
Abstract
Despite expressing many key risk genes, the role of microglia in late-onset Alzheimer’s disease pathophysiology is somewhat ambiguous, with various phenotypes reported to be either harmful or protective. Herein, we review some key findings from clinical and animal model investigations, discussing the role of microglial genetics in mediating perturbations from homeostasis. We note that impairment to protective phenotypes may include prolonged or insufficient microglial activation, resulting in dysregulated metabolomic (notably lipid-related) processes, compounded by age-related inflexibility in dynamic responses. Insufficiencies of mouse genetics and aggressive transgenic modelling imply severe limitations in applying current methodologies for aetiological investigations. Despite the shortcomings, widely used amyloidosis and tauopathy models of the disease have proven invaluable in dissecting microglial functional responses to AD pathophysiology. Some recent advances have brought modelling tools closer to human genetics, increasing the validity of both aetiological and translational endeavours.
Similar content being viewed by others


Background
The first descriptions of microgliosis (termed ‘rod cells’ or ‘granule cells’) were made in the 19th and early 20th Centuries by Rudolf Virchow, Franz Nissl, and Alois Alzheimer (amongst others; see [1]). In a series of publications in 1919, Pío del Río-Hortega identified that these cells are a microglial phenotype, which transition during injury, describing morphological changes and key functions and hypothesising their mesodermal origin (see [1]). Notably, in his initial description of the pathology that adopted his name, Alois Alzheimer described aberrant glial phenotypes [2]. However, research into Alzheimer’s disease (AD) pathogenesis focussed primarily on neurons for over 100 years after this observation.
Within the past two decades, genome-wide association study (GWAS) findings have identified key genetic risk variants for late-onset AD (LOAD) that are expressed exclusively or highly in microglia [3,4,5,6,7]. Initial studies of microglial biology using mouse models of amyloid pathology reported reductions in amyloid burden and preservation of synapse-associated extracellular matrix (perineuronal net) following microglial depletion. However, the amelioration of amyloid pathology was associated with reduced compaction of AD-associated amyloid-β (Aβ) deposits, resulting in increased diffuse plaques and dystrophic neurites [8]. Similarly, genetic methods of targeted microglial depletion have been reported to increase the size of Aβ deposits [9], strengthening the suggestion of a protective microglial function in limiting or compacting Aβ aggregates. Thus, interpreting microglial modulation of AD pathophysiology requires a holistic assessment of pathological consequences at multiple levels rather than isolated readouts.
Recent advances in single-cell RNA sequencing (scRNAseq) have allowed extensive transcript phenotyping of brain cells, uncovering key gene signatures of microglia in AD pathology. Notably, disease-associated microglia (DAM) was described as a responsive phenotype to amyloid pathology in the 5XFAD mouse model, with key features validated in human AD brains [10, 11]. These plaque-localised cells are characterised by a two-stage activation programme, culminating in TREM2-dependent phenotypic expression of CD11c and LPL (amongst others). Interestingly, the first stage of DAM transition is negatively regulated by BACE1 [12], demonstrating the unique glial cell-type specific functions of this enzyme, despite a clearly established mechanistic role in Alzheimer’s amyloid pathology as the major secretase responsible for amyloidogenic processing of amyloid precursor protein in neurons.
Other scRNAseq and single-nucleus RNAseq (snRNAseq) analyses have uncovered extensive characterisation of unique cellular states in microglial subpopulations, which transition in response to pathology. Amyloid-responsive microglia (ARM) was characterised by the expression of CD163 [13]. Reactive microglia in CK-p25 mice (which overexpress the cyclin-dependent kinase 5 cleavage product p25 in the postnatal forebrain, triggering AD-like neurodegeneration, atrophy, gliosis, and phosphorylation of endogenous tau [14]) have been grouped into type I and type II interferon-responsive phenotypes [15]. Microglial NF-κB signalling was identified as a central mediator of tau pathology in PS19 mice [16] (however, the failure to use appropriate Cx3cr1CreERT2 mice as Cx3cr1 haploinsufficiency controls [see below] in this study raises scepticism about its conclusions). A white-matter-specific signature has been reported in microglia from aged mouse brains, which again is dependent on TREM2 [17]. However, the commonalities of these various transcriptomic datasets have led to speculation that they, in fact, describe the same phenotype, with subtle differences emerging from inconsistent clustering algorithms [18]. Indeed, the homogeneity of activation signatures in these heterogenous cells does not appear to reflect the huge differences in pathological insults implemented in identifying them.
Additionally, there is growing evidence that single-cell transcriptome datasets identified in mouse models of AD-related pathogenesis and neurodegeneration offer poor insight into microglial responses in human AD [19,20,21]. Whilst post-mortem interval periods may confound the dynamic microglial responses reported in human samples, the validity of modelling slow, age-related diseases with aggressive, transgene-induced pathology requires unbiased evaluation. Indeed, microglia from human AD brains have been reportedly more aligned with the IRF8-reactive phenotype, characterised by increased expression of several homeostatic genes, notably TMEM119, CX3CR1, and P2RY12 [21]. Further, the finding that cell-specific transcriptional responses are most dramatic in the early stages of AD may suggest age-related differences in cellular flexibility, implying that modelling this disease in relatively young rodents has certain drawbacks. Thus, shifting towards knock-in models with later disease onset and slower progression may offer more efficacy in both aetiological and translational endeavours. It is also worth noting that half of mouse microglia survive the animal’s lifespan, and proliferation is three times higher during amyloid pathology [22]. In contrast, the human brain microglia may last two decades [23], which is only a fraction of the human lifespan. Therefore, whilst mice present a short-lived model of human disease, their microglia are old relative to human turnover rates.
Microglial genes in AD - risk versus prevalence
High-risk and low prevalence
An obvious disparity exists between the significance and prevalence of genetic AD risk among the ~75 risk loci that have been identified [24, 25]. Most notably, mutations in three genes (APP, PSEN1, PSEN2), which cause early-onset AD (EOAD), are extremely rare in populations. These three genes are ubiquitously expressed and play important functions in neurons and other cell types. As pathogenic mutations are found in only 5% of EOAD patients [26], one may hypothesise the involvement of gene-gene or gene-environment interactions in this aggressive manifestation of AD pathology. APOE ε4 decreases the age of symptom onset in LOAD but interestingly delays symptoms in EOAD [27]. Additionally, the APOE ε3 R136S (Christchurch) variant (APOE-Ch) has been found to protect against an EOAD PSEN1 mutation, delaying cognitive impairment and reducing tau pathology despite high levels of Aβ [28, 29]. Similarly, a rare variant in RELN (H3447R) has recently been associated with delayed onset of EOAD symptoms in an individual with reduced tau pathology and high levels of Aβ [30]. In mouse models, the loss of Reln accelerates both amyloid and tau pathologies [31, 32], suggesting that the protective H3447R variant may increase reelin function. However, this has yet to be determined experimentally. Further, atypical presentations of AD – predominantly affecting visual, language, and motor functions, amongst others – often have early onset (<65 years) and aggressive progressions [33,34,35]. Knowledge of the risk factors for these relatively uncommon conditions is so sparse that they are grouped with AD based primarily on behavioural symptoms (despite divergent progression patterns). Whilst EOAD is not the focus of this review, current knowledge of microglial involvement in atypical variants will be addressed later.
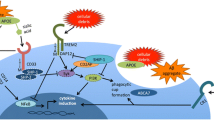
Many genes identified in recent years by GWAS underscore the involvement of microglia-specific functions in AD pathophysiology (Fig. 1). Rare variants that confer a high risk for LOAD include TREM2 and PLD3. These three genes play a role in lipid regulation but have well-established functions specific to AD proteinopathies (Fig. 2).

Microglial protein functions in Alzheimer’s disease pathology. Circular representation of microglial genes stratified based on their Alzheimer’s risk and prevalence. The sequence identities between the human and mouse proteins are indicated. The key cellular and pathology-related functions associated with each protein in Alzheimer’s pathology are annotated. Created with BioRender.com

Detailed schematic of microglial protein functions in Alzheimer’s disease pathology. The relationships between key microglial gene products with pathology-related pathways affected in Alzheimer’s disease are indicated. The colour shading of protein names indicates the abundance of the corresponding transcripts in human myeloid cells relative to all brain cells (quantified by single-cell RNAseq [36]). Numbers indicate cited references that support the functional associations depicted in the schematics. Created with BioRender.com
The microglial surface receptor TREM2 is critical for microglial signal transduction. It regulates a protective microglial response to amyloid deposition in 5XFAD [37] and APP/PS2 [38] mice. Whilst TREM2 binds directly to oligomeric Aβ and facilitates degradation [39], the efficiency of Aβ internalisation by microglia is augmented when Aβ is complexed to lipoproteins [40]. In addition to its requirement for the phenotypic transition from Stage I to Stage II DAM [10], TREM2 is also necessary for the local proliferation of microglia near amyloid deposits [41]. Therefore, the whole activation programme of microglia appears to be dependent on the TREM2 checkpoint, with dysfunction in this surface receptor leaving microglia insensitive to adverse conditions in the amyloid microenvironment.
The role of TREM2 in tauopathy is less clear, with evidence that it both ameliorates [42, 43] and facilitates [44] tau pathology in different models (hTau & AAV-P301L versus PS19 transgenic, respectively). These seemingly contradictory findings may demonstrate a biphasic function at different stages of the disease, with TREM2 protective in the early stages of tau pathogenesis but degenerative in more advanced stages. Indeed, a similar early/late pathology biphasic effect for TREM2 has been reported in amyloid pathogenesis [45], which may reflect age-related alterations to microglial functionality.
A recent study used a combined model of AD-isolated tau seeds injected into 5XFAD mouse brains [46]. Chronic administration of a TREM2-activating antibody increased activation of microglia proximal to plaques. Astoundingly, whilst amyloid burden was unaffected, aspects of tau pathology increased. These data suggest a role for microglial TREM2 in mediating cross-talk between neuronal amyloid and tau pathologies. It also supports our aforementioned hypothesis of a protective role for TREM2 in the early stages of disease (early tau pathology mentioned above), with detrimental consequences in later stages, and suggests a dominance in TREM2 responding to tau over amyloid. In light of age as the strongest risk factor for LOAD, it is worth mentioning that TREM2 deficiency reduced neuronal loss in normal older mice and downregulated microglial activation and immune transcripts [47]. TREM2 loss has also been reported to increase the density of dendritic spines and cognition [48]. Together, these data suggest that whilst TREM2 function may be specifically beneficial during the early stages of AD pathology, it has detrimental consequences outside these early responses.
TREM2 interacts with lipidated APOE, regulating Aβ internalisation by microglia [40]. Deletion of Apoe and Trem2 inhibited microglial transition into an amyloid-responsive phenotype, characterised by expression of CD163 [13]. CD163 has an anti-inflammatory function in peripheral macrophages, scavenging pro-inflammatory ligands (notably haemoglobin) [49]. In a mouse model of atherosclerosis, deficiency of both CD163 and APOE increased macrophage expression of proinflammatory and lipid content and increased plaque size [50]. In human AD brains, CD163-expressing microglia were largely CD68-positive [51], supporting the notion that CD68-expressing microglia are anti-inflammatory.
PLD3 encodes phospholipase D3 (PLD3), an endolysosomal ssDNA exonuclease involved in nucleic acid-driven inflammatory signalling. A rare variant in PLD3 (V232M) reportedly doubles the risk of developing LOAD [52]. PLD3 transcripts are increased in microglia from human AD and mouse models of amyloid and tau pathologies (APPNL-G-F/NL-G-F and rTg4510, respectively) [53]. Whilst PLD3 function in microglia is yet to be determined, its neuronal expression appears to play a protective role by regulating ER stress, nucleotide signalling, and lysosomal function, which are impaired in the V232M variant [54, 55]. Although the loss of PLD3 or the expression of the V232M variant was found to elevate Aβ production in cultured cells [52, 55], there was no change in Aβ production or deposition in young Pld3 KO mice [56].
APOE is perhaps in its own category of high-risk and high prevalence, with the frequency of the ε4 allele between 19.2-36.7% in AD patients and 8.9-19% in control populations [57]. APOE codes for a core component of plasma lipoproteins, which function to transport and deliver lipids from one tissue or cell type to another. APOE is highly expressed in astrocytes in the healthy human brain, but its expression is dysregulated in human AD brains in a cell-specific manner – it is upregulated in microglia and downregulated in astrocytes [20]. This finding highlights the dangers of inferring cell-specific expression changes from bulk tissue analyses. Microglial APOE facilitates migration towards Aβ deposits and phagocytosis, which is impaired in APOE risk variant ε4 [58]. In APP/PS1 mice, the loss of Apoe expression (whole-body knock-out) impaired compaction of amyloid deposits, reduced microglia activation, and increased amyloid deposit-associated neuritic dystrophy [59], producing a similar effect to that of microglial depletion mentioned above [8, 60]. Whilst microglial-specific deletion of Apoe in 5XFAD mice increased the size of Aβ deposits, there was no change in total Aβ load [61], again suggesting that plaque compaction may be a vital function of microglia. APOE regulates microglial activation in the PS19 mouse model of tauopathy, which is reported to mechanistically drive degeneration in this model [62].
High prevalence and low-risk
Common gene variants (e.g., BIN1, PICALM, CLU, CD33, ABCA7, SPI1) typically confer a lower risk for developing LOAD (Fig. 1) [63,64,65,66]. The BIN1 locus harbours the second-most significant risk for LOAD [67]. The BIN1 gene encodes multiple isoforms (at least 9 in the brain) of an adaptor protein expressed in neurons, oligodendrocytes, and microglia [68]. It is clear that in the AD brain, there is a decrease in the abundance of neuronal isoforms and an increase in ubiquitous and glial isoforms, which correspond well with the cellular changes in AD [68]. Correcting for neuronal cell numbers suggests that BIN1 expression is protective against AD [69]. Notably, BIN1-related SNPs associated with LOAD did not alter BIN1 expression levels in the human brain [69]. Neuronal BIN1 expression has been shown to facilitate region-specific tau pathophysiology in PS19 mice [70]. Apparently, it has the opposite effect in cultured neurons (in the absence of extrinsic cues from other CNS cell types; see below), where the loss of BIN1 expression was found to promote cell-to-cell pathology propagation [71]. These findings suggest that isolated cell-autonomous signalling alone does not account for the full repertoire of LOAD genetic risk from BIN1. Microglial BIN1 expression is necessary for proinflammatory phenotypic transition and appears to form a reciprocal regulatory relationship with transcription factors PU.1 (SPI1) and IRF1 [72]. A microglia-specific enhancer upstream of BIN1 was identified in human induced pluripotent stem cell (iPSC)-derived microglia through transposase-accessible chromatin sequencing and chromatin immunoprecipitation sequencing, and its functionality was confirmed by introducing a large deletion [73]. However, whether LOAD GWAS SNPs alter microglial BIN1 expression has yet to be ascertained. Thus, whether BIN1 SNPs influence AD pathophysiology through microglial function remains to be established. Moreover, the precise role of microglial BIN1 in AD-specific pathologies is yet to be determined.
PICALM encodes phosphatidylinositol binding clathrin assembly protein involved in clathrin-mediated endocytosis. As with BIN1, PICALM is expressed in diverse cell types in the central nervous system (CNS), including endothelial cells, neurons, and microglia [74]. The protein products of both of these genes interact with clathrin (although the microglial BIN1 isoforms lack the clathrin-interacting domain [72, 75]), and interestingly, both were found only to pose a significant risk in the absence of the APOE ε4 allele [76]. Conversely, the risk for PICALM (and one for CLU) on episodic memory was also augmented by APOE genotype in LOAD patients [77]. Additionally, a review of APOE-associated polygenic risk highlights consistent findings of associations with several LOAD genes [78]. Therefore, a full meta-analysis is required to validate these polygenic risks. Functionally, PICALM has been shown to regulate endocytosis of γ-secretase and Aβ42 production [79]. A protective variant of PICALM (rs3851179A) increases its gene expression [80], suggesting that PICALM function is generally protective. Concordantly, Picalm expression in microglia is induced by anti-inflammatory IL4 treatment [81]. Whilst the loss of PICALM reduced Aβ42 production in mouse brains [79] and H4 neuroglioma cells [82], PICALM haploinsufficiency has been reported to increase tau pathology in Tg30 mice [83]. Therefore, PICALM function in AD may be biphasic, conferring risk in early stages (Aβ deposition) but protection in more advanced pathology (tau hyperphosphorylation).
CLU codes for the secreted lipid carrier protein clusterin (ApoJ) predominantly expressed in astrocytes. Clusterin has been proposed to contribute to chronic inflammation and neurotoxicity through microglial activation [84]. Genetic ablation studies in the PDAPP model suggest that clusterin promotes fibrillar Aβ deposition [85, 86]. Clusterin directly binds Aβ, and this interaction is not affected by clusterin lipidation [87]. However, lipidation of clusterin allows it to be bound by TREM2, facilitating microglial internalisation of Aβ [40]. Loss of clusterin expression was found to exacerbate tau pathology in an AAV-TauP301L expression-based in vivo model, and direct interaction of clusterin with tau (0N4R isoform) was observed to reduce tau filament formation in a cell-free system [88]. However, another study reported that clusterin facilitates tau seeding (2N4R isoform) by stabilising oligomeric tau seeds in a cell-based seeding assay [89]. An intracellular form of clusterin was found to more readily interact with both tau and BIN1 in AD brains [90]. Additional investigations on extracellular tau interaction with clusterin (which is mostly extracellular) and cell-type specific functional characterisations of intracellular interactions between clusterin and tau are needed to understand how rare AD-associated CLU variants may modulate later stages of disease progression.
CD33 is a transmembrane protein that inhibits microglial clearance of Aβ [91]. Its transcript and protein expression levels are higher in the brains of individuals with AD [91], and increased expression was related to greater cognitive decline [69]. The CD33 minor rs3865444T allele has been reported to protect against AD (compared to the major G allele) [91, 92]. The risk allele rs3865444C increases surface levels of CD33 in peripheral monocytes and increases the inclusion of exon 2 (encoding its extracellular immunoglobulin V-set domain). The protective (minor) rs3865444A allele reduces the inclusion of exon 2 [93]. Interestingly, deletion of the CD33 gene exon 2 or ablation of CD33 expression led to increased inflammatory response and phagocytosis of Aβ peptides [94].
ABCA7 codes for a ubiquitously expressed ATP-binding cassette transporter, which transports a range of molecules and compounds (including amino acids, peptides, hormones, and lipids) across the plasma membrane (see [95]). The loss of ABCA7 function has been associated with increased AD risk [96,97,98]. Targeted deletion of Abca7 alleles affects brain lipid homeostasis and significantly increases cerebral amyloid burden in APP/PS1 amyloidosis model [99]. However, it is unclear if the increase in amyloid deposition pertains to impaired microglial function. Furthermore, ABCA7 expression in mice is required for adequate microglial inflammatory activation in response to LPS challenge [100].
SPI1 codes for the transcription factor PU.1 – a master regulator of the microglial phenotype [101]. Reduced PU.1 expression has been associated with delayed AD onset [102]. Proinflammatory [lipopolysaccharide (LPS)-stimulated] upregulation of SPI1 is BIN1-dependent [72], highlighting a regulatory relationship between these two risk genes. In vitro, Spi1 knockdown ameliorates inflammatory responses and oxidative stress and increases lipid metabolism [103]. These data suggest that PU.1-mediated inflammatory response is detrimental in the context of AD pathology.
INPP5D (also known as SHIP-1) is an inositol polyphosphate-5-phosphatase whose expression in microglia increases with the progression of amyloid pathology in LOAD selectively in microglia near Aβ deposits [104]. SHIP-1 expression or function negatively regulates phagocytosis in microglia and peripheral macrophages [105, 106]. In one study, Inpp5d haploinsufficiency was found to reduce amyloid pathology in the 5XFAD model [107]. However, additional studies using the 5XFAD or the APP/PS1 model found the inverse effect on amyloid burden when Inpp5d expression was ablated in microglia using the Cx3cr1CreER driver [108, 109]. Microglial Inpp5d deficiency increased the density of microglia near Aβ deposits, altered plaque-associated microglial gene expression signature, promoted amyloid encapsulation and engulfment by microglia, and protected against Aβ-induced neuronal dystrophy [108, 109]. One caveat to note in the report by Samuels et al. [109] is that this study compared 5XFAD/Inpp5dΔMG mice with 5XFAD/Inpp5dfl/fl controls but failed to consider that these two groups also differ by Cx3cr1 expression. The Cx3cr1CreER driver line used as the driver to generate Inpp5dΔMG mice expresses the Cre-ER fusion protein from endogenous Cx3cr1 promoter/enhancer elements such that the Cre insertion knocks out endogenous CX3CR1 expression from the knock-in allele. As discussed below, the loss of CX3CR1 expression affects microglial responses to the amyloid pathology [110, 111]. Thus, additional studies comparing 5XFAD/Inpp5dΔMG mice with 5XFAD/Cx3cr1CreER controls or using an alternate microglial driver, such as the TMEM119-CreERT, are needed to clarify the full extent to which Inpp5d function relates to microglial reactivity towards Aβ deposits and cerebral amyloid burden. Collectively, these studies show that whilst INPP5D confers some level of LOAD risk, several key functions of this gene have yet to be identified.
ABI3 codes for Abelson interactor family member 3, a protein highly expressed in microglia implicated in endocytosis and phagocytosis. The ABI3 locus contains an AD risk variant (rs616338) associated with immune responses [112, 113]. Abi3 deletion in 5XFAD mice increases amyloid burden, reduces microglial localisation to Aβ deposits, and impairs microglial migration and phagocytosis [114]. However, no association was found in human AD patients between the ABI3 risk variant and amyloid load (or other endophenotypes) [113].
Other microglial genes implicit in Alzheimer’s pathology
In addition to AD risk genes, several genes exclusively expressed in microglia have been implicated in microglial responses to AD pathophysiology (Fig. 1). We have briefly discussed a few of them in this section.
The fractalkine receptor C-X3-C Motif Chemokine Receptor 1, encoded by CX3CR1, is specifically expressed by microglia within the human and mouse brain [74, 115]. CX3CR1 is crucial for the clearance of myelin debris (and subsequently remyelination) in cuprizone-treated mice [116]. Independent transcriptomics studies observed reduced Cx3cr1 expression in homeostatic microglia as an early response to Aβ pathology in 5XFAD mice [10, 11, 21]. However, an increase in CX3CR1 protein levels has been observed in this model [117]. Moreover, CX3CR1 transcript and protein expression are increased in the human brain cortex of patients with AD [21, 117]. Additionally, CX3CR1 regulates microglial responses to Aβ deposition; however, the direction of effect is divergent at different stages of pathogenesis. In the absence of synaptic loss and cognitive impairment, APP/PS1 (4 months) and R1.40 (24 months) mice display reduced Aβ aggregation and microglial localisation at deposits with the loss of Cx3cr1 in a gene dose-dependent manner [110]. Interestingly, although Cx3cr1 ablation in the 5XFAD model resulted in fewer Aβ deposits at 4 months of age, there was an acceleration of Aβ deposition by 6 months, concomitant with dysregulation of microglial activation, impaired phagocytic function, and tau phosphorylation [111]. Tau binds to CX3CR1 to facilitate its phagocytosis by microglia [118], with Cx3cr1 deletion increasing tau pathology [118, 119]. Conversely, overexpression of CX3CL1 (fractalkine; the CX3CR1 ligand expressed by neurons) ameliorated tau pathology in PS19 mice [120]. Thus, the CX3CL1/CX3CR1 signalling pathway profoundly influences Alzheimer’s Aβ and tau pathogenesis.
ITGAX (integrin subunit alpha X) codes for CD11c – a key marker of activated microglia proximal to Aβ depositions in AD brains and mouse models [10, 121]. Increased expression of CD11c (as well as CD11a and CD11b) was first observed in AD brains over 30 years ago [121]. CD11c dimerises with CD18 (encoded by integrin subunit beta 2; ITGB2) to form complement receptor 4 (CR4). The related complement receptor 3 (CD11b/CD18) is the dominant receptor for phagocytosis of opsonised apoptotic cells [122] and pathogens by myeloid and lymphoid cells. In macrophages and dendritic cells, CR4 may play a minor role in phagocytosis, dependent on the substrate, although it is dispensable for pathogen phagocytosis in dendritic cells. There is little known of CR4 functions specific to microglia; however, its functions in peripheral immune cells predict that CR4 may have similar roles in microglia. Given the constitutive expression of CD11b in microglia, the specific upregulation of CD11c in amyloid deposit-associated microglia does not necessarily imply an increase in their phagocytic capacity. However, CD11c is thought to facilitate cell adhesion by directly binding to cell adhesion molecules. Importantly, CR4 has been shown to play a key role in adhesion to fibrinogen [123]; elevated fibrinogen in plasma is related to AD risk [124]. Fibrinogen deposits have been reported in 5XFAD and J20 mouse brains, mediating microglial elimination of dendritic spines [125]. Fibrinogen also directly binds with Aβ, and this interaction promotes the aggregation of fibrinogen and Aβ fibril formation [126]. Thus, CD11c (CR4) expression in AD microglia may feasibly increase their anchoring to dendritic spines and mediate synapse elimination. Interestingly, a recent study found that CD11c+ve microglia are divided into two subtypes by osteopontin expression [127]. Osteopontin (coded by SPP1) inhibited the compaction of diffuse plaques, Aβ phagocytosis, and degradation but facilitated the production of TNFα. CD11c+ve;osteopontin+ve microglia were associated with increased neurodegeneration and inflammation, whilst CD11c+ve;osteopontin-ve microglia were designated anti-inflammatory and neuroprotective [127].
It must also be noted that CR1 (CD35) locus contains important AD-related SNPs [63, 64, 66] although it is unclear whether these relate to microglia, as CR1 expression is low in all brain cells, with no transcript-level change observed in AD [36]. CR1 encodes a type I membrane protein that controls complement activation. There is some evidence that activation of primary microglia by exposure to LPS or Aβ may increase CR1 protein levels and may modulate phagocytic substrate preference [128]. Still, since the majority of the AD-associated CR1 SNPs do not localise to exons, how CR1 influences AD risk is poorly understood.
IFITM3 codes for restriction factor interferon-induced transmembrane protein 3, a key regulator of cytokine production during viral infection [129]. IFITM3 in neurons and astrocytes has been shown to modulate the APP-cleaving γ-secretase enzyme [130]. IFITM3 expression is elevated in the cortex of patients with LOAD, and the 5XFAD mouse brain [130], and IFITM3 gene networks are enriched in hippocampi and entorhinal cortices of AD patients [131]. The loss of IFITM3 expression significantly reduces amyloid burden in the 5XFAD model [130]. Interestingly, IFITM3 upregulation in microglia during inflammation is dependent on the LOAD risk gene BIN1 [72]. IFITM3 is important for lysosome acidification [132], suggesting the AD risk conferred by these two genes in microglia relates to proteolytic degradation after phagocytosis rather than uptake itself.
CLEC7A (Dectin-1) is a C-type lectin pattern recognition receptor, which initiates immune responses to fungal infection mediated through SYK [133]. CLEC7A expression is upregulated in microglia associated with Aβ deposits in mouse models and human AD [10, 11, 134]. Additionally, Clec7a is increased in microglia sorted from aged mice [135]. In the microglia of 5XFAD mice, CLEC7A reportedly activates SYK, increasing compaction and phagocytosis of Aβ [136]. However, as discussed above, using Sykfl/fl animals in this study rather than Cx3cr1CreER mice (control for Cx3cr1 haploinsufficiency and induced Cre expression) makes it difficult to infer these findings conclusively as SYK-mediated.
Common signalling and functions for Alzheimer’s risk genes
Whilst the data regarding microglial genes in LOAD aetiology is far from clear, there are common indications that impaired activation responses may facilitate or augment pathology in stage-specific manners. Both immunity and lipid metabolism have been highlighted as functions of common LOAD risk genes [7, 25, 137, 138] (Fig. 2). The regulation of communication in these systems largely depends on surface receptors, which account for a noticeable fraction of LOAD risk genes. The following section discusses the roles of inflammation and lipids, their relationship, and how surface receptors may mediate disease risk during pathogenesis.
Immune responses
As the primary immune cells of the brain, it is perhaps unsurprising that microglial dysfunction is linked to immune processes. However, the direction of immune dysfunction is uncertain. Initial studies speculated that microglia facilitate Aβ production and aggregation and that plaque-localised inflammatory microglia may attack an otherwise homeostatic CNS, initiating degeneration of neurons [139]. However, more recent findings indicate that microglial immune responses are dependent on several LOAD risk genes [10, 37, 45, 72, 84, 100], implying that loss-of-function variants leave microglia unable to respond to the proinflammatory demands of adverse conditions sufficiently. Whether these challenges result from homeostatic processes (i.e., metabolic waste, cellular debris, ‘normal’ apoptosis), acute challenges to homeostasis (e.g., traumatic brain injury, infection, hypoxia), or chronic dyshomeostasis (e.g., metabolic disorders, peripheral inflammation, pollution, diet) remains to be determined and may be indecipherable using inherently variable population-based samples. In this vein, it is worth noting that antenatal hypoxia causes increased expression of microglial activation genes at 2 months, and this effect is augmented in 5XFAD mice [140]. Thus, the interaction between genetic predisposition and CNS challenges (at least during development) may have a long-term impact on microglial phenotypes.
The receptor for advanced glycation end products (RAGE) can directly bind Aβ [141, 142]. Microglial overexpression of RAGE has been shown to increase the inflammatory response and Aβ aggregation in transgenic mouse models of amyloid pathology [143], whilst microglia cultured from AD patients’ brains show an exaggerated response to Aβ exposure [144]. Apparently, RAGE mediates the transportation of Aβ from the cell surface to mitochondria, promoting inflammasome formation [145].
Peripheral administration of endotoxin (LPS) in wild-type mice replicates much of the microglial proteomic changes seen in 5XFAD mice [146]; however, there is evidence that both proinflammatory and anti-inflammatory cytokines are increased in human AD brains [147]. This implies that modelling amyloid pathology in mice with an aggressive younger onset of pathology may only replicate the proinflammatory responses of microglia in human LOAD and not the reparative programmes. Whether this results from the manner of transgene expression (i.e., overexpression of human cDNAs bearing multiple FAD-linked mutations) or the insufficiencies of endogenous mouse genetics remains to be determined. Genetic variants affecting cytokine expression have been reviewed elsewhere [148].
Whilst it is well documented that TLR4 mediates proinflammatory signalling following LPS challenge [149], the role of TLR4 in AD pathophysiology is unclear. It has been observed that the loss of TLR4 function results in blunted microglial activation and increased amyloid pathology in 9-month APP/PS1 mice [150, 151], whilst younger (5-month) mice showed impaired microglial activation but no change in Aβ deposition [151]. However, others have found the TLR4 minor allele (resulting in the D299G mutation), which reduces monocyte inflammatory responses to LPS, is more common in control subjects than patients with LOAD [152]. It is plausible that this discrepancy relates to tau pathology in humans with LOAD but is absent in APP/PS1 mice. Indeed, aggregates of hyperphosphorylated tau activate inflammatory responses via TLR4 [153]. However, a complete elucidation of the role of TLR4 in tau pathology is necessary to understand the role of this receptor at different stages of pathology.
As discussed above, pathological microglial activation depends on TREM2 function. Interestingly, despite its role in mediating Stage II DAM phenotypic transition, TREM2 is an important mediator of anti-inflammatory signalling in microglia in vitro [81]. Moreover, it is worth noting that the direction of differential expression change for several DAM genes can be opposite in vitro (cultured primary microglia) compared to microglia in vivo [72], highlighting the differences between these experimental systems.
Whilst functional studies into how microglia modulate pathology are only starting to emerge, there are consistent data that surface receptor expression or localisation is integral. As discussed above, several risk genes code for transmembrane proteins, including TREM2, ABCA7, and CD33. Many surface proteins (CD11c, CLEC7A, TREM2, AXL, B2M, CD9) were identified as ‘markers’ of the DAM phenotype [10]. BIN1 facilitates the surface localisation of CD11c without impacting transcript levels [72]. TREM2 glycosylation, affected by AD-associated variant R47H, facilitates its localisation at the cell surface [154, 155]. Interestingly, in addition to these findings, there is evidence that other microglial surface receptors are involved in pathology, either in harmful or protective mechanisms. As a possible important dysregulation in AD pathology, the mis-localisation of surface receptors warrants in-depth investigation. Indeed, microglial receptors present an exciting area for future research (particularly as drug targets in pharmaceutical lead identification); however, a more comprehensive understanding of the extent of surface mis-localisation is necessary to comprehend disease risk at a protein level.
The complement system has already been alluded to, with CR4 (CD11c/CD18) and CD3 (CD11b/CD18) regulating key cell-specific functions in microglia (adhesion and phagocytosis, respectively) and CD11c+ve microglia emerging in response to amyloid pathology. Additionally, CD88, the complement component 5a receptor 1, is upregulated by microglia associated with Aβ deposits [156] and dystrophic p-tau-laden neurites [157]. Its function facilitates pathology in the Tg2576 mouse model of amyloid pathology [158]. In the mouse brain white matter, CD11c-expressing microglia with reduced levels of CD11b have been observed in response to demyelination [159], suggesting a shift from phagocytosis in this phenotype. Indeed, these microglia are antigen-presenting cells that recruit T cells [159]. Activation of CD8+ve T cells has been observed in patients with AD [160], and T cell infiltration has been reported in transgenic mouse models of aggressive amyloid pathology [161]. A protective allele of PLCG2 induces antigen presentation gene expression in microglia, promoting CNS infiltration of CD8+ve T cells [162]. Functional studies reported that CD8+ve T-cells infiltrating the brain inhibit the proinflammatory activity of microglia and limit amyloid pathology in the 5XFAD model [163], but in the context of tauopathy in the PS19 model, CD8+ve T-cells drive neurodegeneration [164]. Thus, the involvement of peripheral cell infiltration in AD pathology (including the possible involvement of mast cells, as discussed below) is poorly understood and represents an exciting area for future research.
Lipid metabolism and signalling
Microglia are ‘metabolically flexible’, readily switching to glutaminolysis and fatty acid oxidation to facilitate surveillance (process motility) in the absence of glucose [165]. However, inflammatory activation shifts metabolic programming in microglia, suppressing efficient oxidative phosphorylation [166, 167] in favour of inefficient glycolysis [166] (known as the Warburg effect). In activated macrophages, glycolysis feeds the biosynthesis of triacylglycerol, which accumulates in lipid droplets (LD) [168]. Hypoxia induces LD formation in macrophages [169], suggesting that this phenotype may emerge as a stress response, although the induction mechanisms need elucidation.
Alois Alzheimer observed that “many glial cells show adipose saccules” [2]. Recently, LD formation was reported in microglia proximal to amyloid plaques [170]; however, CD11c+ve microglia associated with Aβ plaques upregulate key genes involved in lipid metabolism [10, 171]. Thus, the balance of lipid accumulation and metabolism in pathological contexts requires mechanistic clarification, with microglial responses likely heterogeneous and phenotype-specific.
The differences in LD composition further highlight the heterogeneous phenomenon of LD formation. Impaired degradation of cholesterol from phagocytosed myelin (in TREM2-deficient microglia) results in the accumulation of cholesteryl esters in LD [172], which are absent from the triacylglycerol-laden LD in ageing microglia [173]. Triacylglycerol containing LD possibly form an energy reserve in ageing cells. Myelin-induced cholesterol storage may serve for activation-induced alterations in dynamic local membrane composition (e.g., lipid raft formation). However, the need for studies to identify the functional roles of cholesterol in inflammatory responses makes it difficult to interpret these findings mechanistically.
Cholesterol regulates APOE trafficking of Aβ in microglia, with reduced cholesterol promoting trafficking to lysosomes and subsequent degradation [174]. However, whilst investigations of membrane lipids have shed some light on protein processing, it is clear that lipid dysregulation in AD extends beyond the cell-autonomous level. Additionally, whilst microglia do not express the enzymes necessary for cholesterol conversion into progesterone [175], progesterone and derived hormones profoundly affect microglial inflammatory response programmes [176]. Indeed, progesterone reduced proinflammatory stimulation of TNFα, iNOS, NFκB, and mitogen-activated protein kinase (MAPK)-38 in cultured BV2 cells [177]. Similarly, oestrogen reduced MHC-II+ve microglia following stab injury [178], increased microglial phagocytosis of apoptotic cells and fluorescent beads [179, 180], limited inflammatory release of TNFα, NO2, and superoxide [179,180,181], limited inflammatory expression of iNOS and TLR4 [180, 181], and shifted MAPK phosphorylation from p38-MAPK to p42/44-MAPK [180, 181]. The blunting of microglial inflammatory response in this instance appears to be protective, with oestrogen depletion resulting in decreased microglial clearance of Aβ [182]. There is also evidence that oestrogen protects against tau pathology [183, 184], although it’s difficult to attribute these data specifically to microglia. Testosterone also reduced MHC-II+ve microglia following stab injury [178]. The testosterone metabolite dihydrotestosterone inhibited microglial secretion of several proinflammatory molecules via inhibition of TLR4 signalling [185]. However, compared to oestrogen studies, investigations into the effect of testosterone on microglial activation are remarkably sparse.
The progressive loss of specific lipids in ageing and AD brains has been known for decades (see [186]). The cingulate and temporal cortices are particularly vulnerable to age-related changes in lipid composition [187, 188], and these regions show decreased glucose metabolism in APOE ɛ4 carriers [189, 190], suggesting that alleles of this lipoprotein may drive region-specificity of dysfunction and pathogenesis. Reciprocally, lipidation of APOE regulates microglial activation, which is impaired in ɛ4 variant (relative to ɛ3) [58]. Microglial-like cells differentiated from APOE ɛ4-expressing iPSCs accumulate lipids [191], notably during ageing [173] and in response to neuronal signals [192], demonstrating an allele-specific inflammatory response. As Apoe expression is associated with anti-inflammatory phagocytic microglia in mice [193], these data suggest that ɛ4 induces a destructive proinflammatory phenotype. However, the range of specific functions and molecular interactions of each APOE variant is yet to be fully clarified.
Low-density lipoprotein receptor (LDLR) is a major receptor that binds APOE/lipoproteins to transport cholesterol and triglycerides in the brain and periphery. Although both APOE and LDLR levels are highest in astrocytes, their signalling in microglia regulates LPS-induced inflammatory response [194]. Additionally, whereas LDLR facilitates APOE endocytosis, Ldlr deletion increased CSF APOE levels in transgenic human APOE ɛ3 (210%) and ɛ4 (380%), but not ɛ2 mice [195]; however, cortical APOE levels were only subtly changed in ɛ3 mice, suggesting clearance may not holistically explain the CSF finding. Lipidation of APOE affects its conformation, subsequently affecting binding to receptors and to Aβ (reviewed in [196]). Interestingly, deletion of Apoe or the overexpression of LDLR protects against tau pathology in PS19 mice by suppressing microglial activation of Apoe expression [197]. Thus, inhibiting APOE-LDLR interactions may prove an efficacious therapeutic strategy against LOAD.
TREM2 presents another LOAD risk gene with important lipid-specific implications. TREM2 regulates lipid metabolism in peripheral macrophages, with genetic ablation resulting in increased body fat, insulin, and cholesterol [198]. The transition into Stage II DAM phenotype – characterised by the upregulation of lipid-metabolising genes – is TREM2-dependent [10, 11]. However, AD patients with partial loss-of-function TREM2-R47H variant express increased levels of genes involved in lipid metabolism [21]. Studies in mouse models have shown that TREM2 binds lipids associated with Aβ [37] – as well as lipid-associated risk factors APOE and CLU [40] – mediating the microglial response to pathology. TREM2 also binds directly to Aβ species; oligomeric Aβ blocks the TREM2-APOE interaction [199], suggesting competitive TREM2 binding.
LPL codes for lipoprotein lipase, which hydrolyses triglycerides [200]. Loss of Lpl in microglia impairs lipid uptake, induces a switch to a proinflammatory signature [201] and causes LD accumulation [202]. LPL is upregulated in Aβ plaque-localised microglia, which show internalised Aβ [10] (and also facilitates Aβ phagocytosis in astrocytes [203]). Abca7 deletion in mice caused dysregulation of ceramides, sphingomyelins, and hexosylceramides in the absence of pathology [204]. ABCA7 is stabilised by HDL-associated lipoproteins to augment phagocytic function [205]. Transcription factor MEF2C, which has a role in fat deposition in the periphery, limits microglial proinflammatory response [206] and is inhibited in 5XFAD mice [207]. Additionally, a weighted co-expression network analysis of gene, lipid, and protein modules in human AD patient datasets found PLCG2, CR1, MEF2C, and ABCA7 (as well as non-microglial genes IL34, FERMT2, and ANKRD31) to be associated with lipid modules [208]. Cell biology studies are needed to understand how these risk genes modulate microglial lipid homeostasis and immune responses. Interestingly, sphingolipids regulate the neuronal secretion of Aβ-containing exosomes, which are internalised and cleared by microglia [209]. Sphingolipids such as ceramide are known to be enriched in exosomes, and ceramide levels are increased in AD brain [210], suggesting that exosomal release by neurons may present a mechanism for amyloid aggregation. Potential non-pathological functions of Aβ-containing exosomes in extrinsic signalling from neurons to microglia remain to be investigated.
Leukotrienes are largely synthesised in neurons, but microglia regulate their synthesis through an unknown non-cell autonomous mechanism [211]. Leukotriene B4 is synthesised by microglia in response to intracerebral haemorrhage, signalling neutrophil infiltration and autocrine microglial activation [212]. In the 5XFAD model, treatment with the leukotriene receptor antagonist montelukast reduced neuroinflammation and CD8+ve T-cell infiltration and improved cognitive functions. However, a longitudinal study of individuals using leukotriene receptor antagonists found an association with a slower decline in clinical AD progression but observed no impact on memory performance in patients with AD or MCI and cognitively normal individuals [213].
Omega-3 polyunsaturated fatty acids reduce the microglial inflammatory response in vitro; however, they also increase phagocytosis of Aβ42 peptides [214]. This finding implies that Aβ phagocytosis is a function of homeostatic microglia (in much the same way as phagocytosis of cell debris), suggesting that chronic activation may impair amyloid clearance. Interestingly, maternal deficiency of omega-3 (during gestation and lactation) in rodents increased microglial phagocytosis and the destruction of synapses [215]. It is, therefore, possible that the over-activation of microglia causes a shift from phagocytosis of Aβ to the destruction of synapses, and that this change in the target may be modulated by dietary lipid intake. Similarly, resolvins – derivatives of omega-3 fatty acids – reduce the inflammatory upregulation and secretion of cytokines in microglia [216, 217], inducing an IL4-expressing anti-inflammatory phenotype [218]. However, further studies are required to dissect the specific molecular mechanisms that govern phagocytosis substrate preferences.
Phagocytosis
Functionally, phagocytosis has been proposed as a mechanism dysregulated by LOAD risk genes [5]. As several LOAD-related risk genes code for phagocytic receptors (TREM2, CD33) or mediators (PLCG2, PILRA) [5], this function is likely involved in pathogenesis at some stage of the disease. Early studies of Aβ phagocytosis by microglia observed poor degradation within phagosomes, fuelling hypotheses that different microglia subtypes are engaged in amyloid deposition versus removal [219, 220]. Indeed, early studies considered microglial interactions with amyloid deposits in transgenic mice to be extracellular, with in vivo phagocytosis an unlikely phenomenon [221]. However, recent studies have disproved this notion [222, 223] and also revealed the unexpected finding that microglial phagocytosis regulated by TAM receptor tyrosine kinases Axl and Mer facilitated dense-core plaque formation [224]. Thus, phagocytosis may not be a mechanism for Aβ degradation within the microglia. Interestingly, one study reported that physiological levels of soluble Aβ42 peptides undergo extracellular degradation by microglia-derived insulin-degrading enzyme and are not phagocytosed by mouse brain microglia both in vitro and in vivo, [225]. However, these intriguing findings need to be validated to ascertain the significance of this mechanism in AD pathophysiology.
Mouse brain microglia that had phagocytosed Aβ in vivo contained lower levels of the synaptic marker PSD95; however, they had a higher capacity for synaptosome phagocytosis when cultured [223]. Additionally, these cells expressed high levels of HIF1A, hypothesised to mediate transcription of proinflammatory DAM genes (SPP1, CCL3) in response to NFκB signalling [223]. It must be noted that phagocytosis of apoptotic neurons involves combined specialised efforts by microglia and astrocytes [226]. How AD pathology affects this intricate intercellular relationship is unclear; however, astrocytes have been proposed to compensate for the loss of microglial phagocytosis by undertaking this function [227].
Age
Importantly, age cannot be overlooked as the strongest risk factor for LOAD. Despite the aforementioned differences in microglial lipid homeostasis and LD composition during ageing and amyloid pathology, the proinflammatory state of aged microglia remains a dysfunctional phenotype [173]. Indeed, several ‘DAM genes’ are upregulated in microglia isolated from aged mouse brains, including Apoe, Lpl, Spp1, Itgax, and Clec7a [135]. Aged microglia display impaired (reparative) response to IL-4 after injury [228]. Live imaging of retinal microglia showed slower process motility and reduced ramifications in aged mice [229]. Additionally, aged microglia did not present hyper-ramified morphology in response to ATP; instead, they became less dynamic by retracting processes (although not full amoeboid phenotype) and displaying a slower morphological transition [229]. Microglia cultured from aged mice secrete higher levels of proinflammatory cytokines (such as IL6 and TNFα) and display impaired phagocytosis of Aβ42 [230]. The senescence-associated secretory phenotype of microglia has been described as an irrevocable cell cycle arrest, resulting in the secretion of inflammatory cytokines [231]. This phenotype develops in aged microglia [232], microglia that undergo excessive proliferation during amyloid pathology [233], and microglia that have phagocytosed neurons containing tau aggregates [234]. Removal of senescent microglia reduces age-associated inflammation and cognitive decline [232], whereas early prevention of proliferation appears to offer some protection against amyloid pathogenesis [233]. Whilst these findings demonstrate age-associated impairments in microglial homeostatic and inflammatory functions, whether these impairments play an aetiological role in AD pathophysiology remains largely undetermined.
Modelling Alzheimer’s genes in microglia in vivo and in vitro
Whilst mice are the most common animals used as disease models in AD research, it is worth noting that the protein homology of mouse and human amino acid sequences for LOAD risk factors is largely inadequate. This is particularly notable when considering microglia, where one finds less than 70% homology for over half of microglial-enriched proteins (see Fig.1 and [235]). Additionally, transcript profiles of LOAD risk genes in mouse microglia fail to match the human signatures [236]. This implies the insufficiency of murine genetics to fully model human disease, highlighting the necessity for combination modelling to include human cell types.
There has been a recent surge of interest in adopting methods of human iPSC differentiation into microglial-like cells (iMG). The ability to differentiate multiple brain cell types from the same pluripotent lines has uncovered cell-type-specific enhancer-promoter interactions, including a microglia-specific enhancer for BIN1 expression [73]. However, inconsistencies in differentiation methodology induce variability in outcomes between groups, such as the finding of iMG cells expressing the neuronal BIN1 isoform 1 [73], which others have reported absent in microglia [72, 75]. Microglial culture conditions, in general, have been shown to have phenotypic effects on these environmentally sensitive cells. For instance, the presence of albumin affects microglia morphology in culture [237], whilst TGF-β1, TGFβ-2, CSF-1, and cholesterol sustain healthy microglia [237, 238]. Interestingly, the expression of several LOAD risk genes in iMG was affected by TGF-β1 supplementation [238], further highlighting the need to standardise differentiation protocols between research groups.
A few groups have attempted to further replicate human microglia in situ by implanting iMG cells into the brains of microglia-deficient mice [239,240,241,242,243] as well as microglia-depleted slice cultures [244]. The xenografted cells (in immunodeficient mice expressing human CSF1 on a Rag2/Il2rg deficient background) appear to replicate much of the transcriptional signature of human brain microglia [239,240,241]. Whilst presenting an exciting alternative to genetically pure mouse models, the full efficacy of this chimeric approach (as well as its cost-effectiveness) has yet to be fully established within basic and translational neurodegeneration research. Indeed, whilst non-autonomous signalling is maintained with this approach, the interspecies nature of this chimeric modelling strategy suggests some limitations in interpreting cell-extrinsic mechanisms. Nonetheless, this presents a vital tool that may prove invaluable in elucidating LOAD genetic risks in translational settings.
As mentioned earlier, discrepancies between scRNAseq findings in transgenic mouse models and human AD [19] raise doubt about the validity of modelling the slow progression of age-related AD with aggressive transgene expression in young rodents. Additionally, targeted cell-specific gene manipulation in mice commonly relies on Cre-driven recombination systems. Currently, only a few pan-microglial Cre lines are widely used in research. A set of Cx3cr1-driven lines was developed by replacing the genomic locus with the Cre or CreERT2 coding sequences, thereby losing endogenous Cx3cr1 expression from the modified allele. The aforementioned effects of reduced Cx3cr1 expression on microglial phenotype and AD pathology demonstrate the limitation of this system. As an alternative, a Tmem119-driven line was developed to express Cre without affecting its endogenous expression [245]. However, there have been criticisms of the Tmem119CreER2 driver line that Tmem119 is not expressed by all microglia and is also expressed by some peripheral cells [246]. Additionally, with one exception, these microglial Cre driver lines are tamoxifen-inducible. In light of the aforementioned effects of oestrogen on AD pathology, the utility of tamoxifen raises a compounding set of complications. Thus, more suitable systems for microglial Cre expression are urgently needed to facilitate reverse-genetic manipulations in this important cell type.
It must also be noted that mRNA abundance is often a poor predictor of protein levels. For example, a comparison of protein and mRNA abundance in 384 individuals in the Religious Orders Study and the Rush Memory and Aging Project revealed only 37% of the proteins showed minimally significant concordance with transcript levels [247]. Interestingly, the transcripts most highly upregulated during LPS challenge (notably an NFκB gene network) are repressed translationally by ribosomal binding protein SRSF3 [248], which is expressed in all brain cell types. Thus, whilst many studies have characterised transcriptomic changes in both mouse and human microglia, the potential discrepancy between RNA and protein levels in microglial response to pathological challenges has yet to be characterised.
Microglial non-autonomous functions
As the ‘sentinels’ of the brain, microglia are extremely sensitive to communication from other cell types; arguably, their primary function is to receive extrinsic signals and respond appropriately. Over the years, numerous studies that used cultured microglial cells focused on identifying cell-intrinsic signalling mechanisms. The recent popularity of scRNAseq has augmented this trend by revealing microglial subtype transcript signatures and phenotypes associated with autonomous cell-specific signalling mechanisms. However, this approach does not elucidate the complexity of the CNS, which is functionally reliant on extrinsic communication between different CNS cell types. There is a growing interest in studying extrinsic communication between CNS cell types by utilising co-culture systems that enable non-autonomous signalling to be investigated whilst maintaining cell-specific resolution at a functional level.
The most obvious form of extrinsic signalling from microglia is the release of cytokines during inflammatory responses [149, 249] and in response to AD-like pathologies [16, 249, 250]. Microglial TNFα triggers apoptosis of neurons [251]. IL-1β signals leukocyte and monocyte infiltration via endothelial and ventricular cells, respectively, with neurogenesis also mediated by signalling through endothelial cells [252]. Astoundingly, increases in both IL-1β and IL-10 have been reported to precede Aβ deposition in 5XFAD mice [253], suggesting that this may function as an early preventative mechanism, which becomes chronically dysregulated in AD. Additionally, microglia may respond to neurotoxic signals by releasing fragmented mitochondria, causing proinflammatory activation of astrocytes [254]. As mentioned above, there is also evidence that T cell infiltration in amyloid and tau pathology is regulated by microglia, and this extrinsic signalling to peripheral cells modulates the extent of pathology and neurodegeneration [163, 164]. In addition to destructive non-autonomous mechanisms, microglia exert trophic effects on the CNS. Short-term exposure of microglia to low dose Aβ42 increases BDNF [255], which facilitates synapse formation [256], although long-term exposure abrogated the effect and induced a proinflammatory response [255]. Additionally, IL-1β activation of astrocytes has been associated with neuroprotection against NMDA-induced excitotoxicity [257].
The non-autonomous signalling from other CNS cell types to microglia must also be discussed. For instance, the aforementioned effects of steroid hormones require their synthesis in astrocytes and neurons [175, 258] or by peripheral cells. Glutamate release by stressed neurons activates microglia, reciprocally triggering neuronal apoptosis [251]. Additionally, the entire phenomena of Aβ- and tau-initiated microglial activation essentially comprise neuron-to-microglia extrinsic signals.
The CX3CR1 receptor (expressed specifically by microglia in the CNS) has only one ligand – fractalkine (CX3CL1) – which is predominantly expressed by neurons, either surface-bound or secreted [259]. Fractalkine is cleaved by metalloproteinases, including ADAM10 [260]. In response to excitotoxic signalling, cleavage occurs hours before the neuron dies [261], indicating that neurons alert microglia to ongoing damage and impending catastrophe. Interestingly, fractalkine can be cleaved from the neuronal membrane by microglial-produced cathepsin S in response to neuronal injury [262], demonstrating the complexity of microglia-neuron intercommunication. CX3CR1 signalling limits inflammatory responses to LPS [263], suggesting the neuronal release of fractalkine initiates a reparative microglial programme. This aligns with the aforementioned finding that CX3CR1 is downregulated during the response to Aβ deposition [10, 11, 21], characterised by exacerbated inflammation [264, 265].
IL33 production by astrocytes stimulates microglial phagocytosis of synapses during development [266]. Both IL33 and its receptor (IL1RL1) are expressed in mast cells [267], which have been observed to increase in numbers in AD patients’ brains [268] and readily infiltrate adult rodent brains. Secretion of tryptase by mast cells induces a proinflammatory phenotype in cultured microglia [269]. Intriguingly, TNF increases mast cell production of IL4 [270], implying a feed-forward mechanism of perpetuated proinflammatory signalling between mast cells and microglia. It must also be mentioned that PLX3397 – used to deplete microglia via inhibition of CSF1R– also depletes mast cells by inhibiting c-Kit. Thus, the findings from microglial depletion studies which utilise this molecule must be regarded in the context of non-cell specificity.
Atypical manifestations of Alzheimer’s disease pathology
Atypical manifestations of AD include visual, linguistic, and motor impairments [35] and may account for 38% of young-onset AD [271]. Despite the earlier onset, patients presenting with atypical symptoms do not commonly carry the ε4 allele of APOE [272]. Indeed, the APOE ε3/ε3 genotype has been reported to account for 59% of young-onset AD patients [271]. Posterior cortical atrophy is the most common atypical variant of AD, with a young age of onset [273]. Variants within several microglial-related LOAD genetic loci have been associated with posterior cortical atrophy, including APOE/TOMM40, CR1, ABCA7, and BIN1 [34]. Investigation of how these genes may influence atypical AD pathophysiology would elucidate the region-specific vulnerability of AD pathologies and how microglia impact these regional variations.
The fact that such little has been elucidated about atypical young-onset AD cases highlights the multi-aetiological nature of AD pathology as a whole. Whilst dramatic effort has been made to identify cell-specific mechanisms in more common AD manifestations, increased clinical assessment and banking of patient samples for genomics research is necessary to make significant progress in understanding the mechanistic role microglia play in atypical AD.
Conclusions
Whilst much recent attention has been given to the heterogeneity of microglial phenotypes, the problems posed by this phenomenon are compounded by the non-uniformity of AD progression. In this light, careful consideration must be given to the stage of the disease being modelled, and interpretations limited to the relative temporal progression in human disease. This is especially true of translational endeavours, in which an efficacious therapeutic intervention in early stages may prove destructive at later stages, and vice versa.
The common themes emerging from current knowledge are that (1) several LOAD risk gene variants dysregulate microglial proinflammatory and anti-inflammatory responses, and (2) ‘normal’ lipid metabolism and signalling mechanisms are impaired. The insufficient inflammatory response of LOAD-risk microglia suggests that, whilst inflammation is commonly observed in AD patients, this has perpetuated from an inability to meet ‘homeostatic’ requirements (i.e., clearance of apoptotic cells and metabolic products) or an inability to appropriately respond to an initiating insult (e.g., traumatic injury, transient ischaemic attack, hypoxia). Whilst trends in research have led to variations in findings (sometimes conflictingly so), advances in research tools continue to provide new methodologies for answering the complex questions posed by a heterogeneous cell type in a non-uniform disease.
Availability of data and materials
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- GWAS:
-
Genome-wide association study
- LOAD:
-
Late-onset Alzheimer’s disease
- scRNAseq:
-
Single-cell RNA sequencing
- snRNAseq:
-
Single-nucleus RNA seq
- DAM:
-
Disease-associated microglia
- ARM:
-
Amyloid-responsive microglia
- EOAD:
-
Early-onset Alzheimer’s disease
- CNS:
-
Central nervous system
- CR4:
-
Complement receptor 4
- CR1:
-
Complement receptor 1
- CX3CR1:
-
C-X3-C Motif Chemokine Receptor 1
- TAG:
-
Triacylglycerol
- LD:
-
Lipid droplets
- MAPK:
-
Mitogen-activated protein kinase
- iPSC:
-
Induced pluripotent stem cells
- LDLR:
-
Low-density lipoprotein receptor
- LPS:
-
Lipopolysaccharide
- RAGE:
-
Receptor for advanced glycation end products
- iMG:
-
Microglial-like cells
References
Sierra A, de Castro F, Del Rio-Hortega J, Rafael Iglesias-Rozas J, Garrosa M, Kettenmann H. The “Big-Bang” for modern glial biology: Translation and comments on Pio del Rio-Hortega 1919 series of papers on microglia. Glia. 2016;64:1801–40.
Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde.” Clin Anat. 1995;8:429–31.
Efthymiou AG, Goate AM. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener. 2017;12:43.
Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D, Rongve A, Borte S, Winsvold BS, Drange OK, et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet. 2021;53:1276–82.
Podlesny-Drabiniok A, Marcora E, Goate AM. Microglial phagocytosis: a disease-associated process emerging from Alzheimer’s disease genetics. Trends Neurosci. 2020;43:965–79.
Schwabe T, Srinivasan K, Rhinn H. Shifting paradigms: The central role of microglia in Alzheimer’s disease. Neurobiol Dis. 2020;143: 104962.
Villegas-Llerena C, Phillips A, Garcia-Reitboeck P, Hardy J, Pocock JM. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr Opin Neurobiol. 2016;36:74–81.
Casali BT, MacPherson KP, Reed-Geaghan EG, Landreth GE. Microglia depletion rapidly and reversibly alters amyloid pathology by modification of plaque compaction and morphologies. Neurobiol Dis. 2020;142: 104956.
Zhao R, Hu W, Tsai J, Li W, Gan WB. Microglia limit the expansion of beta-amyloid plaques in a mouse model of Alzheimer’s disease. Mol Neurodegener. 2017;12:47.
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(1276–1290): e1217.
Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell. 2018;173:1073–81.
Singh N, Benoit MR, Zhou J, Das B, Davila-Velderrain J, Kellis M, Tsai LH, Hu X, Yan R. BACE-1 inhibition facilitates the transition from homeostatic microglia to DAM-1. Sci Adv. 2022;8:1286.
Nguyen AT, Wang K, Hu G, Wang X, Miao Z, Azevedo JA, Suh E, Van Deerlin VM, Choi D, Roeder K, et al. APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol. 2020;140:477–93.
Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–83.
Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, De Jager PL, Ransohoff RM, Regev A, Tsai LH. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 2017;21:366–80.
Wang C, Fan L, Khawaja RR, Liu B, Zhan L, Kodama L, Chin M, Li Y, Le D, Zhou Y, et al. Microglial NF-kappaB drives tau spreading and toxicity in a mouse model of tauopathy. Nat Commun. 1969;2022:13.
Safaiyan S, Besson-Girard S, Kaya T, Cantuti-Castelvetri L, Liu L, Ji H, Schifferer M, Gouna G, Usifo F, Kannaiyan N, et al. White matter aging drives microglial diversity. Neuron. 2021;109(1100–1117): e1110.
Chen Y, Colonna M. Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? J Exp Med. 2021;218:e20202717.
Alsema AM, Jiang Q, Kracht L, Gerrits E, Dubbelaar ML, Miedema A, Brouwer N, Hol EM, Middeldorp J, van Dijk R, et al. Profiling microglia from Alzheimer’s disease donors and non-demented elderly in acute human postmortem cortical tissue. Front Mol Neurosci. 2020;13:134.
Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570:332–7.
Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med. 2020;26:131–42.
Fuger P, Hefendehl JK, Veeraraghavalu K, Wendeln AC, Schlosser C, Obermuller U, Wegenast-Braun BM, Neher JJ, Martus P, Kohsaka S, et al. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat Neurosci. 2017;20:1371–6.
Reu P, Khosravi A, Bernard S, Mold JE, Salehpour M, Alkass K, Perl S, Tisdale J, Possnert G, Druid H, Frisen J. The lifespan and turnover of microglia in the human brain. Cell Rep. 2017;20:779–84.
Bellenguez C, Kucukali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, Naj AC, Campos-Martin R, Grenier-Boley B, Andrade V, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. 2022;54:412–36.
Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77:43–51.
Cacace R, Sleegers K, Van Broeckhoven C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 2016;12:733–48.
Polsinelli AJ, Lane KA, Manchella MK, Logan PE, Gao S, Apostolova LG. APOE epsilon4 is associated with earlier symptom onset in LOAD but later symptom onset in EOAD. Alzheimers Dement. 2023;19:2212–7.
Arboleda-Velasquez JF, Lopera F, O’Hare M, Delgado-Tirado S, Marino C, Chmielewska N, Saez-Torres KL, Amarnani D, Schultz AP, Sperling RA, et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25:1680–3.
Sepulveda-Falla D, Sanchez JS, Almeida MC, Boassa D, Acosta-Uribe J, Vila-Castelar C, Ramirez-Gomez L, Baena A, Aguillon D, Villalba-Moreno ND, et al. Distinct tau neuropathology and cellular profiles of an APOE3 Christchurch homozygote protected against autosomal dominant Alzheimer’s dementia. Acta Neuropathol. 2022;144:589–601.
Lopera F, Marino C, Chandrahas AS, O’Hare M, Villalba-Moreno ND, Aguillon D, Baena A, Sanchez JS, Vila-Castelar C, Ramirez Gomez L, et al. Resilience to autosomal dominant Alzheimer’s disease in a Reelin-COLBOS heterozygous man. Nat Med. 2023;29:1243–52.
Kocherhans S, Madhusudan A, Doehner J, Breu KS, Nitsch RM, Fritschy JM, Knuesel I. Reduced Reelin expression accelerates amyloid-beta plaque formation and tau pathology in transgenic Alzheimer’s disease mice. J Neurosci. 2010;30:9228–40.
Marckx AT, Fritschle KE, Calvier L, Herz J. Reelin changes hippocampal learning in aging and Alzheimer’s disease. Behav Brain Res. 2021;414: 113482.
Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10:785–96.
Schott JM, Crutch SJ, Carrasquillo MM, Uphill J, Shakespeare TJ, Ryan NS, Yong KX, Lehmann M, Ertekin-Taner N, Graff-Radford NR, et al. Genetic risk factors for the posterior cortical atrophy variant of Alzheimer’s disease. Alzheimers Dement. 2016;12:862–71.
Graff-Radford J, Yong KXX, Apostolova LG, Bouwman FH, Carrillo M, Dickerson BC, Rabinovici GD, Schott JM, Jones DT, Murray ME. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. 2021;20:222–34.
Srinivasan K, Friedman BA, Etxeberria A, Huntley MA, van der Brug MP, Foreman O, Paw JS, Modrusan Z, Beach TG, Serrano GE, Hansen DV. Alzheimer’s patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Rep. 2020;31: 107843.
Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160:1061–71.
Meilandt WJ, Ngu H, Gogineni A, Lalehzadeh G, Lee SH, Srinivasan K, Imperio J, Wu T, Weber M, Kruse AJ, et al. Trem2 deletion reduces late-stage amyloid plaque accumulation, elevates the Abeta42:Abeta40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer’s mouse model. J Neurosci. 2020;40:1956–74.
Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, Sun Y, Zhu B, Pina-Crespo JC, Zhang M, et al. TREM2 is a receptor for beta-amyloid that mediates microglial function. Neuron. 2018;97(1023–1031): e1027.
Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 2016;91:328–40.
Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016;213:667–75.
Zhu B, Liu Y, Hwang S, Archuleta K, Huang H, Campos A, Murad R, Pina-Crespo J, Xu H, Huang TY. Trem2 deletion enhances tau dispersion and pathology through microglia exosomes. Mol Neurodegener. 2022;17:58.
Bemiller SM, McCray TJ, Allan K, Formica SV, Xu G, Wilson G, Kokiko-Cochran ON, Crish SD, Lasagna-Reeves CA, Ransohoff RM, et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol Neurodegener. 2017;12:74.
Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, Robinson GO, Anderson E, Colonna M, Holtzman DM. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci U S A. 2017;114:11524–9.
Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, Landreth GE. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J Neurosci. 2017;37:637–47.
Jain N, Lewis CA, Ulrich JD, Holtzman DM. Chronic TREM2 activation exacerbates Abeta-associated tau seeding and spreading. J Exp Med. 2023;220:e20220654.
Linnartz-Gerlach B, Bodea LG, Klaus C, Ginolhac A, Halder R, Sinkkonen L, Walter J, Colonna M, Neumann H. TREM2 triggers microglial density and age-related neuronal loss. Glia. 2019;67:539–50.
Qu W, Li L. Loss of TREM2 confers resilience to synaptic and cognitive impairment in aged mice. J Neurosci. 2020;40:9552–63.
Etzerodt A, Moestrup SK. CD163 and inflammation: biological, diagnostic, and therapeutic aspects. Antioxid Redox Signal. 2013;18:2352–63.
Gutierrez-Munoz C, Mendez-Barbero N, Svendsen P, Sastre C, Fernandez-Laso V, Quesada P, Egido J, Escola-Gil JC, Martin-Ventura JL, Moestrup SK, Blanco-Colio LM. CD163 deficiency increases foam cell formation and plaque progression in atherosclerotic mice. FASEB J. 2020;34:14960–76.
Pey P, Pearce RK, Kalaitzakis ME, Griffin WS, Gentleman SM. Phenotypic profile of alternative activation marker CD163 is different in Alzheimer’s and Parkinson’s disease. Acta Neuropathol Commun. 2014;2:21.
Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, Harari O, Norton J, Budde J, Bertelsen S, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature. 2014;505:550–4.
Sobue A, Komine O, Hara Y, Endo F, Mizoguchi H, Watanabe S, Murayama S, Saito T, Saido TC, Sahara N, et al. Microglial gene signature reveals loss of homeostatic microglia associated with neurodegeneration of Alzheimer’s disease. Acta Neuropathol Commun. 2021;9:1.
Demirev AV, Song HL, Cho MH, Cho K, Peak JJ, Yoo HJ, Kim DH, Yoon SY. V232M substitution restricts a distinct O-glycosylation of PLD3 and its neuroprotective function. Neurobiol Dis. 2019;129:182–94.
Van Acker ZP, Perdok A, Hellemans R, North K, Vorsters I, Cappel C, Dehairs J, Swinnen JV, Sannerud R, Bretou M, et al. Phospholipase D3 degrades mitochondrial DNA to regulate nucleotide signaling and APP metabolism. Nat Commun. 2023;14:2847.
Fazzari P, Horre K, Arranz AM, Frigerio CS, Saito T, Saido TC, De Strooper B. PLD3 gene and processing of APP. Nature. 2017;541:E1–2.
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. Effects of age, sex, and ethnicity on the association between Apolipoprotein E Genotype and Alzheimer disease: a meta-analysis. JAMA. 1997;278:1349–56.
Fitz NF, Nam KN, Wolfe CM, Letronne F, Playso BE, Iordanova BE, Kozai TDY, Biedrzycki RJ, Kagan VE, Tyurina YY, et al. Phospholipids of APOE lipoproteins activate microglia in an isoform-specific manner in preclinical models of Alzheimer’s disease. Nat Commun. 2021;12:3416.
Ulrich JD, Ulland TK, Mahan TE, Nystrom S, Nilsson KP, Song WM, Zhou Y, Reinartz M, Choi S, Jiang H, et al. ApoE facilitates the microglial response to amyloid plaque pathology. J Exp Med. 2018;215:1047–58.
Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, Zhang Y, Spevak W, Lin J, Phan NY, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun. 2019;10:3758.
Henningfield CM, Arreola MA, Soni N, Spangenberg EE, Green KN. Microglia-specific ApoE knock-out does not alter Alzheimer’s disease plaque pathogenesis or gene expression. Glia. 2022;70:287–302.
Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, Hoyle R, Holtzman DM. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med. 2019;216:2546–61.
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93.
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9.
Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–41.
Corneveaux JJ, Myers AJ, Allen AN, Pruzin JJ, Ramirez M, Engel A, Nalls MA, Chen K, Lee W, Chewning K, et al. Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum Mol Genet. 2010;19:3295–301.
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51:414–30.
De Rossi P, Buggia-Prevot V, Clayton BL, Vasquez JB, van Sanford C, Andrew RJ, Lesnick R, Botte A, Deyts C, Salem S, et al. Predominant expression of Alzheimer’s disease-associated BIN1 in mature oligodendrocytes and localization to white matter tracts. Mol Neurodegener. 2016;11:59.
Karch CM, Jeng AT, Nowotny P, Cady J, Cruchaga C, Goate AM. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS ONE. 2012;7:e50976.
Ponnusamy M, Wang S, Yuksel M, Hansen MT, Blazier DM, McMillan JD, Zhang X, Dammer EB, Collier L, Thinakaran G: Loss of forebrain BIN1 attenuates hippocampal pathology and neuroinflammation in a tauopathy model. Brain. 2023;146(4):1561–79.
Calafate S, Flavin W, Verstreken P, Moechars D. Loss of Bin1 promotes the propagation of tau pathology. Cell Rep. 2016;17:931–40.
Sudwarts A, Ramesha S, Gao T, Ponnusamy M, Wang S, Hansen M, Kozlova A, Bitarafan S, Kumar P, Beaulieu-Abdelahad D, et al. BIN1 is a key regulator of proinflammatory and neurodegeneration-related activation in microglia. Mol Neurodegener. 2022;17:33.
Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science. 2019;366:1134–9.
Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89:37–53.
Taga M, Petyuk VA, White C, Marsh G, Ma Y, Klein HU, Connor SM, Kroshilina A, Yung CJ, Khairallah A, et al. BIN1 protein isoforms are differentially expressed in astrocytes, neurons, and microglia: neuronal and astrocyte BIN1 are implicated in tau pathology. Mol Neurodegener. 2020;15:44.
Gharesouran J, Rezazadeh M, Khorrami A, Ghojazadeh M, Talebi M. Genetic evidence for the involvement of variants at APOE, BIN1, CR1, and PICALM loci in risk of late-onset Alzheimer’s disease and evaluation for interactions with APOE genotypes. J Mol Neurosci. 2014;54:780–6.
Barral S, Bird T, Goate A, Farlow MR, Diaz-Arrastia R, Bennett DA, Graff-Radford N, Boeve BF, Sweet RA, Stern Y, et al. Genotype patterns at PICALM, CR1, BIN1, CLU, and APOE genes are associated with episodic memory. Neurology. 2012;78:1464–71.
Fan J, Tao W, Li X, Li H, Zhang J, Wei D, Chen Y, Zhang Z. The contribution of genetic factors to cognitive impairment and dementia: Apolipoprotein E gene, gene interactions, and polygenic risk. Int J Mol Sci. 2019;20:1177.
Kanatsu K, Morohashi Y, Suzuki M, Kuroda H, Watanabe T, Tomita T, Iwatsubo T. Decreased CALM expression reduces Abeta42 to total Abeta ratio through clathrin-mediated endocytosis of gamma-secretase. Nat Commun. 2014;5:3386.
Parikh I, Fardo DW, Estus S. Genetics of PICALM expression and Alzheimer’s disease. PLoS ONE. 2014;9:e91242.
Liu W, Taso O, Wang R, Bayram S, Graham AC, Garcia-Reitboeck P, Mallach A, Andrews WD, Piers TM, Botia JA, et al. Trem2 promotes anti-inflammatory responses in microglia and is suppressed under pro-inflammatory conditions. Hum Mol Genet. 2020;29:3224–48.
Thomas RS, Henson A, Gerrish A, Jones L, Williams J, Kidd EJ. Decreasing the expression of PICALM reduces endocytosis and the activity of beta-secretase: implications for Alzheimer’s disease. BMC Neurosci. 2016;17:50.
Ando K, De Decker R, Vergara C, Yilmaz Z, Mansour S, Suain V, Sleegers K, de Fisenne MA, Houben S, Potier MC, et al. Picalm reduction exacerbates tau pathology in a murine tauopathy model. Acta Neuropathol. 2020;139:773–89.
Xie Z, Harris-White ME, Wals PA, Frautschy SA, Finch CE, Morgan TE. Apolipoprotein J (clusterin) activates rodent microglia in vivo and in vitro. J Neurochem. 2005;93:1038–46.
DeMattos RB, O’Dell MA, Parsadanian M, Taylor JW, Harmony JA, Bales KR, Paul SM, Aronow BJ, Holtzman DM. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2002;99:10843–8.
DeMattos RB, Cirrito JR, Parsadanian M, May PC, O’Dell MA, Taylor JW, Harmony JA, Aronow BJ, Bales KR, Paul SM, Holtzman DM. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron. 2004;41:193–202.
Calero M, Tokuda T, Rostagno A, Kumar A, Zlokovic B, Frangione B. GHISO J: Functional and structural properties of lipid-associated apolipoprotein J (clusterin). Biochemical Journal. 1999;344:375–83.
Wojtas AM, Carlomagno Y, Sens JP, Kang SS, Jensen TD, Kurti A, Baker KE, Berry TJ, Phillips VR, Castanedes MC, et al. Clusterin ameliorates tau pathology in vivo by inhibiting fibril formation. Acta Neuropathol Commun. 2020;8:210.
Yuste-Checa P, Trinkaus VA, Riera-Tur I, Imamoglu R, Schaller TF, Wang H, Dudanova I, Hipp MS, Bracher A, Hartl FU. The extracellular chaperone Clusterin enhances Tau aggregate seeding in a cellular model. Nat Commun. 2021;12:4863.
Zhou Y, Hayashi I, Wong J, Tugusheva K, Renger JJ, Zerbinatti C. Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule-associated protein Tau in Alzheimer’s disease. PLoS ONE. 2014;9: e103187.
Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–43.
Dos Santos LR, Pimassoni LHS, Sena GGS, Camporez D, Belcavello L, Trancozo M, Morelato RL, Errera FIV, Bueno MRP, de Paula F. Validating GWAS variants from microglial genes implicated in Alzheimer’s disease. J Mol Neurosci. 2017;62:215–21.
Raj T, Ryan KJ, Replogle JM, Chibnik LB, Rosenkrantz L, Tang A, Rothamel K, Stranger BE, Bennett DA, Evans DA, et al. CD33: increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum Mol Genet. 2014;23:2729–36.
Wissfeld J, Nozaki I, Mathews M, Raschka T, Ebeling C, Hornung V, Brustle O, Neumann H. Deletion of Alzheimer’s disease-associated CD33 results in an inflammatory human microglia phenotype. Glia. 2021;69:1393–412.
Vasiliou V, Vasiliou K, Nebert DW. Human ATP-binding cassette (ABC) transporter family. Hum Genomics. 2009;3:281–90.
Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, Sulem P, Magnusson OT, Gudjonsson SA, Unnsteinsdottir U, et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet. 2015;47:445–7.
Ma FC, Wang HF, Cao XP, Tan CC, Tan L, Yu JT. Meta-Analysis of the association between variants in ABCA7 and Alzheimer’s disease. J Alzheimers Dis. 2018;63:1261–7.
Le Guennec K, Nicolas G, Quenez O, Charbonnier C, Wallon D, Bellenguez C, Grenier-Boley B, Rousseau S, Richard AC, Rovelet-Lecrux A, et al. ABCA7 rare variants and Alzheimer disease risk. Neurology. 2016;86:2134–7.
Sakae N, Liu CC, Shinohara M, Frisch-Daiello J, Ma L, Yamazaki Y, Tachibana M, Younkin L, Kurti A, Carrasquillo MM, et al. ABCA7 Deficiency accelerates Amyloid-beta generation and Alzheimer’s neuronal pathology. J Neurosci. 2016;36:3848–59.
Aikawa T, Ren Y, Yamazaki Y, Tachibana M, Johnson MR, Anderson CT, Martens YA, Holm ML, Asmann YW, Saito T, et al. ABCA7 haplodeficiency disturbs microglial immune responses in the mouse brain. Proc Natl Acad Sci U S A. 2019;116:23790–6.
Rustenhoven J, Smith AM, Smyth LC, Jansson D, Scotter EL, Swanson MEV, Aalderink M, Coppieters N, Narayan P, Handley R, et al. PU.1 regulates Alzheimer’s disease-associated genes in primary human microglia. Mol Neurodegener. 2018;13:44.
Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, Harari O, Bertelsen S, Fairfax BP, Czajkowski J, et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci. 2017;20:1052–61.
Pimenova AA, Herbinet M, Gupta I, Machlovi SI, Bowles KR, Marcora E, Goate AM. Alzheimer’s-associated PU.1 expression levels regulate microglial inflammatory response. Neurobiol Dis. 2021;148:105217.
Tsai AP, Lin PB, Dong C, Moutinho M, Casali BT, Liu Y, Lamb BT, Landreth GE, Oblak AL, Nho K. INPP5D expression is associated with risk for Alzheimer’s disease and induced by plaque-associated microglia. Neurobiol Dis. 2021;153: 105303.
Cox D, Dale BM, Kashiwada M, Helgason CD, Greenberg S. A regulatory role for Src homology 2 domain-containing inositol 5’-phosphatase (SHIP) in phagocytosis mediated by Fc gamma receptors and complement receptor 3 (alpha(M)beta(2); CD11b/CD18). J Exp Med. 2001;193:61–71.
Pedicone C, Fernandes S, Dungan OM, Dormann SM, Viernes DR, Adhikari AA, Choi LB, De Jong EP, Chisholm JD, Kerr WG. Pan-SHIP1/2 inhibitors promote microglia effector functions essential for CNS homeostasis. J Cell Sci. 2020;133:jcs238030.
Lin PB, Tsai AP, Soni D, Lee-Gosselin A, Moutinho M, Puntambekar SS, Landreth GE, Lamb BT, Oblak AL: INPP5D deficiency attenuates amyloid pathology in a mouse model of Alzheimer's disease. Alzheimers Dement. 2023;19(6):2528–37.
Castranio EL, Hasel P, Haure-Mirande JV, Ramirez Jimenez AV, Hamilton BW, Kim RD, Glabe CG, Wang M, Zhang B, Gandy S, et al. Microglial INPP5D limits plaque formation and glial reactivity in the PSAPP mouse model of Alzheimer’s disease. Alzheimers Dement. 2023;19:2239–52.
Samuels JD, Moore KA, Ennerfelt HE, Johnson AM, Walsh AE, Price RJ, Lukens JR: The Alzheimer's disease risk factor INPP5D restricts neuroprotective microglial responses in amyloid beta-mediated pathology. Alzheimers Dement. 2023;19(11):4908–21.
Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am J Pathol. 2010;177:2549–62.
Puntambekar SS, Moutinho M, Lin PB, Jadhav V, Tumbleson-Brink D, Balaji A, Benito MA, Xu G, Oblak A, Lasagna-Reeves CA, et al. CX3CR1 deficiency aggravates amyloid driven neuronal pathology and cognitive decline in Alzheimer’s disease. Mol Neurodegener. 2022;17:47.
Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, Kunkle BW, Boland A, Raybould R, Bis JC, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 2017;49:1373–84.
Olive C, Ibanez L, Farias FHG, Wang F, Budde JP, Norton JB, Gentsch J, Morris JC, Li Z, Dube U, et al. Examination of the effect of rare variants in TREM2, ABI3, and PLCG2 in LOAD through multiple phenotypes. J Alzheimers Dis. 2020;77:1469–82.
Karahan H, Smith DC, Kim B, Dabin LC, Al-Amin MM, Wijeratne HRS, Pennington T, di VianaPrisco G, McCord B, Lin PB, et al. Deletion of Abi3 gene locus exacerbates neuropathological features of Alzheimer’s disease in a mouse model of Abeta amyloidosis. Sci Adv. 2021;7:3954.
Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–47.
Lampron A, Larochelle A, Laflamme N, Prefontaine P, Plante MM, Sanchez MG, Yong VW, Stys PK, Tremblay ME, Rivest S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med. 2015;212:481–95.
Gonzalez-Prieto M, Gutierrez IL, Garcia-Bueno B, Caso JR, Leza JC, Ortega-Hernandez A, Gomez-Garre D, Madrigal JLM. Microglial CX3CR1 production increases in Alzheimer’s disease and is regulated by noradrenaline. Glia. 2021;69:73–90.
Bolos M, Llorens-Martin M, Perea JR, Jurado-Arjona J, Rabano A, Hernandez F, Avila J. Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol Neurodegener. 2017;12:59.
Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68:19–31.
Fan Q, He W, Gayen M, Benoit MR, Luo X, Hu X, Yan R. Activated CX3CL1/Smad2 signals prevent neuronal loss and Alzheimer’s tau pathology-mediated cognitive dysfunction. J Neurosci. 2020;40:1133–44.
Akiyama H, McGeer PL. Brain microglia constitutively express beta-2 integrins. J Neuroimmunol. 1990;30:81–93.
Morelli AE, Larregina AT, Shufesky WJ, Zahorchak AF, Logar AJ, Papworth GD, Wang Z, Watkins SC, Falo LD Jr, Thomson AW. Internalization of circulating apoptotic cells by splenic marginal zone dendritic cells: dependence on complement receptors and effect on cytokine production. Blood. 2003;101:611–20.
Sandor N, Lukacsi S, Ungai-Salanki R, Orgovan N, Szabo B, Horvath R, Erdei A, Bajtay Z. CD11c/CD18 Dominates adhesion of human monocytes, macrophages and dendritic cells over CD11b/CD18. PLoS ONE. 2016;11: e0163120.
van Oijen M, Witteman JC, Hofman A, Koudstaal PJ, Breteler MM. Fibrinogen is associated with an increased risk of Alzheimer disease and vascular dementia. Stroke. 2005;36:2637–41.
Merlini M, Rafalski VA, Rios Coronado PE, Gill TM, Ellisman M, Muthukumar G, Subramanian KS, Ryu JK, Syme CA, Davalos D, et al. Fibrinogen induces microglia-mediated spine elimination and cognitive impairment in an Alzheimer’s disease model. Neuron. 2019;101(1099–1108): e1096.
Cortes-Canteli M, Zamolodchikov D, Ahn HJ, Strickland S, Norris EH. Fibrinogen and altered hemostasis in Alzheimer’s disease. J Alzheimers Dis. 2012;32:599–608.
Qiu Y, Shen X, Ravid O, Atrakchi D, Rand D, Wight AE, Kim HJ, Liraz-Zaltsman S, Cooper I, Schnaider Beeri M, Cantor H. Definition of the contribution of an Osteopontin-producing CD11c(+) microglial subset to Alzheimer’s disease. Proc Natl Acad Sci U S A. 2023;120: e2218915120.
Crehan H, Hardy J, Pocock J. Blockage of CR1 prevents activation of rodent microglia. Neurobiol Dis. 2013;54:139–49.
Stacey MA, Clare S, Clement M, Marsden M, Abdul-Karim J, Kane L, Harcourt K, Brandt C, Fielding CA, Smith SE, et al. The antiviral restriction factor IFN-induced transmembrane protein 3 prevents cytokine-driven CMV pathogenesis. J Clin Invest. 2017;127:1463–74.
Hur JY, Frost GR, Wu X, Crump C, Pan SJ, Wong E, Barros M, Li T, Nie P, Zhai Y, et al. The innate immunity protein IFITM3 modulates gamma-secretase in Alzheimer’s disease. Nature. 2020;586:735–40.
Vavougios GD, Nday C, Pelidou SH, Gourgoulianis KI, Stamoulis G, Doskas T, Zarogiannis SG. Outside-in induction of the IFITM3 trafficking system by infections, including SARS-CoV-2, in the pathobiology of Alzheimer’s disease. Brain Behav Immun Health. 2021;14: 100243.
Wee YS, Roundy KM, Weis JJ, Weis JH. Interferon-inducible transmembrane proteins of the innate immune response act as membrane organizers by influencing clathrin and v-ATPase localization and function. Innate Immun. 2012;18:834–45.
Drummond RA, Saijo S, Iwakura Y, Brown GD. The role of Syk/CARD9 coupled C-type lectins in antifungal immunity. Eur J Immunol. 2011;41:276–81.
Hemonnot-Girard AL, Meersseman C, Pastore M, Garcia V, Linck N, Rey C, Chebbi A, Jeanneteau F, Ginsberg SD, Lachuer J, et al. Comparative analysis of transcriptome remodeling in plaque-associated and plaque-distant microglia during amyloid-beta pathology progression in mice. J Neuroinflammation. 2022;19:234.
Shi L, Rocha M, Zhang W, Jiang M, Li S, Ye Q, Hassan SH, Liu L, Adair MN, Xu J, et al. Genome-wide transcriptomic analysis of microglia reveals impaired responses in aged mice after cerebral ischemia. J Cereb Blood Flow Metab. 2020;40:S49–66.
Ennerfelt H, Frost EL, Shapiro DA, Holliday C, Zengeler KE, Voithofer G, Bolte AC, Lammert CR, Kulas JA, Ulland TK, Lukens JR. SYK coordinates neuroprotective microglial responses in neurodegenerative disease. Cell. 2022;185(4135–4152): e4122.
Robinson M, Lee BY, Hane FT. Recent progress in Alzheimer’s disease research, part 2: genetics and epidemiology. J Alzheimers Dis. 2017;57:317–30.
Murdock MH, Tsai LH. Insights into Alzheimer’s disease from single-cell genomic approaches. Nat Neurosci. 2023;26:181.
Marx F, Blasko I, Pavelka M, Grubeck-Loebenstein B. The possible role of the immune system in Alzheimer’s disease. Exp Gerontol. 1998;33:871–81.
Shen G, Hu S, Zhao Z, Zhang L, Ma Q. Antenatal Hypoxia accelerates the onset of Alzheimer’s disease pathology in 5xFAD mouse model. Front Aging Neurosci. 2020;12:251.
Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–91.
Du Yan S, Zhu H, Fu J, Yan SF, Roher A, Tourtellotte WW, Rajavashisth T, Chen X, Godman GC, Stern D, Schmidt AM. Amyloid-beta peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:5296–301.
Fang F, Lue LF, Yan S, Xu H, Luddy JS, Chen D, Walker DG, Stern DM, Yan S, Schmidt AM, et al. RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 2010;24:1043–55.
Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stern DM, Yan SD. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: identification of a cellular activation mechanism. Exp Neurol. 2001;171:29–45.
Sbai O, Djelloul M, Auletta A, Ieraci A, Vascotto C, Perrone L. AGE-TXNIP axis drives inflammation in Alzheimer’s by targeting Abeta to mitochondria in microglia. Cell Death Dis. 2022;13:302.
Rangaraju S, Dammer EB, Raza SA, Gao T, Xiao H, Betarbet R, Duong DM, Webster JA, Hales CM, Lah JJ, et al. Quantitative proteomics of acutely-isolated mouse microglia identifies novel immune Alzheimer’s disease-related proteins. Mol Neurodegener. 2018;13:34.
Taipa R, das Neves SP, Sousa AL, Fernandes J, Pinto C, Correia AP, Santos E, Pinto PS, Carneiro P, Costa P, et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol Aging. 2019;76:125–32.
Su F, Bai F, Zhang Z. Inflammatory cytokines and Alzheimer’s disease: a review from the perspective of genetic Polymorphisms. Neurosci Bull. 2016;32:469–80.
Hines DJ, Choi HB, Hines RM, Phillips AG, MacVicar BA. Prevention of LPS-induced microglia activation, cytokine production and sickness behavior with TLR4 receptor interfering peptides. PLoS ONE. 2013;8: e60388.
Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 2006;129:3006–19.
Song M, Jin J, Lim JE, Kou J, Pattanayak A, Rehman JA, Kim HD, Tahara K, Lalonde R, Fukuchi K. TLR4 mutation reduces microglial activation, increases Abeta deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J Neuroinflammation. 2011;8:92.
Minoretti P, Gazzaruso C, Vito CD, Emanuele E, Bianchi M, Coen E, Reino M, Geroldi D. Effect of the functional toll-like receptor 4 Asp299Gly polymorphism on susceptibility to late-onset Alzheimer’s disease. Neurosci Lett. 2006;391:147–9.
Meng JX, Zhang Y, Saman D, Haider AM, De S, Sang JC, Brown K, Jiang K, Humphrey J, Julian L, et al. Hyperphosphorylated tau self-assembles into amorphous aggregates eliciting TLR4-dependent responses. Nat Commun. 2022;13:2692.
Shirotani K, Hatta D, Wakita N, Watanabe K, Iwata N. The role of TREM2 N-glycans in trafficking to the cell surface and signal transduction of TREM2. J Biochem. 2022;172:347–53.
Park JS, Ji IJ, Kim DH, An HJ, Yoon SY. The Alzheimer’s disease-associated R47H Variant of TREM2 has an altered Glycosylation pattern and protein stability. Front Neurosci. 2016;10:618.
Ager RR, Fonseca MI, Chu SH, Sanderson SD, Taylor SM, Woodruff TM, Tenner AJ. Microglial C5aR (CD88) expression correlates with amyloid-beta deposition in murine models of Alzheimer’s disease. J Neurochem. 2010;113:389–401.
Fonseca MI, McGuire SO, Counts SE, Tenner AJ. Complement activation fragment C5a receptors, CD88 and C5L2, are associated with neurofibrillary pathology. J Neuroinflammation. 2013;10:25.
Gomez-Arboledas A, Carvalho K, Balderrama-Gutierrez G, Chu SH, Liang HY, Schartz ND, Selvan P, Petrisko TJ, Pan MA, Mortazavi A, Tenner AJ. C5aR1 antagonism alters microglial polarization and mitigates disease progression in a mouse model of Alzheimer’s disease. Acta Neuropathol Commun. 2022;10:116.
Remington LT, Babcock AA, Zehntner SP, Owens T. Microglial recruitment, activation, and proliferation in response to primary demyelination. Am J Pathol. 2007;170:1713–24.
Lueg G, Gross CC, Lohmann H, Johnen A, Kemmling A, Deppe M, Groger J, Minnerup J, Wiendl H, Meuth SG, Duning T. Clinical relevance of specific T-cell activation in the blood and cerebrospinal fluid of patients with mild Alzheimer’s disease. Neurobiol Aging. 2015;36:81–9.
Ferretti MT, Merlini M, Spani C, Gericke C, Schweizer N, Enzmann G, Engelhardt B, Kulic L, Suter T, Nitsch RM. T-cell brain infiltration and immature antigen-presenting cells in transgenic models of Alzheimer’s disease-like cerebral amyloidosis. Brain Behav Immun. 2016;54:211–25.
Claes C, England WE, Danhash EP, Kiani Shabestari S, Jairaman A, Chadarevian JP, Hasselmann J, Tsai AP, Coburn MA, Sanchez J, et al. The P522R protective variant of PLCG2 promotes the expression of antigen presentation genes by human microglia in an Alzheimer’s disease mouse model. Alzheimers Dement. 2022;18:1765–78.
Su W, Saravia J, Risch I, Rankin S, Guy C, Chapman NM, Shi H, Sun Y, Kc A, Li W, et al. CXCR6 orchestrates brain CD8(+) T cell residency and limits mouse Alzheimer’s disease pathology. Nat Immunol. 2023;24:1735–47.
Chen X, Firulyova M, Manis M, Herz J, Smirnov I, Aladyeva E, Wang C, Bao X, Finn MB, Hu H, et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023;615:668–77.
Bernier LP, York EM, Kamyabi A, Choi HB, Weilinger NL, MacVicar BA. Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat Commun. 2020;11:1559.
Hu Y, Mai W, Chen L, Cao K, Zhang B, Zhang Z, Liu Y, Lou H, Duan S, Gao Z. mTOR-mediated metabolic reprogramming shapes distinct microglia functions in response to lipopolysaccharide and ATP. Glia. 2020;68:1031–45.
Cheng J, Zhang R, Xu Z, Ke Y, Sun R, Yang H, Zhang X, Zhen X, Zheng LT. Early glycolytic reprogramming controls microglial inflammatory activation. J Neuroinflammation. 2021;18:129.
Rosas-Ballina M, Guan XL, Schmidt A, Bumann D. Classical activation of macrophages leads to lipid droplet formation without de novo fatty acid synthesis. Front Immunol. 2020;11:131.
Bostrom P, Magnusson B, Svensson PA, Wiklund O, Boren J, Carlsson LM, Stahlman M, Olofsson SO, Hulten LM. Hypoxia converts human macrophages into triglyceride-loaded foam cells. Arterioscler Thromb Vasc Biol. 2006;26:1871–6.
Claes C, Danhash EP, Hasselmann J, Chadarevian JP, Shabestari SK, England WE, Lim TE, Hidalgo JLS, Spitale RC, Davtyan H, Blurton-Jones M. Plaque-associated human microglia accumulate lipid droplets in a chimeric model of Alzheimer’s disease. Mol Neurodegener. 2021;16:50.
Kamphuis W, Kooijman L, Schetters S, Orre M, Hol EM. Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochim Biophys Acta. 2016;1862:1847–60.
Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, et al. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron. 2020;105(837–854): e839.
Marschallinger J, Iram T, Zardeneta M, Lee SE, Lehallier B, Haney MS, Pluvinage JV, Mathur V, Hahn O, Morgens DW, et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci. 2020;23:194–208.
Lee CY, Tse W, Smith JD, Landreth GE. Apolipoprotein E promotes beta-amyloid trafficking and degradation by modulating microglial cholesterol levels. J Biol Chem. 2012;287:2032–44.
Gottfried-Blackmore A, Sierra A, Jellinck PH, McEwen BS, Bulloch K. Brain microglia express steroid-converting enzymes in the mouse. J Steroid Biochem Mol Biol. 2008;109:96–107.
Aryanpour R, Pasbakhsh P, Zibara K, Namjoo Z, Beigi Boroujeni F, Shahbeigi S, Kashani IR, Beyer C, Zendehdel A. Progesterone therapy induces an M1 to M2 switch in microglia phenotype and suppresses NLRP3 inflammasome in a cuprizone-induced demyelination mouse model. Int Immunopharmacol. 2017;51:131–9.
Lei B, Mace B, Dawson HN, Warner DS, Laskowitz DT, James ML. Anti-inflammatory effects of progesterone in lipopolysaccharide-stimulated BV-2 microglia. PLoS ONE. 2014;9: e103969.
Barreto G, Veiga S, Azcoitia I, Garcia-Segura LM, Garcia-Ovejero D. Testosterone decreases reactive astroglia and reactive microglia after brain injury in male rats: role of its metabolites, oestradiol and dihydrotestosterone. Eur J Neurosci. 2007;25:3039–46.
Loiola RA, Wickstead ES, Solito E, McArthur S. Estrogen promotes pro-resolving microglial behavior and phagocytic cell clearance through the actions of annexin A1. Front Endocrinol (Lausanne). 2019;10:420.
Bruce-Keller AJ, Keeling JL, Keller JN, Huang FF, Camondola S, Mattson MP. Antiinflammatory effects of estrogen on microglial activation. Endocrinology. 2000;141:3646–56.
Wu SY, Chen YW, Tsai SF, Wu SN, Shih YH, Jiang-Shieh YF, Yang TT, Kuo YM. Estrogen ameliorates microglial activation by inhibiting the Kir2.1 inward-rectifier K(+) channel. Sci Rep. 2016;6:22864.
Yue X, Lu M, Lancaster T, Cao P, Honda S, Staufenbiel M, Harada N, Zhong Z, Shen Y, Li R. Brain estrogen deficiency accelerates Abeta plaque formation in an Alzheimer’s disease animal model. Proc Natl Acad Sci U S A. 2005;102:19198–203.
Li R, He P, Cui J, Staufenbiel M, Harada N, Shen Y. Brain endogenous estrogen levels determine responses to estrogen replacement therapy via regulation of BACE1 and NEP in female Alzheimer’s transgenic mice. Mol Neurobiol. 2013;47:857–67.
Guglielmotto M, Manassero G, Vasciaveo V, Venezia M, Tabaton M, Tamagno E. Estrogens inhibit amyloid-beta-mediated paired helical filament-like conformation of tau through antioxidant activity and miRNA 218 regulation in htau mice. J Alzheimers Dis. 2020;77:1339–51.
Yang L, Zhou R, Tong Y, Chen P, Shen Y, Miao S, Liu X. Neuroprotection by dihydrotestosterone in LPS-induced neuroinflammation. Neurobiol Dis. 2020;140: 104814.
Farooqui AA, Liss L, Horrocks LA. Neurochemical aspects of Alzheimer’s disease: involvement of membrane phospholipids. Metab Brain Dis. 1988;3:19–35.
Palmer AM, Burns MA. Selective increase in lipid peroxidation in the inferior temporal cortex in Alzheimer’s disease. Brain Res. 1994;645:338–42.
Mota-Martorell N, Andres-Benito P, Martin-Gari M, Galo-Licona JD, Sol J, Fernandez-Bernal A, Portero-Otin M, Ferrer I, Jove M, Pamplona R. Selective brain regional changes in lipid profile with human aging. Geroscience. 2022;44:763–83.
Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996;334:752–8.
Small GW, Ercoli LM, Silverman DH, Huang SC, Komo S, Bookheimer SY, Lavretsky H, Miller K, Siddarth P, Rasgon NL, et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2000;97:6037–42.
Sienski G, Narayan P, Bonner JM, Kory N, Boland S, Arczewska AA, Ralvenius WT, Akay L, Lockshin E, He L, et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci Transl Med. 2021;13:eaaz4564.
Victor MB, Leary N, Luna X, Meharena HS, Scannail AN, Bozzelli PL, Samaan G, Murdock MH, von Maydell D, Effenberger AH, et al. Lipid accumulation induced by APOE4 impairs microglial surveillance of neuronal-network activity. Cell Stem Cell. 2022;29(1197–1212): e1198.
Rangaraju S, Dammer EB, Raza SA, Rathakrishnan P, Xiao H, Gao T, Duong DM, Pennington MW, Lah JJ, Seyfried NT, Levey AI. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegener. 2018;13:24.
Pocivavsek A, Burns MP, Rebeck GW. Low-density lipoprotein receptors regulate microglial inflammation through c-Jun N-terminal kinase. Glia. 2009;57:444–53.
Fryer JD, Demattos RB, McCormick LM, O’Dell MA, Spinner ML, Bales KR, Paul SM, Sullivan PM, Parsadanian M, Bu G, Holtzman DM. The low density lipoprotein receptor regulates the level of central nervous system human and murine apolipoprotein E but does not modify amyloid plaque pathology in PDAPP mice. J Biol Chem. 2005;280:25754–9.
Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2: a006312.
Shi Y, Andhey PS, Ising C, Wang K, Snipes LL, Boyer K, Lawson S, Yamada K, Qin W, Manis M, et al. Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms. Neuron. 2021;109(2413–2426): e2417.
Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell. 2019;178(686–698): e614.
Lessard CB, Malnik SL, Zhou Y, Ladd TB, Cruz PE, Ran Y, Mahan TE, Chakrabaty P, Holtzman DM, Ulrich JD, et al. High-affinity interactions and signal transduction between Abeta oligomers and TREM2. EMBO Mol Med. 2018;10:9027.
Taskinen MR, Kuusi T. Enzymes involved in triglyceride hydrolysis. Baillieres Clin Endocrinol Metab. 1987;1:639–66.
Bruce KD, Gorkhali S, Given K, Coates AM, Boyle KE, Macklin WB, Eckel RH. Lipoprotein Lipase is a feature of alternatively-activated microglia and may facilitate lipid uptake in the CNS during demyelination. Front Mol Neurosci. 2018;11:57.
Loving BA, Tang M, Neal MC, Gorkhali S, Murphy R, Eckel RH, Bruce KD. Lipoprotein Lipase regulates microglial lipid droplet accumulation. Cells. 2021;10:198.
Nishitsuji K, Hosono T, Uchimura K, Michikawa M. Lipoprotein lipase is a novel amyloid beta (Abeta)-binding protein that promotes glycosaminoglycan-dependent cellular uptake of Abeta in astrocytes. J Biol Chem. 2011;286:6393–401.
Dehghan A, Pinto RC, Karaman I, Huang J, Durainayagam BR, Ghanbari M, Nazeer A, Zhong Q, Liggi S, Whiley L, et al. Metabolome-wide association study on ABCA7 indicates a role of ceramide metabolism in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2022;119: e2206083119.
Tanaka N, Abe-Dohmae S, Iwamoto N, Fitzgerald ML, Yokoyama S. Helical apolipoproteins of high-density lipoprotein enhance phagocytosis by stabilizing ATP-binding cassette transporter A7. J Lipid Res. 2010;51:2591–9.
Deczkowska A, Matcovitch-Natan O, Tsitsou-Kampeli A, Ben-Hamo S, Dvir-Szternfeld R, Spinrad A, Singer O, David E, Winter DR, Smith LK, et al. Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat Commun. 2017;8:717.
Xue F, Tian J, Yu C, Du H, Guo L. Type I interferon response-related microglial Mef2c deregulation at the onset of Alzheimer’s pathology in 5xFAD mice. Neurobiol Dis. 2021;152: 105272.
Xu J, Bankov G, Kim M, Wretlind A, Lord J, Green R, Hodges A, Hye A, Aarsland D, Velayudhan L, et al. Integrated lipidomics and proteomics network analysis highlights lipid and immunity pathways associated with Alzheimer’s disease. Transl Neurodegener. 2020;9:36.
Yuyama K, Sun H, Mitsutake S, Igarashi Y. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-beta by microglia. J Biol Chem. 2012;287:10977–89.
Dinkins MB, Wang G, Bieberich E. Sphingolipid-enriched extracellular vesicles and Alzheimer’s disease: a decade of research. J Alzheimers Dis. 2017;60:757–68.
Michael J, Unger MS, Poupardin R, Schernthaner P, Mrowetz H, Attems J, Aigner L. Microglia depletion diminishes key elements of the leukotriene pathway in the brain of Alzheimer’s Disease mice. Acta Neuropathol Commun. 2020;8:129.
Hijioka M, Futokoro R, Ohto-Nakanishi T, Nakanishi H, Katsuki H, Kitamura Y. Microglia-released leukotriene B(4) promotes neutrophil infiltration and microglial activation following intracerebral hemorrhage. Int Immunopharmacol. 2020;85: 106678.
Xiong LY, Ouk M, Wu CY, Rabin JS, Lanctot KL, Herrmann N, Black SE, Edwards JD, Swardfager W. Leukotriene receptor antagonist use and cognitive decline in normal cognition, mild cognitive impairment, and Alzheimer’s dementia. Alzheimers Res Ther. 2021;13:147.
Hjorth E, Zhu M, Toro VC, Vedin I, Palmblad J, Cederholm T, Freund-Levi Y, Faxen-Irving G, Wahlund LO, Basun H, et al. Omega-3 fatty acids enhance phagocytosis of Alzheimer’s disease-related amyloid-beta42 by human microglia and decrease inflammatory markers. J Alzheimers Dis. 2013;35:697–713.
Madore C, Leyrolle Q, Morel L, Rossitto M, Greenhalgh AD, Delpech JC, Martinat M, Bosch-Bouju C, Bourel J, Rani B, et al. Essential omega-3 fatty acids tune microglial phagocytosis of synaptic elements in the mouse developing brain. Nat Commun. 2020;11:6133.
Rey C, Nadjar A, Buaud B, Vaysse C, Aubert A, Pallet V, Laye S, Joffre C. Resolvin D1 and E1 promote resolution of inflammation in microglial cells in vitro. Brain Behav Immun. 2016;55:249–59.
Xu ZZ, Berta T, Ji RR. Resolvin E1 inhibits neuropathic pain and spinal cord microglial activation following peripheral nerve injury. J Neuroimmune Pharmacol. 2013;8:37–41.
Li L, Wu Y, Wang Y, Wu J, Song L, Xian W, Yuan S, Pei L, Shang Y. Resolvin D1 promotes the interleukin-4-induced alternative activation in BV-2 microglial cells. J Neuroinflammation. 2014;11:72.
Wisniewski HM, Barcikowska M, Kida E. Phagocytosis of beta/A4 amyloid fibrils of the neuritic neocortical plaques. Acta Neuropathol. 1991;81:588–90.
Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol. 1992;84:225–33.
Stalder M, Deller T, Staufenbiel M, Jucker M. 3D-Reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol Aging. 2001;22:427–34.
Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61:797–805.
Grubman A, Choo XY, Chew G, Ouyang JF, Sun G, Croft NP, Rossello FJ, Simmons R, Buckberry S, Landin DV, et al. Transcriptional signature in microglia associated with Abeta plaque phagocytosis. Nat Commun. 2021;12:3015.
Huang Y, Happonen KE, Burrola PG, O’Connor C, Hah N, Huang L, Nimmerjahn A, Lemke G. Microglia use TAM receptors to detect and engulf amyloid beta plaques. Nat Immunol. 2021;22:586–94.
Fu H, Liu B, Li L, Lemere CA. Microglia do not take up soluble amyloid-beta peptides, but partially degrade them by secreting insulin-degrading enzyme. Neuroscience. 2020;443:30–43.
Damisah EC, Hill RA, Rai A, Chen F, Rothlin CV, Ghosh S, Grutzendler J. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci Adv. 2020;6:eaba3239.
Konishi H, Okamoto T, Hara Y, Komine O, Tamada H, Maeda M, Osako F, Kobayashi M, Nishiyama A, Kataoka Y, et al. Astrocytic phagocytosis is a compensatory mechanism for microglial dysfunction. EMBO J. 2020;39: e104464.
Fenn AM, Hall JC, Gensel JC, Popovich PG, Godbout JP. IL-4 signaling drives a unique arginase+/IL-1beta+ microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in IL-4Ralpha after traumatic spinal cord injury. J Neurosci. 2014;34:8904–17.
Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, Wong WT. Age-related alterations in the dynamic behavior of microglia. Aging Cell. 2011;10:263–76.
Njie EG, Boelen E, Stassen FR, Steinbusch HW, Borchelt DR, Streit WJ. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol Aging. 2012;33(195):e191–e112.
Matsudaira T, Nakano S, Konishi Y, Kawamoto S, Uemura K, Kondo T, Sakurai K, Ozawa T, Hikida T, Komine O, et al. Cellular senescence in white matter microglia is induced during ageing in mice and exacerbates the neuroinflammatory phenotype. Commun Biol. 2023;6:665.
Zhang X, Pearsall VM, Carver CM, Atkinson EJ, Clarkson BDS, Grund EM, Baez-Faria M, Pavelko KD, Kachergus JM, White TA, et al. Rejuvenation of the aged brain immune cell landscape in mice through p16-positive senescent cell clearance. Nat Commun. 2022;13:5671.
Hu Y, Fryatt GL, Ghorbani M, Obst J, Menassa DA, Martin-Estebane M, Muntslag TAO, Olmos-Alonso A, Guerrero-Carrasco M, Thomas D, et al. Replicative senescence dictates the emergence of disease-associated microglia and contributes to Abeta pathology. Cell Rep. 2021;35: 109228.
Brelstaff JH, Mason M, Katsinelos T, McEwan WA, Ghetti B, Tolkovsky AM, Spillantini MG. Microglia become hypofunctional and release metalloproteases and tau seeds when phagocytosing live neurons with P301S tau aggregates. Sci Adv. 2021;7:4980.
Hasselmann J, Blurton-Jones M. Human iPSC-derived microglia: A growing toolset to study the brain’s innate immune cells. Glia. 2020;68:721–39.
Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, et al. An environment-dependent transcriptional network specifies human microglia identity. Science. 2017;356:eaal3222.
Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, Barres BA. Diverse requirements for microglial survival, specification, and function revealed by defined-medium cultures. Neuron. 2017;94(759–773): e758.
Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C, et al. iPSC-derived human microglia-like cells to study neurological diseases. Neuron. 2017;94(278–293): e279.
Mancuso R, Van Den Daele J, Fattorelli N, Wolfs L, Balusu S, Burton O, Liston A, Sierksma A, Fourne Y, Poovathingal S, et al. Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat Neurosci. 2019;22:2111–6.
Svoboda DS, Barrasa MI, Shu J, Rietjens R, Zhang S, Mitalipova M, Berube P, Fu D, Shultz LD, Bell GW, Jaenisch R. Human iPSC-derived microglia assume a primary microglia-like state after transplantation into the neonatal mouse brain. Proc Natl Acad Sci U S A. 2019;116:25293–303.
Xu R, Li X, Boreland AJ, Posyton A, Kwan K, Hart RP, Jiang P. Human iPSC-derived mature microglia retain their identity and functionally integrate in the chimeric mouse brain. Nat Commun. 2020;11:1577.
Fattorelli N, Martinez-Muriana A, Wolfs L, Geric I, De Strooper B, Mancuso R. Stem-cell-derived human microglia transplanted into mouse brain to study human disease. Nat Protoc. 2021;16:1013–33.
Jin M, Ma Z, Jiang P. Generation of iPSC-based human-mouse microglial brain chimeras to study senescence of human microglia. STAR Protoc. 2022;3: 101847.
Ogaki A, Ikegaya Y, Koyama R. Replacement of mouse microglia with Human Induced Pluripotent Stem Cell (hiPSC)-derived microglia in mouse organotypic slice cultures. Front Cell Neurosci. 2022;16: 918442.
Kaiser T, Feng G: Tmem119-EGFP and Tmem119-CreERT2 Transgenic Mice for Labeling and Manipulating Microglia. eNeuro. 2019;6(4):ENEURO.0448-18.2019.
Vankriekelsvenne E, Chrzanowski U, Manzhula K, Greiner T, Wree A, Hawlitschka A, Llovera G, Zhan J, Joost S, Schmitz C, et al. Transmembrane protein 119 is neither a specific nor a reliable marker for microglia. Glia. 2022;70:1170–90.
Tasaki S, Xu J, Avey DR, Johnson L, Petyuk VA, Dawe RJ, Bennett DA, Wang Y, Gaiteri C. Inferring protein expression changes from mRNA in Alzheimer’s dementia using deep neural networks. Nat Commun. 2022;13:655.
Boutej H, Rahimian R, Thammisetty SS, Beland LC, Lalancette-Hebert M, Kriz J. Diverging mRNA and protein networks in activated microglia reveal SRSF3 suppresses translation of highly upregulated innate immune transcripts. Cell Rep. 2017;21:3220–33.
Meda L, Cassatella MA, Szendrei GI, Otvos L Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–50.
Oblak AL, Lin PB, Kotredes KP, Pandey RS, Garceau D, Williams HM, Uyar A, O’Rourke R, O’Rourke S, Ingraham C, et al. Comprehensive evaluation of the 5XFAD mouse model for preclinical testing applications: a MODEL-AD study. Front Aging Neurosci. 2021;13: 713726.
Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008;28:2221–30.
Liu X, Nemeth DP, McKim DB, Zhu L, DiSabato DJ, Berdysz O, Gorantla G, Oliver B, Witcher KG, Wang Y, et al. Cell-type-specific interleukin 1 receptor 1 signaling in the brain regulates distinct neuroimmune activities. Immunity. 2019;50(317–333): e316.
Boza-Serrano A, Yang Y, Paulus A, Deierborg T. Innate immune alterations are elicited in microglial cells before plaque deposition in the Alzheimer’s disease mouse model 5xFAD. Sci Rep. 2018;8:1550.
Joshi AU, Minhas PS, Liddelow SA, Haileselassie B, Andreasson KI, Dorn GW 2nd, Mochly-Rosen D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci. 2019;22:1635–48.
Caruso GI, Spampinato SF, Costantino G, Merlo S, Sortino MA. SIRT1-dependent upregulation of BDNF in human microglia challenged with abeta: an early but transient response rescued by melatonin. Biomedicines. 2021;9:466.
Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596–609.
Todd L, Palazzo I, Suarez L, Liu X, Volkov L, Hoang TV, Campbell WA, Blackshaw S, Quan N, Fischer AJ. Reactive microglia and IL1beta/IL-1R1-signaling mediate neuroprotection in excitotoxin-damaged mouse retina. J Neuroinflammation. 2019;16:118.
Zwain IH, Yen SS. Neurosteroidogenesis in astrocytes, oligodendrocytes, and neurons of cerebral cortex of rat brain. Endocrinology. 1999;140:3843–52.
Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–901.
Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102:1186–95.
Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci. 2000;20:RC87.
Clark AK, Yip PK, Malcangio M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J Neurosci. 2009;29:6945–54.
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–24.
Ardestani PM, Evans AK, Yi B, Nguyen T, Coutellier L, Shamloo M. Modulation of neuroinflammation and pathology in the 5XFAD mouse model of Alzheimer’s disease using a biased and selective beta-1 adrenergic receptor partial agonist. Neuropharmacology. 2017;116:371–86.
Lopez-Gonzalez I, Schluter A, Aso E, Garcia-Esparcia P, BellenAnsoleaga FLL, Carmona M, Moreno J, Fuso A, Portero-Otin M, et al. Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: correlations with plaques, tangles, and oligomeric species. J Neuropathol Exp Neurol. 2015;74:319–44.
Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, Liddelow SA, Nguyen PT, Nakao-Inoue H, Dorman LC, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 2018;359:1269–73.
Tung HY, Plunkett B, Huang SK, Zhou Y. Murine mast cells secrete and respond to interleukin-33. J Interferon Cytokine Res. 2014;34:141–7.
Maslinska D, Laure-Kamionowska M, Maslinski KT, Gujski M, Maslinski S. Distribution of tryptase-containing mast cells and metallothionein reactive astrocytes in human brains with amyloid deposits. Inflamm Res. 2007;56(Suppl 1):S17-18.
Zhang S, Zeng X, Yang H, Hu G, He S. Mast cell tryptase induces microglia activation via protease-activated receptor 2 signaling. Cell Physiol Biochem. 2012;29:931–40.
Zhang H, Yang H, He S. TNF increases expression of IL-4 and PARs in mast cells. Cell Physiol Biochem. 2010;26:327–36.
Balasa M, Gelpi E, Antonell A, Rey MJ, Sanchez-Valle R, Molinuevo JL, Llado A. Neurological Tissue Bank/University of Barcelona/Hospital Clinic NTBUBHCCG: clinical features and APOE genotype of pathologically proven early-onset Alzheimer disease. Neurology. 2011;76:1720–5.
van der Flier WM, Pijnenburg YA, Fox NC, Scheltens P. Early-onset versus late-onset Alzheimer’s disease: the case of the missing APOE varepsilon4 allele. Lancet Neurol. 2011;10:280–8.
Crutch SJ, Lehmann M, Schott JM, Rabinovici GD, Rossor MN, Fox NC. Posterior cortical atrophy. Lancet Neurol. 2012;11:170–8.
Acknowledgements
None.
Funding
A.S. is the recipient of the BrightFocus Foundation fellowship A2023009F. Research in G.T.’s laboratory is supported by NIH grants AG077610, AG079141, AG063175, and AG056061.
Author information
Authors and Affiliations
Contributions
A.S. wrote the first draft of the manuscript. G.T. and A.S. edited the manuscript and prepared the figures.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent.
Competing interests
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sudwarts, A., Thinakaran, G. Alzheimer’s genes in microglia: a risk worth investigating. Mol Neurodegeneration 18, 90 (2023). https://doi.org/10.1186/s13024-023-00679-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-023-00679-4