
Abstract
The transient receptor potential melastatin subfamily member 2 (TRPM2), a thermo and reactive oxygen species (ROS) sensitive Ca2+-permeable cation channel has a vital role in surviving the cell as well as defending the adaptability of various cell groups during and after oxidative stress. It shows higher expression in several cancers involving breast, pancreatic, prostate, melanoma, leukemia, and neuroblastoma, indicating it raises the survivability of cancerous cells. In various cancers including gastric cancers, and neuroblastoma, TRPM2 is known to conserve viability, and several underlying mechanisms of action have been proposed. Transcription factors are thought to activate TRPM2 channels, which is essential for cell proliferation and survival. In normal physiological conditions with an optimal expression of TRPM2, mitochondrial ROS is produced in optimal amounts while regulation of antioxidant expression is carried on. Depletion of TRPM2 overexpression or activity has been shown to improve ischemia–reperfusion injury in organ levels, reduce tumor growth and/or viability of various malignant cancers like breast, gastric, pancreatic, prostate, head and neck cancers, melanoma, neuroblastoma, T-cell and acute myelogenous leukemia. This updated and comprehensive review also analyzes the mechanisms by which TRPM2-mediated Ca2+ signaling can regulate the growth and survival of different types of cancer cells. Based on the discussion of the available data, it can be concluded that TRPM2 may be a unique therapeutic target in the treatment of several types of cancer.
Video Abstract
Similar content being viewed by others


Introduction
The transient receptor potential melastatin 2 (TRPM2) channel, under the transient receptor potential (TRP) ion channel superfamily, is engaged in a range of physiological and pathophysiological processes in a variety of cells. Genomic study of Drosophila first recognized the trp gene through visual transduction mutation [1, 2]. TRPM2, a decade’s sought-after ion channel, also formerly called TRPC7 and LTRPC-2, a nucleotide-sensing TRP channel enables the influx of Ca2+ and Na+ and increases the cytosolic Ca2+ concentration, [Ca2+]cyt. It also works as a lysosomal Ca2+ release channel [3] involving, under certain situations, cell death. TRPM2 channel is expressed in liver cells [4], blood cells, immunocytes, pancreatic cells, microglia [5,6,7] and brain cells [8]. In addition, extracellular application of oxidants e.g., H2O2 [9,10,11,12], tert-butyl hydroperoxide and dithionite, second messenger arachidonic acid [10], intracellular ADP-ribose (ADPR) [11], and oxidative stress e.g., H2O2 [9,10,11,12], nicotinamide adenine dinucleotide (NAD) [6] can act as an activator of TRPM2 channel gating. The physiological role of TRPM2 is very less understood. TRPM2 channels may be involved in insulin secretion [3, 10, 13,14,15,16,17]; it also may facilitate portions of the responses to tumor necrosis factor alpha (TNF-α), in the cells of the immune system [18], cell motility, cell death [19]. Miller [20] showed that it is related to the amyloid-beta protein toxicity, in the brain. However, the molecular basis of the contribution of the TRPM2 channel to these cellular and pathophysiological processes remains unclear. As the TRPM2 channel undergoes ROS-mediated activation, and the oxidation process is involved in a variety of diseases and clinical problems, the TRPM2 channel may be a possible therapeutic target for those disease states. This review aims to focus on recent findings in TRPM2 Ca2+ permeable channel in IR injury and cancers, and its activation mechanism by ROS in different cell types and its associated implications.
Review methodology
This updated review analyzed the pharmacological studies that included the mechanisms of how oxidative stress modulates TRPM2, Ca2+ signaling mediated by TRPM2, regulation mechanisms in ischemia and in various types of cancer. For this purpose, searches were performed in specialized databases such as Web of Science, Pubmed/MedLine, ScienceDirect, and TRIP Database using the following MeSH terms: “Humans”, “Oxidative Stress”, “TRPM Cation Channels/physiology”, “Calcium/metabolism”, “Cell Survival”, “Humans”, “Molecular Targeted Therapy”,”Oxidative Stress”, “TRPM Cation Channels/genetics”, “TRPM Cation Channels/metabolism”, “Transcriptional Activation”, “Neoplasms/genetics”. The most relevant data were summarized in a table and a figure.
RPM2 channel: a brief overview
Structure and properties
The pharmacology and structural studies of TRPM2 remain less developed. It belongs to the TRPM subfamily, which takes part of the TRPM-homology section (around 700 amino acids) within the N-terminus. The human TRPM2 gene, situated in chromosome 21q22.3, comprises 32 exons and spans about 90 kb [21]. The TRP protein has six putative transmembranes forming the sensor domain by helices S1-S4 [22] along with a pore area within the fifth and sixth transmembrane; besides it gathers in homo- or hetero-tetramers form channels [23]. As it undergoes tetramerization, the putative S5 and S6 segments of the TRPM2 channel can form a central ion-conducting pore. The bacterial KcsA potassium channel has a similar type of ion channel pore structure [24]. The C-terminal end of TRPM2 has a 39% sequence similarity to NUDT9 (Nudix (nucleoside diphosphate linked moiety X-type motif 9). Between the NUDT9 domain (comprising some 300 amino acids) and the Nudix box is the catalytic domain containing 22 amino acids. Eisfeld and Luckhoff [25] suggest that the NUDT9 domain, probably, may provide long-term binding of ADPR which can be important for channel gating. Kuhn and Luckhoff [26] reported that one particular amino acid (Asn-1326) can play a critical role in the binding area for ADPR (adenosine diphosphate (ADP)–ribose) gating of TRPM2. In addition, cytoplasmic N-terminally located TRPM homology regions may be involved for the oligomerization of channels or in regulating transport to the plasma membrane [22]. Although TRPM2 and TRPM8 channels are structurally 42% alike [27], they are highly different in their biological activities. The single-channel characteristics of TRPM2 are distinctive as the channel shows extensive opening times [28, 29]. Like TRPM6/7, TRPM2 is also familiar as a ‘coenzyme’ due to its double actions in the ion channel as well as the C-terminal enzyme domain [30, 31]. However, the proteolytic action of TRPM2’s Nudix box assumes to be tremendously small or the role of the TRPM2 channel is abolished [25].
Physiological functions of TRPM2
The physiological role of TRPM2, although it is expressed in various tissues and first identified in 1998, is not very well understood. However, some propositions are available regarding the physiological function of the TRPM2 channel. It might be involved in insulin secretion [13]; it may arbitrate parts of the reactions to TNF-α, within immune cells [32]. In addition, H2O2-induced TRPM2 channels contribute to alloxan-induced diabetes mellitus [33]. It is also proposed that within the brain it may cause toxicity of amyloid-beta which is a protein related to Alzheimer’s disease [20]. In addition, the channel may be also involved in cell motility and cell death [19]. A recent study revealed that mice having TRPM2- deficiency were tremendously prone to infection with Listeria monocytogenes (Lm), showing an ineffective intrinsic immune response. Therefore, to survive and control the bacterial burden of Lm infection, TRPM2 may play a crucial role [34].
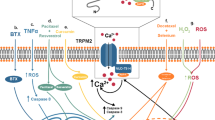
TRPM2 is one of several Ca2+ entry channels in mammalian cells. Ca2+ plays a very essential role as an intracellular messenger. It is a second messenger and an important regulator of cellular metabolism [35,36,37,38,39] (Fig. 1). Cytoplasmic Ca2+ regulates certain key cellular systems. More than one hundred neurotransmitters and hormones, which work on their corresponding receptors and receptor channels transmit their signals by changing intracellular calcium concentration. In cells, Ca2+ does work from minutes to hours to guide gene transcription and cell proliferation. Ca2+ passes via TRPM2 triggered by ROS and may prompt chemokine assembly in monocytes which consequently may exacerbate inflammatory neutrophil penetration [40]. In cancers or other disease conditions, the expression of TRPM2 ion channels along with others is significantly altered causing impaired intracellular calcium homeostasis. While normal intracellular calcium concentration/homeostasis is essential for cellular metabolisms, altered calcium homeostasis may play a critical role in the development and progression of many detrimental diseases [41]. The plasma membrane of mammalian cells possesses different types of Ca2+ entry channels that control the downhill diffusion of Ca2+ entry into cells [42].

Schematic representation of cellular calcium homeostasis and cellular location of TRPM2 channels. Abbreviations: Transient receptor potential melastatin 2 (TRPM2), Adenosine Diphosphate (ADP), phosphates (Pi), Adenosine triphosphate (ATP)
Types of calcium channels according to their gating mechanisms:
-
1)
Voltage-dependent like N, L, T, P, and Q types of calcium channels;
-
2)
Mechanically-gate activated like Stretch activated, non-selective calcium channels;
-
3)
Ligand-gated activated like:
-
i)
External ligands such as neurotransmitters (nicotine acetylcholine receptor)
-
ii)
Intracellular ligands such as IP3, Ca2+, ATP (IP3 receptor or store-operated channels [43].
-
i)
After depletion of Ca2+from the key store of Ca2+ (endoplasmic reticulum) of mammalian cells, a special plasma membrane Ca2+ channel, the CRAC channel is triggered to adjust gradually the extent of calcium in the endoplasmic reticulum. CRAC (Ca2+ release activated Ca2+) channel are the best characterized SOCE channels (store-operated Ca2+ entry channels) with well-studied electrophysiological properties [44]. The putative location of TRPM2 has been indicated in Fig. 1.
Mechanism of activation of TRPM2 channel
Activation mechanism of TRPM2 channel by oxidative stress
Oxidative stress is generally mediated by extreme disclosure of cells to reactive oxygen or nitrogen species, generated by the ischemic attack, radioactivity, seizure, shock, etc. Experience with oxidative stress prompts apoptotic-like late death of neurons as well as decay in cell culturing, facilitated through increased Ca2+, malfunction of mitochondria as well as stimulation of poly[adenosine diphosphate ribose (ADPR)] polymerase (PARP) because of DNA injury [45, 46]. On this matter, one possibly vital calcium influx way can be the stimulation of Na+ and Ca2+ penetrable TRP channels. The cell damage produced by ischemia followed by reperfusion is mainly for superoxide anions [47]. These superoxide anions will produce H2O2 which helps further produce ADPR in the nucleus. H2O2 and superoxide anions are potential members of ROS [10]. ROS, one class of molecules and ions has the potential for the destruction of cells. They have extremely reactive features and are, generally, produced from aerobic metabolism through the mitochondrial electron transport chain resulting from exposure to extracellular mediators and cases, like ionizing emission, cytotoxic medicines, ischemia–reperfusion injury, and hypoxia-reoxygenation [48,49,50]. Nicotinamide adenine dinucleotide (NAD+) can support the TRPM2 opening over ROS stimulus [51]. Presumably, the transformation from NAD+ to ADP-ribose can trigger this opening [11, 52]. But surprisingly, NAD was not active in TRPM2 transduced HEK-293 cells [53]. It has been suggested that, in the case of cardiomyocytes, stimulation of the TRPM2 channel, as well as poly(ADP-ribose) polymerase, is expected to be engaged in oxidative stress-prompted cell death [54]. In cancer, the production of ROS by mitochondria promotes cellular oncogenesis by shaping signaling pathways and specific transcription factors [55]. TRPM2 maintains the bioenergy of cancer cells by maintaining mitochondrial activity and increased ROS production [38, 56]. TRPM2 inhibition decreases ROS production by modulating Nrf2, decreasing NADH and NADPH production, thus causing cancer cell death [57]. Recent studies have also shown that TRPM2 preserves cell viability in the case of other non-tumor cells, protecting them from ischemia–reperfusion injury [58, 59]. According to recent studies it is evident that TRPM2 plays a fundamental role for cardiac myocyte bioenergetics, limits oxidative stress, preserves mitochondrial function and protects the heart (as well as other tissues) from I/R insult. On the other hand, acting through similar mechanisms, TRPM2 is an important player for reducing tumour growth and the survival of cancer cells, also protecting cells from the toxicity of some anticancer drugs (i.e. doxorubicin) [60]. Therefore, by reducing mitochondrial dysfunctions, decreasing free radicals production, TRPM2 channels can protect cardiomyocytes and the heart from I/R injuries [59]. Glutamine consumption is substantially increased in many types of cancer compared to other amino acids, representing a characteristic of malignancy. Glutaminolysis is an alternative energy source for cancer cells and it is a key nutrient for several metabolic processes leading to: ATP generation, redox homeostasis, intracellular antioxidant stock maintenance, and macromolecular synthesis. Regulation of glutaminolysis is determined by oncogenic signals [61]. Recent studies have shown that TRPM2 inhibition decreases glutamine production, thus decreasing antioxidant cofactors and the antioxidant response, increasing cancer cell death and reducing tumor growth [7, 57].
In neuronal cells, TRPM2 channels can be associated with neuronal destruction triggered by means of oxidants, amyloid β-peptide along with tumor necrosis element alpha. Activation of the TRPM2 channel can be influenced by different types of endogenous factors or proteins. The opening of the TRPM2 channel in response to oxidative stress may depend upon the stimulation of DNA restoration enzyme poly (ADP-ribose) polymerase [12]. They also hypothesized that PARP enzyme action may be a crucial element of the pathway relating to oxidative stress with TRPM2 instigation. Zhang, Tong et al. [62] reported that TRPM2 can be quickly tyrosine phosphorylated after the stimulation with H2O2 or TNF-α and phosphorylation is of paramount importance for the activation of the channel; the protein tyrosine phosphatase – L1 (PTPL1) also is associated with the modulation of TRPM2 activation. TRPM2 can be a target for dephosphorylation and inactivation by PTPL1. The molecular mechanism that may influence tyrosine phosphorylation of TRPM2 is not clearly understood yet. Presumably, the pathway may include phosphorylation of tyrosine in the NUDT9-H domain of TRPM2, phosphorylation of calmodulin-binding sites, or phosphorylation of sites that may influence the tertiary structure of TRPM2. Superoxide anions and H2O2 may trigger cellular death through various mechanisms. H2O2, probably, does not activate MAPK signaling and superoxide anions-induced apoptosis is dependent on JNK activity. H2O2 and/or other ROS can release Ca2+ from the intracellular Ca2+ pools and activate suppressors of cytokine signaling (SOCs). Also, during oxidative stress, H2O2 may enter the cytosol by an unidentified mechanism. Using the Fenton reaction, altogether H2O2 and Fe3+ can generate hydroxyl radical (OH–) within the cytosol of cells. TRPM2, also, maybe straightly stimulated by H2O2 and OH– radicals [63]. Interestingly, in HEK293/hTRPM2 cells, the TRPM2 channels have been revealed to be engaged in H2O2- activated cell death in Ca2+ independent mechanism [64].
Attachment of ligand to G protein-linked receptors might result in ADPR production. Receptor stimulation also increases within the intracellular Ca2+ concentration through IP3- mediated Ca2+ transportation from stores. The arrival of Ca2+ into the cell by TRPM2 delivers a positive response augmentation of TRPM2 stimulation. In a similar mechanism connected to that followed by H2O2, the pro-diabetic medication alloxan, [25] leads to TRPM2 triggering. Moreover, the nitric oxide synthase (NOS) enzyme is stimulated by diacylglycerol (DAG) through arachidonic acid. Nitric oxide (NO) radicals can be derived from L-arginine through NOS. Furthermore, NO may trigger TRPM2 channels [63, 65]. By applying the activation mechanism, TRPM2 can be a potential candidate for gene therapy. For a detailed activation mechanism of TRPM2 during oxidative stress, readers are requested to read through some of the recent reviews indicated here [66,67,68]. It has been reported that in glioblastoma cells, cell death by Ca2+ elevation after H2O2 usage could be facilitated by the insertion of TRPM2 into A172 cells [69].
ADPR role in regulation of the TRPM2 channel
The inhibitory function of a small unite variant of TRPM2 on its extended, pore-creating isoform has been stated for TRPM2 [70, 71]. An important regulatory mechanism has been reported through the particular stimulation of TRPM2 channels by intracellular ADP-ribose (ADPR). It has been recommended to be the prime gating mechanism of TRPM2 [71, 72]. TRPM2 channel action is controlled by various cytosolic aspects, involving cyclic ADPR (cADPR), nicotinamide adenine dinucleotide phosphate (NAADP), Ca2+, calmodulin (CaM) as well as adenosine monophosphate (AMP) [73]. In addition, currents of TRPM2 are disabled when intracellular Ca2+ content decreases under 100 nM compared to extracellular Ca2+ content [73]. TRPM2 is triggered by the formation of ADPR in response to oxidative stress, which attaches to the C-terminus of TRPM2, leading to open the channel [74]. ADP-ribose polymers, the source of ADPR, are combined by poly-ADP–ribose polymerase (PARP) along with hydrolyzed through poly-ADP–ribose glycohydrolase. The breaking of NAD into nicotinamide and ADP-ribose may be catalyzed by PARP enzymes after binding to oxidatively-impaired DNA [75]. PARP-reliant pathway leading to TRPM2 stimulation has been established by the usage of PARP inhibitors and these were capable to curb the H2O2-induced TRPM2 triggering [12]. In addition to that nuclear pathway, ROS can stimulate a mitochondrial pathway which consequences the assembly of ADP–ribose in mitochondria as well as in the relief of ADP–ribose to the cytosol, wherever it plays as a second messenger contributing to TRPM2 gating [52]. After gating by ADP-ribose, Ca2+ enters into the cells via TRPM2 which may have effective response augmentation of TRPM2 stimulation. Additionally, the triggering of TRPM2 through ADP-ribose is accelerated at elevated [Ca2+]i indicating a positive feedback regulation of TRPM2 [76]. Other recent studies suggest that activation of TRPM2 may cause cell damage and it is expected to be engaged in various signaling pathways which may result in cell death responding to oxidative stress. Cytosolic Ca2+ enhances TRPM2 gating or opening by ADPR. ADPR, probably the most potent physiological activator of TRPM2, and Ca2+ in a concerted way may act as an important messenger system mediating Ca2+ influx [77, 78]. After attaching the intracellular ADPR to the NUDT9-H domain at the C-terminal, the TRPM2 channel may be activated [72]. It has been reported that endogenous ADPR concentrations in leukocytes are substantially high for activating TRPM2 when increased intracellular Ca2+ concentration, [Ca2+]i is present, but perhaps not in resting [Ca2+] [25].
Effect of hypoxia/anoxia and reoxygenation on TRPM2 channel
Several researchers proposed that oxidative stress is increased by revealing cells or tissues to hypoxia. Anoxic incubation of rat liver mitochondria showed decreased free [Ca2+] in the mitochondrial matrix, probably, implying that during anoxia Ca2+ may be released into the cytoplasm of the cells. The mitochondrial respiratory chains produce most ROS in cells [79]. The level of electron movement by respiratory chain complexes controls significantly the Mitochondrial ROS manufacturing. Currently, it has been proved that in hypoxic situations, the mitochondrial respiratory chain can produce nitric oxide (NO), which can give rise to additional reactive nitrogen species [80]. In acute insults e.g., hypoxic ischemia, it has been recommended that the subsequent cell fate together with necrosis, apoptosis, or survival rely on an intracellular “Ca2+ setpoint” [81]. In hypoxia, TRPM2 and TRPM7 altogether have a crucial function in neuronic cell death [82] proposing that Ca2+ entered into cells through TRPM2 or TRPM7 caused the toxicity; TRPM2 was modulated by intracellular free radicals and conducted toxic levels of Ca2+ [47].
TRPM2 in ischemic reperfusion injury
Ischemic reperfusion mainly occurs in the organ when normal blood flow in that organ suddenly or slowly stops, and after some periods, blood flow is restored. Temporary interruption of blood flow causing organ ischemia is a common occurrence during various surgical procedures. Ischemic stroke is caused by interruption of cerebral blood flow due to an obstruction and the main risk factors for ischemic, hemorrhagic and transient stroke are due to the sedentary lifestyle that generates overweight, the chronic consumption of alcohol or narcotics (cocaine, methamphetamines). Pathologies that favor the occurrence of transient ischemic, ischemic or hemorrhagic accidents are represented by heart diseases (heart failure, congenital heart malformations, heart infections, heart rhythm disorders), obstructive sleep apnea syndrome [83]. Recent studies showed the involvement of TRPM2 in pathogenesis factors of ischemic stroke as follows: by reducing oxidative stress in cardiomyocytes, TRPM2 reduces inflammation and stimulates the remodeling of the atria; it models endothelial dysfunction by deregulating the influx of calcium ions, mediates the damaging effects of ROS on the endothelium and favors hypertension; induces the death of pancreatic β cells that secrete insulin, thus promoting the development of diabetes; promotes the aggregation of platelets and favors the appearance of vascular thrombus [84,85,86]. In the case of hepatic ischemia–reperfusion (IR) injury, it may be considered a clinically important pathological disorder complicating liver surgery and transplantation. IR can be classified as warm IR injury as well as cold storing reperfusion injury. The hepatic surgical procedure, liver replacement, or certain kinds of liver injury can have clinical relevance with warm IR. Besides, at the time of organ preservation, cold storing reperfusion injury can occur, before liver transplantation [87]. It has been considered that long periods of ischemia may cause a drop in intracellular ATP levels, slow down active transport, and cause membrane depolarization [88]. After re-establishing the flow of oxygen and blood, reperfusion increases the damage induced by ischemia [89, 90]. Cellular cytoplasmic and mitochondrial Ca2+ concentrations have shown to be raised instantly following the onset of reperfusion [91]. Elevated stages of cytoplasmic Ca2+ have involvement in cell injury as well as death by triggering a series of Ca2+-dependent enzymes, also involving various proteases and phospholipases. Concurrently mitochondrial Ca2+ overfilling elicits a change in CsA-sensitive mitochondrial permeability transition pores (PTP) causing mitochondrial dysfunction and helping the formation of apoptosomes. In the liver, naturally occurring acidosis has been shown to delay the onset of necrotic cell death; but reperfusion normalizes the intracellular pH and the defense of acidosis vanishes. It has been suggested that intracellular acidosis also inhibits TRPM2 (pKa 6.7) [92]. In some cancerous cell lines, some evidence also suggests that altered expression of TRPM2, including some other TRP proteins, may have roles in the progression of metastatic liver cancers and hepatocellular carcinoma [43]. Overall, this information provides sufficient evidence that intracellular Ca2+ and TRPM2 may play a significant role in pathological conditions of the liver and other cell types. Furthermore, pharmacological inhibitors or genetically depletion of TRPM2 channels demonstrated significant protective effects in kidney I/R injury [93], cardiac I/R injury [58], and neuronal I/R injury [94].
TRPM2 in different types of cancer
TRPM2 in neuroblastoma
Neuroblastoma is an embryonic tumor derived from the sympathetic nervous system that occurs at the level of special nerve cells called neuroblasts [95]. Normally, these immature nerve cells transform into functional mature cells. In the case of neuroblastoma, the neuroblasts do not mature normally, but turn into cancer cells [96, 97]. TRPM2 has been revealed to cause neuroblastoma proliferation which is a non-CNS tumor of childhood as well as chemotherapy sensitivity. The leading negative small splice variant TRPM2-S first inhibited TRPM2 in expression in the cells of neuroblastoma. The inhibited TRPM2 concluded a considerably amplified vulnerability to death of the cell, prompted through low content (50–100 μM) of H2O2 [98] as well as doxorubicin [38, 71]. Mouse xenografts, using human neuroblastoma cells with expression of TRPM2-L or TRPM2-S proved the capability of TRPM2 to augment the progression of neuroblastoma tumors [71]. In neuroblastoma cells where TRPM2 was depleted, tumor development in xenografts considerably declined as well as the sensitivity of doxorubicin increased.
TRPM2 in triple-negative breast cancer
Triple-negative breast cancer is extremely threatening, having the worst consequence, among the three main molecular types of breast cancer. However, therapy also is unsuccessful in a considerable proportion of patients suffering from estrogen-receptor-positive breast cancer, the most common class of breast cancer. 2-aminoethoxydiphenyl borate (2-APB) is known to act as a general inhibitor of several plasma membranes along with organellar ion channels involving TRPM2 [99]. TRPM2 displayed a defensive action in human breast adenocarcinoma cell lines to minimize DNA destruction, where cell proliferation declined and DNA damage climbed up significantly through pharmacological cessation of TRPM2 including 2-APB or TRPM2 mRNA silencing [100]. In the case of both triple-negative as well as estrogen-receptor-positive breast cancer, the inhibition of TRPM2 caused the rise of DNA destruction and cytotoxicity, like neuroblastoma [101]. TRPM2 was situated within the nucleus of breast adenocarcinoma but not limited to that place; about 40–45% of TRPM2 was in the nucleus while the remaining part was in subcellular sections involving the cytoplasm. The mechanisms of action of TRPM2 within the nucleus were assumed to facilitate DNA restoration through nuclear TRPM2 or elevation of the influx of nuclear calcium which was required to be investigated again. ROS content was not observed; however, the elevated oxidative stress content noticed within TRPM2-depleted neuroblastoma cells recommends the possibility to be a probable mechanism for an explanation of elevated DNA damage in breast cancer after the inhibition of TRPM2. On the other hand, within noncancerous breast epithelial cells (MCF-10A), TRPM2 was not located in the nucleus or TRPM2 blockage was detected to have a role in proliferation. These findings recommend that pointing of TRPM2 might be a synergetic method to improve the medical care of chemotherapy-resistant patients with breast cancer, resembling that proposed in neuroblastoma. Other TRPM channels are known to have a function in the case of breast cancer multiplying, movement, and invasion involving TRPM7 and TRPM8 [38, 102]. In what way do TRPM channels interpose their distinct properties and also whether their actions overlay or combine or TRPM2 are regions for imminent investigation.
TRPM2 in lung cancer
Lung cancer results from the uncontrolled growth of abnormal cells in the lungs that do not perform the function of normal lung cells. TRPM2 is highly expressed in lung cancer [38, 103]. In non-small cell lung cancer (NSCLC), TRPM2-AS a long non-coding RNA, antisense transcript of TRPM2 was observed to be overexpressed. Besides, greater expression levels are connected with higher tumor size, progressive TNM stage, as well as reduced patient survival [104]. Cell multiplication and amplified apoptosis were considerably lowered after silencing of TRPM2-AS with siRNA (small interfering RNA). The advanced investigation will be essential to recognize the function of lncRNAs (long non-coding RNAs) including TRPM2 in cell multiplication and survival of patients as well as the consequence of expression and role of TRPM2.
TRPM2 in digestive cancers
Oral malignant tumor or oral cancer appears as a lesion on the oral mucosa, and is caused by the division and chaotic development of cells; it can develop in any oro-maxillo-facial area, but it appears most often in the area of the tongue and floor of the mouth. In human tongue carcinoma specimens and cell lines, TRPM2 expression has been reported to be increased [105]. Treatment through 0.5 or 1 mM H2O2 amplified apoptosis in SCC9 cells of tongue carcinoma. Moreover, the breakdown of TRPM2 with siRNA augmented apoptosis, lowered survival, and hindered the movement of SCC9 cells. The subcellular location of TRPM2 was not similar in cancerous and non-cancerous cells as a considerable extent of TRPM2 protein is located in the cancer cell’s nucleus. Although in TRPM2 KO cells the processes of cell death were not discovered fully, it was not dependent on the p53-p21 pathway. The outcome is that TRPM2 has a role in the survival and movement of SCC cancer cells along with in head and neck cancers, it can be a possible therapeutic object [38, 105].
Gastric cancer begins when cancer cells form in the inner lining of the stomach. These cancer cells can grow into a tumor, and cancer usually develops slowly over several years. TRPM2 expression within tumors has a negative correlation with the overall survival of patients suffering from gastric cancer. After down-regulation of TRPM2 with shRNA in AGS and MKN-45, two gastric cancer cell lines, the cells developed slowly, and the proportion of apoptotic cells raised [38, 105]. Mitochondrial role expressed through oxygen intake amounts and ATP assembly was considerably declined in TRPM2 diminished cells as well as COX 4.1 and 4.2 expression and BNIP3 were lowered, which was described in neuroblastoma [71]. Autophagy was also declined, with a lowered amount of autophagy related genes (ATGs) involving ATG3, ATG5, ATG6, ATG7, and ATG12 and reduced transformation of LC3-I to LC3-II. Decreased autophagy led to the gathering of impaired mitochondria, lowered cellular bioenergetics, and augmented ROS, causing death of the cell. TRPM2 controlled autophagy by an mTOR (mammalian target of rapamycin) independently but JNK (Jun N-terminal Kinase) signaling reliant pathway, facilitated through regulation of ATGs, BNIP3 as well as JNK stimulation. Apoptotic properties of together paclitaxel and doxorubicin were higher in TRPM2 exhausted cells, indicating that TRPM2 conserves the survival of cells where inhibition raises sensitivities of chemotherapy and proposing this as a therapeutic attitude to increase the cell death of gastric tumors.
TRPM2 expression has a considerable role in gastric cancer cells’ bioenergetics and survival, according to confirmation from current numerous investigations [38, 66, 106, 107]. Using molecular and functional assays, one study has demonstrated that downregulated TRPM2 significantly prevents the movement and invasion capacities of gastric cancer cells, including a significant decline in the expression of metastatic markers. Besides, reduced Akt (protein kinase B) and augmented PTEN (phosphatase tensin homologue) actions were connected with the consequences. Moreover, silencing of TRPM2 concluded the deregulated metastatic markers and lost the tumor growing capacity of AGS gastric cancer cells within NOD/SCID mice. Altogether, the known outcomes offer convincing information on the vital role of TRPM2 to modulate the gastric cancer cell invasion presumably by monitoring the PTEN/Akt pathway [108]. In a recent study, Almasi et al. demonstrated that the expression of TRPM2 might be associated with gastric cancer [106]. By authors, two shRNA (short hairpin RNAs) were administered against TRPM2 to lower the expression and role in gastric cancer, AGS, and MKN45 [109]. Outcomes proved that TRPM2 is practically expressed like a plasma membrane ion channel which is penetrable to Ca2+ in gastric cancer cells along with its impediment lowered cell bioenergetics, inhibited cell invasion, and declined cell survival. Further, these consequences were established in vivo through a SCID mouse model, and the decline of TRPM2 was directed to a decreased growth of tumor. The authors recognized that to facilitate gastric cancer survival TRPM2 functions through JNK-dependent as well as mTOR-independent pathways of autophagy [25, 109]. Later, Almasi et al. stated that the efficacy of chemotherapy drugs, paclitaxel and doxorubicin, is increased by the inhibition of TRPM2 which proposes that inhibition of TRPM2 in combination with established chemotherapeutics might be an effective approach to treat gastric cancer [109]. In addition, the authors stated that there is a correlation with the lower overall survival rate of patients particularly in late/advanced phases thus demonstrating the probability of possible function of TRPM2 as a prognostic biomarker for the late phase of gastric cancer [38, 109]. Likewise, further investigations have reported that elevated expression of TRPM5 was related to lower survival in patients suffering from gastric cancer [110]. However, additional research are required to confirm the significance of TRPM2 and TRPM5 in the survival and clinical outcomes of patients with gastric cancer.
TRPM2 in prostate cancer
TRPM2 plays important role in the proliferation of prostate cancer cells [38, 111]. After depletion of TRPM2 with siRNA, the progress of prostate cancer, without non-cancerous cells, was lowered. Within non-cancerous cells, TRPM2 was located in the plasma membrane as well as in the cytoplasm, without in the nucleus. However, a major quantity of TRPM2 was located in the nucleus and the non-nuclear fraction of prostate cancer cells. The role of TRPM2 in the nucleus of cancer cells is unknown. These outcomes recommend that the reduced TRPM2 can be a therapeutic way to regulate prostate cancer development.
TRPM2 in leukemia
In Jurkat cells firmly explicitly empty vector or Bcl-2, TRPM2 inhibition with N-(p- amylcinnamoyl) anthranilic acid (ACA) subsequently irradiation reduced phosphorylation of CAMKII along with closed radiation-prompted phosphorylation-dependent deactivation of cdc2 [38, 102]. Altogether ACA and clotrimazole, the nonspecific TRPM2 inhibitors, elevated cell death. Besides, TRPM2 breakdown considerably reduced the number of cells blocked in G2/M and declined viability. This finding proposes that irradiation excites Ca2+ entrance by TRPM2 that is excessive in Bcl-2 up-regulated T Cell leukemia cells, also donates to deactivation of G2/M cell cycle arrest, cdc2, and cell survival. Also, TRPM2 impediment discharge cells from G2/M arrest, leading to cell death. Those results recommend that the inhibited TRPM2 can be a therapeutic attitude to raise sensitivity within T-cell leukemia towards radiation. The expression pattern of TRPM2 ion channels in various cell lines is shorted in Table 1.
Potential pharmacological inhibitors of TRPM2 channel
TRPM2 channel is potentially regulated through a variety of factors, such as [Ca2+]cyt, H2O2, cADPR (Cyclic adenosine 5′-diphosphate ribose), NAADP (Nicotinic acid adenine dinucleotide 2’-phosphate), and extra- and intracellular pH of the cells [19, 92]. It has been suggested the activation of poly (ADPR) polymerase causes the exposure of TRPM2 channels through H2O2 [12]. Poly (ADPR) polymerase is a universally expressed enzyme which catalyzes the break of NAD+ into nicotinamide and ADPR. Besides, TRPM2 action is escalated at increased [Ca2+]i [76]. The range of active pharmacological modulators of TRPM2 is limited to the TRPM2 channel. Some of the pharmacological inhibitors identified so far are not selective blockers. A small amount of TRPM2 channel blockers have been recognized along with seemed to be cell-specific. For example, N-(p-amylcinnomoyl) anthranilic acid (ACA), the IP3R (inositol 1,4,5-trisphosphate receptor) inhibitor 2-aminoethoxydiphenyl borate or PLC inhibitor flufenamic acid (FFA) did not block the ADPR-induced Ca2+ influx within hippocampal cells of the rat. However, ACA and FFA curbed the entrance of Ca2+ in rat preliminary striatal cells [120]. Either ADPR or H2O2 can gate the TRPM2 channels in neuronal cells. It suggests that the accurate connection between TRPM2 channel stimulation as well as cell death now remains undetermined [120]. Determination of the ion channel structures is important for understanding the mechanisms of gating, ion permeation, and selectivity [99]. Proper structural knowledge of the channel can also play an important role in the development of selective inhibitors. Recent studies suggest that flufenamic acid [121], clotrimazole, econazole [12], ACA [15, 122], 2-APB [99], antioxidants, glycohydrolase inhibitors, PARP inhibitors, chlorpromazine AMP, 8 Br ADPR can act as non-selective inhibitors of TRPM2 channel. It has been known that intracellular acidosis may inhibit TRPM2 (pKa 6.7) [66, 92, 112, 113]. To know the details of the effects of TRPM2 channel inhibitors in mammalian cells, readers are requested to some recent comprehensive reviews [66, 123, 124]. Moreover, as CD38 signals to TRPM2 via ADPR [125] and arouses Ca2+ influx through TRPM2, therefore, another possibility of pharmacologic inhibition of TRPM2 is the inhibition of receptor-mediated activation of CD38 [25].
Conclusion and future perspectives
A lot of work has been done on the activation of TRPM2 channels by ROS for different cell types including neuronal cells, liver cells, pancreatic, and cardiac cells. Now it is well-known that TRPM2 shows a crucial function in Ca2+ entry into different cell types subjected to ischemic reperfusion injury. Inhibitors of Ca2+ entry through TRPM2 have been shown to prevent IR injury in case of heart, liver, brain and/or kidney. Because it has been recognized that ROS-initiated stimulation of TRPM2 takes part together with acute and chronic liver injury, considerable additional investigation is required to explain the mechanisms engaged as well as the situations under which pharmacological hindrance of TRPM2 can be a selective clinical approach to diminish ROS-initiated liver injury. In the case of cancers, the expression pattern of TRPM2 in various kinds of cancer suggests that TRPM2 can stimulate tumor survival. Inhibited TRPM2 has been known to increase cell death along with augmenting sensitivity to chemotherapeutic agents including doxorubicin in several malignancies with neuroblastoma [71, 98], gastric cancer [109], T cell leukemia [102], triple-negative breast cancer cell lines [101]. The prevalence of statistics in cancer models confirms the idea that TRPM2 expression as well as its role has an essential part in conserving the viability of cancer cells- which provides an important therapeutic opportunity. Further research on the roles of TRPM2 channels in cancer and the evaluation of the pharmacological inhibition of TRPM2 in cancers may conceivably lead to improved and selective treatment regimens in the near future.
Availability of data and materials
Not Applicable.
References
Montell C, Rubin GM. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron. 1989;2(4):1313–23. https://doi.org/10.1016/0896-6273(89)90069-X.
Minke B. The history of the Drosophila TRP channel: the birth of a new channel superfamily. J Neurogenet. 2010;24(4):216–33. https://doi.org/10.3109/01677063.2010.514369.
Lange I, Yamamoto S, Partida-Sanchez S, Mori Y, Fleig A, Penner R. TRPM2 functions as a lysosomal Ca2+-release channel in beta cells. Sci Signal. 2009;2(71):ra23. https://doi.org/10.1126/scisignal.2000278.
Perraud A-L, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411(6837):595–9. http://www.nature.com/nature/journal/v411/n6837/suppinfo/411595a0_S1.html.
Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, et al. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol. 2004;286(1):C129–37. https://doi.org/10.1152/ajpcell.00331.2003.
Perraud AL, Schmitz C, Scharenberg AM. TRPM2 Ca2+ permeable cation channels: from gene to biological function. Cell Calcium. 2003;33(5–6):519–31. https://doi.org/10.1016/S0143-41600300057-5.
Yıldızhan K, Nazıroğlu M. Glutathione depletion and Parkinsonian neurotoxin MPP(+)-induced TRPM2 channel activation play central roles in oxidative cytotoxicity and inflammation in microglia. Mol Neurobiol. 2020;57(8):3508–25. https://doi.org/10.1007/s12035-020-01974-7.
Ishii M, Shimizu S, Hara Y, Hagiwara T, Miyazaki A, Mori Y, et al. Intracellular-produced hydroxyl radical mediates H2O2-induced Ca2+ influx and cell death in rat beta-cell line RIN-5F. Cell Calcium. 2006;39(6):487–94. https://doi.org/10.1016/j.ceca.2006.01.013.
Hecquet CM, Ahmmed GU, Vogel SM, Malik AB. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ Res. 2008;102(3):347–55. https://doi.org/10.1161/CIRCRESAHA.107.160176.
Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9(1):163–73.
Kolisek M, Beck A, Fleig A, Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell. 2005;18(1):61–9. https://doi.org/10.1016/j.molcel.2005.02.033.
Fonfria E, Marshall IC, Benham CD, Boyfield I, Brown JD, Hill K, et al. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br J Pharmacol. 2004;143(1):186–92. https://doi.org/10.1038/sj.bjp.0705914.
Togashi K, Hara Y, Tominaga T, Higashi T, Konishi Y, Mori Y, et al. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006;25(9):1804–15. https://doi.org/10.1038/sj.emboj.7601083.
Qian F, Huang P, Ma L, Kuznetsov A, Tamarina N, Philipson LH. TRP genes: candidates for nonselective cation channels and store-operated channels in insulin-secreting cells. Diabetes. 2002;51(Suppl 1):S183–9.
Bari MR, Akbar S, Eweida M, Kuhn FJ, Gustafsson AJ, Luckhoff A, et al. H2O2-induced Ca2+ influx and its inhibition by N-(p-amylcinnamoyl) anthranilic acid in the beta-cells: involvement of TRPM2 channels. J Cell Mol Med. 2009;13(9B):3260–7. https://doi.org/10.1111/j.1582-4934.2009.00737.x.
Inamura K, Sano Y, Mochizuki S, Yokoi H, Miyake A, Nozawa K, et al. Response to ADP-ribose by activation of TRPM2 in the CRI-G1 insulinoma cell line. J Membr Biol. 2003;191(3):201–7. https://doi.org/10.1007/s00232-002-1057-x.
Du J, Xie J, Yue L. Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc Natl Acad Sci U S A. 2009;106(17):7239–44. https://doi.org/10.1073/pnas.0811725106.
Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med. 2008;14(7):738–47. http://www.nature.com/nm/journal/v14/n7/suppinfo/nm1758_S1.html.
Sumoza-Toledo A, Penner R. TRPM2: a multifunctional ion channel for calcium signalling. J Physiol. 2011;589(Pt 7):1515–25. https://doi.org/10.1113/jphysiol.2010.201855.
Miller BA. The role of TRP channels in oxidative stress-induced cell death. J Membr Biol. 2006;209(1):31–41. https://doi.org/10.1007/s00232-005-0839-3.
Nagamine K, Kudoh J, Minoshima S, Kawasaki K, Asakawa S, Ito F, et al. Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics. 1998;54(1):124–31. https://doi.org/10.1006/geno.1998.5551.
Gaudet R. Structural Insights into the Function of TRP Channels. 2007. doi: NBK5266 [bookaccession].
Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A. 2002;99(11):7461–6. https://doi.org/10.1073/pnas.102596199.
Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280(5360):69–77.
Eisfeld J, Luckhoff A. Trpm2. Handb Exp Pharmacol. 2007;179:237–52. https://doi.org/10.1007/978-3-540-34891-7_14.
Kuhn FJ, Luckhoff A. Sites of the NUDT9-H domain critical for ADP-ribose activation of the cation channel TRPM2. J Biol Chem. 2004;279(45):46431–7. https://doi.org/10.1074/jbc.M407263200.
Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, et al. A TRP channel that senses cold stimuli and menthol. Cell. 2002;108(5):705–15.
Heiner I, Eisfeld J, Halaszovich CR, Wehage E, Jungling E, Zitt C, et al. Expression profile of the transient receptor potential (TRP) family in neutrophil granulocytes: evidence for currents through long TRP channel 2 induced by ADP-ribose and NAD. Biochem J. 2003;371(Pt 3):1045–53. https://doi.org/10.1042/BJ20021975.
Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, et al. Immunocyte Ca2+ influx system mediated by LTRPC2. Science. 2001;293(5533):1327–30. https://doi.org/10.1126/science.1062473.
Scharenberg AM. TRPM2 and TRPM7: channel/enzyme fusions to generate novel intracellular sensors. Pflugers Arch. 2005;451(1):220–7. https://doi.org/10.1007/s00424-005-1444-0.
Cahalan MD. Cell biology. Channels as enzymes. Nature. 2001;411(6837):542–3. https://doi.org/10.1038/35079231.
Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med. 2008;14(7):738–47. https://doi.org/10.1038/nm1758.
Herson PS, Ashford ML. Activation of a novel non-selective cation channel by alloxan and H2O2 in the rat insulin-secreting cell line CRI-G1. J Physiol. 1997;501(Pt 1):59–66.
Knowles H, Heizer JW, Li Y, Chapman K, Ogden CA, Andreasen K, et al. Transient Receptor Potential Melastatin 2 (TRPM2) ion channel is required for innate immunity against Listeria monocytogenes. Proc Natl Acad Sci U S A. 2011;108(28):11578–83. https://doi.org/10.1073/pnas.1010678108.
Wang Y, Tao A, Vaeth M, Feske S. Calcium regulation of T cell metabolism. Curr Opin Physiol. 2020;17:207–23. https://doi.org/10.1016/j.cophys.2020.07.016.
Vlahakis A, Lopez Muniozguren N, Powers T. Calcium channel regulator Mid1 links TORC2-mediated changes in mitochondrial respiration to autophagy. J Cell Biol. 2016;215(6):779–88. https://doi.org/10.1083/jcb.201605030.
Viola HM, Adams AM, Davies SMK, Fletcher S, Filipovska A, Hool LC. Impaired functional communication between the L-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart. Proc Natl Acad Sci. 2014;111(28):E2905–14. https://doi.org/10.1073/pnas.1402544111.
Miller BA. TRPM2 in cancer. Cell Calcium. 2019;80:8–17. https://doi.org/10.1016/j.ceca.2019.03.002.
Rimessi A, Pedriali G, Vezzani B, Tarocco A, Marchi S, Wieckowski MR, et al. Interorganellar calcium signaling in the regulation of cell metabolism: a cancer perspective. Semin Cell Dev Biol. 2020;98:167–80. https://doi.org/10.1016/j.semcdb.2019.05.015.
Yamamoto S, Takahashi N, Mori Y. Chemical physiology of oxidative stress-activated TRPM2 and TRPC5 channels. Prog Biophys Mol Biol. 2010;103(1):18–27. https://doi.org/10.1016/j.pbiomolbio.2010.05.005.
Bagur R, Hajnóczky G. Intracellular Ca(2+) sensing: its role in calcium homeostasis and signaling. Mol Cell. 2017;66(6):780–8. https://doi.org/10.1016/j.molcel.2017.05.028.
Brini M, Carafoli E. Calcium signalling: a historical account, recent developments and future perspectives. Cell Mol Life Sci. 2000;57(3):354–70.
Rychkov GY, Barritt GJ. Expression and function of TRP channels in liver cells. Adv Exp Med Biol. 2011;704:667–86. https://doi.org/10.1007/978-94-007-0265-3_35.
Feske S. CRAC channelopathies. Pflugers Arch. 2010;460(2):417–35. https://doi.org/10.1007/s00424-009-0777-5.
Akyuva Y, Nazıroğlu M, Yıldızhan K. Selenium prevents interferon-gamma induced activation of TRPM2 channel and inhibits inflammation, mitochondrial oxidative stress, and apoptosis in microglia. Metab Brain Dis. 2021;36(2):285–98. https://doi.org/10.1007/s11011-020-00624-0.
Osmanlıoğlu H, Yıldırım MK, Akyuva Y, Yıldızhan K, Nazıroğlu M. Morphine induces apoptosis, inflammation, and mitochondrial oxidative stress via activation of TRPM2 channel and nitric oxide signaling pathways in the hippocampus. Mol Neurobiol. 2020;57(8):3376–89. https://doi.org/10.1007/s12035-020-01975-6.
Yıldızhan K, Nazıroğlu M. NMDA receptor activation stimulates hypoxia-induced TRPM2 channel activation, mitochondrial oxidative stress, and apoptosis in neuronal cell line: modular role of memantine. Brain Res. 2023;1803:148232. https://doi.org/10.1016/j.brainres.2023.148232.
Penna C, Rastaldo R, Mancardi D, Raimondo S, Cappello S, Gattullo D, et al. Post–conditioning induced cardioprotection requires signaling through a redox–sensitive mechanism, mitochondrial ATP–sensitive K+ channel and protein kinase C activation. Basic Res Cardiol. 2006;101(2):180–9. https://doi.org/10.1007/s00395-006-0584-5.
Alloatti G, Penna C, Comità S, Tullio F, Aragno M, Biasi F, et al. Aging, sex and NLRP3 inflammasome in cardiac ischaemic disease. Vascul Pharmacol. 2022;145:107001. https://doi.org/10.1016/j.vph.2022.107001.
Penna C, Femminò S, Caldera F, Rubin Pedrazzo A, Cecone C, Alfì E, et al. Cyclic nigerosyl-nigerose as oxygen nanocarrier to protect cellular models from hypoxia/reoxygenation injury: implications from an in vitro model. Int J Mol Sci. 2021;22(8):4208. https://doi.org/10.3390/ijms22084208.
Syed Mortadza SA, Wang L, Li D, Jiang L-H. TRPM2 channel-mediated ROS-sensitive Ca2+ signaling mechanisms in immune cells. Front Immunol. 2015;6:407. https://doi.org/10.3389/fimmu.2015.00407.
Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem. 2005;280(7):6138–48. https://doi.org/10.1074/jbc.M411446200.
Wehage E, Eisfeld J, Heiner I, Jungling E, Zitt C, Luckhoff A. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J Biol Chem. 2002;277(26):23150–6. https://doi.org/10.1074/jbc.M112096200.
Yang KT, Chang WL, Yang PC, Chien CL, Lai MS, Su MJ, et al. Activation of the transient receptor potential M2 channel and poly(ADP-ribose) polymerase is involved in oxidative stress-induced cardiomyocyte death. Cell Death Differ. 2006;13(10):1815–26. https://doi.org/10.1038/sj.cdd.4401813.
Leone GM, Candido S, Lavoro A, Vivarelli S, Gattuso G, Calina D, et al. Clinical relevance of targeted therapy and immune-checkpoint inhibition in lung cancer. Pharmaceutics. 2023;15(4):1252.
Zhang X-M, Song Y, Zhu X-Y, Wang W-J, Fan X-L, El-Aziz TMA. MITOCHONDRIA: the dual function of the transient receptor potential melastatin 2 channels from cytomembrane to mitochondria. Int J Biochem Cell Biol. 2023;157:106374. https://doi.org/10.1016/j.biocel.2023.106374.
Bao L, Festa F, Freet CS, Lee JP, Hirschler-Laszkiewicz IM, Chen S-J, et al. The human transient receptor potential melastatin 2 ion channel modulates ROS through Nrf2. Sci Rep. 2019;9(1):14132. https://doi.org/10.1038/s41598-019-50661-8.
Miller BA, Wang J, Hirschler-Laszkiewicz I, Gao E, Song J, Zhang XQ, et al. The second member of transient receptor potential-melastatin channel family protects hearts from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2013;304(7):H1010–22. https://doi.org/10.1152/ajpheart.00906.2012.
Miller BA, Hoffman NE, Merali S, Zhang XQ, Wang J, Rajan S, et al. TRPM2 channels protect against cardiac ischemia-reperfusion injury: role of mitochondria. J Biol Chem. 2014;289(11):7615–29. https://doi.org/10.1074/jbc.M113.533851.
Yıldızhan K, Huyut Z, Altındağ F. Involvement of TRPM2 channel on doxorubicin-induced experimental cardiotoxicity model: protective role of selenium. Biol Trace Elem Res. 2023;201(5):2458–69. https://doi.org/10.1007/s12011-022-03377-2.
Jin L, Alesi GN, Kang S. Glutaminolysis as a target for cancer therapy. Oncogene. 2016;35(28):3619–25. https://doi.org/10.1038/onc.2015.447.
Zhang W, Tong Q, Conrad K, Wozney J, Cheung JY, Miller BA. Regulation of TRP channel TRPM2 by the tyrosine phosphatase PTPL1. Am J Physiol Cell Physiol. 2007;292(5):C1746–58. https://doi.org/10.1152/ajpcell.00569.2006.
Naziroglu M. New molecular mechanisms on the activation of TRPM2 channels by oxidative stress and ADP-ribose. Neurochem Res. 2007;32(11):1990–2001. https://doi.org/10.1007/s11064-007-9386-x.
Wilkinson JA, Scragg JL, Boyle JP, Nilius B, Peers C. H2O 2-stimulated Ca2+ influx via TRPM2 is not the sole determinant of subsequent cell death. Pflugers Arch. 2008;455(6):1141–51. https://doi.org/10.1007/s00424-007-0384-2.
Yoshizumi M, Tsuchiya K, Tamaki T. Signal transduction of reactive oxygen species and mitogen-activated protein kinases in cardiovascular disease. J Med Invest. 2001;48:11–24.
Ali ES, Rychkov GY, Barritt GJ. TRPM2 non-selective cation channels in liver injury mediated by reactive oxygen species. Antioxidants (Basel). 2021;10(8):1243. https://doi.org/10.3390/antiox10081243.
Ji D, Luo Z-W, Ovcjak A, Alanazi R, Bao M-H, Feng Z-P, et al. Role of TRPM2 in brain tumours and potential as a drug target. Acta Pharmacol Sin. 2022;43(4):759–70. https://doi.org/10.1038/s41401-021-00679-4.
Hantute-Ghesquier A, Haustrate A, Prevarskaya N, Lehen’kyi V. TRPM family channels in cancer. Pharmaceuticals (Basel). 2018;11(2):58. https://doi.org/10.3390/ph11020058.
Ishii M, Oyama A, Hagiwara T, Miyazaki A, Mori Y, Kiuchi Y, et al. Facilitation of H2O2-induced A172 human glioblastoma cell death by insertion of oxidative stress-sensitive TRPM2 channels. Anticancer Res. 2007;27(6B):3987–92.
Zhang Y, Hoon MA, Chandrashekar J, Mueller KL, Cook B, Wu D, et al. Coding of sweet, bitter, and umami tastes: different receptor cells sharing similar signaling pathways. Cell. 2003;112(3):293–301. https://doi.org/10.1016/s0092-8674(03)00071-0.
Chen SJ, Hoffman NE, Shanmughapriya S, Bao L, Keefer K, Conrad K, et al. A splice variant of the human ion channel TRPM2 modulates neuroblastoma tumor growth through hypoxia-inducible factor (HIF)-1/2α. J Biol Chem. 2014;289(52):36284–302. https://doi.org/10.1074/jbc.M114.620922.
Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411(6837):595–9. https://doi.org/10.1038/35079100.
Starkus J, Beck A, Fleig A, Penner R. Regulation of TRPM2 by extra- and intracellular calcium. J Gen Physiol. 2007;130(4):427–40. https://doi.org/10.1085/jgp.200709836.
Rah SY, Kwak JY, Chung YJ, Kim UH. ADP-ribose/TRPM2-mediated Ca2+ signaling is essential for cytolytic degranulation and antitumor activity of natural killer cells. Sci Rep. 2015;5:9482. https://doi.org/10.1038/srep09482.
de Murcia G, Menissierde Murcia J. Poly(ADP-ribose) polymerase: a molecular nick-sensor. Trends Biochem Sci. 1994;19(4):172–6.
McHugh D, Flemming R, Xu SZ, Perraud AL, Beech DJ. Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J Biol Chem. 2003;278(13):11002–6. https://doi.org/10.1074/jbc.M210810200.
Kuhn FJ, Heiner I, Luckhoff A. TRPM2: a calcium influx pathway regulated by oxidative stress and the novel second messenger ADP-ribose. Pflugers Arch. 2005;451(1):212–9. https://doi.org/10.1007/s00424-005-1446-y.
Heiner I, Eisfeld J, Warnstedt M, Radukina N, Jungling E, Luckhoff A. Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem J. 2006;398(2):225–32. https://doi.org/10.1042/BJ20060183.
Tirichen H, Yaigoub H, Xu W, Wu C, Li R, Li Y. Mitochondrial reactive oxygen species and their contribution in chronic kidney disease progression through oxidative stress. Front Physiol. 2021;12:627837. https://doi.org/10.3389/fphys.2021.627837.
Poyton RO, Ball KA, Castello PR. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol Metab. 2009;20(7):332–40. https://doi.org/10.1016/j.tem.2009.04.001.
Choi DW. Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci. 1995;18(2):58–60.
Aarts MM, Tymianski M. TRPMs and neuronal cell death. Pflugers Arch. 2005;451(1):243–9. https://doi.org/10.1007/s00424-005-1439-x.
Ortiz-Garcia J, Gomez CR, Schneck MJ, Biller J. Recent advances in the management of transient ischemic attacks. Fac Rev. 2022;11:19. https://doi.org/10.12703/r/11-19.
Wang Q, Liu N, Ni YS, Yang JM, Ma L, Lan XB, et al. TRPM2 in ischemic stroke: structure, molecular mechanisms, and drug intervention. Channels (Austin). 2021;15(1):136–54. https://doi.org/10.1080/19336950.2020.1870088.
Zong P, Lin Q, Feng J, Yue L. A systemic review of the integral role of TRPM2 in ischemic stroke: from upstream risk factors to ultimate neuronal death. Cells. 2022;11(3):491. https://doi.org/10.3390/cells11030491.
Xu J, Zhang W, Dong J, Cao L, Huang Z. A new potential strategy for treatment of ischemic stroke: targeting TRPM2–NMDAR association. Neurosci Bull. 2022. https://doi.org/10.1007/s12264-022-00971-1.
Teoh NC. Hepatic ischemia reperfusion injury: contemporary perspectives on pathogenic mechanisms and basis for hepatoprotection-the good, bad and deadly. J Gastroenterol Hepatol. 2011;26(Suppl 1):180–7. https://doi.org/10.1111/j.1440-1746.2010.06584.x.
van Wijk SJ, Hageman GJ. Poly(ADP-ribose) polymerase-1 mediated caspase-independent cell death after ischemia/reperfusion. Free Radic Biol Med. 2005;39(1):81–90. https://doi.org/10.1016/j.freeradbiomed.2005.03.021.
Muriel P. Role of free radicals in liver diseases. Hepatol Int. 2009. https://doi.org/10.1007/s12072-009-9158-6.
Britton RS, Bacon BR. Role of free radicals in liver diseases and hepatic fibrosis. Hepatogastroenterology. 1994;41(4):343–8.
Nieuwenhuijs VB, De Bruijn MT, Padbury RT, Barritt GJ. Hepatic ischemia-reperfusion injury: roles of Ca2+ and other intracellular mediators of impaired bile flow and hepatocyte damage. Dig Dis Sci. 2006;51(6):1087–102. https://doi.org/10.1007/s10620-006-8014-y.
Du J, Xie J, Yue L. Modulation of TRPM2 by acidic pH and the underlying mechanisms for pH sensitivity. J Gen Physiol. 2009;134(6):471–88. https://doi.org/10.1085/jgp.200910254.
Gao G, Wang W, Tadagavadi RK, Briley NE, Love MI, Miller BA, et al. TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J Clin Investig. 2014;124(11):4989–5001.
Ye M, Yang W, Ainscough JF, Hu X, Li X, Sedo A, et al. TRPM2 channel deficiency prevents delayed cytosolic Zn2+ accumulation and CA1 pyramidal neuronal death after transient global ischemia. Cell Death Dis. 2014;5(11):e1541.
Pudela C, Balyasny S, Applebaum MA. Nervous system: embryonal tumors: neuroblastoma. Atlas Genet Cytogenet Oncol Haematol. 2020;24(7):284–90. https://doi.org/10.4267/2042/70771.
Yıldızhan K, Nazıroğlu M. Protective role of selenium on MPP(+) and homocysteine-induced TRPM2 channel activation in SH-SY5Y cells. J Recept Signal Transduct Res. 2022;42(4):399–408. https://doi.org/10.1080/10799893.2021.1981381.
Ahlatcı A, Yıldızhan K, Tülüce Y, Bektaş M. Valproic acid attenuated PTZ-induced oxidative stress, inflammation, and apoptosis in the SH-SY5Y cells via modulating the TRPM2 channel. Neurotox Res. 2022;40(6):1979–88. https://doi.org/10.1007/s12640-022-00622-3.
Chen SJ, Zhang W, Tong Q, Conrad K, Hirschler-Laszkiewicz I, Bayerl M, et al. Role of TRPM2 in cell proliferation and susceptibility to oxidative stress. Am J Physiol Cell Physiol. 2013;304(6):C548–60. https://doi.org/10.1152/ajpcell.00069.2012.
Togashi K, Inada H, Tominaga M. Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB). Br J Pharmacol. 2008;153(6):1324–30. https://doi.org/10.1038/sj.bjp.0707675.
Hopkins MM, Feng X, Liu M, Parker LP, Koh DW. Inhibition of the transient receptor potential melastatin-2 channel causes increased DNA damage and decreased proliferation in breast adenocarcinoma cells. Int J Oncol. 2015;46(5):2267–76. https://doi.org/10.3892/ijo.2015.2919.
Koh DW, Powell DP, Blake SD, Hoffman JL, Hopkins MM, Feng X. Enhanced cytotoxicity in triple-negative and estrogen receptor-positive breast adenocarcinoma cells due to inhibition of the transient receptor potential melastatin-2 channel. Oncol Rep. 2015;34(3):1589–98. https://doi.org/10.3892/or.2015.4131.
Klumpp D, Misovic M, Szteyn K, Shumilina E, Rudner J, Huber SM. Targeting TRPM2 channels impairs radiation-induced cell cycle arrest and fosters cell death of T cell leukemia cells in a Bcl-2-dependent manner. Oxid Med Cell Longev. 2016;2016:8026702. https://doi.org/10.1155/2016/8026702.
Park YR, Chun JN, So I, Kim HJ, Baek S, Jeon JH, et al. Data-driven analysis of TRP channels in cancer: linking variation in gene expression to clinical significance. Cancer Genomics Proteomics. 2016;13(1):83–90.
Huang C, Qin Y, Liu H, Liang N, Chen Y, Ma D, et al. Downregulation of a novel long noncoding RNA TRPM2-AS promotes apoptosis in non-small cell lung cancer. Tumour Biol. 2017;39(2):1010428317691191. https://doi.org/10.1177/1010428317691191.
Zhao LY, Xu WL, Xu ZQ, Qi C, Li Y, Cheng J, et al. The overexpressed functional transient receptor potential channel TRPM2 in oral squamous cell carcinoma. Sci Rep. 2016;6:38471. https://doi.org/10.1038/srep38471.
Almasi S, Kennedy BE, El-Aghil M, Sterea AM, Gujar S, Partida-Sánchez S, et al. TRPM2 channel-mediated regulation of autophagy maintains mitochondrial function and promotes gastric cancer cell survival via the JNK-signaling pathway. J Biol Chem. 2018;293(10):3637–50. https://doi.org/10.1074/jbc.M117.817635.
Ali ES, Rychkov GY, Barritt GJ. Targeting Ca(2+) signaling in the initiation, promotion and progression of hepatocellular carcinoma. Cancers (Basel). 2020;12(10):2755. https://doi.org/10.3390/cancers12102755.
Almasi S, Sterea AM, Fernando W, Clements DR, Marcato P, Hoskin DW, et al. TRPM2 ion channel promotes gastric cancer migration, invasion and tumor growth through the AKT signaling pathway. Sci Rep. 2019;9(1):4182. https://doi.org/10.1038/s41598-019-40330-1.
Sterea AM, Egom EE, El Hiani Y. TRP channels in gastric cancer: new hopes and clinical perspectives. Cell Calcium. 2019;82:102053. https://doi.org/10.1016/j.ceca.2019.06.007.
Maeda T, Suzuki A, Koga K, Miyamoto C, Maehata Y, Ozawa S, et al. TRPM5 mediates acidic extracellular pH signaling and TRPM5 inhibition reduces spontaneous metastasis in mouse B16-BL6 melanoma cells. Oncotarget. 2017;8(45):78312.
Zeng X, Sikka SC, Huang L, Sun C, Xu C, Jia D, et al. Novel role for the transient receptor potential channel TRPM2 in prostate cancer cell proliferation. Prostate Cancer Prostatic Dis. 2010;13(2):195–201. https://doi.org/10.1038/pcan.2009.55.
Kheradpezhouh E, Ma L, Morphett A, Barritt GJ, Rychkov GY. TRPM2 channels mediate acetaminophen-induced liver damage. Proc Natl Acad Sci U S A. 2014;111(8):3176–81. https://doi.org/10.1073/pnas.1322657111.
Kheradpezhouh E, Barritt GJ, Rychkov GY. Curcumin inhibits activation of TRPM2 channels in rat hepatocytes. Redox Biol. 2016;7:1–7. https://doi.org/10.1016/j.redox.2015.11.001.
Bao L, Chen SJ, Conrad K, Keefer K, Abraham T, Lee JP, et al. Depletion of the human ion channel TRPM2 in neuroblastoma demonstrates its key role in cell survival through modulation of mitochondrial reactive oxygen species and bioenergetics. J Biol Chem. 2016;291(47):24449–64. https://doi.org/10.1074/jbc.M116.747147.
Bauer I, Grozio A, Lasigliè D, Basile G, Sturla L, Magnone M, et al. The NAD+-dependent histone deacetylase SIRT6 promotes cytokine production and migration in pancreatic cancer cells by regulating Ca2+ responses. J Biol Chem. 2012;287(49):40924–37. https://doi.org/10.1074/jbc.M112.405837.
Huang C, Qin Y, Liu H, Liang N, Chen Y, Ma D, et al. Downregulation of a novel long noncoding RNA TRPM2-AS promotes apoptosis in non–small cell lung cancer. Tumor Biol. 2017;39(2):1010428317691191. https://doi.org/10.1177/1010428317691191.
Masumoto K, Tsukimoto M, Kojima S. Role of TRPM2 and TRPV1 cation channels in cellular responses to radiation-induced DNA damage. Biochim Biophys Acta. 2013;1830(6):3382–90. https://doi.org/10.1016/j.bbagen.2013.02.020.
Li A, Yang J, Zhang T, Li L, Li M. Long noncoding RNA TRPM2-AS promotes the growth, migration, and invasion of retinoblastoma via miR-497/WEE1 axis. Front Pharmacol. 2021;12:592822. https://doi.org/10.3389/fphar.2021.592822.
Çiğ B, Yildizhan K. Resveratrol diminishes bisphenol A-induced oxidative stress through TRPM2 channel in the mouse kidney cortical collecting duct cells. J Recept Signal Transduct Res. 2020;40(6):570–83. https://doi.org/10.1080/10799893.2020.1769657.
Naziroglu M. TRPM2 cation channels, oxidative stress and neurological diseases: where are we now? Neurochem Res. 2011;36(3):355–66. https://doi.org/10.1007/s11064-010-0347-4.
Hill K, Benham CD, McNulty S, Randall AD. Flufenamic acid is a pH-dependent antagonist of TRPM2 channels. Neuropharmacology. 2004;47(3):450–60. https://doi.org/10.1016/j.neuropharm.2004.04.014.
Kraft R, Grimm C, Frenzel H, Harteneck C. Inhibition of TRPM2 cation channels by N-(p-amylcinnamoyl)anthranilic acid. Br J Pharmacol. 2006;148(3):264–73. https://doi.org/10.1038/sj.bjp.0706739.
Zhang H, Zhao S, Yu J, Yang W, Liu Z, Zhang L. Medicinal chemistry perspective of TRPM2 channel inhibitors: where we are and where we might be heading? Drug Discov Today. 2020;25(12):2326–34. https://doi.org/10.1016/j.drudis.2020.09.039.
Li J, Gao Y, Bao X, Li F, Yao W, Feng Z, et al. TRPM2: a potential drug target to retard oxidative stress. Front Biosci (Landmark Ed). 2017;22:1427–38.
Pfister M, Ogilvie A, da Silva CP, Grahnert A, Guse AH, Hauschildt S. NAD degradation and regulation of CD38 expression by human monocytes/macrophages. Eur J Biochem. 2001;268(21):5601–8. https://doi.org/10.1046/j.1432-1033.2001.02495.x.
Acknowledgements
Not Applicable.
Funding
Not Applicable.
Author information
Authors and Affiliations
Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas. That is revising or critically reviewing the article; giving final approval of the version to be published; agreeing on the journal to which the article has been submitted; and, confirming to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ali, E.S., Chakrabarty, B., Ramproshad, S. et al. TRPM2-mediated Ca2+ signaling as a potential therapeutic target in cancer treatment: an updated review of its role in survival and proliferation of cancer cells. Cell Commun Signal 21, 145 (2023). https://doi.org/10.1186/s12964-023-01149-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01149-6