
Abstract
Background
Isolated hypothalamic-pituitary Langerhans cell histiocytosis (HPLCH) is very rare. We investigated the clinicopathological characteristics, endocrine function changes, BRAFV600E mutations and treatments of isolated HPLCH.
Methods
We identified seven patients with isolated HPLCH by reviewing the clinical and pathological files in our hospital from 2007 to 2015. The clinical characteristics of the seven patients were retrospectively reviewed, especially the endocrine function changes. Immunostaining and mutation profiling of BRAFV600E were performed.
Results
The seven HPLCH patients included three men and four women, aged 9–47 years. All patients presented with symptoms of central diabetes insipidus (CDI), and four displayed anterior pituitary hypofunction as well. Magnetic resonance imaging showed hypothalamic-pituitary axis involvement in all patients. There was no evidence for the involvement of other organs in all seven patients. Langerhans cell histiocytosis was confirmed by neuroendoscopic procedures, and immunohistochemical staining showed that all cases (7/7) were positive for CD68, CD1a, Langerin, and S-100. The BRAFV600E mutation was detected in three of the six cases (3/6). Six patients had follow-up information; all received desmopressin acetate and high-dose corticosteroid therapy, and two patients received radiotherapy.
Conclusions
Our study indicated that all patients with isolated HPLCH had CDI as the earliest symptom, and more than half of the patients had anterior pituitary deficiencies. The BRAFV600E mutation is a common genetic change in HPLCH patients. Treatment of HPLCH patients is difficult, and the progressive loss of endocrine function is irreversible in most cases.
Similar content being viewed by others
Background
Langerhans cell histiocytosis (LCH) is characterized by the idiopathic proliferation of specialized bone marrow-derived Langerhans cells and mature eosinophils. LCH can affect any organ or system and may be systemic or localized [1, 2]. Patients with isolated hypothalamic-pituitary (HP) LCH are very rare, although patients with multisystemic LCH often show pituitary gland involvement [3–7]. Among the endocrine regions, LCH is frequently found in the HP region, resulting in diabetes insipidus (DI), the most common endocrine anomaly [8–12]. Anterior pituitary involvement also occurs as a result of the disease process. However, anterior pituitary dysfunction is not invariably associated with abnormal HP region imaging, and it is almost always encountered in patients with multisystemic disease who show DI and HP pathology on magnetic resonance imaging (MRI) [6, 8]. The anterior pituitary endocrine function changes in isolated LCH limited to the HP region have been poorly studied.
Recently, LCH patients were shown to have a high frequency of BRAFV600E mutations and to respond to RAF inhibitors, suggesting that LCH is more likely a neoplastic than a reactive disorder. The BRAFV600E mutations are present in approximately 25–60 % of LCH cases [13–20], but this mutation has not been reported in isolated HPLCH in previous publications.
Several studies on LCH patients, which examined relatively small numbers of patients, have provided information on the evolution of pituitary dysfunction as well as the morphological changes in the HP region [3–7]. However, no large studies have examined patients with isolated HPLCH and assessed the pituitary function without interference from other organs. In the current study, we retrospectively studied seven patients with isolated HPLCH in our hospital from 2007 to 2015 and analysed their clinical and pathological features, endocrine function changes, BRAFV600E mutations, and treatment outcomes.
Methods
Patients and specimens
We reviewed all surgical biopsy or resection records in the Peking Union Medical College Hospital from January 1, 2007 to December 31, 2015 and identified a total of seven cases with isolated HPLCH. The patients’ medical records, including patient complaints, brain MRI findings, evaluation of anterior pituitary function, evaluation of other organs, and treatment, were collected and reviewed. No patient had a history of LCH. The HP regions of the patients were evaluated by MRI scans before and after treatment. The pre-treatment basal levels of growth hormone (GH), insulin-like growth factor-1 (IGF1), adrenocorticotropic hormone (ACTH), cortisol, free thyroxine (FT4), thyroid-stimulating hormone (TSH), prolactin (PRL), luteinizing hormone (LH), follicle-stimulating hormone (FSH), oestradiol, and testosterone were measured early in the morning in conjunction with plasma and urine osmolality tests. A water deprivation test was performed to assess vasopressin deficiency. In four patients with suspected pituitary dysfunction, dynamic pituitary function tests were employed, including the insulin tolerance test for assessments of GH and/or ACTH/cortisol reserves, thyrotropin-releasing hormone and gonadotropin-releasing hormone stimulation tests, and insulin-induced hypoglycaemia test. The pituitary functions of the patients were evaluated endocrinologically at least once per year. Methods used to evaluate other organs included a B ultrasound, computed tomography (CT), full-body bone scans, and a bone marrow biopsy. In addition, two patients received positron-emission tomography/computerized tomography (PET/CT). All patients received endoscopic transnasal transsphenoidal biopsy or resection of the HP regions. All samples were fixed in 10 % neutral buffered formalin, routinely processed, and embedded in paraffin. Haematoxylin-eosin-stained sections were observed using optical microscopy and reviewed independently by three experienced pathologists. The pathological diagnosis of LCH was based on the World Health Organization (WHO) criteria for LCH [1, 2]. We collected follow-up data from outpatient follow-ups.
Immunostaining
Immunostaining of CD1a (EP3622, 1:200; ZSJQ-BIO, Beijing, China), S-100 (polyclonal, prediluted; ZSJQ-BIO, Beijing, China), Langerin (12D6, prediluted; ZSJQ-BIO, Beijing, China), and CD68 (KP-1, 1:100; Dako, Glostrup, Denmark) was performed for all seven cases according to the manufacturer’s instructions. Immunostaining was performed on 4 μm-thick unstained sections cut from representative formalin-fixed paraffin-embedded (FFPE) blocks. For all markers, positive controls and negative controls were used. For CD68, signals appearing as tan particles in the cytoplasm were considered positive. For S-100, tan particles in the nucleus or cytoplasm were considered positive. For CD1a, tan particles in the membrane were considered positive. For Langerin, tan particles in the membrane or cytoplasm were considered positive.
Detection of BRAFV600E mutations
Genomic DNA from six cases of HPLCH (the tissue of the last case was not sufficient for further study) was extracted from freshly cut FFPE tissue sections using a QIAamp DNA Mini Kit (Qiagen, Germany) according to the manufacturer’s instructions. The area of the highest histiocyte density was identified through haematoxylin-eosin staining, and tissue from this area in unstained sections was scraped for DNA extraction. The extracted DNA was then quantified using a Qubit dsDNA BR assay (Life Technologies, USA). DNA from all six cases of LCH was successfully amplified. Analysis of BRAFV600E mutations was performed with a BRAF V600E Mutation Detection Kit (Amoy Diagnostics Co., Ltd., China) according to the manufacturer’s instructions. Real-time quantitative polymerase chain reaction (QPCR) was carried out on an Applied Biosystems7500 QPCR Platform (Life Technologies, USA). The cycling conditions for quality control (QC) runs and for mutation assays were as follows: 5 min incubation at 95 °C, followed by 15 cycles of 95 °C for 25 s, 64 °C for 20 s, and 72 °C for 20 s, and finally 31 cycles of 95 °C for 25 s, 60 °C for 35 s, and 72 °C for 20 s. Fluorescence was measured at 60 °C. Mutation data were interpreted according to the kit manual after curve analysis and calculation of ΔCt values.
Results
Clinical data
The seven cases of isolated HPLCH (Table 1) included three men and four women, aged 9–47 years. All seven patients had symptoms before the diagnosis, and the major complaint was polyuria and polydipsia (7/7). In addition, one female patient had secondary amenorrhoea (1/7), and one child patient showed growth retardation (1/7). No patients had visual field defects. All patients had no history of the smoking and drinking. The interval between onset of symptoms and diagnosis was 1–108 months. Before the LCH diagnosis by pathology, all seven patients had been diagnosed with central diabetes insipidus (CDI), and a mass was found in the HP region by MRI. Chest CT scans, abdominal B ultrasound, and full-body bone scans were performed in all seven cases, but no positive findings were obtained. Two patients received PET/CT scans, and there were no positive findings except in the HP region.
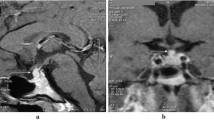
MRI showed pituitary stalk thickening in all seven patients, which was moderate (3.0–7 mm) in three patients including one child, and marked (>7 mm) in four patients including two children. In two cases, the lesion extended to the hypothalamus, and in the other two cases, the anterior pituitary was clearly involved. Optic chiasma involvement was not observed. All patients received gadolinium-enhanced MRI scans, and all showed enhancement (Fig. 1a, b). There were no hyperintense signals on T1WI in the posterior pituitary lobe in all seven patients. PET/CT was carried in two patients, and high uptake was observed in the pituitary stalk. The detailed results of the patients’ MRI scans of the HP region are listed in Table 2.

a. MRI scan of the lesion (arrow) in a 9-year-old child (case 1) before diagnosis demonstrated that the upper pituitary stalk was thickened, and a well-circumscribed and regular homogeneous enhanced mass was observed (sagittal T1 contrast-enhanced images). b. MRI scan of the lesion in case 1 at 11 months of follow-up demonstrated that the pituitary stalk (arrow) was fine and the homogeneous enhanced mass had regressed, but the anterior pituitary narrowed (sagittal T1 contrast-enhanced images)
Anterior pituitary function
Four patients had anterior pituitary deficiency. Three of the four patients showed whole anterior pituitary deficiency, and the other patient had GH deficiency (GHD) and hypogonadism before diagnosis but developed whole anterior pituitary deficiency in the following 4 months. Three patients had normal anterior pituitary function before diagnosis, and anterior pituitary function was also normal in two patients in the following 2 and 5 months, respectively. Data on the anterior pituitary function of the third patient was absent (details in Table 3).
Pathological and immunohistochemical findings
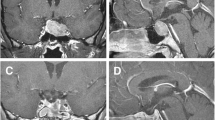
Surgery of the HP region was performed in all cases and was conclusive in all cases. Six patients received a biopsy, and one patient received a complete mass resection. Macroscopically, the maximum diameter of the samples ranged from 3 to 10 mm, and all tissues were yellow or white. Microscopically, all lesions consisted of accumulated mononuclear histiocytic cells. The lesions in all seven cases were predominantly composed of histiocytic cells and a variable fraction of eosinophils (Fig. 2a). The nuclei of the histiocytic cells were characteristic slightly eccentric, ovoid, or reniform, with linear grooves and inconspicuous nucleoli. The cytoplasm was abundant and pale to eosinophilic. Mitotic figures were seldom observed, and no necrosis was found in all cases. Eosinophils infiltration was easily observed. Touton giant cells were absent in all cases. Immunohistochemical staining for CD1a (Fig. 2b), Langerin (Fig. 2c), S-100 (Fig. 2d), and CD68 was positive in the histiocytic cells of all 7 LCH cases (7/7).

a. The lesion was predominantly composed of histiocytic cells and eosinophils (haematoxylin and eosin stain, 200×). b. The histiocytic cells were immunopositive for CD1a (400×). c. The histiocytic cells were immunopositive for Langerin (400×). d. The histiocytic cells were immunopositive for S-100 (400×)
BRAF V600E gene status
BRAF V600E mutations were found in three cases (3/6) of HPLCH using QPCR in 6 successfully amplified cases (Fig. 3a, b).

a. The point mutation V600E of BRAF was identified in case 2 by the QPCR method. b. No BRAF mutation was found in case 5 by the QPCR method
Treatment and outcomes
Six cases had follow-up data (range 2–74 months, details in Table 3). All patients received desmopressin acetate and high-dose corticosteroid therapy. In four patients with anterior pituitary deficiency, three were also treated with Letrox, and one patient received testosterone replacement therapy. Two patients with whole anterior pituitary deficiency received focal radiotherapy of the HP regions (total dose: 30.5 Gy). One patient underwent surgical resection after CyberKnife radiosurgery (details not available) in another hospital. No patients received chemotherapy. During the follow-up, no patients developed extrapituitary involvement, except one patient who died and had suspected liver involvement, but no direct proof, 74 months after resection. Among the five remaining patients, three patients survived with diseases, and two patients who received RT survived without diseases. One patient was lost to follow-up after biopsy.
Discussion
Langerhans cell histiocytosis (LCH) is a rare heterogeneous histiocytic disorder, and the dominant sites of involvement are bone and adjacent soft tissue, liver, spleen and bone marrow [1, 2, 15]. The central nervous system (CNS) may also be involved [2, 21]. In patients with LCH in the HP region, most cases are extended from osseous foci in multifocal or disseminated LCH, but unifocal or multifocal infiltrates can occur primarily within or even restricted to the HP region and other sites, such as the infundibulum, optic chiasm, and choroid plexus [2–7, 22, 23]. However, isolated HPLCH without the involvement of other organs is a rare condition, and there are few reports on the various characteristics of this disease state, including the evaluation of pituitary function, especially anterior pituitary function, the BRAFV600E mutation status, and other clinicopathological features. Here, we reviewed seven cases of isolated LCH limited to the HP region and investigated the clinicopathological characteristics, endocrine function changes, BRAFV600E mutations and treatment of the lesions.
The aetiology of the histiocytic lesions is largely unknown [1, 2]. Although the underlying aetiology of the condition has long been enigmatic, recent studies have suggested that LCH is due to clonal neoplastic proliferation of myeloid-derived precursor cells, with a high frequency of somatic oncogenic BRAFV600E mutations in 25–60 % of LCH patients [13–20]. In CNS, BRAF alterations are found in variable frequencies across a wide spectrum of diverse neoplasms, such as various glial and glioneuronal tumours, craniopharyngiomas, LCH and brain metastases [16]. No association between the BRAFV600E mutation and survival has been found in LCH, but several case reports described an impressive response to a BRAF inhibitor in patients with BRAF V600E-mutated LCH [19]. As an independent condition, the data on the BRAF V600E mutation in isolated HPLCH is unclear, and one reason for this may be the low number of cases. In this report, we examined seven cases of isolated HPLCH, and we had sufficient quantities of tissue for mutation detection in six cases. The results of our study showed that BRAFV600E was mutated in 50 % of patients with HPLCH, suggesting that the mutation rate in isolated HPLCH is consistent with the reported rates in LCH involving other sites. Because the follow-up time for some patients was short in our study, the relationship of BRAFV600E mutation and survival was not analysed. Smoking is an important factor in primary LCH of the lung [17], but all seven patients had no history of smoking in our study, suggesting that smoking is not related to HPLCH.
Clinically, LCH can occur at any age but typically occurs in the paediatric population [1, 6, 10]. The most common neurological signs of LCH are CDI with or without associated signs of hypothalamic dysfunction, increased intracranial pressure, cranial nerve palsies, and seizures [2]. Isolated CDI may be an early diagnostic signature in patients with an abnormal lesion in the HP region and no past medical history, and the association of polyuria-polydipsia suggests that a complete endocrine evaluation and a meticulous MRI examination of the HP region should be performed [2, 4, 6–10]. Patients with unifocal disease are usually older children or adults [1]. In our cohort, four patients were children aged 9–15 years, and three patients were adults. Our study had slightly more children with isolated HPLCH than adults. There was no specific gender preference in our study, which had slightly more females than males. The most common sign was CDI (polyuria and polydipsia, 100 %), and other symptoms included secondary amenorrhoea (1/7) and growth retardation (1/7).
While CDI is the most common HP symptom, anterior pituitary deficiencies are less common. Several studies have shown that most patients with HP lesions had one or more anterior pituitary deficiencies, and hormone deficiencies may be present at diagnosis or appear gradually during the course of disease [2–8, 11]. Regular monitoring of these patients is recommended. Growth hormone deficiency is the most frequent disease-related anterior pituitary deficiency and is found in up to 50 % of patients, followed by gonadotrophin deficiency, but ACTH and TSH are relatively less common; moderately elevated PRL levels are infrequently found and are attributed to pituitary stalk involvement [8]. Established deficiencies almost never recover over time. In our cohort, more than a half of the patients with HPLCH had anterior pituitary deficiencies at diagnosis with a mean interval from the first symptoms to diagnosis of 42 months. Three patients were diagnosed with normal anterior pituitary functions, and the intervals from the first symptoms to diagnosis were less than 2 months for all of them. Our study suggested that the interval from the first symptoms to diagnosis may be related to the anterior pituitary function, and the lesions progressed with time. All patients had posterior pituitary deficiencies at diagnosis, and polyuria and polydipsia were the first symptom in all patients; thus, a MRI scan is necessary for patients presenting with polyuria and polydipsia.
MRI changes in patients with LCH of CNS include the following: 1) intracranial, extra-axial changes of the hypothalamic-pituitary region (50 %), meninges (29 %) or choroid plexus (6 %); 2) intracranial, intra-axial changes of white matter and gray matter (36 %); and 3) cerebral atrophy (8 %) [2]. There are several studies on the MRI changes in HPLCH, and pituitary stalk thickening can be observed in LCH in the HP regions [4, 6]. In our cohort, the MRI features of patients with HPLCH included the following characteristics: 1) a single lesion on the HP region and involvement of the pituitary stalk; 2) isointensity signals on both T1WI and T2WI in most cases; 3) homogeneous enhancement in most cases; 4) relatively clear borders in most cases. Although these are characteristic MRI findings, it is difficult to distinguish HPLCH from other diseases, such as germinoma or lymphocytic thyroiditis.
An accurate pathological diagnosis of LCH is necessary for further treatment and follow-up depending on the classic pathomorphological and immunohistochemical features. Recently, the use of neuroendoscopic biopsy for suprasellar masses has increased, and with this technique, it is possible to obtain an accurate pathological diagnosis [24], although sometimes the pathological diagnosis is difficult due to small specimens with prominent sampling artefacts and haemorrhage [25]. Distinguishing this condition from other diseases that can cause CDI, such as craniopharyngioma, germinoma, sarcoidosis, hypophysitis, and tuberculosis, is essential [2, 10, 26], and the pathological signature with both CD1a-positive, Langerin-positive, and S-100-positive infiltrating histiocytes and increased presentation of eosinophils has made the differential diagnosis relatively easy. In addition, differentiation from other non-Langerhans cell histiocytoses, such as Erdheim-Chester disease or Rosai-Dorfman disease, is also important. Although S-100 positive cells can be found in Erdheim-Chester disease (ECD) and Rosai-Dorfman disease, CD1a is always negative in the same histiocytes [1, 2]. Notably, the BRAFV600E mutation was detected in more than 60 % of patients with ECD [27], but this mutation could not distinguish ECD from LCH because it was detected in approximately 25–60 % of the latter samples [13–20]. In our cohort, the seven isolated HPLCH cases that we assessed had been confirmed by pathomorphological changes and immunohistochemical findings during neuroendoscopic transnasal transsphenoidal surgery, including six cases by biopsy and one by resection, and all cases were simultaneously positive for CD1a, Langerin and S-100. In addition, we evaluated the status of other organs in all patients, which is required to exclude the multisystemic lesions, and there was no evidence of other system involvement in all patients.
The overall 5-year survival rate of LCH patients is 88 %. Patients with unifocal LCH have an excellent prognosis and a high long-term survival rate (99 %) and may spontaneously recover or require minimal treatment [1, 28]. In our cohort, one patient died of liver disease, but we had no evidence of liver involvement with LCH; the other six patients were still alive, and further follow-up should be performed. No lesions were spontaneously recovered in our study. The treatments for LCH involving the CNS, such as conservative treatment, surgical intervention, chemotherapy and radiotherapy, were dependent on the lesion location, size, number and symptoms, and high-dose corticosteroid therapy was also used to control the inflammatory process. Additionally, CyberKnife radiosurgery was performed in some patients [2, 21, 28–30]. However, the treatment of HPLCH patients is controversial because localized radiotherapy and chemotherapy were associated with tumour regression and stabilization but did not appear to prevent the evolution or deterioration of LCH-associated endocrine deficiency [3–7, 30]. In our cohort, although remission was observed in the lesions of two patients after radiotherapy (30.5 Gy) and the lesion of another patient who received surgical resection after CyberKnife radiosurgery failure, the patients’ quality of life was irreversibly impaired because they had irreversible permanent sequelae, panpituitary hormone deficiency, and needed to receive goal-directed hormone replacement therapy and monitor hormone level changes permanently. The anterior pituitary function of another patient who received high-dose corticosteroid therapy only also progressed from GH and LH-FSH deficiency to panpituitary hormone deficiency. Our study suggested that the treatment of patients with HPLCH is a difficult problem, and the deterioration of the endocrine function appeared to be unrelated to lesion regression; thus, diagnosis and treatment as early as possible are essential. In addition, monitoring the whole body to assess multisystemic involvement is also very important. Moreover, for patients with BRAFV600E mutations, new targeted therapies, such as vemurafenib, may be used because the results have been encouraging in select progressive patients with LCH [17, 31]. Recently, Nakagawa et al. [12] reported two relapsed females with LCH limited to the pituitary stalk and found that chemotherapy with cytarabine may be useful for preventing anterior pituitary hormone deficiency and neurodegenerative disease for new-onset CDI with LCH, but the further studies are needed to confirm these results.
Our study has some limitations as it is based on a small group of selected patients due to the low incidence of this condition. Notably, the follow-up time is short in our study because neuroendoscopic biopsy is a new technique in recent years, and it will be necessary to follow-up the patients over a longer period of time.
Conclusions
Our study indicated that some cases of isolated LCH involve only the hypothalamic-pituitary region, and all patients had CDI as the earliest symptom, with more than half having anterior pituitary deficiencies. The BRAFV600E mutation is a common genetic change in HPLCH patients. The treatment of HPLCH patients is difficult, and the loss of endocrine function is irreversible in most cases. For patients with the BRAFV600E mutation, new targeted therapy may be used when the lesion is progressing.
Abbreviations
- ACTH:
-
Adrenocorticotropic hormone
- AP:
-
Anterior pituitary
- CDI:
-
Central diabetes insipidus
- CNS:
-
Central nervous system
- CT:
-
Computed tomography
- E2:
-
Oestradiol
- ECD:
-
Erdheim-Chester disease
- FSH:
-
Follicle-stimulating hormone
- FSH:
-
Follicle-stimulating hormone
- FT4:
-
Free thyroxine
- GH:
-
Growth hormone
- GHD:
-
Growth hormone deficiency
- HP:
-
Hypothalamic-pituitary
- IGF1:
-
Insulin-like growth factor-1
- LCH:
-
Langerhans cell histiocytosis
- LCH:
-
Langerhans cell histiocytosis
- LH:
-
Luteinizing hormone
- LH:
-
Luteinizing hormone
- MRI:
-
Magnetic resonance imaging
- PRL:
-
Prolactin
- QPCR:
-
Quantitative polymerase chain reaction
- RT:
-
Radiotherapy
- T:
-
Testosterone
- TSH:
-
Thyroid-stimulating hormone
References
Jaffe R, Weiss LM, Facchetti F. Tumours derived from Langerhans cells. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO classification of tumours the haematopoietic and lymphoid tissues. Lyon: IARC Press; 2008. p. 358–60.
Paulus W, Perry A. Histiocytic tumours. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO classification of tumours the central nervous system. Lyon: IARC Press; 2007. p. 193–6.
Fahrner B, Prosch H, Minkov M, Krischmann M, Gadner H, Prayer D, et al. Long-term outcome of hypothalamic pituitary tumors in Langerhans cell histiocytosis. Pediatr Blood Cancer. 2012;58(4):606–10.
Kaltsas GA, Powles TB, Evanson J, Plowman PN, Drinkwater JE, Jenkins PJ, et al. Hypothalamo-pituitary abnormalities in adult patients with langerhans cell histiocytosis: clinical, endocrinological, and radiological features and response to treatment. J Clin Endocrinol Metab. 2000;85(4):1370–6.
Imashuku S, Kudo N, Kaneda S, Kuroda H, Shiwa T, Hiraiwa T, et al. Treatment of patients with hypothalamic-pituitary lesions as adult-onset Langerhans cell histiocytosis. Int J Hematol. 2011;94(6):556–60.
Kurtulmus N, Mert M, Tanakol R, Yarman S. The pituitary gland in patients with Langerhans cell histiocytosis: a clinical and radiological evaluation. Endocrine. 2015;48(3):949–56.
Toro Galván S, Planas Vilaseca A, Michalopoulou Alevras T, Torres Díaz A, Suárez Balaguer J, Villabona Artero C. Endocrine changes in histiocytosis of the hypothalamic-pituitary axis. Endocrinol Nutr. 2015;62(2):72–9.
Makras P, Kaltsas G. Langerhans cell histiocytosis and pituitary function. Endocrine. 2015;48(3):728–9.
García Gallo MS, Martínez MP, Abalovich MS, Gutiérrez S, Guitelman MA. Endocrine manifestations of Langerhans cell histiocytosis diagnosed in adults. Pituitary. 2010;13(4):298–303.
Marchand I, Barkaoui MA, Garel C, Polak M, Donadieu J, Writing Committee. Central diabetes insipidus as the inaugural manifestation of Langerhans cell histiocytosis: natural history and medical evaluation of 26 children and adolescents. J Clin Endocrinol Metab. 2011;96(9):E1352–60.
Keates-Baleeiro J, Rincon M. Morbid obesity as early manifestation of occult hypothalamic-pituitary LCH with delay in treatment. Case Rep Oncol Med. 2015;2015:915716.
Nakagawa S, Shinkoda Y, Hazeki D, Imamura M, Okamoto Y, Kawakami K, et al. Central diabetes insipidus as a very late relapse limited to the pituitary stalk in Langerhans cell histiocytosis. J Pediatr Endocrinol Metab. 2016;29(7):873–8.
Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116(11):1919–23.
Satoh T, Smith A, Sarde A, Lu HC, Mian S, Trouillet C, et al. B-RAF mutant alleles associated with Langerhans cell histiocytosis, a granulomatous pediatric disease. PLoS One. 2012;7(4):e33891.
Sahm F, Capper D, Preusser M, Meyer J, Stenzinger A, Lasitschka F, et al. BRAFV600E mutant protein is expressed in cells of variable maturation in Langerhans cell histiocytosis. Blood. 2012;120(12):e28–34.
Berghoff AS, Preusser M. BRAF alterations in brain tumours: molecular pathology and therapeutic opportunities. Curr Opin Neurol. 2014;27(6):689–96.
Roden AC, Hu X, Kip S, Parrilla Castellar ER, Rumilla KM, Vrana JA, et al. BRAF V600E expression in Langerhans cell histiocytosis: clinical and immunohistochemical study on 25 pulmonary and 54 extrapulmonary cases. Am J Surg Pathol. 2014;38(4):548–51.
Hyman DM, Diamond EL, Vibat CR, Hassaine L, Poole JC, Patel M, et al. Prospective blinded study of BRAFV600E mutation detection in cell-free DNA of patients with systemic histiocytic disorders. Cancer Discov. 2015;5(1):64–71.
Haroche J, Cohen-Aubart F, Emile JF, Arnaud L, Maksud P, Charlotte F, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121(9):1495–500.
Go H, Jeon YK, Huh J, Choi SJ, Choi YD, Cha HJ, et al. Frequent detection of BRAF (V600E) mutations in histiocytic and dendritic cell neoplasms. Histopathology. 2014;65(2):261–72.
Willis B, Ablin A, Weinberg V, Zoger S, Wara WM, Matthay KK. Disease course and late sequelae of Langerhans’ cell histiocytosis: 25- year experience at the University of California, San Francisco. J Clin Oncol. 1996;14(7):2073–82.
Belen D, Colak A, Ozcan O. CNS involvement of Langerhans cell histiocytosis: report of 23 surgically treated cases. Neurosurg Rev. 1996;19(4):247–52.
Rodriguez-Pereira C, Borras-Moreno JM, Pesudo-Martinez JV, Vera-Roman JM. Cerebral solitary Langerhans cell histiocytosis: report of two cases and review of the literature. Br J Neurosurg. 2005;19(2):192–7.
Jian F, Bian L, Sun S, Yang J, Chen X, Chen Y, et al. Surgical biopsies in patients with central diabetes insipidus and thickened pituitary stalks. Endocrine. 2014;47(1):325–35.
Noh S, Kim DS, Kim J, Kim SH. Langerhans cell histiocytosis in endoscopic biopsy: marked pinching artifacts by endoscopy. Brain Tumor Pathol. 2011;28(3):285–9.
Liu W, Wang L, Liu M, Li G. Pituitary morphology and function in 43 children with central diabetes insipidus. Int J Endocrinol. 2016;2016:6365830.
Cao XX, Sun J, Li J, Zhong DR, Niu N, Duan MH, et al. Evaluation of clinicopathologic characteristics and the BRAF V600E mutation in Erdheim-Chester disease among Chinese adults. Ann Hematol. 2016;95(5):745–50.
Grana N. Langerhans cell histiocytosis. Cancer Control. 2014;21(4):328–34.
Hong WC, Murovic J, Gibbs I, Vogel H, Chang SD. Pituitary stalk Langerhans cell histiocytosis treated with CyberKnife radiosurgery. Clin Neurol Neurosurg. 2013;115(5):573–7.
Rosenzweig KE, Arceci RJ, Tarbell NJ. Diabetes insipidus secondary to Langerhans’ cell histiocytosis: is radiation therapy indicated? Med Pediatr Oncol. 1997;29(1):36–40.
Bubolz AM, Weissinger SE, Stenzinger A, Arndt A, Steinestel K, Brüderlein S, et al. Potential clinical implications of BRAF mutations in histiocytic proliferations. Oncotarget. 2014;5(12):4060–70.
Acknowledgements
Not applicable.
Funding
This work was supported by the Foundation of Pathologic Research Centre of the China Academy of Medical Sciences (Project No. 2015PT320009). The funders had no role in the design of the study, and collection, analysis, and interpretation of the data and in writing the manuscript.
Availability of data and materials
Slides and raw data files available upon request. Send inquiries to the corresponding author.
Authors’ contributions
HZ and LT analysed the cases, performed the QPCR and drafted the manuscript. LZ was the main pathologist involved in the cases. PF was responsible for the clinical data and evaluated the pituitary functions. SJ was the radiotherapy doctor who treated the patients. MW performed the surgical operations. LJ was responsible for the MRI data. ZDC generated the figures. ZDR was the main editor of the body of the text and the main pathologist involved in the cases. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent for publication of theirs clinical details and/or clinical images was obtained from the patients or their parents. Copies of the consent forms are available for review by the Editor of this journal.
Ethics approval and consent to participate
The ethics committee of the Peking Union Medical Collage Hospital specifically approved this study (No. S-K133), and patients or their parents provided written informed consent to participate in the study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Huo, Z., Lu, T., Liang, Z. et al. Clinicopathological features and BRAFV600E mutations in patients with isolated hypothalamic-pituitary Langerhans cell histiocytosis. Diagn Pathol 11, 100 (2016). https://doi.org/10.1186/s13000-016-0548-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13000-016-0548-5