membrane binds to not only the oxygenase domain but also the reductase domain of eNOS. Binding of Cav-1 to the eNOS reductase domain inhibits the combination of heme iron with eNOS, and prevents electron transfer from the reductase. In eNOS the Cav-1 binding motif lies between the heme and the CaM binding domains adjacent to a glutamate residue (Glu-361), a necessary site for the binding of L-arginine [42–44]. Thus, Cav-1 binding to eNOS inhibits the trafficking and activation of eNOS. Two new-found proteins, eNOS interacting protein (NOSIP) and eNOS traffic inducer (NOSTRIN), have been confirmed to interact with eNOS and Cav-1 to form a ternary complex. The complex then induces eNOS migration from the plasma membrane and significantly reduces eNOS activity [45–47].
Present studies show that eNOS phosphorylation occurs in its serine, threonine and tyrosine residues. Within these residues, the phosphorylation of the Thr495, Ser633 and Ser1177 sites have the most significant influences on eNOS activity. The phosphorylation of the Thr495 site prevents CaM binding to eNOS, thereby reducing eNOS activity [48], while the phosphorylation of the Ser1177 site enhances the catalytic ability of eNOS through inhibiting the separation of CaM and eNOS, and increases the internal electron transfer rate of eNOS; the phosphorylation of the Ser633 site also enhances eNOS activity [49–51]. The threonine protein kinase (Akt/PKB) is an important determinant of the phosphorylation of the eNOS Ser1177 site, which is involved in the basic activation of eNOS and its agonist activation [52, 53].
Akt mainly resides in the cytoplasm inactively; translocation to the cell membrane induces its activation and the phosphorylation of eNOS [54]. It is directly controlled by the phosphatidylinositol-3 kinase serine (PI3K)-dependent phosphorylation pathway. PI3K recruits Akt to cell membrane and phosphorylates it [55]. Bradykinin and 5-hydroxytryptamine (5-HT) act with membrane receptors, leading to the increase of cytoplasmic Ca2+ concentration and activation of CaM. It first promotes eNOS uncoupling with Cav-1; eNOS then couples with CaM to form an eNOS/CaM complex and translocates to the cytoplasmic membrane. After translocation, the eNOS/CaM complex induces phosphorylation of eNOS amino acid residues at the Ser1177 site and dephosphorylation at the Thr495 site through the Akt/PKB pathway, resulting in eNOS activation [56–58].
Meanwhile, after cytoplasmic Ca2+ concentration increases, Ca2+/calmodulin- dependent protein kinase kinase-β (CaMKKβ), an alternative upstream kinase for adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) and a key molecule of the AMPK signaling pathway, induces the phosphorylation of AMPK at the Thr172 site. Upon activation, AMPK promotes the phosphorylation of eNOS at the Ser1177 and/or Ser633 sites, enhances eNOS activity and promotes NO synthesis. It is restrained by Compound C, a specific inhibitor of AMPK [59–63]. Interestingly, eNOS Ser1177 phosphorylation has dual functions after ischemic stroke. eNOS expression, eNOS Ser1177 phosphorylation and eNOS monomer formation is increased in the acute phase, but Ser1177 phosphorylation is markedly decreased in the later phases of ischemia. Uncoupling of eNOS is significantly increased in this phase. Because eNOS dysfunction contributes to secondary injury and inhibits tissue repair, dephosphorylation and monomer formation of eNOS may become a potential therapeutic target of cerebral ischemia in the later phases [64]. The Ser1177 site may be the more important targeting mechanism compared with the Ser633 site.
Furthermore, eNOS can be activated by either shear stress through phosphorylation of Ser1177 via the PI3K/Akt pathway [50], or by high density lipoprotein (HDL) modification by Akt kinase and mitogen-activated protein kinase (MAPK) [65].
In summary, the Ser1177and Ser633 sites may be potential therapeutic target sites. Regulating the phosphorylation of these two sites and the eNOS activation are regulatory mechanisms for CBF.
The candidates for eNOS targeted therapy
Recent studies have demonstrated that statins have a potential to enhance the bioavailability of endothelium-derived NO, principally by up-regulating and activating eNOS. This is mediated by diverse signaling, such as Rho/ROCK, PI3K/Akt, caveolin, ERK 1/2, and so on [66, 67]. Similarly, atorvastatin and simvastatin could increase eNOS expression by inhibiting Rho-kinase and increasing Ras-mediated activation of the PI3K-Akt and ERK1/2-RSK signaling pathways, respectively [68, 69]. Additionally, the Rho-kinase inhibitor fasudil ameliorates brain tissue injury via rapid augmentation of eNOS phosphorylation, suppressing eNOS dephosphorylation and monomerization in the ischemic brain [64].
Conclusion
NO is an important mediator in the regulation of CBF. However, the NO signaling pathway influences the evolution of multiple aspects of secondary brain injury and interacts with many other signaling pathways, which makes it difficult to translate results from animal models to patients. A systematic review of NO donor administration during animal stroke models demonstrates an overall improvement in CBF and a decrease in infarction volume [70]. However, whether these results can be translated into patients with cerebral ischemia/reperfusion injury (e.g. ischemic stroke, cardiac arrest) has not yet been tested [71]. It is also likely that many of the neuroprotective effects of NO donors are the result of not only the improvement of CBF, but also the NO donors’ metabolic and cellular effects.
Therefore, it is difficult to use direct administration of NO donors to achieve the expected therapeutic efficacy. As a key enzyme of NO synthase, eNOS becomes a potential therapeutic target for the prevention and treatment of cerebrovascular diseases. There is numerous evidence to suggest that NO derived from eNOS is neuroprotective after an acquired brain injury. The modulation of eNOS during and/or following ischemic injury often represents a futile compensatory mechanism due to a significant decrease in NO bioavailability, coupled with a dramatic increase in the ROS levels that further neutralize NO [19]. Thus, the modulation of eNOS used in combination with an antioxidant may be significantly more effective. A better understanding of the roles of eNOS might lead to the development of exciting new pharmacotherapies to mitigate secondary brain injury, with the aim of reducing the morbidity and mortality from cerebrovascular diseases.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- 5-HT:
-
5-hydroxytryptamine
- Ach:
-
acetylcholine
- Akt:
-
protein kinase B
- Akt/PKB:
-
threonine protein kinase
- AMP:
-
adenosine 5′-monophosphate
- AMPK:
-
adenosine 5′-monophosphate (AMP)-activated protein kinase
- AP-1:
-
activator protein-1
- AP-2:
-
activator protein-2
- BH4:
-
tetrahydrobiopterin
- BMECs:
-
brain microvascular endothelial cells
- CaM:
-
calmodulin
- CaMKKβ:
-
Ca2+/calmodulin-dependent protein kinase kinase-β
- Cav-1:
-
caveolin-1
- CBF:
-
cerebral blood flow
- cGMP:
-
cyclic guanosine monophosphate
- DCI:
-
delayed cerebral ischemia
- eNOS:
-
endothelial nitric oxide synthase
- ET-1:
-
endothelin-1
- FAD:
-
flavin adenine dinucleotide
- FMN:
-
flavin mononucleotide
- Foxo-1:
-
Forkhead box O 1
- GC:
-
guanylate cyclase
- GTP:
-
guanosine triphosphate
- HDL:
-
high density lipoprotein
- KLF2:
-
Krűppel-like Factor 2
- MAPK:
-
mitogen-activated protein kinase
- MCAO:
-
middle cerebral artery occlusion
- NF-1:
-
neurofibromin 1
- NF-κB:
-
nuclear factor-κB
- NO:
-
nitric oxide
- NOSIP:
-
eNOS interacting protein
- NOSTRIN:
-
eNOS traffic inducer
- PI3K:
-
phosphatidylinositol-3 kinaseserine
- ROS:
-
reactive oxygen species
- SAH:
-
subarachnoid hemorrhage
- SNP:
-
sodium nitroprusside
- TBI:
-
traumatic brain injury
- VSMCs:
-
vascular smooth muscle cells
References
Anggard EE. The endothelium--the body’s largest endocrine gland? J Endocrinol. 1990;127(3):371–5.
Katusic ZS, Austin SA. Endothelial nitric oxide: protector of a healthy mind. Eur Heart J. 2014;35(14):888–94. doi:10.1093/eurheartj/eht544.
Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468(7321):232–43. doi:10.1038/nature09613.
Gao YZ, Zhang JJ, Liu H, Wu GY, Xiong L, Shu M. Regional cerebral blood flow and cerebrovascular reactivity in Alzheimer’s disease and vascular dementia assessed by arterial spinlabeling magnetic resonance imaging. Curr Neurovasc Res. 2013;10(1):49–53.
Sabayan B, Jansen S, Oleksik AM, van Osch MJ, van Buchem MA, van Vliet P, et al. Cerebrovascular hemodynamics in Alzheimer’s disease and vascular dementia: a meta-analysis of transcranial Doppler studies. Ageing Res Rev. 2012;11(2):271–7. doi:10.1016/j.arr.2011.12.009.
Jellinger KA. Pathology and pathogenesis of vascular cognitive impairment-a critical update. Front Aging Neurosci. 2013;5:17. doi:10.3389/fnagi.2013.00017.
Andrew PJ, Mayer B. Enzymatic function of nitric oxide synthases. Cardiovasc Res. 1999;43(3):521–31.
Drake CT, Iadecola C. The role of neuronal signaling in controlling cerebral blood flow. Brain Lang. 2007;102(2):141–52. doi:10.1016/j.bandl.2006.08.002.
Toda N, Ayajiki K, Okamura T. Cerebral blood flow regulation by nitric oxide: recent advances. Pharmacol Rev. 2009;61(1):62–97. doi:10.1124/pr.108.000547.
Broos K, Feys HB, De Meyer SF, Vanhoorelbeke K, Deckmyn H. Platelets at work in primary hemostasis. Blood Rev. 2011;25(4):155–67. doi:10.1016/j.blre.2011.03.002.
Atochin DN, Huang PL. Endothelial nitric oxide synthase transgenic models of endothelial dysfunction. Pflugers Arch. 2010;460(6):965–74. doi:10.1007/s00424-010-0867-4.
Beckman JS. Understanding peroxynitrite biochemistry and its potential for treating human diseases. Arch Biochem Biophys. 2009;484(2):114–6. doi:10.1016/j.abb.2009.03.013.
Katusic ZS, d’Uscio LV, Nath KA. Vascular protection by tetrahydrobiopterin: progress and therapeutic prospects. Trends Pharmacol Sci. 2009;30(1):48–54. doi:10.1016/j.tips.2008.10.003.
Mintun MA, Lundstrom BN, Snyder AZ, Vlassenko AG, Shulman GL, Raichle ME. Blood flow and oxygen delivery to human brain during functional activity: theoretical modeling and experimental data. Proc Natl Acad Sci U S A. 2001;98(12):6859–64. doi:10.1073/pnas.111164398.
Ho JJ, Man HS, Marsden PA. Nitric oxide signaling in hypoxia. J Mol Med. 2012;90(3):217–31. doi:10.1007/s00109-012-0880-5.
Takuwa H, Matsuura T, Bakalova R, Obata T, Kanno I. Contribution of nitric oxide to cerebral blood flow regulation under hypoxia in rats. J Physiol Sci. 2010;60(6):399–406. doi:10.1007/s12576-010-0108-9.
Srivastava K, Bath PM, Bayraktutan U. Current therapeutic strategies to mitigate the eNOS dysfunction in ischaemic stroke. Cell Mol Neurobiol. 2012;32(3):319–36. doi:10.1007/s10571-011-9777-z.
Vellimana AK, Milner E, Azad TD, Harries MD, Zhou ML, Gidday JM, et al. Endothelial nitric oxide synthase mediates endogenous protection against subarachnoid hemorrhage-induced cerebral vasospasm. Stroke. 2011;42(3):776–82. doi:10.1161/STROKEAHA.110.607200.
Khurana VG, Sohni YR, Mangrum WI, McClelland RL, O’Kane DJ, Meyer FB, et al. Endothelial nitric oxide synthase gene polymorphisms predict susceptibility to aneurysmal subarachnoid hemorrhage and cerebral vasospasm. J Cereb Blood Flow Metab. 2004;24(3):291–7. doi:10.1097/01.WCB.0000110540.96047.C7.
Starke RM, Kim GH, Komotar RJ, Hickman ZL, Black EM, Rosales MB, et al. Endothelial nitric oxide synthase gene single-nucleotide polymorphism predicts cerebral vasospasm after aneurysmal subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2008;28(6):1204–11. doi:10.1038/jcbfm.2008.11.
Robertson CS, Gopinath SP, Valadka AB, Van M, Swank PR, Goodman JC. Variants of the endothelial nitric oxide gene and cerebral blood flow after severe traumatic brain injury. J Neurotrauma. 2011;28(5):727–37. doi:10.1089/neu.2010.1476.
Cobbs CS, Fenoy A, Bredt DS, Noble LJ. Expression of nitric oxide synthase in the cerebral microvasculature after traumatic brain injury in the rat. Brain Res. 1997;751(2):336–8.
Lundblad C, Grande PO, Bentzer P. Hemodynamic and histological effects of traumatic brain injury in eNOS-deficient mice. J Neurotrauma. 2009;26(11):1953–62. doi:10.1089/neu.2009.0955.
Tan XL, Xue YQ, Ma T, Wang X, Li JJ, Lan L, et al. Partial eNOS deficiency causes spontaneous thrombotic cerebral infarction, amyloid angiopathy and cognitive impairment. Mol Neurodegener. 2015;10:24. doi:10.1186/s13024-015-0020-0.
Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, et al. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab. 1996;16(5):981–7. doi:10.1097/00004647-199609000-00023.
Morikawa E, Moskowitz MA, Huang Z, Yoshida T, Irikura K, Dalkara T. L-arginine infusion promotes nitric oxide-dependent vasodilation, increases regional cerebral blood flow, and reduces infarction volume in the rat. Stroke. 1994;25(2):429–35.
Salom JB, Orti M, Centeno JM, Torregrosa G, Alborch E. Reduction of infarct size by the NO donors sodium nitroprusside and spermine/NO after transient focal cerebral ischemia in rats. Brain Res. 2000;865(2):149–56.
Cui X, Chopp M, Zacharek A, Zhang C, Roberts C, Chen J. Role of endothelial nitric oxide synthetase in arteriogenesis after stroke in mice. Neuroscience. 2009;159(2):744–50. doi:10.1016/j.neuroscience.2008.12.055.
Gertz K, Priller J, Kronenberg G, Fink KB, Winter B, Schrock H, et al. Physical activity improves long-term stroke outcome via endothelial nitric oxide synthase-dependent augmentation of neovascularization and cerebral blood flow. Circ Res. 2006;99(10):1132–40. doi:10.1161/01.RES.0000250175.14861.77.
Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, et al. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9(11):1370–6. doi:10.1038/nm948.
Fabian RH, Perez-Polo JR, Kent TA. Perivascular nitric oxide and superoxide in neonatal cerebral hypoxia-ischemia. Am J Physiol Heart Circ Physiol. 2008;295(4):H1809–14. doi:10.1152/ajpheart.00301.2007.
Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998;95(16):9220–5.
Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111(8):1201–9. doi:10.1172/JCI14172.
Stuehr DJ. Mammalian nitric oxide synthases. Biochim Biophys Acta. 1999;1411(2–3):217–30.
Nishida K, Harrison DG, Navas JP, Fisher AA, Dockery SP, Uematsu M, et al. Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J Clin Invest. 1992;90(5):2092–6. doi:10.1172/JCI116092.
Awolesi MA, Sessa WC, Sumpio BE. Cyclic strain upregulates nitric oxide synthase in cultured bovine aortic endothelial cells. J Clin Invest. 1995;96(3):1449–54. doi:10.1172/JCI118181.
Ohno M, Gibbons GH, Dzau VJ, Cooke JP. Shear stress elevates endothelial cGMP. Role of a potassium channel and G protein coupling. Circulation. 1993;88(1):193–7.
Liao JK, Zulueta JJ, Yu FS, Peng HB, Cote CG, Hassoun PM. Regulation of bovine endothelial constitutive nitric oxide synthase by oxygen. J Clin Invest. 1995;96(6):2661–6. doi:10.1172/JCI118332.
Goetz RM, Morano I, Calovini T, Studer R, Holtz J. Increased expression of endothelial constitutive nitric oxide synthase in rat aorta during pregnancy. Biochem Biophys Res Commun. 1994;205(1):905–10. doi:10.1006/bbrc.1994.2750.
Sessa WC, Pritchard K, Seyedi N, Wang J, Hintze TH. Chronic exercise in dogs increases coronary vascular nitric oxide production and endothelial cell nitric oxide synthase gene expression. Circ Res. 1994;74(2):349–53.
Ju H, Venema VJ, Marrero MB, Venema RC. Inhibitory interactions of the bradykinin B2 receptor with endothelial nitric-oxide synthase. J Biol Chem. 1998;273(37):24025–9.
Bucci M, Gratton JP, Rudic RD, Acevedo L, Roviezzo F, Cirino G, et al. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat Med. 2000;6(12):1362–7. doi:10.1038/82176.
Garcia-Cardena G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, et al. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem. 1997;272(41):25437–40.
Ghosh S, Gachhui R, Crooks C, Wu C, Lisanti MP, Stuehr DJ. Interaction between caveolin-1 and the reductase domain of endothelial nitric-oxide synthase. Consequences for catalysis. J Biol Chem. 1998;273(35):22267–71.
Dedio J, Konig P, Wohlfart P, Schroeder C, Kummer W, Muller-Esterl W. NOSIP, a novel modulator of endothelial nitric oxide synthase activity. FASEB J. 2001;15(1):79–89. doi:10.1096/fj.00-0078com.
Zimmermann K, Opitz N, Dedio J, Renne C, Muller-Esterl W, Oess S. NOSTRIN: a protein modulating nitric oxide release and subcellular distribution of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2002;99(26):17167–72. doi:10.1073/pnas.252345399.
Schilling K, Opitz N, Wiesenthal A, Oess S, Tikkanen R, Muller-Esterl W, et al. Translocation of endothelial nitric-oxide synthase involves a ternary complex with caveolin-1 and NOSTRIN. Mol Biol Cell. 2006;17(9):3870–80. doi:10.1091/mbc.E05-08-0709.
Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88(11):E68–75.
McCabe TJ, Fulton D, Roman LJ, Sessa WC. Enhanced electron flux and reduced calmodulin dissociation may explain “calcium-independent” eNOS activation by phosphorylation. J Biol Chem. 2000;275(9):6123–8.
Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399(6736):601–5. doi:10.1038/21224.
Gallis B, Corthals GL, Goodlett DR, Ueba H, Kim F, Presnell SR, et al. Identification of flow-dependent endothelial nitric-oxide synthase phosphorylation sites by mass spectrometry and regulation of phosphorylation and nitric oxide production by the phosphatidylinositol 3-kinase inhibitor LY294002. J Biol Chem. 1999;274(42):30101–8.
Bauer PM, Fulton D, Boo YC, Sorescu GP, Kemp BE, Jo H, et al. Compensatory phosphorylation and protein-protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric-oxide synthase. J Biol Chem. 2003;278(17):14841–9. doi:10.1074/jbc.M211926200.
Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399(6736):597–601. doi:10.1038/21218.
Gonzalez E, Kou R, Lin AJ, Golan DE, Michel T. Subcellular targeting and agonist-induced site-specific phosphorylation of endothelial nitric-oxide synthase. J Biol Chem. 2002;277(42):39554–60. doi:10.1074/jbc.M207299200.
Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem Sci. 1997;22(7):267–72.
Simoncini T, Genazzani AR, Liao JK. Nongenomic mechanisms of endothelial nitric oxide synthase activation by the selective estrogen receptor modulator raloxifene. Circulation. 2002;105(11):1368–73.
Gao F, Gao E, Yue TL, Ohlstein EH, Lopez BL, Christopher TA, et al. Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: the roles of PI3-kinase, Akt, and endothelial nitric oxide synthase phosphorylation. Circulation. 2002;105(12):1497–502.
Thomas SR, Chen K, Keaney Jr JF. Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. J Biol Chem. 2002;277(8):6017–24. doi:10.1074/jbc.M109107200.
Ma Y, Yang F, Wang Y, Du Z, Liu D, Guo H, et al. CaMKKbeta is involved in AMP-activated protein kinase activation by baicalin in LKB1 deficient cell lines. PLoS One. 2012;7(10):e47900. doi:10.1371/journal.pone.0047900.
Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, et al. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443(3):285–9.
Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2(1):9–19. doi:10.1016/j.cmet.2005.05.009.
Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2(1):21–33. doi:10.1016/j.cmet.2005.06.005.
Chen Z, Peng IC, Sun W, Su MI, Hsu PH, Fu Y, et al. AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circ Res. 2009;104(4):496–505. doi:10.1161/CIRCRESAHA.108.187567.
Yagita Y, Kitagawa K, Oyama N, Yukami T, Watanabe A, Sasaki T, et al. Functional deterioration of endothelial nitric oxide synthase after focal cerebral ischemia. J Cereb Blood Flow Metab. 2013;33(10):1532–9. doi:10.1038/jcbfm.2013.112.
Mineo C, Yuhanna IS, Quon MJ, Shaul PW. High density lipoprotein-induced endothelial nitric-oxide synthase activation is mediated by Akt and MAP kinases. J Biol Chem. 2003;278(11):9142–9. doi:10.1074/jbc.M211394200.
Balakumar P, Kathuria S, Taneja G, Kalra S, Mahadevan N. Is targeting eNOS a key mechanistic insight of cardiovascular defensive potentials of statins? J Mol Cell Cardiol. 2012;52(1):83–92. doi:10.1016/j.yjmcc.2011.09.014.
Merla R, Ye Y, Lin Y, Manickavasagam S, Huang MH, Perez-Polo RJ, et al. The central role of adenosine in statin-induced ERK1/2, Akt, and eNOS phosphorylation. Am J Physiol Heart Circ Physiol. 2007;293(3):H1918–28. doi:10.1152/ajpheart.00416.2007.
Ito D, Ito O, Mori N, Muroya Y, Cao PY, Takashima K, et al. Atorvastatin upregulates nitric oxide synthases with Rho-kinase inhibition and Akt activation in the kidney of spontaneously hypertensive rats. J Hypertens. 2010;28(11):2278–88. doi:10.1097/HJH.0b013e32833e0924.
Cheng WH, Ho WY, Chang CF, Lu PJ, Cheng PW, Yeh TC, et al. Simvastatin induces a central hypotensive effect via Ras-mediated signalling to cause eNOS up-regulation. Br J Pharmacol. 2013;170(4):847–58. doi:10.1111/bph.12317.
Willmot M, Gray L, Gibson C, Murphy S, Bath PM. A systematic review of nitric oxide donors and L-arginine in experimental stroke; effects on infarct size and cerebral blood flow. Nitric Oxide. 2005;12(3):141–9. doi:10.1016/j.niox.2005.01.003.
Roberts BW, Mitchell J, Kilgannon JH, Chansky ME, Trzeciak S. Nitric oxide donor agents for the treatment of ischemia/reperfusion injury in human subjects: a systematic review. Shock. 2013;39(3):229–39. doi:10.1097/SHK.0b013e31827f565b.
Acknowledgements
The authors are supported by the National Natural Science Foundation of China (81503279 and 81202653) and Natural Science Foundation of Tianjin City (15JCYBJC54800).
We would like to thank Mr. Shaoqian Yue for assistance in the preparation of this manuscript and arranging the references.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JZ and XF conceived and designed the review. JZ, WS and LL drafted the manuscript. XF revised the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhu, J., Song, W., Li, L. et al. Endothelial nitric oxide synthase: a potential therapeutic target for cerebrovascular diseases. Mol Brain 9, 30 (2016). https://doi.org/10.1186/s13041-016-0211-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-016-0211-9