
Abstract
RNA methylation modification plays a crucial role as an epigenetic regulator in the oncogenesis of hepatocellular carcinoma (HCC). Numerous studies have investigated the molecular mechanisms underlying the methylation of protein-coding RNAs in the progression of HCC. Beyond their impact on mRNA, methylation modifications also influence the biological functions of non-coding RNAs (ncRNAs). Here, we present an advanced and comprehensive overview of the interplay between methylation modifications and ncRNAs in HCC, with a specific focus on their potential implications for the tumor immune microenvironment. Moreover, we summarize promising therapeutic targets for HCC based on methylation-related proteins. In the future, a more profound investigation is warranted to elucidate the effects of ncRNA methylation modifications on HCC pathogenesis and devise valuable intervention strategies.
Video Abstract
Similar content being viewed by others

Background
Hepatocellular carcinoma (HCC) ranks as the sixth most prevalent malignancy globally and one of the leading causes of cancer-related mortality [1, 2]. Projections indicate that by 2025, more than 1 million individuals will be diagnosed with liver cancer annually, with HCC accounting for over 90% of the cases [3]. The relative five-year survival rate for HCC is a mere 18%, underscoring its substantial burden on public health [4]. The incidence of HCC is influenced by many factors, such as geographic location, demographic attributes, and lifestyle choices [5]. Prominent among these factors are viral hepatitis and alcohol consumption, which increase the risk of HCC development [6]. Over the recent decades, the global incidence of HCC attributed to viral hepatitis has decreased owing to universal coverage of the hepatitis B virus (HBV) vaccine and increased accessibility to efficacious antiviral therapies for both HBV and hepatitis C virus (HCV) infections [7,8,9]. However, the prominence of nonalcoholic fatty liver disease and non-alcoholic steatohepatitis as culprits for HCC is gradually escalating, which is particularly evident within the United States [10, 11]. Early-stage HCC can be potentially cured by local ablation, surgical excision, and liver transplantation [12,13,14]. For medium-stage HCC, transarterial interventions such as transarterial chemoembolization (TACE) prove to be valuable therapeutic avenues [15]. For advanced HCC, receptor tyrosine kinase inhibitors and immunotherapy offer efficacious courses of treatment [16]. Despite strides in multidisciplinary treatment strategies, the prognosis for HCC remains suboptimal due to challenges such as early surgical recurrence and resistance to targeted therapies. Therefore, a critical imperative lies in unraveling the intricate molecular mechanisms underpinning HCC progression, aimed at identifying novel biomarkers and therapeutic targets.
Non-coding RNAs (ncRNAs) are a class of RNAs that do not participate in protein synthesis, accounting for over 90% of the RNA content in the human genome [17, 18]. Typically, ncRNAs are categorized based on their length, structure, and cellular localization. Among these, microRNAs (miRNAs), long ncRNAs (lncRNAs), and circular RNAs (circRNAs) represent three principal categories of ncRNAs, each with distinct biological functions. MiRNAs are a small fragment of RNA, approximately 20–25 nucleotide long, and can bind to complementary sequences on messenger RNA (mRNA), thereby degrading them through cleavage, destabilization, or inhibition of translation into proteins [19,20,21,22]. Conversely, both lncRNAs and circRNAs consist of more than 200 nucleotides. LncRNAs engage not only in epigenetic modifications and the cis or trans regulation of gene transcription but also participate in post-transcriptional regulation, affecting mRNA splicing, stability, and translation [23, 24]. CircRNAs, characterized by their covalently closed circular structure that enhances stability, serve as miRNA or protein sponges and regulate transcriptional variability as well as parental gene expression [25,26,27]. Over the past decade, the discovery of tens of thousands of ncRNAs has notably transformed researchers’ comprehension of gene expression regulation and disease progression. In particular, numerous studies of ncRNA biology have underscored their pivotal roles during HCC tumorigenesis, where they function as either oncogenic agents or tumor suppressors [28,29,30]. For instance, microRNA-93-5p has been identified as binding to the 3′-untranslated region (UTR) of mitogen-activated protein kinase kinase 2, thereby promoting HCC progression [31]. Another study revealed that circRNA-5692, acting as a miR-328-5p sponge, enhances DAB2IP expression, thereby inhibiting HCC growth [32]. Additionally, the hypoxia-induced lncRNA DACT3-AS1 has been found to facilitate the interaction between HDAC2 and FOXA3, thereby upregulating PKM2 expression and promoting the metastasis of HCC [33].
There is a burgeoning interest in elucidating the role of epigenetic regulation in the pathogenesis of HCC, with a particular focus on RNA methylation modifications. RNA methylation refers to the chemical modification phenomenon wherein methyl groups are selectively added to the methyl adenine of RNA under the catalysis of methyltransferases [34, 35]. This modification has been observed across various RNA categories, including mRNA, transfer RNA (tRNA), ribosomal RNA, small nuclear RNA, miRNA, lncRNA, and circRNA [36,37,38]. Among the most prevalent RNA methylation modifications in mammals are N6-methyladenosine (m6A), N1-methyladenosine (m1A), 5-methylcytosine (m5C), pseudouridine (Ψ), and 7-methylguanine (m7G) [39,40,41,42]. At the post-transcriptional level, RNA methylation dynamically and reversibly regulates RNA stability, localization, transport, cleavage, and translation [43,44,45]. This intricate control is executed by a trio of functional methylation-related proteins: methyltransferases (“writers”), demethylases (“erasers”), and methylated reading proteins (“readers”). In recent years, the role of RNA methylation modifications in the oncogenesis of HCC has been progressively garnering attention. For instance, one study reported that methyltransferase-like 3 (METTL3) mediates m6A modification of FOXO3 mRNA, thereby influencing the drug tolerance of HCC to sorafenib [46]. Another study revealed that the methylated reading protein Aly/REF export factor (ALYREF) regulates m5C methylation levels in target RNA, consequently facilitating the progression of liver cancer [47]. Moreover, the m1A methyltransferase complex hnRNPK/TRMT61A has been shown to augment m1A methylation of tRNA, thereby regulating cholesterol metabolism in liver cancer stem cells [48]. This mechanism, in turn, propels liver tumorigenesis through the Hedgehog signaling pathway. Thus, the development of novel drugs targeting methylation-related proteins may provide novel avenues for the therapy of HCC.
Numerous studies have delved into the mechanisms underlying the methylation of protein-coding RNAs in HCC. In addition to mRNA, methylation modifications also affect the metabolism and functioning of ncRNAs, thereby playing a role in the proliferation, invasion, and drug resistance in HCC cells. In this review, we summarize the interactions between methylation modifications and ncRNAs in HCC, highlighting recent advancements in this burgeoning field of research. This review focuses on the potential implications of m6A and m5C methylation modifications in miRNAs, lncRNAs, and circRNAs. Additionally, it probes into the regulatory mechanisms exerted by miRNAs, lncRNAs, and circRNAs on m6A and m5C methylation-related proteins. Ultimately, we discuss the present status and the future outlook for targeting RNA methylation-related proteins in the treatment of HCC.
Primary ncRNA methylation modifications in HCC
Common RNA methylation modifications, including m6A, m5C, m1A, and Ψ, have been identified in ncRNAs. Here, we primarily summarize the m6A, m5C and m1A methylation modifications in ncRNAs.
m6A methylation of ncRNAs
The m6A methylation was first reported in 1974 [49,50,51]. It involves the methylation of the sixth nitrogen atom on the RNA molecule, catalyzed by methyltransferase enzymes. Subsequently, specific “reader” proteins bind to and recognize these methylation sites. Demethylation of these sites is achieved through the actions of demethylase enzymes. Currently known m6A methyltransferases include METTL3, METTL14, METTL5, METTL16, zinc finger CCCH-type containing 13 (ZC3H13), zinc finger CCHC domain-containing protein 4 (ZCCHC4), Wilms tumor 1-associated protein (WTAP), KIAA1429, and RBM15/15B [52, 53]. Notably, fat mass and obesity-associated protein (FTO) and alkylation repair homolog protein 5 (ALKBH5) are pivotal RNA demethylases [54, 55]. “Reader” proteins primarily include YTH N6-methyladenine RNA binding protein 1 (YTHDF1), YTHDF2, YTHDF3, insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1), IGF2BP2, heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1), and heterogeneous nuclear ribonucleoprotein C (hnRNPC) (Fig. 1A) [56].

Overview of the m6A and m5C methylation modification in ncRNA. A m6A methylation was catalyzed by methyltransferase enzymes, including METTL3, METTL14, METTL5, METTL16, ZC3H13, ZCCHC4, WTAP, KIAA1429, and RBM15/15B. FTO and ALKBH5 participate in the demethylation process. Specific “reader” protein, such as YTHDF1-3, IGF2BP1-2, HNRNPA2B1, hnRNPC, bind to and recognize these methylation sites. B The enzymatic catalysis of m5C methylation involves primarily the NSUN1–NSUN7 and DNMT2. The TET1-TET3 and ALKBH1 can facilitate m5C demethylation. ALYREF and YBX1 are two “reader” proteins for m5C-modified ncRNA. Created with BioRender.com
The m6A modification not only regulates the shear, transport, stability, and degradation of ncRNAs but also modulates cellular functions by altering their expression [57]. This modification can affect the maturation of miRNAs. In the case of lncRNAs, m6A modification may regulate the function of lncRNAs by regulating the structure of local lncRNAs, thereby inducing the binding of RNA-binding proteins [58]. In addition, m6A modifications might play a role in shaping the interplay between RNA and DNA, particularly at specific DNA sites, by influencing the triple helix structure of lncRNAs. Moreover, m6A can promote the export of circRNAs to the cytoplasm, driving circRNA translation and mediating circRNA degradation [59,60,61].
m5C methylation of ncRNAs
In the 1970s, researchers discovered m5C modifications, wherein “m5C” signifies the addition of a methyl group to the fifth carbon atom of the cytosine base in RNA [51, 62]. This m5C methylation modification is prevalent in both mRNA and ncRNA. The enzymatic catalysis of m5C methylation involves primarily the NOL1/NOP2/SUN domain (NSUN) family (NSUN1–NSUN7) and DNA methyltransferase 2 (DNMT2) [63]. Acting as “erasers” to facilitate m5C demethylation in RNA, the ten-eleven translocation (TET) family (TET1-TET3) and ALKBH1 play crucial roles [62]. The functional significance of RNA modification predominantly relies on “readers.“ In this context, ALYREF and Y-box binding protein 1 (YBX1) are two recognition proteins for m5C-modified mRNA (Fig. 1B). These proteins exert biological effects by identifying and binding to m5C sites.
As a reversible epigenetic modification, the m5C modification of ncRNA affects the molecular trajectory of the modified ncRNA, playing a vital role in multiple biological processes, including ncRNA stability regulation, protein binding, and transcriptional regulation [64]. For example, the methyltransferase NSUN2 can interact with the lncRNA NKILA, thereby increasing its m5C levels and facilitating its interaction with YBX1 [65]. In addition, a previous study uncovered the substantial presence of m5C sites on circRNAs in human HCC tissues. The distinctive distribution pattern of m5C modifications on circRNA in HCC exhibited correlations with specific metabolic pathways [66]. Therefore, the exploration of m5C methylation of ncRNA assumes considerable significance, offering insights into the underlying mechanisms of disease pathogenesis and progression.
m1A methylation of ncRNAs
The m1A methylation, a critical internal RNA modification that has gradually received attention from researchers since it was first reported in 1961 [67], occurs on the first nitrogen atom of adenosine in RNA molecule. m1A has been determined in diversified RNA types, including tRNA, rRNA, mRNA, and lncRNA [40]. Similar to the dynamic modification of m6A and m5C, m1A is also regulated by functional methylation-related proteins called “writers”, “erasers”, and “readers”. The “writers” mainly includes tRNA methyltransferase 6 (TRMT6), TRMT61A, TRMT61B, TRMT10C, and nucleomethylin (NML) [68, 69]. ALKBH1, ALKBH3, ALKBH7, and FTO compose the “erasers” of m1A [70,71,72,73]. The “readers”, namely YTHDF1/2/3 and YTHDC1, are responsible for recognizing and binding to the m1A site [74].
m1A modification is considered to participate in the pre-RNA processing, regulate the structure and stability of RNA [72, 75]. Additionally, recent studies have shown that m1A affect the process of translation via modifications in tRNA [70], rRNA, and mRNA. By function, Wu et al. revealed that m1A demethylase ALKBH3 could regulate the glycose metabolism of tumor cells in a demethylation activity-dependent pattern [76]. Specifically, the m1A regulates the translation of ATP5D through YTHDF1/eRF3, and the m1A negatively regulates the mRNA stability of E2F1, thereby initiating the transcription of ATP5D. Another study demonstrated that TRMT61A mediates the m1A modification of tRNA, accelerates the translation of various key proteins after activation of CD4+T cells, and ensures the rapid immune response of CD4+T cells [77]. These studies broaden the understanding of the biological behavior of m1A-regulated RNA, and provide a theoretical basis for the development of novel tumor intervention strategies based on m1A modification. However, the study of biological function of m1A modification is still in its infancy. The interaction between m1A modification and ncRNA in HCC biology need to be further studied.
Biological function of methylation modification in ncRNAs in HCC
A growing body of evidence suggests that the methylation of ncRNAs plays a crucial role in the development of HCC. Here, we mainly summarize the biological functions associated with m6A and m5C methylation modifications of miRNAs, lncRNAs, and circRNAs in HCC (Table 1).
Biological function of methylation modifications in miRNAs
METTL14, a pivotal active component of the m6A methyltransferase complex, primarily localizes within the nucleus. Studies have demonstrated a notable downregulation of METTL14 expression in liver cancer tissues [101, 102]. Depletion of METTL14 has been shown to bolster the metastatic capacity of HCC cells, correlating with unfavorable overall survival rates among patients with HCC. MiR-126 is a tumor suppressor and plays a vital role in tumor metastasis. Research has shown that miR-126 functions as a downstream effector of METTL14 [78]. Moreover, overexpression of METTL14 facilitates its interaction with the microprocessor protein DGCR8, thereby enhancing the transformation of primary miR-126 into mature miRNA through an m6A-dependent mechanism, ultimately inhibiting HCC metastasis.
YBX1, alternatively referred to as YB1, serves as a “reader” protein for m5C modifications. YB1 is a carcinogen, and its elevated expression is closely linked to a dismal prognosis for HCC [103]. Liu et al. found that YB1 impedes the maturation of miR-205 and miR-200b by interacting with DGCR8, DICER, TUT4, and TUT1 [79]. Consequently, this engagement accentuates the expression of ZEB1, thereby facilitating the migration and invasion of HCC.
TET1, an “eraser” enzyme for m5C modifications, exhibits diminished expression within HCC tissues [104]. The absence of TET1 has been correlated with an unfavorable prognosis in HCC. The upregulation of TET1 inhibits the proliferation, migration, and invasion of HCC cells. Sun et al. found that TET1 activates miR-34a through demethylation of the 272/380 bp region within the miR-34a promoter. BACH1, a downstream target of miR-34a, mediates the P53 signaling pathway [80]. This intricate mechanism underscores how TET1 curtails HCC tumor genesis and metastasis through modulation of the miR-34a/BACH1/p53 axis, consequently fostering autophagy and apoptosis.
Biological function of methylation modifications in lncRNAs
METTL3 is the core protein of the m6A methyltransferase complex and plays a key role in m6A modifications of lncRNAs (Fig. 2) [105]. As elucidated by Wu et al., METTL3 mediates m6A modification of the lncRNA MEG3, subsequently leading to the downregulation of MEG3 [81]. This in turn exerts a notable influence on the malignant behavior of HCC through the MEG3/miR-544b/BTG2 axis. As established by a previous study, the lncRNA LINC00958 is associated with lipogenesis and is overexpressed in HCC. Moreover, the upregulation of LINC00958 is associated with an unfavorable overall survival prognosis. The m6A modification catalyzed by METTL3 leads to the upregulation of LINC00958 expression by reinforcing its transcriptional stability [82]. Consequently, LINC00958 operates as a molecular sponge for miR-3619-5p, culminating in the upregulation of hepatoma-derived growth factor (HDGF). This signaling cascade contributes to adipogenesis and the progression of HCC. Lnc-CTHCC, a conserved cancer-testis lncRNA, is highly expressed in testis and HCC. Xia et al. showed that METTL3-mediated m6A modifications in LCC-CTHCC are recognized by IGF2BP1 and IGF2BP3, crucially stabilizing LCC-CTHCC and increasing its expression in HCC [83]. Lnc-CTHCC can directly bind hnRNPK, thereby activating YAP1 transcription and, in turn, fostering the growth and metastasis of HCC. Additionally, both METTL3 and YTHDF2 mediate m6A modifications of LINC01273. Furthermore, LINC01273 increases sorafenib resistance in HCC by regulating the miR-600/METTL3 axis [84]. Targeting the LINC01273/miR-600/METTL3 signaling pathway emerges as a potential novel therapeutic strategy for effectively managing patients with sorafenib-resistant HCC.

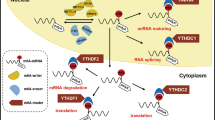
The molecular mechanism of METTL3 mediated m6A modifications in lncRNAs. METTL3 mediated m6A modifications could stabilize lncRNAs, such as lncRNA MEG3, LINC00958, LCC-CTHCC, and LINC01273, followed by regulate the downstream effectors, participating in multiple biological processes of HCC, including proliferation, migration, lipogenesis, metastasis, sorafenib resistance. Created with BioRender.com
METTL14, METTL16, and ZCCHC4 are crucial m6A methyltransferases. IGF2BP2 primarily functions as the “reader” protein. LncRNA ARHGAP5-AS1 is significantly overexpressed in HCC, with its m6A modification orchestrated by METTL14 and IGF2BP2, enhancing its stability. Liu et al. [85]. revealed that ARHGAP5-AS1 interacts with the oncoprotein CSDE1, orchestrating the regulation of carcinogenic RNA and driving the malignant tendencies of HCC. METTL16 is overexpressed in HCC, a factor that contributes to HCC tumorigenesis. METTL16 mediates the m6A modification of RAB11B-AS1, consequently diminishing the stability of its transcript and leading to the downregulation of RAB11B-AS1 expression [86]. Functioning as an RNA-binding protein, ZCCHC4 exhibits abnormal upregulation in HCC and is associated with a poor prognosis. In vitro and in vivo experiments have shown that ZCCHC4 enhances the chemotherapy resistance in HCC cells against DNA-damaging agents. Moreover, its interaction with the lncRNA AL133467.2 impedes apoptosis induced by DNA damage [87]. NSUN2, an m5C RNA methyltransferase, participates in the regulation of cell proliferation and metastasis and is upregulated in various tumors [42]. Sun et al. found that NSUN2 mediated m5C modification of lncRNA H19, thereby augmenting the stability of this lncRNA [88]. Through specific binding to the oncoprotein G3BP1, the m5C-modified lncRNA H19 triggers the accumulation of MYC, subsequently fostering poor differentiation of HCC.
Distinct RNA-binding proteins, known as “reader” proteins, play pivotal roles in executing specific biological functions of methylated RNA. The primary functions of these “reader” proteins primarily include the specific binding to the m6A methylation region, attenuation of homologous RNA-binding protein interactions, and modulation of RNA secondary structures to influence protein–RNA interactions. As elucidated by Chen et al., YTHDF2 orchestrates the m6A modification of lncAY, leading to the upregulation of BMI1 expression in HCC [89]. This, in turn, triggers activation of the Wnt/β-catenin signaling pathway, thereby contributing to HCC progression. YTHDF2 further engages in m6A-dependent splicing of lncFAL [90]. The resultant lncFAL then engages with ferroptosis suppressant protein 1 (FSP1), competitively thwarting TRIM69-mediated polyubiquitination degradation of FSP1 and thereby diminishing ferroptosis susceptibility. Moreover, IGF2BP1 specifically binds to the HCC-associated lncRNA HULC and plays a crucial role in RNA metabolism regulation [91]. Mechanistically, IGF2BP1 recruits the CCR4-NOT deadenylase complex, subsequently instigating the degradation of lncRNA HULC.
ALKBH5, a key demethylase of m6A, is downregulated in HCC. Chen et al. validated that ALKBH5 is a tumor suppressor factor in HCC, with its diminished expression correlating with poor overall survival in patients with HCC [106]. Ahati et al. further demonstrated ALKBH5’s ability to curtail m6A enrichment, resulting in the upregulation of the lncRNA NEAT1’s expression [92]. This ALKBH5-mediated lncRNA NEAT1 then acts as a sponge for miR-214, promoting the proliferation and migration of HCC cells. Moreover, m6A-modified LINC01468 is dependent on ALKBH5, which can enhance the stability of LINC01468 and upregulate its expression [93]. Upregulated LINC01468, in turn, interacts with SHIP2, enhancing CUL4A-associated degradation and enabling the activation of the PI3K/AKT/mTOR signaling pathway, thereby fueling lipogenesis and HCC progression.
Biological function of methylation modification in circRNAs
CircRNAs are a distinctive form of closed circular ncRNAs, primarily produced by variable shear processing of pre-Mrna [107]. Notable for their stability and conservation, circRNAs have garnered increasing attention owing to their potential significance in diverse biological contexts. In the progression of HCC, accumulating evidence underscores the pivotal role of circRNA methylation [108,109,110]. Wu et al. revealed that hsa_circ_0058493 is highly expressed in HCC and positively correlates to a poor prognosis [94]. Moreover, METTL3 mediates the m6A methylation level of hsa_circ_0058493, with YTHDC1 facilitating its translocation from the nucleus to the cytoplasm, thereby modulating intracellular localization. Mechanistically, m6A-modified hsa_circ_0058493 was demonstrated to promote HCC development through the METTL3/hsa_circ_0058493/YTHDC1 axis. In addition, m6A-modified circHPS5HCC is highly expressed in HCC tissues, contributing to epithelial–mesenchymal transition (EMT) and the induction of cancer stem-like cell phenotypes. Mechanistically, METTL3 mediates circHPS5 generation, and YTHDC1 accelerates circHPS5 output from the cytoplasm [95]. Acting as a miR-370 sponge, circHPS5 influences the expression of HMGA2, thereby promoting the tumorigenesis of HCC. circ-ARL3, an HBV-associated circRNA, promotes malignant phenotypes in HBV-associated HCC. METTL3 enhances the m6A modification degree of circ-ARL3, and the binding of YTHDC1 and circ-ARL3 accelerates reverse splicing and biogenesis of circ-ARL3 [96]. Functioning as a miR-1305 sponge, circ-ARL3 further accentuates HBV-associated HCC tumor formation. Moreover, METTL3 mediates the effects of hsa_circ_0008583 on the behavior of HCC cells [97]. Furthermore, hsa_circ_0008583 accelerates the development of HCC via the miR-1301-3p/METTL3 axis.
KIAA1429 is a crucial component of the m6A methyltransferase complex and plays a vital role in m6A modification [111]. Elevated expression of KIAA1429 in HCC tissues was observed, correlating with poorer prognoses. KIAA1429 was found to enhance the development of HCC through m6A-dependent GATA3 post-transcriptional modification [112]. Additionally, KIAA1429 regulates the expression of circDLC1. Further studies have shown that circDLC1 interacts with the RNA-binding protein HuR interactions, thereby inhibiting the interplay between HuR and MMP1 mRNAs and subsequently curtailing HCC proliferation and motility [98]. Except for KIAA1429, Liu et al. revealed the involvement of ALKBH5 and METTL3 in binding to circ-CCT3, mediating its m6A modification. Upregulated in HCC, circ-CCT3 was proposed to function as a miR-378-3p sponge, thereby elevating FLT-1 expression and intensifying HCC proliferation, invasion, and migration [99]. Another study highlighted ALKBH5’s role in circCPSF6 demethylation, with YTHDF2 recognizing and destabilizing it [100]. M6A-modified circCPSF6 was shown to competitively bind with PCBP2, attenuating its interaction with YAP1 mRNA, thus activating YAP1 and sustaining tumorigenicity and metastasis of HCC.
NcRNAs regulate methylation-related proteins in HCC
Both m6A and m5C modifications play pivotal roles in mediating the methylation modification of ncRNA. In turn, ncRNAs also modulate the expression levels of methylation-related proteins, thus affecting the occurrence and development of HCC. Here, we generalize the recent advancements pertaining to the regulatory effects of miRNAs, lncRNAs, and circRNAs on methylation-related proteins in HCC (Table 2).
MiRNAs regulate methylation-related proteins in HCC
MiRNAs can regulate oncogenes and tumor suppressors during the progression of liver cancer [134,135,136]. They are also implicated in liver cancer metastasis, immune modulation, and chemotherapy resistance, among other processes. Notably, miRNAs also regulate the expression levels of methylation-related proteins, thereby influencing the tumorigenesis of HCC through intricate mechanisms. WTAP is notably overexpressed in HCC tissues and exhibits a positive correlation with a poor prognosis in patients with HCC. Liu et al. found that miR-1395p attenuates WTAP expression by targeting its 3′-UTR, thereby inhibiting the growth of HCC [113]. The miR-139-5p/WTAP axis also governs HCC development by modulating EMT. miR-1275 directly targets IGF2BP1, IGF2BP2, and IGF2BP3. The overexpression of miR-1275 inhibits the expression levels of these IGF2BPs, thereby mitigating the malignant behavior of HCC [114]. In parallel, another study demonstrated that phytochemicals, including Tamarix articulata, quercetin, and epigallocatechin gallate, significantly increase the expression of miR-1275, thereby inhibiting the mRNA expression of IGF2BP1 and IGF2BP3 [115]. This suggests that the miRNA/IGF axis could serve as a novel mechanism for these phytochemicals to exert their anti-tumor effects. Zhou et al. found that miR-188-5p can bind to the 3′-UTR of hnRNPA2B1 and regulate the expression of hnRNPA2B1 [116]. Endoplasmic reticulum stress can facilitate sorafenib resistance in HCC through the miR-188-5p/hnRNPA2B1/PKM2 axis.
ALYREF is dysregulated in HCC, and its overexpression is closely linked to a poor prognosis in HCC. Xue et al. found that miR-4666a-5p and miR-6124 are potential regulators of ALYREF, suggesting their significant involvement in the epigenetic regulation of HCC [117]. Han et al. found that miR-3190 downregulates ALKBH5 expression in bone-metastasized HCC [118]. Reduced ALKBH5 levels facilitate HCC progression by regulating gene expression through both m6A-dependent and non-dependent pathways. TET2, crucial for hematopoietic stem cell self-renewal, exhibits low expression in HCC, inhibiting stemness and metastasis of HCC cells. Alcohol consumption further decreases the expression of TET2 in HCC. Further studies indicate that miR-22-3p directly targets TET2, and chronic alcohol intake instigates HCC tumor formation via the β-catenin/miR-22-3p/TET2 axis [119]. The expression of TET1 is decreased in HCC, which may have tumor suppressive effects. Lin et al. found that MiR-29b inhibits HCC metastasis by targeting TET1 [120]. Another study showed that miR-520b curbs HCC cell proliferation through the 3′UTR of TET1 mRNA [121]. These findings indicate the potential of targeting TET1 as a therapeutic strategy for the treatment of HCC.
LncRNAs regulate methylation-related proteins in HCC
LncRNAs have also been reported to interact with methylation-related proteins, thereby influencing their functionality [137]. As expounded by Zhou et al., LINC00839 acts as a sponge for miR-144-3p, effectively downregulating the activity of miR-144-3p [122]. This, in turn, leads to elevated expression of WTAP, fostering the malignant phenotype within HCC cells. Bo et al. found that the lncRNA ILF3-AS1 can recruit METTL3, thereby elevating the m6A modification level of ILF3 [123]. In addition, the lncRNA ILF3-AS1 enhances the interaction between ILF3 mRNA and IGF2BP1, thereby contributing to the malignancy observed in HCC. A few methylation-related proteins are under the regulation of more than one miRNA or lncRNA. Studies have reported that miR503HG interacts with HNRNPA2B1, effectively curbing NF-κB signaling by modulating the ubiquitination status of HNRNPA2B1, consequently restraining HCC metastasis [124]. Moreover, lncRNA-UC002MBe-2 also engages with HNRNPA2B1, facilitating AKT inactivation and p21 induction [125]. This interaction plays a role in the suppressive impact of trecomycin on hepatoma cells. In addition, distinct lncRNAs exhibit varying effects on methylation-related proteins. In p53-deficient-HCC cells, Linc01612 mediates the ubiquitination and degradation of YBX1 through physical interaction with YBX1, thus exerting its biological functions [126]. In addition, Zhao et al. found that the IncRNA AWPPH binds to the YBX1 protein, subsequently enabling YBX1 to activate SNAIL1 or PIK3CA [127]. This activation, in turn, fosters the growth and metastasis of HCC.
CircRNAs regulate methylation-related proteins in HCC
CircRNAs and methylation modifications play a significant role in the progression of HCC, with their underlying mechanisms extensively documented. circRERE is highly expressed in HCC, and its downregulation significantly increases the m6A levels of GBX2, thereby promoting the upregulation of the methyltransferase ZC3H13 [128]. This cascade expedites the proliferation and invasion of HCC cells. A recently identified rt-circRNA, rtcisE2F, is highly expressed in HCC and liver tumor-initiating cells (TICs). Chen et al. found that rtcisE2F regulates the interactions between IGF2BP2 and YTHDF2 with E2F6/E2F3 mRNAs [129]. Consequently, this triggers the self-renewal of TICs through the Wnt/β-catenin pathway, contributing to the onset and metastasis of HCC. The circRNA hsa_circ_0062682 is upregulated in HCC and recognized as a carcinogenic determinant. The YBX1 protein was identified to bind with circ_0062682. However, the precise downstream molecular mechanisms of this circ_0062682 and YBX1 interplay on HCC progression remain a subject warranting further exploration [130].
Under specific circumstances, circRNAs can act as sponges for miRNAs, thus further modulating the expression of downstream target genes. For instance, circDLG1, also upregulated in HCC, exhibits correlation to a poor prognosis. circDLG1 functions as a sponge for miR-141-3p, thereby regulating the expression of WTAP and inhibiting the progression of HCC [131]. Furthermore, Chi et al. constructed a regulatory network of circRNA-miRNA-m6A RNA methylation regulators and unveiled that circMAP2K4 interacts with hsa-miR-139-5p, promoting the expression of YTHDF1 and thereby facilitating HCC cell proliferation [132]. Another study revealed that the downregulation of circGPR137B or the upregulation of miR-4739 is correlated to poor prognosis in patients with HCC. CircGPR137B localizes with miR-4739 in the cytoplasm, where it functions as a sponge, up-regulating the expression of its target protein, FTO [133]. In turn, FTO mediates m6A demethylation of ciGPR137B and elevates its expression. This establishes a feedback loop comprising the circGPR137B/miR-4739/FTO axis, implicated in the development of HCC.
Effect of interactions between ncRNAs and methylations in Tumor immune microenvironment (TIME) of HCC
The TIME of HCC is highly heterogeneous, posing a considerable challenge to liver cancer immunotherapy. Epigenetic modifications have been extensively studied in the context of HCC and exert substantial influence on the TIME. Therefore, we further summarized the effects of the interplay between ncRNA and methylation on the TIME in HCC (Table 3). This endeavor is geared towards offering novel avenues for the enhancement of HCC immunotherapy strategies.
Effect of interactions between miRNAs and methylations in TIME of HCC
ZC3H13, an m6A writer, exhibits down-regulation in HCC tissues and could serve as a tool for evaluating the prognosis of patients with HCC. Wu et al. have identified miR-362-3p/miR-425-5p as upstream regulators of ZC3H13 capable of suppressing its expression. Subsequent investigations have revealed that ZC3H13 within HCC might contribute to disease progression by augmenting the infiltration of immune cells, specifically CD4+ T cells, macrophages, neutrophils, and dendritic cells, especially neutrophils [138]. In addition, ZC3H13 is closely linked to the expression of the immune checkpoint PD-L1, implying its involvement in regulating the immune microenvironment of HCC. Coincidentally, Lin et al. found that over 30 miRNAs contribute to the regulation of m6A methylation-related proteins, especially miR-142. Moreover, the m6A methylation of RNA affects the expression of PD-L1 and is closely linked to the infiltration of immune cells such as T follicular helper cells, activated NK cells, and regulatory T cells [139]. Among these, M1 macrophages stand out for their role in tumor suppression. Wang et al. demonstrated that M1 macrophage-derived exosomes transmit miR-628-5p to HCC cells, subsequently repressing the expression of METTL14 [140]. METTL14, in turn, facilitates the m6A modification of circFUT8, promoting its translocation from the nucleus to the cytoplasm, wherein M1 macrophages regulate the cirfut8/miR-552-3p/CHMP4B pathway, thereby inhibiting HCC progression (Fig. 3). miR4458HG is a carcinogen in HCC. Ye et al. found that miR4458HG binds to the m6A reader IGF2BP2, thereby enhancing the mRNA stability of IGF2BP2-mediated targets HK2 and SLC2A1 [141]. This alteration leads to a modification in the glycolytic response of HCC. In addition, HCC-derived miR4458HG exerts control over ARG1 expression and promotes the polarization of tumor-associated macrophages. Collectively, the intricate interplay of miRNAs and methylation may affect the progression of HCC by regulating the immune microenvironment.

The effect of interactions between lncRNAs and methylations in tumor immune microenvironment of HCC. Created with BioRender.com
Effect of interactions between lncRNAs and methylations in TIME of HCC
The lipopolysaccharide (LPS) originating from intestinal bacteria can induce the upregulation of PD-L1 in HCC cells, thereby orchestrating T cell inhibition, thus affecting the development of HCC. Peng et al. revealed that LPS promotes the m6A methylation of the lncRNA MIR155HG by regulating the expression of METTL14, subsequently stabilizing the lncRNA MIR155HG [142]. MIR155HG regulates the expression of PD-L1 via the miR-223/STAT1 axis and assumes a key role in the immune evasion mechanisms within HCC. A pivotal challenge in effective cancer treatment lies in curtailing tumor recurrence subsequent to the expansion of tumor-initiating stem-like cells. Identifying effective combinations of TIC specificity is a promising strategy for prolonging survival in patients with HCC. Chen et al. demonstrated that the incorporation of immune checkpoint inhibitors yields an additional reduction in recurrence rates and extends the survival of patient-derived xenograft mice. Combination therapy with FDA-approved drugs that can inhibit the lncRNA MIR22HG reduces many toll-like receptors and stemness genes, and this downregulation induces PTEN and TET2, ultimately culminating in the loss of TIC self-renewal properties [143]. However, a more robust body of evidence is imperative to establish direct causality between lncRNA interactions and methylation and their collective influence on the immune microenvironment of HCC.
Effects of interactions between circRNAs and methylation in TIME of HCC
Exosome-derived circRNAs can influence the immune escape of HCC by engaging in intercellular communication. Hu et al. showed that the level of circCCAR1 in exosomes from patients with HCC is increased [144]. WTAP facilitates the m6A modification of circCCAR1 by binding to IGF2BP3. As a consequence, circCCAR1 contributes to the impairment of CD8+ T cell function by stabilizing PD-1 protein, consequently fostering resistance to anti-PD-L1 immunotherapy. Another recently identified circRNA, circRHBDD1, was found to be highly expressed in patients with HCC who exhibited an anti-PD-1 response, which limited the effectiveness of anti-PD-1 therapy. Cai et al. found that circRHBDD1 recruits YTHDF1, thereby expediting the translation of PIK3R1 through m6A modification of PIK3R1 mRNA [145]. This, in turn, activates the PI3K/AKT signaling pathway, thereby promoting the progression of HCC. Inhibition of circRHBDD1 has been postulated to enhance the efficacy of anti-PD-1 therapy in immunodeficient mice. In addition, the downregulation of circTRIM33-12 in HCC is significantly associated with a poor prognosis. Mechanistically, circTRIM33-12 functions as a sponge for miR-191, thus facilitating heightened TET1 expression [146]. The consequent upregulation of TET1 operates in a manner that opposes oncogenic gene expression, inhibiting HCC proliferation, metastasis, and immune evasion. These findings illustrate the potential of circTRIM33-12 as a novel therapeutic target for HCC.
RNA methylation-related proteins may serve as therapeutic targets for HCC
To date, it has been established that RNA methylation affects diverse aspects of HCC, such as its progression, immune microenvironment, and drug sensitivity. Most methylation-related proteins are dysregulated in HCC and play a key role in the development of HCC. Targeting methylation-related proteins holds great promise for the treatment of HCC.
Targeting “writer” proteins for the treatment of HCC
Multiple studies have underscored METTL3’s potential as a therapeutic target in HCC, operating through diverse mechanisms. Silencing METTL3 has been shown to heighten HCC sensitivity to chemotherapy by impeding the m6A modification of p53 mRNA [147]. Furthermore, METTL3 fosters HCC metastasis by establishing a positive feedback loop with STAT3 [148]. Wang et al. have also reported that the METTL3 inhibitor STM2457 targets the epidermal growth factor receptor (EGFR) to improve the sensitivity of HCC to lenvatinib therapy [149]. This suggests that METTL3 might hold promise as a countermeasure against lenvatinib resistance in HCC. METTL14 targets EGFR, regulate the PI3K/AKT pathway, and inhibits the progression of HCC cells [150]. In addition, METTL14 may participate in the malignant development of HCC by mediating m6A methylation of cysteine sulfinic acid decarboxylase and glutamic-oxalotransaminase 2 [151]. Such findings suggest that targeted regulation of METTL14 could emerge as a novel avenue for the treatment of HCC. WTAP is significantly overexpressed in HCC, facilitating m6A modification that propels HCC progression via the ETS1-p21/p27 axis [152]. Therefore, inhibiting the expression of WTAP stands as a potential avenue for enhancing HCC prognosis. ZC3H13 may participate in transcriptional dysregulation or the JAK/STAT pathway in HCC [153]. Its expression is significantly associated with lymphocytes and immunomodulators. The upregulation of ZC3H13 inhibits the progression of HCC, thus designating it as a prospective therapeutic target for HCC.
Targeting “reader” proteins for the treatment of HCC
HNRNPC plays a pivotal role in the development of HCC. Knocking down HNRNPC has been demonstrated to inhibit the proliferation, migration, and invasion of HCC cells via the Ras/MAPK signaling pathway [154]. Therefore, HNRNPC emerges as a potential therapeutic target for patients with HCC. YTHDF1 may enhance the malignant phenotype by promoting EMT and activating the AKT/glycogen synthase kinase-3 beta/beta-catenin signaling cascade [155]. Silencing YTHDF1 significantly inhibits the proliferation, invasion, and migration of HCC cells. In HCC, YTHDF2 may regulates the regulation of tumor-associated macrophage polarization, T cell dysfunction induction, and activation of T regulatory cells, thereby intricately influencing the course of HCC progression [156]. Hence, targeting either YTHDF1 or YTHDF2 presents a novel avenue for devising strategies for HCC treatment. A study showed that small nucleotide polymorphisms within YTHDC2 and FTO are significantly correlated to the prognosis of patients with HCC treated with TACE, suggesting that they may be potential targets to enhance treatment approaches for patients with unresectable HCC [157].
Targeting “eraser” proteins for the treatment of HCC
FTO mediates IL-17 receptor A to regulate both inflammation and the transformation towards carcinogenesis in HCC [158]. Precisely targeting FTO may prevent HCC development, particularly in patients with hepatitis. Additionally, Xiao et al. reported that administering FTO-inhibiting nanomedicine to tumor-infiltrating dendritic cells proves advantageous in fostering HCC immunotherapy, especially when paired with immune checkpoint blockade post-HCC thermal ablation [159]. As demethylases, the ALKB family participates in the development of HCC. Research indicates that ALKBH1/2/3/4/7 is markedly highly expressed in HCC tissues and is correlated with the infiltration of immune cells such as CD8+ T cells, CD4+ T cells, and macrophages [160]. This suggests that the ALKB family may be a potential therapeutic target for HCC. Moreover, Chen et al. unveiled that ALKBH5 suppresses the expression of the oncoprotein LYPD1 in HCC in an M6A-dependent manner [106]. Furthermore, Qu et al. found that ALKBH5 deletion significantly inhibits the growth and migration of HBV-associated HCC cells [161]. HBx-ALKBH5 interactions might establish a positive feedback loop, contributing to the genesis of HBV-induced liver cancer. Consequently, targeting ALKBH5 surfaces as a promising avenue for managing HBV-induced HCC.
Conclusions and perspectives
Accumulating evidence underscores the interplay between ncRNAs and methylation modifications in the development of HCC. On the one hand, RNA methylation modifications affect various facets of ncRNA, encompassing transcription, splicing, processing, translation, localization, and stability. These modifications affect biological processes such as proliferation, migration, invasion, apoptosis, and EMT in HCC. On the other hand, ncRNAs can also, in turn, regulate the expression levels of RNA methylation-related proteins, thereby exerting downstream molecular effects that shape the progression of HCC. In addition, the interaction between ncRNA and methylation-related proteins assumes a k ey role in orchestrating the immune microenvironment of HCC. Certain methylated modulators have shown potential to heighten HCC’s sensitivity to targeted therapies or chemotherapy and hold promise as viable therapeutic targets for HCC. However, existing research pertaining to the intricate interplay between ncRNAs and methylation modifications only scratches the surface. Future endeavors demand a more profound exploration to elucidate and validate the potential implications of ncRNA methylation modifications on HCC pathogenesis, consequently furnishing more precise strategies for HCC treatment.
Availability of data materials
Data sharing is not applicable to this article as no new data were created or analysed in this study. Figures were created by Biorender (https://biorender.com/).
Abbreviations
- HCC:
-
hepatocellular carcinoma
- ncRNAs:
-
non-coding RNAs
- HBV:
-
hepatitis B virus
- HCV:
-
hepatitis C virus
- TACE:
-
transarterial chemoembolization
- miRNAs:
-
microRNAs
- lncRNAs:
-
long ncRNAs
- circRNAs:
-
circular RNAs
- mRNA:
-
messenger RNA
- UTR:
-
3′-untranslated region
- tRNA:
-
transfer RNA
- m6A:
-
N6-methyladenosine
- m1A:
-
N1-methyladenosine
- m5C:
-
5-methylcytosine
- Ψ:
-
pseudouridine
- m7G:
-
7-methylguanine
- METTL3:
-
methyltransferase-like 3
- ALYREF:
-
Aly/REF export factor
- ZC3H13:
-
zinc finger CCCH-type containing 13
- ZCCHC4:
-
zinc finger CCHC domain-containing protein 4
- WTAP:
-
Wilms tumor 1-associated protein
- FTO:
-
fat mass and obesity-associated protein
- ALKBH5:
-
alkylation repair homolog protein 5
- YTHDF1:
-
YTH N6-methyladenine RNA binding protein 1
- IGF2BP1:
-
insulin-like growth factor 2 mRNA-binding protein 1
- HNRNPA2B1:
-
heterogeneous nuclear ribonucleoprotein A2/B1
- hnRNPC:
-
heterogeneous nuclear ribonucleoprotein C
- NSUN:
-
NOL1/NOP2/SUN domain
- DNMT2:
-
DNA methyltransferase 2
- TET:
-
ten-eleven translocation
- YBX1:
-
Y-box binding protein 1
- FSP1:
-
ferroptosis suppressor protein 1
- EMT:
-
epithelial–mesenchymal transition
- TIME:
-
tumor immune microenvironment
- LPS:
-
lipopolysaccharide
- EGFR:
-
epidermal growth factor receptor
- HDGF:
-
hepatoma-derived growth factor
- NML:
-
nucleomethylin
- TRMT6:
-
tRNA methyltransferase 6
References
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. Cancer J Clin. 2023;73(1):17–48.
Rumgay H, Arnold M, Ferlay J, Lesi O, Cabasag CJ, Vignat J, et al. Global burden of primary Liver cancer in 2020 and predictions to 2040. J Hepatol. 2022;77(6):1598–606.
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nature reviews. Disease Primers. 2021;7(1):6.
Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet (London England). 2022;400(10360):1345–62.
Chen Z. Valuing the Prevention and Treatment of Liver Disease to Promote Human Wellbeing. Infect Microbes Dis. 2022;4(2):47–8.
Fujiwara N, Friedman SL, Goossens N, Hoshida Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J Hepatol. 2018;68(3):526–49.
Chiang CJ, Yang YW, You SL, Lai MS, Chen CJ. Thirty-year outcomes of the national Hepatitis B immunization program in Taiwan. JAMA. 2013;310(9):974–6.
Dave S, Park S, Murad MH, Barnard A, Prokop L, Adams LA et al. Comparative effectiveness of Entecavir Versus Tenofovir for preventing Hepatocellular Carcinoma in patients with chronic Hepatitis B: a systematic review and Meta-analysis. Hepatology (Baltimore, Md.). 2021;73(1):68–78.
Kanwal F, Kramer JR, Asch SM, Cao Y, Li L, El-Serag HB. Long-term risk of Hepatocellular Carcinoma in HCV patients treated with Direct Acting Antiviral agents. Hepatology (Baltimore MD). 2020;71(1):44–55.
Kanwal F, Kramer JR, Mapakshi S, Natarajan Y, Chayanupatkul M, Richardson PA, et al. Risk of Hepatocellular Cancer in patients with non-alcoholic fatty Liver Disease. Gastroenterology. 2018;155(6):1828–1837e1822.
Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty Liver Disease demonstrates an exponential increase in burden of Disease. Hepatology (Baltimore MD). 2018;67(1):123–33.
Tabrizian P, Jibara G, Shrager B, Schwartz M, Roayaie S. Recurrence of hepatocellular cancer after resection: patterns, treatments, and prognosis. Ann Surg. 2015;261(5):947–55.
Mehta N, Guy J, Frenette CT, Dodge JL, Osorio RW, Minteer WB, et al. Excellent outcomes of liver transplantation following down-staging of Hepatocellular Carcinoma to within Milan Criteria: a Multicenter Study. Clinical gastroenterology and hepatology: the official clinical practice. J Am Gastroenterol Assoc. 2018;16(6):955–64.
Yu J, Yu XL, Han ZY, Cheng ZG, Liu FY, Zhai HY, et al. Percutaneous cooled-probe microwave versus radiofrequency ablation in early-stage hepatocellular carcinoma: a phase III randomised controlled trial. Gut. 2017;66(6):1172–3.
Li QJ, He MK, Chen HW, Fang WQ, Zhou YM, Xu L, et al. Hepatic arterial infusion of Oxaliplatin, Fluorouracil, and Leucovorin Versus Transarterial Chemoembolization for large Hepatocellular Carcinoma: a Randomized Phase III Trial. J Clin Oncology: Official J Am Soc Clin Oncol. 2022;40(2):150–60.
Brown ZJ, Tsilimigras DI, Ruff SM, Mohseni A, Kamel IR, Cloyd JM, et al. Management of Hepatocellular Carcinoma: a review. JAMA Surg. 2023;158(4):410–20.
Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816.
Adams BD, Parsons C, Walker L, Zhang WC, Slack FJ. Targeting noncoding RNAs in Disease. J Clin Investig. 2017;127(3):761–71.
Pritchard CC, Cheng HH, Tewari M. MicroRNA profiling: approaches and considerations. Nat Rev Genet. 2012;13(5):358–69.
Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610.
Catalanotto C, Cogoni C, Zardo G. MicroRNA in Control of Gene expression: an overview of Nuclear functions. Int J Mol Sci. 2016;17(10):1712.
Diener C, Keller A, Meese E. Emerging concepts of miRNA therapeutics: from cells to clinic. Trends Genet. 2022;38(6):613–26.
Herman AB, Tsitsipatis D, Gorospe M. Integrated lncRNA function upon genomic and epigenomic regulation. Mol Cell. 2022;82(12):2252–66.
Huang Z, Zhou JK, Peng Y, He W, Huang C. The role of long noncoding RNAs in hepatocellular carcinoma. Mol Cancer. 2020;19(1):77.
Xue C, Li G, Zheng Q, Gu X, Bao Z, Lu J, et al. The functional roles of the circRNA/Wnt axis in cancer. Mol Cancer. 2022;21(1):108.
Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20(11):675–91.
Zhou WY, Cai ZR, Liu J, Wang DS, Ju HQ, Xu RH. Circular RNA: metabolism, functions and interactions with proteins. Mol Cancer. 2020;19(1):172.
Huang XY, Huang ZL, Huang J, Xu B, Huang XY, Xu YH, et al. Exosomal circRNA-100338 promotes hepatocellular carcinoma Metastasis via enhancing invasiveness and angiogenesis. J Experiment Clin Cancer Res. 2020;39(1):20.
Zhang B, Bao W, Zhang S, Chen B, Zhou X, Zhao J, et al. LncRNA HEPFAL accelerates ferroptosis in hepatocellular carcinoma by regulating SLC7A11 ubiquitination. Cell Death Dis. 2022;13(8):734.
Komoll RM, Hu Q, Olarewaju O, von Döhlen L, Yuan Q, Xie Y, et al. MicroRNA-342-3p is a potent tumour suppressor in hepatocellular carcinoma. J Hepatol. 2021;74(1):122–34.
Shi X, Liu TT, Yu XN, Balakrishnan A, Zhu HR, Guo HY, et al. microRNA-93-5p promotes hepatocellular carcinoma progression via a microRNA-93-5p/MAP3K2/c-Jun positive feedback circuit. Oncogene. 2020;39(35):5768–81.
Liu Z, Yu Y, Huang Z, Kong Y, Hu X, Xiao W, et al. CircRNA-5692 inhibits the progression of hepatocellular carcinoma by sponging mir-328-5p to enhance DAB2IP expression. Cell Death Dis. 2019;10(12):900.
Wang L, Li B, Bo X, Yi X, Xiao X, Zheng Q. Hypoxia-induced LncRNA DACT3-AS1 upregulates PKM2 to promote Metastasis in hepatocellular carcinoma through the HDAC2/FOXA3 pathway. Exp Mol Med. 2022;54(6):848–60.
Zhou W, Wang X, Chang J, Cheng C, Miao C. The molecular structure and biological functions of RNA methylation, with special emphasis on the roles of RNA methylation in autoimmune Diseases. Crit Rev Clin Lab Sci. 2022;59(3):203–18.
Zheng HX, Zhang XS, Sui N. Advances in the profiling of N(6)-methyladenosine (m(6)A) modifications. Biotechnol Adv. 2020;45:107656.
Motorin Y, Helm M. RNA nucleotide methylation: 2021 update. Wiley interdisciplinary reviews. RNA. 2022;13(1):e1691.
Xue C, Chu Q, Zheng Q, Jiang S, Bao Z, Su Y, et al. Role of main RNA modifications in cancer: N(6)-methyladenosine, 5-methylcytosine, and pseudouridine. Signal Transduct Target Therapy. 2022;7(1):142.
Xiong Q, Zhang Y, Small. RNA modifications: regulatory molecules and potential applications. J Hematol Oncol. 2023;16(1):64.
Sendinc E, Shi Y. RNA m6A methylation across the transcriptome. Mol Cell. 2023;83(3):428–41.
Li J, Zhang H, Wang H. N(1)-methyladenosine modification in cancer biology: current status and future perspectives. Comput Struct Biotechnol J. 2022;20:6578–85.
Lin S, Liu Q, Lelyveld VS, Choe J, Szostak JW, Gregory RI. Mettl1/Wdr4-Mediated m(7)G tRNA methylome is required for normal mRNA translation and embryonic stem cell Self-Renewal and differentiation. Mol Cell. 2018;71(2):244–255e245.
Hu Y, Chen C, Tong X, Chen S, Hu X, Pan B, et al. NSUN2 modified by SUMO-2/3 promotes gastric cancer progression and regulates mRNA m5C methylation. Cell Death Dis. 2021;12(9):842.
Shi H, Chai P, Jia R, Fan X. Novel insight into the regulatory roles of diverse RNA modifications: re-defining the bridge between transcription and translation. Mol Cancer. 2020;19(1):78.
Boo SH, Kim YK. The emerging role of RNA modifications in the regulation of mRNA stability. Exp Mol Med. 2020;52(3):400–8.
Boulias K, Greer EL. Biological roles of adenine methylation in RNA. Nature reviews. Genetics. 2023;24(3):143–60.
Lin Z, Niu Y, Wan A, Chen D, Liang H, Chen X, et al. RNA m(6) a methylation regulates sorafenib resistance in Liver cancer through FOXO3-mediated autophagy. EMBO J. 2020;39(12):e103181.
Xue C, Gu X, Zheng Q, Shi Q, Yuan X, Su Y, et al. ALYREF mediates RNA m(5)C modification to promote hepatocellular carcinoma progression. Signal Transduct Target Therapy. 2023;8(1):130.
Wang Y, Wang J, Li X, Xiong X, Wang J, Zhou Z, et al. N(1)-methyladenosine methylation in tRNA drives liver tumourigenesis by regulating cholesterol metabolism. Nat Commun. 2021;12(1):6314.
Zhang H, Shi X, Huang T, Zhao X, Chen W, Gu N, et al. Dynamic landscape and evolution of m6A methylation in human. Nucleic Acids Res. 2020;48(11):6251–64.
Oerum S, Meynier V, Catala M, Tisné C. A comprehensive review of m6A/m6Am RNA methyltransferase structures. Nucleic Acids Res. 2021;49(13):7239–55.
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA. 1974;71(10):3971–5.
Meyer KD, Jaffrey SR. Rethinking m(6)a readers, writers, and Erasers. Annu Rev Cell Dev Biol. 2017;33:319–42.
Li J, Yao H, Huang J, Li C, Zhang Y, Xu R, et al. METTL3 promotes prostatic hyperplasia by regulating PTEN expression in an m(6)A-YTHDF2-dependent manner. Cell Death Dis. 2022;13(8):723.
Wei J, Yu X, Yang L, Liu X, Gao B, Huang B, et al. FTO mediates LINE1 m(6)a demethylation and chromatin regulation in mESCs and mouse development. Science. 2022;376(6596):968–73.
Zhou J, Zhang X, Hu J, Qu R, Yu Z, Xu H et al. m(6)A demethylase ALKBH5 controls CD4(+) T cell pathogenicity and promotes autoimmunity. Sci Adv. 2021;7(25):eabg0470.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–95.
Huang H, Weng H, Chen J. M(6)a modification in Coding and non-coding RNAs: roles and therapeutic implications in Cancer. Cancer Cell. 2020;37(3):270–88.
Shaath H, Vishnubalaji R, Elango R, Kardousha A, Islam Z, Qureshi R, et al. Long non-coding RNA and RNA-binding protein interactions in cancer: experimental and machine learning approaches. Sem Cancer Biol. 2022;86(Pt 3):325–45.
Chen RX, Chen X, Xia LP, Zhang JX, Pan ZZ, Ma XD, et al. N(6)-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver Metastasis. Nat Commun. 2019;10(1):4695.
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 2017;27(5):626–41.
Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L, et al. N6-Methyladenosine modification controls circular RNA immunity. Mol Cell. 2019;76(1):96–109e109.
Zhang Q, Liu F, Chen W, Miao H, Liang H, Liao Z, et al. The role of RNA m(5)C modification in cancer Metastasis. Int J Biol Sci. 2021;17(13):3369–80.
Bohnsack KE, Höbartner C, Bohnsack MT. Eukaryotic 5-methylcytosine (m5C) RNA methyltransferases: mechanisms, Cellular functions, and links to Disease. Genes. 2019;10(2):102.
Nombela P, Miguel-López B, Blanco S. The role of m(6)A, m(5)C and Ψ RNA modifications in cancer: novel therapeutic opportunities. Mol Cancer. 2021;20(1):18.
Zheng H, Zhu M, Li W, Zhou Z, Wan X. M(5) C and m(6) a modification of long noncoding NKILA accelerates cholangiocarcinoma progression via the mir-582-3p-YAP1 axis. Liver Int. 2022;42(5):1144–57.
He Y, Zhang Q, Zheng Q, Yu X, Guo W. Distinct 5-methylcytosine profiles of circular RNA in human hepatocellular carcinoma. Am J Translational Res. 2020;12(9):5719–29.
Dunn DB. The occurrence of 1-methyladenine in ribonucleic acid. Biochim Biophys Acta. 1961;46:198–200.
Safra M, Sas-Chen A, Nir R, Winkler R, Nachshon A, Bar-Yaacov D, et al. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature. 2017;551(7679):251–5.
Waku T, Nakajima Y, Yokoyama W, Nomura N, Kako K, Kobayashi A, et al. NML-mediated rRNA base methylation links ribosomal subunit formation to cell proliferation in a p53-dependent manner. J Cell Sci. 2016;129(12):2382–93.
Liu F, Clark W, Luo G, Wang X, Fu Y, Wei J, et al. ALKBH1-Mediated tRNA demethylation regulates translation. Cell. 2016;167(3):816–828e816.
Li X, Xiong X, Wang K, Wang L, Shu X, Ma S, et al. Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat Chem Biol. 2016;12(5):311–6.
Zhang LS, Xiong QP, Peña Perez S, Liu C, Wei J, Le C, et al. ALKBH7-mediated demethylation regulates mitochondrial polycistronic RNA processing. Nat Cell Biol. 2021;23(7):684–91.
Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC, et al. Differential m(6)A, m(6)A(m), and m(1)a demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell. 2018;71(6):973–985e975.
Dai X, Wang T, Gonzalez G, Wang Y. Identification of YTH Domain-containing proteins as the readers for N1-Methyladenosine in RNA. Anal Chem. 2018;90(11):6380–4.
Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016;530(7591):441–6.
Wu Y, Chen Z, Xie G, Zhang H, Wang Z, Zhou J, et al. RNA m(1)A methylation regulates glycolysis of cancer cells through modulating ATP5D. Proc Natl Acad Sci U S A. 2022;119(28):e2119038119.
Liu Y, Zhou J, Li X, Zhang X, Shi J, Wang X, et al. tRNA-m(1)a modification promotes T cell expansion via efficient MYC protein synthesis. Nat Immunol. 2022;23(10):1433–44.
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology (Baltimore MD). 2017;65(2):529–43.
Liu X, Chen D, Chen H, Wang W, Liu Y, Wang Y, et al. YB1 regulates miR-205/200b-ZEB1 axis by inhibiting microRNA maturation in hepatocellular carcinoma. Cancer Commun (London England). 2021;41(7):576–95.
Sun X, Zhu H, Cao R, Zhang J, Wang X. BACH1 is transcriptionally inhibited by TET1 in hepatocellular carcinoma in a microRNA-34a-dependent manner to regulate autophagy and inflammation. Pharmacol Res. 2021;169:105611.
Wu J, Pang R, Li M, Chen B, Huang J, Zhu Y. m6A-Induced LncRNA MEG3 suppresses the Proliferation, Migration and Invasion of Hepatocellular Carcinoma Cell through miR-544b/BTG2 signaling. OncoTargets Therapy. 2021;14:3745–55.
Zuo X, Chen Z, Gao W, Zhang Y, Wang J, Wang J, et al. M6A-mediated upregulation of LINC00958 increases lipogenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. J Hematol Oncol. 2020;13(1):5.
Xia A, Yuan W, Wang Q, Xu J, Gu Y, Zhang L, et al. The cancer-testis lncRNA lnc-CTHCC promotes hepatocellular carcinogenesis by binding hnRNP K and activating YAP1 transcription. Nat Cancer. 2022;3(2):203–18.
Kong H, Sun J, Zhang W, Zhang H, Li H. Long intergenic non-protein coding RNA 1273 confers sorafenib resistance in hepatocellular carcinoma via regulation of methyltransferase 3. Bioengineered. 2022;13(2):3108–21.
Liu J, Zhang N, Zeng J, Wang T, Shen Y, Ma C, et al. N(6) -methyladenosine-modified lncRNA ARHGAP5-AS1 stabilises CSDE1 and coordinates oncogenic RNA regulons in hepatocellular carcinoma. Clin Translational Med. 2022;12(11):e1107.
Dai YZ, Liu YD, Li J, Chen MT, Huang M, Wang F, et al. METTL16 promotes hepatocellular carcinoma progression through downregulating RAB11B-AS1 in an m(6)A-dependent manner. Cell Mol Biol Lett. 2022;27(1):41.
Zhu H, Chen K, Chen Y, Liu J, Zhang X, Zhou Y, et al. RNA-binding protein ZCCHC4 promotes human cancer chemoresistance by disrupting DNA-damage-induced apoptosis. Signal Transduct Target Therapy. 2022;7(1):240.
Sun Z, Xue S, Zhang M, Xu H, Hu X, Chen S, et al. Aberrant NSUN2-mediated m(5)C modification of H19 lncRNA is associated with poor differentiation of hepatocellular carcinoma. Oncogene. 2020;39(45):6906–19.
Chen MH, Fu LS, Zhang F, Yang Y, Wu XZ. LncAY controls BMI1 expression and activates BMI1/Wnt/β-catenin signaling axis in hepatocellular carcinoma. Life Sci. 2021;280:119748.
Yuan J, Lv T, Yang J, Wu Z, Yan L, Yang J, et al. HDLBP-stabilized lncFAL inhibits ferroptosis vulnerability by diminishing Trim69-dependent FSP1 degradation in hepatocellular carcinoma. Redox Biol. 2022;58:102546.
Hämmerle M, Gutschner T, Uckelmann H, Ozgur S, Fiskin E, Gross M et al. Posttranscriptional destabilization of the liver-specific long noncoding RNA HULC by the IGF2 mRNA-binding protein 1 (IGF2BP1). Hepatology (Baltimore, Md.). 2013;58(5):1703–12.
Yeermaike A, Gu P, Liu D, Nadire T. LncRNA NEAT1 sponges miR-214 to promoted Tumor growth in hepatocellular carcinoma. Mammalian Genome: Official Journal of the International Mammalian Genome Society. 2022;33(3):525–33.
Wang H, Wang Y, Lai S, Zhao L, Liu W, Liu S, et al. LINC01468 drives NAFLD-HCC progression through CUL4A-linked degradation of SHIP2. Cell Death Discovery. 2022;8(1):449.
Wu A, Hu Y, Xu Y, Xu J, Wang X, Cai A, et al. Methyltransferase-like 3-Mediated m6A methylation of Hsa_circ_0058493 accelerates Hepatocellular Carcinoma Progression by binding to YTH Domain-Containing protein 1. Front cell Dev Biology. 2021;9:762588.
Rong D, Wu F, Lu C, Sun G, Shi X, Chen X, et al. m6A modification of circHPS5 and hepatocellular carcinoma progression through HMGA2 expression. Molecular therapy. Nucleic Acids. 2021;26:637–48.
Rao X, Lai L, Li X, Wang L, Li A, Yang Q. N(6) -methyladenosine modification of circular RNA circ-ARL3 facilitates Hepatitis B virus-associated hepatocellular carcinoma via sponging miR-1305. IUBMB Life. 2021;73(2):408–17.
Wang F, Xie Z, Zhang N, Ding H, Xiong K, Guo L, et al. Has_circ_0008583 modulates hepatocellular carcinoma progression through the miR-1301-3p/METTL3 pathway. Bioengineered. 2022;13(1):1185–97.
Liu H, Lan T, Li H, Xu L, Chen X, Liao H, et al. Circular RNA circDLC1 inhibits MMP1-mediated Liver cancer progression via interaction with HuR. Theranostics. 2021;11(3):1396–411.
Liu H, Jiang Y, Lu J, Peng C, Ling Z, Chen Y, et al. M(6)A-modification regulated circ-CCT3 acts as the sponge of miR-378a-3p to promote hepatocellular carcinoma progression. Epigenetics. 2023;18(1):2204772.
Chen Y, Ling Z, Cai X, Xu Y, Lv Z, Man D, et al. Activation of YAP1 by N6-Methyladenosine-modified circCPSF6 drives malignancy in Hepatocellular Carcinoma. Cancer Res. 2022;82(4):599–614.
Liu X, Qin J, Gao T, Li C, Chen X, Zeng K, et al. Analysis of METTL3 and METTL14 in hepatocellular carcinoma. Aging. 2020;12(21):21638–59.
Gu Z, Du Y, Zhao X, Wang C, Diagnostic. Therapeutic, and Prognostic Value of the m(6)a writer complex in Hepatocellular Carcinoma. Front Cell Dev Biology. 2022;10:822011.
Tao Z, Ruan H, Sun L, Kuang D, Song Y, Wang Q, et al. Targeting the YB-1/PD-L1 Axis to Enhance Chemotherapy and Antitumor Immunity. Cancer Immunol Res. 2019;7(7):1135–47.
Wang P, Yan Y, Yu W, Zhang H. Role of ten-eleven translocation proteins and 5-hydroxymethylcytosine in hepatocellular carcinoma. Cell Prolif. 2019;52(4):e12626.
Liu HT, Zou YX, Zhu WJ, Sen L, Zhang GH, Ma RR, et al. lncRNA THAP7-AS1, transcriptionally activated by SP1 and post-transcriptionally stabilized by METTL3-mediated m6A modification, exerts oncogenic properties by improving CUL4B entry into the nucleus. Cell Death Differ. 2022;29(3):627–41.
Chen Y, Zhao Y, Chen J, Peng C, Zhang Y, Tong R, et al. ALKBH5 suppresses malignancy of hepatocellular carcinoma via m(6)A-guided epigenetic inhibition of LYPD1. Mol Cancer. 2020;19(1):123.
Kristensen LS, Jakobsen T, Hager H, Kjems J. The emerging roles of circRNAs in cancer and oncology. Nat Rev Clin Oncol. 2022;19(3):188–206.
Zhang L, Hou C, Chen C, Guo Y, Yuan W, Yin D, et al. The role of N(6)-methyladenosine (m(6)A) modification in the regulation of circRNAs. Mol Cancer. 2020;19(1):105.
Dong ZR, Ke AW, Li T, Cai JB, Yang YF, Zhou W, et al. CircMEMO1 modulates the promoter methylation and expression of TCF21 to regulate hepatocellular carcinoma progression and sorafenib treatment sensitivity. Mol Cancer. 2021;20(1):75.
Zhang C, Cui H, Huang C, Kong F, Yang Q, Miao P et al. Interactions of circRNAs with methylation: an important aspect of circRNA biogenesis and function (review). Mol Med Rep. 2022;25(5).
Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, et al. The role of m6A modification in the biological functions and Diseases. Signal Transduct Target Therapy. 2021;6(1):74.
Lan T, Li H, Zhang D, Xu L, Liu H, Hao X, et al. KIAA1429 contributes to Liver cancer progression through N6-methyladenosine-dependent post-transcriptional modification of GATA3. Mol Cancer. 2019;18(1):186.
Liu W, Gao X, Chen X, Zhao N, Sun Y, Zou Y, et al. Mir-139-5p loss-mediated WTAP activation contributes to Hepatocellular Carcinoma Progression by promoting the epithelial to mesenchymal transition. Front Oncol. 2021;11:611544.
Fawzy IO, Hamza MT, Hosny KA, Esmat G, El Tayebi HM, Abdelaziz AI. miR-1275: a single microRNA that targets the three IGF2-mRNA-binding proteins hindering Tumor growth in hepatocellular carcinoma. FEBS Lett. 2015;589(17):2257–65.
Shaalan YM, Handoussa H, Youness RA, Assal RA, El-Khatib AH, Linscheid MW, et al. Destabilizing the interplay between miR-1275 and IGF2BPs by Tamarix articulata and quercetin in hepatocellular carcinoma. Nat Prod Res. 2018;32(18):2217–20.
Zhou B, Lu D, Wang A, Cui J, Zhang L, Li J, et al. Endoplasmic reticulum stress promotes sorafenib resistance via miR-188-5p/hnRNPA2B1-mediated upregulation of PKM2 in hepatocellular carcinoma. Molecular therapy. Nucleic Acids. 2021;26:1051–65.
Xue C, Zhao Y, Li G, Li L. Multi-omic analyses of the m(5)C Regulator ALYREF reveal its essential roles in Hepatocellular Carcinoma. Front Oncol. 2021;11:633415.
Han S, Xue L, Wei Y, Yong T, Jia W, Qi Y, et al. Bone Lesion-Derived Extracellular Vesicles Fuel Prometastatic Cascades in Hepatocellular Carcinoma by Transferring ALKBH5-Targeting miR-3190-5p. Adv Sci (Weinh). 2023;10(17):e2207080.
Chen D, Yan Y, Wang X, Li S, Liu Y, Yu D, et al. Chronic alcohol exposure promotes HCC stemness and Metastasis through β-catenin/miR-22-3p/TET2 axis. Aging. 2021;13(10):14433–55.
Lin LL, Wang W, Hu Z, Wang LW, Chang J, Qian H. Negative feedback of miR-29 family TET1 involves in hepatocellular cancer. Med Oncol. 2014;31(12):291.
Zhang W, Lu Z, Gao Y, Ye L, Song T, Zhang X. MiR-520b suppresses proliferation of hepatoma cells through targeting ten-eleven translocation 1 (TET1) mRNA. Biochem Biophys Res Commun. 2015;460(3):793–8.
Zhou X, Chang Y, Zhu L, Shen C, Qian J, Chang R. LINC00839/miR-144-3p/WTAP (WT1 Associated protein) axis is involved in regulating hepatocellular carcinoma progression. Bioengineered. 2021;12(2):10849–61.
Bo C, Li N, He L, Zhang S, An Y. Long non-coding RNA ILF3-AS1 facilitates hepatocellular carcinoma progression by stabilizing ILF3 mRNA in an m(6)A-dependent manner. Hum Cell. 2021;34(6):1843–54.
Wang H, Liang L, Dong Q, Huan L, He J, Li B, et al. Long noncoding RNA miR503HG, a prognostic indicator, inhibits Tumor Metastasis by regulating the HNRNPA2B1/NF-κB pathway in hepatocellular carcinoma. Theranostics. 2018;8(10):2814–29.
Chen T, Gu C, Xue C, Yang T, Zhong Y, Liu S, et al. LncRNA-uc002mbe.2 interacting with hnRNPA2B1 mediates AKT deactivation and p21 Up-Regulation Induced by Trichostatin in Liver Cancer cells. Front Pharmacol. 2017;8:669.
Liu P, Zhong Q, Song Y, Guo D, Ma D, Chen B, et al. Long noncoding RNA Linc01612 represses hepatocellular carcinoma progression by regulating miR-494/ATF3/p53 axis and promoting ubiquitination of YBX1. Int J Biol Sci. 2022;18(7):2932–48.
Zhao X, Liu Y, Yu S. Long noncoding RNA AWPPH promotes hepatocellular carcinoma progression through YBX1 and serves as a prognostic biomarker. Biochimica et biophysica acta. Mol Basis Disease. 2017;1863(7):1805–16.
Lin YH, Zhang BY, Chen ZS. circRERE regulates the expression of GBX2 through miR-1299 and ZC3H13/N(6)-methyladenosine (m(6)A) to promote growth and invasion of hepatocellular carcinoma cells. J Biosci. 2022;47:52.
Chen Z, Huang L, Wang K, Zhang L, Zhong X, Yan Z, et al. rtcisE2F promotes the self-renewal and Metastasis of liver tumor-initiating cells via N(6)-methyladenosine-dependent E2F3/E2F6 mRNA stability. Sci China Life Sci. 2022;65(9):1840–54.
Razpotnik R, Vidmar R, Fonović M, Rozman D, Režen T. Circular RNA hsa_circ_0062682 binds to YBX1 and promotes oncogenesis in Hepatocellular Carcinoma. Cancers. 2022;14(18):4524.
Wang Q, Yu W, Wang T, Huang C, Circular RNA. circDLG1 contributes to HCC progression by regulating the miR-141-3p/WTAP axis. Funct Integr Genom. 2023;23(2):179.
Chi F, Cao Y, Chen Y. Analysis and validation of circRNA-miRNA network in regulating m(6)a RNA methylation modulators reveals CircMAP2K4/miR-139-5p/YTHDF1 Axis Involving the Proliferation of Hepatocellular Carcinoma. Front Oncol. 2021;11.
Liu L, Gu M, Ma J, Wang Y, Li M, Wang H, et al. CircGPR137B/miR-4739/FTO feedback loop suppresses tumorigenesis and Metastasis of hepatocellular carcinoma. Mol Cancer. 2022;21(1):149.
Zhou Y, Ren H, Dai B, Li J, Shang L, Huang J, et al. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to Tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J Experiment Clin Cancer Res. 2018;37(1):324.
Yin S, Jin W, Qiu Y, Fu L, Wang T, Yu H. Solamargine induces hepatocellular carcinoma cell apoptosis and autophagy via inhibiting LIF/miR-192-5p/CYR61/Akt signaling pathways and eliciting immunostimulatory Tumor microenvironment. J Hematol Oncol. 2022;15(1):32.
Zhao J, Li H, Zhao S, Wang E, Zhu J, Feng D, et al. Epigenetic silencing of miR-144/451a cluster contributes to HCC progression via paracrine HGF/MIF-mediated TAM remodeling. Mol Cancer. 2021;20(1):46.
Chen Y, Lin Y, Shu Y, He J, Gao W. Interaction between N(6)-methyladenosine (m(6)A) modification and noncoding RNAs in cancer. Mol Cancer. 2020;19(1):94.
Wu S, Liu S, Cao Y, Chao G, Wang P, Pan H. Downregulation of ZC3H13 by miR-362-3p/miR-425-5p is associated with a poor prognosis and adverse outcomes in hepatocellular carcinoma. Aging. 2022;14(5):2304–19.
Lin Y, Yao Y, Wang Y, Wang L, Cui H. PD-L1 and Immune Infiltration of m(6)a RNA methylation regulators and its miRNA regulators in Hepatocellular Carcinoma. Biomed Res Int. 2021;5516100.
Wang L, Yi X, Xiao X, Zheng Q, Ma L, Li B. Exosomal mir-628-5p from M1 polarized macrophages hinders m6A modification of circFUT8 to suppress hepatocellular carcinoma progression. Cell Mol Biol Lett. 2022;27(1):106.
Ye Y, Wang M, Wang G, Mai Z, Zhou B, Han Y, et al. lncRNA miR4458HG modulates hepatocellular carcinoma progression by activating m6A-dependent glycolysis and promoting the polarization of tumor-associated macrophages. Cell Mol Life Sci. 2023;80(4):99.
Peng L, Pan B, Zhang X, Wang Z, Qiu J, Wang X, et al. Lipopolysaccharide facilitates immune Escape of hepatocellular carcinoma cells via m6A modification of lncRNA MIR155HG to upregulate PD-L1 expression. Cell Biol Toxicol. 2022;38(6):1159–73.
Chen CL, Hernandez JC, Uthaya Kumar DB, Machida T, Tahara SM, El-Khoueiry A, et al. Profiling of Circulating Tumor Cells for Screening of Selective Inhibitors of Tumor-Initiating Stem-Like Cells. Adv Sci (Weinh). 2023;10(14):e2206812.
Hu Z, Chen G, Zhao Y, Gao H, Li L, Yin Y, et al. Exosome-derived circCCAR1 promotes CD8 + T-cell dysfunction and anti-PD1 resistance in hepatocellular carcinoma. Mol Cancer. 2023;22(1):55.
Cai J, Chen Z, Zhang Y, Wang J, Zhang Z, Wu J, et al. CircRHBDD1 augments metabolic rewiring and restricts immunotherapy efficacy via m(6)a modification in hepatocellular carcinoma. Mol Therapy Oncolytics. 2022;24:755–71.
Zhang PF, Wei CY, Huang XY, Peng R, Yang X, Lu JC, et al. Circular RNA circTRIM33-12 acts as the sponge of MicroRNA-191 to suppress hepatocellular carcinoma progression. Mol Cancer. 2019;18(1):105.
Ke W, Zhang L, Zhao X, Lu Z. p53 m(6)a modulation sensitizes hepatocellular carcinoma to apatinib through apoptosis. Apoptosis. 2022;27(5–6):426–40.
Liu B, Cao J, Wu B, Hao K, Wang X, Chen X, et al. METTL3 and STAT3 form a positive feedback loop to promote cell Metastasis in hepatocellular carcinoma. Cell Commun Signal. 2023;21(1):121.
Wang L, Yang Q, Zhou Q, Fang F, Lei K, Liu Z, et al. METTL3-m(6)A-EGFR-axis drives lenvatinib resistance in hepatocellular carcinoma. Cancer Lett. 2023;559:216122.
Shi Y, Zhuang Y, Zhang J, Chen M, Wu S. METTL14 inhibits Hepatocellular Carcinoma Metastasis through regulating EGFR/PI3K/AKT signaling pathway in an m6A-Dependent manner. Cancer Manage Res. 2020;12:13173–84.
Li Z, Li F, Peng Y, Fang J, Zhou J. Identification of three m6A-related mRNAs signature and risk score for the prognostication of hepatocellular carcinoma. Cancer Med. 2020;9(5):1877–89.
Chen Y, Peng C, Chen J, Chen D, Yang B, He B, et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol Cancer. 2019;18(1):127.
Wu S, He G, Liu S, Cao Y, Geng C, Pan H. Identification and validation of the N6-methyladenosine RNA methylation regulator ZC3H13 as a novel prognostic marker and potential target for hepatocellular carcinoma. Int J Med Sci. 2022;19(4):618–30.
Hu J, Cai D, Zhao Z, Zhong GC, Gong J. Suppression of Heterogeneous Nuclear Ribonucleoprotein C inhibit Hepatocellular Carcinoma Proliferation, Migration, and Invasion via Ras/MAPK signaling pathway. Front Oncol. 2021;11:659676.
Bian S, Ni W, Zhu M, Song Q, Zhang J, Ni R, et al. Identification and validation of the N6-Methyladenosine RNA methylation Regulator YTHDF1 as a novel prognostic marker and potential target for Hepatocellular Carcinoma. Front Mol Biosci. 2020;7:604766.
Shao XY, Dong J, Zhang H, Wu YS. Systematic analyses of the role of the reader protein of N (6)-Methyladenosine RNA methylation, YTH Domain Family 2, in Liver Hepatocellular Carcinoma. Front Mol Biosci. 2020;7:577460.
Liu J, Wang D, Zhou J, Wang L, Zhang N, Zhou L, et al. N6-methyladenosine reader YTHDC2 and eraser FTO may determine hepatocellular carcinoma prognoses after transarterial chemoembolization. Arch Toxicol. 2021;95(5):1621–9.
Gan X, Dai Z, Ge C, Yin H, Wang Y, Tan J, et al. FTO promotes liver inflammation by suppressing m6A mRNA methylation of IL-17RA. Front Oncol. 2022;12:989353.
Xiao Z, Li T, Zheng X, Lin L, Wang X, Li B, et al. Nanodrug enhances post-ablation immunotherapy of hepatocellular carcinoma via promoting dendritic cell maturation and antigen presentation. Bioactive Mater. 2023;21:57–68.
Peng B, Yan Y, Xu Z. The bioinformatics and experimental analysis of AlkB family for prognosis and immune cell infiltration in hepatocellular carcinoma. PeerJ. 2021;9:e12123.
Qu S, Jin L, Huang H, Lin J, Gao W, Zeng Z. A positive-feedback loop between HBx and ALKBH5 promotes hepatocellular carcinogenesis. BMC Cancer. 2021;21(1):686.
Acknowledgements
Not applicable.
Funding
This work was supported by the Fundamental Research Funds for the Central Universities (2022ZFJH003), the Shandong Provincial Laboratory Project (SYS202202), and the Research Project of Jinan Micro-ecological Biomedicine Shandong Laboratory (JNL-2022001A, JNL-2022009B, and JNL-2022047D).
Author information
Authors and Affiliations
Contributions
Lanjuan Li conceived this study and critically reviewed and edited the manuscript. Qingmiao Shi, Qingfei Chu, and Yifan Zeng wrote the original draft and prepared the tables. Xin Yuan, Jinzhi Wang, and Yaqi Zhang collected the literature. Chen Xue contributes to the visualization of the image. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shi, Q., Chu, Q., Zeng, Y. et al. Non-coding RNA methylation modifications in hepatocellular carcinoma: interactions and potential implications. Cell Commun Signal 21, 359 (2023). https://doi.org/10.1186/s12964-023-01357-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01357-0