
Abstract
Liver sinusoidal endothelial cells (LSECs) are highly specialized endothelial cells that represent the interface between blood cells on one side and hepatocytes on the other side. LSECs not only form a barrier within the hepatic sinus, but also play important physiological functions such as regulating hepatic vascular pressure, anti-inflammatory and anti-fibrotic. Pathologically, pathogenic factors can induce LSECs capillarization, that is, loss of fenestra and dysfunction, which are conducive to early steatosis, lay the foundation for the progression of metabolic dysfunction-associated fatty liver disease (MAFLD), and accelerate metabolic dysfunction-associated steatohepatitis (MASH) and liver fibrosis. The unique localization, phenotype, and function of LSECs make them potential candidates for reducing liver injury, inflammation, and preventing or reversing fibrosis in the future.
Similar content being viewed by others

Background
Despite the substantial gains in our understanding of non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH) over the past 2 decades, there has been some dissatisfaction with the terminology “non-alcoholic” which overemphasizes “alcohol” and underemphasizes the root cause of this liver disease, namely, the predisposing metabolic risk factors. As a potential remedy, a name change from NAFLD to MAFLD has been proposed [1] and endorsed by international expert consensus [2,3,4]. MAFLD affects about a quarter of the world’s adult population [2, 5, 6]. The subtype of MAFLD can be characterized as MASH, which is a potentially progressive liver disease that can lead to liver cirrhosis and hepatocellular carcinoma (HCC) [7,8,9]. MAFLD is associated with obesity, insulin resistance and other metabolic abnormalities, collectively referred to as metabolic syndrome [10,11,12]. Current views on the pathogenesis of MAFLD focus on the response of hepatocytes to insulin resistance and lipotoxicity, with immune system and HSCs activation considered secondary events [7].
Hepatic microcirculation environment is mainly composed of LSECs, hepatic stellate cells (HSCs) and Kupffer cells (KCs) [13, 14]. Hepatocytes are arranged in hexagonal lobules, separated from the thin-walled LSECs by the Disse space, where HSCs is located [15]. LSECs assemble hepatic sinusoids, which are characterized by the lack of basement membrane (BM), open fenestration and no diaphragms, forming a permeable barrier [16, 17]. Monocyte-derived resident macrophages, known as KCs, reside in the hepatic sinusoid and are the first line of defense of the liver immune system [18]. LSECs represent the interface between blood and other liver tissues, accounting for about 15 to 20% of the number of hepatocytes, but its role in liver disease has not yet been elucidated and valued [19]. This Review focuses on the physiological function of LSECs and its role in the pathological progress of MAFLD.
The research process of LSECs
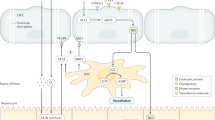
In 1970, Eddie Wisse first observed the rat hepatic sinusoidal fenestration with a transmission electron microscope [20]. This new visualization enabled LSECs to differentiate from other types of cells, including KCs and other vascular endothelial cells [21]. At that time, it was thought that the major part of the transport and exchange of fluid, solutes and particles between the blood and the space of Disse occurred through these open fenestrae [22], that was, the liver was the central organ for lipoprotein metabolism [23]. And the mainstream view was that the change of actin cytoskeleton might play a key role in development of fenestrae [24]. In 1997, Shah et al. proved that LSECs were the main source of liver NO, so they were involved in the regulation of hepatic vascular pressure [25]. In 2000 and beyond, Limmer et al. found that LSECs had immunomodulatory function, that was, they mediated lymphocyte recruitment to the liver and inhibited inflammatory T cell activity to form immune tolerance to circulating soluble antigens [26, 27].These findings laid a solid foundation for subsequent research. In 2003, the concept of capillarization was proposed [28], and this pathological change of LSECs could induce KCs polarization to pro-inflammatory phenotype [29] and HSCs activation, accelerating the progression of MAFLD [15]. MAFLD was closely related to the balance of lipid metabolism, it was then observed that in addition to the LSECs fenestration, the high endocytosis of LSECs was also beneficial to the rapid removal of blood borne ligands, thus maintaining lipid homeostasis [30], and the loss of LSECs transport function could accelerate disease progression. In recent years, LSECs have been gradually discovered to be involved in the progression of many other liver diseases, such as MASH [31], liver fibrosis [15] and HCC [32] by regulating fenestration, secretion of adhesion molecules and vascular secretion signals [33], in which fenestration loss was recognized to be the earliest event [15]. Nowadays, the number of studies on LSECs has increased dramatically, and the research has become more rigorous and meticulous. In 2019, three different groups of endothelial cells were identified by single cell RNA sequencing of human liver, which laid a foundation for future subgroup research and functional refinement of hepatic endothelium [34]. In addition, LSECs specific marker genes have not been introduced before, and the latest research has found that Oit3 was a promising marker gene targeting LSECs, providing a valuable model for studying the complexity of LSECs in liver diseases [35] (Fig. 1).

Schedule of LSECs morphology and function research. The graph shows the main findings of LSECs in the progress of MAFLD. The line chart reflects the total number of publications targeting LSECs in recent years. The research content pointed by the arrows provide a basis for us to understand the morphological structure (lower box) and physiological or pathological function of LSECs (upper box). In the past few decades, publications in this field have rapidly increased, and some targets related to LSECs have entered clinical research through basic trials, all of which are discussed in more detail in this text
Unique features of LSECs
LSECs form blood sinuses with fenestrations
The diameters of sinusoidal fenestrations in normal individuals are between 107+/-1.5 nm [36], and the diameters along the sinusoidal direction vary with oxygen concentration [37]. Fenestration is not unique to hepatic endothelial cells, but also seen in endothelial cells of other organs, such as pancreas [38], kidney [39], spleen [40], bone marrow [41], and even tumor vascular system [42]. However, unlike other endothelial cell groups, liver fenestration lacks a septum or basal layer and is grouped into organized sieve plates, making LSECs highly permeable [39]. Dietary lipids present in circulation must be transported through the hepatic sinusoids for tissue metabolism [43].LSECs are the main channel for two-way lipid exchange between blood and liver parenchyma, allowing the effective transfer of lipoproteins, chylous particle residues and other macromolecules from sinusoidal blood to the Disse space, where they are absorbed by hepatocytes [23, 44]. Secondly, LSECs regulate lipid transfer through its high endocytosis, from cellular components such as collagen and hyaluronic acid to acetylated low-density lipoprotein (LDL) cholesterol, immune complexes and exogenous antigens to maintain the balance of lipid metabolism in the liver [45, 46].
LSECs capillarization
Capillarization is characterized by deposition of ectopic BM, formation of continuous microvascular endothelial cell layer and increased expression of VE-cadherin, which are the initial pathological changes related to MAFLD [47]. The triggers of capillarization have not been fully determined, but excessive dietary micronutrients (including lipids, carbohydrates and intestinal microbial derivatives) may play a role. A study on the addition of 25 different macronutrients and energy to 15-month-old mice showed that the distribution of macronutrients was related to the change of fenestration, porosity was negatively correlated with dietary fat intake, and the fenestration diameter was negatively correlated with protein or carbohydrate intake [48]. In vitro studies have shown that fenestration exclusion occurs after excessive lipid exposure. For example, exposure of human primary LSECs to oxidized low-density lipoprotein (ox-LDL) increased the lectin-like ox-LDL receptor 1 expression at both the mRNA and protein levels in a dose- and time-dependent manner. Ox-LDL stimulation increased reactive oxygen species (ROS) generation and NF-κB activation, upregulated endothelin-1 (ET-1) and caveolin 1 expression, downregulated endothelial nitric oxide synthase (eNOS) expression and reduced the fenestra diameter and porosity [49]. Fenestration is also related to the metabolic changes of intestinal microorganisms caused by diet. The changes of several fatty acid levels (C16:0, C19:0 and C20:4) caused by the higher abundance of Firmicutes and reduced abundance of Bacteroidetes are significantly negatively correlated with the number of fenestrations [48]. In addition, lipopolysaccharide (LPS) may also play a role in inducing the arrangement of fenestrations. A study of rats injected intravenously with LPS for 7 days showed that the diameter and number of fenestrations decreased, and the porosity could be reduced to 40% of the control group [50].
In addition to the phenotypic changes induced by diet, the fenestration arrangement phenomenon may also be related to the signal changes of LSECs itself or crosstalk between the surrounding cells. Hedgehog (Hh) signaling is an important component in the regulation of vasculogenesis, which increases during liver injury and influences the function of liver cells including cholangiocytes, hepatocytes, HSCs and LSECs [51, 52]. The abnormal activation of Hh signal in LSECs is accompanied by the capillarization that was associated with increased expression of inducible nitric oxide synthase (iNOS), eNOS, vascular endothelial growth factor (VEGF)-R1 and ET-1 in vitro [53]. Inhibition of Hh pathway in vivo and in vitro can alleviate early steatosis and fibrosis of liver by promoting fenestration recovery, helping to clear plasma chylous particle residues and antagonizing HSCs activation [54]. In addition, there is evidence that LSECs and HSCs maintain each other’s phenotypic differentiation. LSECs fenestration is maintained by NO downstream of VEGF-A secreted by hepatocytes or HSCs [16, 55, 56]. VEGF-A stimulates NO release from eNOS in LSECs. NO in turn acts through soluble guanylate cyclase (sGC), conversion of guanosine triphosphate to cyclic guanosinc monophosphate (cGMP), and stimulation of protein kinase G (PKG), which can then phosphorylate protein targets. In addition to the VEGF-A-stimulated NO pathway, maintenance of the LSECs phenotype also requires an NO-independent pathway, which remains to be characterized [15]. When VEGF-A/NO secretion decreases, the LSECs fenestration closes significantly and the gatekeeper function is lost, indicating that VEGF-A signaling plays a crucial role in maintaining LSECs differentiation [15]. Bone morphogenetic protein 9 (BMP9) is another circulating factor produced by HSCs, which plays a key role in vascular quiescence. In BMP9 knockout mice, the expression of LSECs terminal differentiation markers (Lyve1, Stab1, Stab2, Ehd3, Cd209b, eNOS, MAF, PLVAP) decreased, the number of basal layer deposition increased and permeable fenestrations decreased significantly [57] (Fig. 2).

The pathological mechanism of capillarization in LSECs. Excessive intake of dietary fat, protein or carbohydrates, as well as changes of intestinal microorganisms or LPS can induce a decrease in the diameter and number of LSECs fenestrations. In addition, ox-LDL exposure increased the lectin-like ox-LDL receptor 1 expression at both the mRNA and protein levels, increased ROS generation and NF-κB activation, and then upregulated ET-1 and caveolin 1 expression, downregulated eNOS expression, thus reducing the fenestra diameter and porosity. Abnormal activation of Hh signal in LSECs is accompanied by increased expression of iNOS, eNOS, VEGF-R1 and ET-1, as well as LSECs capillarization in vitro. Equally important, when the disruption of the local endocrine environment leads to a decrease in the secretion of VEGF-A by hepatocytes and HSCs, NO downstream of eNOS induces a significant closure of the LSECs fenestration through the sGC/cGMP/PKG pathway. Lastly, decreased secretion of BMP9 generated by HSCs also significantly increases the number of basal layer deposition and reduces number of fenestrae by downregulating LSECs terminal differentiation markers. This capillarization of LSECs is accompanied by the loss of gatekeeper function, which leads to the activation of HSCs
Hepatic blood flow regulation
The hepatic sinusoid has a dual blood supply, receiving blood flow from the portal vein (70%) and the hepatic artery (30%) [17]. Blood pressure is balanced in the sinuses and the blood is drained to the hepatic vein and inferior vena cava [58]. The circadian rhythm of hepatic blood flow caused by digestion changes significantly, but the pressure gradient of hepatic vein in normal individuals is kept at 4mmHg or lower, demonstrating the precise regulation of hepatic vascular tension [59]. Intrahepatic shear stress is considered to be the main driving factor of hepatic blood flow regulation [60]. In the liver, as in other vascular beds, endothelial cells can produce vasodilators in response to increased shear stress to lower blood pressure [61]. LSECs are the main source of NO in normal liver, which is generated through shear stress activated eNOS [25]and induce downregulation of vasoconstrictor molecules, including ET-1 [62]. Other molecules released by LSECs that regulate blood flow include the vasodilator CO and metabolites of cyclooxygenase (COX) pathway (thromboxane A2, prostacyclin) [63], all of which act paracrine on HSCs in the Disse space, causing blood pressure to drop. Unlike most vascular beds, blood flow is mainly regulated by smooth muscle cells, which play a limited role in the liver because they are rarely present in the portal vein [64]. Hepatic blood flow regulation is mainly achieved through the contraction of the tentacle-like structure of perivascular HSCs. Healthy LSECs keep HSCs at rest, thus inhibiting its vasoconstriction [65].
Cell interaction and immune regulation
The sieve pore properties of sinusoidal endothelium expose the liver to microbes and food antigens that constantly come from the gastrointestinal tract through the portal vein [66,67,68]. The liver needs to ensure that it does not cause excessive inflammatory activation while eliminating invasive pathogens [69,70,71]. The first site of exposure to these antigens occurs in the hepatic sinusoid. KCs and LSECs are important participants in absorbing and eliminating soluble antigens entering through the portal vein and in determining the nature of any immune response triggered by such antigens [39, 72]. LSECs induce CD4 T cell tolerance through direct cell contact, which leads to interferon-γ or interleukin-17 release inhibition. This lasting inhibition depends on the interaction of co-stimulatory molecules (CD40, CD80, CD86), major histocompatibility complex (MHC) II molecules and programmed death ligand 1 (PD-L1) on LSECs with programmed death 1 (PD-1) and other receptors on CD4 T cells, and is induced by IL-10 abundantly produced in the liver, e.g., by KCs or HSCs [27]. In addition, the interaction between LSECs and CD8 T cells can also induce liver immune tolerance. Using bone-marrow chimeras and a novel transgenic mouse model (Tie2-H-2 K(b) mice) with endothelial cell-specific MHC I expression, it was found that LSECs preferentially absorbed systemically circulating antigen, resulting in cross-presentation on MHC class I molecules, which in turn led to rapid retention and cellular tolerance of antigen-specific native CD8 T cells in the liver [73]. Using electron microscopy, a study demonstrated that liver resident lymphocytes as well as circulating native CD8 T cells made direct contact with hepatocytes through cytoplasmic extensions penetrating the endothelial fenestrations that perforate the LSECs [74]. This unique interaction is significant in inducing liver immune tolerance. However, there is a dynamic regulation of LSECs on the induction of tolerance to CD8 T cells. Although LSECs cross-presentation at low-antigen concentrations resulted in tolerance, they induced CD 8 T cells differentiated into effector T cells at high-antigen concentrations or upon viral infection of LSECs, which can induce the activation of liver immune response [75, 76]. Meanwhile, LSECs also play a certain role in eliminating translocating gut bacteria [71]. KCs are important protective barriers as they engulf translocated microbial products and live bacteria with the promotion of commensal-derived D-lactic acid [77]. LSECs perceive microorganisms and allow KCs and natural killer T cells to locate the area around the portal vein as entry points for invading microorganisms [78]. This immune zonation in the liver protects against translocating gut bacteria [78]. These results highlight specific mechanisms by which LSECs governs the balance between tolerance and immunity.
Maintain HSCs quiescence
The dedifferentiation of LSECs plays a key role in the process of HSCs activation and fibrosis [79,80,81]. Activation of HSCs, which transdifferentiates from resting, vitamin A storage cells into proliferative, fibrotic myofibroblasts, has now been identified as a major driver of fibrosis in mice and human liver injury [82]. In vitro studies have shown that differentiated LSECs can prevent HSCs activation and promote the return of activated HSC (aHSC) to quiet HSC (qHSC) through VEGF-A-stimulated NO production, but LSECs will lose this effect during dedifferentiation or capillarization [65, 83],
The role of LSECs during hepatic steatosis
The capillarization of LSECs occurs in the very early stage of MAFLD, before the establishment of steatosis [84, 85], and before the appearance of activated KCs and HSCs [29]. The phenotypic changes of LSECs promote the development of liver steatosis by preventing the release of very-low-density lipoprotein (VLDL) from hepatocytes to the hepatic sinusoid, resulting in an increase in the storage of total cholesterol (TC) and triglycerides (TG) in the liver [84].In addition, by preventing chylous particle residues into hepatocytes, it leads to a significant increase in plasma cholesterol triglyceride and LDL levels, as well as the production of de novo lipids in the liver, which promote hyperlipidemia and early pathological changes of MAFLD [86].
Previous studies have shown that severe steatosis without inflammation or fibrosis can induce portal hypertension and hyperdynamic circulation, accompanied by intrahepatic vascular hyperreactivity and extrahepatic vascular hyporeactivity [87]. At the same time, the increase of intrahepatic vascular resistance driven by vascular dysfunction may also promote the progress of MAFLD through intralobular hypoxia, and the increase of portal pressure is positively correlated with the severity of steatosis [87,88,89]. It has been determined that the mechanisms leading to increased portal resistance include mechanical factors, which are a direct result of fibrosis deposition, and other factors associated with endothelial dysfunction, insufficient production of NO in the liver, increased production of vasoconstrictors, changes in microvascular structure and increased contraction of HSCs [90,91,92,93,94].
Intact endothelial cells play a crucial role in vascular tension, as most endogenous vasodilators and constrictors act through mechanisms involving endothelial cells [87]. Endothelial dysfunction, characterized by reduced response to the vasodilator acetylcholine and impaired endothelial NO production, is known to exist in liver cirrhosis and is believed to be one of the causes of cirrhosis-associated portal hypertension [88, 93]. Thromboxane A2 (TXA2) is a vasoconstrictor derived from arachidonic acid through COX and thromboxane synthase, and acts through receptors on smooth muscle cells and HSCs. It is also related to the pathogenesis of increased intrahepatic resistance in liver cirrhosis [95, 96]. The overexpression of COX and the increase of TXA2 and ET-1 production in the perfusion model of MAFLD rats resulted in sinusoidal curve, hemodynamic changes and increased vasoconstriction [97]. Equally important, steatosis animals showed significant changes in liver microvascular structure, and the classical arrangement of sine waves arranged in parallel and separated by the regular trabeculae of hepatocytes was replaced by completely chaotic irregular and flat vascular patterns. These vessels have many interconnected pipes and the presence of multiple “bubbles”, that is, circular extended sine waves at the blind end. This may represent vascular blockage or local leakage due to normal wall destruction or neovascularization [88]. In addition, by using spontaneous immortalized cell lines of HSC origin, studies have shown that HSCs in liver cirrhosis enhance the wall coverage of sinusoidal vessels, and because of the contractile nature of HSCs, this “pathological sinusoidal remodeling” process further promotes high resistance, contractile sinusoidal angiogenesis [98,99,100]. In fact, by recruiting and activating HSCs to the vascular wall, and extending the tentacle-like structure that surrounds the vascular lumen and the adjacent LSECs, HSCs have the ability to adjust themself around the vascular lumen in an effective way to achieve these pathological changes [101, 102] (Fig. 3).

LSECs related pathological mechanism of hepatic steatosis. Normal hepatic sinusoids receive dual blood flow from the portal vein and hepatic artery, and the fenestration of hepatic sinusoids allows the absorption of TC, TG and LDL from the bloodstream into hepatocytes, as well as the release of VLDL from hepatocytes to the bloodstream. However, in the very early stage of MAFLD, the phenotypic changes of LSECs cause an increase in the storage of TC and TG in the liver by preventing the release of VLDL from hepatocytes and by preventing chylous particle residues and LDL into hepatocytes, leading to hyperlipidemia and hepatic steatosis. At the same time, insufficient NO production and increased production of COX, TXA2 and ET-1 in the liver lead to hemodynamic changes and increased vasoconstriction, which induce intralobular hypoxia to promote the progression of MAFLD. Equally important, the hepatic microvascular structure of steatosis changes significantly and the parallel sinusoidal waves are replaced by completely irregular and flat vascular patterns. HSCs recruited into the Disse space aggravates endothelial dysfunction and portal hypertension by enhancing the wall coverage of sinusoidal vessels and contraction, thereby inducing the progression of MAFLD
LSECs and MASH
Up to 20 -30% of patients with liver steatosis will develop MASH, and more than 30% of patients with MASH may develop cirrhosis or HCC [103]. In the local immune environment of MASH, in addition to the traditional immune cells such as KCs, dendritic cells, lymphocytes, neutrophils and mast cells, LSECs also play a strong immune function when the liver is under severe pressure [104,105,106]. LSECs express effective scavenger proteins that can clear waste from the bloodstream, making them highly sensitive sentinel cells in the liver [107,108,109]. Meanwhile, LSECs have a unique fenestration phenotype, with a window structure that facilitates the recruitment of immune cells from the blood to liver parenchymal cells, all of which are involved in the pathological process of MASH [110].
In physiological conditions and early stages of MAFLD, LSECs are known for its anti-inflammatory effects [111]. However, during NALFD progression, LSECs then acquire pro-inflammatory phenotypes and functions, which in turn exacerbates MASH progression [84]. Pro-inflammatory phenotype of LSECs referred to in this review as LSECs endotheliopathy manifested by the release of pro-inflammatory mediators including cytokines and chemokines, aberrant expressions of adhesion molecules, acquisition of angiogenic properties, the loss of fenestrae and the formation of basement membranes [112]. There are several potential candidate mediators, including products derived from visceral adipose tissue, such as ox-LDL, palmitate and adipokines, that can induce LSECs endotheliopathy. In vitro studies showed that stimulation of LSECs with ox-LDL and palmitate activated NF-kB and TLR-4, respectively [49, 113, 114]. In addition, in the case of metabolic syndrome, the circulating concentrations of several adipokines, including TNF-a and IL-6, increase in the portal vein and may lead to LSECs endotheliopathy [115]. The increase of intestinal permeability and plasma concentration of LPS may also contribute to LSECs endotheliopathy [116,117,118]. The LSECs endotheliopathy during MASH is characterized by progressive overexpression of adhesion molecules, including intercellular cell adhesion molecule-1, vascular cell adhesion molecule-1 (VCAM-1) and vascular adhesion protein 1, as observed in the MASH mouse model [119,120,121,122,123,124]. LSECs also produced many pro-inflammatory mediators in MASH, including TNF-α, IL-6, IL-1 and CCL2 [119, 125], after recognizing pro-inflammatory mediators through PRRs and SRs. White blood cells homing to the liver and adhering to LSECs is a key element in the pathogenesis of MASH, and it is also a strictly regulated multi-step process [84]. VCAM-1 is a member of the cell adhesion molecule immunoglobulin superfamily, which is mainly expressed on the surface of endothelial cells and regulates the firm adhesion between leukocytes and endothelial cells [126]. Lipotoxic stress enhances the expression of VCAM-1 in LSECs through MLK3/P38 signaling, while inhibition of VCAM-1 can inhibit adhesion and transendothelial migration of monocytes across LSECs (from wild-type mice fed a high-fat diet and from ob/ob obese mice) and improves liver inflammation [31, 124].
In addition, MASH is associated with LSECs autophagy deficiency. The number of LSECs with autophagy vacuoles in MASH patients is half of that in normal subjects [127]. In mice fed a high-fat diet or treated with carbon tetrachloride, the LSECs of endothelial autophagy deficiency induced by IL-6 and TNF-α in the liver show upregulation of genes related to inflammatory pathways (CCL2, CCL5, CD68, VCAM-1), hepatocyte apoptosis (lytic Caspase-3) and peri-sinusoidal fibrosis. Consistently, autophagy defects enhance the expression of inflammation-related genes (CCL2, CCL5, IL-6 and VCAM-1), apoptosis (lytic Caspase-3) and endothelial-interstitial transformation characteristics (α-SMA, TGF-β1, COLLA2 expression) by inhibiting AMPKα in LSECs cell lines [127]. At the same time, Notch/eNOS signal pathway plays an important role in the progression of MASH. Notch activation aggravates MASH by inhibiting eNOS transcription in methionine-choline-deficient diet-induced MASH mouse models, while pharmacological activation of eNOS can reduce liver inflammation and lipid deposition caused by Notch activation [128] (Fig. 4).

Ox-LDL, palmitate, adipokines (TNF-a and IL-6) and LPS lead to inflammatory phenotypes in LSECs, which induce LSECs to produce pro-inflammatory mediators in MASH, including TNF-α, IL-6, IL-1 and CCL2. Lipotoxic stress can enhance the VCAM-1 expression of LSECs through MLK3/P38 signal transduction, and stimulate monocytes to gather in the liver through fenestrae and activate into a pro-inflammatory state. In addition, IL-6 and TNF-α in local microenvironment down-regulate autophagy and increase the expression of CCL2, CCL5, IL-6, Caspase-3, α-SMA and TGF-β1 by inhibiting AMPKα, and activation of Notch signal in damaged LSECs inhibits eNOS transcription, which aggravates liver inflammation and fibrosis, and then promotes the progression of MASH. MOMF: monocyte-derived macrophages
LSECs and liver fibrosis
LSECs and HSCs maintain their respective differentiation phenotypes, but the maintenance of LSECs differentiation requires the secretion of VEGF-A by HSCs and hepatocytes to function through the NO/sGC/cGMP pathway and the NO-independent pathway [55]. Meanwhile, differentiated LSECs can prevent HSCs activation and induce aHSC to reverse to resting state, preventing disease progression or promoting fibrosis regression during sustained injury [15]. But when LSECs undergo dedifferentiation or “capillarization” the protective effect disappears [65]. Using rats transplanted with transgenic enhanced green fluorescent protein-positive BM to identify the LSECs mediator that maintains HSCs quiescence. The study shows that capillarization is due to repair of injured LSECs by BM endothelial progenitors that engraft but fail to fully mature. Lack of maturation of BM-derived LSECs is due to cell autonomous pathways that inhibit the nitric oxide pathway [80]. Heparin-binding epidermal growth factor (HB-EGF) is a signal to keep HSCs still [129]. Undifferentiated LSECs cannot shed HB-EGF from the cytoplasmic membrane, thus creating an environment conducive to liver fibrosis [80].
Hh pathway is a key signaling pathway that regulates cell fate decision-making, covering proliferation, differentiation, migration, and apoptosis [130,131,132]. It plays a crucial role in histogenesis during fetal development [132]. In the process of inducing LSECs spontaneous capillarity in vitro, Hh signal is activated, while inhibition of Hh pathway reduces Hh ligands, mesenchymal genes and capillarization markers, indicating that Hh pathway can regulate LSECs capillarization in vitro. In vivo, Hh signal activation promotes the formation of LSECs tubular structure and the accumulation of hepatic myofibroblasts, while blocking Hh pathway can prevent LSECs capillary formation and liver fibrosis [133, 134]. In addition, studies have shown that after bile duct ligation, the expression of adipocyte-fatty acid binding protein (A-FABP) in mouse LSECs is induced, and the gene ablation or pharmacological inhibition of A-FABP can reduce ligation or carbon tetrachloride-induced liver fibrosis in mice. In terms of mechanism, elevated A-FABP promotes LSECs capillarization by activating Hh signal transduction, which in turn impairs the gatekeeper function of LSECs to HSCs activation. In addition, LSECs-derived A-FABP acts directly on HSCs in a paracrine manner, which enhances the trans-activation of TGF-β1 by activating c-Jun N-terminal kinase (JNK)/c-Jun signal transduction, thus aggravating liver fibrosis induced by HSCs activation [54]. It is suggested that Hh signal pathway is a crucial pathway in liver fibrosis.
Organ specific cytokines derived from LSECs, also known as “vascular secretion factors,” are involved in liver development, homeostasis, and disease pathogenesis [135,136,137]. LSECs secrete BMP-2/6 to regulate iron metabolism by controlling the secretion of ferritin in hepatocytes [138]. Secreted Wnt-2/9b regulates liver growth and maturation, metabolic liver zoning and liver regeneration [139, 140]. Transcription factor GATA4 is the main regulator of LSECs during early liver development, which controls embryonic stem cell migration and fetal hematopoiesis [47]. The genetic defect of Gata4 in adult mouse LSECs leads to continuous endothelial dedifferentiation and peri-sinusoidal liver fibrosis, which involves downregulation of BMP-2 and Wnt-2 signaling, activation of transcription factor MYC, and de novo expression of hepatic stellate cell activating cytokine platelet-derived growth factor (PDGF) subunit B. This suggests that endothelial GATA4 prevents perisinusoidal liver fibrosis by inhibiting MYC activation and fibrogenic vascular secretion signal transduction at the chromatin level [33, 47]. In addition, the vascular secretion signals of LSECs are correlated with an increase in mechanical tensile strength, making them highly suitable for sensing stiffness and generating vascular secretion programs that regulate liver fibrosis and portal hypertension [141, 142]. Glycolytic enzymes, especially phosphofructokinase 1 isoform P (PFKP), are enriched in specific focal adhesion proteins isolated from gel-fixed LSECs, and stiffness causes PFKP recruitment to plaques, which is parallel to the increase in glycolysis. Mechanically, glycolysis promotes CXCL1 expression through nuclear pore changes and increased NF-kB translocation. The secretion of CXCL1 induces neutrophil infiltration, which in turn promotes early liver fibrosis and portal hypertension [141, 143,144,145].
Notch signal of endothelial cells plays an important role in the regulation of liver homeostasis [146,147,148]. In current research, endothelial Notch activation destroys liver homeostasis by weakening eNOS/sGC signal transduction, resulting in reduced fenestration, increased BM and increased liver fibrosis, while pharmacological activation of sGC can reverse the phenotype of LSECs dedifferentiation [149]. In addition, autophagy maintains LSECs homeostasis, and the pharmacological or genetic downregulation of endothelial autophagy can induce cell dysfunction and decrease of intrahepatic NO, leading to oxidative stress in the body and aggravating liver fibrosis [127, 150]. In the early stage of liver diseases, autophagy flux helps to maintain endothelial phenotype and protect LSECs from oxidative stress. Therefore, selectively enhancing autophagy of LSECs in the early stage may be an attractive method to change the course of the disease and prevent fibrosis progression [150] (Fig. 5).

LSECs related pathological mechanism of liver fibrosis. When the stiffness of LSECs increases due to mechanical stretching, PFKP recruits specific macular adhesion proteins, and the increased glycolysis promotes CXCL1 expression and neutrophil infiltration, which in turn promote hepatic portal hypertension and early fibrosis. HB-EGF is the signal to keep HSCs still, but undifferentiated LSECs cannot shed HB-EGF from the cytoplasmic membrane, thus creating an environment conducive to HSCs activation. In addition, the down-regulation of LSECs autophagy caused by chronic liver injury leads to cellular dysfunction and the decrease of intrahepatic NO, leading to oxidative stress in vivo. Elevated A-FABP promotes the capillarization of LSECs by activating Hh signal transduction, hence impairing the gatekeeper function of LSECs. The derived A-FABP also acts directly on HSCs in a paracrine manner, which enhances the transactivation of TGF-β1 by activating JNK/c-Jun signal transduction, thus aggravating liver fibrosis caused by HSCs activation. Notch signal of endothelial cells plays an important role in the regulation of liver homeostasis. Endothelial Notch activation destroys liver homeostasis by weakening eNOS/sGC signal transduction, resulting in a decrease in fenestration and an increase in BM. In addition, Gata4 inhibition leads to down-regulation of BMP-2 and Wnt-2 signals, activation of MYC and ab initio expression of PDGFB, which in turn aggravate the activation of HSCs and perisinusoidal liver fibrosis
LSECs and HCC
MAFLD is already the fastest growing cause of HCC in the USA, France and the UK [9]. Inflammation is a key factor in the progression of HCC [151, 152]. Liver tumor is the result of proliferative and invasive characteristics of precancerous lesions, which are caused by genetic and epigenetic changes developed in the context of persistent inflammatory liver damage [153]. HCC induces phenotypic changes in peripheral LSECs, which helps to reduce the anti-tumor immune response [154]. On the other hand, capillarized LSECs can participate in angiogenesis, coagulation and fibrinolysis events during tumorigenesis [155], indicating the role of LSECs in tumor vascular remodeling during HCC progression [156].
PD-L1 and PD-L2, as well as costimulatory molecules CD80 and CD86, are expressed by LSECs as part of their antigen-presenting function [157]. These structures constitute ligands for immune checkpoint PD1 and cytotoxic T lymphocyte antigen 4 (CTLA4) in T cells, respectively [158,159,160]. In the interaction, the activation of T cells is inhibited and a state of tolerance differentiation promoted by locally produced IL-10 is obtained [157]. Although LSECs increase the expression of CD151, which regulates the activity of VCAM-1 and cooperates with T cell to recruit [161]. However, the overexpression of PD-L1 in LSECs during HCC [162, 163] leads to the inhibition of T cell function and limits T cell anti-tumor activity [164]. In addition, circulating fatty acid binding protein 4 (FABP4) levels are elevated in non-HCC or MAFLD patients and are associated with liver inflammation and fibrosis [165]. Recently, it has been proven that there may also be a correlation between FABP4 and the progression of HCC. LSECs exposed to high concentrations of glucose, insulin or VEGF-A can induce hepatocyte proliferation by releasing FABP4. In mice fed a high-fat diet, downregulation of FABP4 inhibited the growth of HCC [166]. Therefore, it can be speculated that the FABP4 from LSECs is helpful to the development of HCC [84].
In the same line, the peritumoral endothelial cells isolated from HCC patients proliferate more in the culture with IL-6 and soluble IL-6 receptor. IL-6 binds to the IL-6 receptor and then triggers the Janus kinase associated with the receptor, stimulating phosphorylation and activating signal transducer and activator of transcription 3 to initiate downstream angiogenesis, which leads to highly vascularized tumors and development of tumor occurrence. In the process of tumor occurrence, a large number of macrophages exist in the peritumoral liver tissue. IL-6 and IL-6 receptor are secreted by peri-tumor endothelial cells and macrophages, respectively [167, 168]. These data indicate that peritumoral endothelial cells play a major role in the progression of HCC [169] (Fig. 6).

LSECs related pathological mechanism of HCC. The PD-L1/2 and CD80/86 expressed on LSECs constitute the ligands of PD1 and CTLA4 in T cells, respectively. In the interaction, the activation of T cells is inhibited and a state of tolerance differentiation promoted by locally produced IL-10 is obtained. In the microenvironment, LSECs exposed to high concentration of glucose, insulin or VEGF-A can induce hepatocyte proliferation and promote HCC growth by releasing FABP4. In addition, IL-6 and IL-6 receptor secreted by peritumoral endothelial cells and macrophages lead to highly vascularized tumors, which simultaneously aggravate the development of tumor occurrence
Treatment of LSECs in liver diseases
The high prevalence and clinical importance of MAFLD have surfaced [170, 171]. However, no effective treatment strategy for MAFLD has been found. Improving diet quality and increasing physical activity are the only management methods available at present [172,173,174,175,176]. From a liver-centered point of view, in order to avoid over-diagnosis and over-treatment, general practitioners should focus on the diagnosis and treatment of MASH patients with moderate to severe fibrosis [177]. Cell adhesion molecules such as integrin β 1 (ITG β 1) and VCAM-1 play a key role in the development of MASH. Under lipotoxic stress, ITG β 1 is released from hepatocytes as a cargo of extracellular vesicles and mediates the adhesion of monocytes to LSECs, which is an important step in liver inflammation [178]. VCAM-1 is released from LSECs, increasing the number and adhesion of proinflammatory monocytes in the liver, thereby promoting the progress of MASH [179]. In MASH mouse model, blocking ITG β 1 or VCAM-1 can reduce liver inflammation, injury and fibrosis [31, 180]. Therefore, the inhibition of cell adhesion molecules may be a new therapeutic strategy for MASH [31, 178].
Statins up-regulate Kruppel-like factor 2 (KLF2) in LSECs [181]. KLF2 induces vascular protection and HSCs inactivation through paracrine mechanism mediated by KLF2/NO/sGC signal, thus improving hepatic endothelial function and hepatic fibrosis in the experimental model of liver cirrhosis [182]. Peroxisome proliferator-activated receptor α (PPARα) is a ligand-activated transcription factor that regulates genes related to vascular tension, oxidative stress and fibrogenesis, and is related to the development of and portal hypertension [183, 184]. Fenofibrate can reduce the production of COX-1 and thromboxane in rats, increase the bioavailability of NO in LSECs, reduce portal pressure and hepatic fibrosis in cirrhotic rats, and improve hepatic endothelial dysfunction by activating PPAR α [185, 186]. A fairly short-term (6-month) study conducted by Belfort and colleagues confirmed that pioglitazone can reverse MASH in a short period of time [187]. In addition, a meta-analysis of all available randomized trials showed that pioglitazone was beneficial in patients with advanced fibrosis [187,188,189,190].These findings confirm the hypothesis that the improvement of metabolic damage may help slow the progression of fibrosis [191]. PX20606 (PX), a novel non-steroidal selective farnesate X receptor (FXR) agonist, can induce hepatic sinusoidal vasodilation (CTH, DDAH1, eNOS and GCH1 up-regulated) and reduce intrahepatic vasoconstriction (ET-1 and p-Moesin down-regulated). In liver cirrhosis, PX can improve endothelial dysfunction and normalize the overexpression of VEGF-A, PDGF and angiopoietin. In human LSECs, PX treatment can significantly reduce portal vein pressure by inhibiting hepatic sinusoid remodeling [192].
During the development of liver fibrosis, capillarization of LSECs restricts the exchange of substances between blood and the Disse space, further accelerating the activation of HSCs and the process of fibrosis [193, 194].The Disse space therapy drugs are often ignored, which remains the main bottleneck of HSCs targeted therapy for liver fibrosis. However, this challenge can be preliminarily addressed by pretreatment with soluble guanylate cyclase stimulator, Riociguat, and then targeted delivery of insulin growth factor 2 receptor-mediated antifibrotic drug JQ1 by peptide nanoparticles (IGNP). Riociguat can reverse sinusoidal capillarization and maintain relatively normal LSECs porosity, thereby promoting the transport of IGNP-JQ1 through the sinusoidal endothelial wall and its accumulation in the Disse space. IGNP-JQ1 is then selectively absorbed by aHSCs, inhibiting its proliferation and reducing collagen deposition in the liver. This combination strategy can significantly eliminate the fibrosis induced by carbon tetrachloride or methionine-deficient diet in MASH mice. The strategy of restoring the LSECs fenestration through Riociguat represents a promising treatment for liver fibrosis [195, 196] (Fig. 7).

Treatment of LSECs in liver diseases. ITG β1 from hepatocytes and VCAM-1 from LSECs mediate the recruitment and adhesion of monocytes to LSECs in the development of MASH, so blocking ITG β1 or VCAM-1 can reduce liver inflammation and may be a new therapeutic strategy for MASH. Statins up-regulate KLF2 in LSECs. KLF2 induces HSCs inactivation through paracrine mechanism mediated by KLF2/NO/sGC signal, thus improving hepatic endothelial function and hepatic fibrosis. Fenofibrate can reduce the production of COX-1 and thromboxane, increase the bioavailability of NO in LSECs, reduce hepatic fibrosis and improve hepatic endothelial dysfunction by activating PPARα. In addition, pioglitazone is also beneficial in patients with advanced fibrosis. PX is a novel non-steroidal selective FXR agonist, which can induce hepatic sinusoidal vasodilation (CTH, DDAH1, eNOS and GCH1 up-regulated) and reduce intrahepatic vasoconstriction (ET-1, p-Moesin, VEGF-A, PDGF and angiopoietin down-regulated), thus improving endothelial dysfunction. Newly discovered, Riociguat can reverse sinusoidal capillarization and maintain relatively normal LSECs porosity, thereby promoting the transport of IGNP-JQ1 through the sinusoidal endothelial wall, which is then selectively absorbed by aHSCs in the Disse space to reduce collagen deposition in the liver. Regarding the treatment of HCC, Camrelizumab and Temlimumab /Lpilimumab can target PD1 and CTLA4 on T cells separately, thus inhibiting the secretion of IL-10 and immune tolerance
Blocking drugs targeting new angiogenesis, cell proliferation, cell survival or cell movement signaling pathways is a consideration strategy in liver tumor treatment [197]. In clinical trials of HCC (Table 1), first-line drugs Sorafenib [198] or Lenvatinib [199], as well as second-line drugs Regorafenib [200] or Cabozantinib [201], have been shown to improve clinical outcomes [202]. In addition, LSECs express ligands related to immune checkpoints that inhibit or stimulate T cell responses. Therefore, monoclonal antibodies targeting PD1, Camrelizumab, can also alleviate objective symptoms in patients and improve survival rate [203, 204]. Monoclonal antibodies targeting CTLA4, such as Temlimumab and Lpilimumab, are being studied in HCC patients, and both drugs are currently in phase III clinical trials (NCT03412773) [205].
Conclusions
As the gatekeeper of liver dynamic balance, the importance of LSECs in MAFLD and derived diseases is often ignored. This is closely related to the characteristics of these cells, as the research conclusions of LSECs are partly based on in vitro experiments. However, LSECs rapidly dedifferentiates after isolation and cultivation, resulting in differences between the in vitro model and the actual pathological mechanism. New experimental methods, such as adding specific matrix and biomechanical stimulation, are needed to maintain the differentiation characteristics of LSECs in vitro and improve the credibility of the model, so as to explore the potential of LSECs in the prevention and treatment of MAFLD.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- LSECs:
-
Liver sinusoidal endothelial cells
- MAFLD:
-
Metabolic dysfunction-associated fatty liver disease
- MASH:
-
Metabolic dysfunction-associated steatohepatitis
- NAFLD:
-
Non-alcoholic fatty liver disease
- NASH:
-
Non-alcoholic steatohepatitis
- HCC:
-
Hepatocellular carcinoma
- HSCs:
-
Hepatic stellate cells
- KCs:
-
Kupffer cells
- BM:
-
Basement membrane
- LDL:
-
Low-density lipoprotein
- ox-LDL:
-
Oxidized low-density lipoprotein
- ROS:
-
Reactive oxygen species
- ET-1:
-
Endothelin-1
- eNOS:
-
Endothelial nitric oxide synthase
- LPS:
-
Lipopolysaccharide
- Hh:
-
Hedgehog
- iNOS:
-
Inducible nitric oxide synthase
- VEGF:
-
Vascular endothelial growth factor
- sGC:
-
Soluble guanylate cyclase
- cGMP:
-
Cyclic guanosinc monophosphate
- PKG:
-
Protein kinase G
- BMP9:
-
Bone morphogenetic protein 9
- COX:
-
Cyclooxygenase
- MHC:
-
Major histocompatibility complex
- PD-L1:
-
Programmed death ligand 1
- PD-1:
-
Programmed death 1
- aHSC:
-
Activated HSC
- qHSC:
-
Quiet HSC
- VLDL:
-
Very-low-density lipoprotein
- TC:
-
Total cholesterol
- TG:
-
Triglycerides
- TXA2:
-
Thromboxane A2
- VCAM-1:
-
Vascular cell adhesion molecule-1
- MOMF:
-
Monocyte-derived macrophages
- HB-EGF:
-
Heparin-binding epidermal growth factor
- A-FABP:
-
Adipocyte-fatty acid binding protein
- JNK:
-
Jun N-terminal kinase
- PDGF:
-
Platelet-derived growth factor
- PFKP:
-
Phosphofructokinase 1 isoform P
- CTLA4:
-
Cytotoxic T lymphocyte antigen 4
- FABP4:
-
Fatty acid binding protein 4
- ITG β 1:
-
Integrin β 1
- KLF2:
-
Kruppel-like factor 2
- PPARα:
-
Peroxisome proliferator-activated receptorα
- PX:
-
PX20606
- FXR:
-
Farnesate X receptor
- IGNP:
-
Peptide nanoparticles
References
Younossi ZM, Rinella ME, Sanyal AJ, Harrison SA, Brunt EM, Goodman Z, Cohen DE, Loomba R. From NAFLD to MAFLD: implications of a premature change in terminology. Hepatology. 2021;73(3):1194–8.
Eslam M, Sanyal AJ, George J. MAFLD: A Consensus-Driven proposed nomenclature for metabolic Associated fatty liver disease. Gastroenterology. 2020;158(7):1999–2014. e1991.
Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, Zelber-Sagi S, Wai-Sun Wong V, Dufour JF, Schattenberg JM, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020;73(1):202–9.
Shaltout I, Alkandari H, Fouad Y, Hamed AE. Arabic Association for the Study of Diabetes and Metabolism (AASD) endorsing the MAFLD definition of fatty liver disease. J Hepatol. 2022;76(3):739–40.
Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, Bugianesi E. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11–20.
Sarin SK, Kumar M, Eslam M, George J, Al Mahtab M, Akbar SMF, Jia J, Tian Q, Aggarwal R, Muljono DH, et al. Liver diseases in the Asia-Pacific region: a Lancet Gastroenterology & Hepatology Commission. Lancet Gastroenterol Hepatol. 2020;5(2):167–228.
Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908–22.
Younossi ZM. Non-alcoholic fatty liver disease - A global public health perspective. J Hepatol. 2019;70(3):531–44.
Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2021;18(4):223–38.
Åberg F, Byrne CD, Pirola CJ, Männistö V, Sookoian S. Alcohol consumption and metabolic syndrome: clinical and epidemiological impact on liver disease. J Hepatol. 2023;78(1):191–206.
Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S. Nonalcoholic steatohepatitis: a review. JAMA. 2020;323(12):1175–83.
Targher G, Corey KE, Byrne CD, Roden M. The complex link between NAFLD and type 2 diabetes mellitus - mechanisms and treatments. Nat Rev Gastroenterol Hepatol. 2021;18(9):599–612.
Fernández-Iglesias A, Gracia-Sancho J. How to face Chronic Liver Disease: the Sinusoidal Perspective. Front Med (Lausanne). 2017;4:7.
Kumar S, Duan Q, Wu R, Harris EN, Su Q. Pathophysiological communication between hepatocytes and non-parenchymal cells in liver injury from NAFLD to liver fibrosis. Adv Drug Deliv Rev. 2021;176:113869.
Xie G, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC, Gaarde WA, Deleve LD. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology. 2012;142(4):918–e927916.
Marrone G, Shah VH, Gracia-Sancho J. Sinusoidal communication in liver fibrosis and regeneration. J Hepatol. 2016;65(3):608–17.
Gracia-Sancho J, Caparrós E, Fernández-Iglesias A, Francés R. Role of liver sinusoidal endothelial cells in liver diseases. Nat Rev Gastroenterol Hepatol. 2021;18(6):411–31.
Gracia-Sancho J, Marrone G, Fernández-Iglesias A. Hepatic microcirculation and mechanisms of portal hypertension. Nat Rev Gastroenterol Hepatol. 2019;16(4):221–34.
Maslak E, Gregorius A, Chlopicki S. Liver sinusoidal endothelial cells (LSECs) function and NAFLD; NO-based therapy targeted to the liver. Pharmacol Rep. 2015;67(4):689–94.
Braet F, Vanbesien J, De Zanger R, Wisse E. Ageing of the liver sieve and pseudocapillarisation. Lancet. 2002;360(9340):1171–2.
Wisse E. An electron microscopic study of the fenestrated endothelial lining of rat liver sinusoids. J Ultrastruct Res. 1970;31(1):125–50.
Wisse E, De Zanger RB, Charels K, Van Der Smissen P, McCuskey RS. The liver sieve: considerations concerning the structure and function of endothelial fenestrae, the sinusoidal wall and the space of Disse. Hepatology. 1985;5(4):683–92.
Hilmer SN, Cogger VC, Fraser R, McLean AJ, Sullivan D, Le Couteur DG. Age-related changes in the hepatic sinusoidal endothelium impede lipoprotein transfer in the rat. Hepatology. 2005;42(6):1349–54.
Yokomori H, Yoshimura K, Funakoshi S, Nagai T, Fujimaki K, Nomura M, Ishii H, Oda M. Rho modulates hepatic sinusoidal endothelial fenestrae via regulation of the actin cytoskeleton in rat endothelial cells. Lab Invest. 2004;84(7):857–64.
Shah V, Haddad FG, Garcia-Cardena G, Frangos JA, Mennone A, Groszmann RJ, Sessa WC. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J Clin Invest. 1997;100(11):2923–30.
Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, Momburg F, Arnold B, Knolle PA. Efficient presentation of exogenous antigen by liver endothelial cells to CD8 + T cells results in antigen-specific T-cell tolerance. Nat Med. 2000;6(12):1348–54.
Carambia A, Frenzel C, Bruns OT, Schwinge D, Reimer R, Hohenberg H, Huber S, Tiegs G, Schramm C, Lohse AW, et al. Inhibition of inflammatory CD4 T cell activity by murine liver sinusoidal endothelial cells. J Hepatol. 2013;58(1):112–8.
Xu B, Broome U, Uzunel M, Nava S, Ge X, Kumagai-Braesch M, Hultenby K, Christensson B, Ericzon BG, Holgersson J, et al. Capillarization of hepatic sinusoid by liver endothelial cell-reactive autoantibodies in patients with cirrhosis and chronic hepatitis. Am J Pathol. 2003;163(4):1275–89.
Miyao M, Kotani H, Ishida T, Kawai C, Manabe S, Abiru H, Tamaki K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab Invest. 2015;95(10):1130–44.
Øie CI, Appa RS, Hilden I, Petersen HH, Gruhler A, Smedsrød B, Hansen JB. Rat liver sinusoidal endothelial cells (LSECs) express functional low density lipoprotein receptor-related protein-1 (LRP-1). J Hepatol. 2011;55(6):1346–52.
Furuta K, Guo Q, Pavelko KD, Lee JH, Robertson KD, Nakao Y, Melek J, Shah VH, Hirsova P, Ibrahim SH. Lipid-induced endothelial vascular cell adhesion molecule 1 promotes nonalcoholic steatohepatitis pathogenesis. J Clin Invest 2021, 131(6).
Kawai H, Osawa Y, Matsuda M, Tsunoda T, Yanagida K, Hishikawa D, Okawara M, Sakamoto Y, Shimagaki T, Tsutsui Y, et al. Sphingosine-1-phosphate promotes tumor development and liver fibrosis in mouse model of congestive hepatopathy. Hepatology. 2022;76(1):112–25.
Winkler M, Staniczek T, Kürschner SW, Schmid CD, Schönhaber H, Cordero J, Kessler L, Mathes A, Sticht C, Neßling M, et al. Endothelial GATA4 controls liver fibrosis and regeneration by preventing a pathogenic switch in angiocrine signaling. J Hepatol. 2021;74(2):380–93.
Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, Pessaux P, Baumert TF. Grün D: A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572(7768):199–204.
Li ZW, Ruan B, Yang PJ, Liu JJ, Song P, Duan JL, Wang L. Oit3, a promising hallmark gene for targeting liver sinusoidal endothelial cells. Signal Transduct Target Ther. 2023;8(1):344.
Wisse E, Jacobs F, Topal B, Frederik P, De Geest B. The size of endothelial fenestrae in human liver sinusoids: implications for hepatocyte-directed gene transfer. Gene Ther. 2008;15(17):1193–9.
Wisse E, De Zanger RB, Jacobs R, McCuskey RS. Scanning electron microscope observations on the structure of portal veins, sinusoids and central veins in rat liver. Scan Electron Microsc 1983(Pt 3):1441–52.
Milici AJ, L’Hernault N, Palade GE. Surface densities of diaphragmed fenestrae and transendothelial channels in different murine capillary beds. Circ Res. 1985;56(5):709–17.
Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol. 2018;15(9):555–67.
Steiniger BS. Human spleen microanatomy: why mice do not suffice. Immunology. 2015;145(3):334–46.
Bautz F, Rafii S, Kanz L, Möhle R. Expression and secretion of vascular endothelial growth factor-A by cytokine-stimulated hematopoietic progenitor cells. Possible role in the hematopoietic microenvironment. Exp Hematol. 2000;28(6):700–6.
Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156(4):1363–80.
Teratani T, Tomita K, Suzuki T, Furuhashi H, Irie R, Hida S, Okada Y, Kurihara C, Ebinuma H, Nakamoto N, et al. Free cholesterol accumulation in liver sinusoidal endothelial cells exacerbates acetaminophen hepatotoxicity via TLR9 signaling. J Hepatol. 2017;67(4):780–90.
Cogger VC, Hilmer SN, Sullivan D, Muller M, Fraser R, Le Couteur DG. Hyperlipidemia and surfactants: the liver sieve is a link. Atherosclerosis. 2006;189(2):273–81.
Blomhoff R, Drevon CA, Eskild W, Helgerud P, Norum KR, Berg T. Clearance of acetyl low density lipoprotein by rat liver endothelial cells. Implications for hepatic cholesterol metabolism. J Biol Chem. 1984;259(14):8898–903.
Carpenter B, Lin Y, Stoll S, Raffai RL, McCuskey R, Wang R. VEGF is crucial for the hepatic vascular development required for lipoprotein uptake. Development. 2005;132(14):3293–303.
Géraud C, Koch PS, Zierow J, Klapproth K, Busch K, Olsavszky V, Leibing T, Demory A, Ulbrich F, Diett M, et al. GATA4-dependent organ-specific endothelial differentiation controls liver development and embryonic hematopoiesis. J Clin Invest. 2017;127(3):1099–114.
Cogger VC, Mohamad M, Solon-Biet SM, Senior AM, Warren A, O’Reilly JN, Tung BT, Svistounov D, McMahon AC, Fraser R, et al. Dietary macronutrients and the aging liver sinusoidal endothelial cell. Am J Physiol Heart Circ Physiol. 2016;310(9):H1064–1070.
Zhang Q, Liu J, Liu J, Huang W, Tian L, Quan J, Wang Y, Niu R. oxLDL induces injury and defenestration of human liver sinusoidal endothelial cells via LOX1. J Mol Endocrinol. 2014;53(2):281–93.
Dobbs BR, Rogers GW, Xing HY, Fraser R. Endotoxin-induced defenestration of the hepatic sinusoidal endothelium: a factor in the pathogenesis of cirrhosis? Liver 1994;14(5):230–3.
Ingham PW, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nat Rev Genet. 2006;7(11):841–50.
Ingham PW, Nakano Y, Seger C. Mechanisms and functions of hedgehog signalling across the metazoa. Nat Rev Genet. 2011;12(6):393–406.
Xie G, Choi SS, Syn WK, Michelotti GA, Swiderska M, Karaca G, Chan IS, Chen Y, Diehl AM. Hedgehog signalling regulates liver sinusoidal endothelial cell capillarisation. Gut. 2013;62(2):299–309.
Wu X, Shu L, Zhang Z, Li J, Zong J, Cheong LY, Ye D, Lam KSL, Song E, Wang C, et al. Adipocyte fatty acid binding protein promotes the Onset and Progression of Liver Fibrosis via mediating the crosstalk between Liver Sinusoidal endothelial cells and hepatic stellate cells. Adv Sci (Weinh). 2021;8(11):e2003721.
DeLeve LD, Wang X, Hu L, McCuskey MK, McCuskey RS. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am J Physiol Gastrointest Liver Physiol. 2004;287(4):G757–763.
Yamane A, Seetharam L, Yamaguchi S, Gotoh N, Takahashi T, Neufeld G, Shibuya M. A new communication system between hepatocytes and sinusoidal endothelial cells in liver through vascular endothelial growth factor and flt tyrosine kinase receptor family (Flt-1 and KDR/Flk-1). Oncogene. 1994;9(9):2683–90.
Desroches-Castan A, Tillet E, Ricard N, Ouarné M, Mallet C, Belmudes L, Couté Y, Boillot O, Scoazec JY, Bailly S, et al. Bone morphogenetic protein 9 is a paracrine factor Controlling Liver Sinusoidal endothelial cell fenestration and protecting against hepatic fibrosis. Hepatology. 2019;70(4):1392–408.
HEPATIC, blood-flow. Lancet. 1953;1(6756):385.
Lee SS, Hadengue A, Moreau R, Sayegh R, Hillon P, Lebrec D. Postprandial hemodynamic responses in patients with cirrhosis. Hepatology. 1988;8(3):647–51.
Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75(3):519–60.
Gracia-Sancho J, Russo L, García-Calderó H, García-Pagán JC, García-Cardeña G, Bosch J. Endothelial expression of transcription factor kruppel-like factor 2 and its vasoprotective target genes in the normal and cirrhotic rat liver. Gut. 2011;60(4):517–24.
Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA, editors. Jr. : Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116(1):49–58.
Fernandez M. Molecular pathophysiology of portal hypertension. Hepatology. 2015;61(4):1406–15.
Wirz W, Antoine M, Tag CG, Gressner AM, Korff T, Hellerbrand C, Kiefer P. Hepatic stellate cells display a functional vascular smooth muscle cell phenotype in a three-dimensional co-culture model with endothelial cells. Differentiation. 2008;76(7):784–94.
Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48(3):920–30.
Pabst O, Hornef MW, Schaap FG, Cerovic V, Clavel T, Bruns T. Gut-liver axis: barriers and functional circuits. Nat Rev Gastroenterol Hepatol. 2023;20(7):447–61.
Hsu CL, Schnabl B. The gut-liver axis and gut microbiota in health and liver disease. Nat Rev Microbiol 2023.
Scott A. Gut-liver axis: menace in the microbiota. Nature. 2017;551(7681):S94–5.
Szabo G. Gut-liver axis in alcoholic liver disease. Gastroenterology. 2015;148(1):30–6.
Tilg H, Adolph TE, Trauner M. Gut-liver axis: pathophysiological concepts and clinical implications. Cell Metab. 2022;34(11):1700–18.
Hsu CL, Schnabl B. The gut-liver axis and gut microbiota in health and liver disease. Nat Rev Microbiol. 2023;21(11):719–33.
Casey LM, Hughes KR, Saunders MN, Miller SD, Pearson RM, Shea LD. Mechanistic contributions of Kupffer cells and liver sinusoidal endothelial cells in nanoparticle-induced antigen-specific immune tolerance. Biomaterials. 2022;283:121457.
von Oppen N, Schurich A, Hegenbarth S, Stabenow D, Tolba R, Weiskirchen R, Geerts A, Kolanus W, Knolle P, Diehl L. Systemic antigen cross-presented by liver sinusoidal endothelial cells induces liver-specific CD8 T-cell retention and tolerization. Hepatology. 2009;49(5):1664–72.
Warren A, Le Couteur DG, Fraser R, Bowen DG, McCaughan GW, Bertolino P. T lymphocytes interact with hepatocytes through fenestrations in murine liver sinusoidal endothelial cells. Hepatology. 2006;44(5):1182–90.
Schurich A, Berg M, Stabenow D, Böttcher J, Kern M, Schild HJ, Kurts C, Schuette V, Burgdorf S, Diehl L, et al. Dynamic regulation of CD8 T cell tolerance induction by liver sinusoidal endothelial cells. J Immunol. 2010;184(8):4107–14.
Böttcher JP, Knolle PA, Stabenow D. Mechanisms balancing tolerance and immunity in the liver. Dig Dis. 2011;29(4):384–90.
McDonald B, Zucoloto AZ, Yu IL, Burkhard R, Brown K, Geuking MB, McCoy KD. Programing of an Intravascular Immune Firewall by the gut microbiota protects against Pathogen dissemination during infection. Cell Host Microbe. 2020;28(5):660–e668664.
Gola A, Dorrington MG, Speranza E, Sala C, Shih RM, Radtke AJ, Wong HS, Baptista AP, Hernandez JM, Castellani G, et al. Commensal-driven immune zonation of the liver promotes host defence. Nature. 2021;589(7840):131–6.
Schölzel K, Schildberg FA, Welz M, Börner C, Geiger S, Kurts C, Heikenwälder M, Knolle PA, Wohlleber D. Transfer of MHC-class-I molecules among liver sinusoidal cells facilitates hepatic immune surveillance. J Hepatol. 2014;61(3):600–8.
Maretti-Mira AC, Wang X, Wang L, DeLeve LD. Incomplete differentiation of engrafted bone marrow endothelial progenitor cells initiates hepatic fibrosis in the rat. Hepatology. 2019;69(3):1259–72.
de Haan W, Dheedene W, Apelt K, Décombas-Deschamps S, Vinckier S, Verhulst S, Conidi A, Deffieux T, Staring MW, Vandervoort P, et al. Endothelial Zeb2 preserves the hepatic angioarchitecture and protects against liver fibrosis. Cardiovasc Res. 2022;118(5):1262–75.
Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397–411.
DeLeve LD. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology. 2015;61(5):1740–6.
Hammoutene A, Rautou PE. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol. 2019;70(6):1278–91.
Kawashita E, Ozaki T, Ishihara K, Kashiwada C, Akiba S. Endothelial group IVA phospholipase A(2) promotes hepatic fibrosis with sinusoidal capillarization in the early stage of non-alcoholic steatohepatitis in mice. Life Sci. 2022;294:120355.
Herrnberger L, Hennig R, Kremer W, Hellerbrand C, Goepferich A, Kalbitzer HR, Tamm ER. Formation of fenestrae in murine liver sinusoids depends on plasmalemma vesicle-associated protein and is required for lipoprotein passage. PLoS ONE. 2014;9(12):e115005.
Van der Graaff D, Kwanten WJ, Couturier FJ, Govaerts JS, Verlinden W, Brosius I, D’Hondt M, Driessen A, De Winter BY, De Man JG, et al. Severe steatosis induces portal hypertension by systemic arterial hyporeactivity and hepatic vasoconstrictor hyperreactivity in rats. Lab Invest. 2018;98(10):1263–75.
Francque S, Laleman W, Verbeke L, Van Steenkiste C, Casteleyn C, Kwanten W, Van Dyck C, D’Hondt M, Ramon A, Vermeulen W, et al. Increased intrahepatic resistance in severe steatosis: endothelial dysfunction, vasoconstrictor overproduction and altered microvascular architecture. Lab Invest. 2012;92(10):1428–39.
Mendes FD, Suzuki A, Sanderson SO, Lindor KD, Angulo P. Prevalence and indicators of portal hypertension in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2012;10(9):1028–e10331022.
Rockey DC, Shah V. Nitric oxide biology and the liver: report of an AASLD research workshop. Hepatology. 2004;39(1):250–7.
Kawada N, Klein H, Decker K. Eicosanoid-mediated contractility of hepatic stellate cells. Biochem J. 1992;285(Pt 2):367–71.
Pinzani M, Failli P, Ruocco C, Casini A, Milani S, Baldi E, Giotti A, Gentilini P. Fat-storing cells as liver-specific pericytes. Spatial dynamics of agonist-stimulated intracellular calcium transients. J Clin Invest. 1992;90(2):642–6.
Shah V, Toruner M, Haddad F, Cadelina G, Papapetropoulos A, Choo K, Sessa WC, Groszmann RJ. Impaired endothelial nitric oxide synthase activity associated with enhanced caveolin binding in experimental cirrhosis in the rat. Gastroenterology. 1999;117(5):1222–8.
Suematsu M, Goda N, Sano T, Kashiwagi S, Egawa T, Shinoda Y, Ishimura Y. Carbon monoxide: an endogenous modulator of sinusoidal tone in the perfused rat liver. J Clin Invest. 1995;96(5):2431–7.
Gracia-Sancho J, Laviña B, Rodríguez-Vilarrupla A, García-Calderó H, Bosch J, García-Pagán JC. Enhanced vasoconstrictor prostanoid production by sinusoidal endothelial cells increases portal perfusion pressure in cirrhotic rat livers. J Hepatol. 2007;47(2):220–7.
Miller AM, Masrorpour M, Klaus C, Zhang JX. LPS exacerbates endothelin-1 induced activation of cytosolic phospholipase A2 and thromboxane A2 production from Kupffer cells of the prefibrotic rat liver. J Hepatol. 2007;46(2):276–85.
Bravo M, Raurell I, Barberá A, Hide D, Gil M, Estrella F, Salcedo MT, Augustin S, Genescà J, Martell M. Synergic effect of atorvastatin and ambrisentan on sinusoidal and hemodynamic alterations in a rat model of NASH. Dis Model Mech 2021, 14(5).
Thabut D, Shah V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: new targets for the treatment of portal hypertension? J Hepatol. 2010;53(5):976–80.
Klein S, Van Beuge MM, Granzow M, Beljaars L, Schierwagen R, Kilic S, Heidari I, Huss S, Sauerbruch T, Poelstra K, et al. HSC-specific inhibition of rho-kinase reduces portal pressure in cirrhotic rats without major systemic effects. J Hepatol. 2012;57(6):1220–7.
Kharbanda KK, Rogers DD 2nd, Wyatt TA, Sorrell MF, Tuma DJ. Transforming growth factor-beta induces contraction of activated hepatic stellate cells. J Hepatol. 2004;41(1):60–6.
Semela D, Das A, Langer D, Kang N, Leof E, Shah V. Platelet-derived growth factor signaling through ephrin-b2 regulates hepatic vascular structure and function. Gastroenterology. 2008;135(2):671–9.
Rombouts K, Knittel T, Machesky L, Braet F, Wielant A, Hellemans K, De Bleser P, Gelman I, Ramadori G, Geerts A. Actin filament formation, reorganization and migration are impaired in hepatic stellate cells under influence of trichostatin A, a histone deacetylase inhibitor. J Hepatol. 2002;37(6):788–96.
Younossi ZM, Loomba R, Rinella ME, Bugianesi E, Marchesini G, Neuschwander-Tetri BA, Serfaty L, Negro F, Caldwell SH, Ratziu V, et al. Current and future therapeutic regimens for nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2018;68(1):361–71.
Huby T, Gautier EL. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat Rev Immunol. 2022;22(7):429–43.
Peiseler M, Schwabe R, Hampe J, Kubes P, Heikenwälder M, Tacke F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease - novel insights into cellular communication circuits. J Hepatol. 2022;77(4):1136–60.
Tripathi DM, Rohilla S, Kaur I, Siddiqui H, Rawal P, Juneja P, Kumar V, Kumari A, Naidu VGM, Ramakrishna S et al. Immunonano-lipocarrier-mediated liver sinusoidal endothelial cell-specific RUNX1 inhibition impedes Immune Cell infiltration and hepatic inflammation in Murine Model of NASH. Int J Mol Sci 2021, 22(16).
Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol. 2018;15(6):349–64.
Gao B, Jeong WI, Tian Z. Liver: an organ with predominant innate immunity. Hepatology. 2008;47(2):729–36.
Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology. 1990;11(1):74–80.
Rodriguez-Vita J, Morales-Ruiz M. Down the liver sinusoidal endothelial cell (LSEC) hole. Is there a role for lipid rafts in LSEC fenestration? Hepatology. 2013;57(3):1272–4.
Carambia A, Freund B, Schwinge D, Heine M, Laschtowitz A, Huber S, Wraith DC, Korn T, Schramm C, Lohse AW, et al. TGF-β-dependent induction of CD4⁺CD25⁺Foxp3⁺ tregs by liver sinusoidal endothelial cells. J Hepatol. 2014;61(3):594–9.
Furuta K, Tang X, Islam S, Tapia A, Chen ZB, Ibrahim SH. Endotheliopathy in the metabolic syndrome: mechanisms and clinical implications. Pharmacol Ther. 2023;244:108372.
Matsumoto M, Zhang J, Zhang X, Liu J, Jiang JX, Yamaguchi K, Taruno A, Katsuyama M, Iwata K, Ibi M, et al. The NOX1 isoform of NADPH oxidase is involved in dysfunction of liver sinusoids in nonalcoholic fatty liver disease. Free Radic Biol Med. 2018;115:412–20.
Sutter AG, Palanisamy AP, Lench JH, Esckilsen S, Geng T, Lewin DN, Cowart LA, Chavin KD. Dietary Saturated Fat promotes development of hepatic inflammation through toll-like receptor 4 in mice. J Cell Biochem. 2016;117(7):1613–21.
Fontana L, Eagon JC, Trujillo ME, Scherer PE, Klein S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes. 2007;56(4):1010–3.
Wu J, Meng Z, Jiang M, Zhang E, Trippler M, Broering R, Bucchi A, Krux F, Dittmer U, Yang D, et al. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology. 2010;129(3):363–74.
Harte AL, da Silva NF, Creely SJ, McGee KC, Billyard T, Youssef-Elabd EM, Tripathi G, Ashour E, Abdalla MS, Sharada HM, et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J Inflamm (Lond). 2010;7:15.
Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palù G, Martines D. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2007;292(2):G518–525.
Lefere S, Van de Velde F, Hoorens A, Raevens S, Van Campenhout S, Vandierendonck A, Neyt S, Vandeghinste B, Vanhove C, Debbaut C, et al. Angiopoietin-2 promotes pathological angiogenesis and is a therapeutic target in Murine nonalcoholic fatty liver disease. Hepatology. 2019;69(3):1087–104.
Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39(5):1286–96.
Dela Peña A, Leclercq I, Field J, George J, Jones B, Farrell G. NF-kappaB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology. 2005;129(5):1663–74.
Weston CJ, Shepherd EL, Claridge LC, Rantakari P, Curbishley SM, Tomlinson JW, Hubscher SG, Reynolds GM, Aalto K, Anstee QM, et al. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J Clin Invest. 2015;125(2):501–20.
Tomita K, Tamiya G, Ando S, Ohsumi K, Chiyo T, Mizutani A, Kitamura N, Toda K, Kaneko T, Horie Y, et al. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut. 2006;55(3):415–24.
Miyachi Y, Tsuchiya K, Komiya C, Shiba K, Shimazu N, Yamaguchi S, Deushi M, Osaka M, Inoue K, Sato Y, et al. Roles for cell-cell adhesion and contact in obesity-Induced hepatic myeloid cell Accumulation and glucose intolerance. Cell Rep. 2017;18(11):2766–79.
Feder LS, Todaro JA, Laskin DL. Characterization of interleukin-1 and interleukin-6 production by hepatic endothelial cells and macrophages. J Leukoc Biol. 1993;53(2):126–32.
Taooka Y, Chen J, Yednock T, Sheppard D. The integrin alpha9beta1 mediates adhesion to activated endothelial cells and transendothelial neutrophil migration through interaction with vascular cell adhesion molecule-1. J Cell Biol. 1999;145(2):413–20.
Hammoutene A, Biquard L, Lasselin J, Kheloufi M, Tanguy M, Vion AC, Mérian J, Colnot N, Loyer X, Tedgui A, et al. A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J Hepatol. 2020;72(3):528–38.
Fang ZQ, Ruan B, Liu JJ, Duan JL, Yue ZS, Song P, Xu H, Ding J, Xu C, Dou GR, et al. Notch-triggered maladaptation of liver sinusoidal endothelium aggravates nonalcoholic steatohepatitis through endothelial nitric oxide synthase. Hepatology. 2022;76(3):742–58.
DeLeve LD. Liver sinusoidal endothelial cells and liver regeneration. J Clin Invest. 2013;123(5):1861–6.
Briscoe J, Thérond PP. The mechanisms of hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14(7):416–29.
Sekulic A, Von Hoff D. Hedgehog pathway inhibition. Cell. 2016;164(5):831.
Omenetti A, Choi S, Michelotti G, Diehl AM. Hedgehog signaling in the liver. J Hepatol. 2011;54(2):366–73.
Francis H, Bohanan J, Alpini G. Hedgehog signalling and LSEC capillarisation: stopping this one in its tracks. Gut. 2012;61(9):1243–4.
Philips GM, Chan IS, Swiderska M, Schroder VT, Guy C, Karaca GF, Moylan C, Venkatraman T, Feuerlein S, Syn WK, et al. Hedgehog signaling antagonist promotes regression of both liver fibrosis and hepatocellular carcinoma in a murine model of primary liver cancer. PLoS ONE. 2011;6(9):e23943.
Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature. 2016;529(7586):316–25.
Ding BS, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z, Mittal V, Kobayashi H, Shido K, Lyden D, et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature. 2010;468(7321):310–5.
Kostallari E, Shah VH. Angiocrine signaling in the hepatic sinusoids in health and disease. Am J Physiol Gastrointest Liver Physiol. 2016;311(2):G246–251.
Koch PS, Olsavszky V, Ulbrich F, Sticht C, Demory A, Leibing T, Henzler T, Meyer M, Zierow J, Schneider S, et al. Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood. 2017;129(4):415–9.
Leibing T, Géraud C, Augustin I, Boutros M, Augustin HG, Okun JG, Langhans CD, Zierow J, Wohlfeil SA, Olsavszky V, et al. Angiocrine wnt signaling controls liver growth and metabolic maturation in mice. Hepatology. 2018;68(2):707–22.
Wang B, Zhao L, Fish M, Logan CY, Nusse R. Self-renewing diploid Axin2(+) cells fuel homeostatic renewal of the liver. Nature. 2015;524(7564):180–5.
Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol. 2014;15(12):802–12.
Duscher D, Maan ZN, Wong VW, Rennert RC, Januszyk M, Rodrigues M, Hu M, Whitmore AJ, Whittam AJ, Longaker MT, et al. Mechanotransduction and fibrosis. J Biomech. 2014;47(9):1997–2005.
Greuter T, Yaqoob U, Gan C, Jalan-Sakrikar N, Kostallari E, Lu J, Gao J, Sun L, Liu M, Sehrawat TS, et al. Mechanotransduction-induced glycolysis epigenetically regulates a CXCL1-dominant angiocrine signaling program in liver sinusoidal endothelial cells in vitro and in vivo. J Hepatol. 2022;77(3):723–34.
del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP. Stretching single talin rod molecules activates vinculin binding. Science. 2009;323(5914):638–41.
Ross TD, Coon BG, Yun S, Baeyens N, Tanaka K, Ouyang M, Schwartz MA. Integrins in mechanotransduction. Curr Opin Cell Biol. 2013;25(5):613–8.
Romeo S. Notch and nonalcoholic fatty liver and fibrosis. N Engl J Med. 2019;380(7):681–3.
Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, Pellicoro A, Ridgway RA, Seo SS, Spee B, Van Rooijen N, et al. Macrophage-derived wnt opposes notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med. 2012;18(4):572–9.
Pajvani UB, Qiang L, Kangsamaksin T, Kitajewski J, Ginsberg HN, Accili D. Inhibition of Notch uncouples akt activation from hepatic lipid accumulation by decreasing mTorc1 stability. Nat Med. 2013;19(8):1054–60.
Duan JL, Ruan B, Yan XC, Liang L, Song P, Yang ZY, Liu Y, Dou KF, Han H, Wang L. Endothelial notch activation reshapes the angiocrine of sinusoidal endothelia to aggravate liver fibrosis and blunt regeneration in mice. Hepatology. 2018;68(2):677–90.
Ruart M, Chavarria L, Campreciós G, Suárez-Herrera N, Montironi C, Guixé-Muntet S, Bosch J, Friedman SL, Garcia-Pagán JC, Hernández-Gea V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J Hepatol. 2019;70(3):458–69.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–7.
Matsuzaki K, Murata M, Yoshida K, Sekimoto G, Uemura Y, Sakaida N, Kaibori M, Kamiyama Y, Nishizawa M, Fujisawa J, et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology. 2007;46(1):48–57.
Villanueva A. Hepatocellular Carcinoma. N Engl J Med. 2019;380(15):1450–62.
Kin M, Torimura T, Ueno T, Inuzuka S, Tanikawa K. Sinusoidal capillarization in small hepatocellular carcinoma. Pathol Int. 1994;44(10–11):771–8.
Lisman T, Luyendyk JP. Platelets as modulators of Liver diseases. Semin Thromb Hemost. 2018;44(2):114–25.
Thomann S, Weiler SME, Marquard S, Rose F, Ball CR, Tóth M, Wei T, Sticht C, Fritzsche S, Roessler S, et al. YAP orchestrates heterotypic endothelial cell communication via HGF/c-MET signaling in Liver Tumorigenesis. Cancer Res. 2020;80(24):5502–14.
Pinato DJ, Guerra N, Fessas P, Murphy R, Mineo T, Mauri FA, Mukherjee SK, Thursz M, Wong CN, Sharma R, et al. Immune-based therapies for hepatocellular carcinoma. Oncogene. 2020;39(18):3620–37.
Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer. 2019;18(1):155.
Walker LSK. The link between circulating follicular helper T cells and autoimmunity. Nat Rev Immunol. 2022;22(9):567–75.
Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443(7109):350–4.
Wadkin JCR, Patten DA, Kamarajah SK, Shepherd EL, Novitskaya V, Berditchevski F, Adams DH, Weston CJ, Shetty S. CD151 supports VCAM-1-mediated lymphocyte adhesion to liver endothelium and is upregulated in chronic liver disease and hepatocellular carcinoma. Am J Physiol Gastrointest Liver Physiol. 2017;313(2):G138–49.
Knolle PA, Wohlleber D. Immunological functions of liver sinusoidal endothelial cells. Cell Mol Immunol. 2016;13(3):347–53.
Wu K, Kryczek I, Chen L, Zou W, Welling TH. Kupffer cell suppression of CD8 + T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 2009;69(20):8067–75.
Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, Eppolito C, Qian F, Lele S, Shrikant P, et al. Tumor-infiltrating NY-ESO-1-specific CD8 + T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A. 2010;107(17):7875–80.
Milner KL, van der Poorten D, Xu A, Bugianesi E, Kench JG, Lam KS, Chisholm DJ, George J. Adipocyte fatty acid binding protein levels relate to inflammation and fibrosis in nonalcoholic fatty liver disease. Hepatology. 2009;49(6):1926–34.
Laouirem S, Sannier A, Norkowski E, Cauchy F, Doblas S, Rautou PE, Albuquerque M, Garteiser P, Sognigbé L, Raffenne J, et al. Endothelial fatty liver binding protein 4: a new targetable mediator in hepatocellular carcinoma related to metabolic syndrome. Oncogene. 2019;38(16):3033–46.
Xu J, Lin H, Wu G, Zhu M, Li M. IL-6/STAT3 is a Promising Therapeutic Target for Hepatocellular Carcinoma. Front Oncol. 2021;11:760971.
Soresi M, Giannitrapani L, D’Antona F, Florena AM, La Spada E, Terranova A, Cervello M, D’Alessandro N, Montalto G. Interleukin-6 and its soluble receptor in patients with liver cirrhosis and hepatocellular carcinoma. World J Gastroenterol. 2006;12(16):2563–8.
Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, Gupta S, Moore J, Wrobel MJ, Lerner J, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359(19):1995–2004.
Wang XJ, Malhi H. Nonalcoholic fatty liver disease. Ann Intern Med. 2018;169(9):Itc65–80.
Neuschwander-Tetri BA. Therapeutic Landscape for NAFLD in 2020. Gastroenterology. 2020;158(7):1984–98. e1983.
Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313(22):2263–73.
Rinella ME, Sanyal AJ. Management of NAFLD: a stage-based approach. Nat Rev Gastroenterol Hepatol. 2016;13(4):196–205.
Rodriguez B, Torres DM, Harrison SA. Physical activity: an essential component of lifestyle modification in NAFLD. Nat Rev Gastroenterol Hepatol. 2012;9(12):726–31.
Romero-Gómez M, Zelber-Sagi S, Trenell M. Treatment of NAFLD with diet, physical activity and exercise. J Hepatol. 2017;67(4):829–46.
Mehal WZ. The Gordian knot of dysbiosis, obesity and NAFLD. Nat Rev Gastroenterol Hepatol. 2013;10(11):637–44.
Rowe IA. Too much medicine: overdiagnosis and overtreatment of non-alcoholic fatty liver disease. Lancet Gastroenterol Hepatol. 2018;3(1):66–72.
Guo Q, Furuta K, Lucien F, Gutierrez Sanchez LH, Hirsova P, Krishnan A, Kabashima A, Pavelko KD, Madden B, Alhuwaish H, et al. Integrin β(1)-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J Hepatol. 2019;71(6):1193–205.
Ross EA, Douglas MR, Wong SH, Ross EJ, Curnow SJ, Nash GB, Rainger E, Scheel-Toellner D, Lord JM, Salmon M, et al. Interaction between integrin alpha9beta1 and vascular cell adhesion molecule-1 (VCAM-1) inhibits neutrophil apoptosis. Blood. 2006;107(3):1178–83.
Nachit M, Montemagno C, Clerc R, Ahmadi M, Briand F, Bacot S, Devoogdt N, Serdjebi C, Ghezzi C, Sulpice T, et al. Molecular imaging of liver inflammation using an anti-VCAM-1 nanobody. Nat Commun. 2023;14(1):1062.
Guixé-Muntet S, de Mesquita FC, Vila S, Hernández-Gea V, Peralta C, García-Pagán JC, Bosch J, Gracia-Sancho J. Cross-talk between autophagy and KLF2 determines endothelial cell phenotype and microvascular function in acute liver injury. J Hepatol. 2017;66(1):86–94.
Marrone G, Russo L, Rosado E, Hide D, García-Cardeña G, García-Pagán JC, Bosch J, Gracia-Sancho J. The transcription factor KLF2 mediates hepatic endothelial protection and paracrine endothelial-stellate cell deactivation induced by statins. J Hepatol. 2013;58(1):98–103.
Hamed AM, El-Kharashi OA, Boctor SS, Abd-Elaziz LF. Potential involvement of PPAR α activation in diminishing the hepatoprotective effect of fenofibrate in NAFLD: Accuracy of non- invasive panel in determining the stage of liver fibrosis in rats. Biomed Pharmacother. 2017;85:68–78.
Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest. 2006;116(3):571–80.
Rodríguez-Vilarrupla A, Laviña B, García-Calderó H, Russo L, Rosado E, Roglans N, Bosch J, García-Pagán JC. PPARα activation improves endothelial dysfunction and reduces fibrosis and portal pressure in cirrhotic rats. J Hepatol. 2012;56(5):1033–9.
Boyer-Diaz Z, Aristu-Zabalza P, Andrés-Rozas M, Robert C, Ortega-Ribera M, Fernández-Iglesias A, Broqua P, Junien JL, Wettstein G, Bosch J, et al. Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J Hepatol. 2021;74(5):1188–99.
Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355(22):2297–307.
Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, Tio F, Hardies J, Darland C, Musi N, et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes Mellitus: a Randomized Trial. Ann Intern Med. 2016;165(5):305–15.
Aithal GP, Thomas JA, Kaye PV, Lawson A, Ryder SD, Spendlove I, Austin AS, Freeman JG, Morgan L, Webber J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135(4):1176–84.
Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362(18):1675–85.
Musso G, Cassader M, Paschetta E, Gambino R. Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: a Meta-analysis. JAMA Intern Med. 2017;177(5):633–40.
Schwabl P, Hambruch E, Seeland BA, Hayden H, Wagner M, Garnys L, Strobel B, Schubert TL, Riedl F, Mitteregger D, et al. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J Hepatol. 2017;66(4):724–33.
Haubrich WS. Disse of the space of Disse. Gastroenterology. 2004;127(6):1684.
Orrego H, Blendis LM, Crossley IR, Medline A, Macdonald A, Ritchie S, Israel Y. Correlation of intrahepatic pressure with collagen in the Disse space and hepatomegaly in humans and in the rat. Gastroenterology. 1981;80(3):546–56.
Li F, Zhao Y, Cheng Z, Wang Y, Yue Y, Cheng X, Sun J, Atabakhshi-Kashi M, Yao J, Dou J, et al. Restoration of Sinusoid Fenestrae followed by targeted Nanoassembly Delivery of an anti-fibrotic Agent improves treatment efficacy in liver fibrosis. Adv Mater. 2023;35(17):e2212206.
Zhang LF, Wang XH, Zhang CL, Lee J, Duan BW, Xing L, Li L, Oh YK, Jiang HL. Sequential Nano-Penetrators of Capillarized Liver sinusoids and Extracellular Matrix barriers for Liver Fibrosis Therapy. ACS Nano. 2022;16(9):14029–42.
Sarcognato S, García-Lezana T, Villanueva A. Mechanisms of action of drugs effective in Hepatocellular Carcinoma. Clin Liver Dis (Hoboken). 2019;14(2):62–5.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90.
Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, Baron A, Park JW, Han G, Jassem J, et al. Lenvatinib versus Sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–73.
Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66.
Kelley RK, Verslype C, Cohn AL, Yang TS, Su WC, Burris H, Braiteh F, Vogelzang N, Spira A, Foster P, et al. Cabozantinib in hepatocellular carcinoma: results of a phase 2 placebo-controlled randomized discontinuation study. Ann Oncol. 2017;28(3):528–34.
Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15(10):599–616.
Qin S, Ren Z, Meng Z, Chen Z, Chai X, Xiong J, Bai Y, Yang L, Zhu H, Fang W, et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: a multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 2020;21(4):571–80.
Wang F, Qin S, Sun X, Ren Z, Meng Z, Chen Z, Chai X, Xiong J, Bai Y, Yang L, et al. Reactive cutaneous capillary endothelial proliferation in advanced hepatocellular carcinoma patients treated with camrelizumab: data derived from a multicenter phase 2 trial. J Hematol Oncol. 2020;13(1):47.
Xu W, Liu K, Chen M, Sun JY, McCaughan GW, Lu XJ, Ji J. Immunotherapy for hepatocellular carcinoma: recent advances and future perspectives. Ther Adv Med Oncol. 2019;11:1758835919862692.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China, (Grant NO.82274470 and NO.81974567).
Author information
Authors and Affiliations
Contributions
QH and HD conceived the paper. QH, FL and DW wrote the article. YG, HD, YG and WH revised the Figures and reviewed the article. GY, XS provided guidance on the revision of the paper. All authors contributed to the writing of this manuscript and approved the final version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors are consent for publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
He, Q., He, W., Dong, H. et al. Role of liver sinusoidal endothelial cell in metabolic dysfunction-associated fatty liver disease. Cell Commun Signal 22, 346 (2024). https://doi.org/10.1186/s12964-024-01720-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-024-01720-9