
Abstract
Breast cancer exhibits the highest global incidence among all tumor types. Regardless of the type of breast cancer, metastasis is a crucial cause of poor prognosis. Anoikis, a form of apoptosis initiated by cell detachment from the native environment, is an outside-in process commencing with the disruption of cytosolic connectors such as integrin-ECM and cadherin-cell. This disruption subsequently leads to intracellular cytoskeletal and signaling pathway alterations, ultimately activating caspases and initiating programmed cell death. Development of an anoikis-resistant phenotype is a critical initial step in tumor metastasis. Breast cancer employs a series of stromal alterations to suppress anoikis in cancer cells. Comprehensive investigation of anoikis resistance mechanisms can inform strategies for preventing and regressing metastatic breast cancer. The present review first outlines the physiological mechanisms of anoikis, elucidating the alterations in signaling pathways, cytoskeleton, and protein targets that transpire from the outside in upon adhesion loss in normal breast cells. The specific anoikis resistance mechanisms induced by pathological changes in various spatial structures during breast cancer development are also discussed. Additionally, the genetic loci of targets altered in the development of anoikis resistance in breast cancer, are summarized. Finally, the micro-RNAs and targeted drugs reported in the literature concerning anoikis are compiled, with keratocin being the most functionally comprehensive.
Video Abstract
Similar content being viewed by others


Introduction
The World Health Organization's International Agency for Research on Cancer (IARC) posted the latest data on global cancer burden in 2020. Revealing a rapid increase in breast cancer (BC) cases to 2.26 million. This accounted for approximately 11.7% of all new cancer patients, establishing BC as the most prevalent cancer worldwide [1]. BC exhibits significant heterogeneity, with some cases presenting slow growth and favorable prognosis, while others are highly aggressive and rapidly metastasize to other organs [2].
Molecular subtypes of BC include Luminal A, Luminal B, human epidermal growth factor receptor2 (HER2) overexpression, normal breast-like, and Basal-like or Triple Negative cancer (TNBC) [3]. This molecular sub-classification could improve BC treatment techniques to a large extent, including guiding the delivery of targeted therapies such as hormone therapy (e.g., Toremifene) and HER2-targeted therapy (e.g., Pertuzumab) [4]. Intrinsic subtypes of BC have been used in research settings for more than two decades [5], with ongoing efforts to refine differentiation among BC subtypes [6]. Evidence suggests that BC cells may interconvert between different disease subtypes, indicating the potential coexistence of multiple BC subtypes within a single tumor [7]. Regardless of the subtypes, metastasis significantly reduces patients’ survival rate [8]. A statistical analysis of all BC patients diagnosed in the US between 2009 and 2015 showed 5-year survival rates of 98% for stage I, 92% for stage II, 75% for stage III and 27% for stage IV [9].
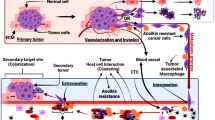
Metastasis initiation occurs when local tumor cells detach from the extracellular matrix(ECM) at the site of origin [10]. Loss of contact with the ECM or other cells induces anoikis, a specific form of programmed cell death and a subtype of apoptosis [11]. Anoikis is a pivotal mechanism to inhibit cell colonization and growth in the new stromal environment [12]. Tumor cells develop a survival phenotype that allows them to bypass anoikis upon ECM detachment, migrate to other organs, and repopulate to form metastatic tumors (Fig. 1) [13]. Anoikis resistance is a prerequisite and a significant indicator of tumor cell metastatic potential. Preserving anoikis functional integrity is an essential means of preventing metastasis, necessitating a deeper understanding of tumor cell resistance mechanisms to anoikis [14].

When normal breast cells are detached from their natural environment, this triggers changes in the corresponding structures in the cell membrane, which, by inhibiting intracellular pro-survival signalling pathways and triggering changes in the cytoskeleton, eventually causes the cells to undergo physiological anoikis. When breast cancer cells are detached from tumour tissue, the cells are altered from receptors on the cell membrane, resulting in significant activation of the pro-survival pathway and changes to the cytoskeleton, allowing the tumour cells to develop anoikis resistance after detachment from their native environment, thus allowing them to metastasise and continue to grow elsewhere
Studying anoikis resistance mechanisms in BC is important because mammary gland epithelial cells adhere to laminin-rich basement membranes via integrins, rather than interacting with collagen I(COLI) [15]. Catheter space filling is a feature of many early BC lesions [16], and anoikis resistance facilitates vitro 3D ductal filling in breast follicular epithelial structures [17], which is a critical process in BC distant metastasis.
Overall, anoikis is a specific form of programmed cell death induced by the loss of cell contact with the extracellular matrix and other cells, and a subtype of apoptosis that still induces cell death via the traditional apoptotic pathway. This review examines the activation of anoikis by adhesion-initiating signals in both normal and metastatic tumor cells, which trigger a series of changes in intracellular pathways, proteins, cytoskeleton, and genetic material. A comprehensive understanding of anoikis resistance mechanisms in BC cells may provide potential opportunities for the prevention and treatment of metastatic cancer.
Main text
Physiological anoikis (Fig. 2)

Detachment of normal breast cells from their native environment triggers activation of adhesion mediators on the cell membrane, inhibition of survival pathways (e.g., PI3K-AKT, MEK-ERK), and simultaneous alteration of the cytoskeletal structure, triggering a BCL2 family protein response, activation of the mitochondrial pathway of apoptosis, and ultimately the release of caspases, triggering anpikis
ECM-The cradle of cell growth
ECM provides adhesion support for cells and regulates cellular physiological behavior through signaling [18]. ECM detachment causes a range of metabolic changes, including glucose uptake defective, the pentose phosphate pathway(PPP) flux decreases, cellular ATP levels lower, and reactive oxygen species (ROS) increases [19]. Proteolytic cleavage, integral proteins, or changes in microenvironment can also activate potential TGF-β [20], activating classical Smad signaling [21], and non-Smad signaling pathways including MAP/Erk, TβRI induced Shc phosphorylation, as well as Ras, Rho, Rac, and CDC42 small GTases, further promote anoikis development [22].
Cell membrane alterations
Cells express various cell adhesion molecules (CAM) that mediate cell–cell or cell-ECM contacts (Fig. 3) [23]. The cell-ECM linkage is a focal adhesion, which relies on integral protein-actin interactions [24]. The adhesion belt is responsible for cell–cell contacts, with the main proteins involved in adhesion band junctions being cadherin and actin [25]. These CAMs are typically transmembrane proteins consisting of three structural domains: an extracellular structural domain responsible for ligand binding, a transmembrane structural domain, and a cytoplasmic tail attached to the actin cytoskeleton by a protein complex such as an enzyme or kinase [26]. Many transmembrane proteins, such as growth factor receptor (GFR), are also present in the cell membrane and are subject to changes in cell-ECM or cell–cell contact status [27].

CAMs associated with anoikis are described here, where integrins mediate cell-ECM adhesion, E-cadherin and P-cadherin mediate cell–cell adhesion, and some cell membrane receptors such as TβR I/II and EGFR also affect anoikis when cells lose their adhesion
Integrin-messengers of cell and ECM
Integrins are transmembrane αβ heterodimers [28]. These cell surface glycoproteins mediate cell-ECM interactions and dynamic adhesion [29]. The α and β subunits of integrins typically include a large extracellular structural domain, a transmembrane helix, and a short cytoplasmic tail [30].
The extracellular structural domain of integrins binds to specific ECM proteins, such as collagen, vitronectin, fibronectin, and other proteins [29]. Disassociation of cells from ECM can lead to the activation of integrins, resulting in a shift of integrin legs from an inactive leaning-together state to an active elongated detached state [31]. Long-range conformational changes in extracellular structural domains cause intracellular protein reactions that trigger the reorganization of ligand binding sites [32]. Through the recruitment and aggregation of integrins, intracellular proteins bind directly or indirectly to integrin tails, forming a specific structure of focal adhesion [33]. Integrin tails do not have intrinsic enzymatic activity. However, adherent spots contain many protein kinases and scaffolding proteins, and some adherent spot proteins (e.g., talin) can bind actin and thus have specific effects [34].
The intracellular signaling pathway triggered by integral proteins has two main functions: to organize the actin cytoskeleton and to regulate cellular behavior [35]. Integrins regulate the cytoskeleton by directly binding actin proteins, including synuclein, nuclein and filament proteins [36]. Integrins activate their regulated signaling pathways by phosphorylating integrin-related kinase (ILK), proto-oncogene tyrosine protein kinase (Src) and focal adhesion kinase (FAK), thereby regulating cellular behavior [37].
Cadherin-messengers of cells and cells
Cadherins are responsible for cell–cell adhesion and include type I, II, and III/atypical cadherins, all of which are expressed in the mammary gland [38]. The most important E-cadherin is a type I cadherin, a membrane glycoprotein located at the cell adhesion junctions, which anchors epithelial cells to each other and is essential for the adhesion of adjacent epithelial cells [39]. In normal breasts, monolayer epithelial cap cells of terminal buds also express P-cadherin, which is important to the branching process of breast ducts [40]. At the adhesion junctions, the cadherin cytoplasmic tail provides binding sites for p120-, α-, γ-, and β-catenin, facilitating connections between the signaling pathways and actin cytoskeleton [41]. Anoikis is induced by cadherin when cell–cell adhesion is broken.
Epidermal Growth Factor Receptor (EGFR)
In human mammary epithelial cells, ECM exposure is the main factor in EGFR expression and downstream signaling activation [42]. Activated EGFRs implicate downstream molecules in the response, such as Janus-activated kinase (JAK), Ras, phospholipase Cγ (PLCγ) and phosphatidylinositol 3-kinase (PI3K) [43]. EGFR and integrins are functionally coupled [44]. Loss of integrin-ECM adhesion leads to downregulation of EGFR expression and inhibition of downstream EGFR molecules, [42], which synergically induced anoikis.
Cytoplasm
ECM detachment in mammary epithelial cells promotes changes in CAM and protein receptors on the cell membrane, resulting in reduced intracellular EGFR, PI3K/Akt and Mek/Erk signals, which are transmitted to the mitochondria and affect cell survival [45]. The mitochondria play a critical role in anoikis [46]. The mitochondrial intermembrane space (IMS) contains many key pro-apoptotic factors, such as cytochrome c. When incoming survival signals to the mitochondria are reduced, pro-apoptotic factors are released into the cytoplasm, thereby triggering anoikis [47].
Cytoplasmic plaque
The cytoplasmic region contains several cytoplasmic patches, which are multimolecular protein complexes. Cytoplasmic plaques are involved in building membrane protein scaffolds and cytoskeletons, regulating polarity, and transmitting signals [48]. Cytoplasmic plaque components of the cell-ECM and cell–cell are each distinct.
Focal adhesion-cytoplasmic plaque
Focal adhesion is the cytoplasmic part of integrins and the site of proteoglycan-mediated adhesion to the actin cytoskeleton [34]. It contains various kinases, such as focal adhesion kinase(FAK) and Src, and serine/threonine kinase ILK [49]. FAK is a multifunctional protein that integrates signals sensed by integrin or growth factor receptors, which are then transduced into the cel [50]. Src interacts with FAK and facilitates FAK phosphorylation and activation [51]. ILK connects integrins to the actin cytoskeleton by interacting with pilings and parvins [52]. It also binds to phosphatidylinositol 3,4,5-triphosphate(PIP3) and affects the downstream signaling pathway [53].
Adhesion belt-cytoplasmic plaque
The intracellular structural domain of cadherin is linked to actin fibers through protein-mediated connection in the cytoplasmic plaque, which contains β-catenin, α-catenin, p120-catenin, etc. [54]. P120-catenin is a substrate for Src and several receptor tyrosine kinases, and interacts with the proximal membrane domain of cadherin to direct cadherin aggregation and cell motility [55]. β-catenin binds to α-catenin, which connects cadherin to actin cytoskeleton [56]. The cadherin protein linker complex enhances adhesion by linking α-catenin to actin cytoskeleton. α-catenin in its monomeric form binds to calmodulin-linkerin complex via β-catenin, while the homodimeric form of α-catenin binds to F-actin [57]. Cadherin regulates the expression of GFR, PI3K and ERBB4 through catenins [58].
Signaling pathway
PI3K-AKT signaling pathway
FAK, cadherin and EGFR are all upstream activation conditions of PI3K [59]. PI3K phosphorylates the plasma membrane lipid substrate PIP2, generating PIP3 [60]. PIP3 then recruits AKT and 3-phosphatidylinositol-dependent protein kinase 1 (PDK1) to the plasma membrane via the PH structural domain, phosphorylating AKT and PDK1 at Ser and Thr, respectively [61]. Activated AKT phosphorylates target proteins on the cell membrane, which then lose their attachment to the cell membrane and enter the cytoplasm [62]. It further implicates the downstream response of the Bcl2 protein family, which regulates anoikis by controlling mitochondrial permeability [63].
NF-κB signaling pathway
Akt can undergo proteasome-mediated degradation and release NF-κB through IkB kinase (IKK) phosphorylation and inhibition of IκB [64]. IKK supports the translocation of NF-κB to the nucleus by phosphorylating IκB-α, and enhances relA transcriptional activation by phosphorylating the relA activation domain, ultimately upregulating NF-κB target genes Bcl2 and bcl-xl expression, and further triggering anoikis [65].
Mek/Erk signaling pathway
ECM-cell junctions elevate intracellular ROS levels via integrins, subsequently activating tyrosine kinase Src. Redox regulation of Src mediates integration independent EGFR trans-phosphorylation [66]. Activated Src elicits EGFR downstream signaling (AKT and ERK) in a ligand-independent manner, ultimately leading to Bim downregulation [67]. RAF was identified as the first direct effector of Ras and an upstream kinase of MEK [68]. Ras binds to Raf, and Raf is further phosphorylated to activate ERK1 and ERK2 mitogen-activated protein kinases [69]. Activated ERK1/2 can target mitochondria, enhance ATP synthase activity, maintain mitochondrial membrane potential, inactivate Bad, and reduce cytochrome c release [42]. The second best Ras effector is PI3K, further enhancing the pro-anoikis effect [70].
Cytoskeleton
Focal adhesion (FA) proteins, which contribute to the establishment and maintenance of integrin-cytoskeleton junctions, can be divided into four categories: (I) integrin-binding proteins that directly bind to actin, such as α-actinin, talin, and fine filament proteins; (II) integrin-binding proteins indirectly associated with the cytoskeleton, including kindlin, core scaffold ILK, plectin, and FAK [71]; (III) non-integrin-binding actin-binding proteins such as nucleoporins; (IV) modulators and signaling molecules that regulate various protein interactions [72]. FA sequesters the BH3 structural domain proteins Bim and Bmf near the membrane. Bmf interacts with dynein light chain 2 (DLC2) of the MYO5/myosin V complex, attaching to actin filaments. Integrin-mediated cell detachment disrupts the actin state, causing Bmf to be released from the cytoskeleton and promoting anoikis [73].
Protein
Bcl-2 protein family
The Bcl-2 protein family includes both pro- and anti-apoptotic members. The BH3-only proteins Bim, tBid and Puma activate Bax and promote anoikis, whereas Bcl-2, Bcl-XL and Mcl-1 suppress Bax activation and anoikis [74]. Mcl-1 is known to prevent anoikis by isolating BH3-only proteins(e.g., Bim and tBid) in mitochondria [75]. Anoikis is highly dependent on the mitochondrial pathway, with Bax serving as a crucial effector of this process [76]. The structure of Bax determines its active state and significantly impacts the intrinsic anoikis pathway [77]. Anti-anoikis agents BCL-2 and BCL-XL sequester BH3 structural domain-only molecules in a stable mitochondrial complex, preventing the activation of Bax and Bak [78]. However, Bad can counteract the anti-anoikis function of Bcl-2 by competing for its BH3 binding domain, indirectly inducing Bax/Bak activation [79]. Bid and Bim directly promote the formation of Bax/Bak oligomers, and the activation of Bax/Bak increases outer mitochondrial membrane permeability, leading to the release of soluble proteins, including cytochrome c, from the intermembrane space into the cytoplasm [80].
Caspases
Cytochrome c forms apoptosomes by complexing with procaspase-9, apoptotic protease-activating factor-1 (Apaf-1) and dATP [12, 81]. Apaf-1 binding and free caspase-9 maintain a homeostatic equilibrium and activate caspase-9 [82]. Further activation of effector caspases (such as caspase-3, -6 and -7 in mammals) results in the cleavage of specific substrates and promotes cell disassembly [83].
Anoikis resistance in BC (Fig. 4)

Breast cancer cells detached from tumor tissue have an anoikis-resistant phenotype with multiple mechanisms of altered activation of survival pathways, cytoskeletal remodeling, cellular deformation, and inhibition of mitochondrial apoptotic pathways, resulting in a anoikis-resistant phenotype (red indicates high expression in breast cancer, blue indicates low expression in breast cancer)
Tumor cells resist anoikis through multiple mechanisms (Fig. 5): extracellular factors such as degradation and remodeling of ECM, significant expression of ECM, and transforming growth factor β; cytosolic factors including continuous alteration of integrins, calmodulin, and altered expression profiles of EGFR and ERBB2; intracellular factors involving the alteration of intracytoplasmic components (e.g., FAK, Src, connexins), abnormal activation of pathways (e.g., PI3K-Akt, Mek/Erk, NF-κB), and expression profiles of anoikis-related proteins (BCL2 family, P53, zinc catalase, ferritin). Additionally, constant changes in the cytoskeleton, miRNA, and reactive oxygen species (ROS) are crucial for tumor cells to counteract anoikis.

The various factors that contribute to the development of anoikis resistance in BC are outlined
Extracellular
ECM: An important tissue barrier for tumor metastasis
ECM is a major component of the tumor microenvironment, regulating numerous pathways in cancer cells, including PI3K/AKT, ERK, Src-FAK, and Rho-GTPases [84]. BC progression necessitates extensive degradation and remodeling of ECM [85]. During tumor progression, stromal EMT deposition increases, altering the chemical composition and mechanical properties of ECM [86]. Cancer cells' invasive capabilities are further enhanced by the mechanical stretching of the collagen matrix or increased matrix stiffness [87].
Invasive BC cells confer anoikis resistance to myofibroblasts during tissue remodeling [88]. Downregulation of tropomyosin-1 (TM1) in BC promotes stress fiber assembly [89], which may disrupt the microfilament structure, thereby enhancing BC cell resistance to anoikis [90].
Abnormal matrix metalloproteinase (MMP) activity is frequently observed in tumors. MMP activation correlates with BC survival signals, with higher MMP activity associated with greater cell migration and metastatic capacity [91]. For example, MMP-11 is overexpressed in many lobular carcinoma cells [92]; MMP-2 is activated on the αvβ3 integrin and its downstream ERK signaling pathway [93]; MMP-7 restores insulin-like growth factor-I (IGF-I) mediated phosphorylation of IGF-IR and activation of Akt [94]; MMP-9 is overexpressed in BC and activates TGF-β/SMAD signaling [95].
Epithelial-mesenchymal transition (EMT)
EMT enhances metastasis in epithelial carcinomas and leads to cytoskeletal changes that are a prerequisite for the development of anoikis resistance [96]. EMT results in the downregulation of proteins that maintain polarized epithelium, such as occludin, E-cadherin, and claudins, and an increase in mesenchymal proteins (e.g., vimentin, N-cadherin, and smooth muscle actin) [97]. ECM mediates EMT effects through various cell signaling molecules, with Wnt, TGFβ, and Notch ligands playing central roles [98]. Zinc finger E-box binding homeobox 1 (ZEB1) is the master regulator of EMT program [99]. Grainyhead-like2 (GRHL2) and Thyroid Hormone Receptor Interacting Protein 12 (TRIP12) inhibit ZEB1 expression by repressing the ZEB1 promoter and ZEB1 gene, respectively, thereby suppressing TGF-β or Twist-induced or spontaneous EMT [100]. ZEB1 depletion rescues EMT behavior. Loss of CCN6, which is widely found in BC, increases IGF-1 levels in the ECM and activates IGF-1R signaling, leading to EMT [101]. EMT-induced loss of cell polarity in metastatic cancer cells leads to downregulation of the Hippo pathway [102]. The ubiquitin-like modifier-activating enzyme 6 (UBA6) in the human genome initiates ubiquitination through ATP-dependent activation of ubiquitin and inhibits EMT [103]. The tumor protein P53-inducible protein 11 (TP53I11) also inhibites EMT in vitro [104].
TGF-β is upregulated in BC and promotes a malignant phenotype
TGF-β superfamily members promote advanced cancers while suppressing early events that may lead to cancer [105]. Elevated local or systemic TGF-β levels are typical indicators of metastatic BC and are associated with reduced tumor cell responsiveness to its suppressive function [106]. TGF-β and EGF can synergistically promote malignant phenotypes, such as EMT, anoikis resistance and metastasis, and TGF-β also increases the expression of EGFR [107]. TGF-β is shown to increase the expression of EGFR by stimulating the expression and secretion of multiple ECM components, such as collagen I (COL1A1) and fibronectin (FN1) in stromal fibroblasts, and ECM cross-linking enzymes such as lysyl oxidase (LOX) in BC cells, enhancing the anoikis resistance effect of BC [108].
Cytomembrane
Integrin-ECM interactions promote breast cancer cell survival following matrix stripping
Integrin expression profiles change during cell transformation from normal cells to tumor cells and subsequently to tumor progression [109]. To circumvent anoikis and initiate proliferation, tumor cells must maintain continuous interactions with the extracellular matrix (ECM) through surface receptors such as integrins [110]. Integrin function is influenced by various factors, including the ECM, cell membrane surface receptors, intracellular proteins, and the cytoskeleton.
Collagen XIII, which is highly expressed in breast cancer(BC), induces β1 integrin activation in the ECM [111]. The Rho GTPase regulator D4-GDI, exhibiting increased expression in BC, inhibits β1 expression upon silencing [112]. This leads to Rac1 activation and translocation from cytoplasmic lysates to the cell membrane region, which in turn positively regulates cancer cell migration [113]. α5 is a key signal for Src and ErbB2 conduction in the low-adhesion state and is also an essential mediator for blocking Bim-promoted anoikis [17]. GPI-anchored glycoprotein CEA and carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6) are overexpressed in BC [114], mediating their biological effects through enhanced integrin α5β1-fibronectin interactions [115]. The α5β3 integrin specifically recognizes the arginine-glycine-aspartate (RGD) motif in the ECM and is antagonized by the RGD peptide, which holds promise as a drug carrier for targeted therapy in metastatic BC due to its specific recognition of integrins [93]. Loss of Ferritin (FER) on the cell membrane surface increases α6β1 integral protein expression and adhesion of BC cells to collagen I and laminin [116]. Elevated Semaphorin-7a (SEMA7A) protein is observed in BC, promoting anoikis resistance through activating α6β1 integrin and pAKT [117].
cadherin
E-cadherin inhibits tumor metastasis and exhibits downregulated expression in BC
Nearly 90% of infiltrating BC cases demonstrate downregulated or lost E-cadherin expression [118]. Loss of E-cadherin triggers EMT and is accompanied by increased cell motility, invasiveness and resistance to anoikis [119]. Protein tyrosine kinase 6 (PTK6) is expressed in approximately 70% of TNBCs, and its downregulation restores E-cadherin levels [120]. Pax-5, a member of the Paired Box x (Pax) gene family, is commonly expressed in 97% of breast samples. It binds and induces E-cadherin gene expression, inhibiting and reversing EMT in BC [121]. Zinc finger transcription factor (SLUG) is consistently overexpressed in invasive basal BC [122], and also inhibits E-cadherin [123]. E-cadherin cytoplasmic tail protein p120-, α-, γ- and β-linked proteins link cadherins to actin filaments, establishing strong cell–cell adhesion Mutations in these genes often occur in cancer cells, promoting the progression of anoikis resistance [124].
P-cadherin promotes the progression of BC
P-cadherin is frequently expressed in BC near oxygenated, vascular and hypoxic zones. Its overexpression leads to the activation of FAK, AKT and Src kinases, as well as reduced oxidative stress in stromal isolated BC cells by upregulating carbon flux through the pentose phosphate pathway [125]. P-cadherin activates the heterodimeric α6β4 integrin [125]. Moreover, p-cadherin expression diminishes the invasion inhibitory function of E-cadherin by disrupting the cell membrane E-cadherin/p120-linked protein complexes [126].
EGFR Overexpression in BC promotes anoikis resistance
EGFR overexpression is observed across all BC subtypes and is higher in the more aggressive TNBC and inflammatory breast cancer (IBC) [127]. EGFR overexpression signals the Mek-Erk pathway and inhibits anoikis by blocking Bim expression [42, 128]. It also indirectly activates Src and EGFR-induced survival signaling due to the dramatically increased ROS levels resulting from the loss of adhesion [129]. Depletion of pre-mRNA processing factor 4 kinase (PRP4K) leads to diminished degradation of EGFR [130]. Recombinant human tyrosine kinase ErbB-2 (also known as HER2 or Neu) stabilizes EGFR in ECM isolated cells by activating the ERK/Sprouty2 (Spry2) pathway [128]. Cells overexpressing EGFR can escape the adjustment of integrin deficiency [131].
ERBB2 is a member of the EGFR family and amplified in 20–30% of human BC [132]. Overexpression of ERBB2 promotes anoikis resistance [42], leading to the filling of the ductal lumen of BC and polar disruption of vesicle-like structures in the breast epithelium [133]. It also simultaneously maintains EGFR expression and EGF-induced signaling allowing cell survival after ECM detachment [17, 42, 134]. Hypoxia-inducible factor-1 (HIF-1) is one mechanism by which cancer cells overexpressing ERBB2 achieve three-dimensional culture growth, anchorage independence and anoikis resistance [135]. ErbB2 driven interferon regulatory factor 6 (Irf6) is downregulated in highly invasive BC cell lines but upregulated in less invasive cell lines, and ErbB2 can downregulate the pro-apoptotic protein Irf6 [136]. Mucin4 (MUC4) interacts directly with the extracellular structural domain of ErbB2 [137], and phosphorylation of ErbB2 is induced by tyrosine residues 1139 and 1248 to isolate ErbB2 and other ErbB receptors, initiating downstream signaling cascades in polarized epithelial cells [138]. PDK1 overexpression significantly enhances the ability of ERBB2 to form tumors [139].
Intracellular
Cytoplasmic plaque
Focal adhesion-cytoplasmic plaque
Elevated FAK expression in early breast carcinogenesis correlates with poorer OS in BC [140]. Integral protein or FAK activation impedes the ability of death-associated protein kinase (DAPK) to increase p53 levels, which are frequently hypermethylated in DNA and lost in various tumor types [141]. Tumor necrosis factor (TNF) and TNF receptor-associated factor 2 (TRAF2) synergistically function with focal adhesion (FA) signaling, and TRAF2 upregulation in BC [142], promoting cellular resistance to anoikis [143]. Peptidyl-tRNA hydrolase 2 (PTRH2; Bit-1; Bit1) exhibits significant downregulation in advanced BC tissues compared to normal breast epithelial and ductal carcinoma in situ (DCIS) tissues [144]. PTRH2 complexes with FAK at the cytosol to promote PI3K expression, it complexes with AES/TLE to induce anoikis and inhibit EMT [145].
Proto-oncogene c-Src is widely overexpressed in BC [146], promoting anoikis resistance in HER2 + BC cells in an integrin-dependent manner [17]. Protein tyrosine phosphatase 1B (PTP1B) levels increase significantly in BC tissues [147], and PTP1B is the primary phosphatase that dephosphorylates c-Src, thereby controlling its kinase activity in BC cell lines [148]. The metabolic enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 (PFKFB4) is overexpressed in BC [149], and activates src-3, which in turn activates the estrogen receptor (ER) [150].
ILK expression is upregulated in human tumors and tumor cell lines [151], and its function is required for TGFβ-1-induced EMT in mammary epithelial cells [152]. β-parvin specifically binds ILK [153], is down-regulated in BC [154], and its overexpression inhibits ILK kinase activity [154]. Rictor, a component of the mTORC2 complex, regulates ILK's ability to promote Akt phosphorylation [155]. DOC-2/hDab2 (DOC-2) inhibits ILK activity through an Akt-independent pathway [156].
Adhesion belt-cytoplasmic plaque
In over 50% of BC, β-catenin signaling is upregulated through multiple pathways [157]. Stable β-catenin induces mammary tumorigenesis in mice, functioning as a crucial part of classical Wnt signaling and binding to the cytoplasmic tail of E-cadherin, thereby negatively regulating E-cadherin [158]. β-catenin directly interacts with c-erbB-2 protein and EGFR, playing a crucial role in the tumor signaling pathway [159]. Cadherin system inactivation may increase β-catenin, which activates the Wnt signaling pathway [160]. β-Catenin target genes cyclin D1 and c-myc are also upregulated [161].
P120-catenin is reduced or lost in early BC [162], but it accumulates in the cytoplasm and nucleus rather than being degraded [163]. The transition from a complete loss of protein expression to accumulation is a significant characteristic of p120-catenin. Loss of E-cadherin leads to reduced adhesion and p120-catenin translocation to the cytoplasm and nucleus. P120-catenin may function as a tumor suppressor or metastasis promoter [164], displaying full oncogenic properties in the absence of anchoring [165]. p120-catenin can indirectly activate the Rho/Rock signaling pathway by binding to the Rho function inhibitor Mrip [58]. It can also directly bind RhoA and act as a Rho-GDP dissociation inhibitor (RhoGDI), thereby inhibiting stress fiber-mediated contractility and increasing tumor cell motility [166]. Nuclear p120-catenin controls the anchorage-independent upregulation of Wnt11 by repressing Kaiso-mediated transcription, thereby promoting RhoA activation [167].α-catenin inhibits tumor progression [168], and is downregulated in BC with a more metastatic phenotype [169]. Its loss can lead to lower BC survival rates [170]. α-catenin controls formic acid-dependent radial actin filament formation [171] and also competes with the Arp2/3 complex to bind actin, thereby inhibiting actin branching [172]. Additionally, α-catenin enhances the binding of p120-catenin to E-cadherin, making the connection more stable. The loss of α-catenin leads to mislocalization and aggregation of E-cadherin, which predominantly remains in the plasma membrane [173]. Deletion of α-catenin activates both Rho- and Rock-dependent actin contraction [58]. Furthermore, α-catenin inhibits tumorigenesis by interacting with IκBα [169].
Signaling pathway
Alterations in the PI3K-Akt signaling pathway are prevalent in BC
Gene amplification encoding PI3K or Akt, or mutations in pathway components, may result in constitutive activation of the PI3K-Akt pathway. Approximately 25% to 30% of BC cases contain PIK3CA mutations [174].
Cellular Retinol Binding Protein 1 (CRBP-1) suppresses the PI3K/Akt survival pathway in a retinoic acid receptor-dependent manner. CRBP-1 downregulation is associated with a malignant phenotype in BC, and CRBP-1 inhibits p85 phosphorylation at Y688 [175]. A series of proteins with increased expression in BC, including γ-catenin, PDK1, tyrosine protein kinase receptor A (TrkA), and Ephrin type-A receptor 2 (EphA2), positively regulate the PI3K-AKT pathway and impair anoikis function in BC [176]. Overexpression of PDK1 protein and its mRNA, recruited by PIP3, elevates PDK1 gene copy number, and enhances AKT and downstream pathway activity [139, 177]. TrkA protein is overexpressed in a large cohort of clinically relevant BC [177]. TrkA and downstream AKT signaling modulate cell growth and viability [178]. EphA2, a receptor tyrosine kinase and guanine nucleotide exchange factor for GTPase RhoG, interacts with Ephexin4 to activate RhoG and PI3K downstream of EphA2 [179].
Phosphatase and tensin homologs (PTEN) absent on chromosome 10 are frequently mutated in BC, inhibiting Akt activation [180]. PTEN interacts with FAK, reducing FAK phosphorylation and activity [181]. Heterozygous deletion of PTEN activates the PI3K/Akt and MAPK pathways, while pure deletion of PTEN expression results in the activation of both, thus conferring BC anoikis resistance [182]. WNT5A, a member of the WNT family, exhibits down-regulated in BC expression and inhibits ERK1/2 activity in BC cells [183].
NF-κB Signaling expression is significantly upregulated in BC
I-κB kinase (IKK), a core regulator of NF-κB signaling, is associated with tumorigenesis through its three isoforms (α, β, and ε) [184]. The Deleted in Breast Cancer 1 (DBC1) protein interacts with IKK-β as a cofactor, stimulating the phosphorylation of nuclear relA (serine-536), promoting transcriptional activation of NF-κB target genes, and upregulating the expression of target genes that protect against anoikis (e.g., c-FLIP and Bcl-xl) [65, 185]. IKKε is amplified and overexpressed in approximately 30% of BC cases [186], and activates classical NF-kB signaling by directly phosphorylating the RelA/p65 subunit [187].
In BC cells detached from the ECM, NF-κB activity was found to be increased sixfold compared to normal breast cells. RelA and NF-κB1 are transcription factors responsible for upregulating neurotrophic receptor TrkB and its ligand neurotrophic factor 3 (NTF3) [188]. Etoposide induced 2.4 (EI24) attenuates NF-κB activity by binding to complex I component TRAF2 and inducing its lysosome-dependent degradation. EI24 has been identified as an oncogene and significantly downregulated in invasive cancers [189].
MEK/ERK Signaling pathway is significantly activated in BC
ERK signaling activation is observed in more than 85% of cancers and is directly attributable to genetic alterations in its upstream activators, including BRAF, Ras, and RTK [190]. Ras-related protein Rab25 activates intracellular signaling pathways, and Rab25 knockdown reduces phospho-ERK1/2 levels and promotes BC cell proliferation [191]. Multiple Ras signaling pathways contribute to breast EMT, with the ERK signaling pathway potentially being a key component downstream of EGFR activation during tumorigenesis [192].
Protein kinase C theta (PRKCQ/PKCθ) activates ERK in a kinase activity-dependent manner [193]. Sustained ERK signaling activation upregulates T-cell death-associated gene 51 (TDAG51), which inhibits ERK-mediated mammary EMT [194]. Reduced phosphoprotein expression in astrocytes-15 (PEA15) in metastatic BC cells leads to ERK1/2 binding and altered ERK1/2 cellular localization and target preference [195]. Caveolin-1 (Cav-1) expression is downregulated in BC, inhibiting stromal invasion and blocking laminin-dependent activation of ERK1/2 [196].
Upon ECM attachment loss, p38ERK is activated and recruited into the high molecular weight mitochondrial complex, a necessary signal for Bax translocation to the mitochondria and Bax activation. P38ERK targeting the outer mitochondrial membrane (OMM) drives anoikis [197]. ECM attachment loss also results in the release of PTRH2 from mitochondria into the cytoplasm. Increased ERK levels and decreased ERK-directed phosphatase activity disrupt mitochondrial Bit1 [144], and loss of Bit1 expression leads to increased ERK activation [145, 198]. Bit1 expression was reduced in higher grade BC with positive lymph node metastases, compared to normal breast tissue or invasive low BC [145].
AMPK Signaling pathway is actively expressed in BC
Stromal deprivation activates AMPK activity through LKB1 and Ca2 + /cadherin-dependent protein kinase kinase (CaMKK) [199], and further by upregulating the Akt phosphatase pleckstrin-homology (PH)-domain leucine-rich-repeat protein phosphatases (PHLPP2) while inhibiting AKT activity. PAMPK high/pAkt low state impairs both autophagy and metastasis [200]. AMPK directly affects metabolic enzymes (such as G6PD, ACC, and HMG-CoA reductase) and plays a key role in regulating growth and metabolism [201]. AMPK contributes to anti-anoikis by phosphorylating phosphoprotein 15 kDa (PEA15) and inhibiting mTORC1 [202] as well as Acetyl-CoA carboxylase (ACC) to maintain NADPH homeostasis [199].
Protein
Bcl-2 Protein Family Alterations Inhibit Anoikis
Expression of several pro-apoptotic BH3-only proteins of the Bcl-2 family is downregulated following ECM detachment, resulting in an intrinsic cell death cascade. Hypoxia contributes to blocking the expression of pro-apoptotic BH3 family members, such as Bim and Bmf, in epithelial cells [203]. Chemokine receptors CCR7 and CXCR4 are highly expressed in BC [204]. Activation of CCR4 and CCR7 by their respective chemokine ligands CXCL12, CCL19/CCL21 downregulates pro-apoptotic Bmf and upregulates pro-survival proteins Bcl-2 and Bcl-xL, specifically reducing the sensitivity of metastatic BC cells to anoikis [205].
Zinc-finger protein binds specifically to DNA, RNA, and DNA-RNA sequences to inhibit anoikis
ZNF367 is significantly upregulated in BC, interacts with the chromatin remodeling protein Brahma-related gene 1 (BRG1), and transcriptionally activates Citron (CIT) and TP53. It represses the Hippo pathway and activates Yes-associated protein 1 (YAP1), causing anoikis resistance [206]. ZnF protein E4F transcription factor 1 (E4F1) is involved in DNA repair, with E4F1 binding to the catalytic subunit BRG1/SMARCA4 and mediating its recruitment to DNA damage together with Poly(ADP-ribose) (PAR) polymerase-1 (PARP-1) [207]. ZNF367 is an upstream transcription factor of kinesin superfamily proteins 15 (KIF15), highly expressed in BC tissues and regulates mitosis during cellular processes [208]. Additionally, the expression of integrin α 3 (ITGA3) is also regulated by ZNF367 [209].
Tumor protein p53 (TP53) is significantly altered in BC
TP53 can mediate anoikis by upregulating the transcription of pro-apoptotic Bcl-2 family members, including PUMA, Noxa and Bax [210]. TP53 is highly mutated in BC [211], with mutant p53-R273H inhibiting BMF via AKT [212], and mutant p53P151S causes increased tumor growth and metastasis [213]. TP53-inducible protein 11 (TP53I11) is a transcriptional target of TP53. Deletion of TP53I11 promotes AKT/m-TOR and AMPK pathways, respectively, subsequently reducing m-TOR activation and consequently maintaining energy homeostasis in BC cells [214], promoting cell survival after ECM isolation. HIF1α is a key mediator of cellular adaptation to hypoxia [215], and TP53I11 overexpression inhibits HIF1α expression and affects EMT and metastasis [104]. YAP positively regulates p53 family members, YAP protein is significantly reduced in BC, and knockdown of YAP short hairpin RNA inhibits anoikis [216]. Transcription factor p73 is a homolog of p53 and can be expressed as a pro- or anti-apoptotic isoform. Conversely, p73 is stably expressed in cancer and can replace defective p53, inducing anoikis [217].
Ferritin (FER) promotes BC cell migration
FER protein is significantly elevated in metastatic BC cells and is closely associated with the development of anoikis resistance mechanisms [116]. FER promotes BC cell migration and inhibits anchorage-dependence, leading to increased formation of distant metastases [218]. The primary mechanism of action involves inhibiting cellular morphological changes, including the regulation of actin stress fiber and focal adhesion formation [219]. FER deficiency increases α6β1 integrin expression and collagen I(COL1) adhesion and laminin in BC cells [220]. Consequently, FER controls the recirculation of α6-integrins in metastatic BC cells [116], and downregulates the synthesis of laminin-binding glycans, reducing cell adhesion to laminin [221].
Abnormal actin presence as a main feature of malignant tumors
Cytoskeletal proteins induce anoikis in BC cells. Restoration of stress fiber networks can be achieved by enhancing the expression of key cytoskeletal proteins, thereby regulating focal adhesion activity and sensitizing tumor cells to anoikis [89]. Osteopontin (OPN) is highly expressed in various cancers and increases the potential for cell invasion. Bone bridging proteins have two forms corresponding to two functions: the soluble form favors invasiveness, and the aggregated form favors adhesion [222]. KIAA0100 protein is upregulated in BC and acts as a microtubule binding protein to stabilize the MT structure [223]. KIF18A is overexpressed in human BC. The inhibition of Kif18A stabilizes MT at the leading edge, inducing inactivation of the PI3K-Akt signaling pathway, reducing cancer cell migration and proliferation [224]. High expression of ubiquitin-binding enzyme E2S (UBE2S) in BC disrupts the actin cytoskeleton [225]. BC metastasis suppressor 1 (BRMS1) decreases cytoskeletal reorganization and cell adhesion protrusion formation [226]. Histone deacetylase 6 (HDAC6) is located in the cytoplasm, upregulated in multiple cancers, binds to MT and the actin cytoskeleton, and regulates cell adhesion [227].
Gene localization analysis
The major structures and components of BC anoikis resistance were targeted, with a focus on studying their loci on chromosomes (Fig. 6). We aimed to find out if there are genes at similar loci undergo cascading alterations and therefore affect BC anoikis resistance. This analysis was conducted by accessing the NCBI (National Center for Biotechnology Information; https://www.ncbi.nlm.nih.gov/) database, obtaining gene locus information, including chromosome numbers and the starting and ending points of the genes on the chromosomes, and using the "RIdeogram" R package to map the loci for presentation. According to our results, E-cadherin and P-cadherin were located at 16q22.1, and P-cadherin was located in the upstream of E-cadherin [228]. Long-arm heterozygous deletion (LOH) of chromosome 16 occurs in at least half of breast tumors, and the smallest region of overlap (SRO) is found in the region of 16q22.1 [229], suggesting the presence of tumor suppressor genes (TSG) in this region of the BC. E-cadherin fits this profile [230], while P-cadherin is associated with poor survival [231]. Further exploration is warranted to determine if cascade alterations occur at the genetic level. Similarly, MMP-11 (22q11.23) and ERK (22q11.22), both located on chromosome 22q11, are highly expressed in BC. Copy number gains on BC chromosome 22q11 were more frequent in the Chinese cohort than in the Cancer Genome Atlas [232], and gains in the 22q11.2 region were shown to contribute to triggering EMT. The summary of genetic loci associated with BC anoikis resistance provided here is intended to inform studies of metastatic BC at the chromosomal level.

Chromosomal location of the major associated proteins of anoikis resistance in BC. Blue indicates low density of gene distribution, red indicates high density of gene distribution
Micro RNA (miRNA)
MicroRNAs (miRNAs) are small non-protein-coding RNAs, approximately 18 to 25 nucleotides in length, that are predicted to regulate the expression of over 90% of genes at the post-transcriptional level. These miRNAs influence various cellular and molecular processes, including anoikis, angiogenesis, tumorigenesis, tumor growth, cell migration, metabolic pathways, and signal transduction [233]. The results (Table 1) indicate that miR-630, miR-223, miR-200c, miR-148b, miR-18a, miR-124, miRNA-200b, miR-491-5p, miR-6744-5p, and miRNA-188-3p were down-regulated in BC. Conversely, miR-23b, miR-27b, miR-522, miR-200a, and miR-181a were up-regulated in BC, leading to enhanced anoikis resistance.
Targeted therapy
Targeted therapy for BC is essential in the treatment of this condition.Controlling BC in situ and eliminating the possibility of metastasis serve as effective strategies for improving treatment outcomes. Restoring BC cell anoikis sensitivity can help achieve this objective. A collection of drugs and compounds reported to influence BC anoikis function has been compiled, including plant extracts and drugs commonly used in clinical practice for other diseases, in addition to conventional BC targeted therapies. These are all summarized in Table 2. Among the compiled results, Goniothalamin (GTN) appears to be the most comprehensive drug for targeting the restoration of anoikis function. Further exploration is required to determine its potential for treating BC.
Conclusion and outlook
Metastasis is a complex biological process and the leading cause of death in BC patients. Tumor cells detach from their original locations, migrate, invade the circulatory and lymphatic systems, and ultimately invade and colonize distal locations, growing and proliferating to form secondary tumors. The ability of cells to survive independently is a critical factor in the metastatic process, as tumor cells with malignant potential can initiate mechanisms to resist anoikis and survive. Cancer cells undergo alterations at various levels, including genes, proteins, cytoskeleton, and cell structure, in order to resist anoikis. Different cancer cells are involved in distinct mechanistic alterations, and multiple mechanisms often collaborate to affect a single cell. The cumulative result of these factors presents a significant challenge for clinical treatment.
Precisely targeting the critical period when anoikis resistance occurs (i.e., after disruption of tumor epithelial cell-ECM interactions and before metastatic spread) is an essential approach for prognostic regression of BC. Current strategies using anoikis sensitizing drugs primarily involve combinatorial algorithms aimed at reducing the activation of alternative signaling pathways and maximizing efficacy. The mechanism by which GTN promotes anoikis in multiple directions requires further investigation.
Promoting the restoration of the anoikis phenotype in tumor cells through modulation of miRNAs is another highly promising approach. MiRNAs can play a role in the clinical treatment of patients with tumor metastasis, in addition to their previous value as predictive molecular markers in clinical work. Consequently, potential anoikis sensitizing drugs and genetic manipulation targeting miRNA families could provide a practical framework for inhibiting tumor metastasis.
Availability of data and materials
Not applicable.
Abbreviations
- BC:
-
Breast Cancer
- HER2:
-
Human Epidermal Growth Factor Receptor2
- ECM:
-
Extracellular Matrix
- PPP:
-
Pentose Phosphate Pathway
- BMP:
-
Bone Morphogenetic Protein
- GFRs:
-
Growth Factor Receptors
- FAK:
-
Focal Adhesion Kinase
- JAK:
-
Janus-activated Kinase
- PI3K:
-
Phosphatidylinositol 3-kinase
- IMS:
-
Intermembrane Space
- KD:
-
Kinase Domain
- PDK1:
-
3-Phosphatidylinositol-dependent Protein Kinase 1
- FA:
-
Focal Adhesion
- TNFR2:
-
Tumor Necrosis Factor Receptor 2
- Apaf-1:
-
Apoptotic protease-activating factor-1
- MMP:
-
Matrix Metalloproteinase
- ZEB1:
-
Zinc finger E-box Binding homeobox 1
- GRHL2:
-
Grainyhead-like2
- TP53I11:
-
Tumor Protein P53-inducible protein 11
- FN1:
-
Fibronectin
- CEACAM6:
-
Carcinoembryonic Antigen-related Cell Adhesion Molecule 6
- FER:
-
Ferritin
- IARC:
-
International Agency for Research on Cancer
- TNBC:
-
Triple Negative cancer
- COLI:
-
Collagen I
- ROS:
-
Reactive Oxygen Species
- CAM:
-
Cell Adhesion Molecules
- ILK:
-
Integrin-related Kinase
- TEB:
-
Terminal Bud
- PLCγ:
-
Phospholipase Cγ
- EGFR:
-
Epidermal Growth Factor Receptor
- PH:
-
Pleckstrin Homology
- PIP3:
-
Phosphatidylinositol 3,4,5-trisphosphate
- IKK:
-
IkB kinase
- TNF:
-
Tumor Necrosis Factor
- DLC2:
-
Dynein Light Chain 2
- TM1:
-
Pro-myosin-1
- IGF-I:
-
Insulin-like Growth factor-I
- UBA6:
-
Ubiquitin-like Modifier-activating Enzyme 6
- TRIP12:
-
Thyroid Hormone Receptor Interacting Protein 12
- LOX:
-
Lysyl Oxidase
- RGD:
-
Arginine-glycine-aspartate
- SEMA7A:
-
Semaphorin-7a
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Tao Z, Shi A, Lu C, Song T, Zhang Z, Zhao J. Breast cancer: epidemiology and etiology. Cell Biochem Biophys. 2015;72(2):333–8.
Yersal O, Barutca S. Biological subtypes of breast cancer: prognostic and therapeutic implications. World J Clin Oncol. 2014;5(3):412–24.
Trayes KP, Cokenakes SEH. Breast cancer treatment. Am Fam Physician. 2021;104(2):171–8.
Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52.
Cserni G. Histological type and typing of breast carcinomas and the WHO classification changes over time. Pathologica. 2020;112(1):25–41.
Yeo SK, Guan JL. Breast cancer: multiple subtypes within a tumor? Trends in cancer. 2017;3(11):753–60.
Liang Y, Zhang H, Song X, Yang Q. Metastatic heterogeneity of breast cancer: molecular mechanism and potential therapeutic targets. Semin Cancer Biol. 2020;60:14–27.
DeSantis CE, Ma J, Gaudet MM, Newman LA, Miller KD, Goding Sauer A, et al. Breast cancer statistics, 2019. CA Cancer J Clin. 2019;69(6):438–51.
Scully OJ, Bay BH, Yip G, Yu Y. Breast cancer metastasis. Cancer Genomics Proteomics. 2012;9(5):311–20.
Kim YN, Koo KH, Sung JY, Yun UJ, Kim H. Anoikis resistance: an essential prerequisite for tumor metastasis. Int J Cell Biol. 2012;2012: 306879.
Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochem Biophys Acta. 2013;1833(12):3481–98.
Simpson CD, Anyiwe K, Schimmer AD. Anoikis resistance and tumor metastasis. Cancer Lett. 2008;272(2):177–85.
Adeshakin FO, Adeshakin AO, Afolabi LO, Yan D, Zhang G, Wan X. Mechanisms for modulating anoikis resistance in cancer and the relevance of metabolic reprogramming. Front Oncol. 2021;11: 626577.
Streuli CH, Gilmore AP. Adhesion-mediated signaling in the regulation of mammary epithelial cell survival. J Mammary Gland Biol Neoplasia. 1999;4(2):183–91.
Craig S, Bui-Mansfield LT, Lusk JD. Radiology pathology conference of brooke army medical center: trichoblastoma of breast. Clin Imaging. 2009;33(4):311–3.
Haenssen KK, Caldwell SA, Shahriari KS, Jackson SR, Whelan KA, Klein-Szanto AJ, et al. ErbB2 requires integrin alpha5 for anoikis resistance via Src regulation of receptor activity in human mammary epithelial cells. J Cell Sci. 2010;123(Pt 8):1373–82.
Hynes RO. Cell adhesion: old and new questions. Trends Cell Biol. 1999;9(12):M33–7.
Davison CA, Durbin SM, Thau MR, Zellmer VR, Chapman SE, Diener J, et al. Antioxidant enzymes mediate survival of breast cancer cells deprived of extracellular matrix. Can Res. 2013;73(12):3704–15.
Buck MB, Knabbe C. TGF-beta signaling in breast cancer. Ann N Y Acad Sci. 2006;1089:119–26.
Yan X, Liu Z, Chen Y. Regulation of TGF-beta signaling by Smad7. Acta Biochim Biophys Sin. 2009;41(4):263–72.
Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118(Pt 16):3573–84.
Garcia-Gonzalez E, Swerlick RA, Lawley TJ. Cell adhesion molecules. Am J Dermatopathol. 1990;12(2):188–92.
Huang MW, Lin CD, Chen JF. Integrin activation, focal adhesion maturation and tumor metastasis. Sheng li xue bao : [Acta physiologica Sinica]. 2021;73(2):151–9.
Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochem Biophys Acta. 2008;1778(3):660–9.
Zhong X, Rescorla FJ. Cell surface adhesion molecules and adhesion-initiated signaling: understanding of anoikis resistance mechanisms and therapeutic opportunities. Cell Signal. 2012;24(2):393–401.
Leof EB. Growth factor receptor signalling: location, location, location. Trends Cell Biol. 2000;10(8):343–8.
Shimaoka M, Springer TA. Therapeutic antagonists and conformational regulation of integrin function. Nat Rev Drug Discovery. 2003;2(9):703–16.
Cox D, Brennan M, Moran N. Integrins as therapeutic targets: lessons and opportunities. Nat Rev Drug Discovery. 2010;9(10):804–20.
Takada Y, Ye X, Simon S. The integrins. Genome Biol. 2007;8(5):215.
Beglova N, Blacklow SC, Takagi J, Springer TA. Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat Struct Biol. 2002;9(4):282–7.
Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–87.
Puklin-Faucher E, Sheetz MP. The mechanical integrin cycle. J Cell Sci. 2009;122(Pt 2):179–86.
Geiger B, Bershadsky A, Pankov R, Yamada KM. Transmembrane crosstalk between the extracellular matrix–cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001;2(11):793–805.
Giancotti FG, Ruoslahti E. Integrin signaling. Science (New York, NY). 1999;285(5430):1028–32.
Calderwood DA, Campbell ID, Critchley DR. Talins and kindlins: partners in integrin-mediated adhesion. Nat Rev Mol Cell Biol. 2013;14(8):503–17.
Legate KR, Montañez E, Kudlacek O, Fässler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol. 2006;7(1):20–31.
Andrews JL, Kim AC, Hens JR. The role and function of cadherins in the mammary gland. Breast Cancer Res. 2012;14(1):203.
Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17(5):548–58.
Albergaria A, Ribeiro AS, Vieira AF, Sousa B, Nobre AR, Seruca R, et al. P-cadherin role in normal breast development and cancer. Int J Dev Biol. 2011;55(7–9):811–22.
Nelson WJ. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans. 2008;36(Pt 2):149–55.
Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, et al. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat Cell Biol. 2003;5(8):733–40.
Normanno N, Campiglio M, Maiello MR, De Luca A, Mancino M, Gallo M, et al. Breast cancer cells with acquired resistance to the EGFR tyrosine kinase inhibitor gefitinib show persistent activation of MAPK signaling. Breast Cancer Res Treat. 2008;112(1):25–33.
Carraway KL 3rd, Sweeney C. Co-opted integrin signaling in ErbB2-induced mammary tumor progression. Cancer Cell. 2006;10(2):93–5.
Horbinski C, Mojesky C, Kyprianou N. Live free or die: tales of homeless (cells) in cancer. Am J Pathol. 2010;177(3):1044–52.
Gilmore AP. Anoikis. Cell Death Differ. 2005;12(Suppl 2):1473–7.
Kamarajugadda S, Cai Q, Chen H, Nayak S, Zhu J, He M, et al. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis. 2013;4(2): e504.
Guillemot L, Paschoud S, Pulimeno P, Foglia A, Citi S. The cytoplasmic plaque of tight junctions: a scaffolding and signalling center. Biochem Biophys Acta. 2008;1778(3):601–13.
Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochem Biophys Acta. 2004;1692(2–3):103–19.
Kleinschmidt EG, Schlaepfer DD. Focal adhesion kinase signaling in unexpected places. Curr Opin Cell Biol. 2017;45:24–30.
Sood AK, Armaiz-Pena GN, Halder J, Nick AM, Stone RL, Hu W, et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J Clin Investig. 2010;120(5):1515–23.
Tsirtsaki K, Gkretsi V. The focal adhesion protein Integrin-Linked Kinase (ILK) as an important player in breast cancer pathogenesis. Cell Adh Migr. 2020;14(1):204–13.
McDonald PC, Fielding AB, Dedhar S. Integrin-linked kinase–essential roles in physiology and cancer biology. J Cell Sci. 2008;121(Pt 19):3121–32.
Garcia MA, Nelson WJ, Chavez N. Cell-cell junctions organize structural and signaling networks. Cold Spring Harb Perspect Biol. 2018;10(4):a029181.
Anastasiadis PZ, Reynolds AB. The p120 catenin family: complex roles in adhesion, signaling and cancer. J Cell Sci. 2000;113(Pt 8):1319–34.
Mo CF, Li J, Yang SX, Guo HJ, Liu Y, Luo XY, et al. IQGAP1 promotes anoikis resistance and metastasis through Rac1-dependent ROS accumulation and activation of Src/FAK signalling in hepatocellular carcinoma. Br J Cancer. 2020;123(7):1154–63.
Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ. Deconstructing the cadherin-catenin-actin complex. Cell. 2005;123(5):889–901.
Schackmann RC, van Amersfoort M, Haarhuis JH, Vlug EJ, Halim VA, Roodhart JM, et al. Cytosolic p120-catenin regulates growth of metastatic lobular carcinoma through Rock1-mediated anoikis resistance. J Clin Investig. 2011;121(8):3176–88.
Guinebault C, Payrastre B, Racaud-Sultan C, Mazarguil H, Breton M, Mauco G, et al. Integrin-dependent translocation of phosphoinositide 3-kinase to the cytoskeleton of thrombin-activated platelets involves specific interactions of p85 alpha with actin filaments and focal adhesion kinase. J Cell Biol. 1995;129(3):831–42.
Carnero A, Paramio JM. The PTEN/PI3K/AKT Pathway in vivo. Cancer Mouse Models Front in Oncol. 2014;4:252.
Milburn CC, Deak M, Kelly SM, Price NC, Alessi DR, Van Aalten DM. Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem J. 2003;375(Pt 3):531–8.
Miricescu D, Totan A, Stanescu S II, Badoiu SC, Stefani C, Greabu M. PI3K/AKT/mTOR signaling pathway in breast cancer: from molecular landscape to clinical aspects. Int J Mol Sci. 2020;22(1):173.
Rahmani M, Nkwocha J, Hawkins E, Pei X, Parker RE, Kmieciak M, et al. Cotargeting BCL-2 and PI3K Induces BAX-Dependent Mitochondrial Apoptosis in AML Cells. Can Res. 2018;78(11):3075–86.
Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401(6748):86–90.
Park SH, Riley Pt, Frisch SM. Regulation of anoikis by deleted in breast cancer-1 (DBC1) through NF-κB. Apoptosis : an international journal on programmed cell death. 2013;18(8):949–62.
Boerner JL, Demory ML, Silva C, Parsons SJ. Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol Cell Biol. 2004;24(16):7059–71.
Giannoni E, Buricchi F, Grimaldi G, Parri M, Cialdai F, Taddei ML, et al. Redox regulation of anoikis: reactive oxygen species as essential mediators of cell survival. Cell Death Differ. 2008;15(5):867–78.
Moelling K, Heimann B, Beimling P, Rapp UR, Sander T. Serine- and threonine-specific protein kinase activities of purified gag-mil and gag-raf proteins. Nature. 1984;312(5994):558–61.
Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418(6901):934.
Cantley LC. The phosphoinositide 3-kinase pathway. Science (New York, NY). 2002;296(5573):1655–7.
Sakai T, Li S, Docheva D, Grashoff C, Sakai K, Kostka G, et al. Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev. 2003;17(7):926–40.
Legate KR, Wickström SA, Fässler R. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 2009;23(4):397–418.
Puthalakath H, Villunger A, O’Reilly LA, Beaumont JG, Coultas L, Cheney RE, et al. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science (New York, NY). 2001;293(5536):1829–32.
Zimmermann KC, Bonzon C, Green DR. The machinery of programmed cell death. Pharmacol Ther. 2001;92(1):57–70.
Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426(6967):671–6.
Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6(6):1389–99.
Yamaguchi H, Wang HG. Bcl-XL protects BimEL-induced Bax conformational change and cytochrome C release independent of interacting with Bax or BimEL. J Biol Chem. 2002;277(44):41604–12.
Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91(5):627–37.
Schimmer AD, Hedley DW, Pham NA, Chow S, Minden MD. BAD induces apoptosis in cells over-expressing Bcl-2 or Bcl-xL without loss of mitochondrial membrane potential. Leuk Lymphoma. 2001;42(3):429–43.
Zhou H, Hou Q, Chai Y, Hsu YT. Distinct domains of Bcl-XL are involved in Bax and Bad antagonism and in apoptosis inhibition. Exp Cell Res. 2005;309(2):316–28.
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–89.
Wu CC, Lee S, Malladi S, Chen MD, Mastrandrea NJ, Zhang Z, et al. The Apaf-1 apoptosome induces formation of caspase-9 homo- and heterodimers with distinct activities. Nat Commun. 2016;7:13565.
Fuentes-Prior P, Salvesen GS. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J. 2004;384(Pt 2):201–32.
Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906.
Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr HA, Delaloye JF, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2011;481(7379):85–9.
Gehler S, Ponik SM, Riching KM, Keely PJ. Bi-directional signaling: extracellular matrix and integrin regulation of breast tumor progression. Crit Rev Eukaryot Gene Expr. 2013;23(2):139–57.
Kostic A, Lynch CD, Sheetz MP. Differential matrix rigidity response in breast cancer cell lines correlates with the tissue tropism. PLoS ONE. 2009;4(7): e6361.
Kim BG, Gao MQ, Choi YP, Kang S, Park HR, Kang KS, et al. Invasive breast cancer induces laminin-332 upregulation and integrin β4 neoexpression in myofibroblasts to confer an anoikis-resistant phenotype during tissue remodeling. Breast Cancer Res. 2012;14(3):R88.
Bharadwaj S, Thanawala R, Bon G, Falcioni R, Prasad GL. Resensitization of breast cancer cells to anoikis by tropomyosin-1: role of Rho kinase-dependent cytoskeleton and adhesion. Oncogene. 2005;24(56):8291–303.
Raval GN, Bharadwaj S, Levine EA, Willingham MC, Geary RL, Kute T, et al. Loss of expression of tropomyosin-1, a novel class II tumor suppressor that induces anoikis, in primary breast tumors. Oncogene. 2003;22(40):6194–203.
Di Nezza LA, Misajon A, Zhang J, Jobling T, Quinn MA, Ostör AG, et al. Presence of active gelatinases in endometrial carcinoma and correlation of matrix metalloproteinase expression with increasing tumor grade and invasion. Cancer. 2002;94(5):1466–75.
Takeuchi T, Adachi Y, Nagayama T, Furihata M. Matrix metalloproteinase-11 overexpressed in lobular carcinoma cells of the breast promotes anoikis resistance. Virchows Arch. 2011;459(3):291–7.
Lino RLB, Dos Santos PK, Pisani GFD, Altei WF, Cominetti MR, Selistre-de-Araújo HS. Alphavbeta3 integrin blocking inhibits apoptosis and induces autophagy in murine breast tumor cells. Biochim Biophys Acta. 2019;1866(12): 118536.
Miyamoto S, Yano K, Sugimoto S, Ishii G, Hasebe T, Endoh Y, et al. Matrix metalloproteinase-7 facilitates insulin-like growth factor bioavailability through its proteinase activity on insulin-like growth factor binding protein 3. Can Res. 2004;64(2):665–71.
Dong H, Diao H, Zhao Y, Xu H, Pei S, Gao J, et al. Overexpression of matrix metalloproteinase-9 in breast cancer cell lines remarkably increases the cell malignancy largely via activation of transforming growth factor beta/SMAD signalling. Cell Prolif. 2019;52(5): e12633.
Berx G, Raspé E, Christofori G, Thiery JP, Sleeman JP. Pre-EMTing metastasis? recapitulation of morphogenetic processes in cancer. Clin Exp Metas. 2007;24(8):587–97.
Na TY, Schecterson L, Mendonsa AM, Gumbiner BM. The functional activity of E-cadherin controls tumor cell metastasis at multiple steps. Proc Natl Acad Sci USA. 2020;117(11):5931–7.
Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016;35(4):645–54.
Zhang P, Sun Y, Ma L. ZEB1: at the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell cycle (Georgetown, Tex). 2015;14(4):481–7.
Lee KK, Rajagopalan D, Bhatia SS, Tirado-Magallanes R, Chng WJ, Jha S. The oncogenic E3 ligase TRIP12 suppresses epithelial-mesenchymal transition (EMT) and mesenchymal traits through ZEB1/2. Cell death discovery. 2021;7(1):95.
Huang W, Pal A, Kleer CG. On how CCN6 suppresses breast cancer growth and invasion. J Cell Commun Signal. 2012;6(1):5–10.
Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012;26(1):54–68.
Liu X, Sun L, Gursel DB, Cheng C, Huang S, Rademaker AW, et al. The non-canonical ubiquitin activating enzyme UBA6 suppresses epithelial-mesenchymal transition of mammary epithelial cells. Oncotarget. 2017;8(50):87480–93.
Xiao T, Xu Z, Zhang H, Geng J, Qiao Y, Liang Y, et al. TP53I11 suppresses epithelial-mesenchymal transition and metastasis of breast cancer cells. BMB Rep. 2019;52(6):379–84.
Elliott RL, Blobe GC. Role of transforming growth factor Beta in human cancer. J Clin Oncol. 2005;23(9):2078–93.
Drabsch Y, ten Dijke P. TGF-β signaling in breast cancer cell invasion and bone metastasis. J Mammary Gland Biol Neoplasia. 2011;16(2):97–108.
Zhao Y, Ma J, Fan Y, Wang Z, Tian R, Ji W, et al. TGF-β transactivates EGFR and facilitates breast cancer migration and invasion through canonical Smad3 and ERK/Sp1 signaling pathways. Mol Oncol. 2018;12(3):305–21.
Taylor MA, Amin JD, Kirschmann DA, Schiemann WP. Lysyl oxidase contributes to mechanotransduction-mediated regulation of transforming growth factor-β signaling in breast cancer cells. Neoplasia (New York, NY). 2011;13(5):406–18.
Moschos SJ, Drogowski LM, Reppert SL, Kirkwood JM. Integrins and cancer. Oncology (Williston Park). 2007;21(9 Suppl 3):13–20.
van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res. 2011;728(1–2):23–34.
Zhang H, Fredericks T, Xiong G, Qi Y, Rychahou PG, Li JD, et al. Membrane associated collagen XIII promotes cancer metastasis and enhances anoikis resistance. Breast Cancer Res. 2018;20(1):116.
Zhang Y, Zhang B. D4-GDI, a Rho GTPase regulator, promotes breast cancer cell invasiveness. Can Res. 2006;66(11):5592–8.
Zuo Y, Shields SK, Chakraborty C. Enhanced intrinsic migration of aggressive breast cancer cells by inhibition of Rac1 GTPase. Biochem Biophys Res Commun. 2006;351(2):361–7.
Lewis-Wambi JS, Cunliffe HE, Kim HR, Willis AL, Jordan VC. Overexpression of CEACAM6 promotes migration and invasion of oestrogen-deprived breast cancer cells. Eur J Cancer (Oxford, England : 1990). 2008;44(12):1770–9.
Ordonez C, Zhai AB, Camacho-Leal P, Demarte L, Fan MM, Stanners CP. GPI-anchored CEA family glycoproteins CEA and CEACAM6 mediate their biological effects through enhanced integrin alpha5beta1-fibronectin interaction. J Cell Physiol. 2007;210(3):757–65.
Ivanova IA, Vermeulen JF, Ercan C, Houthuijzen JM, Saig FA, Vlug EJ, et al. FER kinase promotes breast cancer metastasis by regulating α6- and β1-integrin-dependent cell adhesion and anoikis resistance. Oncogene. 2013;32(50):5582–92.
Rutherford TR, Elder AM, Lyons TR. Anoikis resistance in mammary epithelial cells is mediated by semaphorin 7a. Cell Death Dis. 2021;12(10):872.
Kowalski PJ, Rubin MA, Kleer CG. E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res. 2003;5(6):R217–22.
Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Can Res. 2008;68(10):3645–54.
Ito K, Park SH, Nayak A, Byerly JH, Irie HY. PTK6 inhibition suppresses metastases of triple-negative breast cancer via SNAIL-Dependent E-Cadherin Regulation. Can Res. 2016;76(15):4406–17.
Benzina S, Beauregard AP, Guerrette R, Jean S, Faye MD, Laflamme M, et al. Pax-5 is a potent regulator of E-cadherin and breast cancer malignant processes. Oncotarget. 2017;8(7):12052–66.
Phillips S, Kuperwasser C. SLUG: Critical regulator of epithelial cell identity in breast development and cancer. Cell Adh Migr. 2014;8(6):578–87.
Leong KG, Niessen K, Kulic I, Raouf A, Eaves C, Pollet I, et al. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med. 2007;204(12):2935–48.
Takeichi M. The cadherins: cell-cell adhesion molecules controlling animal morphogenesis. Development (Cambridge, England). 1988;102(4):639–55.
Vieira AF, Ribeiro AS, Dionísio MR, Sousa B, Nobre AR, Albergaria A, et al. P-cadherin signals through the laminin receptor α6β4 integrin to induce stem cell and invasive properties in basal-like breast cancer cells. Oncotarget. 2014;5(3):679–92.
Yang X, Chung JY, Rai U, Esumi N. Cadherins in the retinal pigment epithelium (RPE) revisited: P-cadherin is the highly dominant cadherin expressed in human and mouse RPE in vivo. PLoS ONE. 2018;13(1): e0191279.
Burness ML, Grushko TA, Olopade OI. Epidermal growth factor receptor in triple-negative and basal-like breast cancer: promising clinical target or only a marker? Cancer J (Sudbury, Mass). 2010;16(1):23–32.
Grassian AR, Schafer ZT, Brugge JS. ErbB2 stabilizes epidermal growth factor receptor (EGFR) expression via Erk and Sprouty2 in extracellular matrix-detached cells. J Biol Chem. 2011;286(1):79–90.
Giannoni E, Chiarugi P. Redox circuitries driving Src regulation. Antioxid Redox Signal. 2014;20(13):2011–25.
Corkery DP, Clarke LE, Gebremeskel S, Salsman J, Pinder J, Le Page C, et al. Loss of PRP4K drives anoikis resistance in part by dysregulation of epidermal growth factor receptor endosomal trafficking. Oncogene. 2018;37(2):174–84.
Guo W, Pylayeva Y, Pepe A, Yoshioka T, Muller WJ, Inghirami G, et al. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell. 2006;126(3):489–502.
Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science (New York, NY). 1989;244(4905):707–12.
Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3(9):785–92.
Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461(7260):109–13.
Whelan KA, Schwab LP, Karakashev SV, Franchetti L, Johannes GJ, Seagroves TN, et al. The oncogene HER2/neu (ERBB2) requires the hypoxia-inducible factor HIF-1 for mammary tumor growth and anoikis resistance. J Biol Chem. 2013;288(22):15865–77.
Surette A, Yoo BH, Younis T, Matheson K, Rameh T, Snowdon J, et al. Tumor levels of the mediators of ErbB2-driven anoikis resistance correlate with breast cancer relapse in patients receiving trastuzumab-based therapies. Breast Cancer Res Treat. 2021;187(3):743–58.
Carraway KL 3rd, Rossi EA, Komatsu M, Price-Schiavi SA, Huang D, Guy PM, et al. An intramembrane modulator of the ErbB2 receptor tyrosine kinase that potentiates neuregulin signaling. J Biol Chem. 1999;274(9):5263–6.
Ramsauer VP, Pino V, Farooq A, Carothers Carraway CA, Salas PJ, Carraway KL. Muc4-ErbB2 complex formation and signaling in polarized CACO-2 epithelial cells indicate that Muc4 acts as an unorthodox ligand for ErbB2. Mol Biol Cell. 2006;17(7):2931–41.
Maurer M, Su T, Saal LH, Koujak S, Hopkins BD, Barkley CR, et al. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Can Res. 2009;69(15):6299–306.
Qiao W, Wang W, Liu H, Guo W, Li P, Deng M. Prognostic and clinical significance of focal adhesion kinase expression in breast cancer: a systematic review and meta-analysis. Translational Oncol. 2020;13(11): 100835.
Zhu Y, Li S, Wang Q, Chen L, Wu K, Huang Y, et al. Quantitative and correlation analysis of the DNA methylation and expression of DAPK in breast cancer. PeerJ. 2017;5: e3084.
Zhao ZJ, Ren HY, Yang F, Wang J, Wu GP, Mi XY. Expression, correlation, and prognostic value of TRAF2 and TRAF4 expression in malignant plural effusion cells in human breast cancer. Diagn Cytopathol. 2015;43(11):897–903.
da Silva SD, Xu B, Maschietto M, Marchi FA, Alkailani MI, Bijian K, et al. TRAF2 Cooperates with Focal Adhesion Signaling to Regulate Cancer Cell Susceptibility to Anoikis. Mol Cancer Ther. 2019;18(1):139–46.
Karmali PP, Brunquell C, Tram H, Ireland SK, Ruoslahti E, Biliran H. Metastasis of tumor cells is enhanced by downregulation of Bit1. PLoS ONE. 2011;6(8): e23840.
Corpuz AD, Ramos JW, Matter ML. PTRH2: an adhesion regulated molecular switch at the nexus of life, death, and differentiation. Cell Death Discovery. 2020;6(1):124.
Lou L, Yu Z, Wang Y, Wang S, Zhao Y. c-Src inhibitor selectively inhibits triple-negative breast cancer overexpressed Vimentin in vitro and in vivo. Cancer Sci. 2018;109(5):1648–59.
Yu M, Liu Z, Liu Y, Zhou X, Sun F, Liu Y, et al. PTP1B markedly promotes breast cancer progression and is regulated by miR-193a-3p. FEBS J. 2019;286(6):1136–53.
Hilmarsdottir B, Briem E, Halldorsson S, Kricker J, Ingthorsson S, Gustafsdottir S, et al. Inhibition of PTP1B disrupts cell-cell adhesion and induces anoikis in breast epithelial cells. Cell Death Dis. 2017;8(5): e2769.
Minchenko OH, Ochiai A, Opentanova IL, Ogura T, Minchenko DO, Caro J, et al. Overexpression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4 in the human breast and colon malignant tumors. Biochimie. 2005;87(11):1005–10.
Dasgupta S, Rajapakshe K, Zhu B, Nikolai BC, Yi P, Putluri N, et al. Metabolic enzyme PFKFB4 activates transcriptional coactivator SRC-3 to drive breast cancer. Nature. 2018;556(7700):249–54.
White DE, Cardiff RD, Dedhar S, Muller WJ. Mammary epithelial-specific expression of the integrin-linked kinase (ILK) results in the induction of mammary gland hyperplasias and tumors in transgenic mice. Oncogene. 2001;20(48):7064–72.
Gil D, Ciołczyk-Wierzbicka D, Dulińska-Litewka J, Zwawa K, McCubrey JA, Laidler P. The mechanism of contribution of integrin linked kinase (ILK) to epithelial-mesenchymal transition (EMT). Adv Enzyme Regul. 2011;51(1):195–207.
Sepulveda JL, Wu C. The parvins. Cell Mol Life Sci. 2006;63(1):25–35.
Mongroo PS, Johnstone CN, Naruszewicz I, Leung-Hagesteijn C, Sung RK, Carnio L, et al. Beta-parvin inhibits integrin-linked kinase signaling and is downregulated in breast cancer. Oncogene. 2004;23(55):8959–70.
McDonald PC, Oloumi A, Mills J, Dobreva I, Maidan M, Gray V, et al. Rictor and integrin-linked kinase interact and regulate Akt phosphorylation and cancer cell survival. Can Res. 2008;68(6):1618–24.
Wang SC, Makino K, Xia W, Kim JS, Im SA, Peng H, et al. DOC-2/hDab-2 inhibits ILK activity and induces anoikis in breast cancer cells through an Akt-independent pathway. Oncogene. 2001;20(47):6960–4.
Cowin P, Rowlands TM, Hatsell SJ. Cadherins and catenins in breast cancer. Curr Opin Cell Biol. 2005;17(5):499–508.
Hülsken J, Birchmeier W, Behrens J. E-cadherin and APC compete for the interaction with beta-catenin and the cytoskeleton. J Cell Biol. 1994;127(6 Pt 2):2061–9.
Shibata T, Ochiai A, Kanai Y, Akimoto S, Gotoh M, Yasui N, et al. Dominant negative inhibition of the association between beta-catenin and c-erbB-2 by N-terminally deleted beta-catenin suppresses the invasion and metastasis of cancer cells. Oncogene. 1996;13(5):883–9.
Sousa B, Pereira J, Marques R, Grilo LF, Pereira SP, Sardão VA, et al. P-cadherin induces anoikis-resistance of matrix-detached breast cancer cells by promoting pentose phosphate pathway and decreasing oxidative stress. Biochim Biophys Acta. 2020;1866(12): 165964.
Hatsell S, Rowlands T, Hiremath M, Cowin P. Beta-catenin and Tcfs in mammary development and cancer. J Mammary Gland Biol Neoplasia. 2003;8(2):145–58.
Dillon DA, D’Aquila T, Reynolds AB, Fearon ER, Rimm DL. The expression of p120ctn protein in breast cancer is independent of alpha- and beta-catenin and E-cadherin. Am J Pathol. 1998;152(1):75–82.
Shibata T, Kokubu A, Sekine S, Kanai Y, Hirohashi S. Cytoplasmic p120ctn regulates the invasive phenotypes of E-cadherin-deficient breast cancer. Am J Pathol. 2004;164(6):2269–78.
Thoreson MA, Reynolds AB. Altered expression of the catenin p120 in human cancer: implications for tumor progression. Differentiation. 2002;70(9–10):583–9.
Tenhagen M, Klarenbeek S, Braumuller TM, Hofmann I, van der Groep P, Ter Hoeve N, et al. p120-Catenin Is Critical for the Development of Invasive Lobular Carcinoma in Mice. J Mammary Gland Biol Neoplasia. 2016;21(3–4):81–8.
Noren NK, Liu BP, Burridge K, Kreft B. p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J Cell Biol. 2000;150(3):567–80.
van de Ven RA, Tenhagen M, Meuleman W, van Riel JJ, Schackmann RC, Derksen PW. Nuclear p120-catenin regulates the anoikis resistance of mouse lobular breast cancer cells through Kaiso-dependent Wnt11 expression. Dis Model Mech. 2015;8(4):373–84.
Silvis MR, Kreger BT, Lien WH, Klezovitch O, Rudakova GM, Camargo FD, et al. α-catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci Signal. 2011;4(174):ra33.
Piao HL, Yuan Y, Wang M, Sun Y, Liang H, Ma L. α-catenin acts as a tumour suppressor in E-cadherin-negative basal-like breast cancer by inhibiting NF-κB signalling. Nat Cell Biol. 2014;16(3):245–54.
Benjamin JM, Nelson WJ. Bench to bedside and back again: molecular mechanisms of alpha-catenin function and roles in tumorigenesis. Semin Cancer Biol. 2008;18(1):53–64.
Vasioukhin V, Bauer C, Yin M, Fuchs E. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 2000;100(2):209–19.
Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell. 2005;123(5):903–15.
de Groot JS, Ratze MA, van Amersfoort M, Eisemann T, Vlug EJ, Niklaas MT, et al. αE-catenin is a candidate tumor suppressor for the development of E-cadherin-expressing lobular-type breast cancer. J Pathol. 2018;245(4):456–67.
Lee JW, Soung YH, Kim SY, Lee HW, Park WS, Nam SW, et al. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene. 2005;24(8):1477–80.
Doldo E, Costanza G, Agostinelli S, Tarquini C, Ferlosio A, Arcuri G, et al. Vitamin A, cancer treatment and prevention: the new role of cellular retinol binding proteins. Biomed Res Int. 2015;2015: 624627.
Huang L, Ji H, Yin L, Niu X, Wang Y, Liu Y, et al. High Expression of Plakoglobin Promotes Metastasis in Invasive Micropapillary Carcinoma of the Breast via Tumor Cluster Formation. J Cancer. 2019;10(12):2800–10.
Gagliardi PA, di Blasio L, Orso F, Seano G, Sessa R, Taverna D, et al. 3-phosphoinositide-dependent kinase 1 controls breast tumor growth in a kinase-dependent but Akt-independent manner. Neoplasia (New York, NY). 2012;14(8):719–31.
Griffin N, Marsland M, Roselli S, Oldmeadow C, Attia J, Walker MM, et al. The receptor tyrosine kinase TrkA is increased and targetable in HER2-positive breast cancer. Biomolecules. 2020;10(9):1329.
Zhao P, Jiang D, Huang Y, Chen C. EphA2: A promising therapeutic target in breast cancer. Journal of genetics and genomics = Yi chuan xue bao. 2021;48(4):261–7.
Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9(6):667–76.
Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science (New York, NY). 1998;280(5369):1614–7.
Vitolo MI, Weiss MB, Szmacinski M, Tahir K, Waldman T, Park BH, et al. Deletion of PTEN promotes tumorigenic signaling, resistance to anoikis, and altered response to chemotherapeutic agents in human mammary epithelial cells. Can Res. 2009;69(21):8275–83.
Prasad CP, Chaurasiya SK, Axelsson L, Andersson T. WNT-5A triggers Cdc42 activation leading to an ERK1/2 dependent decrease in MMP9 activity and invasive migration of breast cancer cells. Mol Oncol. 2013;7(5):870–83.
Hsu S, Kim M, Hernandez L, Grajales V, Noonan A, Anver M, et al. IKK-ε coordinates invasion and metastasis of ovarian cancer. Can Res. 2012;72(21):5494–504.
Mawji IA, Simpson CD, Hurren R, Gronda M, Williams MA, Filmus J, et al. Critical role for Fas-associated death domain-like interleukin-1-converting enzyme-like inhibitory protein in anoikis resistance and distant tumor formation. J Natl Cancer Inst. 2007;99(10):811–22.
Shen RR, Hahn WC. Emerging roles for the non-canonical IKKs in cancer. Oncogene. 2011;30(6):631–41.
Wang Y, Lu X, Zhu L, Shen Y, Chengedza S, Feng H, et al. IKK epsilon kinase is crucial for viral G protein-coupled receptor tumorigenesis. Proc Natl Acad Sci USA. 2013;110(27):11139–44.
Howe EN, Cochrane DR, Cittelly DM, Richer JK. miR-200c targets a NF-κB up-regulated TrkB/NTF3 autocrine signaling loop to enhance anoikis sensitivity in triple negative breast cancer. PLoS ONE. 2012;7(11): e49987.
Zhao X, Ayer RE, Davis SL, Ames SJ, Florence B, Torchinsky C, et al. Apoptosis factor EI24/PIG8 is a novel endoplasmic reticulum-localized Bcl-2-binding protein which is associated with suppression of breast cancer invasiveness. Can Res. 2005;65(6):2125–9.
Maik-Rachline G, Seger R. The ERK cascade inhibitors: towards overcoming resistance. Drug Resist Updat. 2016;25:1–12.
Mitra S, Federico L, Zhao W, Dennison J, Sarkar TR, Zhang F, et al. Rab25 acts as an oncogene in luminal B breast cancer and is causally associated with Snail driven EMT. Oncotarget. 2016;7(26):40252–65.
Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers. 2017;9(5):52.
Byerly JH, Port ER, Irie HY. PRKCQ inhibition enhances chemosensitivity of triple-negative breast cancer by regulating Bim. Breast Cancer Res. 2020;22(1):72.
Oberst MD, Beberman SJ, Zhao L, Yin JJ, Ward Y, Kelly K. TDAG51 is an ERK signaling target that opposes ERK-mediated HME16C mammary epithelial cell transformation. BMC Cancer. 2008;8:189.
Glading A, Koziol JA, Krueger J, Ginsberg MH. PEA-15 inhibits tumor cell invasion by binding to extracellular signal-regulated kinase 1/2. Can Res. 2007;67(4):1536–44.
Fiucci G, Ravid D, Reich R, Liscovitch M. Caveolin-1 inhibits anchorage-independent growth, anoikis and invasiveness in MCF-7 human breast cancer cells. Oncogene. 2002;21(15):2365–75.
Owens TW, Valentijn AJ, Upton JP, Keeble J, Zhang L, Lindsay J, et al. Apoptosis commitment and activation of mitochondrial Bax during anoikis is regulated by p38MAPK. Cell Death Differ. 2009;16(11):1551–62.
Yao X, Gray S, Pham T, Delgardo M, Nguyen A, Do S, et al. Downregulation of Bit1 expression promotes growth, anoikis resistance, and transformation of immortalized human bronchial epithelial cells via Erk activation-dependent suppression of E-cadherin. Biochem Biophys Res Commun. 2018;495(1):1240–8.
Sundararaman A, Amirtham U, Rangarajan A. Calcium-oxidant signaling network regulates AMP-activated Protein Kinase (AMPK) Activation upon Matrix Deprivation. J Biol Chem. 2016;291(28):14410–29.
Saha M, Kumar S, Bukhari S, Balaji SA, Kumar P, Hindupur SK, et al. AMPK-Akt double-negative feedback loop in breast cancer cells regulates their adaptation to matrix deprivation. Can Res. 2018;78(6):1497–510.
Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016–23.
Hindupur SK, Balaji SA, Saxena M, Pandey S, Sravan GS, Heda N, et al. Identification of a novel AMPK-PEA15 axis in the anoikis-resistant growth of mammary cells. Breast Cancer Res. 2014;16(4):420.
Whelan KA, Caldwell SA, Shahriari KS, Jackson SR, Franchetti LD, Johannes GJ, et al. Hypoxia suppression of Bim and Bmf blocks anoikis and luminal clearing during mammary morphogenesis. Mol Biol Cell. 2010;21(22):3829–37.
Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410(6824):50–6.
Kochetkova M, Kumar S, McColl SR. Chemokine receptors CXCR4 and CCR7 promote metastasis by preventing anoikis in cancer cells. Cell Death Differ. 2009;16(5):664–73.
Wu X, Zhang X, Yu L, Zhang C, Ye L, Ren D, et al. Zinc finger protein 367 promotes metastasis by inhibiting the Hippo pathway in breast cancer. Oncogene. 2020;39(12):2568–82.
Moison C, Chagraoui J, Caron MC, Gagné JP, Coulombe Y, Poirier GG, et al. Zinc finger protein E4F1 cooperates with PARP-1 and BRG1 to promote DNA double-strand break repair. Proc Natl Acad Sci U S A. 2021;118(11):e2019408118.
Zeng H, Li T, Zhai D, Bi J, Kuang X, Lu S, et al. ZNF367-induced transcriptional activation of KIF15 accelerates the progression of breast cancer. Int J Biol Sci. 2020;16(12):2084–93.
Jain M, Zhang L, Boufraqech M, Liu-Chittenden Y, Bussey K, Demeure MJ, et al. ZNF367 inhibits cancer progression and is targeted by miR-195. PLoS ONE. 2014;9(7): e101423.
Geng Y, Walls KC, Ghosh AP, Akhtar RS, Klocke BJ, Roth KA. Cytoplasmic p53 and activated Bax regulate p53-dependent, transcription-independent neural precursor cell apoptosis. J Histochem Cytochem. 2010;58(3):265–75.
Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70.
Tan BS, Tiong KH, Choo HL, Chung FF, Hii LW, Tan SH, et al. Mutant p53–R273H mediates cancer cell survival and anoikis resistance through AKT-dependent suppression of BCL2-modifying factor (BMF). Cell Death Dis. 2015;6(7): e1826.
Xie TX, Zhou G, Zhao M, Sano D, Jasser SA, Brennan RG, et al. Serine substitution of proline at codon 151 of TP53 confers gain of function activity leading to anoikis resistance and tumor progression of head and neck cancer cells. Laryngoscope. 2013;123(6):1416–23.
Xiao T, Xu Z, Zhou Y, Zhang H, Geng J, Liang Y, et al. Loss of TP53I11 enhances the extracellular matrix-independent survival by promoting activation of AMPK. IUBMB Life. 2019;71(2):183–91.
Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Investig. 2007;117(12):3810–20.
Yuan M, Tomlinson V, Lara R, Holliday D, Chelala C, Harada T, et al. Yes-associated protein (YAP) functions as a tumor suppressor in breast. Cell Death Differ. 2008;15(11):1752–9.
Alsafadi S, Tourpin S, Bessoltane N, Salomé-Desnoulez S, Vassal G, André F, et al. Nuclear localization of the caspase-3-cleaved form of p73 in anoikis. Oncotarget. 2016;7(11):12331–43.
Braun S, Cevatli BS, Assemi C, Janni W, Kentenich CR, Schindlbeck C, et al. Comparative analysis of micrometastasis to the bone marrow and lymph nodes of node-negative breast cancer patients receiving no adjuvant therapy. J Clin Oncol. 2001;19(5):1468–75.
Kao J, Salari K, Bocanegra M, Choi YL, Girard L, Gandhi J, et al. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS ONE. 2009;4(7): e6146.
Brew CT, Aronchik I, Kosco K, McCammon J, Bjeldanes LF, Firestone GL. Indole-3-carbinol inhibits MDA-MB-231 breast cancer cell motility and induces stress fibers and focal adhesion formation by activation of Rho kinase activity. Int J Cancer. 2009;124(10):2294–302.
Arregui C, Pathre P, Lilien J, Balsamo J. The nonreceptor tyrosine kinase fer mediates cross-talk between N-cadherin and beta1-integrins. J Cell Biol. 2000;149(6):1263–74.
He B, Mirza M, Weber GF. An osteopontin splice variant induces anchorage independence in human breast cancer cells. Oncogene. 2006;25(15):2192–202.
Zhong Z, Pannu V, Rosenow M, Stark A, Spetzler D. KIAA0100 modulates cancer cell aggression behavior of MDA-MB-231 through microtubule and heat shock proteins. Cancers. 2018;10(6):180.
Zhang C, Zhu C, Chen H, Li L, Guo L, Jiang W, et al. Kif18A is involved in human breast carcinogenesis. Carcinogenesis. 2010;31(9):1676–84.
Akter KA, Hyodo T, Asano E, Sato N, Mansour MA, Ito S, et al. Erratum to: UBE2S is associated with malignant characteristics of breast cancer cells. Tumour Biol. 2016;37(5):6999.
Khotskaya YB, Beck BH, Hurst DR, Han Z, Xia W, Hung MC, et al. Expression of metastasis suppressor BRMS1 in breast cancer cells results in a marked delay in cellular adhesion to matrix. Mol Carcinog. 2014;53(12):1011–26.
Lee YS, Lim KH, Guo X, Kawaguchi Y, Gao Y, Barrientos T, et al. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Can Res. 2008;68(18):7561–9.
Paredes J, Figueiredo J, Albergaria A, Oliveira P, Carvalho J, Ribeiro AS, et al. Epithelial E- and P-cadherins: role and clinical significance in cancer. Biochem Biophys Acta. 2012;1826(2):297–311.
Cleton-Jansen AM, Callen DF, Seshadri R, Goldup S, McCallum B, Crawford J, et al. Loss of heterozygosity mapping at chromosome arm 16q in 712 breast tumors reveals factors that influence delineation of candidate regions. Can Res. 2001;61(3):1171–7.
Rakha EA, Armour JA, Pinder SE, Paish CE, Ellis IO. High-resolution analysis of 16q22.1 in breast carcinoma using DNA amplifiable probes (multiplex amplifiable probe hybridization technique) and immunohistochemistry. Int J Cancer. 2005;114(5):720–9.
Qi R, Lin J, Chen S, Jiang J, Zhang X, Yao B, et al. Breast cancer prognosis and P-cadherin expression: systematic review and study-level meta-analysis. BMJ Support Palliat Care. 2020;12(e6):e893–905.
Jiang YZ, Ma D, Suo C, Shi J, Xue M, Hu X, et al. Genomic and transcriptomic landscape of triple-negative breast cancers: subtypes and treatment strategies. Cancer Cell. 2019;35(3):428-40.e5.
Heneghan HM, Miller N, Kerin MJ. Circulating microRNAs: promising breast cancer Biomarkers. Breast cancer research : BCR. 2011;13(1):402; author reply 3.
Corcoran C, Rani S, Breslin S, Gogarty M, Ghobrial IM, Crown J, et al. miR-630 targets IGF1R to regulate response to HER-targeting drugs and overall cancer cell progression in HER2 over-expressing breast cancer. Mol Cancer. 2014;13:71.
Gong B, Hu H, Chen J, Cao S, Yu J, Xue J, et al. Caprin-1 is a novel microRNA-223 target for regulating the proliferation and invasion of human breast cancer cells. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2013;67(7):629–36.
Radisky DC. miR-200c at the nexus of epithelial-mesenchymal transition, resistance to apoptosis, and the breast cancer stem cell phenotype. Breast Cancer Res. 2011;13(3):110.
Howe EN, Cochrane DR, Richer JK. Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011;13(2):R45.
Cimino D, De Pittà C, Orso F, Zampini M, Casara S, Penna E, et al. miR148b is a major coordinator of breast cancer progression in a relapse-associated microRNA signature by targeting ITGA5, ROCK1, PIK3CA, NRAS, and CSF1. FASEB J. 2013;27(3):1223–35.
Krutilina R, Sun W, Sethuraman A, Brown M, Seagroves TN, Pfeffer LM, et al. MicroRNA-18a inhibits hypoxia-inducible factor 1α activity and lung metastasis in basal breast cancers. Breast Cancer Res. 2014;16(4):R78.
Lv XB, Jiao Y, Qing Y, Hu H, Cui X, Lin T, et al. miR-124 suppresses multiple steps of breast cancer metastasis by targeting a cohort of pro-metastatic genes in vitro. Chin J Cancer. 2011;30(12):821–30.
Zhang X, Zhang B, Gao J, Wang X, Liu Z. Regulation of the microRNA 200b (miRNA-200b) by transcriptional regulators PEA3 and ELK-1 protein affects expression of Pin1 protein to control anoikis. J Biol Chem. 2013;288(45):32742–52.
Guo J, Luo C, Yang Y, Dong J, Guo Z, Yang J, et al. MiR-491-5p, as a Tumor suppressor, prevents migration and invasion of breast cancer by targeting ZNF-703 to Regulate AKT/mTOR Pathway. Cancer Manag Res. 2021;13:403–13.
Malagobadan S, Ho CS, Nagoor NH. MicroRNA-6744-5p promotes anoikis in breast cancer and directly targets NAT1 enzyme. Cancer Biol Med. 2020;17(1):101–11.
Pei J, Zhang J, Yang X, Wu Z, Sun C, Wang Z, et al. TMED3 promotes cell proliferation and motility in breast cancer and is negatively modulated by miR-188-3p. Cancer Cell Int. 2019;19:75.
Jin L, Wessely O, Marcusson EG, Ivan C, Calin GA, Alahari SK. Prooncogenic factors miR-23b and miR-27b are regulated by Her2/Neu, EGF, and TNF-α in breast cancer. Can Res. 2013;73(9):2884–96.
Tan SM, Kirchner R, Jin J, Hofmann O, McReynolds L, Hide W, et al. Sequencing of captive target transcripts identifies the network of regulated genes and functions of primate-specific miR-522. Cell Rep. 2014;8(4):1225–39.
Yu SJ, Hu JY, Kuang XY, Luo JM, Hou YF, Di GH, et al. MicroRNA-200a promotes anoikis resistance and metastasis by targeting YAP1 in human breast cancer. Clin Cancer Res. 2013;19(6):1389–99.
Taylor MA, Sossey-Alaoui K, Thompson CL, Danielpour D, Schiemann WP. TGF-β upregulates miR-181a expression to promote breast cancer metastasis. J Clin Investig. 2013;123(1):150–63.
Simpson CD, Mawji IA, Anyiwe K, Williams MA, Wang X, Venugopal AL, et al. Inhibition of the sodium potassium adenosine triphosphatase pump sensitizes cancer cells to anoikis and prevents distant tumor formation. Can Res. 2009;69(7):2739–47.
Zandi Z, Kashani B, Bashash D, Poursani EM, Mousavi SA, Chahardoli B, et al. The anticancer effect of the TLR4 inhibition using TAK-242 (resatorvid) either as a single agent or in combination with chemotherapy: A novel therapeutic potential for breast cancer. J Cell Biochem. 2020;121(2):1623–34.
Lauricella M, Ciraolo A, Carlisi D, Vento R, Tesoriere G. SAHA/TRAIL combination induces detachment and anoikis of MDA-MB231 and MCF-7 breast cancer cells. Biochimie. 2012;94(2):287–99.
Kim JB, Yu JH, Ko E, Lee KW, Song AK, Park SY, et al. The alkaloid Berberine inhibits the growth of Anoikis-resistant MCF-7 and MDA-MB-231 breast cancer cell lines by inducing cell cycle arrest. Phytomedicine. 2010;17(6):436–40.
Karalis TT, Heldin P, Vynios DH, Neill T, Buraschi S, Iozzo RV, et al. Tumor-suppressive functions of 4-MU on breast cancer cells of different ER status: regulation of hyaluronan/HAS2/CD44 and specific matrix effectors. Matrix Biol. 2019;78–79:118–38.
Li CL, Huang CW, Ko CJ, Fang SY, Ou-Yang FU, Pan MR, et al. Curcumol suppresses triple-negative breast cancer metastasis by attenuating anoikis resistance via inhibition of Skp2-mediated transcriptional addiction. Anticancer Res. 2020;40(10):5529–38.
Zheng H, Li Y, Wang Y, Zhao H, Zhang J, Chai H, et al. Downregulation of COX-2 and CYP 4A signaling by isoliquiritigenin inhibits human breast cancer metastasis through preventing anoikis resistance, migration and invasion. Toxicol Appl Pharmacol. 2014;280(1):10–20.
Schempp CM, von Schwarzenberg K, Schreiner L, Kubisch R, Müller R, Wagner E, et al. V-ATPase inhibition regulates anoikis resistance and metastasis of cancer cells. Mol Cancer Ther. 2014;13(4):926–37.
Lim GE, Sung JY, Yu S, Kim Y, Shim J, Kim HJ, et al. Pygenic Acid A (PA) Sensitizes Metastatic Breast Cancer Cells to Anoikis and Inhibits Metastasis In Vivo. Int J Mol Sci. 2020;21(22):8444.
Kim JY, Lee N, Kim YJ, Cho Y, An H, Oh E, et al. Disulfiram induces anoikis and suppresses lung colonization in triple-negative breast cancer via calpain activation. Cancer Lett. 2017;386:151–60.
Braig S, Wiedmann RM, Liebl J, Singer M, Kubisch R, Schreiner L, et al. Pretubulysin: a new option for the treatment of metastatic cancer. Cell Death Dis. 2014;5(1): e1001.
Lakshmi S, Renjitha J, S BS, Priya S. Epoxyazadiradione induced apoptosis/anoikis in triple-negative breast cancer cells, MDA-MB-231, by modulating diverse cellular effects. J Biochem Mol Toxicol. 2021;35(6):1–17.
Wang K, Zhu X, Chen Y, Yin Y, Ma T. Tubeimoside V sensitizes human triple negative breast cancer MDA-MB-231 cells to anoikis via regulating caveolin-1-related signaling pathways. Arch Biochem Biophys. 2018;646:10–5.
Khaw-On P, Pompimon W, Banjerdpongchai R. Goniothalamin Induces Necroptosis and Anoikis in Human Invasive Breast Cancer MDA-MB-231 Cells. Int J Mol Sci. 2019;20(16):3953.
Chen IH, Shih HC, Hsieh PW, Chang FR, Wu YC, Wu CC. HPW-RX40 restores anoikis sensitivity of human breast cancer cells by inhibiting integrin/FAK signaling. Toxicol Appl Pharmacol. 2015;289(2):330–40.
Wang K, Zou P, Zhu X, Zhang T. Ziyuglycoside II suppresses the aggressive phenotype of triple negative breast cancer cells through regulating Src/EGFR-dependent ITGB4/FAK signaling. Toxicol In Vitro. 2019;61:104653.
An H, Kim JY, Oh E, Lee N, Cho Y, Seo JH. Salinomycin promotes anoikis and decreases the CD44+/CD24- stem-like population via inhibition of STAT3 activation in MDA-MB-231 Cells. PLoS ONE. 2015;10(11): e0141919.
Cameron HL, Foster WG. Dieldrin promotes resistance to anoikis in breast cancer cells in vitro. Reproductive toxicology (Elmsford, NY). 2008;25(2):256–62.
Bizjak M, Malavašič P, Dolinar K, Pohar J, Pirkmajer S, Pavlin M. Combined treatment with Metformin and 2-deoxy glucose induces detachment of viable MDA-MB-231 breast cancer cells in vitro. Sci Rep. 2017;7(1):1761.
Chang HW, Wang HC, Chen CY, Hung TW, Hou MF, Yuan SS, et al. 5-azacytidine induces anoikis, inhibits mammosphere formation and reduces metalloproteinase 9 activity in MCF-7 human breast cancer cells. Molecules (Basel, Switzerland). 2014;19(3):3149–59.
Muscella A, Calabriso N, Vetrugno C, Fanizzi FP, De Pascali SA, Storelli C, et al. The platinum (II) complex [Pt(O, O’-acac)(γ-acac)(DMS)] alters the intracellular calcium homeostasis in MCF-7 breast cancer cells. Biochem Pharmacol. 2011;81(1):91–103.
Alipour F, Riyahi N, Safaroghli-Azar A, Sari S, Zandi Z, Bashash D. Inhibition of PI3K pathway using BKM120 intensified the chemo-sensitivity of breast cancer cells to arsenic trioxide (ATO). Int J Biochem Cell Biol. 2019;116: 105615.
Videnović M, Mojsin M, Stevanović M, Opsenica I, Srdić-Rajić T, Šolaja B. Benzothiazole carbamates and amides as antiproliferative species. Eur J Med Chem. 2018;157:1096–114.
Engel N, Falodun A, Kühn J, Kragl U, Langer P, Nebe B. Pro-apoptotic and anti-adhesive effects of four African plant extracts on the breast cancer cell line MCF-7. BMC Complement Altern Med. 2014;14:334.
Muscella A, Calabriso N, Vetrugno C, Urso L, Fanizzi FP, De Pascali SA, et al. Sublethal concentrations of the platinum(II) complex [Pt(O, O’-acac)(gamma-acac)(DMS)] alter the motility and induce anoikis in MCF-7 cells. Br J Pharmacol. 2010;160(6):1362–77.
O’Neill S, Porter RK, McNamee N, Martinez VG, O’Driscoll L. 2-Deoxy-D-Glucose inhibits aggressive triple-negative breast cancer cells by targeting glycolysis and the cancer stem cell phenotype. Sci Rep. 2019;9(1):3788.
Xu R, Yan Y, Zheng X, Zhang H, Chen W, Li H, et al. Aspirin suppresses breast cancer metastasis to lung by targeting anoikis resistance. Carcinogenesis. 2022;43(2):104–14.
Chen KY, Lin JA, Yao HY, Hsu AC, Tai YT, Ho BY. Monascin accelerates anoikis in circulating tumor cells and prevents breast cancer metastasis. Oncol Lett. 2020;20(5):166.
Acknowledgements
Not applicable
Funding
This work was supported by National Natural Science Foundation of China (No. 81972643, No. 82172962), Sichuan Science and Technology Project (2021YJ0201, No. 22023), Basic research project of science and technology strategic cooperation and application of Southwest Medical University of Luzhou Municipal People's Government (2019LZXNYDJ19), Luzhou Science and Technology Planning Project (2017LZXNYD-Z04), and Sichuan Provincial College Student Innovation and Entrepreneurship Training Program(S202110632237).
Author information
Authors and Affiliations
Contributions
QLW, ZGX and YLD conceived and designed the study. YLD, XYZ and YJO wrote the manuscript, and prepared the figures. DLZ, QFY, YQ, XJD, WL and ZPY revised the manuscript. All authors read the manuscript and approved the final draft that was submitted.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
No competing interest exists.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dai, Y., Zhang, X., Ou, Y. et al. Anoikis resistance––protagonists of breast cancer cells survive and metastasize after ECM detachment. Cell Commun Signal 21, 190 (2023). https://doi.org/10.1186/s12964-023-01183-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01183-4