
Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that primarily affects elderly people and constitutes a major source of disability worldwide. Notably, the neuropathological hallmarks of PD include nigrostriatal loss and the formation of intracellular inclusion bodies containing misfolded α-synuclein protein aggregates. Cardinal motor symptoms, which include tremor, rigidity and bradykinesia, can effectively be managed with dopaminergic therapy for years following symptom onset. Nonetheless, patients ultimately develop symptoms that no longer fully respond to dopaminergic treatment. Attempts to discover disease-modifying agents have increasingly been supported by translational molecular imaging concepts, targeting the most prominent pathological hallmark of PD, α-synuclein accumulation, as well as other molecular pathways that contribute to the pathophysiology of PD. Indeed, molecular imaging modalities such as positron emission tomography (PET) and single-photon emission computed tomography (SPECT) can be leveraged to study parkinsonism not only in animal models but also in living patients. For instance, mitochondrial dysfunction can be assessed with probes that target the mitochondrial complex I (MC-I), while nigrostriatal degeneration is typically evaluated with probes designed to non-invasively quantify dopaminergic nerve loss. In addition to dopaminergic imaging, serotonin transporter and N-methyl-D-aspartate (NMDA) receptor probes are increasingly used as research tools to better understand the complexity of neurotransmitter dysregulation in PD. Non-invasive quantification of neuroinflammatory processes is mainly conducted by targeting the translocator protein 18 kDa (TSPO) on activated microglia using established imaging agents. Despite the overwhelming involvement of the brain and brainstem, the pathophysiology of PD is not restricted to the central nervous system (CNS). In fact, PD also affects various peripheral organs such as the heart and gastrointestinal tract – primarily via autonomic dysfunction. As such, research into peripheral biomarkers has taken advantage of cardiac autonomic denervation in PD, allowing the differential diagnosis between PD and multiple system atrophy with probes that visualize sympathetic nerve terminals in the myocardium. Further, α-synuclein has recently gained attention as a potential peripheral biomarker in PD. This review discusses breakthrough discoveries that have led to the contemporary molecular concepts of PD pathophysiology and how they can be harnessed to develop effective imaging probes and therapeutic agents. Further, we will shed light on potential future trends, thereby focusing on potential novel diagnostic tracers and disease-modifying therapeutic interventions.
Similar content being viewed by others
Background
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that mainly affects elderly people [1]. The clinical diagnosis of PD is based on the presence of a constellation of movement abnormalities, including the presence of slowness of movement (bradykinesia), poverty of movement (akinesia), tremor at rest, and muscle stiffness (rigidity). Although the history of parkinsonism dates back to the ancient Indian medical literature [2], the first coherent description of PD-related clinical symptoms was reported in 1817 by James Parkinson in his seminal work ‘An essay on the shaking palsy’ [3, 4]. Notably, the incidence and prevalence of PD have risen rapidly in the past two decades, contributing to a mounting socioeconomic burden [5]. Along this line, epidemiological studies unveiled that the age-standardized global prevalence of PD has more than doubled from 1990–2016, thereby exhibiting the highest growth rate among all major neurological disorders [6]. While underlying causes for this disproportionate growth are not fully understood, the alerting development calls for improved diagnostic and therapeutic measures.
PD has traditionally been classified into two distinct forms, namely, the hereditary form with genetic mutations known to contribute to the pathophysiology of PD and the idiopathic form, which is essentially of unknown origin [7]. In 2004, Braak et. al. introduced a PD staging metric comprising six neuropathological phenotypes that included both, presymptomatic and symptomatic disease stages [8]. Of note, while initial pathological stages included the formation of intracellular inclusion bodies (mainly aggregates of misfolded α-synuclein) in neurons of asymptomatic individuals, more advanced disease stages were characterized by degeneration of dopaminergic neurons in the substantia nigra pars compacta. Many of the prototypical parkinsonian motor signs (see above) are due to the loss of dopamine in the striatum, the primary recipient of nigral efferents. The brain has a remarkable ability to compensate for the loss of nigrostriatal fibers: It is estimated that up to 50–80% of dopaminergic cells in the nigrostriatal system can be lost before motor symptoms manifest [9,10,11]. In later stages, neurodegeneration is observed in the neocortex and other brain regions, leading to additional (non-dopaminergic) symptoms [8, 12].
Although the clinical presentation of PD is dominated by motor symptoms (Fig. 1A), it has become apparent that the majority of patients also display a multitude of non-motor features [13]. Indeed, individuals with prodromal disease may first experience one or several of the following symptoms, including autonomic dysfunction, cognitive impairments, depression, sleep disorders, hyposmia and pain [10, 14,15,16]. Accordingly, PD constitutes a clinical syndrome that affects multiple organs.

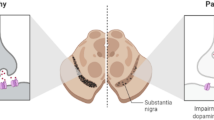
Clinical presentation, pathophysiology and diagnostic imaging in Parkinson’s disease (PD). A Cardinal motor symptoms and non-motor features of PD. B Pathological hallmarks of PD include aggregates of misfolded α-synuclein (Lewy bodies), mitochondrial dysfunction & oxidative stress, infiltration of immune cells and persistent activation of microglia, as well as an increased release of cytokines. The pathophysiology of PD prompts neurodegeneration and dopamine deficiency, which is particularly pronounced in the mammalian striatum. C Translational molecular imaging with positron emission tomography (PET) and single-photon emission computed tomography (SPECT), visualizing striatal degeneration in PD non-invasively. DAT, dopamine transporter.; DDC, DOPA decarboxylase; VMAT2, vesicular monoamine transporter 2. Figure 1C reprinted by permission from the Royal College of Physicians (RCP): Pagano et al., [17] copyright (2016)
Molecular imaging has contributed to a better understanding of a variety of molecular and cellular concepts that have been linked to PD onset and progression, including mitochondrial dysfunction [18, 19], oxidative stress [20, 21], neuroinflammation [22, 23], excitotoxicity [24], genetic mutations [25, 26], failure of protein clearance pathways and abnormal protein aggregation (Fig. 1B) [27]. Amyloid-based imaging has been suggested to aid the clinical evaluation of PD-associated forms of dementia, in particular, to assess whether cognitive decline is occurring in the setting of abnormal Aβ accumulation [28]. With the development of improved sensitivity of detection modalities, PET now allows the assessment of reactive oxygen species (ROS) in vivo [29], however, this application is still in its infancy and future studies will reveal whether ROS-targeted PET is useful in PD patients. Advanced molecular imaging applications have led to considerable progress in our understanding of non-dopaminergic receptor systems, including glutamatergic, cholinergic, serotonergic and adrenergic signaling in PD [10, 30, 31].
PD can be successfully managed for many years following disease onset. Nonetheless, while the oral administration of L-dopa is still considered the cornerstone of PD therapy, chronic L-dopa treatment is associated with several limitations, including drug-induced dyskinesia, motor fluctuations, and hallucinations [32, 33]. Further, PD remains a progressive disease with no curative treatment to date, which ultimately leads to severe disability. Accordingly, the development of improved PD therapy constitutes an unmet medical need and various therapy concepts have been suggested in addition to dopamine substitution. Contemporary efforts aim at introducing novel disease-modifying drugs that harbor potential to delay disease progression.
Innovative concepts involving translational molecular imaging have proven particularly useful for the early detection of PD and can facilitate the identification of pre-symptomatic individuals at risk. Similarly, strenuous efforts have been devoted to the development of molecular probes that would facilitate drug discovery and development by enabling the non-invasive assessment of molecular processes, which may include alterations in receptor expression for target validation and therapy monitoring, drug biodistribution and pharmacokinetics, target engagement experiments, and micro-dosing studies [34,35,36].
Molecular imaging tools have proven valuable, from bench-to-bedside, and may effectively reduce late-stage failures in drug development. Imaging modalities such as positron emission tomography (PET) and single photon-emission tomography (SPECT) can provide real-time information on drug distribution in patients as well as the assessment of target protein abundancy in different organs without physiological disturbance of the tissue microenvironment [37]. Indeed, PET and SPECT have been applied to study PD and were established as reliable clinical tools that provide information on PD onset and progression (Fig. 1C) [38, 39]. In most cases, targeted PET probes can be used for multiple purposes, including pharmacological assessment by means of target engagement studies, as well as for diagnostic purposes when alterations in target abundancy are anticipated. Another key advantage of molecular imaging-guided drug development is the ability to provide effective patient stratification during clinical PD trials [40,41,42,43].
In this review, we discuss key concepts that led to the discovery of translational molecular imaging probes and therapeutic agents for PD. Further, this review sheds light on current developments, thereby discussing potential novel diagnostic tracers and experimental disease-modifying drug candidates.
Targeting α-synuclein
As PD progresses, protein pathology spreads from the brainstem to the substantia nigra pars compacta (SNpc) and then to the forebrain [13]. Although a variety of molecular mechanisms are believed to contribute to PD onset and its irreversible progression, the most extensively studied pathological hallmark of PD is the accumulation of intracellular inclusion bodies (Lewy bodies), that consist of misfolded α-synuclein (αS). Of note, αS is a 140 amino acid long native presynaptic neuronal protein that is encoded by the SNCA gene [44] and is abundantly expressed in the healthy CNS, where it regulates vesicle fusion and neurotransmitter release via molecular interactions with presynaptic soluble N-ethylmaleimide-sensitive attachment protein receptors (SNAREs) [45]. Misfolding of αS leads to the formation of aggregates of the protein, and such aggregates are implicated in a group of overlapping neurodegenerative disorders, designated α-synucleinopathies, which include PD, dementia with Lewy bodies (DLB) and multiple system atrophy (MSA) [46,47,48,49]. Genetic mutations, post-translational modifications (PTMs), and increased αS protein concentrations have been linked to the formation of insoluble αS aggregates [50]. One of the earliest PD-related mutations discovered were within the SNCA gene, which was found to initiate early-onset PD with an autosomal dominant inheritance pattern [51]. Some advances have been made since then by the identification of a series of mutations in the αS gene that were linked to hereditary PD. A30P, E46K, H50Q, G51D, A53E, A53V and A53T are mutations in the αS gene that were linked to hereditary PD [52,53,54,55,56,57,58,59,60]. Similarly, several PTMs, such as phosphorylation, ubiquitination and nitration, have emerged as valuable markers of αS pathology [61,62,63]. Even though today's medications against PD provide symptomatic relief at early disease stages, there is a need to develop drugs that instead target the underlying disease process and thereby can delay or halt disease progression.
There is much interest in non-invasive molecular imaging of αS, with the hope that αS detection could serve as a biomarker for presymptomatic detection of PD, or for monitoring of disease progression. Although the development of αS-targeted probes is still in its infancy, there have been attempts to develop high-affinity ligands with appropriate tracer pharmacokinetics. An overview of selected αS-targeted translational molecular imaging probes is provided in Fig. 2. Initially, probes that were known for their use in Alzheimer’s disease were tested in PD patients. Indeed, amyloid-beta (Aβ)-targeted probes, such as the thioflavin-T derivative, 11C-Pittsburgh compound-B (11C-PIB), and benzoxazole BF227 (18F-BF227), have been suggested to also be useful for the early diagnosis of PD since they not only bind to Aβ, but also to the beta-sheet structures of αS-aggregates [64]. However, recent studies with brain homogenates revealed that 11C-PIB binding was not associated with the presence of Lewy bodies [65, 66]. Similarly, the radiofluorinated amyloid PET ligand, 18F-BF227, did not show meaningful binding to brain homogenates enriched with Lewy bodies [67, 68]. These findings encouraged the development of novel αS-selective PET tracers beyond repurposed Aβ-targeted probes. Along this line, Yu et al. synthesized a series of phenothiazine derivatives and reported the discovery of SIL5, SIL23, and SIL26 as high-affinity ligands for αS fibrils [69]. The same group subsequently reported the synthesis of the 125I-labeled analogue of SIL23, 125I-SIL23, which exhibited specific binding to αS fibrils in postmortem PD brain tissue as well as on brain slices of αS transgenic mice [70]. Despite these encouraging in vitro findings, the further development of 125I-SIL23 was plagued by a high degree of non-specific binding in vivo [70]. Structural derivatization of 125I-SIL23 yielded 11C-SIL5 and 18F-SIL26 with improved selectivity, however, their pharmacokinetic properties suggested the need for further optimization to achieve successful αS imaging in vivo [71]. The development of novel PET imaging agents for PD has been increasingly focusing on the discovery of high-affinity αS-targeted probes with suitable in vivo kinetics and biodistribution, which led to the design of novel scaffolds via high throughput screening [72, 73]. Among these scaffolds, N, N-dibenzyl-cinnamamide [74] and bisquinoline derivatives such as 18F-BQ2 [75] and 11C-MODAG-001 [76] proved to be promising lead candidates with moderate-to-high brain uptake, laying the foundation for the development of suitable clinical candidates. Matsuoka et al., recently reported on the clinical translation of an αS-targeted PET ligand codenamed 18F-SPAL-T-06, which was co-localized with regions known to accumulate glial cytoplasmic inclusion bodies in multiple system atrophy (MSA) patients [77]. Similarly, the biotech company AC Immune has recently announced that their lead αS-targeted PET ligand, 18F-ACI-12589, revealed excellent in vivo performance characteristics, supporting its translation into humans [78]. Future studies will examine the suitability of 18F-ACI-12589 to image αS aggregates in patients with PD or MSA.
Targeting αS with radiolabeled antibodies constitutes a challenge due to the low penetration of antibodies across the blood–brain barrier. To overcome this limitation, more recent examples of αS-targeted PET imaging involve the use of bispecific antibodies that carry a transferrin binding domain, to support blood–brain barrier penetration via receptor-mediated transcytosis, as well as a high-affinity αS binding domain [79]. Probing αS is further complicated by posttranslational modifications, which may affect protein folding and capacity to induce neuronal damage. Indeed, it was found that the serine residue at position 129 (Ser 129) of αS is selectively and extensively phosphorylated in synucleinopathy lesions. Phosphorylation of αS at Ser 129 promoted fibril formation in vitro, pointing towards a key role of this particular modification in the pathogenesis of α- synuclopathies [80], and prompting calls to carefully consider the impact of posttranslational modifications on the pharmacodynamic properties of PET and SPECT imaging probes. It should be noted that dementia is increasingly recognized as a common consequence of PD and around 50% of PD dementia cases develop significant amounts of Aβ plaques or p-tau containing neurofibrillary tangles [81]. As such, the specificity and selectivity of αS-targeted probes over these other proteinopathies is of paramount importance to decrease the likelihood for false positive scans, which constitutes an ongoing challenge for contemporary probe development. Other challenges in tracer development include the notion that αS is not abundantly expressed at early PD stages, requiring a highly sensitive detection tool and hampering the development of a suitable αS-targeted imaging probe for early PD diagnosis. Notably, αS aggregate structure, as well as spatial brain distribution and abundance of αS can vary depending on the underlying α-synucleinopathy, adding more layers of complexity, but concurrently providing an opportunity to harness these differences for differential diagnosis. A recent study concluded that the intrinsic structure of αS fibrils determines the characteristics of synucleinopathies [82]. For instance, fibrillar αS assemblies derived from patients with MSA exhibited strong similarities with PD-derived αS fibrils, but were significantly more potent in prompting nigrostriatal neurodegeneration, spread of αS pathology, inflammation and motor deficits, reflecting the aggressive nature of this disease [82]. In sharp contrast, strains from DLB cases displayed very modest neuropathological features. DLB is clinically distinguished from PD dementia by the timing of dementia onset – with individuals that develop dementia up to one year after the onset of motor symptoms considered to suffer from DLB, and individuals developing dementia later than one year following the onset of motor symptoms being assigned to the PD dementia group [83, 84].
Despite these various challenges in the development of αS-targeted PET ligands, a clinically validated probe could be used as a biomarker for disease progression, as well as to monitor therapy response in future trials. Further, other approaches such as real-time quaking-induced conversion (RT-QuIC) have enabled the identification of misfolded αS in the cerebrospinal fluid (CSF) of patients with prodromal and manifest α-synuclopathies with a sensitivity of 90–95% and a specificity of 80–100% [85, 86]. While the latter diagnostic performance is intriguing, limitations of current RT-QuIC applications for the detection of misfolded αS include the invasive nature of lumbar puncturing as well as the lack of information on spatial distribution of Lewy bodies across different brain regions. Finally, given the involvement of peripheral organs in PD, more recent findings indicated that colon, skin, gastrointestinal and submandibular gland biopsies may constitute valid alternatives to the brain as a source of pathological αS for diagnostic purposes [87,88,89,90,91,92]. A recent study by Wang et al. concluded that αS seeding activity from skin samples can serve as a novel biomarker for antemortem diagnoses of synucleinopathies [93]. Employing RT-QuIC analysis to assess 41 antemortem skin biopsies from patients with PD and controls, the authors found that posterior cervical and leg skin biopsy tissues provided a diagnostic sensitivity and specificity for the detection synucleinopathies in PD patients of 95% (95% CI, 77–100) and 100% (95% CI, 84–100), respectively. Along this line, phosphorylated αS depositions within autonomic nerve terminals of biopsied skin samples have been detected by immunohistochemistry (IHC) and immunofluorescence (IF) microscopy in living PD patients. Although a variety of immunohistochemical assays have been reported, the majority of studies were conducted at single centers without blinded and independent replication. However, in a recent study by Donadio et al., skin biopsies have been validated across laboratories, showing a high degree of reproducibility [94]. In most reported cases, peripheral biopsies were successfully used to discriminate PD patients from healthy subjects. Larger multicenter trials are warranted to unleash the full diagnostic potential of peripheral PD biopsies. Further, molecular imaging probes with tailored pharmacokinetic properties may facilitate the non-invasive assessment of pathological αS in peripheral organs such as the colon or submandibular gland.

Molecular imaging in Parkinson’s disease (PD). Selected biological targets that have been harnessed for positron emission tomography (PET) and single photon-emission tomography (SPECT) imaging in PD. These targets have been used to allow diagnostic imaging as well as to facilitate drug discovery & development and include the translocator protein 18 kDa (TSPO), which is upregulated in activated microglia, mitochondrial complex I (MC-I) to assess mitochondrial function, various α-synuclein and amyloid-β (Aβ)-targeted probes, radiolabeled dopamine & serotonin transporter substrates & ligands of dopamine receptors, probes targeting the vesicular monoamine transporter 2 (VMAT2) and attempts to visualize autophagy as well as cytokines such as the tumor necrosis factor α (TNF-α). Given that these molecular variables are critical to the pathophysiology of PD, most of them have been suggested as therapeutic targets
The availability of molecular imaging tools to visualize αS has the potential to support emerging therapeutic concepts that are focused on attenuating the formation of misfolded αS aggregates, through reduction of αS production, inhibition of αS aggregation, increased αS degradation, or inhibition of uptake of extracellular αS into neurons [95]. For instance, reduced αS expression can be achieved by the silencing of the SNCA gene via small interfering RNA (siRNA). Studies have established the potential neuroprotective effects of siRNAs in rodent models of α-synucleinopathies, particularly upon intracranial administration in hippocampal and cortical regions of the rodent brain [96,97,98]. In a recent study by Mittal et al., β2-adrenoreceptors were found to regulate SNCA transcription via histone 3 lysine 27 acetylation of its promoter and enhancers [99]. Along this line, individuals treated with β2-adrenoreceptor agonists had a significantly reduced risk of developing PD (OR = 0.66, 95% CI, 0.58 to 0.76) [99]. Further, studies have highlighted the potential of heat shock proteins (HSPs) [100, 101]. Of note, HSPs serve as molecular chaperons, capable of preventing the aggregation of misfolded proteins such as pathological αS. It was found that the small heat-shock proteins, αB-crystallin and Hsp27, prevent αS aggregation in vitro via transient molecular interactions that were dependent on the kinetics of αS aggregation [101]. Similarly, Hsp70 was found to reduce the amount of aggregated αS in vitro and in vivo, in an α-synucleopathy mouse model overexpressing Hsp70, protecting it from αS-dependent neurotoxicity. Further, first-generation epitope vaccines that were generated against B cell antigenic determinants of human αS and produced high titers of anti-human αS antibodies attenuated Lewy bodies pathology in brain specimens from DLB cases [102]. Active and passive immunization against α-syn have shown neuroprotective activity via attenuation of αS aggregation in transgenic mouse models of PD [103,104,105]. In an initial clinical trial, the humanized IgG1 monoclonal antibody, prasinezumab, prompted a 96.5% reduction of soluble serum αS with good tolerability and favorable safety profile in patients [106]. However, results of a phase 2 clinical trial (NCT03100149) showed that the drug has limited effects on disease progression [107]. To date, interesting exploratory concepts of αS-targeted therapy involve the development of monoclonal antibodies against αS, which promote its degradation [105]. Further, the anti-αS antibody, Lu-AF82422, has proven to inhibit seeding of αS in preclinical studies [108] and may constitute a promising candidate for the treatment of α-synucleinopathies in future clinical trials. Of note, drug development efforts in Alzheimer’s disease revealed that immunotherapies directed at aggregated forms of Aβ, with the particular aim not to target monomeric Aβ, proved to be highly promising [109, 110]. Similar attempts to design and develop antibodies that specifically target aggregated oligomeric forms of αS are currently emerging [111,112,113], and future studies will reveal their therapeutic potential. Given their neurotoxic properties, oligomeric αS forms are considered a particularly attractive target for passive immunization strategies in PD [114]. As such, several examples have been reported in which antibodies against oligomeric αS triggered a reduction in αS accumulation in studies with transgenic murine models of synucleinopathies, as recently reviewed by Vaikath et al. [115]. Similar to the examples from PET ligand development, bispecific antibodies that undergo transferrin receptor-mediated transcytosis have also been developed for therapeutic purposes [116]. This concept has been leveraged to advance Aβ-targeted antibodies to AD patients and has found application in preclinical PD studies, as evidenced by the reduction of αS pathology in transgenic L61 mice following treatment with the bispecific antibody, RmAbSynO2-scFv8D3 [117]. It is anticipated that similar efforts will be used in clinical trials with PD patients in the near future. Overall, αS-targeted therapy in PD has been extensively investigated in preclinical models and a steadily accumulating body of clinical evidence seems to underscore the potential value of development of αS-targeted therapy.
Mitochondrial and lysosomal dysfunction
The link between parkinsonism and mitochondrial dysfunction was initially established when the neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), was reported to trigger acute parkinsonism in humans [118], with similar observations in rodents and non-human primates [119,120,121,122]. These observations gave rise to concerted research into the role of mitochondrial dysfunction in PD. Of note, MPTP is a pro-drug that rapidly passes through the blood–brain barrier. Once in the brain, it is oxidized to the active compound, 1-methyl-4-pyridinium (MPP +). MPP + is a substrate of the dopamine transporter, thus leading to MPP + accumulation in dopaminergic cells, where the compound inhibits respiration at mitochondrial complex I (MC-I) of the electron transport chain [123,124,125]. Abnormal mitochondrial dynamics, including fission and fusion, was found to prompt dopaminergic nerve loss, thus providing a potential mechanism of MPTP-induced parkinsonism [126]. Later studies showed that many PD-associated gene mutations directly or indirectly lead to mitochondrial dysfunction, [127,128,129] which leads to the formation of reactive oxygen species (ROS), exposing the respective neurons to persistent oxidative stress [47, 57, 58]. Although MC-I deficiencies are the primary source of ROS in PD [125], other factors including auto-oxidation of dopamine [130] and neuroinflammation also contribute to oxidative stress [21]. Given (1) the implications of mitochondrial dysfunction and oxidative stress in PD and (2) the critical role of MC-I in maintaining redox homeostasis, targeting MC-I may constitute a promising diagnostic and therapeutic approach in PD. Indeed, MC-I selective PET ligands such as 18F-BCPP-EF and 18F-flurpiridaz have been developed and validated in humans [131, 132]. These tools can be readily harnessed to facilitate the discovery of MC-I-targeted therapeutic agents, thereby harboring the potential to provide a novel disease-modifying concept in PD. It should be mentioned, however, that 18F-flurpiridaz is mainly used for myocardial perfusion imaging due to the perfusion-dependent tracer kinetics in the myocardium [133, 134]. It is currently unclear whether 18F-flurpiridaz can be leveraged for mapping MC-1 in the mammalian brain. Beyond the role of MC-I in mitochondrial dysfunction, mutations in PINK1, the gene encoding for PTEN-induced putative kinase 1 (PINK1), are associated with mitochondrial dysfunction in PD, whereas patients who are heterozygous for mutations in PINK1 present with reduced intensity on 18F-fluoro-levodopa PET compared with controls [135].
Lysosomal dysfunction has been increasingly recognized as another key determinant in the pathophysiology of PD. Along this line, the lysosomal hydroxylase glucocerebrosidase (GCase), encoded by the GBA gene, is known to exhibit pathogenic mutations that can cause hereditary PD [136]. GCase and αS form a bidirectional pathologic loop where GCase deficiency leads to a decline of lysosomal proteolysis that affects αS, while increased levels of αS attenuate the function of GCase in neuronal lysosomes [137, 138]. Targeting GCase as a therapeutic strategy for PD has been suggested. Ambroxol is considered a potentially disease-modifying agent that proved to increase brain GCase activity and decrease αS in experimental parkinsonism, prompting its clinical translation. Indeed, ambroxol has recently been advanced to clinical trials for the treatment of PD (NCT02941822) and PD-associated dementia (NCT02914366) [136, 139].
Similar to GCase, leucine-rich repeat kinase 2 (LRRK2) is involved in hereditary PD [140]. Genetic and biochemical data unveiled that hyperactivity of LRRK2 is linked to PD progression and thus it has been hypothesized that lowering its activity may exhibit disease-modifying effects in PD [141]. While attempts to develop LRRK2-targeted molecular imaging probes have been reported, a clinically validated PET or SPECT tracer is currently lacking [142,143,144]. Nonetheless, the recently reported radiofluorinated probe, 18F-PF-06455943, exhibited favorable performance characteristics in rodents and non-human primates, rendering it a promising candidate for clinical translation [145]. The availability of such a tool would facilitate the development of LRRK2-targeted therapeutics in the pipeline. Although LRRK2 kinase inhibitors have proven protective in experimental parkinsonism and have been suggested for clinical translation [146], the mechanisms by which mutant LRRK2 contributes to PD pathophysiology are not fully understood. LRRK2 does not form aggregates in the affected PD brain, however, the pathological function of mutated LRRK2 may involve common pathways with αS. For instance, mice that were transgenic for mutant human LRRK2 and overexpressed mutant human SNCA exhibited an enhanced deposition of α-synuclein, as compared with control mice that overexpressed mutant human SNCA alone [147]. In contrast, removal of the endogenous Lrrk2 gene limited the detrimental effects of mutant α-synuclein, reducing protein deposition and neuronal loss in experimental parkinsonism [148]. Studies assessing PD-related changes in LRRK2 levels are limited. Nonetheless, a study that used laser microdissection to isolate dopaminergic neurons from the substantia nigra pars compacta of subjects with idiopathic PD and controls suggested that LRRK2 mRNA levels were attenuated in PD patients [149]. These results warrant confirmation in larger trials and may support the notion that LRRK2 could serve as a disease biomarker that can be targeted with molecular imaging probes. While the molecular mechanism underlying a beneficial effect of LRRK2 inhibitors in PD are not entirely understood, an LRRK2-induced attenuation of autophagy has recently been proposed [150]. Among the LRRK2 inhibitors in clinical development, DNL201 and DNL151 were well tolerated in a phase I study in healthy volunteers [146]. Further, DNL201 was evaluated in a phase Ib trial assessing drug safety and pharmacokinetics in 28 patients with early PD. DNL201 was generally well tolerated and demonstrated a robust cerebrospinal fluid penetration [151]. Other LRRK2 inhibitors such as MLi-2 and PF-06685360 have shown encouraging preclinical efficacy, however, there are no reports on human use of these agents to date [152]. Another approach to suppress LRRK2 activity is through the use of anti-sense oligonucleotides (ASOs) that can decrease LRRK2 expression. Indeed, the recently reported ASO, BIIB094, prompted a decrease of αS inclusions in mice, and its safety, tolerability and pharmacokinetic profile are currently being evaluated in humans (NCT03976349) [153, 154].
While PD has been linked to dysfunctional autophagy, attempts to target autophagy by translational molecular imaging probes have been scarce. The histone deacetylase 6 (HDAC6) constitutes a potential biological target to non-invasively assess autophagy. Indeed, HDAC6 was found to orchestrate autophagosome maturation [155] and prevent neurodegeneration by enhancing autophagy when the ubiquitin–proteasome system is impaired [156]. HDAC6 was further found to participate in PD pathophysiology [157], and can be targeted using 18F-EKZ-001 PET, as recently shown in a first-in-human study [158]. Indeed, 18F-EKZ-001 was well tolerated and exhibited favorable tracer pharmacokinetics, including rapid brain uptake and retention in cortical regions with high HDAC6 abundancy [158]. This imaging tool can be harnessed for clinical target engagement studies, facilitating the development of HDAC6 modulators in the pipeline. Moreover, future studies will reveal whether HDAC6-based molecular imaging is useful to assess the integrity of the autophagy machinery in PD.
Neuroinflammation in Parkinson’s disease
Neuroinflammatory mechanisms that contribute to the neuronal degeneration in PD include, but are not limited to, microglial activation [159], excessive cytokine release, and lymphocytic CNS infiltration (Fig. 3) [160]. The latter triggers deleterious events that ultimately result in cytokine receptor-mediated apoptosis, which, in turn, may contribute to the dopaminergic cell death and PD progression [22]. Microglia represent 10–20% of all glial cells in the brain and act as a natural defense mechanism against pathogens and as key mediators of immune response in the brain [161, 162]. However, excessive microglial activation following mitochondrial dysfunction and oxidative stress can ultimately result in neuronal cell death. Upon activation, microglial cells upregulate the peripheral benzodiazepine receptor (translocator protein 18 kDa, TSPO) – an essential component of the mitochondrial membrane that mediates functions such as cholesterol transport, mitochondrial respiration, apoptosis, and cell proliferation [163]. Although the underlying trigger of increased TSPO expression in activated microglia is not fully understood, TSPO is believed to be a key modulator of microglial activity [164]. TSPO has been used as a biomarker for neuroinflammation and a variety of clinically validated imaging probes for TSPO have been developed (Fig. 2). The isoquinoline derivative, 11C-PK11195, was the first TSPO PET ligand that proved to be clinically useful for imaging neuroinflammation [165, 166]. PET studies indicated that there is pronounced microglial activation in various regions of the PD brain, as evidenced by studies with 11C-PK11195 [167, 168]. However, due to the limitations of 11C-PK11195, which include high non-specific binding, low brain penetration, and high plasma protein binding, the clinical use of 11C-PK11195 is hampered [169, 170]. Second generation radiotracers with superior imaging characteristics have subsequently been developed, including several probes with variable chemical structures, such as 11C-PBR28, 18F-FEPPA, 11C-DAA1106 [171,172,173,174,175,176,177,178,179]. One critical drawback of these tracers is their sensitivity to polymorphism in the TSPO gene, thus affecting their binding properties and leading to increased inter-individual variability. The latter shortcoming prompted the development of third generation TSPO-targeted probes such as the tricyclic indole, 18F-GE180 [180]. The ability of 18F-GE180 to bind selectively and with high affinity to TSPO is critical for the assessment of neuroinflammation in disease states, providing significantly better TSPO imaging signal to noise ratios and lower nonspecific binding, as compared to 11C-PK11195 [181, 182]. Notably, it was recently suggested that substantial discrepancies between in vitro and in vivo sensitivity to TSPO gene polymorphism might further complicate TSPO PET ligand development, as evidenced by studies involving the translational work on quinazoline-2-carboxamide derivative, 11C-ER176 [183], of which a radiofluorinated analogue (18F-3b) has been reported. [184] Future clinical trials are warranted to assess the utility of 18F-3b and 18F-GE180 to visualize neuroinflammatory processes in PD.

Established vs. experimental therapies in Parkinson’s disease (PD). While current drug PD therapy is primarily symptomatic and attempts to restore dopaminergic function, many lines of experimental research focus currently on the development of potentially disease-modifying concepts. The latter involve various targets that can be visualized using translational molecular imaging probes, thus providing the opportunity to conduct imaging-guided preclinical and clinical studies in PD. Among the promising concepts, preventing the formation of inclusion bodies by targeting misfolded α-synuclein, attenuating neuroinflammation via anti-tumor necrosis factor α (TNF-α) antibodies and glucagon-like peptide 1 (GLP-1) activation, as well as stimulation of autophagy with leucine-rich repeat kinase 2 (LRRK2) inhibitors have been suggested
Other mechanisms by which microglial function can be attenuated include the activation of glucagon-like peptide-1 (GLP-1) receptor and the inhibition of inflammatory cytokines such as the tumor necrosis factor α (TNFα) [185, 186]. In a recent single-center, randomized, double-blind and placebo-controlled trial, patients with moderate idiopathic PD received the GLP-1 receptor agonist, exenatide, once weekly for 48 weeks in addition to their regular medication, prompting a significant improvement of their clinical scores, as compared to the placebo group [185]. These results not only suggested that exenatide may warrant further study as a therapy, but also provided the opportunity to pursue imaging-guided clinical trials with radiolabeled exenatide analogs, which have recently entered the clinical arena [187,188,189]. While anti-TNFα antibody therapy with agents such as with infliximab or adalimumab has been established for peripheral inflammatory diseases [190], these agents have not yet been extensively studied as potential therapeutics in PD. In the wake of previous examples of bispecific anti-TNFα antibodies, it is conceivable that a bispecific anti-TNFα antibody, that allows receptor-mediated transcytosis, may be of particular interest for therapy studies in PD patients. Further, the notion that 64Cu-etanercept was shown to rapidly accumulate in the choroid plexus and the CSF within the cerebral ventricles of a living rat after peripheral administration [191] points towards a potential utility of the probe to visualize TNFα in the central nervous system. The latter may lay the foundation for the assessment of TNFα abundancy in live PD patients in the near future, as well as for the potential assessment of target engagement by anti-TNFα drugs in the PD brain.
Triggering receptor expressed on myeloid cells 2 (TREM2) is heavily implicated in glial function and axonal myelination [192]. Genetic variants of TREM2 were linked to PD, however, the potential role of TREM2 on the formation and aggregation of pathological αS is poorly understood. In a recent study involving BV2 microglial cells and TREM2 knock-out mice, it was demonstrated that TREM2 deficiency aggravates αS-related neuropathology [193]. Further, soluble TREM2 levels were found to be elevated in PD subgroups presenting with increased levels of tau in the CSF [194]. Further studies to identify molecular pathways linking TREM2 with αS are required to pave the way for TREM2-targeted novel therapeutic concepts in PD. Similarly, molecular imaging probes directed at TREM2 [195] hold promise to serve as research tools, thereby shedding light on the role of TREM2 in PD pathophysiology.
Dysregulation of neuroreceptor systems
To date, most of the routinely used antiparkinsonian medications target dopamine deficiency. While dopaminergic pharmacological treatment is presumed not to considerably affect disease progression, it has enabled sustained symptom control and ground-breaking improvements in the quality of life for PD patients [13]. An overview of the contemporary arsenal of validated and selected experimental PD drugs is provided in Fig. 3.
While progressive degeneration of dopaminergic neurons is evident in PD, experimental and clinical studies have highlighted the involvement of other neuroreceptor systems, including the glutamatergic, serotonergic, adrenergic and cholinergic system [10, 196]. For instance, it has been suggested that a well-defined balance between central dopaminergic and cholinergic signaling is required for movement control and that efforts to restore that balance, with anticholinergic agents, would result in improvement of motor symptoms in PD patients [197, 198]. Approved anticholinergics provide symptomatic relief for tremor and to some extent for rigidity, however, they exhibit significant side effects (such as memory disturbances or somnolence), which makes them unusable in most elderly parkinsonian patients. Although dopaminergic therapy has largely supplanted the use of anticholinergic drugs, they are still used in clinical practice, particularly in younger PD patients with prominent tremor [199,200,201,202].
Aside from the CNS implications of the cholinergic system in PD, there is a solid body of evidence supporting its role in autonomic dysfunction in peripheral organs, which is observed in most PD patients and substantially affects their quality of life. Peripheral PD symptoms (cholinergic or non-cholinergic) include gastrointestinal dysfunction, cardiovascular dysregulation, urinary disturbances, sexual dysfunction, and thermoregulatory changes [203, 204]. Intriguingly, αS pathology has recently been linked to autonomic denervation [205,206,207,208]. Cardiac autonomic denervation has the potential to allow differential diagnosis between PD and other forms of parkinsonism, thus rendering it a useful biomarker for diagnostic imaging [209]. A summary of contemporary molecular imaging concepts involving dysregulated neuroreceptor systems in PD is provided in Fig. 2.
Dopamine receptors
Postsynaptic dopamine receptors are divided into five subtypes, namely, dopamine receptors 1–5 (D1-D5). While D1 and D5 are coupled to G stimulatory sites and activate adenylyl cyclase, D2-D4 couple to G inhibitory sites, thereby inhibiting adenylyl cyclase [210]. Dopamine receptors are typically upregulated in early PD, which can be quantified by dopamine receptor PET [211, 212]. A major obstacle of D1-targeted PET imaging has been the challenging development of selective probes. While 11C-SCH23390 was initially identified as D1 PET ligand and was advanced to humans [213], the broad use of this tracer was hampered due to the limited subtype-selectivity. A close derivative, 11C-SCH39166 exhibited improved selectivity, however, at the expense of reduced binding affinity towards D1 [214]. Further, another PET ligand code-named, 11C-NNC112, was suggested for quantitative assessment of dopamine receptors, [215] but was demonstrated to have off-target binding at 5-HT2A receptors in humans [216]. Tamagnan et al. reported on the development of a potentially promising D1-targeted probe codenamed 18F-MNI-968, which was administered to non-human primates and humans [217]. Although preliminary data was deemed promising, further studies are currently ongoing to elucidate the potential of 18F-MNI-968 for subtype-selective imaging of D1 receptors. With regard to D2 receptor imaging, 11C-raclopride has been widely used. This agent competes with endogenous dopamine, thereby proving to be a useful tool for qualitative assessment of D2 receptor density [213]. Alternative D2-targeted PET probes such as 18F-fallypride are considered valid alternatives with improved binding affinities at D2 receptors [218].
These dopamine receptor-targeted probes are not currently in routine use for the diagnosis of PD because of potential interactions of dopamine replacement therapy with tracer-receptor binding, and the complexity of such analyses related to the dopamine receptor autoregulation in response to the loss of dopaminergic neurons. However, the imaging tools are valuable to facilitate the development of improved dopamine receptor agonists. Indeed, these tools have been harnessed to assess target occupancy by endogenous dopamine, or dopamine released in response to levodopa therapy, and various synthetic agonists across different species, thereby advancing our understanding of the biologic effects of dopaminergic therapy [219,220,221,222,223].
Various dopamine receptor agonists have been developed and shown to attenuate PD motor symptoms. Dopamine receptor agonists can be divided in two major classes, namely, ergot- and non-ergot derivatives. Because the use of ergot derivatives has been associated with pleuropulmonary and cardiac valvular fibrosis [13], currently used dopamine receptor agonists belong to the non-ergot family [224]. Examples include pramipexole, ropinirole, rotigotine, apomorphine and piribedil [225]. Although, they were initially licensed for synergistic administration with levodopa at advanced PD stages, their application as monotherapy has also proven useful in PD [224, 226]. In conclusion, targeting dopamine receptors constitutes a compelling example of how translational molecular imaging probes can be exploited to provide imaging-supported PD therapy.
DOPA decarboxylase and dopamine transporters
In the mammalian brain, DOPA decarboxylase (DDC) facilitates the transformation of levodopa to dopamine [227]. Levodopa is the L-isomer of the naturally occurring amino acid, D,L-dihydroxy-phenylalanine [228]. Unlike dopamine, levodopa crosses the blood–brain barrier, thus allowing peripheral administration of the drug. The paramount therapeutic value of levodopa in PD was recognized when the enzyme that converts levodopa to dopamine, DDC, was found to be present in high amounts in the mammalian brain [229, 230]. For more than half a century, levodopa has been the first-line treatment for PD [231,232,233]. Indeed, levodopa therapy is remarkably effective in attenuating primary motor symptoms of PD patients.
Upon oral administration of levodopa, it undergoes an extensive first-pass metabolism and rapid plasma clearance, allowing for only 1% of the administered dose to enter the brain [234]. This limitation can be overcome by co-administration of peripheral DDC inhibitors that do not enter the central nervous system (CNS). In fact, the latter was shown to substantially increase CNS availability of levodopa, thus reducing the substantial peripheral side effects of levodopa treatment, and improving its central efficacy [234,235,236,237,238]. Moreover, administration of levodopa via inhalation (CVT-301) [239] or continuous subcutaneous infusion [240,241,242] – mainly to circumvent the first-pass effect – have been suggested to maintain a steady prodrug supply to the brain. Contemporary levodopa treatment regimens include the co-administration of a peripheral DDC inhibitor, carbidopa or benserazide, which inhibits the peripheral decarboxylation of levodopa, thus reducing peripheral toxicity (nausea) and allowing greater CNS bioavailability of levodopa [13]. To further improve the availability of dopamine in the brain of parkinsonian patients, inhibitors of levodopa (and dopamine) metabolism via catechol-ortho-methyl-transferase (COMT) can be used. COMT inhibitors such as entacapone, tolcapone and opicapone have been introduced as an adjunct to levodopa-based combination therapies [243, 244]. As opposed to DDC and COMT inhibitors, which primarily lead to inhibition of peripheral levodopa metabolism, monoamine oxidase B (MAO-B) inhibitors prevent the degradation of dopamine in the CNS [245]. As such, synaptically released dopamine can be cleared by oxidation via MAO-B, and pharmacological modulation of MAO-B with rasagiline, selegiline or safinamide has been shown to increase synaptic dopamine concentrations, thus rendering them useful as an adjunct to levodopa therapy, and also as early-stage monotherapies in PD [246,247,248,249].
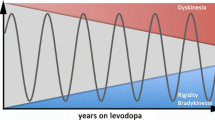
Chronic levodopa therapy can be associated with the development of troublesome motor complications, specifically levodopa-induced dyskinesias (LID) and motor response fluctuations, including sudden wearing-off effects [250, 251]. The underlying mechanisms for the development of dyskinesias with chronic levodopa replacement therapy are not yet fully understood, however, non-physiological pulsatile dopamine receptor stimulation in the striatum is considered to be an important factor [13, 252]. Based on this concept, drug delivery approaches that are less encumbered by pulsatile changes in brain dopamine levels, including sustained-release formulations and continuous levodopa release through implanted pumps or intestinal gels have been used effectively to prevent or ameliorate the development levodopa-induced dyskinesias [253, 254].
Translational molecular imaging offers an effective way to evaluate the biodistribution of levodopa when administered via different application routes, such as those mentioned above. Similar to levodopa, the radiofluorinated analog, 6-18F-fluoro-levodopa (18F-FDOPA), is trapped in the brain by DDC-mediated biotransformation, rendering 18F-FDOPA a suitable tool to study drug biodistribution, dopamine storage capacity, as well as DDC activity and the loss of dopaminergic neurons in living subjects [255,256,257,258]. Similar to levodopa, 18F-FDOPA readily crosses the blood brain barrier through neuronal amino acid transporters and is taken up by presynaptic terminals of dopaminergic neurons via the dopamine transporter (DAT), where it is metabolized to 18F-dopamine by DDC and stored in synaptic vesicles (Fig. 2). [257, 259] 18F-FDOPA PET has been used to assess the degree of nigrostriatal dopaminergic nerve loss in PD patients [260, 261]. In combination with a clinical history, 18F-FDOPA is considered a reliable diagnostic tool for the early diagnosis of PD. In a recent clinical trial encompassing 56 patients with suspected PD, 18F-FDOPA was found to be highly specific and sensitive for the diagnosis of PD, which led to approval of its use for this purpose by the US Food and Drug Administration (FDA) in 2019 [262].
Other PET tracers that are considered substrates of DAT are typically employed for the assessment of presynaptic dopaminergic neurons in the nigrostriatal system [38]. In line with histopathological evidence, it was found that the uptake of DAT-targeted probes in the striatum is substantially reduced in PD patients, as compared to controls. Selected examples of reported DAT PET radioligands include 11C-WIN 35,428 (11C-CFT) [263], 18F-CFT [264], 11C-PE2I [265] and 11C-altropane [266]. All of these PET probes are highly sensitive to striatal dopaminergic denervation in PD. Similarly, DAT-targeted SPECT radioligands have proven useful for PD diagnosis. Particularly, 123I-ioflupane (the ligand used commercially for DaTscans) and 123I-β-CIT have been broadly used to support the assessment of parkinsonism [267]. While carbon-11 and fluorine-18 labeled DAT-targeted PET ligands typically provide higher image resolution than iodine-123 labeled SPECT tracer, the latter exhibit a longer physical half-life, which facilitates the logistics and distribution to facilities without on-site tracer production. In conclusion, DAT imaging with either PET or SPECT is the most widely applied functional imaging tool for the diagnostic work-up and can be considered a valuable tool for PD diagnosis in clinical routine [268,269,270].
Vesicular monoamine transporter 2
While vesicular monoamine transporter 1 (VMAT1) is mainly expressed in the periphery, the central nervous system is endowed with a high abundancy of vesicular monoamine transporter 2 (VMAT2) [271]. There are several PET tracers available to image the distribution of VMAT2. The potentially most prominent VMAT2-targeted probes belong to the class of tetrabenazines and include tritium-labeled methoxytetrabenazine for preclinical applications, as well as 11C-dihydrotetrabenazine (11C-DTBZ) for preclinical and clinical PET imaging. 3H-methoxytetrabenazine was successfully used to map VMAT2 density in rodents [272]. Similarly, 11C-DTBZ has been used in humans to monitor the integrity of striatal monoaminergic nerve terminals [273]. To improve the half-live of 11C-DTBZ, a radiofluorinated analog, 18F-DTBZ (18F-AV-133), was developed and tested in PD patients [274]. VMAT2 binding, as determined by 18F-DTBZ PET, correlated significantly with clinical disease severity and duration [274, 275]. Collectively, these results suggested that 18F-DTBZ PET is a powerful tool to measure dopaminergic degeneration and monitor disease progression in PD patients.
Serotonergic and glutamatergic system
Although most reported PET and SPECT tracers for use in PD are geared towards evaluating the function or integrity of the dopaminergic system, several non-dopaminergic targets have recently been suggested as molecular biomarkers in PD. While dopaminergic markers allow for the diagnosis of PD at intermediate disease stages (Braak stages ≥ 3), it is possible that non-dopaminergic biomarkers could identify PD-related pathophysiological changes prior to the onset of clinical symptoms (Braak stages 1–2). The serotonergic system has been implicated in PD-associated depression, which typically develops early in the course of disease. Nonetheless, given the relatively high prevalence of depression in the general population, it remains to be seen whether serotonergic changes in PD-related depression can be distinguished from those in non-PD-related depression. Early observations included the low levels of 5-hydroxyindolacetic acid, a serotonin metabolite, in the cerebrospinal fluid, as well as the reduced cortical 5-HT1A receptors, in (symptomatic) PD patients suffering from depression [276]. Investigating the serotonergic system in PD patients was facilitated by the availability of serotonin transporter (SERT) PET ligands such as 11C-DASB [277] and 11C-WAY100635 [165, 278, 279]. Recently, serotonergic brain imaging with 11C-DASB has proven effective in identifying individuals at risk to develop PD, prior to the development of dopaminergic pathology and motor symptoms, which highlights the potential of imaging the serotonergic system for risk-stratification [280]. To date, pimavanserin, a serotonin 5-HT2A and 5-HT2C receptor ligand with antipsychotic effects, is approved by the FDA for the treatment of hallucinations and delusions associated with PD psychosis [281]. Further, conventional antidepressant therapy with selective serotonin reuptake inhibitors (SSRIs) and selective noradrenaline reuptake inhibitors (SNRI) are commonly used in PD patients suffering from depression.
Glutamate receptors (GluRs) are multimeric ligand-gated cation channels that mediate excitatory neurotransmission in the CNS. Given the role of glutamate receptors in modulating neurotransmission in the basal ganglia, it was suggested that drugs acting at these receptors may have antiparkinsonian effects [282]. GluRs are classified into ionotropic (iGluRs) and metabotropic glutamate receptors (mGluRs). IGluRs can be further subdivided into N-methyl-d-aspartate (NMDA), amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate (KA) receptors, and the family of mGluRs constitutes 8 different subtypes, mGluR1-8 [279, 283]. Several NMDA antagonists, including competitive antagonists, noncompetitive antagonists, and glycine site antagonists were found to reverse muscle rigidity in rodent models of PD [282, 284]. Anti-parkinsonism effects of NMDA antagonists were also observed in other animal models of PD, including MPTP-treated non-human primates [285, 286]. Along this line, co-administration of NMDA antagonists with levodopa was found to potentiate the levodopa effect and decrease LID [282, 287]. Similarly, amantadine, a drug with non-competitive NMDA receptor antagonist properties, ameliorates LID [288]. Due to the possible side effects that arise from global inhibition of NMDA receptors, recent drug discovery efforts are focused on the identification of subtype-selective NMDA antagonists, for instance by targeting the GluN2B subunit of the NMDA receptor, which is believed to be a key contributor to excitotoxicity-induced neuronal cell death [289,290,291]. The recent breakthroughs in translational molecular imaging of GluN2B-containing NMDA receptors may help with the development of subtype-selective NMDA antagonists for the treatment of PD [292,293,294,295,296,297,298,299,300]. The most advanced class of GluN2B-targeted PET probes is based on a benzazepine scaffold and includes the carbon-11 derivative, (R)-11C-Me-NB1 [295, 300], which has recently been translated to humans, as well as the radiofluorinated analogs, (R)-18F-OF-Me-NB1 [292, 298], and (R)/(S)-18F-OF-NB1 [293, 296], which have recently been advanced to non-human primate studies and exhibited favorable performance characteristics, providing effective tools for the non-invasive quantification of GluN2B-carrying NMDA receptors.
Cardiac sympathetic denervation
Cardiac sympathetic denervation in PD patients was first observed more than two decades ago, when Goldstein et al. used PET imaging to visualize the significant loss of adrenergic nerve terminals in the left ventricle [301]. Since then, numerous studies have corroborated the loss of adrenergic neurons in the myocardium of PD patients. Although autonomic function is impaired in various organs, adrenergic denervation is particularly pronounced in the heart [207]. Indeed, some PD patients present with evidence of cardiac adrenergic denervation early in the disease course. Thus, in a review by Sakakibara et al., the authors concluded that cardiac sympathetic imaging with 123I-metaiodobenzylguanidine (MIBG) could harbor potential for the diagnosis of pre-motor PD, in patients presenting with (non-specific) symptoms such as autonomic dysfunction, mild memory impairments, sleep disorder, visual hallucinations and depression/anxiety [302]. In addition to this potential diagnostic use, the detection of autonomic dysfunction may also have prognostic value: Autonomic dysfunction early in the course of symptomatic PD may presage a fast rate of progression [303, 304]. Mechanistic studies to explain these findings are needed. Of note, the norepinephrine derivative, droxidopa, constitutes a prodrug for the management of neurogenic orthostatic hypotension in PD patients, indicating that adrenergic signaling is a valid target for the treatment of peripheral symptoms in PD [305].
Cardiac imaging with 123I-MIBG can also be used to discriminate PD from MSA and progressive supranuclear palsy because the latter two conditions show only modest reduction in cardiac 123I-MIBG uptake, and greater sympathetic ganglion dysfunction [306, 307]. Prospective clinical trials that are adequately powered are warranted to accurately assess the diagnostic and prognostic value of cardiac sympathetic imaging in early PD as well as to identify ideal subpopulations for cardiac 123I-MIBG-based differential diagnosis of parkinsonism.
Concluding remarks
Notwithstanding the significant progress in disease management, PD remains a major source of global disability, with no current cure. The main challenge in contemporary drug discovery is the identification of disease-modifying agents. Attempts to target αS are particularly promising, given the established role of αS across all stages of disease pathology. Therapeutic efficacy of αS-directed antibodies in the pipeline could potentially be monitored with a clinically validated αS-targeted probe, which holds promise to deliver an improved disease prognosis, however, their clinical development is still at an early stage and larger clinical trials that are supported by αS-specific and selective PET ligands with appropriate in vivo performance characteristics are currently pending. Beyond αS, molecular imaging with PET and SPECT has been established for the assessment of nigrostriatal loss, particularly with probes that are substrates of the dopamine transporter and serve to quantify dopaminergic nerve loss in clinical routine. More exploratory imaging approaches, that are currently mainly applied for research purposes in PD, include the assessment mitochondrial dysfunction, as well as dysregulation of the serotonergic and glutamatergic systems. To assess the role of neuroinflammation in PD patients, validated TSPO-targeted probes are employed due to their capacity to detect activated microglia. While the availability of a diverse arsenal of selective imaging probes is undoubtedly helpful as a research toolbox, further studies are required to determine the true potential of these exploratory imaging approaches to support drug discovery and improve the current standard of care for PD patients. There is an urgent need for early biomarkers that support diagnosis and patient recruitment. Further, disease progression markers are needed to facilitate the evaluation of therapeutic efficacy to improve the contemporary management of PD. Translational molecular imaging holds promise to advance the diagnostic landscape and contribute to an improved clinical decision-making. Tailored therapeutic strategies, based on individual patient scans, may help to define optimal management concepts and lead to the best possible treatment for every PD patient in the future.
Availability of data and materials
Not applicable.
Abbreviations
- 11C-DTBZ:
-
11C-Dihydrotetrabenazine
- 11C-PIB:
-
11C-Pittsburgh compound-B
- 11C-CFT:
-
11C-WIN 35,428
- MIBG:
-
123I-Metaiodobenzylguanidine
- MPP + :
-
1-Methyl-4-pyridinium
- 18F-FDOPA:
-
6-18F-fluoro-levodopa
- αS:
-
α-Synuclein
- AMPA:
-
Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- 18F-BF227:
-
Benzoxazole BF227
- COMT:
-
Catechol ortho methyl transferase
- CNS:
-
Central nervous system
- DLB:
-
Dementia with Lewy bodies
- DDC:
-
DOPA decarboxylase
- D1:
-
Dopamine receptor type I
- D2:
-
Dopamine receptor type II
- DAT:
-
Dopamine transporter
- GLP-1:
-
Glucagon-like peptide-1
- GCase:
-
Glucocerebrosidase
- GluRs:
-
Glutamate receptors
- HDAC6:
-
Histone deacetylase 6
- KA receptors:
-
Kainate receptors
- LRRK2:
-
Leucine-rich repeat kinase 2
- MC-I:
-
Mitochondrial complex I
- MAO-B:
-
Monoamine oxidase B inhibitors
- MSA:
-
Multiple system atrophy
- MPTP:
-
N-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NMDA:
-
N-methyl-d-aspartate
- PD:
-
Parkinson’s disease
- translocator protein 18 kDa, TSPO:
-
Peripheral benzodiazepine receptor
- PET:
-
Positron emission tomography
- PTMs:
-
Post-translational modifications
- SPECT:
-
Single-photon emission computed tomography
- SNAREs:
-
Soluble N-ethylmaleimide-sensitive attachment protein receptors
- SNpc:
-
Substantia nigra pars compacta
- TNF:
-
Tumor necrosis factor
- FDA:
-
US Food and Drug Administration
- VMAT1:
-
Vesicular monoamine transporter 1
- VMAT2:
-
Vesicular monoamine transporter 2
References
Bloem BR, Okun MS, Klein C. Parkinson’s disease. The Lancet. 2021;397:2284–303.
Ovallath S, Deepa P. The history of parkinsonism: descriptions in ancient Indian medical literature. Mov Disord. 2013;28:566–8.
Parkinson J. An Essay on the Shaking Palsy. 1817. J Neuropsychiatry Clin Neurosci. 2002;14:223–36.
Goetz CG. The history of Parkinson’s disease: early clinical descriptions and neurological therapies. Cold Spring Harb Perspect Med. 2011;1:a008862–a008862.
Nussbaum RL, Ellis CE. Alzheimer’s disease and Parkinson’s disease. N Engl J Med. 2003;348:1356–64.
Collaborators GN. Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18:459–80.
Papapetropoulos S, Adi N, Ellul J, Argyriou AA, Chroni E. A prospective study of familial versus sporadic Parkinson’s disease. Neurodegener Dis. 2007;4:424–7.
Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318:121–34.
Masato A, Plotegher N, Boassa D, Bubacco L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol Neurodegener. 2019;14:35.
Sveinbjornsdottir S. The clinical symptoms of Parkinson’s disease. J Neurochem. 2016;139:318–24.
Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol. 2010;67:715–25.
Braak H, Bohl JR, Müller CM, Rüb U, de Vos RAI, Del Tredici K. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov Disord. 2006;21:2042–51.
Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag A-E, Lang AE. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013.
Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79:368–76.
Solari N, Bonito-Oliva A, Fisone G, Brambilla R. Understanding cognitive deficits in Parkinson’s disease: lessons from preclinical animal models. Learn Mem. 2013;20:592–600.
Schneider F, Althaus A, Backes V, Dodel R. Psychiatric symptoms in Parkinson’s disease. Eur Arch Psychiatry Clin Neurosci. 2008;258:55–9.
Pagano G, Niccolini F, Politis M. Imaging in Parkinson’s disease Clin Med (Lond). 2016;16:371–5.
Schapira AHV, Gu M, Taanman JW, Tabrizi SJ, Seaton T, Cleeter M, Cooper JM. Mitochondria in the etiology and pathogenesis of parkinson’s disease. Ann Neurol. 1998;44:S89–98.
Park J-S, Davis RL, Sue CM. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr Neurol Neurosci Rep. 2018;18(5):21.
Jenner P, Olanow CW. Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology. 1996;47:161S.
Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR. Oxidative stress and Parkinson’s disease. Front Neuroanatomy. 2015;9:91.
Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? The Lancet Neurology. 2009;8:382–97.
Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegener. 2015;4:19.
Ambrosi G, Cerri S, Blandini F. A further update on the role of excitotoxicity in the pathogenesis of Parkinson’s disease. J Neural Transm (Vienna, Austria : 1996). 2014;121:849–59.
van der Brug MP, Singleton A, Gasser T, Lewis PA. Parkinson’s disease: From human genetics to clinical trials. Sci Transl Med. 2015;7:205ps220.
Funayama M, Nishioka K, Li Y, Hattori N. Molecular genetics of Parkinson's disease: Contributions and global trends. J Hum Genet. 2022. https://doi.org/10.1038/s10038-022-01058-5.
McNaught KS, Olanow CW. Protein aggregation in the pathogenesis of familial and sporadic Parkinson’s disease. Neurobiol Aging. 2006;27:530–45.
Akhtar RS, Xie SX, Brennan L, Pontecorvo MJ, Hurtig HI, Trojanowski JQ, Weintraub D, Siderowf AD. Amyloid-Beta Positron Emission Tomography Imaging of Alzheimer’s Pathology in Parkinson’s Disease Dementia. Mov Disord Clin Pract. 2016;3:367–75.
Murphy MP, Bayir H, Belousov V, Chang CJ, Davies KJA, Davies MJ, Dick TP, Finkel T, Forman HJ, Janssen-Heininger Y, et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat Metab. 2022;4:651–62.
Chaudhuri KR, Schapira AH. Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 2009;8:464–74.
Braak H, Braak E. Pathoanatomy of Parkinson’s disease. J Neurol. 2000;247:II3–10.
Rascol O, Payoux P, Ory F, Ferreira JJ, Brefel-Courbon C, Montastruc JL. Limitations of current Parkinson’s disease therapy. Ann Neurol. 2003;53(Suppl 3):S3-12; discussion S12-5.
Armstrong MJ, Okun MS. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA. 2020;323:548–60.
Verger A, Grimaldi S, Ribeiro MJ, Frismand S, Guedj E. Single Photon Emission Computed Tomography/Positron Emission Tomography Molecular Imaging for Parkinsonism: A Fast-Developing Field. Ann Neurol. 2021;90:711–9.
Nerella SG, Singh P, Sanam T, Digwal CS. PET Molecular Imaging in Drug Development: The Imaging and Chemistry Perspective. Front Med (Lausanne). 2022;9:812270.
Lu FM, Yuan Z. PET/SPECT molecular imaging in clinical neuroscience: recent advances in the investigation of CNS diseases. Quant Imaging Med Surg. 2015;5:433–47.
Ametamey SM, Honer M, Schubiger PA. Molecular imaging with PET. Chem Rev. 2008;108:1501–16.
Zhu L, Ploessl K, Kung HF. PET/SPECT imaging agents for neurodegenerative diseases. Chem Soc Rev. 2014;43:6683–91.
Pavese N, Brooks DJ. Imaging neurodegeneration in Parkinson’s disease. Biochem Biophys Acta. 2009;1792:722–9.
Matthews DC, Lerman H, Lukic A, Andrews RD, Mirelman A, Wernick MN, Giladi N, Strother SC, Evans KC, Cedarbaum JM, Even-Sapir E. FDG PET Parkinson’s disease-related pattern as a biomarker for clinical trials in early stage disease. Neuroimage Clin. 2018;20:572–9.
Meyer PT, Frings L, Rücker G, Hellwig S. (18)F-FDG PET in Parkinsonism: Differential Diagnosis and Evaluation of Cognitive Impairment. J Nucl Med. 2017;58:1888–98.
Garraux G, Phillips C, Schrouff J, Kreisler A, Lemaire C, Degueldre C, Delcour C, Hustinx R, Luxen A, Destée A, Salmon E. Multiclass classification of FDG PET scans for the distinction between Parkinson’s disease and atypical parkinsonian syndromes. Neuroimage Clin. 2013;2:883–93.
Mudali D, Teune LK, Renken RJ, Leenders KL, Roerdink JB. Classification of Parkinsonian syndromes from FDG-PET brain data using decision trees with SSM/PCA features. Comput Math Methods Med. 2015;2015:136921.
Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet. 2014;46:989–93.
Wassouf Z, Schulze-Hentrich JM. Alpha-synuclein at the nexus of genes and environment: the impact of environmental enrichment and stress on brain health and disease. J Neurochem. 2019;150:591–604.
Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501.
Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L. Pathological roles of α-synuclein in neurological disorders. Lancet Neurol. 2011;10:1015–25.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40.
Wakabayashi K, Tanji K, Odagiri S, Miki Y, Mori F, Takahashi H. The Lewy Body in Parkinson’s Disease and Related Neurodegenerative Disorders. Mol Neurobiol. 2013;47:495–508.
Benskey MJ, Perez RG, Manfredsson FP. The contribution of alpha synuclein to neuronal survival and function - Implications for Parkinson’s disease. J Neurochem. 2016;137:331–59.
Polymeropoulos MH. Mutation in the -Synuclein Gene Identified in Families with Parkinson’s Disease. Science. 1997;276:2045–7.
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–8.
Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, et al. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–73.
Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord. 2013;28:811–3.
Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, Proukakis C, Quinn N, Lees AJ, Hardy J, et al. α-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013;125:753–69.
Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Pöyhönen M, Paetau A. A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging. 2014;35:2180.e2181-2180.e2185.
Lesage S, Anheim M, Letournel F, Bousset L, Honoré A, Rozas N, Pieri L, Madiona K, Dürr A, Melki R, et al. G51D α-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome. Ann Neurol. 2013;73:459–71.
Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, Schapira AH. A novel α-synuclein missense mutation in Parkinson disease. Neurology. 2013;80:1062.
Phillipson OT. Alpha-synuclein, epigenetics, mitochondria, metabolism, calcium traffic, & circadian dysfunction in Parkinson’s disease. An integrated strategy for management. Ageing Res Rev. 2017;40:149–67.
Yoshino H, Hirano M, Stoessl AJ, Imamichi Y, Ikeda A, Li Y, Funayama M, Yamada I, Nakamura Y, Sossi V, et al. Homozygous alpha-synuclein p.A53V in familial Parkinson’s disease. Neurobiol Aging. 2017;57(248):e247-248 e212.
Chavarría C, Souza JM. Oxidation and nitration of α-synuclein and their implications in neurodegenerative diseases. Arch Biochem Biophys. 2013;533:25–32.
Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson’s disease. Science. 2001;293:263–9.
Paleologou KE, Oueslati A, Shakked G, Rospigliosi CC, Kim HY, Lamberto GR, Fernandez CO, Schmid A, Chegini F, Gai WP, et al. Phosphorylation at S87 Is Enhanced in Synucleinopathies, Inhibits -Synuclein Oligomerization, and Influences Synuclein-Membrane Interactions. J Neurosci. 2010;30:3184–98.
Shah M, Seibyl J, Cartier A, Bhatt R, Catafau AM. Molecular imaging insights into neurodegeneration: focus on alpha-synuclein radiotracers. J Nucl Med. 2014;55:1397–400.
Ye L, Velasco A, Fraser G, Beach TG, Sue L, Osredkar T, Libri V, Spillantini MG, Goedert M, Lockhart A. In vitro high affinity alpha-synuclein binding sites for the amyloid imaging agent PIB are not matched by binding to Lewy bodies in postmortem human brain. J Neurochem. 2008;105:1428–37.
Fodero-Tavoletti MT, Smith DP, McLean CA, Adlard PA, Barnham KJ, Foster LE, Leone L, Perez K, Cortes M, Culvenor JG, et al. In vitro characterization of Pittsburgh compound-B binding to Lewy bodies. J Neurosci. 2007;27:10365–71.
Levigoureux E, Lancelot S, Bouillot C, Chauveau F, Verdurand M, Verchere J, Billard T, Baron T, Zimmer L. Binding of the PET radiotracer [(1)(8)F]BF227 does not reflect the presence of alpha-synuclein aggregates in transgenic mice. Curr Alzheimer Res. 2014;11:955–60.
Fodero-Tavoletti MT, Mulligan RS, Okamura N, Furumoto S, Rowe CC, Kudo Y, Masters CL, Cappai R, Yanai K, Villemagne VL. In vitro characterisation of BF227 binding to alpha-synuclein/Lewy bodies. Eur J Pharmacol. 2009;617:54–8.
Yu L, Cui J, Padakanti PK, Engel L, Bagchi DP, Kotzbauer PT, Tu Z. Synthesis and in vitro evaluation of alpha-synuclein ligands. Bioorg Med Chem. 2012;20:4625–34.
Bagchi DP, Yu L, Perlmutter JS, Xu J, Mach RH, Tu Z, Kotzbauer PT. Binding of the Radioligand SIL23 to α-Synuclein Fibrils in Parkinson Disease Brain Tissue Establishes Feasibility and Screening Approaches for Developing a Parkinson Disease Imaging Agent. PLoS ONE. 2013;8(2):e55031.
Zhang X, Jin H, Padakanti P, Li J, Yang H, Fan J, Mach R, Kotzbauer P, Tu Z. Radiosynthesis and in Vivo Evaluation of Two PET Radioligands for Imaging α-Synuclein. Appl Sci. 2014;4(1):66–78.
Ferrie JJ, Lengyel-Zhand Z, Janssen B, Lougee MG, Giannakoulias S, Hsieh CJ, Pagar VV, Weng CC, Xu H, Graham TJA, et al. Identification of a nanomolar affinity alpha-synuclein fibril imaging probe by ultra-high throughput in silico screening. Chem Sci. 2020;11:12746–54.
Miranda-Azpiazu P, Svedberg M, Higuchi M, Ono M, Jia Z, Sunnemark D, Elmore CS, Schou M, Varrone A. Identification and in vitro characterization of C05–01, a PBB3 derivative with improved affinity for alpha-synuclein. Brain Res. 2020;1749:147131.
Chen YF, Bian J, Zhang P, Bu LL, Shen Y, Yu WB, Lu XH, Lin X, Ye DY, Wang J, Chu Y. Design, synthesis and identification of N, N-dibenzylcinnamamide (DBC) derivatives as novel ligands for alpha-synuclein fibrils by SPR evaluation system. Bioorg Med Chem. 2020;28:115358.
Kaide S, Watanabe H, Shimizu Y, Iikuni S, Nakamoto Y, Hasegawa M, Itoh K, Ono M. Identification and Evaluation of Bisquinoline Scaffold as a New Candidate for alpha-Synuclein-PET Imaging. ACS Chem Neurosci. 2020;11:4254–61.
Kuebler L, Buss S, Leonov A, Ryazanov S, Schmidt F, Maurer A, Weckbecker D, Landau AM, Lillethorup TP, Bleher D, et al. [(11)C]MODAG-001-towards a PET tracer targeting alpha-synuclein aggregates. Eur J Nucl Med Mol Imaging. 2021;48:1759–72.
Matsuoka K, Ono M, Takado Y, Hirata K, Endo H, Ohfusa T, Kojima T, Yamamoto T, Onishi T, Orihara A, et al. High-Contrast Imaging of α-Synuclein Pathologies in Living Patients with Multiple System Atrophy. Movement Disorders. 2022;37(10):2159–61. https://doi.org/10.1002/mds.29186.
Capotosti F, Vokali E, Molette J, Ravache M, Delgado C, Kocher J, Pittet L, Dimitrakopoulos IK, Di-Bonaventura I, Touilloux T, et al. The development of [18F]ACI-12589, a high affinity and selective alpha-synuclein radiotracer, as a biomarker for Parkinson’s disease and other synucleinopathies. Alzheimers Dement. 2021;17:e053943.
Roshanbin S, Xiong M, Hultqvist G, Söderberg L, Zachrisson O, Meier S, Ekmark-Lewén S, Bergström J, Ingelsson M, Sehlin D, Syvänen S. In vivo imaging of alpha-synuclein with antibody-based PET. Neuropharmacology. 2022;208:108985.
Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–4.
Irwin DJ, Lee VM, Trojanowski JQ. Parkinson’s disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci. 2013;14:626–36.
Van der Perren A, Gelders G, Fenyi A, Bousset L, Brito F, Peelaerts W, Van den Haute C, Gentleman S, Melki R, Baekelandt V. The structural differences between patient-derived α-synuclein strains dictate characteristics of Parkinson’s disease, multiple system atrophy and dementia with Lewy bodies. Acta Neuropathol. 2020;139:977–1000.
McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47:1113–24.
Lippa CF, Duda JE, Grossman M, Hurtig HI, Aarsland D, Boeve BF, Brooks DJ, Dickson DW, Dubois B, Emre M, et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology. 2007;68:812–9.
van Rumund A, Green AJE, Fairfoul G, Esselink RAJ, Bloem BR, Verbeek MM. α-Synuclein real-time quaking-induced conversion in the cerebrospinal fluid of uncertain cases of parkinsonism. Ann Neurol. 2019;85:777–81.
Iranzo A, Fairfoul G, Ayudhaya ACN, Serradell M, Gelpi E, Vilaseca I, Sanchez-Valle R, Gaig C, Santamaria J, Tolosa E, et al. Detection of α-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: a longitudinal observational study. Lancet Neurol. 2021;20:203–12.
Lee JM, Derkinderen P, Kordower JH, Freeman R, Munoz DG, Kremer T, Zago W, Hutten SJ, Adler CH, Serrano GE, Beach TG. The Search for a Peripheral Biopsy Indicator of α-Synuclein Pathology for Parkinson Disease. J Neuropathol Exp Neurol. 2017;76:2–15.
Antelmi E, Donadio V, Incensi A, Plazzi G, Liguori R. Skin nerve phosphorylated α-synuclein deposits in idiopathic REM sleep behavior disorder. Neurology. 2017;88:2128–31.
Donadio V, Incensi A, Piccinini C, Cortelli P, Giannoccaro MP, Baruzzi A, Liguori R. Skin nerve misfolded α-synuclein in pure autonomic failure and Parkinson disease. Ann Neurol. 2016;79:306–16.
Donadio V, Incensi A, Rizzo G, Capellari S, Pantieri R, Stanzani Maserati M, Devigili G, Eleopra R, Defazio G, Montini F, et al. A new potential biomarker for dementia with Lewy bodies: Skin nerve α-synuclein deposits. Neurology. 2017;89:318–26.
Gibbons CH, Garcia J, Wang N, Shih LC, Freeman R. The diagnostic discrimination of cutaneous α-synuclein deposition in Parkinson disease. Neurology. 2016;87:505–12.
Visanji NP, Mollenhauer B, Beach TG, Adler CH, Coffey CS, Kopil CM, Dave KD, Foroud T, Chahine L, Jennings D. The Systemic Synuclein Sampling Study: toward a biomarker for Parkinson’s disease. Biomark Med. 2017;11:359–68.
Wang Z, Becker K, Donadio V, Siedlak S, Yuan J, Rezaee M, Incensi A, Kuzkina A, Orrú CD, Tatsuoka C, et al. Skin α-Synuclein Aggregation Seeding Activity as a Novel Biomarker for Parkinson Disease. JAMA Neurol. 2020;78:1–11.
Donadio V, Doppler K, Incensi A, Kuzkina A, Janzen A, Mayer G, Volkmann J, Rizzo G, Antelmi E, Plazzi G, et al. Abnormal α-synuclein deposits in skin nerves: intra- and inter-laboratory reproducibility. Eur J Neurol. 2019;26:1245–51.
Fields CR, Bengoa-Vergniory N, Wade-Martins R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front Mol Neurosci. 2019;12:299–299.
Sapru MK, Yates JW, Hogan S, Jiang L, Halter J, Bohn MC. Silencing of human alpha-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. Exp Neurol. 2006;198:382–90.
Takahashi M, Suzuki M, Fukuoka M, Fujikake N, Watanabe S, Murata M, Wada K, Nagai Y, Hohjoh H. Normalization of Overexpressed α-Synuclein Causing Parkinson’s Disease By a Moderate Gene Silencing With RNA Interference. Molecular Therapy - Nucleic Acids. 2015;4:e241.
Lewis J, Melrose H, Bumcrot D, Hope A, Zehr C, Lincoln S, Braithwaite A, He Z, Ogholikhan S, Hinkle K, et al. In vivo silencing of alpha-synuclein using naked siRNA. Mol Neurodegener. 2008;3:19.
Mittal S, Bjørnevik K, Im DS, Flierl A, Dong X, Locascio JJ, Abo KM, Long E, Jin M, Xu B, et al. β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science. 2017;357:891–8.
Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 Reduces alpha-Synuclein Aggregation and Toxicity. J Biol Chem. 2004;279:25497–502.
Cox D, Selig E, Griffin MDW, Carver JA, Ecroyd H. Small Heat-shock Proteins Prevent α-Synuclein Aggregation via Transient Interactions and Their Efficacy Is Affected by the Rate of Aggregation. J Biol Chem. 2016;291:22618–29.
Ghochikyan A, Petrushina I, Davtyan H, Hovakimyan A, Saing T, Davtyan A, Cribbs DH, Agadjanyan MG. Immunogenicity of epitope vaccines targeting different B cell antigenic determinants of human α-synuclein: Feasibility study. Neurosci Lett. 2014;560:86–91.
Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005;46:857–68.
Sanchez-Guajardo V, Annibali A, Jensen PH, Romero-Ramos M. alpha-Synuclein vaccination prevents the accumulation of parkinson disease-like pathologic inclusions in striatum in association with regulatory T cell recruitment in a rat model. J Neuropathol Exp Neurol. 2013;72:624–45.
Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, Patrick C, Trejo M, Ubhi K, Rohn TT, et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS ONE. 2011;6:e19338.
Schenk DB, Koller M, Ness DK, Griffith SG, Grundman M, Zago W, Soto J, Atiee G, Ostrowitzki S, Kinney GG. First-in-human assessment of PRX002, an anti-alpha-synuclein monoclonal antibody, in healthy volunteers. Mov Disord. 2017;32:211–8.
Pagano G, Taylor KI, Anzures-Cabrera J, Marchesi M, Simuni T, Marek K, Postuma RB, Pavese N, Stocchi F, Azulay JP, et al. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N Engl J Med. 2022;387:421–32.
Fjord-Larsen L, Thougaard A, Wegener KM, Christiansen J, Larsen F, Schrøder-Hansen LM, Kaarde M, Ditlevsen DK. Nonclinical safety evaluation, pharmacokinetics, and target engagement of Lu AF82422, a monoclonal IgG1 antibody against alpha-synuclein in development for treatment of synucleinopathies. MAbs. 2021;13:1994690.
Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537:50–6.
van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, Kanekiyo M, Li D, Reyderman L, Cohen S, et al. Lecanemab in Early Alzheimer’s Disease. N Engl J Med. 2023;388:9–21.
Weihofen A, Liu Y, Arndt JW, Huy C, Quan C, Smith BA, Baeriswyl JL, Cavegn N, Senn L, Su L, et al. Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol Dis. 2019;124:276–88.
Lindström V, Fagerqvist T, Nordström E, Eriksson F, Lord A, Tucker S, Andersson J, Johannesson M, Schell H, Kahle PJ, et al. Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice. Neurobiol Dis. 2014;69:134–43.
Nordström E, Eriksson F, Sigvardson J, Johannesson M, Kasrayan A, Jones-Kostalla M, Appelkvist P, Söderberg L, Nygren P, Blom M, et al. ABBV-0805, a novel antibody selective for soluble aggregated α-synuclein, prolongs lifespan and prevents buildup of α-synuclein pathology in mouse models of Parkinson’s disease. Neurobiol Dis. 2021;161:105543.
Chen SW, Drakulic S, Deas E, Ouberai M, Aprile FA, Arranz R, Ness S, Roodveldt C, Guilliams T, De-Genst EJ, et al. Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc Natl Acad Sci USA. 2015;112:E1994-2003.
Vaikath NN, Hmila I, Gupta V, Erskine D, Ingelsson M, El-Agnaf OMA. Antibodies against alpha-synuclein: tools and therapies. J Neurochem. 2019;150:612–25.
Kariolis MS, Wells RC, Getz JA, Kwan W, Mahon CS, Tong R, Kim DJ, Srivastava A, Bedard C, Henne KR, et al. Brain delivery of therapeutic proteins using an Fc fragment blood-brain barrier transport vehicle in mice and monkeys. Sci Transl Med. 2020;12(545):eaay1359.
Roshanbin S, Julku U, Xiong M, Eriksson J, Masliah E, Hultqvist G, Bergström J, Ingelsson M, Syvänen S, Sehlin D. Reduction of αSYN Pathology in a Mouse Model of PD Using a Brain-Penetrating Bispecific Antibody. Pharmaceutics. 2022;14(7):1412.
Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–80.
Burns RS, Chiueh CC, Markey SP, Ebert MH, Jacobowitz DM, Kopin IJ. A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci USA. 1983;80:4546–50.
Langston JW, Forno LS, Rebert CS, Irwin I. Selective nigral toxicity after systemic administration of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyrine (MPTP) in the squirrel monkey. Brain Res. 1984;292:390–4.
Schneider JS, Denaro FJ. Astrocytic responses to the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in cat and mouse brain. J Neuropathol Exp Neurol. 1988;47:452–8.
Sayre LM. Biochemical mechanism of action of the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicol Lett. 1989;48:121–49.
Tipton KF, Singer TP. Advances in our understanding of the mechanisms of the neurotoxicity of MPTP and related compounds. J Neurochem. 1993;61:1191–206.
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem. 1990;54:823–7.
Greenamyre JT, Sherer TB, Betarbet R, Panov AV. Complex I and Parkinson’s Disease. IUBMB Life. 2001;52:135–41.
Su B, Wang X, Zheng L, Perry G, Smith MA, Zhu X. Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochem Biophys Acta. 2010;1802:135–42.
Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4:600–9.
Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–95.
Bose A, Beal MF. Mitochondrial dysfunction in Parkinson’s disease. J Neurochem. 2016;139(Suppl 1):216–31.
Sulzer D, Zecca L. Intraneuronal dopamine-quinone synthesis: a review. Neurotox Res. 2000;1:181–95.
Mansur A, Comley R, Lewis Y, Middleton L, Huiban M, Guo Q, Passchier J, Tsukada H, Gunn R, Rabiner E. MIND MAPS CONSORTIUM ft: <strong>Imaging of Mitochondrial Complex 1 with <sup>18</sup>F-BCPP-EF in the Healthy Human Brain</strong>. J Nucl Med. 2018;59:1709–1709.
Berman DS, Maddahi J, Tamarappoo BK, Czernin J, Taillefer R, Udelson JE, Gibson CM, Devine M, Lazewatsky J, Bhat G, Washburn D. Phase II safety and clinical comparison with single-photon emission computed tomography myocardial perfusion imaging for detection of coronary artery disease: flurpiridaz F 18 positron emission tomography. J Am Coll Cardiol. 2013;61:469–77.
Bengs S, Warnock GI, Portmann A, Mikail N, Rossi A, Ahmed H, Etter D, Treyer V, Gisler L, Pfister SK, et al. Rest/stress myocardial perfusion imaging by positron emission tomography with (18)F-Flurpiridaz: A feasibility study in mice. J Nucl Cardiol. 2022. https://doi.org/10.1007/s12350-022-02968-9.
Haider A, Bengs S, Portmann A, Rossi A, Ahmed H, Etter D, Warnock GI, Mikail N, Grämer M, Meisel A, et al. Role of sex hormones in modulating myocardial perfusion and coronary flow reserve. Eur J Nucl Med Mol Imaging. 2022;49:2209–18.
Khan NL, Valente EM, Bentivoglio AR, Wood NW, Albanese A, Brooks DJ, Piccini P. Clinical and subclinical dopaminergic dysfunction in PARK6-linked parkinsonism: an 18F-dopa PET study. Ann Neurol. 2002;52:849–53.
Gan-Or Z, Liong C, Alcalay RN. GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Curr Neurol Neurosci Rep. 2018;18:44.
Choi JH, Stubblefield B, Cookson MR, Goldin E, Velayati A, Tayebi N, Sidransky E. Aggregation of alpha-synuclein in brain samples from subjects with glucocerebrosidase mutations. Mol Genet Metab. 2011;104:185–8.
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52.
Migdalska-Richards A, Daly L, Bezard E, Schapira AH. Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann Neurol. 2016;80:766–75.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7.
Chen J, Chen Y, Pu J. Leucine-Rich Repeat Kinase 2 in Parkinson’s Disease: Updated from Pathogenesis to Potential Therapeutic Target. Eur Neurol. 2018;79:256–65.
Chen Z, Shao T, Gao W, Fu H, Collier TL, Rong J, Deng X, Yu Q, Zhang X, Davenport AT, et al. Synthesis and Preliminary Evaluation of [(11) C]GNE-1023 as a Potent PET Probe for Imaging Leucine-Rich Repeat Kinase 2 (LRRK2) in Parkinson’s Disease. ChemMedChem. 2019;14:1580–5.
Malik N, Kornelsen R, McCormick S, Colpo N, Merkens H, Bendre S, Benard F, Sossi V, Schirrmacher R, Schaffer P. Development and biological evaluation of[(18)F]FMN3PA & [(18)F]FMN3PU for leucine-rich repeat kinase 2 (LRRK2) in vivo PET imaging. Eur J Med Chem. 2021;211:113005.
Rideout HJ, Chartier-Harlin MC, Fell MJ, Hirst WD, Huntwork-Rodriguez S, Leyns CEG, Mabrouk OS, Taymans JM. The Current State-of-the Art of LRRK2-Based Biomarker Assay Development in Parkinson’s Disease. Front Neurosci. 2020;14:865.
Chen Z, Chen J, Chen L, Yoo C-H, Rong J, Fu H, Shao T, Coffman K, Steyn SJ, Davenport AT, et al: Imaging Leucine-Rich Repeat Kinase 2 In Vivo with 18F-Labeled Positron Emission Tomography Ligand. J Med Chem. 2022.
Tolosa E, Vila M, Klein C, Rascol O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol. 2020;16:97–107.
Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–27.
Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat Rev Neurosci. 2010;11:791–7.
Simunovic F, Yi M, Wang Y, Macey L, Brown LT, Krichevsky AM, Andersen SL, Stephens RM, Benes FM, Sonntag KC. Gene expression profiling of substantia nigra dopamine neurons: further insights into Parkinson’s disease pathology. Brain. 2009;132:1795–809.
Manzoni C, Lewis PA. LRRK2 and Autophagy. Adv Neurobiol. 2017;14:89–105.
Jennings D, Huntwork-Rodriguez S, Henry AG, Sasaki JC, Meisner R, Diaz D, Solanoy H, Wang X, Negrou E, Bondar VV, et al: Preclinical and clinical evaluation of the LRRK2 inhibitor DNL201 for Parkinson's disease. Sci Transl Med. 2022;14:eabj2658.
West AB. Achieving neuroprotection with LRRK2 kinase inhibitors in Parkinson disease. Exp Neurol. 2017;298:236–45.
Zhao HT, John N, Delic V, Ikeda-Lee K, Kim A, Weihofen A, Swayze EE, Kordasiewicz HB, West AB, Volpicelli-Daley LA. LRRK2 Antisense Oligonucleotides Ameliorate alpha-Synuclein Inclusion Formation in a Parkinson’s Disease Mouse Model. Mol Therapy Nucleic Acids. 2017;8:508–19.
Ntetsika T, Papathoma PE, Markaki I. Novel targeted therapies for Parkinson’s disease. Mol Med. 2021;27:17.
Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. Embo j. 2010;29:969–80.
Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:860–4.
Simões-Pires C, Zwick V, Nurisso A, Schenker E, Carrupt PA, Cuendet M. HDAC6 as a target for neurodegenerative diseases: what makes it different from the other HDACs? Mol Neurodegener. 2013;8:7.
Koole M, Van Weehaeghe D, Serdons K, Herbots M, Cawthorne C, Celen S, Schroeder FA, Hooker JM, Bormans G, de Hoon J, et al. Clinical validation of the novel HDAC6 radiotracer [(18)F]EKZ-001 in the human brain. Eur J Nucl Med Mol Imaging. 2021;48:596–611.
Vila M, Jackson-Lewis V, Guegan C, Wu DC, Teismann P, Choi DK, Tieu K, Przedborski S. The role of glial cells in Parkinson’s disease. Curr Opin Neurol. 2001;14:483–9.
Kurkowska-Jastrzebska I, Wronska A, Kohutnicka M, Czlonkowski A, Czlonkowska A. The inflammatory reaction following 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine intoxication in mouse. Exp Neurol. 1999;156:50–61.
Fu R, Shen Q, Xu P, Luo JJ, Tang Y. Phagocytosis of microglia in the central nervous system diseases. Mol Neurobiol. 2014;49:1422–34.
Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. 2014;32:367–402.
Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discovery. 2010;9:971–88.
Venneti S, Lopresti BJ, Wiley CA. The peripheral benzodiazepine receptor (Translocator protein 18kDa) in microglia: from pathology to imaging. Prog Neurobiol. 2006;80:308–22.
Brooks DJ. Technology Insight: imaging neurodegeneration in Parkinson’s disease. Nat Clin Pract Neurol. 2008;4:267–77.
Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003;106:518–26.
Bartels AL, Willemsen AT, Doorduin J, de Vries EF, Dierckx RA, Leenders KL. [11C]-PK11195 PET: quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism Relat Disord. 2010;16:57–9.
Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, Eggert K, Oertel W, Banati RB, Brooks DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. 2006;21:404–12.
Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol. 2005;57:168–75.
Kobylecki C, Counsell SJ, Cabanel N, Wächter T, Turkheimer FE, Eggert K, Oertel W, Brooks DJ, Gerhard A. Diffusion-weighted imaging and its relationship to microglial activation in parkinsonian syndromes. Parkinsonism Relat Disord. 2013;19:527–32.
Chauveau F, Van Camp N, Dolle F, Kuhnast B, Hinnen F, Damont A, Boutin H, James M, Kassiou M, Tavitian B. Comparative evaluation of the translocator protein radioligands 11C-DPA-713, 18F-DPA-714, and 11C-PK11195 in a rat model of acute neuroinflammation. J Nucl Med. 2009;50:468–76.
Zhang M-R, Maeda J, Furutsuka K, Yoshida Y, Ogawa M, Suhara T, Suzuki K. [18F]FMDAA1106 and [18F]FEDAA1106: two positron-Emitter labeled ligands for peripheral benzodiazepine receptor (PBR). Bioorg Med Chem Lett. 2003;13:201–4.
Zhang M-R, Kida T, Noguchi J, Furutsuka K, Maeda J, Suhara T, Suzuki K. [11C]DAA1106: radiosynthesis and in vivo binding to peripheral benzodiazepine receptors in mouse brain. Nucl Med Biol. 2003;30:513–9.
Maeda J, Suhara T, Zhang MR, Okauchi T, Yasuno F, Ikoma Y, Inaji M, Nagai Y, Takano A, Obayashi S, Suzuki K. Novel peripheral benzodiazepine receptor ligand [11C]DAA1106 for PET: an imaging tool for glial cells in the brain. Synapse. 2004;52:283–91.
Yui J, Hatori A, Kawamura K, Yanamoto K, Yamasaki T, Ogawa M, Yoshida Y, Kumata K, Fujinaga M, Nengaki N, et al. Visualization of early infarction in rat brain after ischemia using a translocator protein (18 kDa) PET ligand [11C]DAC with ultra-high specific activity. Neuroimage. 2011;54:123–30.
Gulyas B, Toth M, Vas A, Shchukin E, Kostulas K, Hillert J, Halldin C. Visualising neuroinflammation in post-stroke patients: a comparative PET study with the TSPO molecular imaging biomarkers [11C]PK11195 and [11C]vinpocetine. Curr Radiopharm. 2012;5:19–28.
Brody AL, Gehlbach D, Garcia LY, Enoki R, Hoh C, Vera D, Kotta KK, London ED, Okita K, Nurmi EL, et al. Effect of overnight smoking abstinence on a marker for microglial activation: a [(11)C]DAA1106 positron emission tomography study. Psychopharmacology. 2018;235:3525–34.
Best L, Ghadery C, Pavese N, Tai YF, Strafella AP. New and Old TSPO PET Radioligands for Imaging Brain Microglial Activation in Neurodegenerative Disease. Curr Neurol Neurosci Rep. 2019;19:24.
Zhang L, Hu K, Shao T, Hou L, Zhang S, Ye W, Josephson L, Meyer JH, Zhang MR, Vasdev N, et al. Recent developments on PET radiotracers for TSPO and their applications in neuroimaging. Acta pharmaceutica Sinica B. 2021;11:373–93.
Unterrainer M, Mahler C, Vomacka L, Lindner S, Havla J, Brendel M, Böning G, Ertl-Wagner B, Kümpfel T, Milenkovic VM, et al. TSPO PET with [18F]GE-180 sensitively detects focal neuroinflammation in patients with relapsing–remitting multiple sclerosis. Eur J Nucl Med Mol Imaging. 2018;45:1423–31.
Wadsworth H, Jones PA, Chau WF, Durrant C, Fouladi N, Passmore J, O’Shea D, Wynn D, Morisson-Iveson V, Ewan A, et al. [18F]GE-180: a novel fluorine-18 labelled PET tracer for imaging Translocator protein 18 kDa (TSPO). Bioorg Med Chem Lett. 2012;22:1308–13.
Alam MM, Lee J, Lee SY. Recent Progress in the Development of TSPO PET Ligands for Neuroinflammation Imaging in Neurological Diseases. Nucl Med Mol Imaging. 2017;51:283–96.
Ikawa M, Lohith TG, Shrestha S, Telu S, Zoghbi SS, Castellano S, Taliani S, Da Settimo F, Fujita M, Pike VW, Innis RB. 11C-ER176, a Radioligand for 18-kDa Translocator Protein, Has Adequate Sensitivity to Robustly Image All Three Affinity Genotypes in Human Brain. J Nucl Med. 2017;58:320–5.
Siméon FG, Lee J-H, Morse CL, Stukes I, Zoghbi SS, Manly LS, Liow J-S, Gladding RL, Dick RM, Yan X, et al. Synthesis and Screening in Mice of Fluorine-Containing PET Radioligands for TSPO: Discovery of a Promising 18F-Labeled Ligand. J Med Chem. 2021;64:16731–45.
Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, Hibbert S, Budnik N, Zampedri L, Dickson J, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1664–75.
Tansey MG, Wallings RL, Houser MC, Herrick MK, Keating CE, Joers V. Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol. 2022;22(11):657–73.
Fujimoto H, Fujita N, Hamamatsu K, Murakami T, Nakamoto Y, Saga T, Ishimori T, Shimizu Y, Watanabe H, Sano K, et al. First-in-Human Evaluation of Positron Emission Tomography/Computed Tomography With [(18)F]FB(ePEG12)12-Exendin-4: A Phase 1 Clinical Study Targeting GLP-1 Receptor Expression Cells in Pancreas. Front Endocrinol (Lausanne). 2021;12:717101.
Luo Y, Pan Q, Yao S, Yu M, Wu W, Xue H, Kiesewetter DO, Zhu Z, Li F, Zhao Y, Chen X. Glucagon-Like Peptide-1 Receptor PET/CT with 68Ga-NOTA-Exendin-4 for Detecting Localized Insulinoma: A Prospective Cohort Study. J Nucl Med. 2016;57:715–20.
Antwi K, Fani M, Nicolas G, Rottenburger C, Heye T, Reubi JC, Gloor B, Christ E, Wild D. Localization of Hidden Insulinomas with 68Ga-DOTA-Exendin-4 PET/CT: A Pilot Study. J Nucl Med. 2015;56:1075–8.
Liu Y, Liu S, Liu L, Gong X, Liu J, Sun L, Liu X, Wu L, Chen L, Wang L, et al. Fine Comparison of the Efficacy and Safety Between GB242 and Infliximab in Patients with Rheumatoid Arthritis: A Phase III Study. Rheumatol Ther. 2022;9:175–89.
Tobinick EL, Chen K, Chen X. Rapid intracerebroventricular delivery of Cu-DOTA-etanercept after peripheral administration demonstrated by PET imaging. BMC Res Notes. 2009;2:28.
Colonna M. TREMs in the immune system and beyond. Nat Rev Immunol. 2003;3:445–53.
Guo Y, Wei X, Yan H, Qin Y, Yan S, Liu J, Zhao Y, Jiang F, Lou H. TREM2 deficiency aggravates α-synuclein-induced neurodegeneration and neuroinflammation in Parkinson’s disease models. Faseb j. 2019;33:12164–74.
Wilson EN, Swarovski MS, Linortner P, Shahid M, Zuckerman AJ, Wang Q, Channappa D, Minhas PS, Mhatre SD, Plowey ED, et al. Soluble TREM2 is elevated in Parkinson’s disease subgroups with increased CSF tau. Brain. 2020;143:932–43.
Meier SR, Sehlin D, Hultqvist G, Syvänen S. Pinpointing Brain TREM2 Levels in Two Mouse Models of Alzheimer’s Disease. Mol Imaging Biol. 2021;23:665–75.
Sanjari Moghaddam H, Zare-Shahabadi A, Rahmani F, Rezaei N. Neurotransmission systems in Parkinson’s disease. Rev Neurosci. 2017;28:509–36.
Tanimura A, Pancani T, Lim SAO, Tubert C, Melendez AE, Shen W, Surmeier DJ. Striatal cholinergic interneurons and Parkinson’s disease. Eur J Neurosci. 2018;47:1148–58.
Aosaki T, Miura M, Suzuki T, Nishimura K, Masuda M. Acetylcholine-dopamine balance hypothesis in the striatum: an update. Geriatr Gerontol Int. 2010;10(Suppl 1):S148-157.
Maurice N, Liberge M, Jaouen F, Ztaou S, Hanini M, Camon J, Deisseroth K, Amalric M, Kerkerian-Le Goff L, Beurrier C. Striatal Cholinergic Interneurons Control Motor Behavior and Basal Ganglia Function in Experimental Parkinsonism. Cell Rep. 2015;13:657–66.
Pisani A, Bernardi G, Ding J, Surmeier DJ. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007;30:545–53.
Ztaou S, Amalric M. Contribution of cholinergic interneurons to striatal pathophysiology in Parkinson’s disease. Neurochem Int. 2019;126:1–10.
DeMaagd G, Philip A. Parkinson’s Disease and Its Management: Part 3: Nondopaminergic and Nonpharmacological Treatment Options. P T. 2015;40:668–79.
Appenzeller O, Goss JE. Autonomic deficits in Parkinson’s syndrome. Arch Neurol. 1971;24:50–7.
Chen Z, Li G, Liu J. Autonomic dysfunction in Parkinson’s disease: Implications for pathophysiology, diagnosis, and treatment. Neurobiol Dis. 2020;134: 104700.
Orimo S, Uchihara T, Nakamura A, Mori F, Kakita A, Wakabayashi K, Takahashi H. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain. 2008;131:642–50.
Phillips RJ, Walter GC, Wilder SL, Baronowsky EA, Powley TL. Alpha-synuclein-immunopositive myenteric neurons and vagal preganglionic terminals: autonomic pathway implicated in Parkinson’s disease? Neuroscience. 2008;153:733–50.
Goldstein DS, Sharabi Y, Karp BI, Bentho O, Saleem A, Pacak K, Eisenhofer G. Cardiac sympathetic denervation preceding motor signs in Parkinson disease. Clin Auton Res. 2007;17:118–21.
Goldstein DS. Dysautonomia in Parkinson’s disease: neurocardiological abnormalities. Lancet Neurol. 2003;2:669–76.
Treglia G, Cason E, Stefanelli A, Cocciolillo F, Di Giuda D, Fagioli G, Giordano A. MIBG scintigraphy in differential diagnosis of Parkinsonism: a meta-analysis. Clin Auton Res. 2012;22:43–55.
Beaulieu J-M, Espinoza S, Gainetdinov RR. Dopamine receptors - IUPHAR Review 13. Br J Pharmacol. 2015;172:1–23.
Seeman P, Niznik HB. Dopamine receptors and transporters in Parkinson’s disease and schizophrenia. FASEB J. 1990;4:2737–44.
Kaasinen V, Ruottinen HM, Någren K, Lehikoinen P, Oikonen V, Rinne JO. Upregulation of putaminal dopamine D2 receptors in early Parkinson’s disease: a comparative PET study with [11C] raclopride and [11C]N-methylspiperone. J Nucl Med. 2000;41:65–70.
Farde L, Halldin C, Stone-Elander S, Sedvall G. PET analysis of human dopamine receptor subtypes using 11C-SCH 23390 and 11C-raclopride. Psychopharmacology. 1987;92(3):278–84.
Halldin C, Farde L, Barnett A, Sedvall G. Synthesis of carbon-11 labelled SCH 39166, a new selective dopamine D-1 receptor ligand, and preliminary PET investigations. Int J Rad Appl Instrum A. 1991;42:451–5.
Halldin C, Foged C, Chou Y-H, Karlsson P, Swahn C-G, Johan S, Sedvall G, Farde L. Carbon-11-NNC 112: A Radioligand for PET Examination of Striatal and Neocortical D<sub>1</sub>-Dopamine Receptors. J Nucl Med. 1998;39:2061.
Ekelund J, Slifstein M, Narendran R, Guillin O, Belani H, Guo N-N, Hwang Y, Hwang D-R, Abi-Dargham A, Laruelle M. In Vivo DA D1 Receptor Selectivity of NNC 112 and SCH 23390. Mol Imag Biol. 2007;9:117–25.
Tamagnan G, Barret O, Alagille D, Carroll V, Madonia J, Constantinescu C, SanDiego C, Papin C, Morley T, Russell D, et al. T156. In vivo characterization of the first agonist dopamine D1 receptors PET imaging tracer [18F]MNI-968 in human. Schizophr Bull. 2018;44:1.
Ray NJ, Miyasaki JM, Zurowski M, Ko JH, Cho SS, Pellecchia G, Antonelli F, Houle S, Lang AE, Strafella AP. Extrastriatal dopaminergic abnormalities of DA homeostasis in Parkinson’s patients with medication-induced pathological gambling: a [11C] FLB-457 and PET study. Neurobiol Dis. 2012;48:519–25.
Nyberg S, Farde L, Eriksson L, Halldin C, Eriksson B. 5-HT2 and D2 dopamine receptor occupancy in the living human brain A PET study with risperidone. Psychopharmacology. 1993;110:265–72.
Chefer SI, Kimes AS, Matochik JA, Horti AG, Kurian V, Shumway D, Domino EF, London ED, Mukhin AG. Estimation of D2-like receptor occupancy by dopamine in the putamen of hemiparkinsonian Monkeys. Neuropsychopharmacology. 2008;33:270–8.
Sahin G, Thompson LH, Lavisse S, Ozgur M, Rbah-Vidal L, Dollé F, Hantraye P, Kirik D. Differential dopamine receptor occupancy underlies L-DOPA-induced dyskinesia in a rat model of Parkinson’s disease. PLoS ONE. 2014;9:e90759.
de la Fuente-Fernández R, Sossi V, Huang Z, Furtado S, Lu JQ, Calne DB, Ruth TJ, Stoessl AJ. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain. 2004;127:2747–54.
Del Bello F, Giannella M, Giorgioni G, Piergentili A, Quaglia W. Receptor Ligands as Helping Hands to L-DOPA in the Treatment of Parkinson’s Disease. Biomolecules. 2019;9(4):142.
Clarke CE, Guttman M. Dopamine agonist monotherapy in Parkinson’s disease. The Lancet. 2002;360:1767–9.
Blandini F, Armentero MT. Dopamine receptor agonists for Parkinson’s disease. Expert Opin Investig Drugs. 2014;23:387–410.
Davie CA. A review of Parkinson’s disease. Br Med Bull. 2008;86:109–27.
Abbott A. Levodopa: the story so far. Nature. 2010;466:S6–7.
Guggenheim M. Dioxyphenylalanin, eine neue Aminosäure aus Vicia faba. Biological Chemistry. 1913;88:276–84.
Holtz P. Dopadecarboxylase. Naturwissenschaften. 1939;27:724–5.
Hornykiewicz O. A brief history of levodopa. J Neurol. 2010;257:249–52.
Neurology TL. Building on 50 years of levodopa therapy. Lancet Neurol. 2016;15:1.
Senek M, Nielsen EI, Nyholm D. Levodopa-entacapone-carbidopa intestinal gel in Parkinson’s disease: A randomized crossover study. Mov Disord. 2017;32:283–6.
Antonini A. Levodopa in the treatment of Parkinson’s disease: an old drug still going strong. Clin Interv Aging. 2010;5:229.
Tambasco N, Romoli M, Calabresi P. Levodopa in Parkinson’s Disease: Current Status and Future Developments. Curr Neuropharmacol. 2018;16:1239–52.
Nutl JG, Fellman JH. Pharmacokinetics of Levodopa. Clin Neuropharmacol. 1984;7:35–50.
Hauser RA. Levodopa: Past, Present, and Future. Eur Neurol. 2009;62:1–8.
Cedarbaum JM. Clinical Pharmacokinetics of Anti-Parkinsonian Drugs. Clin Pharmacokinet. 1987;13:141–78.
Salat D, Tolosa E. Levodopa in the Treatment of Parkinson’s Disease: Current Status and New Developments. J Parkinson’s dis. 2013;3:255–69.
Lipp MM, Batycky R, Moore J, Leinonen M, Freed MI. Preclinical and clinical assessment of inhaled levodopa for OFF episodes in Parkinson’s disease. Sci Transl Med. 2016;8:360ra136.
Ellenbogen A, Stocchi F, Espay A, Poewe W, Oren S, Case R, Olanow CW. Impact of Subcutaneous Levodopa Infusion with ND0612 on Patient Reported Outcomes (4506). Neurology. 2020;94:4506.
Birnberg T, Smania G, Bjornsson M, Jonsson N, Case R, Oren S, Adar L, Karlsson M. Pharmacokinetic analysis of levodopa and carbidopa following subcutaneous infusion: A population pharmacokinetics model (2019). Neurology. 2019;2021:96.
Urso D, Chaudhuri KR, Qamar MA, Jenner P. Improving the Delivery of Levodopa in Parkinson’s Disease: A Review of Approved and Emerging Therapies. CNS Drugs. 2020;34:1149–63.
Müller T. Catechol-O-methyltransferase inhibitors in Parkinson’s disease. Drugs. 2015;75:157–74.
Ferreira JJ, Lees A, Rocha JF, Poewe W, Rascol O, Soares-da-Silva P. Opicapone as an adjunct to levodopa in patients with Parkinson’s disease and end-of-dose motor fluctuations: a randomised, double-blind, controlled trial. Lancet Neurol. 2016;15:154–65.
Schapira AH. Monoamine oxidase B inhibitors for the treatment of Parkinson’s disease: a review of symptomatic and potential disease-modifying effects. CNS Drugs. 2011;25:1061–71.
Fox SH, Katzenschlager R, Lim S-Y, Ravina B, Seppi K, Coelho M, Poewe W, Rascol O, Goetz CG, Sampaio C. The Movement Disorder Society Evidence-Based Medicine Review Update: Treatments for the motor symptoms of Parkinson’s disease. Mov Disord. 2011;26:S2–41.
Group PS. A controlled trial of rasagiline in early Parkinson disease: the TEMPO Study. Arch Neurol. 2002;59:1937–43.
Schapira AH, Fox SH, Hauser RA, Jankovic J, Jost WH, Kenney C, Kulisevsky J, Pahwa R, Poewe W, Anand R. Assessment of Safety and Efficacy of Safinamide as a Levodopa Adjunct in Patients With Parkinson Disease and Motor Fluctuations: A Randomized Clinical Trial. JAMA Neurol. 2017;74:216–24.
Ellis JM, Fell MJ. Current approaches to the treatment of Parkinson’s Disease. Bioorg Med Chem Lett. 2017;27:4247–55.
Bezard E, Brotchie JM, Gross CE. Pathophysiology of levodopa-induced dyskinesia: Potential for new therapies. Nat Rev Neurosci. 2001;2:577–88.
Lee J, Zhu W-M, Stanic D, Finkelstein DI, Horne MH, Henderson J, Lawrence AJ, O’Connor L, Tomas D, Drago J, Horne MK. Sprouting of dopamine terminals and altered dopamine release and uptake in Parkinsonian dyskinaesia. Brain. 2008;131:1574–87.
Olanow CW, Obeso JA, Stocchi F. Continuous dopamine-receptor treatment of Parkinson’s disease: scientific rationale and clinical implications. Lancet Neurol. 2006;5:677–87.
Antonini A, Fung VS, Boyd JT, Slevin JT, Hall C, Chatamra K, Eaton S, Benesh JA. Effect of levodopa-carbidopa intestinal gel on dyskinesia in advanced Parkinson’s disease patients. Mov Disord. 2016;31:530–7.
Poewe W, Antonini A. Novel formulations and modes of delivery of levodopa. Mov Disord. 2015;30:114–20.
Firnau G, Nahmias C, Garnett S. The preparation of [18F]5-fluoro-DOPA with reactor-produced fluorine-18. Int J Appl Radiat Isot. 1973;24:182–4.
Garnett S, Firnau G, Nahmias C, Chirakal R. Striatal dopamine metabolism in living monkeys examined by positron emission tomography. Brain Res. 1983;280:169–71.
Nanni C, Fanti S, Rubello D. 18F-DOPA PET and PET/CT. J Nucl Med. 2007;48:1577–9.
Ribeiro M-J, Vidailhet M, Loc’h C, Dupel C, Nguyen JP, Ponchant M, Dollé F, Peschanski M, Hantraye P, Cesaro P, et al. Dopaminergic Function and Dopamine Transporter Binding Assessed With Positron Emission Tomography in Parkinson Disease. Arch Neurol. 2002;59:580–6.
Weeks RA, Brooks DJ. Positron emission tomography and central neurotransmitter systems in movement disorders. Fundam Clin Pharmacol. 1994;8:503–17.
Garnett ES, Firnau G, Nahmias C. Dopamine visualized in the basal ganglia of living man. Nature. 1983;305:137–8.
Piel M, Vernaleken I, Rosch F. Positron emission tomography in CNS drug discovery and drug monitoring. J Med Chem. 2014;57:9232–58.
Dhawan V, Niethammer M, Lesser M, Pappas K, Hellman M, Fitzpatrick T, Quartarolo L, Bjelke D, Eidelberg D, Chlay T. Prospective FDOPA PET imaging study in human PD :our final step towards NDA approval. J Nucl Med. 2020;61:1565.
Dannals RF, Neumeyer JL, Milius RA, Ravert HT, Wilson AA, Wagner HN Jr. Synthesis of a radiotracer for studying dopamine uptake sites in vivo using PET: 2β-carbomethoxy-3β-(4-fluorophenyl)-[N-11C-methyl]tropane ([11C]CFT or [11C]WIN-35,428). J Labelled Compd Radiopharm. 1993;33:147–52.
Rinne JO, Bergman J, Ruottinen H, Haaparanta M, Eronen E, Oikonen V, Sonninen P, Solin O. Striatal uptake of a novel PET ligand, [18F]beta-CFT, is reduced in early Parkinson’s disease. Synapse. 1999;31:119–24.
Appel L, Jonasson M, Danfors T, Nyholm D, Askmark H, Lubberink M, Sörensen J. Use of 11C-PE2I PET in differential diagnosis of parkinsonian disorders. J Nucl Med. 2015;56:234–42.
Fischman AJ, Bonab AA, Babich JW, Livni E, Alpert NM, Meltzer PC, Madras BK. [(11)C, (127)I] Altropane: a highly selective ligand for PET imaging of dopamine transporter sites. Synapse. 2001;39:332–42.
Seifert KD, Wiener JI. The impact of DaTscan on the diagnosis and management of movement disorders: A retrospective study. Am J Neurodegener Dis. 2013;2:29–34.
Frost JJ, Rosier AJ, Reich SG, Smith JS, Ehlers MD, Snyder SH, Ravert HT, Dannals RF. Positron emission tomographic imaging of the dopamine transporter with 11C-WIN 35,428 reveals marked declines in mild Parkinson’s disease. Ann Neurol. 1993;34:423–31.
Piccini PP. Dopamine transporter: Basic aspects and neuroimaging. Mov Disord. 2003;18:S3–8.
Brooks DJ. Molecular imaging of dopamine transporters. Ageing Res Rev. 2016;30:114–21.
Peter D, Liu Y, Sternini C, De Giorgio R, Brecha N, Edwards R. Differential expression of two vesicular monoamine transporters. J Neurosci. 1995;15:6179–88.
Vander Borght TM, Sima AAF, Kilbourn MR, Desmond TJ, Kuhl DE, Frey KA. [3H]methoxytetrabenazine: A high specific activity ligand for estimating monoaminergic neuronal integrity. Neuroscience. 1995;68:955–62.
Frey KA, Koeppe RA, Kilbourn MR, Vander Borght TM, Albin RL, Gilman S, Kuhl DE. Presynaptic monoaminergic vesicles in Parkinson’s disease and normal aging. Ann Neurol. 1996;40:873–84.
Okamura N, Villemagne VL, Drago J, Pejoska S, Dhamija RK, Mulligan RS, Ellis JR, Ackermann U, O’Keefe G, Jones G, et al. In vivo measurement of vesicular monoamine transporter type 2 density in Parkinson disease with (18)F-AV-133. J Nucl Med. 2010;51:223–8.
Hsiao IT, Weng YH, Hsieh CJ, Lin WY, Wey SP, Kung MP, Yen TC, Lu CS, Lin KJ. Correlation of Parkinson disease severity and 18F-DTBZ positron emission tomography. JAMA Neurol. 2014;71:758–66.
Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol. 2006;5:235–45.
Politis M, Wu K, Loane C, Kiferle L, Molloy S, Brooks DJ, Piccini P. Staging of serotonergic dysfunction in Parkinson’s disease: an in vivo 11C-DASB PET study. Neurobiol Dis. 2010;40:216–21.
Doder M, Rabiner EA, Turjanski N, Lees AJ, Brooks DJ. study CWP: Tremor in Parkinson’s disease and serotonergic dysfunction: an 11C-WAY 100635 PET study. Neurology. 2003;60:601–5.
Fu H, Rong J, Chen Z, Zhou J, Collier T, Liang SH. Positron Emission Tomography (PET) Imaging Tracers for Serotonin Receptors. J Med Chem. 2022;65:10755–808.
Wilson H, Dervenoulas G, Pagano G, Koros C, Yousaf T, Picillo M, Polychronis S, Simitsi A, Giordano B, Chappell Z, et al. Serotonergic pathology and disease burden in the premotor and motor phase of A53T alpha-synuclein parkinsonism: a cross-sectional study. Lancet Neurol. 2019;18:748–59.
Sahli ZT, Tarazi FI. Pimavanserin: novel pharmacotherapy for Parkinson’s disease psychosis. Expert Opin Drug Discov. 2018;13:103–10.
Johnson KA, Conn PJ, Niswender CM. Glutamate receptors as therapeutic targets for Parkinson’s disease. CNS Neurol Disord: Drug Targets. 2009;8:475–91.
Crupi R, Impellizzeri D, Cuzzocrea S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front Mol Neurosci. 2019;12:20.
Karcz-Kubicha M, Lorenz B, Danysz W. GlycineB antagonists and partial agonists in rodent models of Parkinson’s disease–comparison with uncompetitive N-methyl-D-aspartate receptor antagonist. Neuropharmacology. 1999;38:109–19.
Ossowska K. The role of excitatory amino acids in experimental models of Parkinson’s disease. J Neural Transm Park Dis Dement Sect. 1994;8:39–71.
Marino MJ, Valenti O, Conn PJ. Glutamate receptors and Parkinson’s disease: opportunities for intervention. Drugs Aging. 2003;20:377–97.
Marin C, Papa S, Engber TM, Bonastre M, Tolosa E, Chase TN. MK-801 prevents levodopa-induced motor response alterations in parkinsonian rats. Brain Res. 1996;736:202–5.
Sawada H, Oeda T, Kuno S, Nomoto M, Yamamoto K, Yamamoto M, Hisanaga K, Kawamura T, Amantadine Study G. Amantadine for dyskinesias in Parkinson’s disease: a randomized controlled trial. PLoS ONE. 2010;5:e15298.
Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47.
Nash JE, Fox SH, Henry B, Hill MP, Peggs D, McGuire S, Maneuf Y, Hille C, Brotchie JM, Crossman AR. Antiparkinsonian actions of ifenprodil in the MPTP-lesioned marmoset model of Parkinson’s disease. Exp Neurol. 2000;165:136–42.
Steece-Collier K, Chambers LK, Jaw-Tsai SS, Menniti FS, Greenamyre JT. Antiparkinsonian actions of CP-101,606, an antagonist of NR2B subunit-containing N-methyl-d-aspartate receptors. Exp Neurol. 2000;163:239–43.
Smart K, Zheng MQ, Ahmed H, Fang H, Xu Y, Cai L, Holden D, Kapinos M, Haider A, Felchner Z, et al. Comparison of three novel radiotracers for GluN2B-containing NMDA receptors in non-human primates: (R)-[(11)C]NR2B-Me, (R)-[(18)F]of-Me-NB1, and (S)-[(18)F]of-NB1. J Cereb Blood Flow Metab. 2022;42:1398–409.
Ahmed H, Zheng MQ, Smart K, Fang H, Zhang L, Emery PR, Gao H, Ropchan J, Haider A, Tamagnan G, et al. Evaluation of (rac)-, (R)- and (S)-(18)F-OF-NB1 for imaging GluN2B subunit-containing N-methyl-D-aspartate receptors in non-human primates. J Nucl Med. 2022;63(12):1912–8.
Zheng M, Ahmed H, Smart K, Xu Y, Holden D, Kapinos M, Felchner Z, Haider A, Tamagnan G, Carson RE, et al. Characterization in nonhuman primates of (R)-[(18)F]OF-Me-NB1 and (S)-[(18)F]OF-Me-NB1 for imaging the GluN2B subunits of the NMDA receptor. Eur J Nucl Med Mol Imaging. 2022;49:2153–62.
Rischka L, Vraka C, Pichler V, Rasul S, Nics L, Gryglewski G, Handschuh P, Murgaš M, Godbersen GM, Silberbauer LR, et al. First-in-Humans Brain PET Imaging of the GluN2B-Containing N-methyl-d-aspartate Receptor with (R)-(11)C-Me-NB1. J Nucl Med. 2022;63:936–41.
Ahmed H, Wallimann R, Haider A, Hosseini V, Gruber S, Robledo M, Nguyen TAN, Herde AM, Iten I, Keller C, et al. Preclinical Development of (18)F-OF-NB1 for Imaging GluN2B-Containing N-Methyl-d-Aspartate Receptors and Its Utility as a Biomarker for Amyotrophic Lateral Sclerosis. J Nucl Med. 2021;62:259–65.
Ahmed H, Haider A, Varisco J, Stanković M, Wallimann R, Gruber S, Iten I, Häne S, Müller Herde A, Keller C, et al. Structure-Affinity Relationships of 2,3,4,5-Tetrahydro-1H-3-benzazepine and 6,7,8,9-Tetrahydro-5H-benzo[7]annulen-7-amine Analogues and the Discovery of a Radiofluorinated 2,3,4,5-Tetrahydro-1H-3-benzazepine Congener for Imaging GluN2B Subunit-Containing N-Methyl-d-aspartate Receptors. J Med Chem. 2019;62:9450–70.
Haider A, Iten I, Ahmed H, Müller Herder A, Gruber S, Krämer SD, Keller C, Schibli R, Wünsch B, Mu L, Ametamey SM. Identification and Preclinical Evaluation of a Radiofluorinated Benzazepine Derivative for Imaging the GluN2B Subunit of the Ionotropic NMDA Receptor. J Nucl Med. 2018;60:259–66.
Szermerski M, Börgel F, Schepmann D, Haider A, Betzel T, Ametamey SM, Wünsch B. Fluorinated GluN2B Receptor Antagonists with a 3-Benzazepine Scaffold Designed for PET Studies. ChemMedChem. 2018;13:1058–68.
Krämer SD, Betzel T, Mu L, Haider A, Herde AM, Boninsegni AK, Keller C, Szermerski M, Schibli R, Wünsch B, Ametamey SM. Evaluation of (11)C-Me-NB1 as a Potential PET Radioligand for Measuring GluN2B-Containing NMDA Receptors, Drug Occupancy, and Receptor Cross Talk. J Nucl Med. 2018;59:698–703.
Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic Cardioneuropathy in Dysautonomias. N Engl J Med. 1997;336:696–702.
Sakakibara R, Tateno F, Kishi M, Tsuyusaki Y, Terada H, Inaoka T. MIBG myocardial scintigraphy in pre-motor Parkinson’s disease: A review. Parkinsonism Relat Disord. 2014;20:267–73.
De Pablo-Fernandez E, Tur C, Revesz T, Lees AJ, Holton JL, Warner TT. Association of Autonomic Dysfunction With Disease Progression and Survival in Parkinson Disease. JAMA Neurol. 2017;74:970–6.
De Pablo-Fernandez E, Warner TT. Autonomic Dysfunction in Parkinson’s Disease: The Hidden Game Changer? Mov Disord. 2018;33:1028.
Hauser RA, Heritier S, Rowse GJ, Hewitt LA, Isaacson SH. Droxidopa and Reduced Falls in a Trial of Parkinson Disease Patients With Neurogenic Orthostatic Hypotension. Clin Neuropharmacol. 2016;39:220–6.
Braune S, Reinhardt M, Schnitzer R, Riedel A, Lücking CH. Cardiac uptake of [123I]MIBG separates Parkinson’s disease from multiple system atrophy. Neurology. 1999;53:1020–5.
Rascol O, Schelosky L. 123I-metaiodobenzylguanidine scintigraphy in Parkinson’s disease and related disorders. Mov Disord. 2009;24(Suppl 2):S732-741.
Acknowledgements
Figures were created with www.biorender.com.
Funding
We gratefully acknowledge the financial support provided, in part, by NIH grants (MH120197, MH128705, AG063290, AG070060, AG073428, AG074218, AG075444, AG078058, AG079956 and AG080262), Emory Radiology Chair Fund and Emory School of Medicine Endowed Directorship to S.H.L, and the Swiss National Science Foundation AQ7 (Grant number 191836) to A.H.
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Haider, A., Elghazawy, N.H., Dawoud, A. et al. Translational molecular imaging and drug development in Parkinson’s disease. Mol Neurodegeneration 18, 11 (2023). https://doi.org/10.1186/s13024-023-00600-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-023-00600-z