
Abstract
Collagen is the major component of the tumor microenvironment and participates in cancer fibrosis. Collagen biosynthesis can be regulated by cancer cells through mutated genes, transcription factors, signaling pathways and receptors; furthermore, collagen can influence tumor cell behavior through integrins, discoidin domain receptors, tyrosine kinase receptors, and some signaling pathways. Exosomes and microRNAs are closely associated with collagen in cancer. Hypoxia, which is common in collagen-rich conditions, intensifies cancer progression, and other substances in the extracellular matrix, such as fibronectin, hyaluronic acid, laminin, and matrix metalloproteinases, interact with collagen to influence cancer cell activity. Macrophages, lymphocytes, and fibroblasts play a role with collagen in cancer immunity and progression. Microscopic changes in collagen content within cancer cells and matrix cells and in other molecules ultimately contribute to the mutual feedback loop that influences prognosis, recurrence, and resistance in cancer. Nanoparticles, nanoplatforms, and nanoenzymes exhibit the expected gratifying properties. The pathophysiological functions of collagen in diverse cancers illustrate the dual roles of collagen and provide promising therapeutic options that can be readily translated from bench to bedside. The emerging understanding of the structural properties and functions of collagen in cancer will guide the development of new strategies for anticancer therapy.
Similar content being viewed by others

Background
Cancer continues to receive increasing attention from the academic community because it was the third most common cause of death worldwide in 2018. A total of 18.1 million new cancer cases and 9.6 million cancer deaths were evaluated in 2018 [1], and there are predicted to be 1,762,450 additional cancer cases and 606,880 cancer deaths in the United States in 2019 [2]. Despite various cancer-related guidelines for diagnosis, treatment, and follow-up, improving the long-term prognoses of certain cancer patients remains difficult. Cancer treatment strategies with highly effective response rates still need to be explored. An increasing amount of recent research has concentrated on the function of the tumor microenvironment in favoring cancer progression. In addition, cancer cells exhibit multiple hallmarks of cancer progression, including the recruitment of various cells to form a tumor microenvironment [3], which consists of varying functional stromal cell subtypes and matrix protein polymers [4]. The most abundant matrix protein polymers are collagens, which increase tumor tissue stiffness, regulate tumor immunity, and promote metastasis [5, 6]. In addition, extensive collagen deposition is the main pathological characteristic of some cancers, for which sufficient therapeutic applications are lacking, resulting in the poor survival outcomes of patients [7]. Herein, we summarize the current understanding of the key basic and clinical functions of collagen in cancer and provide clues regarding promising treatments for modifying the tumor matrix.
Physiological and physicochemical properties of collagen
Collagen is a type of right-handed helix glycoprotein that contains three homologous or nonhomologous left-handed helix α chains. These α chain amino acid sequences are characterized by glycine–X–Y repeats with or without interruptions, with X and Y most likely being proline or hydroxyproline, and the hydroxyproline content of collagen contributes to its thermal stability [8].
Nascent α chains by different genes are encoded first to compose the N-terminus. The next step of assembly into a three-helix structure begins with the C-terminus of the nascent α chains to form procollagen, which is accompanied by certain chaperone proteins including heat shock protein 47, prolyl-hydroxylase, and protein disulfide isomerase to ensure precise alignment [9]. Hydroxylation and glycosylation in the endoplasmic reticulum are two main modifications that occur after translation, and the hydroxylation modification is regulated by vitamin C and pyruvate metabolism [10, 11]. Then, procollagen is hydrolyzed to form collagen by procollagen N-proteinase and C-proteinase within Ca2+ surrounding the endoplasmic reticulum along with the chaperone heat shock protein 47 and protein disulfide isomerase. This important hydrolysis reaction is the rate-limiting step of collagen biosynthesis. In addition, endopeptidases and metalloproteinases can also excise procollagen at both the N-terminus and C-terminus, and the removed propeptides can conversely regulate the amount of procollagen, further influencing collagen production [12, 13].
Collagen is released into the extracellular matrix (ECM) to form a fibril supramolecular assembly that may start in Golgi-to-membrane carriers after procollagen excision or be localized at the plasma membrane of fibroblasts. The stability of collagen assembly is influenced by intramolecular and intermolecular linkages, particularly covalent linkages, chiefly including lysyl oxidase (LOX) crosslinks [14], glycosylation crosslinks [15], and transglutaminase crosslinks [16], which vary across collagen types.
Different collagens in the ECM are finally degraded by various matrix metalloproteinases (MMPs) belonging to the zinc-dependent endopeptidase family, by proline oxidase, or by sheddases that release the soluble ectodomain of membrane collagens [17, 18].
In general, the 28 known collagen types are classified into four subfamilies on the basis of their supramolecular assemblies, including fibril-forming collagens (I, II, III, V, XI, XXVI, XXVII); fibril-associated collagens with interrupted triple helices (FACITs: IX, XII, XIV, XVI, XIX, XX, XXI, XXII, XXIV), which characteristically link to the surface of collagen fibrils rather than form fibrils by themselves; network-forming collagens (IV, VIII, X), which characteristically generate noncollagenous C-terminal domain dimers and N-terminal 7S domain tetramers; and membrane-anchored collagens (MACITs: XIII, XVII, XXIII, XXV) [19]. Among these types, COLI, COLIII, and COLV are mainly produced by fibroblasts, while COLIV is predominantly expressed by epithelial and endothelial cells. Notably, cancer cells and tumor-associated macrophages also produce collagen under some circumstances [20, 21].
Cancer cells influence collagen formation
During the occurrence and development of cancer, the ECM undergoes structural changes. In cancer cells, the content and distribution of collagen is modified to further coordinate cancer cell biological properties, including various gene mutations, transcription factors, signal transduction pathways, and receptors.
The heterogeneity of mutated genes is one of the major promoters for cancer cell behavior and influences the interaction between cancer cells and ECM components. The mutation of oncogenes, which are mainly divided into tumor suppressor genes and proto-oncogenes, also alters the collagen conditions in the tumor matrix.
The content and architecture of collagen are strongly altered by mutated tumor suppressor genes in cancer cells. The p53 pathway regulates the formation of tumor-associated collagen signature-3, which is referred to as a collagen bundle angled 60° to 90° relative to the cancer border and is indicated by the proliferation and invasion of cancer [22]. Mutated p53 in cancer cells, along with the activation of Janus kinase 2-signal transducers and activators of transcription (STAT) 3 signaling, influences the collagen production response to paracrine stimulation from pancreatic stellate cells [23]. In addition, the effects of collagen resolvents are associated with p53. The extracellular collagen-derived antiangiogenic factor Arresten, which is located in the C-terminal noncollagenous domain of COL4A1, has been linked to p53 activation [24]. The p53 gene upregulated collagen prolyl-hydroxylase to potentiate the production of full-length COL4A1, further enhancing the content of Arresten [25]. Collagen closely interacts with not only p53 but also other tumor suppressor genes associated with cancer processes. Cancer progression can be regulated by deleting a single copy of the phosphate and tension homology deleted on chromosome ten (PTEN) gene or by completely silencing this gene, resulting in the increased recruitment of cancer-associated fibroblasts (CAFs) and production of COL1A1 [26, 27].
Consistently, mutated proto-oncogenes combine with collagen to support cancer progression. Mutant Kras together with the epithelial-mesenchymal transition (EMT) regulator Snail enhanced collagen production by pancreatic cancer stellate cells, and silencing Kras expression markedly decreased COLI deposition in renal fibrosis [28, 29].
Transcription factors can lead to aberrant target gene expression and tumorigenesis, and nuclear factor kappa-B (NF-κB) and STATs mostly participate in collagen expression and organization. For example, COL2A1 was shown to be under the transcriptional control of the NF-κB subunit p65 in sarcomatous [30]. Collagen fibers showed less parallel alignment, less skewed distribution, and more direction variation rather than decreased numbers following combination treatment with the Janus kinase/STAT3 inhibitor AZD1480 and gemcitabine for pancreatic cancer [31].
Cancer cells further communicate with collagen via signaling pathways during the processes of cellular metabolism, proliferation, differentiation, and apoptosis. Transforming growth factor-β (TGF-β)/Smad signaling is a typical component of serine/threonine kinase signal transduction. Accumulating studies have revealed a positive role of TGF-β/Smad signaling in collagen modification. The architecture and mechanics of collagen fibers adjacent to epithelial lesions, rather than abundant bulk collagen, transformed the pancreatic epithelium into stiff fibrotic tissue via nonfunctional Smad4-phosphorylated myosin light chain 2 [32]. TGF-β sometimes reverses cancer cell functions via collagen. Collagen stiffness induced melanoma differentiation through the Yes-associated protein (YAP)/pax3/microphthalmia-associated transcription factor (MITF) axis, but in the presence of fibroblasts, TGF-β suppressed YAP/pax3/MITF expression and induced YAP/transcriptional enhanced associate domain/Smad-driven transcription, leading to dedifferentiation [33].
Other signaling pathways also affect collagen within cancer cells. Crosstalk between TGF-β and the Ras-Raf-mitogen-activated protein kinase (MEK)-extracellular signal-regulated kinase (ERK) signaling pathway increased collagen synthesis along with p38 activation in melanoma cells to promote cancer progression [34]. Overexpression of tRNA Meti increased the production of collagens and collagen-processing enzymes, especially COLII, forming a protumorigenic ECM [35].
Tyrosine kinase receptors are one category of principle collagen-related receptors that are expressed in various cancers. Fibroblast growth factor receptor (FGFR) 4-R388, in which Gly388 in the FGFR4 transmembrane domain was replaced with arginine, regulated the degradation of COLI, COLII, and COLIV by increasing MMP-14 protein expression in prostate cancer cells, especially within the tumor and in the fibrous capsule around the cancer [36]. The effect of epidermal growth factor receptor (EGFR) on collagen remains to be further studied. Collagen was reduced in recurrent breast cancer by combinatorial treatment with FGFR- and EGFR-specific inhibitors, similar to the effect of this treatment on primary tumors [37]. In contrast, c-Met expression, rather than EGFR expression, colocalized with abundant COLI in pulmonary adenocarcinoma [38].
The G protein family receptors, especially small G proteins, including Ras and Rho members, are important in collagen fiber properties and production. G proteins can promote matrix stiffness due to their collagen alignment change. The high stiffness increases nuclear localization of the transcription factor Twist1 by further reducing the expression of the cytoplasmic binding partner Ras-GTPase-activating SH3 domain-binding protein 2 to induce cancer EMT, invasion, and metastasis [39]. Cell division cycle 42, a member of the Rho family, was shown to regulate the thickness and contractility of collagen with the activation of MMP-9 [40]. Rho-associated coiled-coil kinase (ROCK)/Rho signaling may communicate with collagen directly through fibroblasts to regulate cancer cell behavior. At least two interconvertible types of cancer cell migratory motility were shown to be regulated by adhesion to collagen: mesenchymal motility was dependent on integrin and MMPs with Ras-related C3 botulinum toxin substrate 1 (RAC1) signaling and caused cells to appear elongated and bipolar, while amoeboid motility was dependent upon the ROCK/Rho kinase and caused cells to appear round, further leading to myosin-II light chain phosphorylation and actomyosin shrinkage [41]. The ROCK/Rho signaling pathway also influences collagen by other mechanisms. Acetylation of the COL1A1 gene promoter was facilitated by ROCK/Rho signaling pathways in breast cancer cells [42]. In addition, in pancreatic ductal adenocarcinoma (PDAC), collagen impairment via ROCK inhibition was independent of changes in fibroblast proliferation and survival [43]. Notably, the three‐dimensional collagen matrix was remodeled by PDAC cells, possibly by the fusion of ROCK with estrogen receptor (ER) causing increases in MMP-10 and MMP-13 [44].
The influence of collagen on cancer cell behavior
Cellular behavior is controlled by cell signal transduction pathways. Cells accept external signals through receptors and transmit them by cascade, which then transform extracellular signals into intracellular signals, causing physiological cellular reactions that regulate biological activities. Collagen, a component of the ECM, also influences cancer cell behavior (Fig. 1). Cancer cells reversely reshape collagen to form a reinforcing cell-collagen loop, which gradually fosters cancer progression.

The contribution of collagen to cancer cells
Collagen interacts with cancer cells mainly by directly connecting to cancer cell receptors. Discoidin domain receptors (DDRs) are a subfamily of tyrosine kinases that are divided into homologous DDR1 and DDR2 receptors. Collagens closely associate with preferred DDRs, such as COLIV with DDR1 and COLII and COLX with DDR2. A COLIV-DDR1-MMP-9-COLIV feed-forward loop was shown to promote the migration and adhesion of myeloid leukemia cells in bone marrow by activating AKT [45]. DDR1b phosphorylated at Tyr513 by COLI, as opposed to DDR1a, interacted with the signaling adaptor Src homolog 1 to affect focal adhesion kinase (FAK)-related protein-tyrosine kinase, resulting in N-cadherin upregulation in both primary and metastatic PDAC cells to induce EMT [46]. Others further reported that COLI activated DDR2 rather than integrin or TGF-βR to stimulate ERK2 in a Src-dependent manner; activated DDR2 then phosphorylated Snail1 at S82 and S104 and inhibited glycogen synthase kinase (GSK) 3β activity, ultimately contributing to sustained MMP-14 and collagen synthesis in breast cancer [47]. Nevertheless, DDR2 activated by collagen was not conducive to Src homolog 2 domain phosphorylation [48]. Notably, the binding of COL11A1 to both α1β1 integrin and DDR2 to activate the Src-phosphatidylinositol 3-kinase (PI3K)/AKT-NFκB signaling pathways induced the expression of three cisplatin-induced apoptosis inhibitors in ovarian cancer [49].
Adhesion between collagen and cancer cells, such as the adhesion of COLI and COLIV to cancer cells, impacts cancer progression [50]. The cadherin family represents one typical cell adhesion molecule. COLI stimulated E-cadherin upregulation to facilitate the migration of PDAC cells [51]. However, other studies on PDAC have reported the opposite effect of COLI on E-cadherin via different signaling pathways; Smad-interacting protein 1, a member of the small Zfh-1 family that acts as a transcriptional repressor, was induced by COLI to downregulate E-cadherin by simultaneously binding to two defined DNA target sites at E-boxes of the E-cadherin promoter through two zinc-fingers clusters [52]. The COLIV-regulating chemokine (C-C motif) ligand (CCL) 5 and CCL7 were associated with the alteration of E-cadherin to influence EMT, further promoting liver metastasis [53]. COLIV not only promoted a decrease in E-cadherin expression, an increase in N-cadherin expression, and upregulation of Snail1, Snail2, and Sip1 (E-cadherin transcriptional repressors that bind at E-boxes of the E-cadherin promoter) but also induced FAK and ERK1/2 activation in affiliation with TGF-β during EMT, resulting in increased MMP-2 secretion and enhanced cell migration [54]. In addition, the mediation of prostate cancer metastasis by COLXXIII corresponds to changes in OB-cadherin, α-catenin, β-catenin, γ-catenin, vimentin, and galectin-3 protein expression [55].
Integrin, a typical adhesion molecule in cancer cells, often mediates cancer cell behavior, especially when combined with collagen. Integrin comprises two units: α and β. Different types of collagen bind to various integrins in numerous signaling pathways in cancer cells. The binding of integrin to collagen led to the activation of AKT/PI3K signaling, mitogen-activated protein kinase (MAPK) signaling, and Rho family signaling, and the MEK/ERK signaling pathway especially regulated αv integrin subfamily members such as αvβ3 and αvβ5, inducing the proliferation and invasion of squamous cell carcinoma (SCC) cells [56]. Additionally, the deposition of collagen through integrin-regulated ROCK, FAK, and AKT activation inactivated GSK3β and increased the nuclear localization of the mechanotranscription coactivator β-catenin to promote cutaneous SCC progression [57]. Various experiments have further revealed the effects of specific types of collagen in combination with integrin on cancer. COLI is a typical interstitial matrix collagen via integrin to induce cancer cell behavior. Remodeled COLI affected the invasion of ovarian cancer cells by mediating the integrin-PTEN/PI3K/AKT signaling pathway [58]. COLI and α2β1 integrin-promoting cathepsin B-mediated invasiveness were associated with secreted acidic and cysteine-rich proteins in melanoma [59]. The mediation of COLI by α2 integrin led to EMT-like changes, such as downregulated E-cadherin and β-catenin expression, decreased differentiation, increased clonogenicity, and increased colorectal cancer stem cells [60]. The expression of αv integrin response to COLI was enhanced by melanoma cells to promote the upregulation of protein kinase C (PKC) α, thereby relocating endogenous p53 protein [61]. During its adhesion to COLI, mda-9/syntenin at the plasma membrane facilitated processes in the formation of β1 integrin signaling complexes, including the assembly of the integrin-linked kinase (ILK)-PINCH1-α-parvin complex and its translocation to the cell membrane, leading to the activation of AKT, RAC1, and ERK1/2 to promote cancer metastasis [62]. The linearization and matrix compaction of COLI, via β1 integrin-FAK signaling modulated myosin IIA, was exhibited by most radiation-induced breast cancer cells [63]. COLIV accounts for the basement membrane. The high-density COLIV matrix induced the formation of cancer cell invadopodia and actin-rich proteolytic protrusions, which locally degraded collagen via αvβ3 integrin [64, 65]. The COLIV/β1 integrin signaling pathway significantly stimulated Src and ERK phosphorylation, reducing cell stiffness and accelerating cell motility [66]. Ras GTPases, Rac GTPases, PI3K, and PKC participated in melanoma cell migration mediated by COLIV/β1 integrin [67]. In soft-tissue sarcoma, the interplay of COLVI and NG2 triggered PI3K activation through α2β1 integrin, which was associated with adhesion, survival, aggregation, and migration and did not directly influence cell mitosis [68]. Both matrix collagen and basement membrane collagen communicate with integrin to impact cancer cell behavior, and numerous other collagens also bind to integrin to regulate cancer progression. COLXIII in breast cancer [69], COLXVI in glioblastoma [70], and COLXVI in OSCC [71] induce β1 integrin to promote cancer stemness, invasion, and drug resistance. Even collagen glycosylation modulates integrin binding. Galactosylation occurred on the periphery of α2β1 integrin, where it interacted with α1(IV)382–393 but occurred in the middle of α3β1 integrin, where it interacted with α1(IV)531–543 in melanoma cell adhesion [72].
Collagen can stimulate additional signaling pathways in cancer cells to exert various functions. The increased expression of COL1A1 affected the caspase-3/PI3K/AKT pathways to inhibit cell apoptosis in cervical cancer tissues [73]. After the withdrawal of rapamycin treatment, mutated COL1A1 reinforced PI3K–AKT-mammalian target of rapamycin (mTOR) signals in cancer stem cells to sustain the metastatic burden of ERα-positive breast cancer cells; however, lung metastases were independent of mTOR signaling [74]. In addition, increased COLI did not alter primary tumor growth and ERα expression but enhanced circulating cancer cells and metastasizing cancer cells with decreased phospho-STAT5 expression, increased phospho-ERK1/2, and increased phospho-AKT expression; this phenomenon coincided with the formation of invasive protrusions of the primary tumor harboring collagen fibers angled perpendicularly to the tumor mass [75]. However, COLI in non-small cell lung cancer (NSCLC) induced mTOR activation through an AKT-independent pathway, leading to EGFR-tyrosine kinase inhibitor resistance [76]. The Notch3-COL4A2 loop promoted anoikis resistance with a reduction in phosphorylated AKT and ERK 1/2 in ovarian cancer cells [77]. Although both collagen glycation and carbamylation affected the metastasis of cancer cells, glycation caused a more obvious delay in cell adhesion time and deficient actin stress fibers and inhibited the mean cell speed and FAK phosphorylation state more than carbamylation [78]. However, increased collagen in fibrosarcoma tissue inhibited tumor growth and metastasis because the tumor necrosis factor (TNF) receptor 2/p38 MAPK signaling pathway activated collagen expression via gadolinium-containing fullerenol [79]. This distinction implies that different cancer cells facilitate collagen expression to exert inverse effects on cancer progression.
The relationship among exosomes, microRNAs and collagen in cancer
Recent studies have highlighted the relationship among exosomes, microRNAs (miRNAs) and collagen in cancer [80,81,82].
MiRNAs are a class of small, noncoding RNAs that act as epigenetic regulators of various physiological and pathological processes [83, 84]. The deregulation of miRNAs is associated with the initiation and progression of many diseases, such as cardiovascular diseases, infectious diseases, diabetes, central nervous system-related diseases, and cancer [85,86,87,88]. MiRNAs exert their regulatory activity by affecting a variety of physiological and pathological processes in cancer. Several studies have documented the roles of miRNAs in tumor growth, angiogenesis, and metastasis [89, 90], as well as their utility as diagnostic and therapeutic biomarkers [91,92,93]. Regarding the influence of miRNAs on collagen, various miRNAs are regulated in cancer cells to affect distinct functions. Breast cancer cells downregulate miR-196b-5p, which decreases COL1A1 levels, to induce growth and metastasis [94]. Intestinal gastric cancer cells suppress COL1A2 via miR-25 to promote EMT and angiogenesis [95]. MiR-27b-3p and miR-455-3p enhance cancer cell quiescence in response to activated p53 to increase drug resistance and recurrence [96]. The miRNA let-7d inhibits pancreatic stellate cell activation [97] and macrophage infiltration by targeting COL3A1 in renal cell carcinoma [98]. Collagen-related enzymes are also differentially regulated by miRNAs in cancer cells. MiRNA-29a targets heat shock protein 47 in cervical SCC [99], and miR-26a/b affects lysyl oxidase-like (LOXL) 2 and procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 (PLOD2) levels in renal cancer cells [99]. Losartan improves the efficacy of chemotherapy in ovarian cancer partly by inducing antifibrotic miRNAs to normalize the ECM [100].
Exosomes are membrane-enclosed structures that facilitate communication between cancer cells and the ECM to influence cancer cell survival, growth, and metastasis and the immune system [101]. Cancer-derived exosomes induce the formation of CAFs in the collagen matrix to promote EMT [102] and increase the secretion of MMP-14 to regulate collagen [103]. In addition, collagen enhances exosome secretion [104]. Collagen and exosomes form a mutually beneficial feedback loop to promote cancer progression.
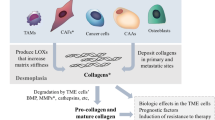
The reciprocity between collagen and cancer cells under hypoxic conditions
Hypoxic conditions, a common condition in collagen-rich ECM, intensify cancer progression based on the interaction between cancer cells and collagen. Hypoxia-inducible factor (HIF)-1, LOX, and MMP participate in the process. COLI fibers are reduced in the hypoxic microenvironment via increased expression of LOX in renal cell carcinoma, decreased expression of MMP-3 in breast cancer, or decreased expression of MMP-1 and MMP-16 in prostate cancer [105, 106]. Collagen can be remodeled by HIF-1 regulating LOXL1, LOXL2, and LOXL4 [107].
Other substances, such as hepatitis transactivator protein X in liver cancer [108], LKB1 via COLIV and β1 integrin in lung cancer [109], and HIF1α-dependent PLOD2 in primary sarcomas and pulmonary metastasis [110], participate with collagen in the HIF/LOX pathway. Hypoxia increased the expression of procollagen-lysine as collagen crosslinker to further enhance collagen fiber size and thus promote cancer metastasis [111]. COLI fibers exhibited covalent crosslinking with prolyl 4-hydroxylase alpha 1 and prolyl 4-hydroxylase alpha 2 in differentiated cell types of triple-negative breast cancer, and the prolyl 4-hydroxylase alpha 1/HIF-1 axis increased chemoresistance [112, 113]. Even mutant p53 regulated the expression of COL7A1 in NSCLC, not by influencing HIF-1 binding to DNA, but rather by inhibiting its transcriptional activity [114].
Hypoxic conditions are closely associated with vessels in cancer. Matrix collagen stiffness, rather than collagen density, alters vascular growth and integrity. Moreover, increased fibril density decreased vessel network formation, while increased interfibril branching improved vessel volume density and formation, which were markedly dependent on the changed temporal and spatial depositions of COLIV [115]. In addition, neovessel branching is associated primarily with collagen crosslinking rather than with collagen content [116]. Notably, the von Hippel–Lindau protein directly regulated COL4A2 assembly independent of HIF-α to restrain cancer angiogenic formation [117]. Further reflecting the relationship between collagen and cancer cells with regard to treatment, improved tumor stromal pO2 levels and the recovery of blood flow were associated with the content and diameter of collagen fibril under imatinib treatment [118].
Interaction between collagen and tumor matrix components
The complex interactions among collagen and matrix proteins within ECM cells contribute to cancer initiation and progression, as shown in Fig. 2.

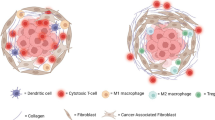
The complexity between collagen and the extracellular matrix. Multiple stromal cells and the extracellular matrix play important roles in stimulating or inhibiting collagen functions via different pathways in cancer progression. Collagen-rich extracellular matrices can bind to other molecules to form dense fibrosis, which induces an anoxic environment and changes the condition of new blood vessels. The behavior of cancer cells is also closely related to collagen. This process also affects the activity and localization of innate and adaptive immune cells
Collagen in cancer tissue accompanies various categories and degrees of myeloid-lineage immune cells, including mast cells, macrophages, and neutrophils, and lymphocytes, including T cells and B cells, to reflect different cancer progression stages [119]. For example, pancreatic cancer stromal compositions were classified as inert, dormant, fibrogenic, or fibrotic based on α-smooth muscle actin (SMA) and COLI expression; these differing stromal compositions individually influenced the levels of CD4+ T cells, CD8+ T cells, macrophages, and neutrophils [120]. Increased infiltration of macrophages and lymphocytes was also observed in subcutaneous adipose collagen in gastrointestinal cancer patients with cachexia [121]. However, increased stromal COL10A1 was accompanied by low numbers of total tumor-infiltrating lymphocytes in ER-positive/EGFR2-positive breast cancer [122].
The location and quantity of collagen with lymphocytes reinforce discrepant cancer progression via distinct signaling pathways. A high-density collagen matrix, rather than a low-density matrix, decreases T cell abundance and cytotoxic T cells but upregulates regulatory T cells [123, 124]. Aligned fibers and collagen density around vascular regions and epithelial cancer cells restricted the migration of T cells and limited their entry into the tumor mass [125]. Moreover, others have further reported that the distribution of collagen with lymphocytes affects cancer progression, but the effects on different cancers remain distinct. COLI was not associated with T cell deficiency in PDAC [126]. However, others noted that the metastatic urothelial cancer response to programmed death-1 and programmed death-ligand 1 treatment involved the movement of CD8+ T cells from the cancer parenchyma to the collagen-rich peritumoral stroma [127]. Nevertheless, contact between infiltrating T cells, Tregs and antitumor T cells with collagen was inhibited by members of the TGF-β pathway to provide antimelanoma-immunity [128, 129].
Furthermore, both adaptive immunity and innate immunity affect collagen. Collagen deposition and linearization are positively related to macrophage activity [130]. Additionally, denatured collagen acts as a strong chemoattractant for macrophages that mediate the promotion of cancer [131]. The possible reasons for these functions of collagen are described below. Macrophages closely interact with stellate cells to influence the collagen matrix [132]. Tumor-associated macrophages orchestrate the deposition, crosslinking, and linearization of collagen fibers, specifically COLI, VI, and XIV, at areas of tumor invasiveness [133]. Although macrophages do not exhibit efficient collagen internalization of mesenchymal origin, those originating from circulating CCR2 monocytes internalize collagen in an MMP-dependent manner, which is mediated by the mannose receptor rather than by β1 integrin [134,135,136]. In the collagen-rich breast cancer environment, coexpression of fibroblast activation protein and heme oxygenase-1 in macrophages resulted from an early innate regenerative response to IL-6 to directly facilitate transendothelial migration [137]. Cyclooxygenase-2 also induced overall collagen deposition and macrophages in early-stage breast cancer [138]. Notably, in the context of an αvβ3 integrin-specific collagen hydrogel, M2-like immunosuppressive macrophages promoted angiogenesis in glioblastoma, while M1-like proinflammatory macrophages suppressed angiogenesis, which was regulated by Src-PI3K-YAP signaling associated with TGF-β1 [139]. IL-10 induces M2 differentiation in heterospheroid macrophages [140]. In contrast, collagen degradation is enhanced not by M2 macrophages but rather by M1 macrophages, as endogenous macrophages are recruited, modified and activated to produce MMPs and hepatic growth factors, thereby enhancing hepatocyte proliferation and the release of TNF-related apoptosis-inducing ligands by natural killer cells, leading to hepatic stellate cell apoptosis [141]. Moreover, there are other important innate immune cells in the ECM that exhibit anticancer immunity or immunosuppressive effects. Collagen deposition can be induced by neurofibromatosis type 1-recruited mast cells, which mediate stem cell factor/c-kit signaling in neurofibromas [142]. Collagen-dense tumors increased granulocyte monocyte-colony stimulating factor, which was associated with neutrophil signaling [143]. However, COLXVII overexpression in cancer cells by activated leukocyte-associated immunoglobulin-like receptor-1 diminished natural killer cell cytotoxic activity [144]. By interaction with collagen in ovarian and breast cancer, tumor-associated dendritic cells expressed the highly receptor CD305/leukocyte-associated immunoglobulin-like receptor-1 [145].
Fibroblasts are another important cell type in the ECM. CAFs cause various collagen deposits to develop into intratumoral fibrosis, resulting in cancer occurrence, differentiation, and invasion [4]. These CAFs can even originate from adipose tissue-derived stromal cells [146]. CAFs are classified as tumor-restraining, tumor-promoting, secretory, or ECM-remodeling cells [147], which also exhibit CAF functions via mutated genes [148], the secreted cytokines IL-1β, TNF-α and NF-κB, inflammatory signals, epigenetic regulation, etc. [6]. Cancer cells can invade collagen with fibroblasts, which are characterized by cell cycle phases and small cancer cell nest formation [149]. Additionally, CAFs exert anticancer and cancer-induced effects to show dual influences for cancer progression [150].
Some ECM proteins, mainly including MMPs, hyaluronic acid, fibronectin, and laminin, regulate cancer cell invasion and migration with collagen. The functions of MMPs with collagen are summarized in Table 1, and MMP activity with contractility necessarily establish the ECM stiffness associated with collagen bulk and distribution [151]. The collagen gel storage modulus with glycosaminoglycans chondroitin sulfate and hyaluronic acid was dependent on both the fiber diameter and network mesh size [152]. When cancer cells encountered stiff collagen fibers at fibronectin-rich invasive fronts, they engaged αvβ1 integrin to recruit vinculin and zyxin to focal adhesions sites in a tension-dependent manner to induce PI3K signaling [153]. Similarly, with increased COLI in the laminin-rich ECM regarded as an early tumor microenvironment, cancer stem cells maintained their endothelial-like gene signatures and secreted high levels of VEGFR-2 in a paracrine and autocrine manner to simulate progression [154]. In contrast, unlike the laminin-rich microenvironment, a COLI Matrigel was sufficient to induce colon cancer mesenchymal gene expression, suppressing hepatocyte nuclear factor 4α and its target genes [155]. Additionally, CAFs initially constructed the ECM network by depositing fibronectin, followed by the preferential interaction of COLI with relaxed fibronectin, which, in turn, limited the stretching and mechanical unfolding of fibronectin, leading to collagen superseding fibronectin [156, 157]. Other types of collagen also have functions. The expression of COLIV was associated with the expression of fibronectin and laminin during central nervous system metastasis [158]. The binding of COLXV, rather than I, III, IV, and V, to fibronectin, laminin, and vitronectin inhibited the adhesion and migration of fibrosarcoma cells [159].
Collagen and clinical applications
Various experiments and clinical data have revealed collagen to be a prognostic factor correlated with cancer differentiation, cancer invasion, lymph node metastasis, and clinical stage in cancer patients (Table 2). Lower COLI and COLIV content is a biomarker of differentiated tumors and proliferation potency of cancer cells [160, 161]. The increased expression of COLIV and COLVI and the collagen structure reflect tumor angiogenesis and glioblastoma progression [162]. Even hypomethylation of the COL17A1 promoter is associated with advanced stage, increased invasion of breast cancer, lung adenocarcinoma, cervical cancer, neck SCC, and lung SCC [163].
Factors other than collagen content are correlated with clinical outcome [164], such as collagen alignment and distribution, which also affect cancer. During tumor progression, collagen exhibits different signatures. Tumor-associated collagen signature 1 (TACS-1) indicates the presence of dense collagen near the cancer, TACS-2 represents collagen fibers parallel to the tumor edge, and TACS-3 depicts radially aligned collagen fibers [165]. TACS-3 was related to cancer cell invasion and poor survival in breast carcinoma and in situ breast ductal carcinoma [166, 167]. TACS-3 was shown to be driven by increased plasminogen activator inhibitor 1 via ERK signaling and promoted the migration of triple-negative breast cancer cells [168]. The elevated density and depth of collagen deposition showed high proliferation and invasion of cancer cells [169, 170]. Although COLI and COLIV are expressed in different tumor stroma compartments in pancreatic cancer tissue, they stimulate proliferation, migration, and antiapoptosis. The main form of COLI was generated by pancreatic stellate cells attributed to cancer cells in an indirect contactable desmoplastic area to activate TGF-β, while COLIV was produced by cancer cells themselves to form an autocrine loop in direct proximity to cancer cells, causing discontinuous basement membrane-like structures that interacted with the COLIV CB3 region and β1 integrin of the cancer cells [171]. However, another study showed that COLV was expressed by pancreatic stellate cells via paracrine loops in PDAC [172]. The desmoplastic reactions in primary cancer were divided into mature, intermediate, and immature based on the presence of keloid-like collagen and myxoid stroma; immature desmoplastic reactions were associated with higher T and N stages, more extensive liver metastasis, and higher recurrence rate than other reaction types [173]. Furthermore, COLVII positively regulates the abundance of the cell polarity markers E-cadherin and B-catenin [174]. The mechanisms underlying these important collagen properties are described below. Among five collagen parameters (alignment, density, width, length, and straightness), increased collagen width is the most powerful parameter for predicting cancer prognosis [175]. The elasticity of the collagen matrix is controlled by fibril bending stiffness rather than by fibril diameter or intrafibrillar crosslinking [176]. Increased collagen fiber alignment, elevated levels of immunoreactive glycosaminoglycans such as heparan sulfate and chondroitin sulfate, and decreased levels of the proteoglycan decorin enhance the stiffness of carcinoma tissues [177]. In contrast, others have notably reported that the distribution of collagen had no effect in the regenerated cervical tissue following excisional cervical intraepithelial neoplasia [178].
Collagen plays an important role in therapy resistance. In esophageal SCC, increased collagen content was associated with chemotherapy resistance via the MAPK and PI3K/AKT pathways [179]. Even at metastatic sites, collagen crosslinking increases tissue stiffness to promote resistance to treatment [180]. Furthermore, increased collagen content was accompanied by increased hyaluronan accumulation, contributing to doxorubicin drug resistance in pancreatic cancer [181]. Specifically, different collagen types exhibit distinct treatment resistances. COLI induced resistance to drugs, such as cisplatin and mitoxantrone, by activating β1 integrin followed by the FAK/PI3K/AKT pathway in ER-positive cancer cells, the MAPK pathway in triple-negative cancer cells, the coexpression of LOX with COL1A2 in ovarian cancer, and the TGF-β1/Smad3-mediated expression of COLI and COLIII in bromocriptine-resistant prolactinoma cells [182,183,184]. Other mechanisms of drug resistance include COLI-induced tau upregulation, resulting in paclitaxel resistance in ovarian carcinoma [185]. The resistance to radiation of renal cancer cells with intact COLI rather than micronized COLI was mediated by apoptosis attenuation rather than cell cycle redistribution via the PI3K/AKT pathway [186]. In addition, other collagens also play important roles in therapy resistance. COL3A1 optimally predicted the absence of a response to neoadjuvant treatment in rectal cancer and the resistance of ovarian cancer cells to topotecan and paclitaxel [187, 188]. High COLVI expression promoted anoikis resistance and affected the response of salivary gland cancer to radiotherapy and colorectal cancer to adjuvant chemotherapy [189,190,191]. The reactions of cancers to antiangiogenic therapy have also been closely associated with COLIV. The reduced binding of collagen to PDAC cell surface receptors promoted resistance to VEGF therapy via TGF-β signaling [192], but antiangiogenic therapy increased intratumoral adenovirus distribution by decreasing COLIV [193]. Another study showed that the correlation of bevacizumab with elevated tumor stiffness was driven by hypoxia, leading to increased hyaluronic acid and sulfated glycosaminoglycan contents without significantly changing collagen deposition [194]. COL11A1 induced chemoresistance and exerted antiapoptosis effects in ovarian cancer cells by mediating the transcriptional activation of NF-κB to upregulate the Twist family [195].
Collagen can also be used for the imaging of targeted sites. Imaging of the collagen content and arrangement is conducive to the assessment of tissue stiffness, metabolism, and drug resistance [196,197,198], and technology to utilize these data is currently under development [199]; for instance, magnetic resonance apparent diffusion coefficient values were used to indicate a negative correlation of collagen with esophageal SCC cells [200]. The label-free Raman spectroscopic measurements used to distinguish radiation-sensitive tumors were based on differences in collagen content [201].
Because collagen is closely associated with clinical outcome, it can also be used in clinical applications (Fig. 3). Collagen can be utilized as a predictor of prognosis and recurrence, in diagnosis, as a therapy resistance biomarker, as a targeted-therapy strategy, and as a drug carrier.

The value of collagen in clinical applications
Collagen-related therapy
Cancer resistance has substantially hindered the ability to control cancer. Therefore, both cancer cells and tumor microenvironment must be treated, and collagen is a potential target. Moreover, collagen has obvious genetic stability, and its spatial structure remains relatively stable. As collagen is basically present in various types of cancer, the treatment value of modifying collagen conditions in cancer is worth exploring.
Collagen can be regulated by different types of inhibitors against biosynthesized processes and distribution arrangements by interfering with collagen biosynthesis enzymes, disturbing cancer cell signaling pathways, mediating ECM components, or directly utilizing collagenases (Table 3). Although many types of inhibitors exist, most have been investigated in only cell and animal experiments. These indirect methods of modifying collagen are also partially attributed to other intricate related molecular mechanisms and signaling pathways. The treatment of collagen in the current study is mainly to target the function of CAFs. While quiescent CAFs are characterized by the presence of lipid droplets loaded with vitamin A in the cytoplasm, these perinuclear lipid droplets disappear and express the activation marker α-SMA after CAF activation [202]. Moreover, low expression of retinoid receptors is associated with poor survival of pancreatic cancer [203]. Therefore, the vitamin A analog all-trans retinoic acid in the treatment of PDAC induces CAF to be at quiescent status and inhibits cancer cell migration and EMT; however, the relevant clinical trials are only stage I in the recruitment state (NCT03307148) [204]. In addition, some studies focus on the interaction of cancer cells with collagen, which regulates collagen by targeting cancer cells. For instance, the development of PDAC is also associated with vitamin D, which affects insulin synthesis and secretion via reduced CYP24A1 in islets but impairs the anti-proliferation in transformed duct cells via increased CYP24A1 [205]. The vitamin D signaling pathway has the functions of anti-proliferation, pro-differentiation, anti-inflammation, pro-apoptosis, and immune regulation. There is also a stage III clinical trial recruiting to study the influence of high-dose vitamin D3 intake on pancreatic cancer surgery outcomes (NCT03472833). Moreover, vitamin D receptor ligand calcipotriol regulates CAFs to reprise the quiescent state [206], and paricalcitol was selected to participate in ongoing clinical trials on PDAC therapy (NCT03883919, NCT03415854). Hydroxychloroquine can also effectively inhibit the proliferation and metabolic activity of fibroblasts for cancerous interstitial fibrosis and inhibit cancer cells autophagy. However, the combination of gemcitabine hydrochloride and nab-paclitaxel plus hydroxychloroquine did not increase overall survival for metastatic pancreatic cancer; thus, the clinical effect still needs to be further explored [207]. Notably, although MMPs degrade collagen, they also induce cancer angiogenesis; thus, they have complex and paradoxical effects on cancer progression. The combination of an anti-MMP-9 antibody and nab-paclitaxel-based standard cytotoxic therapy was shown to decrease COLI and the metastatic burden compared with that achieved with nab-paclitaxel-based standard cytotoxic therapy in PDAC mouse models [208]. Moreover, collagen-targeted treatments have paradoxical effects on drug delivery and treatment efficacy within the same cancer type. In particular, collagenases monotherapy for cancer may have obvious side effects, and it may even exert opposite effects than those intended. Addition of the sonic hedgehog antagonist vismodegib to gemcitabine did not improve the survival outcomes of metastatic pancreatic cancer patients in a phase Ib/II trial [209]; in contrast, halofuginone disrupted collagen barriers to effectively deliver the drug and promote anticancer immunity [210]. While the antifibrotic drug pirfenidone was effective in early-stage liver fibrosis, it did not influence advanced liver fibrosis and initiation-promotion liver cancer [211].
Combining collagen inhibitors and standard therapeutics, such as chemotherapy and radiotherapy, is a promising anticancer strategy. Collagenase combined with trastuzumab via thermosensitive hydrogels exerted an anticancer effect on animals [212]. Nitric oxide activated endogenous MMP-1 and MMP-2 to deplete collagen, and it significantly improved anticancer efficacy while exerting no overt toxicity in animal models [213]. Under hypoxic cancer conditions, hyperbaric oxygen therapy decreased collagen deposition to enhance chemotherapy efficacy and photodynamic upconversion nanophotosensitizer cancer therapy [214].
Preclinical studies on collagen-related therapy partly demonstrated encouraging outcomes. However, few clinical trials have been conducted, and most have focused on signaling pathways or receptors. These indirect collagen-related treatments remain controversial due to the potential effects of other mechanisms. For example, cilengitide inhibiting αvβ3 and αvβ5 integrins to influence the connection with cancer cells and collagen did not show evident clinical benefits [215]. There are major drugs of direct collagen depletion or collagen alignment changes, but their effects are not clear and need to be further explored (Table 4). Confusing and conflicting results for anti-collagen therapy suggest complex collagen properties. As revealed herein, physiological, pathological and clinical analyses reveal the dual roles of collagen. Collagen has essential functions in normal tissues but also plays important roles in cancer progression and clinical outcomes. Collagen depletion results in the activation of residual cancer cells and incomplete ECM and microvessels. Collagen can function as a barrier for certain stages of cancer, but it can also enhance other stages of cancer. In addition, although collagen can be degraded, its decomposition products can continue to function and thus promote cancer angiogenesis and invasion. The interaction of collagen with other components in the ECM also shows dual effects on cancers. CAFs, which mainly produce stromal collagen, express anti-tumor and tumor-promoting effects; epithelial and endothelial cells, which mainly produce the collagen of basement membrane, can not only maintain vascular stability but also promote cancer angiogenesis and contribute to cancer cells penetration into blood vessels. The individual differences, genetic heterogeneity, and epigenetic heterogeneity of cancer patients can also affect the efficacy of drugs in clinical trials. Therefore, balancing the content, crosslinking, alignment, and distribution of collagen may be a reasonable strategy for cancer treatment. Nanoparticles, nanoplatforms, and nanoenzymes exhibit the expected gratifying properties.
Nanoparticles influence the use of collagen for anticancer therapy in animal models, including combinations with other chemotherapy drugs and treatment regimens [216]. Losartan nanoparticles significantly decrease collagen content to improve tumor penetration and tumor treatment efficiency [217], and losartan in combination with photodynamic nanoplatforms suppressed tumor volume in a breast cancer mouse model [218]. The implantation of nanoparticle/losartan-loaded hydrogel enhanced the intratumoral distribution and anticancer effect of nanoparticles in mice [219]. In addition, supplementation with other nanoparticles further orchestrated collagen normalization rather than destruction, thereby improving the survival rates of cancer patients [220]. A near-infrared light irradiation-activated semiconducting polymer nanoenzyme efficiently digested collagen, leading to nanoparticle accumulation in cancer tissue and the consequential improvement of photothermal therapy [221, 222].
Collagen can also act as a drug carrier or a drug targeted site. Hybrid collagen-cell penetrating peptide carriers improved resistance to enzymatic degradation [223]. Treating cancer with the single-chain fragment of cetuximab along with its collagen-binding domain demonstrated effective results [224]. Collagen affinity can also be used to mediate targeted immunotherapy antibodies. Fusion of the collagen-binding protein lumican and cytokines increased the efficacy of systemic immunotherapies in a melanoma model [223]. The therapeutic use of immune checkpoint inhibitors and interleukin-2 by conjugation (for antibodies) or recombinant fusion (for cytokine) to the von Willebrand factor A3 domain (a collagen-binding domain) eradicated tumors and exhibited obvious safety and efficacy in a breast cancer model [225]. Cancer-collagen-targeting immunoconjugate therapy was evidently applicable to anticancer therapy [226].
Conclusions
Cells and molecules in the tumor microenvironment have dual effects on cancer progression. The role of collagen is a double-edged sword in cancer. On the one hand, collagen, cancer cells, other cells, and other matrix molecules mutually form an inter-reinforcing loop. This loop contributes to the development of cancer by inducing cancer cells proliferation, migration, and metastasis. On the other hand, preclinical and clinical studies have demonstrated that collagen may slow the development of cancer cells to some extent under some conditions. In summary, the association of collagen with cancer is only partially understood, and future studies are needed to elucidate detailed collagen biological mechanisms in cancer tissue that can be applied to precisely regulate collagen balance to achieve the maximum benefit of treatment. This new strategy combined with other treatment modalities can ultimately improve patient survival and quality of life.
Availability of data and materials
Not applicable
Abbreviations
- CAFs:
-
cancer-associated fibroblasts
- CCL:
-
chemokine (C-C motif) ligand
- COL:
-
collagen type
- DDR:
-
discoidin domain receptor
- ECM:
-
extracellular matrix
- EGFR:
-
epidermal growth factor receptor
- EMT:
-
epithelial–mesenchymal transition
- ER:
-
estrogen receptor
- ERK:
-
extracellular signal-regulated kinase
- FACITs:
-
fibril-associated collagens with interrupted triple helices
- FAK:
-
focal adhesion kinase
- FGFR:
-
fibroblast growth factor receptor
- GSK:
-
glycogen synthase kinase
- HIF:
-
hypoxia-inducible factor
- LOX:
-
lysyl oxidase
- LOXL:
-
lysyl oxidase-like
- MAPK:
-
mitogen-activated protein kinase
- MEK:
-
mitogen-activated protein kinase
- MITF:
-
microphthalmia-associated transcription factor
- miRNA:
-
microRNA
- mTOR:
-
mammalian target of rapamycin
- MMP:
-
matrix metalloproteinase
- NF-κB:
-
nuclear factor kappa-B
- NSCLC:
-
non-small cell lung cancer
- PDAC:
-
pancreatic ductal adenocarcinoma
- PI3K:
-
phosphatidylinositol 3-kinase
- PKC:
-
protein kinase C
- PLOD2:
-
procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2
- PTEN:
-
phosphate and tension homology deleted on chromosome ten
- RAC1:
-
Ras-related C3 botulinum toxin substrate 1
- SCC:
-
squamous cell carcinoma
- SMA:
-
smooth muscle actin
- STAT:
-
signal transducers and activators of transcription
- TACS:
-
tumor-associated collagen signature
- TGF-β:
-
transforming growth factor-β
- TNF:
-
tumor necrosis factor
- YAP:
-
Yes-associated protein
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, Bassez A, Decaluwe H, Pircher A, Van den Eynde K, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24:1277–89.
Discher DE, Smith L, Cho S, Colasurdo M, Garcia AJ, Safran S. Matrix mechanosensing: from scaling concepts in ‘Omics data to mechanisms in the nucleus, regeneration, and cancer. Annu Rev Biophys. 2017;46:295–315.
Yamauchi M, Barker TH, Gibbons DL, Kurie JM. The fibrotic tumor stroma. J Clin Invest. 2018;128:16–25.
Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, Hostetter G, Shepard HM, Von Hoff DD, Han H. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin Cancer Res. 2015;21:3561–8.
Gu L, Shan T, Ma YX, Tay FR, Niu L. Novel biomedical applications of crosslinked collagen. Trends Biotechnol. 2019;37:464–91.
Ishikawa Y, Rubin K, Bachinger HP, Kalamajski S. The endoplasmic reticulum-resident collagen chaperone Hsp47 interacts with and promotes the secretion of decorin, fibromodulin, and lumican. J Biol Chem. 2018;293:13707–16.
Shi R, Hu W, Zhang Y, Gao S, Smith AH, Ye J, Cai L, Graham LM, Li C. Ascorbate inducible N259 glycans on prolyl 4-hydroxylase subunit alpha1 promote hydroxylation and secretion of type I collagen. Cell Mol Life Sci. 2019;76:3449–64.
Elia I, Rossi M, Stegen S, Broekaert D, Doglioni G, van Gorsel M, Boon R, Escalona-Noguero C, Torrekens S, Verfaillie C, et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature. 2019;568:117–21.
Scietti L, Chiapparino A, De Giorgi F, Fumagalli M, Khoriauli L, Nergadze S, Basu S, Olieric V, Cucca L, Banushi B, et al. Molecular architecture of the multifunctional collagen lysyl hydroxylase and glycosyltransferase LH3. Nat Commun. 2018;9:3163.
Bekhouche M, Colige A. The procollagen N-proteinases ADAMTS2, 3 and 14 in pathophysiology. Matrix Biol. 2015;44–46:46–53.
Tjin G, White ES, Faiz A, Sicard D, Tschumperlin DJ, Mahar A, Kable EPW, Burgess JK. Lysyl oxidases regulate fibrillar collagen remodelling in idiopathic pulmonary fibrosis. Dis Model Mech. 2017;10:1301–12.
Gautieri A, Passini FS, Silvan U, Guizar-Sicairos M, Carimati G, Volpi P, Moretti M, Schoenhuber H, Redaelli A, Berli M, Snedeker JG. Advanced glycation end-products: mechanics of aged collagen from molecule to tissue. Matrix Biol. 2017;59:95–108.
Scarpellini A, Huang L, Burhan I, Schroeder N, Funck M, Johnson TS, Verderio EA. Syndecan-4 knockout leads to reduced extracellular transglutaminase-2 and protects against tubulointerstitial fibrosis. J Am Soc Nephrol. 2014;25:1013–27.
Van Doren SR. Matrix metalloproteinase interactions with collagen and elastin. Matrix Biol. 2015;44–46:224–31.
Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, Lac S, Roques J, Lavaut MN, Berthezene P, et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun. 2017;8:16031.
Ricard-Blum S. The collagen family. Cold Spring Harb Perspect Biol. 2011;3:a004978.
Ohlund D, Lundin C, Ardnor B, Oman M, Naredi P, Sund M. Type IV collagen is a tumour stroma-derived biomarker for pancreas cancer. Br J Cancer. 2009;101:91–7.
Qiu S, Deng L, Liao X, Nie L, Qi F, Jin K, Tu X, Zheng X, Li J, Liu L, et al. Tumor-associated macrophages promote bladder tumor growth through PI3K/AKT signal induced by collagen. Cancer Sci. 2019;110:2110–8.
Kenny TC, Schmidt H, Adelson K, Hoshida Y, Koh AP, Shah N, Mandeli J, Ting J, Germain D. Patient-derived interstitial fluids and predisposition to aggressive sporadic breast cancer through collagen remodeling and inactivation of p53. Clin Cancer Res. 2017;23:5446–59.
Wormann SM, Song L, Ai J, Diakopoulos KN, Kurkowski MU, Gorgulu K, Ruess D, Campbell A, Doglioni C, Jodrell D, et al. Loss of P53 function activates JAK2-STAT3 signaling to promote pancreatic tumor growth, stroma modification, and gemcitabine resistance in mice and is associated with patient survival. Gastroenterology. 2016;151(180–193):e112.
Teodoro JG, Parker AE, Zhu X, Green MR. p53-mediated inhibition of angiogenesis through up-regulation of a collagen prolyl hydroxylase. Science. 2006;313:968–71.
Assadian S, El-Assaad W, Wang XQ, Gannon PO, Barres V, Latour M, Mes-Masson AM, Saad F, Sado Y, Dostie J, Teodoro JG. p53 inhibits angiogenesis by inducing the production of Arresten. Cancer Res. 2012;72:1270–9.
Alvarez-Garcia V, Tawil Y, Wise HM, Leslie NR. Mechanisms of PTEN loss in cancer: It’s all about diversity. Semin Cancer Biol. 2019. https://doi.org/10.1016/j.semcancer.2019.02.001.
Jolly LA, Novitskiy S, Owens P, Massoll N, Cheng N, Fang W, Moses HL, Franco AT. Fibroblast-mediated collagen remodeling within the tumor microenvironment facilitates progression of thyroid cancers driven by BrafV600E and Pten loss. Cancer Res. 2016;76:1804–13.
Shields MA, Ebine K, Sahai V, Kumar K, Siddiqui K, Hwang RF, Grippo PJ, Munshi HG. Snail cooperates with KrasG12D to promote pancreatic fibrosis. Mol Cancer Res. 2013;11:1078–87.
Wang JH, Newbury LJ, Knisely AS, Monia B, Hendry BM, Sharpe CC. Antisense knockdown of Kras inhibits fibrosis in a rat model of unilateral ureteric obstruction. Am J Pathol. 2012;180:82–90.
Yoshida T, Hashimura M, Kuwata T, Matsumoto T, Suzuki E, Tazo Y, Nakajima H, Inukai M, Saegusa M. Transcriptional regulation of the alpha-1 type II collagen gene by nuclear factor B/p65 and Sox9 in the chondrocytic phenotype of uterine carcinosarcomas. Hum Pathol. 2013;44:1780–8.
Nagathihalli NS, Castellanos JA, Shi C, Beesetty Y, Reyzer ML, Caprioli R, Chen X, Walsh AJ, Skala MC, Moses HL, Merchant NB. Signal transducer and activator of transcription 3, mediated remodeling of the tumor microenvironment results in enhanced tumor drug delivery in a mouse model of pancreatic cancer. Gastroenterology. 2015;149(1932–1943):e1939.
Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016;22:497–505.
Miskolczi Z, Smith MP, Rowling EJ, Ferguson J, Barriuso J, Wellbrock C. Collagen abundance controls melanoma phenotypes through lineage-specific microenvironment sensing. Oncogene. 2018;37:3166–82.
Jenkins MH, Croteau W, Mullins DW, Brinckerhoff CE. The BRAF(V600E) inhibitor, PLX4032, increases type I collagen synthesis in melanoma cells. Matrix Biol. 2015;48:66–77.
Clarke CJ, Berg TJ, Birch J, Ennis D, Mitchell L, Cloix C, Campbell A, Sumpton D, Nixon C, Campbell K, et al. The initiator methionine tRNA drives secretion of type II collagen from stromal fibroblasts to promote tumor growth and angiogenesis. Curr Biol. 2016;26:755–65.
Sugiyama N, Varjosalo M, Meller P, Lohi J, Hyytiainen M, Kilpinen S, Kallioniemi O, Ingvarsen S, Engelholm LH, Taipale J, et al. Fibroblast growth factor receptor 4 regulates tumor invasion by coupling fibroblast growth factor signaling to extracellular matrix degradation. Cancer Res. 2010;70:7851–61.
Holdman XB, Welte T, Rajapakshe K, Pond A, Coarfa C, Mo Q, Huang S, Hilsenbeck SG, Edwards DP, Zhang X, Rosen JM. Upregulation of EGFR signaling is correlated with tumor stroma remodeling and tumor recurrence in FGFR1-driven breast cancer. Breast Cancer Res. 2015;17:141.
Chang CC, Hsieh TL, Tiong TY, Hsiao CH, Ji AT, Hsu WT, Lee OK, Ho JH. Regulation of metastatic ability and drug resistance in pulmonary adenocarcinoma by matrix rigidity via activating c-Met and EGFR. Biomaterials. 2015;60:141–50.
Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, Chen AC, Sah RL, Taylor SS, Engler AJ, Yang J. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol. 2015;17:678–88.
Sipes NS, Feng Y, Guo F, Lee HO, Chou FS, Cheng J, Mulloy J, Zheng Y. Cdc42 regulates extracellular matrix remodeling in three dimensions. J Biol Chem. 2011;286:36469–77.
Yamazaki D, Kurisu S, Takenawa T. Involvement of Rac and Rho signaling in cancer cell motility in 3D substrates. Oncogene. 2009;28:1570–83.
Meng C, He Y, Wei Z, Lu Y, Du F, Ou G, Wang N, Luo XG, Ma W, Zhang TC, He H. MRTF-A mediates the activation of COL1A1 expression stimulated by multiple signaling pathways in human breast cancer cells. Biomed Pharmacother. 2018;104:718–28.
Vennin C, Chin VT, Warren SC, Lucas MC, Herrmann D, Magenau A, Melenec P, Walters SN, Del Monte-Nieto G, Conway JR, et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci Transl Med. 2017. https://doi.org/10.1126/scitranslmed.aai8504.
Rath N, Morton JP, Julian L, Helbig L, Kadir S, McGhee EJ, Anderson KI, Kalna G, Mullin M, Pinho AV, et al. ROCK signaling promotes collagen remodeling to facilitate invasive pancreatic ductal adenocarcinoma tumor cell growth. EMBO Mol Med. 2017;9:198–218.
Favreau AJ, Vary CP, Brooks PC, Sathyanarayana P. Cryptic collagen IV promotes cell migration and adhesion in myeloid leukemia. Cancer Med. 2014;3:265–72.
Huang H, Svoboda RA, Lazenby AJ, Saowapa J, Chaika N, Ding K, Wheelock MJ, Johnson KR. Up-regulation of N-cadherin by Collagen I-activated discoidin domain receptor 1 in pancreatic cancer requires the adaptor molecule Shc1. J Biol Chem. 2016;291:23208–23.
Zhang K, Corsa CA, Ponik SM, Prior JL, Piwnica-Worms D, Eliceiri KW, Keely PJ, Longmore GD. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol. 2013;15:677–87.
Iwai LK, Payne LS, Luczynski MT, Chang F, Xu H, Clinton RW, Paul A, Esposito EA, Gridley S, Leitinger B, et al. Phosphoproteomics of collagen receptor networks reveals SHP-2 phosphorylation downstream of wild-type DDR2 and its lung cancer mutants. Biochem J. 2013;454:501–13.
Rada M, Nallanthighal S, Cha J, Ryan K, Sage J, Eldred C, Ullo M, Orsulic S, Cheon DJ. Inhibitor of apoptosis proteins (IAPs) mediate collagen type XI alpha 1-driven cisplatin resistance in ovarian cancer. Oncogene. 2018;37:4809–20.
Oliveira-Ferrer L, Rossler K, Haustein V, Schroder C, Wicklein D, Maltseva D, Khaustova N, Samatov T, Tonevitsky A, Mahner S, et al. c-FOS suppresses ovarian cancer progression by changing adhesion. Br J Cancer. 2014;110:753–63.
Procacci P, Moscheni C, Sartori P, Sommariva M, Gagliano N. Tumor(-)stroma cross-talk in human pancreatic ductal adenocarcinoma: a focus on the effect of the extracellular matrix on tumor cell phenotype and invasive potential. Cells. 2018;7:E158.
Imamichi Y, Konig A, Gress T, Menke A. Collagen type I-induced Smad-interacting protein 1 expression downregulates E-cadherin in pancreatic cancer. Oncogene. 2007;26:2381–5.
Vaniotis G, Rayes RF, Qi S, Milette S, Wang N, Perrino S, Bourdeau F, Nystrom H, He Y, Lamarche-Vane N, Brodt P. Collagen IV-conveyed signals can regulate chemokine production and promote liver metastasis. Oncogene. 2018;37:3790–805.
Espinosa Neira R, Salazar EP. Native type IV collagen induces an epithelial to mesenchymal transition-like process in mammary epithelial cells MCF10A. Int J Biochem Cell Biol. 2012;44:2194–203.
Spivey KA, Chung I, Banyard J, Adini I, Feldman HA, Zetter BR. A role for collagen XXIII in cancer cell adhesion, anchorage-independence and metastasis. Oncogene. 2012;31:2362–72.
Hayashido Y, Kitano H, Sakaue T, Fujii T, Suematsu M, Sakurai S, Okamoto T. Overexpression of integrin alphav facilitates proliferation and invasion of oral squamous cell carcinoma cells via MEK/ERK signaling pathway that is activated by interaction of integrin alphavbeta8 with type collagen. Int J Oncol. 2014;45:1875–82.
Ibbetson SJ, Pyne NT, Pollard AN, Olson MF, Samuel MS. Mechanotransduction pathways promoting tumor progression are activated in invasive human squamous cell carcinoma. Am J Pathol. 2013;183:930–7.
Shen Y, Shen R, Ge L, Zhu Q, Li F. Fibrillar type I collagen matrices enhance metastasis/invasion of ovarian epithelial cancer via beta1 integrin and PTEN signals. Int J Gynecol Cancer. 2012;22:1316–24.
Girotti MR, Fernandez M, Lopez JA, Camafeita E, Fernandez EA, Albar JP, Benedetti LG, Valacco MP, Brekken RA, Podhajcer OL, Llera AS. SPARC promotes cathepsin B-mediated melanoma invasiveness through a collagen I/alpha2beta1 integrin axis. J Invest Dermatol. 2011;131:2438–47.
Kirkland SC. Type I collagen inhibits differentiation and promotes a stem cell-like phenotype in human colorectal carcinoma cells. Br J Cancer. 2009;101:320–6.
Smith SD, Enge M, Bao W, Thullberg M, Costa TD, Olofsson H, Gashi B, Selivanova G, Stromblad S. Protein kinase Calpha (PKCalpha) regulates p53 localization and melanoma cell survival downstream of integrin alphav in three-dimensional collagen and in vivo. J Biol Chem. 2012;287:29336–47.
Hwangbo C, Park J, Lee JH. mda-9/Syntenin protein positively regulates the activation of Akt protein by facilitating integrin-linked kinase adaptor function during adhesion to type I collagen. J Biol Chem. 2011;286:33601–12.
Blockhuys S, Van Rompaye B, De Rycke R, Lambein K, Claes K, Bracke M, De Wagter C, De Wever O. Radiation-induced myosin IIA expression stimulates collagen type I matrix reorganization. Radiother Oncol. 2013;108:162–7.
Artym VV, Swatkoski S, Matsumoto K, Campbell CB, Petrie RJ, Dimitriadis EK, Li X, Mueller SC, Bugge TH, Gucek M, Yamada KM. Dense fibrillar collagen is a potent inducer of invadopodia via a specific signaling network. J Cell Biol. 2015;208:331–50.
Yan T, Zhang A, Shi F, Chang F, Mei J, Liu Y, Zhu Y. Integrin alphavbeta3-associated DAAM1 is essential for collagen-induced invadopodia extension and cell haptotaxis in breast cancer cells. J Biol Chem. 2018;293:10172–85.
Chen SY, Lin JS, Yang BC. Modulation of tumor cell stiffness and migration by type IV collagen through direct activation of integrin signaling pathway. Arch Biochem Biophys. 2014;555–556:1–8.
Hodgson L, Henderson AJ, Dong C. Melanoma cell migration to type IV collagen requires activation of NF-kappaB. Oncogene. 2003;22:98–108.
Cattaruzza S, Nicolosi PA, Braghetta P, Pazzaglia L, Benassi MS, Picci P, Lacrima K, Zanocco D, Rizzo E, Stallcup WB, et al. NG2/CSPG4-collagen type VI interplays putatively involved in the microenvironmental control of tumour engraftment and local expansion. J Mol Cell Biol. 2013;5:176–93.
Zhang H, Fredericks T, Xiong G, Qi Y, Rychahou PG, Li JD, Pihlajaniemi T, Xu W, Xu R. Membrane associated collagen XIII promotes cancer metastasis and enhances anoikis resistance. Breast Cancer Res. 2018;20:116.
Bauer R, Ratzinger S, Wales L, Bosserhoff A, Senner V, Grifka J, Grassel S. Inhibition of collagen XVI expression reduces glioma cell invasiveness. Cell Physiol Biochem. 2011;27:217–26.
Ratzinger S, Grassel S, Dowejko A, Reichert TE, Bauer RJ. Induction of type XVI collagen expression facilitates proliferation of oral cancer cells. Matrix Biol. 2011;30:118–25.
Stawikowski MJ, Aukszi B, Stawikowska R, Cudic M, Fields GB. Glycosylation modulates melanoma cell alpha2beta1 and alpha3beta1 integrin interactions with type IV collagen. J Biol Chem. 2014;289:21591–604.
Liu S, Liao G, Li G. Regulatory effects of COL1A1 on apoptosis induced by radiation in cervical cancer cells. Cancer Cell Int. 2017;17:73.
Shea MP, O’Leary KA, Wegner KA, Vezina CM, Schuler LA. High collagen density augments mTOR-dependent cancer stem cells in ERalpha + mammary carcinomas, and increases mTOR-independent lung metastases. Cancer Lett. 2018;433:1–9.
Barcus CE, O’Leary KA, Brockman JL, Rugowski DE, Liu Y, Garcia N, Yu M, Keely PJ, Eliceiri KW, Schuler LA. Elevated collagen-I augments tumor progressive signals, intravasation and metastasis of prolactin-induced estrogen receptor alpha positive mammary tumor cells. Breast Cancer Res. 2017;19:9.
Yamazaki S, Higuchi Y, Ishibashi M, Hashimoto H, Yasunaga M, Matsumura Y, Tsuchihara K, Tsuboi M, Goto K, Ochiai A, Ishii G. Collagen type I induces EGFR-TKI resistance in EGFR-mutated cancer cells by mTOR activation through Akt-independent pathway. Cancer Sci. 2018;109:2063–73.
Brown CW, Brodsky AS, Freiman RN. Notch3 overexpression promotes anoikis resistance in epithelial ovarian cancer via upregulation of COL4A2. Mol Cancer Res. 2015;13:78–85.
Said G, Guilbert M, Millerot-Serrurot E, Van Gulick L, Terryn C, Garnotel R, Jeannesson P. Impact of carbamylation and glycation of collagen type I on migration of HT1080 human fibrosarcoma cells. Int J Oncol. 2012;40:1797–804.
Liu J, Kang SG, Wang P, Wang Y, Lv X, Liu Y, Wang F, Gu Z, Yang Z, Weber JK, et al. Molecular mechanism of Gd@C82(OH)22 increasing collagen expression: implication for encaging tumor. Biomaterials. 2018;152:24–36.
Fang T, Lv H, Lv G, Li T, Wang C, Han Q, Yu L, Su B, Guo L, Huang S, et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat Commun. 2018;9:191.
Liu X, Cao M, Palomares M, Wu X, Li A, Yan W, Fong MY, Chan WC, Wang SE. Metastatic breast cancer cells overexpress and secrete miR-218 to regulate type I collagen deposition by osteoblasts. Breast Cancer Res. 2018;20:127.
Colden M, Dar AA, Saini S, Dahiya PV, Shahryari V, Yamamura S, Tanaka Y, Stein G, Dahiya R, Majid S. MicroRNA-466 inhibits tumor growth and bone metastasis in prostate cancer by direct regulation of osteogenic transcription factor RUNX2. Cell Death Dis. 2017;8:e2572.
Chen B, Li H, Zeng X, Yang P, Liu X, Zhao X, Liang S. Roles of microRNA on cancer cell metabolism. J Transl Med. 2012;10:228.
Li AL, Chung TS, Chan YN, Chen CL, Lin SC, Chiang YR, Lin CH, Chen CC, Ma N. microRNA expression pattern as an ancillary prognostic signature for radiotherapy. J Transl Med. 2018;16:341.
Rabieian R, Boshtam M, Zareei M, Kouhpayeh S, Masoudifar A, Mirzaei H. Plasminogen activator inhibitor type-1 as a regulator of fibrosis. J Cell Biochem. 2018;119:17–27.
Mirzaei H, Ferns GA, Avan A, Mobarhan MG. Cytokines and MicroRNA in coronary artery disease. Adv Clin Chem. 2017;82:47–70.
Simonian M, Mosallayi M, Mirzaei H. Circulating miR-21 as novel biomarker in gastric cancer: diagnostic and prognostic biomarker. J Cancer Res Ther. 2018;14:475.
Saeedi Borujeni MJ, Esfandiary E, Taheripak G, Codoner-Franch P, Alonso-Iglesias E, Mirzaei H. Molecular aspects of diabetes mellitus: resistin, microRNA, and exosome. J Cell Biochem. 2018;119:1257–72.
Hoey C, Ahmed M, Fotouhi Ghiam A, Vesprini D, Huang X, Commisso K, Commisso A, Ray J, Fokas E, Loblaw DA, et al. Circulating miRNAs as non-invasive biomarkers to predict aggressive prostate cancer after radical prostatectomy. J Transl Med. 2019;17:173.
Lin Y, Chen F, Shen L, Tang X, Du C, Sun Z, Ding H, Chen J, Shen B. Biomarker microRNAs for prostate cancer metastasis: screened with a network vulnerability analysis model. J Transl Med. 2018;16:134.
Keshavarzi M, Sorayayi S, Jafar Rezaei M, Mohammadi M, Ghaderi A, Rostamzadeh A, Masoudifar A, Mirzaei H. MicroRNAs-based imaging techniques in cancer diagnosis and therapy. J Cell Biochem. 2017;118:4121–8.
Gholamin S, Mirzaei H, Razavi SM, Hassanian SM, Saadatpour L, Masoudifar A, ShahidSales S, Avan A. GD2-targeted immunotherapy and potential value of circulating microRNAs in neuroblastoma. J Cell Physiol. 2018;233:866–79.
Jamali L, Tofigh R, Tutunchi S, Panahi G, Borhani F, Akhavan S, Nourmohammadi P, Ghaderian SMH, Rasouli M, Mirzaei H. Circulating microRNAs as diagnostic and therapeutic biomarkers in gastric and esophageal cancers. J Cell Physiol. 2018;233:8538–50.
Zhu X, Rao X, Yao W, Zou X. Downregulation of MiR-196b-5p impedes cell proliferation and metastasis in breast cancer through regulating COL1A1. Am J Transl Res. 2018;10:3122–32.
Lai YH, Chen J, Wang XP, Wu YQ, Peng HT, Lin XH, Wang WJ. Collagen triple helix repeat containing-1 negatively regulated by microRNA-30c promotes cell proliferation and metastasis and indicates poor prognosis in breast cancer. J Exp Clin Cancer Res. 2017;36:92.
La T, Liu GZ, Farrelly M, Cole N, Feng YC, Zhang YY, Sherwin SK, Yari H, Tabatabaee H, Yan XG, et al. A p53-responsive miRNA network promotes cancer cell quiescence. Cancer Res. 2018;78:6666–79.
Asama H, Suzuki R, Hikichi T, Takagi T, Masamune A, Ohira H. MicroRNA let-7d targets thrombospondin-1 and inhibits the activation of human pancreatic stellate cells. Pancreatology. 2019;19:196–203.
Su B, Zhao W, Shi B, Zhang Z, Yu X, Xie F, Guo Z, Zhang X, Liu J, Shen Q, et al. Let-7d suppresses growth, metastasis, and tumor macrophage infiltration in renal cell carcinoma by targeting COL3A1 and CCL7. Mol Cancer. 2014;13:206.
Yamamoto N, Kinoshita T, Nohata N, Yoshino H, Itesako T, Fujimura L, Mitsuhashi A, Usui H, Enokida H, Nakagawa M, et al. Tumor-suppressive microRNA-29a inhibits cancer cell migration and invasion via targeting HSP47 in cervical squamous cell carcinoma. Int J Oncol. 2013;43:1855–63.
Zhao Y, Cao J, Melamed A, Worley M, Gockley A, Jones D, Nia HT, Zhang Y, Stylianopoulos T, Kumar AS, et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc Natl Acad Sci USA. 2019;116:2210–9.
Ramanathan S, Douglas SR, Alexander GM, Shenoda BB, Barrett JE, Aradillas E, Sacan A, Ajit SK. Exosome microRNA signatures in patients with complex regional pain syndrome undergoing plasma exchange. J Transl Med. 2019;17:81.
Yeon JH, Jeong HE, Seo H, Cho S, Kim K, Na D, Chung S, Park J, Choi N, Kang JY. Cancer-derived exosomes trigger endothelial to mesenchymal transition followed by the induction of cancer-associated fibroblasts. Acta Biomater. 2018;76:146–53.
Hakulinen J, Sankkila L, Sugiyama N, Lehti K, Keski-Oja J. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J Cell Biochem. 2008;105:1211–8.
Szvicsek Z, Oszvald A, Szabo L, Sandor GO, Kelemen A, Soos AA, Paloczi K, Harsanyi L, Tolgyes T, Dede K, et al. Extracellular vesicle release from intestinal organoids is modulated by Apc mutation and other colorectal cancer progression factors. Cell Mol Life Sci. 2019;76:2463–76.
Kakkad SM, Solaiyappan M, O’Rourke B, Stasinopoulos I, Ackerstaff E, Raman V, Bhujwalla ZM, Glunde K. Hypoxic tumor microenvironments reduce collagen I fiber density. Neoplasia. 2010;12:608–17.
Di Stefano V, Torsello B, Bianchi C, Cifola I, Mangano E, Bovo G, Cassina V, De Marco S, Corti R, Meregalli C, et al. Major action of endogenous lysyl Oxidase in clear cell renal cell carcinoma progression and collagen stiffness revealed by primary cell cultures. Am J Pathol. 2016;186:2473–85.
Wong CC, Gilkes DM, Zhang H, Chen J, Wei H, Chaturvedi P, Fraley SI, Wong CM, Khoo US, Ng IO, et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci USA. 2011;108:16369–74.
Tse AP, Sze KM, Shea QT, Chiu EY, Tsang FH, Chiu DK, Zhang MS, Lee D, Xu IM, Chan CY, et al. Hepatitis transactivator protein X promotes extracellular matrix modification through HIF/LOX pathway in liver cancer. Oncogenesis. 2018;7:44.
Gao Y, Xiao Q, Ma H, Li L, Liu J, Feng Y, Fang Z, Wu J, Han X, Zhang J, et al. LKB1 inhibits lung cancer progression through lysyl oxidase and extracellular matrix remodeling. Proc Natl Acad Sci USA. 2010;107:18892–7.
Eisinger-Mathason TS, Zhang M, Qiu Q, Skuli N, Nakazawa MS, Karakasheva T, Mucaj V, Shay JE, Stangenberg L, Sadri N, et al. Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discov. 2013;3:1190–205.
Lewis DM, Pruitt H, Jain N, Ciccaglione M, McCaffery JM, Xia Z, Weber K, Eisinger-Mathason TSK, Gerecht S. A feedback loop between hypoxia and matrix stress relaxation increases oxygen-axis migration and metastasis in sarcoma. Cancer Res. 2019;79:1981–95.
Goggins E, Kakkad S, Mironchik Y, Jacob D, Wildes F, Krishnamachary B, Bhujwalla ZM. Hypoxia inducible factors modify collagen I fibers in MDA-MB-231 triple negative breast cancer xenografts. Neoplasia. 2018;20:131–9.
Xiong G, Stewart RL, Chen J, Gao T, Scott TL, Samayoa LM, O’Connor K, Lane AN, Xu R. Collagen prolyl 4-hydroxylase 1 is essential for HIF-1alpha stabilization and TNBC chemoresistance. Nat Commun. 2018;9:4456.
Amelio I, Mancini M, Petrova V, Cairns RA, Vikhreva P, Nicolai S, Marini A, Antonov AA, Le Quesne J, Baena Acevedo JD, et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc Natl Acad Sci USA. 2018;115:E10869–78.
Whittington CF, Yoder MC, Voytik-Harbin SL. Collagen-polymer guidance of vessel network formation and stabilization by endothelial colony forming cells in vitro. Macromol Biosci. 2013;13:1135–49.
Bordeleau F, Mason BN, Lollis EM, Mazzola M, Zanotelli MR, Somasegar S, Califano JP, Montague C, LaValley DJ, Huynh J, et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc Natl Acad Sci USA. 2017;114:492–7.
Kurban G, Duplan E, Ramlal N, Hudon V, Sado Y, Ninomiya Y, Pause A. Collagen matrix assembly is driven by the interaction of von Hippel-Lindau tumor suppressor protein with hydroxylated collagen IV alpha 2. Oncogene. 2008;27:1004–12.
Burmakin M, van Wieringen T, Olsson PO, Stuhr L, Ahgren A, Heldin CH, Reed RK, Rubin K, Hellberg C. Imatinib increases oxygen delivery in extracellular matrix-rich but not in matrix-poor experimental carcinoma. J Transl Med. 2017;15:47.
Coussens LM, Pollard JW. Leukocytes in mammary development and cancer. Cold Spring Harb Perspect Biol. 2011;3:a003285.
Mahajan UM, Langhoff E, Goni E, Costello E, Greenhalf W, Halloran C, Ormanns S, Kruger S, Boeck S, Ribback S, et al. Immune cell and stromal signature associated with progression-free survival of patients with resected pancreatic ductal adenocarcinoma. Gastroenterology. 2018;155(1625–1639):e1622.
Batista ML Jr, Henriques FS, Neves RX, Olivan MR, Matos-Neto EM, Alcantara PS, Maximiano LF, Otoch JP, Alves MJ, Seelaender M. Cachexia-associated adipose tissue morphological rearrangement in gastrointestinal cancer patients. J Cachexia Sarcopenia Muscle. 2016;7:37–47.
Brodsky AS, Xiong J, Yang D, Schorl C, Fenton MA, Graves TA, Sikov WM, Resnick MB, Wang Y. Identification of stromal ColXalpha1 and tumor-infiltrating lymphocytes as putative predictive markers of neoadjuvant therapy in estrogen receptor-positive/HER2-positive breast cancer. BMC Cancer. 2016;16:274.
Kuczek DE, Larsen AMH, Thorseth ML, Carretta M, Kalvisa A, Siersbaek MS, Simoes AMC, Roslind A, Engelholm LH, Noessner E, et al. Collagen density regulates the activity of tumor-infiltrating T cells. J Immunother Cancer. 2019;7:68.
Bougherara H, Mansuet-Lupo A, Alifano M, Ngo C, Damotte D, Le Frere-Belda MA, Donnadieu E, Peranzoni E. Real-time imaging of resident t cells in human lung and ovarian carcinomas reveals how different tumor microenvironments control T lymphocyte migration. Front Immunol. 2015;6:500.
Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, Mami-Chouaib F, Donnadieu E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122:899–910.
Carstens JL, Correa de Sampaio P, Yang D, Barua S, Wang H, Rao A, Allison JP, LeBleu VS, Kalluri R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun. 2017;8:15095.
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–8.
Budhu S, Schaer DA, Li Y, Toledo-Crow R, Panageas K, Yang X, Zhong H, Houghton AN, Silverstein SC, Merghoub T, Wolchok JD. Blockade of surface-bound TGF-beta on regulatory T cells abrogates suppression of effector T cell function in the tumor microenvironment. Sci Signal. 2017;10:eaak9702.
Pinho AV, Van Bulck M, Chantrill L, Arshi M, Sklyarova T, Herrmann D, Vennin C, Gallego-Ortega D, Mawson A, Giry-Laterriere M, et al. ROBO2 is a stroma suppressor gene in the pancreas and acts via TGF-beta signalling. Nat Commun. 2018;9:5083.
Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang ES, Weaver VM. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol (Camb). 2015;7:1120–34.
O’Brien J, Lyons T, Monks J, Lucia MS, Wilson RS, Hines L, Man YG, Borges V, Schedin P. Alternatively activated macrophages and collagen remodeling characterize the postpartum involuting mammary gland across species. Am J Pathol. 2010;176:1241–55.
Shi C, Washington MK, Chaturvedi R, Drosos Y, Revetta FL, Weaver CJ, Buzhardt E, Yull FE, Blackwell TS, Sosa-Pineda B, et al. Fibrogenesis in pancreatic cancer is a dynamic process regulated by macrophage-stellate cell interaction. Lab Invest. 2014;94:409–21.
Afik R, Zigmond E, Vugman M, Klepfish M, Shimshoni E, Pasmanik-Chor M, Shenoy A, Bassat E, Halpern Z, Geiger T, et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J Exp Med. 2016;213:2315–31.
Madsen DH, Ingvarsen S, Jurgensen HJ, Melander MC, Kjoller L, Moyer A, Honore C, Madsen CA, Garred P, Burgdorf S, et al. The non-phagocytic route of collagen uptake: a distinct degradation pathway. J Biol Chem. 2011;286:26996–7010.
Madsen DH, Leonard D, Masedunskas A, Moyer A, Jurgensen HJ, Peters DE, Amornphimoltham P, Selvaraj A, Yamada SS, Brenner DA, et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J Cell Biol. 2013;202:951–66.
Madsen DH, Jurgensen HJ, Siersbaek MS, Kuczek DE, Grey Cloud L, Liu S, Behrendt N, Grontved L, Weigert R, Bugge TH. Tumor-associated macrophages derived from circulating inflammatory monocytes degrade collagen through cellular uptake. Cell Rep. 2017;21:3662–71.
Muliaditan T, Caron J, Okesola M, Opzoomer JW, Kosti P, Georgouli M, Gordon P, Lall S, Kuzeva DM, Pedro L, et al. Macrophages are exploited from an innate wound healing response to facilitate cancer metastasis. Nat Commun. 2018;9:2951.
Esbona K, Inman D, Saha S, Jeffery J, Schedin P, Wilke L, Keely P. COX-2 modulates mammary tumor progression in response to collagen density. Breast Cancer Res. 2016;18:35.
Cui X, Morales RT, Qian W, Wang H, Gagner JP, Dolgalev I, Placantonakis D, Zagzag D, Cimmino L, Snuderl M, et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials. 2018;161:164–78.
Tevis KM, Cecchi RJ, Colson YL, Grinstaff MW. Mimicking the tumor microenvironment to regulate macrophage phenotype and assessing chemotherapeutic efficacy in embedded cancer cell/macrophage spheroid models. Acta Biomater. 2017;50:271–9.
Ma PF, Gao CC, Yi J, Zhao JL, Liang SQ, Zhao Y, Ye YC, Bai J, Zheng QJ, Dou KF, et al. Cytotherapy with M1-polarized macrophages ameliorates liver fibrosis by modulating immune microenvironment in mice. J Hepatol. 2017;67:770–9.
Staser K, Yang FC, Clapp DW. Pathogenesis of plexiform neurofibroma: tumor-stromal/hematopoietic interactions in tumor progression. Annu Rev Pathol. 2012;7:469–95.
Garcia-Mendoza MG, Inman DR, Ponik SM, Jeffery JJ, Sheerar DS, Van Doorn RR, Keely PJ. Neutrophils drive accelerated tumor progression in the collagen-dense mammary tumor microenvironment. Breast Cancer Res. 2016;18:49.
Rygiel TP, Stolte EH, de Ruiter T, van de Weijer ML, Meyaard L. Tumor-expressed collagens can modulate immune cell function through the inhibitory collagen receptor LAIR-1. Mol Immunol. 2011;49:402–6.
Sprague L, Muccioli M, Pate M, Singh M, Xiong C, Ostermann A, Niese B, Li Y, Li Y, Courreges MC, Benencia F. Dendritic cells: in vitro culture in two- and three-dimensional collagen systems and expression of collagen receptors in tumors and atherosclerotic microenvironments. Exp Cell Res. 2014;323:7–27.
Okumura T, Ohuchida K, Kibe S, Iwamoto C, Ando Y, Takesue S, Nakayama H, Abe T, Endo S, Koikawa K, et al. Adipose tissue-derived stromal cells are sources of cancer-associated fibroblasts and enhance tumor progression by dense collagen matrix. Int J Cancer. 2019;144:1401–13.
Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582–98.
Jones CE, Hammer AM, Cho Y, Sizemore GM, Cukierman E, Yee LD, Ghadiali SN, Ostrowski MC, Leight JL. Stromal PTEN regulates extracellular matrix organization in the mammary gland. Neoplasia. 2019;21:132–45.
Miyashita T, Omori T, Nakamura H, Sugano M, Neri S, Fujii S, Hashimoto H, Tsuboi M, Ochiai A, Ishii G. Spatiotemporal characteristics of fibroblasts-dependent cancer cell invasion. J Cancer Res Clin Oncol. 2019;145:373–81.
Gieniec KA, Butler LM, Worthley DL, Woods SL. Cancer-associated fibroblasts-heroes or villains? Br J Cancer. 2019. https://doi.org/10.1038/s41416-019-0509-3.
Keating M, Kurup A, Alvarez-Elizondo M, Levine AJ, Botvinick E. Spatial distributions of pericellular stiffness in natural extracellular matrices are dependent on cell-mediated proteolysis and contractility. Acta Biomater. 2017;57:304–12.
Yang YL, Sun C, Wilhelm ME, Fox LJ, Zhu J, Kaufman LJ. Influence of chondroitin sulfate and hyaluronic acid on structure, mechanical properties, and glioma invasion of collagen I gels. Biomaterials. 2011;32:7932–40.
Miroshnikova YA, Rozenberg GI, Cassereau L, Pickup M, Mouw JK, Ou G, Templeman KL, Hannachi EI, Gooch KJ, Sarang-Sieminski AL, et al. alpha5beta1-Integrin promotes tension-dependent mammary epithelial cell invasion by engaging the fibronectin synergy site. Mol Biol Cell. 2017;28:2958–77.
Biondani G, Zeeberg K, Greco MR, Cannone S, Dando I, Dalla Pozza E, Mastrodonato M, Forciniti S, Casavola V, Palmieri M, et al. Extracellular matrix composition modulates PDAC parenchymal and stem cell plasticity and behavior through the secretome. FEBS J. 2018;285:2104–24.
Vellinga TT, den Uil S, Rinkes IH, Marvin D, Ponsioen B, Alvarez-Varela A, Fatrai S, Scheele C, Zwijnenburg DA, Snippert H, et al. Collagen-rich stroma in aggressive colon tumors induces mesenchymal gene expression and tumor cell invasion. Oncogene. 2016;35:5263–71.
Kubow KE, Vukmirovic R, Zhe L, Klotzsch E, Smith ML, Gourdon D, Luna S, Vogel V. Mechanical forces regulate the interactions of fibronectin and collagen I in extracellular matrix. Nat Commun. 2015;6:8026.
Wang K, Wu F, Seo BR, Fischbach C, Chen W, Hsu L, Gourdon D. Breast cancer cells alter the dynamics of stromal fibronectin-collagen interactions. Matrix Biol. 2017;60–61:86–95.
Chang KC, Lu YC, Lin MJ, Chen HY, Jin YT. Desmoplastic tumour-associated stroma versus neural tissue in central nervous system metastasis: effects of different microenvironments on tumour growth. Histopathology. 2011;59:31–9.
Hurskainen M, Ruggiero F, Hagg P, Pihlajaniemi T, Huhtala P. Recombinant human collagen XV regulates cell adhesion and migration. J Biol Chem. 2010;285:5258–65.
Jang M, Koh I, Lee JE, Lim JY, Cheong JH, Kim P. Increased extracellular matrix density disrupts E-cadherin/beta-catenin complex in gastric cancer cells. Biomater Sci. 2018;6:2704–13.
Wegner CS, Gaustad JV, Andersen LM, Simonsen TG, Rofstad EK. Diffusion-weighted and dynamic contrast-enhanced MRI of pancreatic adenocarcinoma xenografts: associations with tumor differentiation and collagen content. J Transl Med. 2016;14:161.
Mammoto T, Jiang A, Jiang E, Panigrahy D, Kieran MW, Mammoto A. Role of collagen matrix in tumor angiogenesis and glioblastoma multiforme progression. Am J Pathol. 2013;183:1293–305.
Thangavelu PU, Krenacs T, Dray E, Duijf PH. In epithelial cancers, aberrant COL17A1 promoter methylation predicts its misexpression and increased invasion. Clin Epigenetics. 2016;8:120.
Matte BF, Kumar A, Placone JK, Zanella VG, Martins MD, Engler AJ, Lamers ML. Matrix stiffness mechanically conditions EMT and migratory behavior of oral squamous cell carcinoma. J Cell Sci. 2019;132:jcs224360.
Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4:38.
Conklin MW, Gangnon RE, Sprague BL, Van Gemert L, Hampton JM, Eliceiri KW, Bredfeldt JS, Liu Y, Surachaicharn N, Newcomb PA, et al. Collagen alignment as a predictor of recurrence after ductal carcinoma in situ. Cancer Epidemiol Biomarkers Prev. 2018;27:138–45.
Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178:1221–32.
Humphries BA, Buschhaus JM, Chen YC, Haley HR, Qyli T, Chiang B, Shen N, Rajendran S, Cutter A, Cheng YH, et al. Plasminogen activator inhibitor 1 (PAI1) promotes actin cytoskeleton reorganization and glycolytic metabolism in triple-negative breast cancer. Mol Cancer Res. 2019;17:1142–54.
Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326–43.
Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P, Munro J, Schroder E, Zhou J, Brunton VG, et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell. 2011;19:776–91.
Ohlund D, Franklin O, Lundberg E, Lundin C, Sund M. Type IV collagen stimulates pancreatic cancer cell proliferation, migration, and inhibits apoptosis through an autocrine loop. BMC Cancer. 2013;13:154.
Berchtold S, Grunwald B, Kruger A, Reithmeier A, Hahl T, Cheng T, Feuchtinger A, Born D, Erkan M, Kleeff J, Esposito I. Collagen type V promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Cancer Lett. 2015;356:721–32.
Ueno H, Konishi T, Ishikawa Y, Shimazaki H, Ueno M, Aosasa S, Saiura A, Hase K, Yamamoto J. Histologic categorization of fibrotic cancer stroma in the primary tumor is an independent prognostic index in resectable colorectal liver metastasis. Am J Surg Pathol. 2014;38:1380–6.
Dayal JH, Cole CL, Pourreyron C, Watt SA, Lim YZ, Salas-Alanis JC, Murrell DF, McGrath JA, Stieger B, Jahoda C, et al. Type VII collagen regulates expression of OATP1B3, promotes front-to-rear polarity and increases structural organisation in 3D spheroid cultures of RDEB tumour keratinocytes. J Cell Sci. 2014;127:740–51.
Zhou ZH, Ji CD, Xiao HL, Zhao HB, Cui YH, Bian XW. Reorganized collagen in the tumor microenvironment of gastric cancer and its association with prognosis. J Cancer. 2017;8:1466–76.
Sapudom J, Kalbitzer L, Wu X, Martin S, Kroy K, Pompe T. Fibril bending stiffness of 3D collagen matrices instructs spreading and clustering of invasive and non-invasive breast cancer cells. Biomaterials. 2019;193:47–57.
Brauchle E, Kasper J, Daum R, Schierbaum N, Falch C, Kirschniak A, Schaffer TE, Schenke-Layland K. Biomechanical and biomolecular characterization of extracellular matrix structures in human colon carcinomas. Matrix Biol. 2018;68–69:180–93.
Phadnis SV, Atilade A, Bowring J, Kyrgiou M, Young MP, Evans H, Paraskevaidis E, Walker P. Regeneration of cervix after excisional treatment for cervical intraepithelial neoplasia: a study of collagen distribution. BJOG. 2011;118:1585–91.
Senthebane DA, Jonker T, Rowe A, Thomford NE, Munro D, Dandara C, Wonkam A, Govender D, Calder B, Soares NC, et al. The role of tumor microenvironment in chemoresistance: 3D extracellular matrices as accomplices. Int J Mol Sci. 2018;19:E2861.
Liu L, Zhang SX, Liao W, Farhoodi HP, Wong CW, Chen CC, Segaliny AI, Chacko JV, Nguyen LP, Lu M, et al. Mechanoresponsive stem cells to target cancer metastases through biophysical cues. Sci Transl Med. 2017;9:eaan2966.
Li X, Shepard HM, Cowell JA, Zhao C, Osgood RJ, Rosengren S, Blouw B, Garrovillo SA, Pagel MD, Whatcott CJ, et al. Parallel accumulation of tumor hyaluronan, collagen, and other drivers of tumor progression. Clin Cancer Res. 2018;24:4798–807.
Jakubzig B, Baltes F, Henze S, Schlesinger M, Bendas G. Mechanisms of matrix-induced chemoresistance of breast cancer cells-deciphering novel potential targets for a cell sensitization. Cancers (Basel). 2018;10:E495.
Hu B, Mao Z, Jiang X, He D, Wang Z, Wang X, Zhu Y, Wang H. Role of TGF-beta1/Smad3-mediated fibrosis in drug resistance mechanism of prolactinoma. Brain Res. 2018;1698:204–12.
Sterzynska K, Klejewski A, Wojtowicz K, Swierczewska M, Nowacka M, Kazmierczak D, Andrzejewska M, Rusek D, Brazert M, Brazert J, et al. Mutual expression of ALDH1A1, LOX, and collagens in ovarian cancer cell lines as combined CSCs- and ECM-related models of drug resistance development. Int J Mol Sci. 2018;20:E54.
Gurler H, Yu Y, Choi J, Kajdacsy-Balla AA, Barbolina MV. Three-dimensional collagen type I matrix up-regulates nuclear isoforms of the microtubule associated protein tau implicated in resistance to paclitaxel therapy in ovarian carcinoma. Int J Mol Sci. 2015;16:3419–33.
Krasny L, Shimony N, Tzukert K, Gorodetsky R, Lecht S, Nettelbeck DM, Haviv YS. An in vitro tumour microenvironment model using adhesion to type I collagen reveals Akt-dependent radiation resistance in renal cancer cells. Nephrol Dial Transplant. 2010;25:373–80.
Januchowski R, Swierczewska M, Sterzynska K, Wojtowicz K, Nowicki M, Zabel M. Increased expression of several collagen genes is associated with drug resistance in ovarian cancer cell lines. J Cancer. 2016;7:1295–310.
Goncalves-Ribeiro S, Sanz-Pamplona R, Vidal A, Sanjuan X, Guillen Diaz-Maroto N, Soriano A, Guardiola J, Albert N, Martinez-Villacampa M, Lopez I, et al. Prediction of pathological response to neoadjuvant treatment in rectal cancer with a two-protein immunohistochemical score derived from stromal gene-profiling. Ann Oncol. 2017;28:2160–8.
Burnier JV, Wang N, Michel RP, Hassanain M, Li S, Lu Y, Metrakos P, Antecka E, Burnier MN, Ponton A, et al. Type IV collagen-initiated signals provide survival and growth cues required for liver metastasis. Oncogene. 2011;30:3766–83.
Angenendt L, Mikesch JH, Gorlich D, Busch A, Arnhold I, Rudack C, Hartmann W, Wardelmann E, Berdel WE, Stenner M, et al. Stromal collagen type VI associates with features of malignancy and predicts poor prognosis in salivary gland cancer. Cell Oncol (Dordr). 2018;41:517–25.
Rolff HC, Christensen IJ, Vainer B, Svendsen LB, Eefsen RL, Wilhelmsen M, Lund IK, Hoyer-Hansen G, Nielsen HJ, Illemann M, Danish Collaborative Research Group on Colorectal C. The prognostic and predictive value of soluble type IV collagen in colorectal cancer: a retrospective multicenter study. Clin Cancer Res. 2016;22:2427–34.
Aguilera KY, Rivera LB, Hur H, Carbon JG, Toombs JE, Goldstein CD, Dellinger MT, Castrillon DH, Brekken RA. Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma. Cancer Res. 2014;74:1032–44.
Thaci B, Ulasov IV, Ahmed AU, Ferguson SD, Han Y, Lesniak MS. Anti-angiogenic therapy increases intratumoral adenovirus distribution by inducing collagen degradation. Gene Ther. 2013;20:318–27.
Rahbari NN, Kedrin D, Incio J, Liu H, Ho WW, Nia HT, Edrich CM, Jung K, Daubriac J, Chen I, et al. Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases. Sci Transl Med. 2016;8:360ra135.
Wu YH, Huang YF, Chang TH, Chou CY. Activation of TWIST1 by COL11A1 promotes chemoresistance and inhibits apoptosis in ovarian cancer cells by modulating NF-kappaB-mediated IKKbeta expression. Int J Cancer. 2017;141:2305–17.
Wang H, Mislati R, Ahmed R, Vincent P, Nwabunwanne SF, Gunn JR, Pogue BW, Doyley MM. Elastography can map the local inverse relationship between shear modulus and drug delivery within the pancreatic ductal adenocarcinoma microenvironment. Clin Cancer Res. 2019;25:2136–43.
Bennink LL, Li Y, Kim B, Shin IJ, San BH, Zangari M, Yoon D, Yu SM. Visualizing collagen proteolysis by peptide hybridization: from 3D cell culture to in vivo imaging. Biomaterials. 2018;183:67–76.
Golaraei A, Mirsanaye K, Ro Y, Krouglov S, Akens MK, Wilson BC, Barzda V. Collagen chirality and three-dimensional orientation studied with polarimetric second-harmonic generation microscopy. J Biophotonics. 2019;12:e201800241.
Montesi SB, Desogere P, Fuchs BC, Caravan P. Molecular imaging of fibrosis: recent advances and future directions. J Clin Invest. 2019;129:24–33.
Aoyagi T, Shuto K, Okazumi S, Hayano K, Satoh A, Saitoh H, Shimada H, Nabeya Y, Kazama T, Matsubara H. Apparent diffusion coefficient correlation with oesophageal tumour stroma and angiogenesis. Eur Radiol. 2012;22:1172–7.
Paidi SK, Diaz PM, Dadgar S, Jenkins SV, Quick CM, Griffin RJ, Dings RPM, Rajaram N, Barman I. Label-free Raman spectroscopy reveals signatures of radiation resistance in the tumor microenvironment. Cancer Res. 2019;79:2054–64.
Chronopoulos A, Robinson B, Sarper M, Cortes E, Auernheimer V, Lachowski D, Attwood S, Garcia R, Ghassemi S, Fabry B, Del Rio Hernandez A. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat Commun. 2016;7:12630.
Bleul T, Ruhl R, Bulashevska S, Karakhanova S, Werner J, Bazhin AV. Reduced retinoids and retinoid receptors’ expression in pancreatic cancer: a link to patient survival. Mol Carcinog. 2015;54:870–9.
Bynigeri RR, Jakkampudi A, Jangala R, Subramanyam C, Sasikala M, Rao GV, Reddy DN, Talukdar R. Pancreatic stellate cell: Pandora’s box for pancreatic disease biology. World J Gastroenterol. 2017;23:382–405.
Hummel D, Aggarwal A, Borka K, Bajna E, Kallay E, Horvath HC. The vitamin D system is deregulated in pancreatic diseases. J Steroid Biochem Mol Biol. 2014;144(Pt B):402–9.
Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93.
Karasic TB, O’Hara MH, Loaiza-Bonilla A, Reiss KA, Teitelbaum UR, Borazanci E, De Jesus-Acosta A, Redlinger C, Burrell JA, Laheru DA, et al. Effect of Gemcitabine and nab-paclitaxel with or without hydroxychloroquine on patients with advanced pancreatic cancer: a phase 2 randomized clinical trial. JAMA Oncol. 2019. https://doi.org/10.1001/jamaoncol.2019.0684.
Awasthi N, Mikels-Vigdal AJ, Stefanutti E, Schwarz MA, Monahan S, Smith V, Schwarz RE. Therapeutic efficacy of anti-MMP9 antibody in combination with nab-paclitaxel-based chemotherapy in pre-clinical models of pancreatic cancer. J Cell Mol Med. 2019;23:3878–87.
Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, Marsh R, Wallace J, Kozloff M, Rajdev L, et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J Clin Oncol. 2015;33:4284–92.
Elahi-Gedwillo KY, Carlson M, Zettervall J, Provenzano PP. Antifibrotic therapy disrupts stromal barriers and modulates the immune landscape in pancreatic ductal adenocarcinoma. Cancer Res. 2019;79:372–86.
Seniutkin O, Furuya S, Luo YS, Cichocki JA, Fukushima H, Kato Y, Sugimoto H, Matsumoto T, Uehara T, Rusyn I. Effects of pirfenidone in acute and sub-chronic liver fibrosis, and an initiation-promotion cancer model in the mouse. Toxicol Appl Pharmacol. 2018;339:1–9.
Pan A, Wang Z, Chen B, Dai W, Zhang H, He B, Wang X, Wang Y, Zhang Q. Localized co-delivery of collagenase and trastuzumab by thermosensitive hydrogels for enhanced antitumor efficacy in human breast xenograft. Drug Deliv. 2018;25:1495–503.
Dong X, Liu HJ, Feng HY, Yang SC, Liu XL, Lai X, Lu Q, Lovell JF, Chen HZ, Fang C. Enhanced drug delivery by nanoscale integration of a nitric oxide donor to induce tumor collagen depletion. Nano Lett. 2019;19:997–1008.
Li J, Huang J, Ao Y, Li S, Miao Y, Yu Z, Zhu L, Lan X, Zhu Y, Zhang Y, Yang X. Synergizing upconversion nanophotosensitizers with hyperbaric oxygen to remodel the extracellular matrix for enhanced photodynamic cancer therapy. ACS Appl Mater Interfaces. 2018;10:22985–96.
Happold C, Gorlia T, Chinot O, Gilbert MR, Nabors LB, Wick W, Pugh SL, Hegi M, Cloughesy T, Roth P, et al. Does valproic acid or levetiracetam improve survival in glioblastoma? A pooled analysis of prospective clinical trials in newly diagnosed glioblastoma. J Clin Oncol. 2016;34:731–9.
Wang L, Liu X, Zhou Q, Sui M, Lu Z, Zhou Z, Tang J, Miao Y, Zheng M, Wang W, Shen Y. Terminating the criminal collaboration in pancreatic cancer: nanoparticle-based synergistic therapy for overcoming fibroblast-induced drug resistance. Biomaterials. 2017;144:105–18.
Cun X, Ruan S, Chen J, Zhang L, Li J, He Q, Gao H. A dual strategy to improve the penetration and treatment of breast cancer by combining shrinking nanoparticles with collagen depletion by losartan. Acta Biomater. 2016;31:186–96.
Tang Y, Liu Y, Wang S, Tian Y, Li Y, Teng Z, Lu G. Depletion of collagen by losartan to improve tumor accumulation and therapeutic efficacy of photodynamic nanoplatforms. Drug Deliv Transl Res. 2019;9:615–24.
Shen H, Gao Q, Ye Q, Yang S, Wu Y, Huang Q, Wang X, Sun Z. Peritumoral implantation of hydrogel-containing nanoparticles and losartan for enhanced nanoparticle penetration and antitumor effect. Int J Nanomed. 2018;13:7409–26.
Whatcott CJ, Han H, Von Hoff DD. Orchestrating the tumor microenvironment to improve survival for patients with pancreatic cancer: normalization, not destruction. Cancer J. 2015;21:299–306.
Li J, Xie C, Huang J, Jiang Y, Miao Q, Pu K. Semiconducting polymer nanoenzymes with photothermic activity for enhanced cancer therapy. Angew Chem Int Ed Engl. 2018;57:3995–8.
Kolosnjaj-Tabi J, Marangon I, Nicolas-Boluda A, Silva AKA, Gazeau F. Nanoparticle-based hyperthermia, a local treatment modulating the tumor extracellular matrix. Pharmacol Res. 2017;126:123–37.
Ayalew L, Acuna J, Urfano SF, Morfin C, Sablan A, Oh M, Gamboa A, Slowinska K. Conjugation of paclitaxel to hybrid peptide carrier and biological evaluation in jurkat and A549 cancer cell lines. ACS Med Chem Lett. 2017;8:814–9.
Liang H, Li X, Chen B, Wang B, Zhao Y, Zhuang Y, Shen H, Zhang Z, Dai J. A collagen-binding EGFR single-chain Fv antibody fragment for the targeted cancer therapy. J Control Release. 2015;209:101–9.
Ishihara J, Ishihara A, Sasaki K, Lee SS, Williford JM, Yasui M, Abe H, Potin L, Hosseinchi P, Fukunaga K, et al. Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci Transl Med. 2019;11:eaau3259.
Yasunaga M, Manabe S, Tarin D, Matsumura Y. Cancer-stroma targeting therapy by cytotoxic immunoconjugate bound to the collagen 4 network in the tumor tissue. Bioconjug Chem. 2011;22:1776–83.
Blackburn JS, Liu I, Coon CI, Brinckerhoff CE. A matrix metalloproteinase-1/protease activated receptor-1 signaling axis promotes melanoma invasion and metastasis. Oncogene. 2009;28:4237–48.
Devy J, Duca L, Cantarelli B, Joseph-Pietras D, Scandolera A, Rusciani A, Parent L, Thevenard J, Pasco SB, Tarpin M, et al. Elastin-derived peptides enhance melanoma growth in vivo by upregulating the activation of Mcol-A (MMP-1) collagenase. Br J Cancer. 2010;103:1562–70.
Rider L, Oladimeji P, Diakonova M. PAK1 regulates breast cancer cell invasion through secretion of matrix metalloproteinases in response to prolactin and three-dimensional collagen IV. Mol Endocrinol. 2013;27:1048–64.
Elenjord R, Allen JB, Johansen HT, Kildalsen H, Svineng G, Maelandsmo GM, Loennechen T, Winberg JO. Collagen I regulates matrix metalloproteinase-2 activation in osteosarcoma cells independent of S100A4. FEBS J. 2009;276:5275–86.
Freise C, Erben U, Muche M, Farndale R, Zeitz M, Somasundaram R, Ruehl M. The alpha 2 chain of collagen type VI sequesters latent proforms of matrix-metalloproteinases and modulates their activation and activity. Matrix Biol. 2009;28:480–9.
Bates AL, Pickup MW, Hallett MA, Dozier EA, Thomas S, Fingleton B. Stromal matrix metalloproteinase 2 regulates collagen expression and promotes the outgrowth of experimental metastases. J Pathol. 2015;235:773–83.
Koikawa K, Ohuchida K, Ando Y, Kibe S, Nakayama H, Takesue S, Endo S, Abe T, Okumura T, Iwamoto C, et al. Basement membrane destruction by pancreatic stellate cells leads to local invasion in pancreatic ductal adenocarcinoma. Cancer Lett. 2018;425:65–77.
Sandri S, Faiao-Flores F, Tiago M, Pennacchi PC, Massaro RR, Alves-Fernandes DK, Berardinelli GN, Evangelista AF, de Lima Vazquez V, Reis RM, Maria-Engler SS. Vemurafenib resistance increases melanoma invasiveness and modulates the tumor microenvironment by MMP-2 upregulation. Pharmacol Res. 2016;111:523–33.
Mahabir R, Tanino M, Elmansuri A, Wang L, Kimura T, Itoh T, Ohba Y, Nishihara H, Shirato H, Tsuda M, Tanaka S. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol. 2014;16:671–85.
Correia AL, Mori H, Chen EI, Schmitt FC, Bissell MJ. The hemopexin domain of MMP3 is responsible for mammary epithelial invasion and morphogenesis through extracellular interaction with HSP90beta. Genes Dev. 2013;27:805–17.
Wu YH, Chang TH, Huang YF, Huang HD, Chou CY. COL11A1 promotes tumor progression and predicts poor clinical outcome in ovarian cancer. Oncogene. 2014;33:3432–40.
Prakobwong S, Charoensuk L, Hiraku Y, Pinlaor P, Pairojkul C, Mairiang E, Sithithaworn P, Yongvanit P, Khuntikeo N, Pinlaor S. Plasma hydroxyproline, MMP-7 and collagen I as novel predictive risk markers of hepatobiliary disease-associated cholangiocarcinoma. Int J Cancer. 2012;131:E416–24.
Orgaz JL, Pandya P, Dalmeida R, Karagiannis P, Sanchez-Laorden B, Viros A, Albrengues J, Nestle FO, Ridley AJ, Gaggioli C, et al. Diverse matrix metalloproteinase functions regulate cancer amoeboid migration. Nat Commun. 2014;5:4255.
Prakobwong S, Yongvanit P, Hiraku Y, Pairojkul C, Sithithaworn P, Pinlaor P, Pinlaor S. Involvement of MMP-9 in peribiliary fibrosis and cholangiocarcinogenesis via Rac1-dependent DNA damage in a hamster model. Int J Cancer. 2010;127:2576–87.
Cho K, Matsuda Y, Ueda J, Uchida E, Naito Z, Ishiwata T. Keratinocyte growth factor induces matrix metalloproteinase-9 expression and correlates with venous invasion in pancreatic cancer. Int J Oncol. 2012;40:1040–8.
Wilkins-Port CE, Ye Q, Mazurkiewicz JE, Higgins PJ. TGF-beta1 + EGF-initiated invasive potential in transformed human keratinocytes is coupled to a plasmin/MMP-10/MMP-1-dependent collagen remodeling axis: role for PAI-1. Cancer Res. 2009;69:4081–91.
Krantz SB, Shields MA, Dangi-Garimella S, Cheon EC, Barron MR, Hwang RF, Rao MS, Grippo PJ, Bentrem DJ, Munshi HG. MT1-MMP cooperates with Kras(G12D) to promote pancreatic fibrosis through increased TGF-beta signaling. Mol Cancer Res. 2011;9:1294–304.
Borrirukwanit K, Pavasant P, Blick T, Lafleur MA, Thompson EW. High threshold of beta1 integrin inhibition required to block collagen I-induced membrane type-1 matrix metalloproteinase (MT1-MMP) activation of matrix metalloproteinase 2 (MMP-2). Cancer Cell Int. 2014;14:99.
Maquoi E, Assent D, Detilleux J, Pequeux C, Foidart JM, Noel A. MT1-MMP protects breast carcinoma cells against type I collagen-induced apoptosis. Oncogene. 2012;31:480–93.
Moss NM, Liu Y, Johnson JJ, Debiase P, Jones J, Hudson LG, Munshi HG, Stack MS. Epidermal growth factor receptor-mediated membrane type 1 matrix metalloproteinase endocytosis regulates the transition between invasive versus expansive growth of ovarian carcinoma cells in three-dimensional collagen. Mol Cancer Res. 2009;7:809–20.
Tatti O, Gucciardo E, Pekkonen P, Holopainen T, Louhimo R, Repo P, Maliniemi P, Lohi J, Rantanen V, Hautaniemi S, et al. MMP16 mediates a proteolytic switch to promote cell-cell adhesion, collagen alignment, and lymphatic invasion in melanoma. Cancer Res. 2015;75:2083–94.
Rodgers UR, Kevorkian L, Surridge AK, Waters JG, Swingler TE, Culley K, Illman S, Lohi J, Parker AE, Clark IM. Expression and function of matrix metalloproteinase (MMP)-28. Matrix Biol. 2009;28:263–72.
Zou X, Feng B, Dong T, Yan G, Tan B, Shen H, Huang A, Zhang X, Zhang M, Yang P, et al. Up-regulation of type I collagen during tumorigenesis of colorectal cancer revealed by quantitative proteomic analysis. J Proteomics. 2013;94:473–85.
Penet MF, Kakkad S, Pathak AP, Krishnamachary B, Mironchik Y, Raman V, Solaiyappan M, Bhujwalla ZM. Structure and function of a prostate cancer dissemination-permissive extracellular matrix. Clin Cancer Res. 2017;23:2245–54.
Xiong G, Deng L, Zhu J, Rychahou PG, Xu R. Prolyl-4-hydroxylase alpha subunit 2 promotes breast cancer progression and metastasis by regulating collagen deposition. BMC Cancer. 2014;14:1.
Yu Y, Liu D, Liu Z, Li S, Ge Y, Sun W, Liu B. The inhibitory effects of COL1A2 on colorectal cancer cell proliferation, migration, and invasion. J Cancer. 2018;9:2953–62.
Ji J, Zhao L, Budhu A, Forgues M, Jia HL, Qin LX, Ye QH, Yu J, Shi X, Tang ZY, Wang XW. Let-7g targets collagen type I alpha2 and inhibits cell migration in hepatocellular carcinoma. J Hepatol. 2010;52:690–7.
Tamiya M, Kobayashi M, Morimura O, Yasue T, Nakasuji T, Satomu M, Kohei O, Takayuki S, Morishita N, Suzuki H, et al. Clinical significance of the serum crosslinked N-telopeptide of type I collagen as a prognostic marker for non-small-cell lung cancer. Clin Lung Cancer. 2013;14:50–4.
Nurmenniemi S, Koivula MK, Nyberg P, Tervahartiala T, Sorsa T, Mattila PS, Salo T, Risteli J. Type I and III collagen degradation products in serum predict patient survival in head and neck squamous cell carcinoma. Oral Oncol. 2012;48:136–40.
Imamura M, Nishimukai A, Higuchi T, Ozawa H, Yanai A, Miyagawa Y, Murase K, Sakita I, Hatada T, Takatsuka Y, et al. High levels at baseline of serum pyridinoline crosslinked carboxyterminal telopeptide of type I collagen are associated with worse prognosis for breast cancer patients. Breast Cancer Res Treat. 2015;154:521–31.
Ferreira AR, Alho I, Shan N, Matias M, Faria M, Casimiro S, Leitzel K, Ali S, Lipton A, Costa L. N-telopeptide of type i collagen long-term dynamics in breast cancer patients with bone metastases: clinical outcomes and influence of extraskeletal metastases. Oncologist. 2016;21:1418–26.
Ganapathi MK, Jones WD, Sehouli J, Michener CM, Braicu IE, Norris EJ, Biscotti CV, Vaziri SA, Ganapathi RN. Expression profile of COL2A1 and the pseudogene SLC6A10P predicts tumor recurrence in high-grade serous ovarian cancer. Int J Cancer. 2016;138:679–88.
Tarpey PS, Behjati S, Cooke SL, Van Loo P, Wedge DC, Pillay N, Marshall J, O’Meara S, Davies H, Nik-Zainal S, et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat Genet. 2013;45:923–6.
Wang Y, Resnick MB, Lu S, Hui Y, Brodsky AS, Yang D, Yakirevich E, Wang L. Collagen type III alpha1 as a useful diagnostic immunohistochemical marker for fibroepithelial lesions of the breast. Hum Pathol. 2016;57:176–81.
Ando H, Aihara R, Ohno T, Ogata K, Mochiki E, Kuwano H. Prognostic significance of the expression of MUC1 and collagen type IV in advanced gastric carcinoma. Br J Surg. 2009;96:901–9.
Fan HX, Li HX, Chen D, Gao ZX, Zheng JH. Changes in the expression of MMP2, MMP9, and ColIV in stromal cells in oral squamous tongue cell carcinoma: relationships and prognostic implications. J Exp Clin Cancer Res. 2012;31:90.
Nystrom H, Naredi P, Hafstrom L, Sund M. Type IV collagen as a tumour marker for colorectal liver metastases. Eur J Surg Oncol. 2011;37:611–7.
Shimizu A, Kobayashi A, Yokoyama T, Motoyama H, Sakai H, Kitagawa N, Notake T, Shirota T, Fukushima K, Miyagawa SI. Correlation between the serum levels of type IV collagen 7 s domain and the risk of intractable ascites following liver resection for hepatocellular carcinoma: a propensity score-matched analysis. Surgery. 2016;160:1244–55.
Pan Z, Li L, Fang Q, Zhang Y, Hu X, Qian Y, Huang P. Analysis of dynamic molecular networks for pancreatic ductal adenocarcinoma progression. Cancer Cell Int. 2018;18:214.
Miyake M, Morizawa Y, Hori S, Tatsumi Y, Onishi S, Owari T, Iida K, Onishi K, Gotoh D, Nakai Y, et al. Diagnostic and prognostic role of urinary collagens in primary human bladder cancer. Cancer Sci. 2017;108:2221–8.
Nie XC, Wang JP, Zhu W, Xu XY, Xing YN, Yu M, Liu YP, Takano Y, Zheng HC. COL4A3 expression correlates with pathogenesis, pathologic behaviors, and prognosis of gastric carcinomas. Hum Pathol. 2013;44:77–86.
Souza P, Rizzardi F, Noleto G, Atanazio M, Bianchi O, Parra ER, Teodoro WR, Carrasco S, Velosa AP, Fernezlian S, et al. Refractory remodeling of the microenvironment by abnormal type V collagen, apoptosis, and immune response in non-small cell lung cancer. Hum Pathol. 2010;41:239–48.
Hou T, Tong C, Kazobinka G, Zhang W, Huang X, Huang Y, Zhang Y. Expression of COL6A1 predicts prognosis in cervical cancer patients. Am J Transl Res. 2016;8:2838–44.
Zhang B, Zhang C, Yang X, Chen Y, Zhang H, Liu J, Wu Q. Cytoplasmic collagen XIalphaI as a prognostic biomarker in esophageal squamous cell carcinoma. Cancer Biol Ther. 2018;19:364–72.
Moilanen JM, Kokkonen N, Loffek S, Vayrynen JP, Syvaniemi E, Hurskainen T, Makinen M, Klintrup K, Makela J, Sormunen R, et al. Collagen XVII expression correlates with the invasion and metastasis of colorectal cancer. Hum Pathol. 2015;46:434–42.
Galiger C, Loffek S, Stemmler MP, Kroeger JK, Mittapalli VR, Fauth L, Esser PR, Kern JS, Meiss F, Lassmann S, et al. Targeting of cell surface proteolysis of collagen XVII impedes squamous cell carcinoma progression. Mol Ther. 2018;26:17–30.
Liu CC, Lin SP, Hsu HS, Yang SH, Lin CH, Yang MH, Hung MC, Hung SC. Suspension survival mediated by PP2A-STAT3-Col XVII determines tumour initiation and metastasis in cancer stem cells. Nat Commun. 2016;7:11798.
Spivey KA, Banyard J, Solis LM, Wistuba II, Barletta JA, Gandhi L, Feldman HA, Rodig SJ, Chirieac LR, Zetter BR. Collagen XXIII: a potential biomarker for the detection of primary and recurrent non-small cell lung cancer. Cancer Epidemiol Biomarkers Prev. 2010;19:1362–72.
Wang Q, Yu J. MiR-129-5p suppresses gastric cancer cell invasion and proliferation by inhibiting COL1A1. Biochem Cell Biol. 2018;96:19–25.
Ivanovic RF, Viana NI, Morais DR, Silva IA, Leite KR, Pontes-Junior J, Inoue G, Nahas WC, Srougi M, Reis ST. miR-29b enhances prostate cancer cell invasion independently of MMP-2 expression. Cancer Cell Int. 2018;18:18.
Guo Q, Zheng M, Xu Y, Wang N, Zhao W. MiR-384 induces apoptosis and autophagy of non-small cell lung cancer cells through the negative regulation of Collagen alpha-1(X) chain gene. Biosci Rep. 2019. https://doi.org/10.1042/BSR20181523.
D’Aniello C, Cermola F, Palamidessi A, Wanderlingh LG, Gagliardi M, Migliaccio A, Varrone F, Casalino L, Matarazzo MR, De Cesare D, et al. collagen prolyl hydroxylation-dependent metabolic perturbation governs epigenetic remodeling and mesenchymal transition in pluripotent and cancer cells. Cancer Res. 2019;79:3235–50.
Vasta JD, Raines RT. Collagen prolyl 4-hydroxylase as a therapeutic target. J Med Chem. 2018;61:10403–11.
Atkinson A, Renziehausen A, Wang H, Lo Nigro C, Lattanzio L, Merlano M, Rao B, Weir L, Evans A, Matin R, et al. Collagen prolyl hydroxylases are bifunctional growth regulators in melanoma. J Invest Dermatol. 2019;139:1118–26.
Nagaraju GP, Park W, Wen J, Mahaseth H, Landry J, Farris AB, Willingham F, Sullivan PS, Proia DA, El-Hariry I, et al. Antiangiogenic effects of ganetespib in colorectal cancer mediated through inhibition of HIF-1alpha and STAT-3. Angiogenesis. 2013;16:903–17.
Dong H, Luo L, Zou M, Huang C, Wan X, Hu Y, Le Y, Zhao H, Li W, Zou F, Cai S. Blockade of extracellular heat shock protein 90alpha by 1G6-D7 attenuates pulmonary fibrosis through inhibiting ERK signaling. Am J Physiol Lung Cell Mol Physiol. 2017;313:L1006–15.
Bellaye PS, Burgy O, Causse S, Garrido C, Bonniaud P. Heat shock proteins in fibrosis and wound healing: good or evil? Pharmacol Ther. 2014;143:119–32.
Henke A, Franco OE, Stewart GD, Riddick AC, Katz E, Hayward SW, Thomson AA. reduced contractility and motility of prostatic cancer-associated fibroblasts after inhibition of heat shock protein 90. Cancers (Basel). 2016;8:E77.
Duarte BDP, Bonatto D. The heat shock protein 47 as a potential biomarker and a therapeutic agent in cancer research. J Cancer Res Clin Oncol. 2018;144:2319–28.
Zhu J, Xiong G, Fu H, Evers BM, Zhou BP, Xu R. Chaperone Hsp47 drives malignant growth and invasion by modulating an ECM gene network. Cancer Res. 2015;75:1580–91.
Yao Q, Kou L, Tu Y, Zhu L. MMP-responsive ‘Smart’ Drug delivery and tumor targeting. Trends Pharmacol Sci. 2018;39:766–81.
Mohsen A, Collery P, Garnotel R, Brassart B, Etique N, Mohamed Sabry G, Elsherif Hassan R, Jeannesson P, Desmaele D, Morjani H. A new gallium complex inhibits tumor cell invasion and matrix metalloproteinase MMP-14 expression and activity. Metallomics. 2017;9:1176–84.
Trackman PC. Lysyl oxidase isoforms and potential therapeutic opportunities for fibrosis and cancer. Expert Opin Ther Targets. 2016;20:935–45.
Wei MG, Sun W, He WM, Ni L, Yang YY. Ferulic acid attenuates TGF-beta1-induced renal cellular fibrosis in NRK-52E cells by inhibiting Smad/ILK/Snail pathway. Evid Based Complement Alternat Med. 2015;2015:619720.
Cortes E, Lachowski D, Robinson B, Sarper M, Teppo JS, Thorpe SD, Lieberthal TJ, Iwamoto K, Lee DA, Okada-Hatakeyama M, et al. Tamoxifen mechanically reprograms the tumor microenvironment via HIF-1A and reduces cancer cell survival. EMBO Rep. 2019;20:e46557.
Szoka L, Karna E, Hlebowicz-Sarat K, Karaszewski J, Boryczka S, Palka JA. Acetylenic derivative of betulin induces apoptosis in endometrial adenocarcinoma cell line. Biomed Pharmacother. 2017;95:429–36.
Jiang H, Liu X, Knolhoff BL, Hegde S, Lee KB, Jiang H, Fields RC, Pachter JA, Lim KH, DeNardo DG. Development of resistance to FAK inhibition in pancreatic cancer is linked to stromal depletion. Gut. 2019. https://doi.org/10.1136/gutjnl-2018-317424.
Wang Z, Li J, Xiao W, Long J, Zhang H. The STAT3 inhibitor S3I-201 suppresses fibrogenesis and angiogenesis in liver fibrosis. Lab Invest. 2018;98:1600–13.
Zhang Q, Hou X, Evans BJ, VanBlaricom JL, Weroha SJ, Cliby WA: LY2157299 Monohydrate, a TGF-betaR1 Inhibitor, Suppresses Tumor Growth and Ascites Development in Ovarian Cancer. Cancers (Basel) 2018, 10.
Colak S, Ten Dijke P. Targeting TGF-beta signaling in cancer. Trends Cancer. 2017;3:56–71.
Averett C, Bhardwaj A, Arora S, Srivastava SK, Khan MA, Ahmad A, Singh S, Carter JE, Khushman M, Singh AP. Honokiol suppresses pancreatic tumor growth, metastasis and desmoplasia by interfering with tumor-stromal cross-talk. Carcinogenesis. 2016;37:1052–61.
Wang T, Fu X, Jin T, Zhang L, Liu B, Wu Y, Xu F, Wang X, Ye K, Zhang W, Ye L. Aspirin targets P4HA2 through inhibiting NF-kappaB and LMCD1-AS1/let-7 g to inhibit tumour growth and collagen deposition in hepatocellular carcinoma. EBioMedicine. 2019;45:168–80.
Khan S, Ebeling MC, Chauhan N, Thompson PA, Gara RK, Ganju A, Yallapu MM, Behrman SW, Zhao H, Zafar N, et al. Ormeloxifene suppresses desmoplasia and enhances sensitivity of gemcitabine in pancreatic cancer. Cancer Res. 2015;75:2292–304.
Ediriweera MK, Tennekoon KH, Samarakoon SR. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin Cancer Biol. 2019. https://doi.org/10.1016/j.semcancer.2019.05.012.
Jung KH, Zhang J, Zhou C, Shen H, Gagea M, Rodriguez-Aguayo C, Lopez-Berestein G, Sood AK, Beretta L. Differentiation therapy for hepatocellular carcinoma: multifaceted effects of miR-148a on tumor growth and phenotype and liver fibrosis. Hepatology. 2016;63:864–79.
Cortes JE, Gutzmer R, Kieran MW, Solomon JA. Hedgehog signaling inhibitors in solid and hematological cancers. Cancer Treat Rev. 2019;76:41–50.
Abd El-Rahman SS, Fayed HM. Targeting AngII/AT1R signaling pathway by Perindopril inhibits ongoing liver fibrosis in rat. J Tissue Eng Regen Med. 2019. https://doi.org/10.1002/term.2940.
Olsson PO, Gustafsson R, In’t Zandt R, Friman T, Maccarana M, Tykesson E, Oldberg A, Rubin K, Kalamajski S. The tyrosine kinase inhibitor imatinib augments extracellular fluid exchange and reduces average collagen fibril diameter in experimental carcinoma. Mol Cancer Ther. 2016;15:2455–64.
Jiang W, Ji M. Receptor tyrosine kinases in PI3K signaling: The therapeutic targets in cancer. Semin Cancer Biol. 2019. https://doi.org/10.1016/j.semcancer.2019.03.006.
Grither WR, Longmore GD. Inhibition of tumor-microenvironment interaction and tumor invasion by small-molecule allosteric inhibitor of DDR2 extracellular domain. Proc Natl Acad Sci USA. 2018;115:E7786–94.
Aguilera KY, Huang H, Du W, Hagopian MM, Wang Z, Hinz S, Hwang TH, Wang H, Fleming JB, Castrillon DH, et al. Inhibition of discoidin domain receptor 1 reduces collagen-mediated tumorigenicity in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2017;16:2473–85.
Brandsma I, Fleuren EDG, Williamson CT, Lord CJ. Directing the use of DDR kinase inhibitors in cancer treatment. Expert Opin Investig Drugs. 2017;26:1341–55.
Lin Y, Zheng Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin Drug Discov. 2015;10:991–1010.
Kobayashi M, Sawada K, Kimura T. Potential of integrin inhibitors for treating ovarian cancer: a literature review. Cancers (Basel). 2017;9:E83.
Compte M, Harwood SL, Munoz IG, Navarro R, Zonca M, Perez-Chacon G, Erce-Llamazares A, Merino N, Tapia-Galisteo A, Cuesta AM, et al. A tumor-targeted trimeric 4-1BB-agonistic antibody induces potent anti-tumor immunity without systemic toxicity. Nat Commun. 2018;9:4809.
Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. 2019;18:99–115.
Alvarez R, Musteanu M, Garcia-Garcia E, Lopez-Casas PP, Megias D, Guerra C, Munoz M, Quijano Y, Cubillo A, Rodriguez-Pascual J, et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br J Cancer. 2013;109:926–33.
Huang H, Brekken RA. The next wave of stroma-targeting therapy in pancreatic cancer. Cancer Res. 2019;79:328–30.
Sielaff CM, Mousa SA. Status and future directions in the management of pancreatic cancer: potential impact of nanotechnology. J Cancer Res Clin Oncol. 2018;144:1205–17.
Li Y, Hong J, Oh JE, Yoon AR, Yun CO. Potent antitumor effect of tumor microenvironment-targeted oncolytic adenovirus against desmoplastic pancreatic cancer. Int J Cancer. 2018;142:392–413.
Eriksson E, Milenova I, Wenthe J, Stahle M, Leja-Jarblad J, Ullenhag G, Dimberg A, Moreno R, Alemany R, Loskog A. Shaping the tumor stroma and sparking immune activation by CD40 and 4-1BB signaling induced by an armed oncolytic virus. Clin Cancer Res. 2017;23:5846–57.
Acknowledgements
We appreciate the help from other teammates.
Funding
This work was supported by grants from the National Science Foundation for Distinguished Young Scholars of China (81625016), the National Natural Science Foundation of China (81871941, 81872366, 81827807, 81802675, and 81702341), the Outstanding Academic Leader Program of the “Technological Innovation Action Plan” of the Shanghai Science and Technology Commission (18XD1401200), and the Young Talented Specialist Training Program of Shanghai.
Author information
Authors and Affiliations
Contributions
SX and HX contributed equally to this work. They conducted information collection and wrote the manuscript. SX, HX, WW, SL, HL, TL, and WZ made the figures and tables. LL and XY contributed equally to this work. They designed the research, revised the manuscript, and provided valuable suggestions for this study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xu, S., Xu, H., Wang, W. et al. The role of collagen in cancer: from bench to bedside. J Transl Med 17, 309 (2019). https://doi.org/10.1186/s12967-019-2058-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-019-2058-1