
Abstract
The influenza virus (IFV) imposes a considerable health and economic burden globally, requiring a comprehensive understanding of its pathogenic mechanisms. Ferroptosis, an iron-dependent lipid peroxidation cell death pathway, holds unique implications for the antioxidant defense system, with possible contributions to inflammation. This exploration focuses on the dynamic interplay between ferroptosis and the host defense against viruses, emphasizing the influence of IFV infections on the activation of the ferroptosis pathway. IFV causes different types of cell death, including apoptosis, necrosis, and ferroptosis. IFV-induced ferroptotic cell death is mediated by alterations in iron homeostasis, intensifying the accumulation of reactive oxygen species and promoting lipid peroxidation. A comprehensive investigation into the mechanism of ferroptosis in viral infections, specifically IFV, has great potential to identify therapeutic strategies. This understanding may pave the way for the development of drugs using ferroptosis inhibitors, presenting an effective approach to suppress viral infections.
Similar content being viewed by others


Introduction
Influenza virus (IFV), is among the major respiratory viruses that have caused significant mortality and health issues, imposing a significant economic burden on society [1, 2]. The worldwide dissemination of this virus has resulted in numerous outbreaks and pandemics, emphasizing the critical importance of obtaining a comprehensive understanding of its pathogenic mechanisms [3,4,5]. Viruses manipulate diverse cellular metabolic and regulatory pathways to facilitate the progression of the pathogenic process, which can result in different forms of cell death within their target cells [6]. Moreover, cell death is significant in the host reaction to viral infection. Cell death mechanisms have the potential to restrict virus reproduction within the affected host cell and at the same time trigger innate and pro-inflammatory immune reactions against the viral infection [7]. Among different cell death pathways, ferroptosis represents a distinctive form of controlled cell death distinguished by iron-dependent lipid peroxidation and excessive production of reactive oxygen species (ROS) [8, 9].
Ferroptosis, an iron-dependent oxidation-regulated cell death, is characterized by changes in iron balance and overproduction of reactive oxygen and nitrogen species (ROS/RNS) as well as irregular lipid peroxidation (LPO) [10, 11]. Storage of harmful plasma membrane lipid peroxides and initiation of cell lysis represent potential risks associated with ferroptosis and may lead to the developments of pathologies such as neurodegenerative diseases, autoimmune disorders and cancer [12, 13]. Furthermore, there is compelling evidence to suggest that ferroptosis contributes significantly to inflammation [14]. Ferroptosis is distinguished by unique morphological and chemical attributes. It also exhibits variations in the genes involved and regulatory pathways, which distinguishes it from other programmed cell deaths such as apoptosis and necroptosis [15]. It typically induces morphological alterations in the energy-generating organelle of a cell. This results in the development of smaller mitochondria with heightened membrane density, the decrease or elimination of cristae, and the rupture of the outer mitochondrial membrane, distinguishing it from other forms of cell death [16]. Ferroptosis induces significant biochemical alterations, encompassing iron and ROS accumulation, stimulation of the MAPK signaling pathway, suppression of the Xc-system, decreased glutathione (GSH) levels, increased NOX activity, and secretion of arachidonic acid (AA) mediators [17, 18]. In contrast to the biochemical changes observed in ferroptosis, the biochemical modifications in apoptosis, autophagy, and necroptosis consist of DNA fragmentation, elevated lysosomal activity, and diminished ATP levels, respectively [16].
Consequently, this article aims to investigate the dynamic interplay between ferroptosis and host defense mechanisms against IFV infections. By exploring how IFV infection triggers the ferroptosis pathway and leads to inflammation, this study provides novel insights that could inform the development of new antiviral strategies.
Overview of ferroptosis
Role of Iron in ferroptotic processes
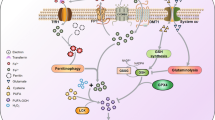
Iron metabolism plays a role in essential cellular processes, including DNA synthesis and mitochondrial respiration. Imbalances in iron homeostasis have the potential to induce cellular damage [19]. An increase in cellular iron accumulation plays a role in ferroptotic death [20]. Regulators of iron metabolism, from iron absorption and storage to release, could have an effect on ferroptosis (Fig. 1) [21].

Exploring how lipid peroxidation, alterations in iron metabolism, and changes in the antioxidant system contribute to ferroptosis. (A) In antioxidant pathway, cystine enters the cell through the Xc-System and converted to cysteine and then GSH in the cell, which activates GPX-4. Ferroptosis inhibited when GPX-4 reduces P-LOOH to PLOH. (B) The Fenton reaction creates highly reactive hydroxyl radicals (OH⋅) by reacting Fe2 + with H2O2, generating Fe3+. This process drives a cycle of oxidative stress, leading to increased free radicals and lipid peroxidation. (C) Lipid peroxidation (LPO) in cell membranes, crucial for ferroptosis, occurs when antioxidant defenses like GPX-4 and GSH are impaired. Long-chain fatty acids, particularly those activated by ACSL4 such as arachidonic acid (AA), contribute to ferroptosis through the formation of AA-acyl Co-A and AA-PE. AA-PE then oxidized by 15-LOX to produce lipid hydroperoxides, which, in the presence of labile Fe2+, generate radicals that damage cellular membranes. Abbreviations; HMOX1: heme oxygenase 1; Lip: lipid iron pool; FPN: Ferroportin; TF: Transferrin; PUFA: polyunsaturated fatty acids; ACSL4: acyl-CoA synthetases 4; LPCTA3: lysophosphatidylcholine acyltransferase 3; ALOX: arachidonate lipoxygenase; GSH: Reduced glutathione; GPX-4: Glutathione peroxidase 4; GSSG: Glutathione disulfide
The redox characteristics of iron involve it in reactions that generate ROS, causing oxidative damage to lipids and potentially resulting in the accumulation of lipid ROS or ferroptosis [22]. Fenton reaction contributes to the generation of extremely reactive hydroxyl radicals (OH⋅) by combining ferrous iron (Fe2+) with hydrogen peroxide (H2O2) and producing Fe3+ (Fig. 1B) [23]. The onset of iron-induced oxidative stress triggers a self-perpetuating cycle, resulting in increased iron release and increased production of free radicals. The proximity of ROS production to membrane phospholipids that peroxidize polyunsaturated fatty acids (PUFAs), causes the production of lipid hydroperoxides and α,β-unsaturated 4-hydroxyaldehydes [24].
Hephaestin is a ferroxidase enzyme, which facilitates the conversion of ferrous iron (Fe2+) to ferric iron (Fe3+), a critical step in the absorption of dietary iron from the small intestine iron exporter (FPN1: Ferroportin 1) and allows the binding of iron to the blood transporter, transferrin (TF) [25, 26]. This process allows the transfer of iron by TF to red blood cell precursors and various tissues [25]. Gao et al., identified transferrin as the active component related to ferroptosis through a four-step fractionation procedure of dialyzed FBS, ultimately purifying a single protein that correlated with lethal activity. Removal of transferrin from FBS reduced cell death, while addition of commercial bovine holotransferrin-induced cell death, confirmed the role of transferrin in ferroptosis [27]. Transferrin receptor 1 (TFR1) facilitates the uptake of TF that transports ferric iron into endosomes through clathrin-mediated endocytosis. The low pH of the endosome causes the release of Fe3+ from transferrin. Prostate epithelial 6-transmembrane antigen 3 (STEAP3) converts released Fe3+ to Fe2+, and divalent metal iron transporter-1 (DMT1) transports ferrous iron to the cytoplasm. Furthermore, Fe2+ can reach cells through ZIP8/ZIP14 (zinc transporters) [28]. In study on the role of TFR2 in ferroptosis, Tong et al. found that elevated TFR2 expression stimulated the production of ROS and LPO. TFR2 also promoted cellular susceptibility to ferroptosis inducers, Erastin and RSL3, while ferrostatin-1 )Fer-1( and DFO (ferroptosis suppressors) effectively mitigated TRF2-mediated LPO, restoring cellular activity [29].
An elevation in cellular labile iron pool (LIP), which contains predominantly Fe2+, is the result of increased heme oxygenase 1 (HMOX1) activity, a reduction in FPN expression, and an increase in TF expression, which potentially leading to ferroptosis (Fig. 1B) [30, 31]. Furthermore, ferritin, which is the cellular iron storage protein, undergoes autophagy-mediated degradation by autolysosomes, elevating intracellular iron. This phenomenon, called ferritinophagy, can cause increased ROS and the onset of ferroptosis [32]. NCOA4 aids in the transport of ferritin to autolysosomes [33]. Mitochondrial ferritin (FtMt) plays a crucial role in storing excess cellular iron. The mitochondrial electron transport chain depends on the crucial cofactors such as heme and iron-sulfur groups, both of which are iron dependent [34]. Wang et al. found that in brains of iron-increased ischemic mice, the absence of FtMt caused more severe damage and showed molecular indicators of ferroptosis, such as accumulated free iron, increased LPO, and exacerbation of GSH inhibition. Consequently, these findings may underline the critical role of FtMt in protecting against ferroptosis [35].
Mechanisms of ferroptosis lipid peroxidation (LPO)
The LPO mechanism involves the binding of oxygen to lipids, resulting in the production of lipid hydroperoxides and peroxyl radicals [36]. The increased ROS from lipids, leading to the cell death through oxidative stress, is specifically identified as ferroptosis [37]. Alterations in the shape and curvature of lipid membranes improve the accessibility of oxidants, accelerating membrane degradation and causing the disappearance of ferroptotic cells [38].
LPO in cell membranes occurs when the activity of key antioxidant defenses, such as glutathione peroxidase 4 (GPX-4) and GSH, is altered. Phospholipids associated with PUFAs are essential for lipid peroxidation, a determinant of ferroptosis (Fig. 1C) [39]. The critical sites in lipids that drive ferroptosis are the bis-allylic carbons, which are vulnerable to attack by free radicals and lipoxygenase enzymes as well as oxygen, due to their adjacency to a double-bonded carbon–carbon pair [40]. Long-chain fatty acids activated by acyl-CoA synthetases (ACSLs) like ACSL4 which interact with arachidonic acid (AA), influence cell sensitivity to ferroptosis [39]. ACSL4 facilitates the binding of AA to generate AA-acyl Co-A, which are then converted to phosphatidylethanolamines (AA-PE) by lysophosphatidylcholine acyltransferase 3 (LPCTA3). 15-lipoxygenase (15-LOX) oxidizes AA-PE and produce lipid hydroperoxides (LOOH), which serve as key factor of ferroptosis (Fig. 1C) [41]. Lipid hydroperoxides (PL-PUFA-OOH) interact with labile Fe2+ (in Fenton chemistry), producing the PL-PUFA-O⋅ radical, which can trigger peroxidation in neighboring phospholipids, causing membrane damage [42]. GPX-4, as an antioxidants, mitigates ferroptosis by reducing and converting lipid hydroperoxides (PL-PUFA-OOH) to lipid alcohols (PL-PUFA-OH) [42]. Additionally, other antioxidants such as coenzyme Q10 and vitamin E interact with peroxyl radicals to produce peroxides. Subsequently, GSH and GPX-4 can detoxify the generated oxidized lipids [36].
Regulatory proteins and pathways
The initial trigger of ferroptosis is the dysfunction of the system Xc−, a membrane antiporter that plays a role in cellular cystine import and glutamate export [37, 43]. The Xc system is formed by the xCT light chain (SLC7A11), which is connected by a disulfide bond to the heavy chain (4F2hc) [44]. The alteration in the Xc system prevents the uptake of cystine, interrupts the biosynthesis of GSH, causing an increase in of lipid reactive oxygen species, triggering oxidative cellular damage, and inducing ferroptosis (Fig. 1A) [37]. Research indicates that SLC7A11 is crucial for suppressing ferroptosis in acinar cells by maintaining both GSH level and ROS balance [45].
A study on regulation of the Xc system revealed that stimulation of sensing stressor, transcription factor 3 (ATF3), can lead to erastin-induced ferroptosis by suppressing the Xc-system and decreasing the level of GSH through direct regulation of the SLC7A11 promoter [46]. Furthermore, silencing BECN1 hinders ferroptosis induced by Xc-system suppressors such as erastin, sulfasalazine, and sorafenib. AMP-activated protein kinase (AMPK) plays a role in BECN1 phosphorylation of at serine, which is vital for the formation of the complex between BECN1 and SLC7A11, as well as lipid peroxidation [47]. Cystine enters the cell through the Xc-System, where it is transformed into cysteine and subsequently contributes to the production of GSH. Within the GPX-4 cycle, P-LOOH (which can be generated by the Fenton reaction) oxidizes GPX-4-SeH to GPX-4-SeOH (Fig. 1A). GSH plays a crucial role in activating GPX-4, thereby preventing its deactivation through the release of GSSG. The reduction of GSSG back to GSH is facilitated by enzymes such as glutathione reductase (GR) and the cofactor NADPH. Ferroptosis is inhibited when P-LOOH is converted to PLOH through the action of GPX-4 [48]. Ferroptosis inhibitors, such as Ferroptosis Suppressor Protein 1 (FSP1) and ubiquinone (CoQ10), work similarly to GSH. The reduction of CoQ10 to ubiquinol by FSP1 transform it into a lipophilic radical scavenger, effectively preventing ferroptosis (Fig. 1A) [49].
The regulation of ferroptosis encompasses various signaling pathways. AMPK inhibits ferroptosis by reducing lipogenesis, while glucose promotes ferroptosis by counteracting AMPK. Furthermore, E-cadherin–NF2–Hippo prevents ferroptosis by reducing YAP-mediated transcription. p53 and BAP1 also play a role in suppressing SLC7A11 transcription, which reduces cystine import and leads to ferroptotic cell death [50].
Currently, multiple drugs have been identified to stimulate ferroptosis, classified into four categories according to their ability to inhibit the Xc-system and GSH, directly inhibit GPX-4, reduce GPX-4 and CoQ10 protein, and induce lipid peroxidation [51]. Class 1 ferroptosis inducers such as Erastin, piperazine erastin, imidazole ketone erastin (IKE), sulfasalazine and sorafenib as well as glutamate target the Xc− system. RSL3, a class 2 inducer, acts on GPX-4. FIN56 and CIL56, as class 3 inducer, reduce the levels of GPX-4 and coQ10, while FINO2 (class 4) induces lipid peroxidation. In contrast, iron chelators (DFO, cyclipirox, deferiprone), lipophilic antioxidants (e.g., vit E, Fer-1, CoQ10), D-PUFA (e.g., D4-arachidonic acid), and LOX inhibitors are ferroptosis suppressors [40].
Mitochondria role in ferroptosis
Mitochondria are thought to be associated with ferroptotic signaling pathways due to their roles in ATP synthesis, ROS production, iron homeostasis, and maintaining redox balance [34]. Several mitochondrial enzyme systems, such as ACSL4, lysophos-phatidylcholine acyltransferase 3 (LPCAT3), and acyl-CoA synthetase family member 2 (ACSF2), are involved in lipid metabolism and play role in ferroptosis [52]. ACSF2 in mitochondria, controls fatty acid activation and synthesis, crucial for lipid peroxidation and erastin-induced ferroptosis. ACSL4 in the outer mitochondrial membrane, is another key pro-ferroptosis gene involved in lipid metabolism [52]. On the outer surface of the inner mitochondrial membrane, the dihydroorotate dehydrogenase (DHODH)-CoQH2 pathway, facilitates the conversion of dihydroorotate (DHO) to orotate (OA) and concurrently regenerates CoQH2 to neutralize lipid radicals, consequently suppress ferroptosis. Mitochondrial GPX4 (mGPX4) and DHODH-CoQH2 separately but both convert CoQ to CoQH2 [53].
Mitochondrial homeostasis is regulated by fission, fusion, and mitophagy, but these processes can become dysregulated. Various mitochondria-regulated molecules, including the fission genes (DRP1 and FIS1), the fusion-related genes (MFN1/2 and OPA1), and the mitophagy-related genes (PINK1 and FUNDC1), play a role in ferroptosis [54]. Inhibiting both ferritinophagy and mitophagy (by double knockdown of NCOA4 and PINK1) nearly prevents O-GlcNAcylation-induced ferroptosis indicating that ferroptosis and mitochondrial dynamics are co-regulated, and any imbalance between them disrupts cellular homeostasis [54]. In interplay between mitochondrial dysfunction and ferroptosis, after cerebral ischemia/reperfusion injury, the NOD LRR pyrin domain-containing protein 3 (NLRP3) inflammasome in microglia was activated by mitochondrial dysfunction, which involves mitochondrial ROS, DNA damage, and cardiolipin release. This activation leads to pyroptosis and other cell deaths, such as ferroptosis [55] Research showed that controlled NLRP3 inflammasome activation is vital for homeostasis. ROS regulates this activation, with elevated ROS causing cell damage via the NLRP3 inflammasome. Moreover, augmented ROS can facilitate viral spread between cells and heighten susceptibility to respiratory viruses [56].
IFV infection reduced total antioxidant capacity (TAC), superoxide dismutase (SOD), GPX, and catalase activities in A549 cells while significantly increasing malondialdehyde (MDA) and ROS levels, indicating induced oxidative stress injury [56]. The IAV polymerase basic 2 (PB2), with its N-terminal mitochondrial targeting sequence, transports IAV into mitochondria, disrupting their fusion and fission balance, leading to fragmentation and superoxide ion leakage. Additionally, the NS1 protein also trigger mitochondrial fragmentation, boosting ROS generation and enhancing IAV replication and pathogenesis [57].
Unveiling links between ferroptosis and host defense mechanisms against viruses
Ferroptosis promotes inflammation and releases damage-associated molecular patterns (DAMPs), such as high-mobility group box 1 (HMGB1) [58]. Intense oxidative stress, caused by ROS, amplifies LPO, initiating ferroptotic cell death. This process releases DAMPs, which activates inflammation through receptors such as the receptor for advanced glycation end products (RAGE) and NF-κB. [59, 60]. Cell stress can induce ATP release, which then activates purinoreceptors and facilitates key survival mechanisms [61]. Researches have highlighted the involvement of extracellular ATP in activating the NLRP3 inflammasome triggered by DAMPs [62]. Moreover, macrophages are crucial in efferocytosis by generating nitric oxide (NO) and ROS, removing apoptotic cells, and secreting anti-inflammatory cytokines [63]. Redox Imbalance and elevated LPO induce inflammation, and proinflammatory cytokines worsen oxidative stress. In the process of ferroptosis, activation of NF-κB increases cytokine release, while inhibition of ferroptosis with Trolox can reduce TNF-α, IL-1β, and IL-6 [64]. Furthermore, LPO alters low-density lipoprotein, promoting inflammation through macrophage polarization. GPX-4, mitigates ferroptosis and inflammation by preventing the oxidation of AA and NF-κB, while increasing inflammatory mediators such as cyclooxygenase 1 (COX1) and 12-lipoxygenase. Furthermore, iron participates in the modulation of the immune system by influencing the polarization of M1 macrophage [59, 65]. IL-6 influences ferritin expression in iron metabolism, promoting inflammation. In degenerative chondrocytes, IL-6 induces LPO and iron imbalance, worsening intervertebral disc injuries [66].
Ferroptosis is also induced in adaptive immune cells, specifically T cells (CD8+), by ACSL4 and CD36, while SLC7A11 and GPX-4 suppress it. Furthermore, ferroptosis can impair IgM secretion from B lymphocytes and Marginal Zone B (MZB) cells by reducing GPX-4 activity. CD36 also plays a role in ferroptosis by uptaking PUFAs [67]. Signaling pathways associated with inflammation may be related to ferroptosis. Kong et al. revealed that interferon-γ induced a reduction in GSH levels, induced cell cycle arrest in the G0/G1 phase, and elevated ROS level and lipid peroxidation leading to ferroptosis. IFNγ by stimulating JAK/STAT cell signaling can negatively regulate the expression of SLC3A2 and SLC7A11 of the Xc-system, highlighting its role in promoting ferroptosis [68]. The TNF-α/IL-6-ERK pathway was also upregulated in ischemia/reperfusion following ferroptosis [69]. The complement system, crucial for host defense against viral infections, is often targeted by viruses that develop mechanisms to evade or deregulate it, thereby elevating infectivity and worsening disease symptoms [70]. A study in a murine model indicates that the Coxsackie virus can induce ferroptosis by altering the complement components, C4b and C3. This study demonstrated that the ferroptosis inhibitor, fer-1, successfully mitigated inflammation in viral infection and prohibited ferroptosis in Coxsackie virus-induced myocarditis [71]. Additionally, several viruses interfere with iron absorption and the antioxidant system, so they require iron for optimal replication of their genome. This leads them to compete with the host for iron uptake. Viruses can enhance cellular iron uptake by downregulating hepcidin expression or increasing DMT expression [9, 72].
Host cells response to increased ROS/LPO accumulation
Enzymes and molecules, both endogenous and exogenous, protect cells from oxidative stress by neutralizing free radicals and metabolizing toxic aldehydes from lipid peroxidation. Endogenous enzymes include GPX, SOD, glutathione S-transferase (GST), catalase, thioredoxin (TRX), thioredoxin peroxidase (TRXP), peroxiredoxin (PRDX), and HMOX1. Nonenzymatic antioxidants include GSH, proteins like ferritin, TF, albumin, ceruloplasmin, and molecules such as uric acid, coenzyme Q, and lipoic acid [73, 74].
Antioxidant mechanisms are regulated by nuclear factor erythroid 2 like 2 (NFE2L2/NRF2) and membrane repair during ferroptosis involves endosomal sorting complexes required for transport (ESCRT-III) activation [74]. Nrf2 binds to antioxidant response elements in gene promoters like GPX4, SLC7A11, HMOX1, and NAD(P)H quinone oxidoreductase 1 to scavenge ROS. It also reduces Fe uptake by downregulating TFR1 and DMT1, and increases GSH synthesis by upregulating glutamate-cysteine ligase [75]. Therefore, activation of the P62-Keap1-NRF2 pathway has been shown to inhibit ferroptosis. In cell peroxidation, Keap1 expression was inhibited, leading to the dissociation of NRF2-Keap1. This allowed NRF2 to enter the nucleus, increase antioxidant protein expression, and decrease ROS and oxidative stress damage [76]. Moreover, Oxidative stress could induction lipid droplet accumulation [77]. Lipid droplets (LDs) regarded as a lipid storage organelle which have antioxidative activity, reducing ROS levels and preventing PUFA peroxidation by sequestering these lipids in their cores, making them less accessible to ROS [78].
Interplay between influenza virus and ferroptosis
Ferroptosis has been implicated in the pathogenesis of several viral infections including IFV, human immunodeficiency virus (HIV), severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and, enteroviruses [79]. Studies have demonstrated a connection between ferroptosis and the reduction of CD4 cells after HIV infection [80, 81]. When primary CD4 cells are infected with wild-type HIV-1, these HIV-infected cells show increased regulated cell death, including ferroptosis. Xiao and colleagues observed that traditional markers of ferroptosis were evident in the CD4 T cells of HIV immune non-responders [81]. These markers included heightened lipid peroxidation within mitochondria and damage to mitochondrial structure (81). Moreover, it has been found that, the HIV-1 transcriptional activator of transcription (Tat) protein boosts ACSL4 expression via exosomes secreted by infected cells. This overexpression results in lipid accumulation, heightening the risk of ferroptosis [82]. Furthermore, HIV can elevate cellular iron levels by utilizing hepcidin-mediated degradation of ferroportin, which in turn enhances HIV transcription, replication, and release [83, 84]. In the context of SARS-CoV-2 infection, various molecular changes contribute to ferroptosis in the lungs [85]. The infection decreases GPX4 expression in Vero cells, weakening lipid repair mechanisms and increasing vulnerability to lipid peroxidation. The structural proteins of SARS-CoV-2 also suppress the NRF2 antioxidant pathway, which normally defends against oxidative damage. Elevated serum ferritin levels in severe COVID-19 cases correlate with disease severity, as ferritin provides iron that promotes ferroptosis. Disruptions in iron metabolism are indicated by altered iron homeostasis proteins and higher levels of reactive iron in the lungs. Additionally, lipid peroxidation has been noted in cardiovascular complications related to COVID-19 and in the heart pacemakers of SARS-CoV-2-infected hamster models [85, 86]. In addition, an in vitro study has shown that enteroviruses, such as coxsackievirus A6, can increase ACSL4 expression, thereby triggering ferroptosis [87]. These observations indicate that ferroptosis is closely associated with various viral infections, which could potentially be a therapeutic target in the future. However, our focus here is on the relationship between ferroptosis and IFV infection.
IFV is known to induce multiple forms of cell death, including apoptosis and necrosis [88,89,90,91]. Recent studies suggest a potential link between IFV and ferroptosis, marked by elevated iron levels, release of ROS, and lipid peroxidation (Fig. 2A) [92]. This intricate relationship between ferroptosis and IFV opens avenues to explore novel therapeutic interventions.

Ferroptosis in viral infections and unravelling the mechanisms underlying ferroptotic cell death induced by influenza virus. (A) Research indicates that the influenza virus can induce ferroptosis in the host by impacting lipid peroxidation, disrupting antioxidant systems like the Xc-, GPX-4, and GSH pathways, and causing mitochondrial dysfunction. (B) During influenza infection, lung epithelial cells and macrophages release IFP35, which drives the cytokine storm. Studies show that IFP35 exacerbates ferroptosis and lung damage, while its knockdown reduces both ferroptosis and lung injury. PGE2 hampers immunity against influenza by suppressing macrophage and T-cell functions. Proanthocyanidins (PAs) reduce lung damage, inflammation, and ferroptosis in influenza-induced acute lung injury (ALI) by lowering IFN-γ levels. Abbreviations; IAV: Influenza A virus; SIV: Swine influenza virus; LPO: lipid peroxidation; Fer-1: Ferrostatin-1; HIF-1-alpha: Hypoxia-inducible factor 1; iNOS: Inducible nitric oxide synthase; VEGF: Vascular endothelial growth factor; MDA: Malondialdehyde
As shown in Table 1, evidence suggests a connection between ferroptosis and the destruction of nasal epithelial cells, which is triggered by inflammation resulting from the influenza virus. IFV H1N1 infection induces alterations in the expression of genes and metabolites associated with ferroptotic cell death in human nasal epithelial progenitor cells (hNEPCs). A notable decrease in the expressions of Nrf2/KEAP1 and GCLC (catalytic subunit of glutamate-cysteine ligase) is observed, along with impaired glutaminolysis (Fig. 2A) [93]. The KEAP1/NRF2 signaling pathway serves as a primary mechanism to protect cells against oxidative stress. NRF2 (nuclear factor erythroid-2-like-2) is recognized not only for its ability to alleviate inflammation and counteract oxidative damage but also for its antiviral properties, particularly to inhibit IFV replication [94]. Liu et al. reported that IFV H1N1 can trigger ferroptosis in hNECs through the NRF2-KEAP1-GCLC signaling pathway [93]. In another study, by using murine lung epithelial cells, it was revealed that IFV triggers ferroptosis. The discerned indicators of influenza-induced ferroptosis encompassed mitochondrial impairment, increased ROS release, elevated intracellular iron levels, decreased GPX-4 gene expression, and increased LPO by the presence of ACSL4. Enhanced activation of hypoxia-inducible factor 1α (HIF-1α), along with the induction of nitric oxide synthase (iNOS) and vascular endothelial growth factor (VEGF) within the HIF-1 signaling pathway were other properties of IFV-mediated ferroptosis [95, 96]. ACSL4, as a crucial factor of ferroptosis, is used for IFV replication process [87]. ACSL4, present in the ER, colocalized with dsRNA (viral replication marker) and calnexin (ER marker) during CV-A6 infection, confirming its participation in the viral replication complex. Furthermore, Kung et al. reported that IFV titers decreased in ACSL4-deficient cells, underscoring the role of ACSL4 as a key host component in the virus replication process [87].
As well, key mechanisms driving ROS production after IAV infection are dysfunctional mitochondria, activated NADPH oxidase, and suppressed antioxidant pathways [57]. Oxidative stress plays a crucial role in the replication of IFV. A study by Liu et al. investigated the function of tripartite motif containing 16 (TRIM16) in relation to oxidative stress triggered by infection with the highly pathogenic H5N1 avian influenza virus [97]. Their findings revealed that, during H5N1 infection, ROS levels in A549 cells reached their highest point 24 h after infection. Additionally, there was a noted decrease in the expression levels of several antioxidant genes, including NRF2, GCLC, GCLM, SLC7A11, NADPH, GPX2, and HO-1. Moreover, they found that elevated levels of the TRIM16 protein can diminish the titer of highly pathogenic avian influenza virus and enhance the expression of antioxidant genes in A549 cells by activating the SQSTM1/NRF2/KEAP1 antioxidant pathway during H5N1 HPAIV infection. Under normal conditions, TRIM16 interacts with NRF2 and facilitates its ubiquitination by promoting K63-linked polyubiquitination. Given that SLC7A11 is closely linked to ferroptosis, TRIM16 might also influence ferroptosis in cells [97]. Oxidative stress intensifies lung injury and acute respiratory distress syndrome, but antioxidants such as GSH precursors can effectively mitigate these effects [98]. It has been suggested that the redox state of host cells can affect viral replication [99]. The replication of IFV is influenced by the redox state of host cells, as evidenced by Bcl-2 expression and GSH levels [100]. Cells with higher Bcl-2 and GSH exhibited reduced viral replication, affecting the expression of late viral proteins (HA and M) and the nuclear-cytoplasmic transport of viral ribonucleoproteins. Reduced HA expression in cells with elevated GSH levels might be related to reducing environments that disrupted disulfide bond formation, hindering proper folding and maturation of HA, and subsequently affecting its translocation and integration into the membrane [100].
Furthermore, it has been observed that ROS generation is necessary to initiate macroautophagy/autophagy in the context of IFV infection. Specifically, the IFV M2 protein triggers the formation of ROS, a crucial step to stimulate macroautophagy/autophagy [101]. Furthermore, IFV H1N1 infection causes oxidative stress and severe lung damage, with ROS generation regulated by superoxide dismutase 1 (SOD1). The study found that SOD1 is reduced during IFV infection, possibly due to enhanced autophagy, which is activated early in infection and contributes to increased ROS [102]. During an infection, autophagy can lead to the accumulation of ROS, which may trigger ferroptosis [9]. Alternatively, autophagy can facilitate ferroptosis by breaking down ferritin [103]. It was found that autophagy plays a role in ferroptosis by targeting and degrading ferritin in fibroblasts and cancer cells. When the genes Atg5 and Atg7, essential for autophagy, were disrupted, there was a reduction in ferroptosis induced by erastin, evidenced by lower levels of intracellular ferrous iron and decreased lipid peroxidation. Notably, NCOA4 emerged as a key receptor responsible for the selective autophagic degradation of ferritin, known as ferritinophagy, which is crucial for ferroptosis. Inhibition of NCOA4 led to reduced ferritin breakdown and a decrease in ferroptosis, whereas overexpression of NCOA4 enhanced ferritin degradation and intensified ferroptosis [103]. It is crucial to emphasize that autophagy often occurs alongside ferroptosis, and these processes are interconnected in various ways [32, 41, 103].
Viruses can use TfR1 as an entry receptor, taking control of iron transport to elevate iron uptake and TfR1 expression. This may contribute to iron accumulation, promoting ferroptosis and intensifying disease severity in later stages of viral infection [8]. Investigations into the entry protein involved in IFV endocytosis showed that lack of TfR1 reduces IFV entry. IFV hemagglutinin exploited TfR1 recycling as a revolving-door process to undergo host endocytosis [104]. IFV H5N1 alters the iron balance of the host cell by degrading the lysosome-associated membrane protein and releasing iron-rich lysosomal contents, which can probably trigger oxidative stress or ferroptosis [105]. During influenza infection, lung epithelial cells and the innate immune system, through macrophages, secrete interferon-induced protein 35 (IFP35), which significantly contributes to the cytokine storm triggered by the IFV infection (Fig. 2B) [106]. Dai et al. found that IFP35 is related to ferroptosis by initiating the ubiquitination and subsequent degradation of Nrf2. In vivo investigations on the involvement of IFP35 in Staphylococcal-induced lung infection demonstrated that IFP35 amplifies ferroptosis, thereby contributing to lung damage. Furthermore, knockdown of IFP35 alleviated both ferroptosis and subsequent lung injury [75]. However, in the case of IFV infection it is unclear that this IFP35 leads to ferroptosis or not and further studies are needed. As well, pro-inflammatory lipid mediators such as prostaglandin E2 (PGE2) which released from ferroptotic cells, may regulate immunity. PGE2 may negatively affect immunity against IFV by suppressing both macrophage antigen presentation and T-cell immune responses (Fig. 2B) [107, 108]. Another research examined the impact of Proanthocyanidins (PAs) on ferroptosis during an IAV infection [109]. In this study, mice were nasally inoculated with IAV to trigger acute lung injury (ALI). The infection notably exacerbated pathological damage and pulmonary edema in the mice. Firstly, the study found that IAV infection led to a reduction in GPX4 and SLC7A11 levels, while simultaneously increasing the mRNA and protein levels of ACSL4. These findings indicate that ferroptosis is heightened in IAV-induced ALI [109]. Secondly, they assess the protective impact of PAs and explore the possible mechanisms against IAV-induced ALI, which will be detailed in the following section.
Lysosomes, crucial in iron metabolism, contain a significant amount of redox-active iron [9]. Lysosomes play an active role in ferroptosis by influencing iron levels within the cell, notably through the disruption of transferrin transport and ferritin degradation. Additionally, lysosomal activity generates ROS, including lipid peroxides. Moreover, lysosomal cathepsin B has been identified as a factor in GSH depletion, and STAT3, a cathepsin B activator, may thus participate in the ferroptosis process [9]. Viral infections, such as H5N1 influenza, can damage lysosomes and release redox-active iron. It was reported that H5N1 neuraminidase can degrade lysosome-associated membrane protein (LAMP), triggering cell death [110]. Degradation of ferritin by released lysosomal enzymes leads to iron accumulation, which can cause lipid peroxidation or trigger ferroptosis [9]. Furthermore, other IFV subtypes, such as H1N1 swine influenza virus (SIV), have been shown to trigger ferroptosis [92]. In the study by Cheng et al., it was found that when A549 cells were infected with the SIV, the infection disrupted the Xc−/GPX4 axis, leading to a reduction in GSH levels and an increase in lipid peroxidation products like MDA (Fig. 2A). The SIV infection caused a significant rise in Fe2+ levels and ROS accumulation within the infected cells. The infection also caused a substantial depletion of GSH, compromising the cells’ antioxidant defenses. As a critical coenzyme for GSH reductase, NADPH is essential for maintaining GSH levels, and its decreased levels were observed in response to SIV infection. As well, in SIV infection, there was a reduction in the levels of SLC7A11, SLC3A2L, and GPX4, along with an increased expression of inflammatory cytokines such as IL-1β, TNF-α, IL-6, and IL-8 [92]. Additionally, they examined the effectiveness of Fer-1 in preventing ferroptosis, which is detailed in the following section. In a separate study aimed at assessing the antiviral properties of metastable iron sulfides (mFeS), researchers noted that IFV can trigger ferroptosis [111]. They observed that IFV causes a depletion of intracellular GSH, an increase in lipid ROS and lipid peroxidation, a reduction in GPX4 levels, and an elevation of intracellular iron during IFV infection. Furthermore, Liproxastatin-1 was found to effectively block the replication of the influenza virus within cells, suggesting that the virus’s replication process depends on cellular ferroptosis [111]. Additionally, The redox state of host cells has been linked to IAV replication, emphasizing the importance of cellular ferroptosis [100]. In this context, the study introduces the concept of Oxeiptosis, a form of ROS-induced cell death independent of known pathways, suggesting an interplay between ROS-induced cell death pathways and their role in limiting harmful inflammation [112].
In conclusion, the studies indicate that IFV significantly relies on ferroptosis to drive inflammation and enhance its replication. IFV infection triggers ferroptosis by causing oxidative stress, which leads to increased levels of reactive oxygen species ROS and intracellular iron. The interplay between ferroptosis and oxidative stress underscores the virus’s strategy to exploit these processes for its benefit. This understanding provides important insights into how IFV exacerbates inflammation and highlights potential avenues for therapeutic strategies aimed at modulating ferroptosis to mitigate the severe effects of influenza infections.
Obligate intracellular pathogens and ferroptosis
Evidence suggests that some pathogens exploit ferroptosis to enhance their replication. For instance, it was reported that intracellular pathogen could modulates host cell ferroptosis to enhance its intracellular replication and release. Brucella trigger ferroptosis in macrophages by suppressing the GPX4-GSH axis late in infection but alleviates it early on by upregulating the GCH1-BH4 axis. Manipulating host cell ferroptosis helps Brucella control its intracellular replication and release [113]. Moreover, a mycobacterial effector, tyrosine phosphatase A secreted by Mycobacterium tuberculosis (Mtb), could induces ferroptosis to enhance Mtb pathogenicity and spread [114]. Kung et al. demonstrated that ACSL4 is indispensable for the replication of Coxsackieviruses [87]. The study revealed that the virus facilitates the generation of viral replicative compartments and virus assembly by recruiting ACSL4. Moreover, the virus enhances lipid peroxidation through ACSL4, triggering ferroptosis in cells and ultimately facilitating viral release. Their research also revealed that inhibitors targeting ferroptosis, specifically ACSL4, such as rosiglitazone and pioglitazone, resulted in a reduction in the viral load of Coxsackievirus. This suggests that ACSL4-mediated ferroptosis stands out as an attractive target for the treatment of viral infections [87]. Moreover, treating macrophages from mice infected with the murine hepatitis virus strain MHV-A59 with a Fer-1 led to reduced viral replication, lower secretion of inflammatory cytokines, and decreased formation of cell syncytia [115]. Furthermore, use of liproxstatin-1 (ferroptosis inhibitor) mitigates lung inflammation and injuries. RNA-seq analysis showed that MHV-A59 infection increases ACSL1 expression, a key ferroptosis gene, independently of TLR4 but dependent on NF-kB [115]. Kan et al. demonstrated that NDV infection leads to the upregulation of p53 expression and concurrent downregulation of SLC7A11 and Gpx4 [116]. This orchestrated regulation during infection induces ferroptosis in Malignant cells, characterized by a notable elevation in the amounts of ROS and lipid peroxides. These findings provide insights into the molecular mechanisms through which NDV influences cellular processes, ultimately promoting ferroptosis in cancerous cells [116].
These examples illustrate how various obligate intracellular pathogens exploit ferroptosis pathways to enhance their replication. Understanding these mechanisms provides valuable insights that could be applied to the study of IFV. Further research into how IFV may similarly manipulate ferroptosis could reveal novel therapeutic targets and strategies to combat influenza infections.
Antiviral activity of ferroptosis inhibitors
Influenza remains a significant global health challenge, causing substantial morbidity and mortality each year. Despite advancements in antiviral therapies and vaccines, the continuous evolution of the influenza virus poses persistent threats, necessitating the exploration of novel therapeutic strategies. One such promising avenue lies in targeting cell death pathways, particularly ferroptosis. Ferroptosis inhibitors have emerged as potent therapeutic candidates in this context (Table 2). Here we describe several ferroptosis inhibitors which have a therapeutic effect against IFV.
JHU-083
JHU-083 is a small molecule compound which acts as an antagonist to glutaminase, effectively blocking glutamine metabolism. This compound has been applied in treating cancers and infections, yielding promising outcomes [117,118,119,120]. In the case of IFV infecrion researchers tested it on mice by administering it through intraperitoneal injection. JHU-083 appears to modulate the ferroptosis caused by H1N1 infection in hNECs [93]. After treatment with JHU-083, levels of AKG and NADPH dropped. The activity of nasal mucosal cells, stimulated by the H1N1 virus, increased under JHU-083 treatment, while lipid peroxidation, intracellular ROS, MDA, 4-HNE, and iron content significantly decreased. Additionally, expression levels of NRF2, KEAP1, HO-1, NQO1, SLC7A11, GPX4, GCLC, and GCLM in nasal mucosal tissue of mice exposed to JHU-083 and H1N1 were reduced. Intracellular levels of MDA and 4-HNE also significantly decreased following JHU-083 treatment. Furthermore, inflammation in the nasal mucosa and lungs was notably reduced, and the inflammatory markers IFN-α, CXCL10, and MCP-1 in nasal lavage fluid of the treated mice were significantly lowered. These findings indicate that JHU-083 effectively inhibits the ferroptosis triggered by the H1N1 virus and can also lessen the intensity of the associated inflammation [93].
Maxing shigan decoction (MXSGD)
Another notable formulation is the Maxing Shigan Decoction (MXSGD), originating from the treatise on Cold-Induced Diseases. This traditional remedy is a prescription for addressing lung ailments and is composed of a blend of various ingredients [121]. In a study led by Huang and colleagues, the effects of MXSGD were initially examined in vitro, to uncover its mechanism of action in ferroptosis triggered by IAV [95]. When cells were exposed to IAV, there was an increase in ROS release and elevated levels of total iron and ferrous ions. However, MXSGD was shown to effectively curb this ROS release and reduce both total iron and ferrous ions. The treatment with MXSGD not only decreased the intracellular viral load but also mitigated ROS levels and iron content, repaired mitochondrial damage, and suppressed cellular ferroptosis as well as the HIF-1 signaling pathway [95]. Furthermore, in the in vivo model revealed that MXSGD significantly improved pulmonary conditions in IAV-infected mice, alleviating congestion, edema, and inflammation, while also inhibiting the expression of ferroptosis-related proteins and the HIF-1 signaling pathway in lung tissues [95].
Tripartite motif containing 16 (TRIM16)
The tripartite-motif (TRIM) family is a major group of potential single-protein RING-finger E3 ubiquitin ligases, playing crucial roles in various cellular signaling pathways and biological functions. Among its members, TRIM16 stands out for its involvement in a range of processes, including cell differentiation, the innate immune response, and tumor suppressing [122,123,124,125,126]. The impact of TRIM16 on viral infections has not been extensively documented. However, Liu et al. conducted a study to investigate how TRIM16 influences oxidative stress caused by infection with the highly pathogenic H5N1 avian influenza virus [97]. In this in vitro study, a significant down-regulation of the TRIM16 protein following infection with the H5N1 subtype of the influenza virus was observed. To investigate how TRIM16 overexpression affects H5N1 replication, A549 cells were infected with the virus 24 h after being transfected with a vector expressing TRIM16. The results showed that virus titers were notably lower in the TRIM16-overexpressed cells compared to those transfected with an empty vector at both 24- and 36-hours post-infection. These findings indicate that TRIM16 overexpression can reduce the H5N1 subtype avian influenza virus titers in A549 cells. Additionally, TRIM16 overexpression enhances the expression of several antioxidant genes, including glutamate–cysteine ligase catalytic subunit (GCLC), glutamate–cysteine ligase modifier subunit (GCLM), HO-1, NADPH, and GPX2. However, this overexpression leads to a significant down-regulation in the mRNA expression level of SLC7A11. These findings suggest that TRIM16 enhances the activity of the SQSTM1/NRF2/KEAP1 pathway. Under normal conditions, TRIM16 modifies NRF2 by increasing its K63-linked poly-ubiquitination.
Proanthocyanidins (PAs)
Proanthocyanidins (PAs) are emerging as a promising option for treating influenza. They are known for their potent ability to neutralize free radicals and their strong antioxidant properties. Additionally, PAs have demonstrated noteworthy biological effects, including antitumor, antiviral, and anti-inflammatory activities [127]. In a recent in vivo study using mice, it was observed that PAs have multiple beneficial effects [109]. Initially, PAs notably improved pathological changes and inflammatory responses in lung tissues affected by IAV-induced acute lung injury (ALI). This was evident from reductions in inflammatory cell infiltration, pulmonary interstitial edema, and protein content in BALF. Moreover, PAs lowered the mRNA levels of inflammatory cytokines such as TGF-β1, IL-1β, IL-6, and TNF-α in the lungs. The expression of IFN-γ and its related chemokines (CXCL9, CXCL10, and CXCL11) was also significantly reduced by PAs, as assessed by both protein and mRNA levels [109] (Fig. 2B). Additionally, PAs appear to protect against lung damage through the TGF-β1/Smad2/3 signaling pathway. On a different note, PAs counteract IAV-induced ALI by inhibiting IFN-γ-driven ferroptosis. It was previously reported that IFN-γ treatment exacerbates glutathione depletion, increases lipid peroxidation, and improves sensitivity to ferroptosis [68]. Additionally, IAV significantly reduced both the mRNA and protein levels of GPX4 and SLC7A11, while boosting the mRNA and protein levels of ACSL4. On the other hand, PA substantially counteracted these changes. PAs effectively reduced MDA production and restored GSH levels in the mice [109].
Ferrostatin-1 (Fer-1)
Blocking the ferroptotic pathway with ferrostatin-1 (Fer-1) is another approach that has been shown to reduce both viral loads and inflammatory responses. According to an in vitro study, Fer-1 enhances cell viability compromised by SIV infection [92]. SIV infection significantly elevated Fe2+ levels in cells, but Fer-1 treatment reduced these elevated Fe2 + levels. Furthermore, SIV infection and erastin treatment led to increased ROS accumulation, whereas Fer-1 treatment mitigated this ROS generation induced by SIV. SIV infection raised MDA levels in A549 cells, but Fer-1 treatment prior to infection significantly lowered these levels. Similarly, the decrease in GSH and NADPH levels in SIV-infected cells was also countered by Fer-1 [92]. Moreover, Fer-1 partially reversed the downregulation of SLC7A11, SLC3A2, and GPX4 expression caused by SIV, indicating that SIV induces ferroptosis by inhibiting the system Xc−/GPX4 axis. Fer-1 also contributed to a reduction in viral titers. SIV-infected cells showed increased mRNA expression of IL-1β, TNF-α, IL-6, and IL-8 at 24 h post-infection, with SIV infection significantly enhancing these cytokine levels. Treatment with Fer-1 substantially reduced the production of these inflammatory cytokines in SIV-infected cells [92].
Metastable iron sulfides (mFeS)
In a separate investigation, researchers have created a novel multi-antiviral compound, metastable iron sulfides (mFeS), which targets multiple influenza A and B virus subtypes [111]. This research introduces a new concept called viral ferroptosis, which leads to a reduction in viral infectivity and pathogenicity both in vitro and in vivo (Fig. 3). The mFeS compounds trigger significant lipid peroxidation and generate hydroxyl radicals (•OH) within the viral envelope, a process that relies on Fe2+. They discovered that mFeS triggered viral ferroptosis against extracellular influenza viruses [111]. Additionally, mFeS compromised the viral lipid envelope structure, causing leakage of the virion’s internal contents. It also promoted viral lipid peroxidation by elevating MDA levels. MFeS effectively degraded HA and NA proteins in a dose-dependent manner. This mechanism of action against IAV is also observed in iron oxide nanozymes (IONzymes). IONzymes effectively inactivate IAV by inducing LPO in the viral envelope, which compromises the integrity of adjacent proteins such as hemagglutinin (HA) and neuraminidase (NA), and matrix protein 1 (M1) [128].
Also, decoction of mFeS (Dc(mFeS)) inhibited host cellular ferroptosis and curtailed intracellular influenza virus replication by restoring sulfur metabolism [111]. Experimental evidence demonstrates that mFeS-coated materials, including facemasks and protective suits, effectively inhibit several IFV subtypes even at low concentrations, demonstrating their potential as antiviral personal protective equipment (PPE) (Fig. 3). In vivo studies with mice administered mFeS&Dc showed no significant adverse effects, affirming its safety and potential therapeutic effects against IFV [111]. Atomization of mFeS further demonstrates its applicability in the treatment of respiratory diseases [129]. Similarly, using IONzymes on a facemask enhances its effectiveness in protecting against three significant IFV subtypes likes H1N1, H5N1, and H7N9 [128]. Altogether, these results demonstrate the effectiveness of ferroptosis inhibitors in the context of IFV infection, highlighting their potential as a novel therapeutic strategy. Further studies are necessary to validate their safety and efficacy particularly in the human population.

Using metastable iron sulfides (mFeS) against IFV infection. This compound offers a novel antiviral approach with broad-spectrum activity against both intracellular and extracellular influenza viruses. By significantly increasing lipid peroxidation and generating hydroxyl radicals (•OH) within the viral envelope—a process reliant on Fe2+—mFeS induces viral ferroptosis. Furthermore, mFeS effectively degrades HA and NA proteins in a dose-dependent manner. In the context of intracellular infection, decoction of mFeS (Dc(mFeS)) inhibits host cellular ferroptosis and curtails intracellular influenza virus replication by restoring sulfur metabolism. Remarkably, personal protective gear infused with mFeS offers substantial antiviral defense. Experimental evidence shows that mFeS-coated materials, including facemasks and protective suits, inhibit several IFV subtypes even at low concentrations
However, it is essential to acknowledge that IFV relies significantly on the host cell’s metabolic processes, especially lipid metabolism, to complete its replication cycle. Lipids and cellular membranes are essential at various points in the IAV lifecycle, including virus-host receptor interactions, membrane fusion, nuclear transport, virion assembly, and the budding process [130, 131]. For example, the IAV assembles and exits from particular regions of the apical plasma membrane that are rich in cholesterol and sphingolipids. These regions, known as “lipid rafts,” serve as the assembly and budding sites for various enveloped viruses [132]. Moreover, IAV structural proteins engage in specific lipid–protein interactions within infected cells. The HA and NA, are transported to the plasma membrane via vesicles rich in cholesterol and sphingomyelin [131]. Also, IAV need cholesterol in their envelopes to efficiently enter host cells [133]. These findings underscore the critical importance of controlled lipid metabolism during the replication process of IAV infection. However, research indicates that IFV can trigger oxidative stress, leading to elevated levels of free radicals or ROS. These ROS can then cause direct damage to lipids through a process known as LPO [134,135,136]. It can be suggested that this alteration in cellular lipids might affect the replication cycle of the IFV, as this virus depends on the host cell’s metabolic processes, particularly lipid metabolism, to complete its life cycle [130, 137, 138]. However, IFV have developed strategies to circumvent this issue. For example, IFV induces the host cells to produce a substantial number of lipid droplets, which may serve as precursors for the virus’s lipid components [139]. This increase in lipid droplets occurred early after the viral infection [140]. Lipid droplets are organelles encapsulated by a phospholipid monolayer and contain lipids such as triacylglycerols, cholesterol esters [139]. These droplets are produced under various conditions and are essential for the replication of positive-strand RNA viruses [139, 141]. Lipid droplets play crucial roles in processes like viral morphogenesis and the assembly and release of viral particles. Moreover, viruses can utilize the breakdown of these lipid reserves as an alternative energy source. Recent findings underscore the LDs’ influence on regulating the host’s immune response [142]. Alternatively, IFV can modify lipid synthesis pathways to its advantage. For instance, it has been observed that the IFV disrupts host cell lipid metabolism by targeting regulatory proteins like the interferon-induced viperin protein and protectin D1 (PD1). The IFV decreases the levels of both viperin and PD1 [142]. Viperin inactivates the enzyme farnesyl pyrophosphate synthase (FPPS), which is crucial for the formation of lipid rafts. The absence of these structures prevents the release of new influenza virions, thereby inhibiting the infection of additional host cells [143]. Suppression of PD1 activates lipoxygenase 12/15, an enzyme integral to the immune response and the production of proinflammatory cytokines such as IL-1β, as well as the formation of ROS by macrophages. The activation of this enzyme has been linked to increased lethality and the severity of influenza infections [142]. Thus, by modulating lipid metabolism, including the downregulation of PD1 and viperin, the influenza virus enhances its replication and pathogenicity. As well, it is reported that, IFV infection stimulates autophagy pathways, as shown by increased activity of either mTORC1 or mTORC2, resulting in the degradation of lipid droplets during the infection [142].
Conclusion
In conclusion, the intricate relationship between ferroptosis and IFV infection underscores the need for a deeper understanding of viral pathogenic mechanisms. The substantial impact of this virus on both worldwide health systems and economies underscores the need for a comprehensive strategy to address its consequences. Ferroptosis, as a cell death pathway by iron-dependent lipid peroxidation, emerges as a crucial player in host defense against viral infections. Influenza virus-induced ferroptosis disrupts iron homeostasis, leading to increased reactive oxygen species and lipid peroxidation, which contribute to cellular damage and inflammation. This link between ferroptosis and IFV pathogenesis highlights the potential for targeted therapeutic interventions. By elucidating the role of ferroptosis in viral infections, particularly IFV, researchers may identify novel strategies to mitigate viral-induced damage.
Furthermore, the development of ferroptosis inhibitors presents a promising avenue for therapeutic intervention against IFV. Targeting ferroptosis may offer a novel approach to suppress viral replication and mitigate inflammation-induced tissue damage. Therefore, leveraging our understanding of ferroptosis in the context of IFV infection has immense potential for the development of effective antiviral therapies. Future research efforts aimed at unraveling the complexities of ferroptosis in viral infections will undoubtedly pave the way for innovative therapeutic interventions that will ultimately improve outcomes for people affected by IFV.
Data availability
No datasets were generated or analysed during the current study.
References
Bolek H, Ozisik L, Caliskan Z, Tanriover MD. Clinical outcomes and economic burden of seasonal influenza and other respiratory virus infections in hospitalized adults. J Med Virol. 2023;95(1):e28153.
Petrova VN, Russell CA. The evolution of seasonal influenza viruses. Nat Rev Microbiol. 2018;16(1):47–60.
Weston S, Frieman MB. Respiratory viruses. Encyclopedia Microbiol. 2019:85.
Schaechter M. Encyclopedia of microbiology. Academic; 2009.
Leung NH. Transmissibility and transmission of respiratory viruses. Nat Rev Microbiol. 2021;19(8):528–45.
Nahand JS, Shojaie L, Akhlagh SA, Ebrahimi MS, Mirzaei HR, Baghi HB, et al. Cell death pathways and viruses: role of microRNAs. Mol Therapy-Nucleic Acids. 2021;24:487–511.
Rex DAB, Keshava Prasad TS, Kandasamy RK. Revisiting regulated cell death responses in viral infections. Int J Mol Sci. 2022;23(13):7023.
Huang R, Wu J, Ma Y, Kang K. Molecular mechanisms of Ferroptosis and its role in viral pathogenesis. Viruses. 2023;15(12):2373.
Wang M-p, Joshua B, Jin N-y, Du S-w, Li C. Ferroptosis in viral infection: the unexplored possibility. Acta Pharmacol Sin. 2022;43(8):1905–15.
Sun S, Shen J, Jiang J, Wang F, Min J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct Target Therapy. 2023;8(1):372.
Fang X, Ardehali H, Min J, Wang F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Reviews Cardiol. 2023;20(1):7–23.
Otasevic V, Vucetic M, Grigorov I, Martinovic V, Stancic A. Ferroptosis in different pathological contexts seen through the eyes of mitochondria. Oxidative Med Cell Longev. 2021;2021:1–16.
Pope LE, Dixon SJ. Regulation of ferroptosis by lipid metabolism. Trends Cell Biol. 2023.
Sun Y, Chen P, Zhai B, Zhang M, Xiang Y, Fang J, et al. The emerging role of ferroptosis in inflammation. Biomed Pharmacother. 2020;127:110108.
Barker KL, Boucher KM, Judson-Torres RL. Label-free classification of apoptosis, ferroptosis and necroptosis using digital holographic cytometry. Appl Sci. 2020;10(13):4439.
Li J, Cao F, Yin H-l, Huang Z-j, Lin Z-t, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–79.
Cui S, Niu K, Xie Y, Li S, Zhu W, Yu L, et al. Screening of potential key ferroptosis-related genes in sepsis. PeerJ. 2022;10:e13983.
Ni S, Yuan Y, Song S, Li X. A double-edged sword with a therapeutic target: iron and ferroptosis in immune regulation. Nutr Rev. 2023;81(5):587–96.
Zhao T, Guo X, Sun Y. Iron accumulation and lipid peroxidation in the aging retina: implication of ferroptosis in age-related macular degeneration. Aging Disease. 2021;12(2):529.
Qu L, He X, Tang Q, Fan X, Liu J, Lin A. Iron metabolism, ferroptosis, and lncRNA in cancer: knowns and unknowns. J Zhejiang University-SCIENCE B. 2022;23(10):844–62.
Chang S, Tang M, Zhang B, Xiang D, Li F. Ferroptosis in inflammatory arthritis: a promising future. Front Immunol. 2022;13:955069.
Rishi G, Huang G, Subramaniam VN, Cancer. The role of iron and ferroptosis. Int J Biochem Cell Biol. 2021;141:106094.
Gleason A, Bush AI. Iron and ferroptosis as therapeutic targets in Alzheimer’s disease. Neurotherapeutics. 2021;18(1):252–64.
Yan N, Zhang J. Iron metabolism, ferroptosis, and the links with Alzheimer’s disease. Front NeuroSci. 2020;13:1443.
Fuqua BK, Lu Y, Darshan D, Frazer DM, Wilkins SJ, Wolkow N, et al. The multicopper ferroxidase hephaestin enhances intestinal iron absorption in mice. PLoS ONE. 2014;9(6):e98792.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308.
Ouyang J, Zhou L, Wang Q. Spotlight on iron and ferroptosis: research progress in diabetic retinopathy. Front Endocrinol. 2023;14.
Tong S, Hong Y, Xu Y, Sun Q, Ye L, Cai J et al. TFR2 regulates ferroptosis and enhances temozolomide chemo-sensitization in gliomas. Exp Cell Res. 2023:113474.
Hassannia B, Vandenabeele P, Berghe TV. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35(6):830–49.
Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY, et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Investig. 2018;128(8):3341–55.
Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10(11):822.
Plays M, Müller S, Rodriguez R. Chemistry and biology of ferritin. Metallomics. 2021;13(5):mfab021.
Javadov S. Mitochondria and ferroptosis. Curr Opin Physiol. 2022;25:100483.
Wang P, Cui Y, Ren Q, Yan B, Zhao Y, Yu P, et al. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis. 2021;12(5):447.
Conrad M, Kagan VE, Bayir H, Pagnussat GC, Head B, Traber MG, et al. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018;32(9–10):602–19.
Su G, Yang W, Wang S, Geng C, Guan X. SIRT1-autophagy axis inhibits excess iron-induced ferroptosis of foam cells and subsequently increases IL-1Β and IL-18. Biochem Biophys Res Commun. 2021;561:33–9.
Agmon E, Solon J, Bassereau P, Stockwell BR. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep. 2018;8(1):5155.
Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med. 2020;152:175–85.
Feng H, Stockwell BR. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol. 2018;16(5):e2006203.
Latunde-Dada GO. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim et Biophys Acta (BBA)-General Subj. 2017;1861(8):1893–900.
Rodencal J, Dixon SJ. A tale of two lipids: lipid unsaturation commands ferroptosis sensitivity. Proteomics. 2023;23(6):2100308.
Liu M-r, Zhu W-t. Pei D-s. System Xc–: a key regulatory target of ferroptosis in cancer. Investig New Drugs. 2021;39(4):1123–31.
Tu H, Tang L-J, Luo X-J, Ai K-L, Peng J. Insights into the novel function of system Xc-in regulated cell death. Eur Rev Med Pharmacol Sci. 2021;25(3).
Pan Z, Van den Bossche J-L, Rodriguez-Aznar E, Janssen P, Lara O, Ates G, et al. Pancreatic acinar cell fate relies on system xC-to prevent ferroptosis during stress. Cell Death Dis. 2023;14(8):536.
Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc–. Cell Death Differ. 2020;27(2):662–75.
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc–activity. Curr Biol. 2018;28(15):2388–99. e5.
Li F-J, Long H-Z, Zhou Z-W, Luo H-Y, Xu S-G, Gao L-C. System Xc–/GSH/GPX4 axis: an important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front Pharmacol. 2022;13:910292.
Tan M, Yin Y, Ma X, Zhang J, Pan W, Tan M, et al. Glutathione system enhancement for cardiac protection: pharmacological options against oxidative stress and ferroptosis. Cell Death Dis. 2023;14(2):131.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–82.
Chen M, Shi Z, Sun Y, Ning H, Gu X, Zhang L. Prospects for anti-tumor mechanism and potential clinical application based on glutathione peroxidase 4 mediated ferroptosis. Int J Mol Sci. 2023;24(2):1607.
Feng F, He S, Li X, He J, Luo L. Mitochondria-mediated ferroptosis in diseases therapy: from molecular mechanisms to implications. Aging Disease. 2024;15(2):714.
Liu Ye, Lu S, Wu L-l, Yang L, Yang L, Wang J. The diversified role of mitochondria in ferroptosis in cancer. Cell Death Dis. 2023;14(8):519.
Li J, Jia Y-c, Ding Y-x, Bai J, Cao F, Li F. The crosstalk between ferroptosis and mitochondrial dynamic regulatory networks. Int J Biol Sci. 2023;19(9):2756.
Tian H-Y, Huang B-Y, Nie H-F, Chen X-Y, Zhou Y, Yang T, et al. The interplay between mitochondrial dysfunction and ferroptosis during ischemia-associated central nervous system diseases. Brain Sci. 2023;13(10):1367.
Huang X, Zhou Y, Li Y, Wang T, Chen Y, Zhou Y et al. Astragaloside IV inhibits inflammation caused by influenza virus via ROS/NLRP3/Caspase-1 signaling pathway. 2024.
Wang L, Cao Z, Wang Z, Guo J, Wen J. Reactive oxygen species associated immunoregulation post influenza virus infection. Front Immunol. 2022;13:927593.
Fernández-García V, González-Ramos S, Martín-Sanz P, Castrillo A, Boscá L. Unraveling the interplay between iron homeostasis, ferroptosis and extramedullary hematopoiesis. Pharmacol Res. 2022:106386.
Yu Y, Yan Y, Niu F, Wang Y, Chen X, Su G, et al. Ferroptosis: a cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discovery. 2021;7(1):193.
Tang D, Kroemer G, Kang R. Ferroptosis in immunostimulation and immunosuppression. Immunol Rev. 2023.
Ahmad S, Ahmad A, Ghosh M, Leslie CC, White CW. Extracellular ATP-mediated signaling for survival in hyperoxia-induced oxidative stress. J Biol Chem. 2004;279(16):16317–25.
Gombault A, Baron L, Couillin I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol. 2013;3:414.
Rochette L, Dogon G, Rigal E, Zeller M, Cottin Y, Vergely C. Interplay between efferocytosis and atherosclerosis. Archives of Cardiovascular Diseases; 2023.
Chen Y, Fang Z-M, Yi X, Wei X, Jiang D-S. The interaction between ferroptosis and inflammatory signaling pathways. Cell Death Dis. 2023;14(3):205.
Zhang X, Ma Y, Lv G, Wang H. Ferroptosis as a therapeutic target for inflammation-related intestinal diseases. Front Pharmacol. 2023;14:1095366.
Deng L, He S, Guo N, Tian W, Zhang W, Luo L. Molecular mechanisms of ferroptosis and relevance to inflammation. Inflamm Res. 2023;72(2):281–99.
Chen X, Kang R, Kroemer G, Tang D. Ferroptosis in infection, inflammation, and immunity. J Exp Med. 2021;218(6):e20210518.
Kong R, Wang N, Han W, Bao W, Lu J. IFNγ-mediated repression of system xc – drives vulnerability to induced ferroptosis in hepatocellular carcinoma cells. J Leukoc Biol. 2021;110(2):301–14.
Yan Q, Zheng W, Jiang Y, Zhou P, Lai Y, Liu C, et al. Transcriptomic reveals the ferroptosis features of host response in a mouse model of Zika virus infection. J Med Virol. 2023;95(1):e28386.
Mellors J, Tipton T, Longet S, Carroll M. Viral evasion of the complement system and its importance for vaccines and therapeutics. Front Immunol. 2020;11:1450.
Yi L, Yang Y, Hu Y, Wu Z, Kong M, Zuoyuan B, et al. Complement components regulates ferroptosis in CVB3 viral myocarditis by interatction with TFRC. Free Radical Biology and Medicine; 2023.
Ilbäck N-G, Frisk P, Mohamed N, Gadhasson I-L, Blomberg J, Friman G. Virus induces metal-binding proteins and changed trace element balance in the brain during the course of a common human infection (coxsackievirus B3) in mice. Sci Total Environ. 2007;381(1–3):88–98.
Pizzimenti S, Ribero S, Cucci MA, Grattarola M, Monge C, Dianzani C, et al. Oxidative stress-related mechanisms in melanoma and in the acquired resistance to targeted therapies. Antioxidants. 2021;10(12):1942.
Kuang F, Liu J, Tang D, Kang R. Oxidative damage and antioxidant defense in ferroptosis. Front cell Dev Biology. 2020;8:586578.
Dai M, Ouyang W, Yu Y, Wang T, Wang Y, Cen M et al. IFP35 aggravates Staphylococcus aureus infection by promoting Nrf2-regulated ferroptosis. J Adv Res. 2023.
Li X, Chen J, Yuan S, Zhuang X, Qiao T. Activation of the P62-Keap1‐NRF2 pathway protects against Ferroptosis in Radiation‐Induced Lung Injury. Oxidative Med Cell Longev. 2022;2022(1):8973509.
Lee J, Homma T, Kurahashi T, Kang ES, Fujii J. Oxidative stress triggers lipid droplet accumulation in primary cultured hepatocytes by activating fatty acid synthesis. Biochem Biophys Res Commun. 2015;464(1):229–35.
Geltinger F, Schartel L, Wiederstein M, Tevini J, Aigner E, Felder TK, et al. Friend or foe: lipid droplets as organelles for protein and lipid storage in cellular stress response, aging and disease. Molecules. 2020;25(21):5053.
Feng S, Tang D, Wang Y, Li X, Bao H, Tang C, et al. The mechanism of ferroptosis and its related diseases. Mol Biomed. 2023;4(1):33.
Cao D, Khanal S, Wang L, Li Z, Zhao J, Nguyen LN, et al. A matter of life or death: productively infected and bystander CD4 T cells in early HIV infection. Front Immunol. 2021;11:626431.
Xiao Q, Yan L, Han J, Yang S, Tang Y, Li Q et al. Metabolism-dependent ferroptosis promotes mitochondrial dysfunction and inflammation in CD4 + T lymphocytes in HIV-infected immune non-responders. EBioMedicine. 2022;86.
Kannan M, Sil S, Oladapo A, Thangaraj A, Periyasamy P, Buch S. HIV-1 Tat-mediated microglial ferroptosis involves the miR-204–ACSL4 signaling axis. Redox Biol. 2023;62:102689.
Xu M, Kashanchi F, Foster A, Rotimi J, Turner W, Gordeuk VR, et al. Hepcidin induces HIV-1 transcription inhibited by ferroportin. Retrovirology. 2010;7:1–16.
Chang H-C, Bayeva M, Taiwo B, Palella FJ Jr, Hope TJ, Ardehali H. High cellular iron levels are associated with increased HIV infection and replication. AIDS Res Hum Retroviruses. 2015;31(3):305–12.
Qiu B, Zandkarimi F, Saqi A, Castagna C, Tan H, Sekulic M, et al. Fatal COVID-19 pulmonary disease involves ferroptosis. Nat Commun. 2024;15(1):3816.
Li Q, Chen Z, Zhou X, Li G, Zhang C, Yang Y. Ferroptosis and multi-organ complications in COVID-19: mechanisms and potential therapies. Front Genet. 2023;14:1187985.
Kung Y-A, Chiang H-J, Li M-L, Gong Y-N, Chiu H-P, Hung C-T, et al. Acyl-coenzyme a synthetase long-chain family member 4 is involved in viral replication organelle formation and facilitates virus replication via ferroptosis. Mbio. 2022;13(1):e02717–21.
Tripathi S, Batra J, Cao W, Sharma K, Patel J, Ranjan P, et al. Influenza a virus nucleoprotein induces apoptosis in human airway epithelial cells: implications of a novel interaction between nucleoprotein and host protein Clusterin. Cell Death Dis. 2013;4(3):e562–e.
Mosavi SZ, Shahsavandi S, Ebrahimi MM, Hatami AR, Sadeghi K, Shahivandi H. Necrotic response to low pathogenic H9N2 influenza virus in chicken hepatoma cells. Jundishapur J Microbiol. 2015;8(1).
Hartmann BM, Albrecht RA, Zaslavsky E, Nudelman G, Pincas H, Marjanovic N, et al. Pandemic H1N1 influenza a viruses suppress immunogenic RIPK3-driven dendritic cell death. Nat Commun. 2017;8(1):1931.
Gannagé M, Dormann D, Albrecht R, Dengjel J, Torossi T, Rämer PC, et al. Matrix protein 2 of influenza a virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. 2009;6(4):367–80.
Cheng J, Tao J, Li B, Shi Y, Liu H. Swine influenza virus triggers ferroptosis in A549 cells to enhance virus replication. Virol J. 2022;19(1):104.
Liu C, Wu X, Bing X, Qi W, Zhu F, Guo N, et al. H1N1 influenza virus infection through NRF2-KEAP1-GCLC pathway induces ferroptosis in nasal mucosal epithelial cells. Free Radic Biol Med. 2023;204:226–42.
Waqas FH, Shehata M, Elgaher WA, Lacour A, Kurmasheva N, Begnini F, et al. NRF2 activators inhibit influenza a virus replication by interfering with nucleo-cytoplasmic export of viral RNPs in an NRF2-independent manner. PLoS Pathog. 2023;19(7):e1011506.
Huang J, Xinyue M, Zexuan L, Zhuolin L, Kangyu W, Zhiying F, et al. Network pharmacology and experimental validation of maxing Shigan decoction in the treatment of influenza virus-induced ferroptosis. Chin J Nat Med. 2023;21(10):775–88.
Lin Z, Liu J, Kang R, Yang M, Tang D. Lipid metabolism in ferroptosis. Adv Biology. 2021;5(8):2100396.
Liu Y, Wei Y, Zhou Z, Gu Y, Pang Z, Liao M, et al. Overexpression of TRIM16 reduces the Titer of H5N1 highly pathogenic avian influenza virus and promotes the expression of antioxidant genes through regulating the SQSTM1-NRF2-KEAP1 Axis. Viruses. 2023;15(2):391.
Zheng Y, Huang Y, Xu Y, Sang L, Liu X, Li Y. Ferroptosis, pyroptosis and necroptosis in acute respiratory distress syndrome. Cell Death Discovery. 2023;9(1):91.
Checconi P, De Angelis M, Marcocci ME, Fraternale A, Magnani M, Palamara AT, et al. Redox-modulating agents in the treatment of viral infections. Int J Mol Sci. 2020;21(11):4084.
Nencioni L, Iuvara A, Aquilano K, Ciriolo MR, Cozzolino F, Rotilio G, et al. Influenza a virus replication is dependent on an antioxidant pathway that involves GSH and Bcl-2. FASEB J. 2003;17(6):758–60.
Wang R, Zhu Y, Lin X, Ren C, Zhao J, Wang F, et al. Influenza M2 protein regulates MAVS-mediated signaling pathway through interacting with MAVS and increasing ROS production. Autophagy. 2019;15(7):1163–81.
Jung KI, Pyo CW, Choi S-Y. Influenza a virus-induced autophagy contributes to enhancement of virus infectivity by SOD1 downregulation in alveolar epithelial cells. Biochem Biophys Res Commun. 2018;498(4):960–6.
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ III, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–8.
Mazel-Sanchez B, Niu C, Williams N, Bachmann M, Choltus H, Silva F, et al. Influenza a virus exploits transferrin receptor recycling to enter host cells. Proc Natl Acad Sci. 2023;120(21):e2214936120.
Dou J, Liu X, Yang L, Huang D, Tan X. Ferroptosis interaction with inflammatory microenvironments: mechanism, biology, and treatment. Biomed Pharmacother. 2022;155:113711.
Yu Y, Xu N, Cheng Q, Deng F, Liu M, Zhu A et al. IFP35 as a promising biomarker and therapeutic target for the syndromes induced by SARS-CoV-2 or influenza virus. Cell Rep. 2021;37(12).
Shi L, Liu Y, Li M, Luo Z. Emerging roles of ferroptosis in the tumor immune landscape: from danger signals to anti-tumor immunity. FEBS J. 2022;289(13):3655–65.
Xu X, Lin D, Tu S, Gao S, Shao A, Sheng J. Is ferroptosis a future direction in exploring cryptococcal meningitis? Front Immunol. 2021;12:598601.
Lv Y-w, Du Y, Ma S-s, Shi Y-c, Xu H-c, Deng L, et al. Proanthocyanidins attenuates ferroptosis against influenza-induced acute lung injury in mice by reducing IFN-γ. Life Sci. 2023;314:121279.
Ju X, Yan Y, Liu Q, Li N, Sheng M, Zhang L, et al. Neuraminidase of influenza a virus binds lysosome-associated membrane proteins directly and induces lysosome rupture. J Virol. 2015;89(20):10347–58.
Miao X, Yin Y, Chen Y, Bi W, Yin Y, Chen S et al. Bidirectionally regulating viral and Cellular Ferroptosis with Metastable Iron Sulfide against Influenza Virus. Adv Sci. 2023:2206869.
Holze C, Michaudel C, Mackowiak C, Haas DA, Benda C, Hubel P, et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol. 2018;19(2):130–40.
Zhang G, Hu H, Yin Y, Tian M, Bu Z, Ding C, et al. Brucella Manipulates Host Cell Ferroptosis to facilitate its intracellular replication and egress in RAW264. 7 Macrophages. Antioxidants. 2024;13(5):577.
Qiang L, Zhang Y, Lei Z, Lu Z, Tan S, Ge P, et al. A mycobacterial effector promotes ferroptosis-dependent pathogenicity and dissemination. Nat Commun. 2023;14(1):1430.
Xia H, Zhang Z, You F. Inhibiting ACSL1-related ferroptosis restrains murine coronavirus infection. Viruses. 2021;13(12):2383.
Kan X, Yin Y, Song C, Tan L, Qiu X, Liao Y et al. Newcastle-disease-virus-induced ferroptosis through nutrient deprivation and ferritinophagy in tumor cells. Iscience. 2021;24(8).
Yamashita AS, da Costa Rosa M, Stumpo V, Rais R, Slusher BS, Riggins GJ. The glutamine antagonist prodrug JHU-083 slows malignant glioma growth and disrupts mTOR signaling. Neuro-oncology Adv. 2021;3(1):vdaa149.
Praharaj M, Shen F, Lee AJ, Zhao L, Nirschl TR, Theodros D et al. Metabolic reprogramming of tumor-associated macrophages using glutamine antagonist JHU083 drives tumor immunity in myeloid-rich prostate and bladder cancers. Cancer Immunol Res. 2024:OF1–22.
Zhang G-Q, Xi C, Ju N-T, Shen C-T, Qiu Z-L, Song H-J et al. Targeting glutamine metabolism exhibits anti-tumor effects in thyroid cancer. J Endocrinol Investig. 2024:1–17.
Bell BJ, Hollinger KR, Deme P, Sakamoto S, Hasegawa Y, Volsky D et al. Glutamine antagonist JHU083 improves psychosocial behavior and sleep deficits in EcoHIV-infected mice. Brain, Behavior, & Immunity-Health. 2022;23:100478.
Xu Y, Bao L, Cao S, Pang B, Zhang J, Zhang Y, et al. Pharmacological effects and mechanism of maxing Shigan decoction in the treatment of Pseudomonas aeruginosa pneumonia. J Ethnopharmacol. 2024;320:117424.
Huang N, Sun X, Li P, Liu X, Zhang X, Chen Q, et al. TRIM family contribute to tumorigenesis, cancer development, and drug resistance. Experimental Hematol Oncol. 2022;11(1):75.
Marshall G, Bell J, Koach J, Tan O, Kim P, Malyukova A, et al. TRIM16 acts as a tumour suppressor by inhibitory effects on cytoplasmic vimentin and nuclear E2F1 in neuroblastoma cells. Oncogene. 2010;29(46):6172–83.
Jena KK, Kolapalli SP, Mehto S, Nath P, Das B, Sahoo PK, et al. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 2018;37(18):e98358.
Roshanazadeh MR, Adelipour M, Sanaei A, Chenane H, Rashidi M. TRIM3 and TRIM16 as potential tumor suppressors in breast cancer patients. BMC Res Notes. 2022;15(1):312.
Munding C, Keller M, Niklaus G, Papin S, Tschopp J, Werner S, et al. The estrogen-responsive B box protein: a novel enhancer of interleukin-1β secretion. Cell Death Differ. 2006;13(11):1938–49.
Qin Z, Liu H-M, Ma Y-X, Wang X-D. Developments in extraction, purification, and structural elucidation of proanthocyanidins (2000–2019). Stud Nat Prod Chem. 2021;68:347–91.
Qin T, Ma R, Yin Y, Miao X, Chen S, Fan K, et al. Catalytic inactivation of influenza virus by iron oxide nanozyme. Theranostics. 2019;9(23):6920.
Leyva-Grado VH, Palese P. Aerosol administration increases the efficacy of oseltamivir for the treatment of mice infected with influenza viruses. Antiviral Res. 2017;142:12–5.
Limsuwat N, Boonarkart C, Phakaratsakul S, Suptawiwat O, Auewarakul P. Influence of cellular lipid content on influenza a virus replication. Arch Virol. 2020;165:1151–61.
Petrich A, Chiantia S. Influenza a virus infection alters lipid packing and surface electrostatic potential of the host plasma membrane. Viruses. 2023;15(9):1830.
Dou D, Revol R, Östbye H, Wang H, Daniels R. Influenza a virus cell entry, replication, virion assembly and movement. Front Immunol. 2018;9:1581.
Heaton NS, Randall G. Multifaceted roles for lipids in viral infection. Trends Microbiol. 2011;19(7):368–75.
Su L-J, Zhang J-H, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxidative Med Cell Longev. 2019;2019(1):5080843.
Liu M, Chen F, Liu T, Chen F, Liu S, Yang J. The role of oxidative stress in influenza virus infection. Microbes Infect. 2017;19(12):580–6.
Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy‐2‐nonenal. Oxidative Med Cell Longev. 2014;2014(1):360438.
Zhou Y, Pu J, Wu Y. The role of lipid metabolism in influenza a virus infection. Pathogens. 2021;10(3):303.
Kawabata K, Sato Y, Kubo T, Tokumura A, Nishi H, Morimoto K. Phospholipid analysis of two influenza a virus-infected cell lines differing in their viral replication kinetics. Arch Virol. 2023;168(5):132.
Episcopio D, Aminov S, Benjamin S, Germain G, Datan E, Landazuri J, et al. Atorvastatin restricts the ability of influenza virus to generate lipid droplets and severely suppresses the replication of the virus. FASEB J. 2019;33(8):9516.
Monson E, Crosse K, Duan M, Chen W, O’shea R, Wakim L, et al. Intracellular lipid droplet accumulation occurs early following viral infection and is required for an efficient interferon response. Nat Commun. 2021;12(1):4303.
Girdhar K, Powis A, Raisingani A, Chrudinová M, Huang R, Tran T, et al. Viruses and metabolism: the effects of viral infections and viral insulins on host metabolism. Annual Rev Virol. 2021;8(1):373–91.
Herrera-Moro Huitron L, De Jesús-González LA, Martínez-Castillo M, Ulloa-Aguilar JM, Cabello-Gutierrez C, Helguera-Repetto C, et al. Multifaceted nature of lipid droplets in viral interactions and Pathogenesis. Microorganisms. 2023;11(7):1851.
Li Y-J, Chen C-Y, Yang J-H, Chiu Y-F. Modulating cholesterol-rich lipid rafts to disrupt influenza a virus infection. Front Immunol. 2022;13:982264.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
A.L and Z.T: Writing original draft, Investigation, Review and editing. OS.A, S.A: Review and editing. A.QN, N.J, R.S: Writing original draft. R.R: Review and editing. J.Y, L.S: Supervision, Review and editing.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Letafati, A., Taghiabadi, Z., Ardekani, O.S. et al. Unveiling the intersection: ferroptosis in influenza virus infection. Virol J 21, 185 (2024). https://doi.org/10.1186/s12985-024-02462-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-024-02462-3