
Abstract
Oncogenic driver mutations have implications that extend beyond cancer cells themselves. Aberrant tumour cell signalling has various effects on the tumour microenvironment and anti-tumour immunity, with important consequences for therapy response and resistance. We provide an overview of how mutant RAS, one of the most prevalent oncogenic drivers in cancer, can instigate immune evasion programs at the tumour cell level and through remodelling interactions with the innate and adaptive immune cell compartments. Finally, we describe how immune evasion networks focused on RAS, and the immune checkpoint molecule PD-L1 can be disrupted through therapeutic intervention, and discuss potential strategies for combinatorial treatment.
Video abstract
Similar content being viewed by others


Introduction
As our understanding of cancer and the tumour microenvironment (TME) has improved, more advanced targeted and refined therapies have been developed, including cancer immunotherapies that leverage the patient’s immune system. For example, immune checkpoint blockade (ICB) is now clinically approved for the treatment of a multitude of different cancer types. Most cancer types employ some form of immune evasion [1,2,3,4]. Mechanistically, this can range from the remodelling of the TME through increased recruitment of Myeloid-Derived Suppressor Cells (MDSC) and T regulatory (Treg) cells, and stromal remodelling with TGF-β induction of Cancer-Associated Fibroblasts (CAFs), to differential cytokine (VEGF, IL-6, IL-8) and checkpoint molecule (PD-L1, B7-H4) expression. In this review, we will focus on tumour-intrinsic immune modulation through oncogenic signalling via RAS and other prominent oncogenes, and how this can affect the immune contexture [5] through differential expression of immune checkpoint molecules and other mechanisms [2, 3, 6,7,8,9]. In particular, we will discuss how our understanding of tumour cell-intrinsic KRAS signalling and PD-L1 expression has evolved in relation to anti-tumour immunity and mechanisms of immune escape, and how this is driving innovation in research and clinical settings.
Immune evasion and oncogenic KRAS—what led us here?
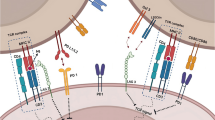
Mutations in RAS are some of the most frequent mutations across all cancer types, with a 15–30% mutation rate across all cancers [7, 9]. Alterations in the KRAS isoform account for 75–85% of RAS-mutant cancers [10, 11], with mutations at G12 accounting for 81% of these [11], and are mainly associated with non-small cell lung cancer (NSCLC), colorectal cancer (CRC), and pancreatic ductal adenocarcinoma (PDAC) [12]. However, KRAS driver mutation and allelic frequency vary between tissue types [13]. KRAS is a GTPase, with activating mutant forms of KRAS favouring the GTP-bound, active state. Such aberrant signalling leads to transcriptional upregulation of intrinsic pro-survival, anti-apoptotic, angiogenic, and proliferative pathways such as PI3K-AKT-mTOR and MAPK, as well as immune evasion mechanisms such as reduced antigen presentation (Fig. 1) [6, 7, 9, 14]. As we discuss below, oncogenic RAS has been associated with significantly enhanced immune checkpoint molecule (ICM) expression and immune evasion programmes, making RAS-mutant tumours promising candidates for ICB.

Immunoevasive signalling pathways in KRAS-mutant cancers. Immune evasion is mediated via MAPK pathway signalling enhanced by mutant KRAS, decreased type I/II IFN signalling, TTP inhibition, and co-mutations (e.g. STK11/KEAP1/TP53/EGFR). The effect of these pathway alterations is represented with cell-autonomous (e.g. increased survival and proliferation) and non-cell-autonomous programmes (e.g. decreased MHC expression, increased PD-L1 expression). Signalling through the TME to immune cells, via free and bound signalling molecules, and the immune evasive effects are also highlighted and attributed to specific signalling pathways (e.g. ICM suppression of CD8 + T cells, and reduced neoantigen detection due to PGE2). Immune cell metabolic reprogramming (IDO-1), increased pro-tumour cytokines and physical remodelling of the TME (MMP-9 and LOX), due to the KRAS signalling demonstrate its central role in tumour immune evasion. Treatments for abating these immune evasive mechanisms are shown. Arrows represent enhancement and flat heads represent inhibition, dotted lines are used to indicate inhibition by small molecule treatments, and red crosses represent pathways that have been blocked due to mutation or alterations in cell signalling
Recent pharmacological research has been focused on targeting the third most common G12 KRAS mutation, KRASG12C (11.9%), which trails behind the most common KRAS mutations, KRASG12D and KRASG12V (29.5% and 23%, respectively) [15]. Interestingly, the KRASG12C variant is associated with a higher response rate to ICB and progression free survival (PFS), perhaps because of a significant co-occurrence of high tumour mutational burden (TMB)/microsatellite unstable status and the potential for an “immune-hot” TME [15,16,17]. Indeed, in both non-small cell lung cancer (NSCLC) and colorectal cancer (CRC), high-TMB smoker patients are more likely to be KRASG12C [15]. This trend towards favourable ICB responses can also be seen with the KRASG12V variant, likely due to the commonality of the KRASG12C and KRASG12V variants attributed to smoking, and consequently a high-TMB [16]. In contrast, KRASG12D, a driver in approximately 40% of PDAC tumours [18], has been noted as having a reduced response rate to immunotherapies, including ICBs [19]. This has been attributed to particularly high levels of GM-CSF production [20], a high degree of mutational intertumoral and intratumoral heterogeneity [21], most KRASG12D-driven NSCLC tumours presenting in more “immune cold” never-smokers [22], and potentially, lower PD-L1 expression in KRASG12D tumours [16]. Furthermore, KRASQ61X, accounting for ~ 7% of KRAS mutant NSCLCs [23], has been associated with decreased TMB and PD-L1 expression [24]. This is significant because the Checkmate 568 trial of NSCLC patients treated with nivolumab plus ipilimumab combination therapy confirmed that a low TMB is an important determinant of treatment failure [25]. However, other studies have found that KRAS-mutation subtype has no significant effect on PFS or OS, instead attributing response to other factors such as co-mutations in TP53, KEAP1, STK11, EGFR and SAMARCA4 [22, 26, 27], highlighting the complexity of this disease and the need for further studies.
The intrinsic and extrinsic mechanisms by which KRAS mutations and tumour PD-L1 expression remodel the immune landscape of the TME have become a focus of research in recent years. An interesting genetic mechanism for enhanced immune suppression was discovered, whereby 3’UTR loss in PD-L1 (CD274) resulted in enhanced expression and more effective immune evasion in multiple cancers, though the precise mechanism for this upregulation was unclear at the time [28]. It was later demonstrated that oncogenic RAS signalling could drive PD-L1 expression in cancer cells. Mechanistically, enhanced PD-L1 mRNA expression is driven through RAS-MEK signalling and the RAS-ROS-p38 axes, phosphorylating TTP and inhibiting TTP binding to AU-rich elements in the PD-L1 mRNA 3’UTR, reducing mRNA degradation [3]. PI3K signalling downstream of RAS has also been implicated in increasing translation of the PD-L1 mRNA transcript, overall leading to coordinate upregulation of mRNA and protein [29] (Fig. 1). Extrinsically, increased KRAS signalling and TTP inhibition have been linked to an altered cytokine/chemokine signature (e.g. IL-8, GM-CSF, CXCL5, IL-10, VEGF, PEG-2), giving rise to the recruitment of myeloid-derived suppressor cells (MDSC) and Tregs, CAF transformation, and increased endothelial Fas ligand expression, which can inhibit CD8 + T cell extravasation (Fig. 1, Fig. 2) [3, 8, 9].

KRAS-mutant tumour immunoevasive signalling in the TME. Tumour-immune and immune-immune interactions within the TME of a KRAS-mutant tumour, including cytokines and chemokines signalling, metabolic deficiencies, immune checkpoint molecules (ICMs), interactions with the fungal microbiome, and remodelling of the extracellular matrix (ECM) and stroma. For example, the main effects of each cell type and decreased/increased characteristics of a KRAS-mutant TME are shown. Arrows represent enhancement and flat heads represent inhibition, dashed lines represent tumour signalling and hard lines represent signalling of the TME, red text indicates immunoevasive signalling and blue text indicates pro-immune signalling. Red crosses represent pathways that have been blocked due to KRAS-driven immune evasive signalling
Clinically, the prospect of combining targeted therapies with ICB is exciting, especially with the recent development and FDA approval of the first-in-class KRASG12C-specific inhibitor, sotorasib (AMG-510) (Fig. 1) [2]. The chemically tractable mutant cysteine of G12C allows for mutant-specific covalent binding to inhibitors, concomitant with an extremely low off-target score and no inhibitory effect on wild-type KRAS or other KRAS-mutant variants [2]. Such mutant-specific compounds can lead to a step-change in both the reduction of associated toxicities and benefit for patient outcomes in KRASG12C-mutant NSCLC and CRC [2]. Interestingly, sotorasib requires an adaptive immune response to provide maximum benefit [2, 7]. In responsive tumours, sotorasib has been seen to encourage a proinflammatory TME and increase T cell infiltration via increased CXCL10/11 (Fig. 1) [2, 9]. ICB is still only effective in a subset of patients [30], so these recent discoveries have provided an indication that such ICB-combination strategies could improve response rates in some contexts. Finally, a unique advantage to covalently modifying the mutant KRASG12C protein with an inhibitor is the potential to specifically target drug-modified KRAS epitopes with antibodies or cell therapies [31], precisely flagging KRAS-mutant tumour cells for immune-mediated destruction.
The TME, PD-L1 and KRAS—Recent innovations in treatment and novel mechanisms
Novel combination therapies for the treatment of KRAS-driven malignancies
Clinical trials are now underway that combine ICB with sotorasib, MEK and ERK inhibitors [2, 8, 9]. Current trends have been focused on understanding the mutational and immune landscape of KRAS-mutant tumours to elucidate potential resistance mechanisms and immune modulatory niches that might be rationally exploited. Clinical trials evaluating MEK inhibition alongside PD-1/L1 therapy (Fig. 1) in KRAS-mutant tumours had limited success, ostensibly due to tumour heterogenicity and MEK inhibition of CD8 + T cell IL-2 production, which is crucial for clonal expansion and leads to an exhausted phenotype, inhibiting the adaptive immune response [14, 32]. Interestingly, in a colon mouse tumour model, a synergistic effect was produced by combining sotorasib alongside both trametinib (MEK inhibitor) and anti-PD-1 therapy, greatly enhancing survival [2]. There is evidence that a pulsatile dosing regimen for MEK inhibitors can alleviate the anti-proliferative effect on CD8 + T cells, actually resulting in enhanced Ki-67 and CD69 detection; hallmarks of successful clonal expansion (Fig. 1) [14]. This is in agreement with a study in mice by Ebert et al., where MEK inhibition with cobimetinib profoundly blocked naïve CD8+ T cell priming, but led to a greater proportion of antigen-specific CD8+ T effector cells within the tumour by abating chronic T cell receptor (TCR) stimulation (Fig. 1), and ultimately resulted in a synergistic effect with PD-L1 ICB [33]. Clinical trials (NCT03581487, NCT03600701) are currently recruiting to evaluate this further in NSCLC patients (Table 1).
Mutant-specific KRAS inhibitors and anti-tumour immunity
With multiple mutant-specific KRASG12C inhibitors entering the clinic (JNJ-74699157, JDQ443, GDC-6036, and LY3499446), many clinical trials are underway to assess combinations of PD-1 pathway ICB and different KRASG12C inhibitors, spanning PDAC, NSCLC and CRC patients in varying stages (KRYSTAL-1, KRYSTAL-7, CodeBreak 100, and CodeBreak 101) (Table 1). The number of possible combinations to test clinically could rapidly expand, which emphasises the need to understand mechanisms of action on the tumour cell, and the interplay between tumour cells and the immune compartment, to identify robust biomarkers of response [15].
For example, recent work by Downward and colleagues showed that KRASG12C inhibition in a KRAS-mutant mouse model of NSCLC suppresses the downstream function of MYC, resulting in up-regulated interferon signalling, leading to reduced tumour immunosuppressive cytokine production, enhanced infiltration and activation of CD8 + T cells, and increased neoantigen presentation by MHCs (Fig. 1, Fig. 2) [7]. Without pharmacological intervention, aberrant KRAS and MYC programs act cooperatively to drive tumorigenesis and extensive remodelling of the TME [34, 35]. Indeed, in vivo models of cancer have also demonstrated how overactive MYC enhances the expression of the ICMs CD47 and PD-L1 [36], as well as the anti-immune signalling molecules IL-23 and CCL9 (Fig. 1, Fig. 2) [37]. Taken together, these signals can cause extensive local remodelling of both the adaptive and innate immune cell compartments through decreased MHC I expression, upregulation of Rae-1 (NKG2D receptor ligand), enhanced angiogenesis by VEGF [36], reduced macrophage recruitment, and PD-L1 dependent T/B/APC/NK cell exclusion (Fig. 1, Fig. 2) [37]. Inhibition of MYC resulted in marked apoptosis and tumour regression [36, 37]. However, combinations of KRASG12C inhibitors with PD-L1 therapy only showed synergy in highly immunogenic tumours, with many still developing secondary resistance following treatment [7, 15, 38].
Modulation of interferon signalling is crucial for KRAS-mutant immune evasion. Indeed, a recent study observed that when a variety of mouse KRASG12C lines (including the immune-hot KPAR and immune-cold KPB6 [39]) were treated with the KRASG12C inhibitor MRTX1257, the transcriptional effects of IFN-γ signalling were enhanced in all cell lines. This resulted in an upregulation of T cell chemo-attractants such as Cxcl9/10/11 and antigen presentation genes including H2-d/k1, Ciita, and B2m (Fig. 1) [7], thus demonstrating how even neoantigen-high KRAS-mutant tumours can escape the adaptive immune response [7, 8, 40].
Resistance mechanisms in KRAS-driven malignancies—how might these be overcome?
KRASG12C combination strategies with ICB could be helpful to forestall the onset of acquired drug resistance. Resistances to KRASG12C inhibitors include increased receptor tyrosine kinase (RTK) feedback reducing GDP occupancy [41], secondary KRAS mutations including G12D, G13, Q61, R68, H95, and Y96 mutations, or amplification of the KRASG12C allele [42]. Combinations of KRASG12C inhibitors with ICB, chemotherapy, anti-EGFR antibodies [43], and pan-KRAS targeting agents (e.g. SOS1 and SHP2 inhibitors) are being investigated (Fig. 1) [7, 44,45,46]. Moreover, the use of new combinations with pan-KRAS inhibitors is already being tested, as are SHP2 inhibitors (SHP2i/RG6433) and SOS1 inhibitors (BAY-293 and BI-3406) in combination with KRASG12C inhibitors [45, 46]. Additionally, trials targeting KRASG12V in advanced epithelial cancer (NCT05389514) and NSCLC (NCT04620330) are taking place with ICB and RAF/MEK inhibitors, respectively (Table 1). Zygosity and location of KRAS mutations have a large effect on KRAS-KRAS dimerization, which is crucial for KRAS downstream signalling and is involved in mechanisms of resistance [47]. Investigations into how oncogenic KRAS mutations and mutant-specific inhibitors affect dimerization are currently being explored as a method for reducing resistance, such as the resistance mutation KRASY96D for sotorasib and MRTX849 treatment [48]. Indeed, resistance by KRAS mutational heterogenicity affecting dimerization provides a good rationale for the use of MEK inhibitors to disrupt this mechanism [47]. Additionally, resistance can be achieved through gain-of-function mutations in epigenetic regulators, producing a profound effect on signalling and tumour metabolism within the TME. Following genetic KRAS silencing, HDAC5 upregulation causes remodelling of the TME via SOCS3-dependent upregulation of CCL2 and CCL7, and recruitment of immunosuppressive tumour-associated macrophages (TAMs) (Fig. 2) [49]. This process is driven through increasing TGFβ signalling and enables KRAS-independent tumour progression [49]. These data highlight the potential for other novel co-treatments to be employed in KRAS-mutant cancers, which may become resistant to KRAS inhibition. For example, CCR2 inhibitors such as PF-04136309 (ORR of 40% in PDAC trials [50]) or epigenetic drugs such as HDAC inhibitors could be leveraged to impede KRAS-independent tumour progression [51].
Immunoediting of neoantigens, reduced HLA expression and inactivation of the IFN-γ pathway are perhaps the most ubiquitous examples of immune evasion in KRAS-mutant PD-L1 + cancers, while acquired resistance to immunotherapies tends to be more nuanced. A poignant example of this is the discovery of an on-target mutation in PD-L1 as a result of tumour challenge with EGFR (cetuximab) and PD-L1 (avelumab) antibodies plus chemotherapy (FOLFOX) [38]. Following therapy, a truncating mutation (PD-L1K162fs) and a new phosphorylation site mutation (PD-L1L88S) emerged, leading to PD-L1 mRNA degradation by nonsense-mediated decay, and causing loss of protein stability and proteasomal degradation, respectively [38]. Treatment cessation led to these mutant subclones declining, indicating that drug holidays may be beneficial in this context [38]. Additional subclones with loss of function (LOF) mutations in JAK proteins were also seen (Fig. 1), further implicating the role of IFN-γ and the adaptive immune response as major determinants of successful anti-tumour responses in KRAS-mutant malignancies [38]. More recently, it was observed that KRAS-mutant pancreatic tumours acquire SMAD4/TGFBR2 LOF mutations (35–50%) as a resistance mechanism to ICB [52]. Compounds which bind target proteins and induce the ubiquitination and subsequent degradation through E3 ligases (Proteolysis Targeting Chimeras, PROTACs) are also being utilised to target PD-L1 in cancer [53, 54], and could serve as a useful, orthogonal treatment avenue to antibody-mediated ICB therapies.
Concurrent mutations with KRAS and immune evasion
Although oncogenic KRAS can orchestrate immune evasion pathways, the response of KRAS-mutant tumours to ICB is still low [~ 25%] [26], with perhaps the most reliable biomarker for response remaining TMB [30]. Hence, multiple studies have identified genomic and phenotypic changes accompanying innate or acquired immunoresistance. KEAP1, STK11, LRP1B and CDKN2A mutations and mismatch repair defects commonly co-occur in KRAS-mutant NSCLC and CRC [15, 55,56,57,58]. In addition, KEAP1 and STK11 LOF mutations have both been linked to immune evasion by reducing the number of tumour-infiltrating lymphocytes (TILs) within the TME (Fig. 1) [59, 60].
KEAP1LOF occurs in ~ 20% of NSCLC, is associated with an immunosuppressive microenvironment, and results in increased levels of NRF2-ARE-driven expression [57, 59, 61]. NRF2 itself has been associated with decreased TILs, increased EGFR and PD-L1 expression, reduced IL-6 and IL-1β expression [62], blocking T cell receptor signalling via COX2/PGE2, and blocking cGAS/STING pathway signalling (Fig. 1) [26, 38, 39, 41, 42]. Indeed, COX2/PEG2 signalling plays an important role in sustained pro-tumour inflammatory pathways and has been observed in BrafV600E and NrasG12D melanoma cells, with inhibition of this pathway promoting anti-tumour immunity and tumour rejection when combined with ICB [63]. STK11LOF occurs in approximately 15% of lung adenocarcinomas and is associated with a lack of tumour PD-L1 expression, enhanced mTOR activity, CD8 + T cells transformation to exhaustion phenotype and reduction of new infiltrates, neutrophil recruitment [64], blocking of cGAS/STING signalling [57, 65], and resistance to ICB in patients with KRAS-mutant NSCLC (Fig. 1) [57, 65, 66]. Interestingly, KEAP1LOF alone increases PD-L1, but when combined with KRAS-mutation, PD-L1 expression is reduced, whereas STK11 mutation is associated with decreased PD-L1 expression regardless of KRAS mutation status [57]. Curiously, both KEAP1 and STK11 mutations are associated with worse prognosis in tumours harbouring KRAS G12C mutations compared to G12V or G12D. KEAP1 and STK11 mutations are associated with positive survival impact in KRAS-WT tumours [55, 57]. This may be due to the reduced Foxp3 expression seen in STK11LOF/KRASWT tumours which impairs the function of Tregs [67], as well as the increased TMB and neoantigen load seen in both STK11LOF and KEAP1LOF tumours driven from aberrant ROS generation [68] and a lack of the KRAS-dependent MHC complex downregulation [34], which would enhance the effect of ICB (Fig. 1). NRF2 modulators/inhibitors (e.g. luteolin, ascorbic acid, tretinoin) might prove helpful in treating KRAS/KEAP-mutant cancers [69] (Fig. 1); 2 clinical trials are currently underway investigating the clinical efficacy of ascorbic acid (NCT03146962) and tretinoin (NCT04919369) in NSCLC and RAS-driven cancers respectively (Table 1). mTOR inhibitors (Fig. 1), while previously unsuccessful in a 2018 study in KRAS-mutant cancers (NCT01347866) (Table 1), have been seen to promote tumour regression in the treatment of KRASG12C inhibitor refractory disease [41], and may yet prove effective by using STK11-mutation status as a surrogate biomarker for pathway activity. Lastly, treatment of Kras/Trp53/Stk11-mutant tumours with Axl inhibitors has recently been shown to reverse STK11LOF-driven resistance to PD-1 ICB in mouse models through enhancing type I IFN production by DCs to promote T cell proliferation (Fig. 2) [70].
Another study analysing the potential non-cell-autonomous effects of oncogenic signalling in KRASG12C-driven NSCLC found significant differentially expressed genes, mainly associated with the regulation of cell proliferation and migration, immune cells and hormone signalling [71]. The most noteworthy of these included the downregulation of VTCN1 (B7-H4) [71]. B7-H4+ tumour status has been previously shown to co-express in KRAS-mutant NSCLC (76%) [72] and other KRAS-mutant cancers such as CRC [73]. B7-H4 acts to increase CD4+ T cell transformation into Tregs and negatively regulates T-cell-mediated immune responses (Fig. 1), although the exact mechanisms for this are still unclear [74, 75]. Interestingly, examples of synergy between B7-H4 and PD-L1 ICB have been reported [76, 77]. Notably, EGFR-mutant NSCLCs express elevated levels of B7-H4 in a MEK/ERK dependent manner [78]. Since ~ 15% of NSCLC patients treated with the KRASG12C inhibitor sotorasib develop resistance by enhancing EGFR signalling [79] and with clinical benefit already seen in KRASG12C CRCs [73], these findings suggest that B7-H4 could be a promising ICM target for KRASG12C inhibitor-resistant NSCLC and CRC, or in combination with KRASG12C inhibitor, anti-EGFR therapy and PD-L1 ICB. With the first phase I clinical trial of the B7-H4 ICB monoclonal antibody FPA150 (NCT03514121) recently completed [80], there is growing potential for new combination therapies (Table 1).
Consequences of aberrant KRAS and PD-L1 signalling on TILs
Oncogenic KRAS modulates both the expression of ICMs and the infiltration of TILs, which are important factors of the anti-tumour immune response. Until recently, there has been less emphasis on investigating the regulation of ICM expression on TILs and how this is influenced by tumour signalling [81]. This issue is complicated as ICM expression on TILs is highly varied between cancers and lymphocyte populations; for example, NSCLC and head and neck squamous cell carcinoma display an increased population of CD45+/CD3+ TILs while other cancer types have a reduction in this population [81]. However, there are some common themes; a higher proportion of CD3+/CD4+/CD25+/FoxP3+ Tregs and reduced NK populations are consistently associated with late-stage malignancies, and T cell expression of Tim3 is highly correlated with LAG3 expression, though negatively correlated with CD28 (Fig. 2) [81]. T cells in advanced vs early-stage tumours displayed increased CD244, PD-1, CTLA4, CD39, PD-L2, LAG3, TIGIT, Tim3 and decreased expression of CD73 (Fig. 2) [81].
As with adaptive immunity, KRAS-linked cytokine expression can induce mechanistic alterations in the innate immune system, thus impeding adaptive immune activation and treatment response [82]. For example, KRAS-mutation status has been linked to PPARδ-CCL2 expression (involved in M2 macrophage transformation and recruitment) [83, 84], reduced NK cell cytotoxicity by increased expression of HLA-E [85] and PD-L1 [86], and epithelial-mesenchymal transition (EMT) [52], a sensitiser for tumours to NK cytotoxicity (Fig. 2). Indeed, KRAS/TP53 co-mutated tumours promote TNF-driven pro-tumour inflammation [87], and increases in NK-specific MHC complexes (MICA/B) [87, 88], while KRAS/MYC co-mutation in lung cancer drives the recruitment of anti-inflammatory macrophages and the blocking of NK infiltration by CCL9 and IL-23 (Fig. 1, Fig. 2) [34]. Collectively, these data support the potential for leveraging the innate immune system, particularly NK cells, in the treatment of KRAS-mutant tumours. Specific pro-immune mutations have been discovered in patients where NK cells harbour FcγR with the rs396991 genotype (FcγR3a), causing heightened affinity to the IgG1 Fc domain by NK cells and a subsequent increase in IgG1-driven antibody-dependent cell cytotoxicity (ADCC) by NK cells when using IgG1 based mAb therapies such as avelumab [38]. Treatments such as adoptive cell transfer [89] are being explored to fill this niche, although no clinical studies have commenced so far.
Metabolic and structural remodelling of the TME by oncogenic KRAS
KRAS drives TME remodelling via several pathways. Enhanced IL-1 and IL-6 expression has been observed in KRAS-mutant tumours [34], which leads to upregulation of IDO-1 in TILs via NF-kB and STAT3 signalling (Fig. 2) [90]. This results in a change in metabolism with a subsequent increase in kynurenine production and removal of tryptophan [35], simultaneously causing exhaustion and enhancing PD-1 expression in CD8 + T cells [8], increasing PD-L1 expression in myeloid cells, and stimulating the transformation of CD4+ T cells to FoxP3+ Tregs (Fig. 2) [91]. This is particularly relevant, as the proportion of CD8+/PD-1+ T cells relative to PD-1+ Tregs is a predictive biomarker for response to PD-1 blockade [92]. Furthermore, PD-1 expression is negatively correlated with the maturation of macrophages and DCs, and is associated with MDSC and TH17 differentiation within the TME (Fig. 2) [93]. TH17 are characterised by their production of IL-17, a proinflammatory cytokine that promotes pro-tumour inflammation and inhibits antitumour immunity through the recruitment of both MDSCs and neutrophils into the TME (Fig. 2) [94].
Oncogenic KRAS signalling can lead to increased expression of IL-8, IL-10, GM-CSF, CXCL1, CXCL12, CCL2 and MIF [7, 83], and decreased IL-18 [34], with the subsequent remodelling of the TME to an “immune-cold” contexture, which is more refractory to immunotherapy (Fig. 2). Moreover, secretion of these immune factors influences the recruitment of neutrophils, reduced inflammation via TH1 and macrophage inhibition, transformation of macrophages to M2-like TAMs [49, 95], triggering of chemotactic factor expression and MDSC infiltration (Fig. 2) [7, 96]. Such factors have already been targeted therapeutically, with adoptive CAR-T cell transfer in combination with IL-10 blockade being explored to combat resistance to anti-PD-L1 therapy by KRAS-driven cytokine expression [97]. Aberrant cytokine/chemokine signalling in KRAS-mutant tumours, such as IL-1β, IL-8, TGF-β and CXCL1 [34, 98], can also result in structural remodelling of the TME extracellular matrix (ECM) by neutrophils, CAFs [99] and M1 macrophages [34] resulting in degradation of ECM by MMP-9 and collagenase IV and the release of bound angiogenic factors such as VEGF [99], as well as cross-linking of ECM collagen fibres by lysyl oxidases (LOX) to enhance matrix stiffness and provide a physical barrier for immune cell infiltration (Fig. 2) [99, 100]. Interestingly, collagen-based discoidin domain receptor 1 (DDR1), CD44 and integrin signalling have been shown to stimulate both the MEK/ERK and PI3K/AKT axis independently of RAS signalling, and cause expression of PD-L1 in response to ECM stiffness (Fig. 2) [98]. Notably, DDR1 inhibition can attenuate KRAS-mutant tumour progression [101]. Taken together, the prospect of targeting TME remodelling is evolving to be a potential therapeutic avenue for reversing oncogene-driven immune evasion and the induced “immune-cold” state.
Preclinical testing of cancer immunotherapies
Historically, preclinical models for testing ICB have been restricted to highly mutated syngeneic mouse cancer cell lines. Although these have proved incredibly useful, their limited genetic diversity has restricted the number of targeted combinations that can be meaningfully tested in mice. In addition, autochthonous models of RAS-driven tumours have fewer mutations and are not responsive to immunotherapies [39]. Boumelha et al. recently developed an immunogenic KRAS-mutant lung cancer model (KPAR) [39] and compared it to a non-immunogenic (KPB6) mouse cancer cell line. Interestingly, expression of human APOBEC3B in KrasLSL−G12D/+/Trp53fl/fl/RosaA3Bi mice with Rag2−/− (KPAR) background failed to produce a TMB/neoantigen-high tumour, but a clonal line from the resulting tumour gave rise to an immunogenic model with MHC-I presentation of the retroviral antigen Emv2; a common tumour antigen in other immunogenic lines (CT26, B16, MC38) [102], as well as human breast cancer and melanoma [103]. When grown in immunocompetent Rag2± mice, the KPAR line displayed markers of increased immunogenicity, such as increased infiltration of TILs, PD-L1+ myeloid cells, and susceptibility to anti-CTLA-4/anti-PD-L1 combination treatment [39]. This model has been used to demonstrate the importance of IFN-γ in the immune response to KRAS-mutant tumours [7]. Of note, orthotopic tumours seeded using the KPAR line displayed differential TIL populations, ICB response and survival rates when compared to tumours initiated by subcutaneous injection, further indicating the similarity between this model and in situ tumours. The importance of orthotopic models in lung [104, 105] and colorectal cancer for the study of immune mechanisms have already been noted [106]. The KPAR model will be a useful addition to LL/2, an alternative mouse tumour model used for lung orthotopic tumour generation, which is extremely immune evasive [107, 108].
Future prospects for immune evasive KRAS tumours
As new instances of KRAS inhibitor/ICB-induced resistance are uncovered, new treatment opportunities are identified. For example, VISTA is an ICM receptor binding VSIG3 with high sequence and functional homology to PD-1. Elevated expression of VISTA on T cells, MDSCs and TAMs are associated with oncogenic KRAS-driven tumours [109] and resistance to anti-PD-1 by ICB [110, 111], identifying VISTA as a promising immunotherapy target. This was supported by the observation that in an acidic TME, as is common in KRAS-mutant tumours due to the enhancement of the Warburg effect [112], VISTA can also bind PSGL-1, another potent inhibitor of T cell activity promoting an exhausted phenotype (Fig. 1) [113]. Recently, the effects of a VISTA mAb (HMBD-002) inhibiting VSIG3 suppression of T cell activity, have been shown to be potentiated by a distinct shift in the TME to a pro-inflammatory phenotype, thus inhibiting tumour growth in vivo in preclinical syngeneic and humanised murine models of colorectal, lung, and breast cancer [114]. Indeed, a clinical trial testing HMBD-002 and pembrolizumab is currently recruiting (NCT05082610), with eagerly awaited results (Table 1). Additionally, preclinical testing of the VISTA-VSIG3/PSGL-1 blocking mAbs BMS-767, and SG7 have demonstrated enhanced T cell activity, though these have not yet progressed to clinical trials [110]. Future prospects for these mAbs potentially include co-treatment with anti-PD-1 ICB and KRASG12C inhibitors in immunotherapy-resistant NSCLC.
The first KRASG12C mutant-specific inhibitors provided significant benefit to patients harbouring KRASG12C-driven malignancies such as NSCLC [2, 7]. The most common KRAS mutation, G12D, is found in 29.5% of all cancers [15], and 40–50% of PDAC tumours [18, 115]. Recently, potent KRASG12D -specific inhibitors have been described (TH-Z816 [115] and MRTX1133 [116]). These compounds also exploit the same switch-II pocket as sotorasib but target the mutant aspartic acid. Interestingly, both MRTX1133 and TH-Z816 rely on the formation of a salt bridge rather than covalently bonding to the aspartic acid residue, with TH-Z816 able to inhibit both GDP and GTP-bound forms of KRAS, a mechanism which could potentially be exploited for other mutant-specific KRAS inhibitors [115, 116]. Both compounds have been shown to elicit significant tumour regression in mouse models. Notably, the regression in MRTX1133 was conducted in immunodeficient CD-1 mice [116], while a humanised C57BL/6 mouse model was used for TH-Z618 and also showed synergy with an anti-PD-L1 therapy [115].
Immune escape of tumour cells is often associated with an “immune-cold” and anti-inflammatory TME. As a result of these immunosuppressive mechanisms, and the invasive nature of tumours into surrounding tissues, microorganisms can often be found within tumours giving rise to an intratumoral microbiome, further altering the immune state of the tumour [117]. The effect of the microbiome in KRAS-mutant cancers has been recently characterised by Alam et al. [118] where they describe how activation of dectin-1 by the fungal microbiome of KRASG12D PDAC is crucial for enhanced expression of IL-33, the principal cytokine for the recruitment of innate lymphoid cells 2 (ILC2) into the TME (Fig. 2). This caused increased IL-13 and IL-5 signalling in the TME, resulting in increased M2 macrophage recruitment and immunosuppression; a process which was abated by antifungal treatment [118]. These changes to the TME enhanced tumour immunosuppression via inhibition of NK cells and enhancement of Tregs and MDSCs in both lung [119] and colorectal malignancies [120] (Fig. 2). Furthermore, infiltration of the gut microbiome is linked to TP53 conversion from tumour-suppressor function to oncogenic in CRC [121], a common co-mutation in KRAS-mutant tumours. Finally, dysbiosis of the microbiome has been linked with increased ICM expression, such as PD-1/L1 and CTLA-4 in solid tumours [122]. The effect of tumour microbiota on immune evasion in KRAS-mutant tumours, and how this is linked to tumour-intrinsic signalling, is an exciting area for future investigation.
Potential toxicities involved in novel combination therapies
Although combinations of ICB with MAPK pathway inhibitors have the potential to be highly effective, they could also have more serious toxicity profiles. MAPK pathway inhibition is generally associated with gastrointestinal (diarrhoea, colitis) and dermal toxicities (rash, paronychia), with rarer cases of cardio-pulmonary (dyspnea, hypoxia, hypotension) and other toxicities (pyrexia, acute inflammation, neurotoxicity) [123,124,125,126]. Toxicities for pan-KRAS inhibitors are unknown, but will likely have widespread activity in normal tissues, similar to MEK and ERK inhibitors [127,128,129]. Likewise, mTOR inhibitors have gastrointestinal and dermal effects, which could become higher grade in combination [130, 131]. Immunotherapies such as ICB, CAR-T cell, CCR2i and IL-10 blockade primarily present with autoimmune-linked toxicities, due to their effect on self-tolerance. Regular toxicities include acute inflammation (colitis, bowel abscesses, rash), cytokine storm, pyrexia, endocrinopathy, hypotension, and neuropathies [132,133,134,135,136,137]. MAPK modulators in combination with immunotherapies could worsen gastrointestinal and dermal toxicities specifically, or increase the frequency of rarer effects such as neuropathies. A phase 3 trial (NCT02908672) combining atezolizumab (ant-PD-L1), cobimetinib (MEKi) and vemurafenib (BRAFi), observed toxicities of diarrhoea, nausea, rash and pyrexia, with some participants experiencing acute inflammation, and peripheral neuropathy (Table 1) [138]. Combinations with DDR1i and AXLi, are predicted to have a similar toxicity profile, with diarrhoea, nausea, rash and inflammation common [139, 140]. DDR1 receptors are confined to the epithelial compartment and DDR1 inhibitors also cause inhibition of the MAPK pathway [139], therefore combining DDR1 inhibitors with MAPK inhibitors could lead to more severe toxicity. In summary, the toxicity profiles of monotherapies can inform the probable toxicities of drug combinations, but careful investigation of how these drugs may interact in patients is warranted in order to increase the success rate of these clinical trials.
Concluding remarks
Our growing understanding of how oncogenic signalling can affect immune evasion strategies has important therapeutic implications in oncology [141]. Targeted therapies can reverse immune evasion programs in KRAS-mutant tumours, clearly operating through cell-autonomous and non-cell-autonomous mechanisms [2, 7], which can be enhanced by combination with ICB. If performed correctly, this strategy could partly ameliorate key limitations of either treatment strategy alone—that is, a limited subset of responders in ICB, and the rapid onset of acquired resistance to targeted agents. The research community is developing more therapeutically relevant preclinical models to test these approaches, and we have highlighted a number of exciting ongoing trials to test these strategies in humans (Table 1). More research will be required to investigate how chemotherapies and targeted therapies influence other important factors, such as TMB [30], intratumoural heterogeneity [142], and the host/tumour microbiome. In the future, a deeper understanding of how the specific constellation of genetic drivers in a patient’s tumour influences the TME will ultimately improve precision medicine approaches and responses to immunotherapies.
Availability of data and materials
All data within this review is fully referenced and publicly available through the referenced unique DOI.
References
Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–63.
Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–23.
Coelho MA, de Carne TS, Rana S, Zecchin D, Moore C, Molina-Arcas M, East P, Spencer-Dene B, Nye E, Barnouin K, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity. 2017;47(1083–1099): e1086.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020;20:662–80.
Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. 2020;10:727–42.
Mugarza E, van Maldegem F, Boumelha J, Moore C, Rana S, Llorian Sopena M, East P, Ambler R, Anastasiou P, Romero-Clavijo P, et al. Therapeutic KRAS(G12C) inhibition drives effective interferon-mediated antitumor immunity in immunogenic lung cancers. Sci Adv. 2022;8:eabm8780.
Tang S, Ning Q, Yang L, Mo Z, Tang S. Mechanisms of immune escape in the cancer immune cycle. Int Immunopharmacol. 2020;86: 106700.
Ward AB, Keeton AB, Chen X, Mattox TE, Coley AB, Maxuitenko YY, Buchsbaum DJ, Randall TD, Zhou G, Piazza GA. Enhancing anticancer activity of checkpoint immunotherapy by targeting RAS. MedComm. 2020;2020(1):121–8.
Downward J. Targeting ras signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22.
Prior IA, Hood FE, Hartley JL. The frequency of Ras mutations in cancer. Can Res. 2020;80:2969–74.
Huang LM, Guo ZX, Wang F, Fu LW. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther. 2021;6:386.
Cook JH, Melloni GEM, Gulhan DC, Park PJ, Haigis KM. The origins and genetic interactions of KRAS mutations are allele- and tissue-specific. Nat Commun. 1808;2021:12.
Choi H, Deng JH, Li S, Silk T, Dong L, Brea EJ, Houghton S, Redmond D, Zhong H, Boiarsky J, et al. Pulsatile MEK inhibition improves anti-tumor immunity and T cell function in murine kras mutant lung cancer. Cell Rep. 2019;27:806.
Salem ME, El-Refai SM, Sha W, Puccini A, Grothey A, George TJ, Hwang JJ, O’Neil B, Barrett AS, Kadakia KC, et al. Landscape of KRAS(G12C), associated genomic alterations, and interrelation with immuno-oncology biomarkers in KRAS-mutated cancers. Jco Precis Oncol. 2022;6:e2100245.
Liu C, Zheng S, Wang Z, Wang S, Wang X, Yang L, Xu H, Cao Z, Feng X, Xue Q, et al. KRAS-G12D mutation drives immune suppression and the primary resistance of anti-PD-1/PD-L1 immunotherapy in non-small cell lung cancer. Cancer Commun (Lond). 2022;42:828–47.
Torralvo J, Friedlaender A, Achard V, Addeo A. The activity of immune checkpoint inhibition in KRAS mutated non-small cell lung cancer: a single centre experience. Cancer Genom Proteom. 2019;16:577–82.
Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2020;17:153–68.
Kubli SP, Berger T, Araujo DV, Siu LL, Mak TW. Beyond immune checkpoint blockade: emerging immunological strategies. Nat Rev Drug Discov. 2021;20:899–919.
Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–47.
Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJ, et al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity. 2018;49(178–193): e177.
El Osta B, Behera M, Kim S, Berry LD, Sica G, Pillai RN, Owonikoko TK, Kris MG, Johnson BE, Kwiatkowski DJ, et al. Characteristics and outcomes of patients with metastatic KRAS-mutant lung adenocarcinomas: the lung cancer mutation consortium experience. J Thorac Oncol. 2019;14:876–89.
Scheffler M, Ihle MA, Hein R, Merkelbach-Bruse S, Scheel AH, Siemanowski J, Bragelmann J, Kron A, Abedpour N, Ueckeroth F, et al. K-ras mutation subtypes in NSCLC and associated co-occuring mutations in other oncogenic pathways. J Thorac Oncol. 2019;14:606–16.
Yang Y, Shen SP, Sun YJ, Husain H, Zhou HY, Lu S, Li ZM. The relationship between different subtypes of KRAS and PD-L1 & tumor mutation burden (TMB) based on next-generation sequencing (NGS) detection in Chinese lung cancer patients. Transl Lung Cancer Res. 2022;11:213.
Ready N, Hellmann MD, Awad MM, Otterson GA, Gutierrez M, Gainor JF, Borghaei H, Jolivet J, Horn L, Mates M, et al. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. Journal of Clinical Oncology. 2019;37:992.
Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, Thai AA, Mascaux C, Couraud S, Veillon R, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol. 2019;30:1321–8.
Shen M, Qi R, Ren J, Lv D, Yang H. Characterization With KRAS mutant is a critical determinant in immunotherapy and other multiple therapies for non-small cell lung cancer. Front Oncol. 2021;11: 780655.
Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, Maeda T, Nagata Y, Kitanaka A, Mizuno S, et al. Aberrant PD-L1 expression through 3 ’-UTR disruption in multiple cancers. Nature. 2016;534:402–6.
Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, et al. Loss of tumor suppressor PTEN function increases B7–H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–8.
Litchfield K, Reading JL, Puttick C, Thakkar K, Abbosh C, Bentham R, Watkins TBK, Rosenthal R, Biswas D, Rowan A, et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell. 2021;184(596–614): e514.
Zhang Z, Rohweder PJ, Ongpipattanakul C, Basu K, Bohn MF, Dugan EJ, Steri V, Hann B, Shokat KM, Craik CS. A covalent inhibitor of K-Ras(G12C) induces MHC class I presentation of haptenated peptide neoepitopes targetable by immunotherapy. Cancer Cell. 2022;40(1060–1069): e1067.
Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang TD, Yang JS, Seestaller-Wehr L, Zhang SY, Hopson C, et al. The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res. 2015;21:1639–51.
Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, Gould SE, Maecker H, Irving BA, Kim JM, et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with pd-l1 checkpoint blockade. Immunity. 2016;44:609–21.
Hamarsheh S, Gross O, Brummer T, Zeiser R. Immune modulatory effects of oncogenic KRAS in cancer. Nat Commun. 2020;11:5439.
Liu M, Wang X, Wang L, Ma X, Gong Z, Zhang S, Li Y. Targeting the IDO1 pathway in cancer: from bench to bedside. J Hematol Oncol. 2018;11:100.
Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gutgemann I, Eilers M, Felsher DW. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–31.
Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, Littlewood TD, Evan GI. Myc cooperates with Ras by programming inflammation and immune suppression. Cell. 2017;171(1301–1315): e1314.
Stein A, Simnica D, Schultheiss C, Scholz R, Tintelnot J, Gokkurt E, von Wenserski L, Willscher E, Paschold L, Sauer M, et al. PD-L1 targeting and subclonal immune escape mediated by PD-L1 mutations in metastatic colorectal cancer. J Immunother Cancer. 2021. https://doi.org/10.1136/jitc-2021-002844.
Boumelha J, de Carne Trecesson S, Law EK, Romero-Clavijo P, Coelho M, Ng K, Mugarza E, Moore C, Rana S, Caswell DR, et al. An immunogenic model of KRAS-mutant lung cancer enables evaluation of targeted therapy and immunotherapy combinations. Cancer Res. 2022. https://doi.org/10.1158/0008-5472.CAN-22-0325.
Newey A, Griffiths B, Michaux J, Pak HS, Stevenson BJ, Woolston A, Semiannikova M, Spain G, Barber LJ, Matthews N, et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class I neoantigen landscape and no increase in neoantigens with interferon or MEK-inhibitor treatment. J Immunother Cancer. 2019;7:1–15.
Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, Sudhakar N, Bowcut V, Baer BR, Ballard JA, et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020;10:54–71.
Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, Johnson ML, Heist RS, Patil T, Riely GJ, et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N Engl J Med. 2021;384:2382–93.
Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, Arena S, Montone M, Mussolin B, Bian Y, et al. EGFR blockade reverts resistance to KRAS(G12C) inhibition in colorectal cancer. Cancer Discov. 2020;10:1129–39.
Fedele C, Li S, Teng KW, Foster CJR, Peng D, Ran H, Mita P, Geer MJ, Hattori T, Koide A, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med. 2021;218:e20201414.
Kwan AK, Piazza GA, Keeton AB, Leite CA. The path to the clinic: a comprehensive review on direct KRAS(G12C) inhibitors. J Exp Clin Cancer Res. 2022;41:27.
Plangger A, Rath B, Hochmair M, Funovics M, Hamilton G. Cytotoxicity of combinations of the pan-KRAS inhibitor BAY-293 against primary non-small lung cancer cells. Transl Oncol. 2021;14: 101230.
Ambrogio C, Kohler J, Zhou ZW, Wang H, Paranal R, Li J, Capelletti M, Caffarra C, Li S, Lv Q, et al. KRAS dimerization impacts MEK inhibitor sensitivity and oncogenic activity of mutant KRAS. Cell. 2018;172(857–868): e815.
Vasta JD, Peacock DM, Zheng QH, Walker JA, Zhang ZY, Zimprich CA, Thomas MR, Beck MT, Binkowski BF, Corona CR, et al. KRAS is vulnerable to reversible switch-II pocket engagement in cells. Nat Chem Biol. 2022;18:596–604.
Hou P, Kapoor A, Zhang Q, Li J, Wu CJ, Li J, Lan Z, Tang M, Ma X, Ackroyd JJ, et al. Tumor microenvironment remodeling enables bypass of oncogenic KRAS dependency in pancreatic cancer. Cancer Discov. 2020;10:1058–77.
Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17:651–62.
Lu Y, Chan YT, Tan HY, Li S, Wang N, Feng Y. Epigenetic regulation in human cancer: the potential role of epi-drug in cancer therapy. Mol Cancer. 2020;19:79.
Ischenko I, D’Amico S, Rao M, Li J, Hayman MJ, Powers S, Petrenko O, Reich NC. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat Commun. 2021;12:1482.
Cheng BB, Ren YC, Cao H, Chen JJ. Discovery of novel resorcinol diphenyl ether-based PROTAC-like molecules as dual inhibitors and degraders of PD-L1. Eur J Med Chem. 2020;199:112377.
Li XY, Pu WC, Zheng QQ, Ai M, Chen S, Peng Y. Proteolysis-targeting chimeras (PROTACs) in cancer therapy. Mol Cancer. 2022;21:99.
de Lima VCC, Corassa M, Saldanha E, Freitas H, Arrieta O, Raez L, Samtani S, Ramos M, Rojas C, Burotto M, et al. STK11 and KEAP1 mutations in non-small cell lung cancer patients: descriptive analysis and prognostic value among Hispanics (STRIKE registry-CLICaP). Lung Cancer. 2022;170:114–21.
Di Federico A, De Giglio A, Parisi C, Gelsomino F. STK11/LKB1 and KEAP1 mutations in non-small cell lung cancer: Prognostic rather than predictive? Eur J Cancer. 2021;157:108–13.
Ricciuti B, Arbour KC, Lin JJ, Vajdi A, Vokes N, Hong LZ, Zhang JJ, Tolstorukov MY, Li YY, Spurr LF, et al. Diminished efficacy of programmed death-(ligand)1 inhibition in STK11- and KEAP1-mutant lung adenocarcinoma is affected by KRAS mutation status. J Thorac Oncol. 2022;17:399–410.
Marinelli D, Mazzotta M, Scalera S, Terrenato I, Sperati F, D’Ambrosio L, Pallocca M, Corleone G, Krasniqi E, Pizzuti L, et al. KEAP1-driven co-mutations in lung adenocarcinoma unresponsive to immunotherapy despite high tumor mutational burden. Ann Oncol. 2020;31:1746–54.
Cheng W, Xu B, Zhang H, Fang S. Lung adenocarcinoma patients with KEAP1 mutation harboring low immune cell infiltration and low activity of immune environment. Thorac Cancer. 2021;12:2458–67.
Li ZQ, Ding B, Xu JX, Mao K, Zhang PF, Xue Q. Relevance of STK11 mutations regarding immune cell infiltration, drug sensitivity, and cellular processes in lung adenocarcinoma. Front Oncol. 2020;10:580027.
Friedmann Angeli JP, Meierjohann S. NRF2-dependent stress defense in tumor antioxidant control and immune evasion. Pigment Cell Melanoma Res. 2021;34:268–79.
Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi H, Nakayama K, Yamamoto M. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. 2016;7:11624.
Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. 2015;162:1257–70.
Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, Buczkowski KA, Liu Y, Awad MM, Denning WL, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016;76:999–1008.
Pons-Tostivint E, Lugat A, Fontenau JF, Denis MG, Bennouna J. STK11/LKB1 modulation of the immune response in lung cancer: from biology to therapeutic impact. Cells. 2021;10:3129.
Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco SE, Gay L, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-Mutant lung adenocarcinoma. Cancer Discov. 2018;8:822–35.
Wu D, Luo Y, Guo W, Niu Q, Xue T, Yang F, Sun X, Chen S, Liu Y, Liu J, et al. Lkb1 maintains Treg cell lineage identity. Nat Commun. 2017;8:15876.
Ricciuti B, Wang X, Alessi JV, Rizvi H, Mahadevan NR, Li YY, Polio A, Lindsay J, Umeton R, Sinha R, et al. Association of high tumor mutation burden in non-small cell lung cancers with increased immune infiltration and improved clinical outcomes of PD-L1 blockade across PD-L1 expression levels. JAMA Oncol. 2022;8:1160–8.
Pouremamali F, Pouremamali A, Dadashpour M, Soozangar N, Jeddi F. An update of Nrf2 activators and inhibitors in cancer prevention/promotion. Cell Commun Signal. 2022;20:100.
Li HY, Liu ZD, Liu LC, Zhang HY, Han CH, Girard L, Park H, Zhang AL, Dong CB, Ye JF, et al. AXL targeting restores PD-1 blockade sensitivity of STK11/LKB1 mutant NSCLC through expansion of TCF1(+) CD8 T cells. Cell Rep Med. 2022;3:100554.
Alcaraz-Sanabria A, Cabanas Morafraile E, Fernandez-Hinojal G, Velasco G, Perez-Segura P, Pandiella A, Gyorffy B, Ocana A. Transcriptomic mapping of non-small cell lung cancer K-RAS p.G12C mutated tumors: identification of surfaceome targets and immunologic correlates. Front Immunol. 2021;12:786069.
Cheng HY, Borczuk A, Janakiram M, Ren XX, Lin J, Assal A, Halmos B, Perez-Soler R, Zang XX. Wide expression and significance of alternative immune checkpoint molecules, B7x and HHLA2, in PD-L1-negative human lung cancers. Clin Cancer Res. 2018;24:1954–64.
Li YX, Liu Y, Zhao N, Yang XJ, Li YQ, Zhai FZ, Zang XX, Cui W. Checkpoint regulator B7x is epigenetically regulated by HDAC3 and mediates resistance to HDAC inhibitors by reprogramming the tumor immune environment in colorectal cancer. Cell Death Dis. 2020;11:753.
John P, Pulanco MC, Galbo PM, Wei Y, Ohaegbulam KC, Zheng DY, Zang XX. The immune checkpoint B7x expands tumor-infiltrating Tregs and promotes resistance to anti-CTLA-4 therapy. Nat Commun. 2022;13:2506.
Wang JY, Wang WP. B7-H4, a promising target for immunotherapy. Cell Immunol. 2020;347:104008.
Altan M, Kidwell KM, Pelekanou V, Carvajal-Hausdorf DE, Schalper KA, Toki MI, Thomas DG, Sabel MS, Hayes DF, Rimm DL. Association of B7–H4, PD-L1, and tumor infiltrating lymphocytes with outcomes in breast cancer. Npj Breast Cancer. 2018;4:40.
Schalper KA, Carvajal-Hausdorf D, McLaughlin J, Altan M, Velcheti V, Gaule P, Sanmamed MF, Chen LP, Herbst RS, Rimm DL. Differential expression and significance of PD-L1, IDO-1, and B7-H4 in human lung cancer. Clin Cancer Res. 2017;23:370–8.
Lu YW, Wu FY, Cao QY, Sun Y, Huang ML, Xiao J, Zhou B, Zhang L. B7–H4 is increased in lung adenocarcinoma harboring EGFR-activating mutations and contributes to immunosuppression. Oncogene. 2022;41:704–17.
Zhao Y, Murciano-Goroff YR, Xue JY, Ang A, Lucas J, Mai TT, Da Cruz Paula AF, Saiki AY, Mohn D, Achanta P, et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature. 2021;599:679–83.
Sachdev JC, Bauer TM, Chawla SP, Pant S, Patnaik A, Wainberg ZA, Inamdar SP, Marina N, Sun S, Schmidt M, et al. Phase 1a/1b study of first-in-class B7-H4 antibody, FPA150, as monotherapy in patients with advanced solid tumors. J Clin Oncol. 2019. https://doi.org/10.1200/JCO.2019.37.15_suppl.2529.
Thelen M, Wennhold K, Lehmann J, Garcia-Marquez M, Klein S, Kochen E, Lohneis P, Lechner A, Wagener-Ryczek S, Plum PS, et al. Cancer-specific immune evasion and substantial heterogeneity within cancer types provide evidence for personalized immunotherapy. Npj Precis Oncol. 2021;5:52.
Kitajima S, Asahina H, Chen T, Guo S, Quiceno LG, Cavanaugh JD, Merlino AA, Tange S, Terai H, Kim JW, et al. Overcoming resistance to dual innate immune and MEK inhibition downstream of KRAS. Cancer Cell. 2018;34(439–452): e436.
Schmall A, Al-Tamari HM, Herold S, Kampschulte M, Weigert A, Wietelmann A, Vipotnik N, Grimminger F, Seeger W, Pullamsetti SS, Savai R. Macrophage and cancer cell cross-talk via CCR2 and CX3CR1 is a fundamental mechanism driving lung cancer. Am J Respir Crit Care Med. 2015;191:437–47.
Liu Y, Deguchi Y, Wei D, Liu F, Moussalli MJ, Deguchi E, Li D, Wang H, Valentin LA, Colby JK, et al. Rapid acceleration of KRAS-mutant pancreatic carcinogenesis via remodeling of tumor immune microenvironment by PPARdelta. Nat Commun. 2022;13:2665.
Naseem M, Cao S, Stintzing S, Battaglin F, Tokunaga R, Puccini A, Berger MD, Soni S, Zhang W, Barzi A, et al. Variation in genetic polymorphisms and gene expression of HLA-E to predict outcomes in metastatic colorectal cancer (mCRC) patients (pts) treated with first-line FOLFIRI/cetuximab: data from the phase III FIRE-3 trial. J Clin Oncol. 2020;38:245–245.
Quatrini L, Mariotti FR, Munari E, Tumino N, Vacca P, Moretta L. The immune checkpoint PD-1 in natural killer cells: expression, function and targeting in tumour immunotherapy. Cancers (Basel). 2020;12:3285.
Datta J, Bianchi A, De Castro SI, Deshpande NU, Cao LL, Mehra S, Singh S, Rafie C, Sun X, Chen X, et al. Distinct mechanisms of innate and adaptive immune regulation underlie poor oncologic outcomes associated with KRAS-TP53 co-alteration in pancreatic cancer. Oncogene. 2022;41:3640–54.
Ding W, Ma Y, Zhu W, Pu W, Zhang J, Qian F, Zhou Y, Deng Y, Guo S, Wang J, Zhou X. MICA (*)012:01 Allele facilitates the metastasis of KRAS-Mutant colorectal cancer. Front Genet. 2020;11:511.
Lanuza PM, Alonso MH, Hidalgo S, Uranga-Murillo I, Garcia-Mulero S, Arnau R, Santos C, Sanjuan X, Santiago L, Comas L, et al. Adoptive NK cell transfer as a treatment in colorectal cancer patients: analyses of tumour cell determinants correlating with efficacy in vitro and in vivo. Front Immunol. 2022;13: 890836.
Meireson A, Devos M, Brochez L. IDO expression in cancer: Different compartment, different functionality? Front Immunol. 2020;11: 531491.
Opitz CA, Somarribas Patterson LF, Mohapatra SR, Dewi DL, Sadik A, Platten M, Trump S. The therapeutic potential of targeting tryptophan catabolism in cancer. Br J Cancer. 2020;122:30–44.
Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, Vitaly K, Itahashi K, Maeda Y, Matsui S, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. 2020;21:1346–58.
Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv. 2020;6:eabd2712.
Zhao J, Chen X, Herjan T, Li X. The role of interleukin-17 in tumor development and progression. J Exp Med. 2020;217:e20190297.
Hu J, Zhao Q, Kong LY, Wang J, Yan J, Xia X, Jia Z, Heimberger AB, Li S. Regulation of tumor immune suppression and cancer cell survival by CXCL1/2 elevation in glioblastoma multiforme. Sci Adv. 2021;7:eabc2511.
Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180:5771–7.
Sullivan KM, Jiang X, Guha P, Lausted C, Carter JA, Hsu C, Labadie KP, Kohli K, Kenerson HL, Daniel SK, et al. Blockade of interleukin 10 potentiates antitumour immune function in human colorectal cancer liver metastases. Gut. 2022;72:325–37.
Huang JC, Zhang LL, Wan DL, Zhou L, Zheng SS, Lin SZ, Qiao YT. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct Target Ther. 2021;6:153.
Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020;11:5120.
Jiang YF, Zhang HY, Wang J, Liu YL, Luo T, Hua H. Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J Hematol Oncol. 2022;15:34.
Ambrogio C, Gomez-Lopez G, Falcone M, Vidal A, Nadal E, Crosetto N, Blasco RB, Fernandez-Marcos PJ, Sanchez-Cespedes M, Ren XM, et al. Combined inhibition of DDR1 and Notch signaling is a therapeutic strategy for KRAS-driven lung adenocarcinoma. Nat Med. 2016;22:270–7.
Ottina E, Levy P, Eksmond U, Merkenschlager J, Young GR, Roels J, Stoye JP, Tuting T, Calado DP, Kassiotis G. Restoration of endogenous retrovirus infectivity impacts mouse cancer models. Cancer Immunol Res. 2018;6:1292–300.
Downey RF, Sullivan FJ, Wang-Johanning F, Ambs S, Giles FJ, Glynn SA. Human endogenous retrovirus K and cancer: Innocent bystander or tumorigenic accomplice? Int J Cancer. 2015;137:1249–57.
Bibby MC. Orthotopic models of cancer for preclinical drug evaluation: advantages and disadvantages. Eur J Cancer. 2004;40:852–7.
Li HY, McSharry M, Bullock B, Nguyen TT, Kwak J, Poczobutt JM, Sippel TR, Heasley LE, Weiser-Evans MC, Clambey ET, Nemenoff RA. The tumor microenvironment regulates sensitivity of murine lung tumors to PD-1/PD-L1 antibody blockade. Cancer Immunol Res. 2017;5:767–77.
Zhao X, Li L, Starr TK, Subramanian S. Tumor location impacts immune response in mouse models of colon cancer. Oncotarget. 2017;8:54775–87.
Mosely SI, Prime JE, Sainson RC, Koopmann JO, Wang DY, Greenawalt DM, Ahdesmaki MJ, Leyland R, Mullins S, Pacelli L, et al. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol Res. 2017;5:29–41.
Tran Chau V, Liu W, de Gerbe Thore M, Meziani L, Mondini M, O’Connor MJ, Deutsch E, Clemenson C. Differential therapeutic effects of PARP and ATR inhibition combined with radiotherapy in the treatment of subcutaneous versus orthotopic lung tumour models. Br J Cancer. 2020;123:762–71.
Papanicolau-Sengos A, Pabla S, Dy GK, Ernstoff MS, Puzanov I, Conroy JM, Nesline M, Glenn ST, Burgher B, Andreas J, et al. Correlation of lung cancer mutational profile with immune profile. J Clin Oncol. 2018. https://doi.org/10.1200/JCO.2018.36.5_suppl.146.
Yuan L, Tatineni J, Mahoney KM, Freeman GJ. VISTA: a mediator of quiescence and a promising target in cancer immunotherapy. Trends Immunol. 2021;42:209–27.
Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, Zhao H, Chen J, Chen H, Efstathiou E, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med. 2017;23:551–5.
Kawada K, Toda K, Sakai Y. Targeting metabolic reprogramming in KRAS-driven cancers. Int J Clin Oncol. 2017;22:651–9.
Tinoco R, Carrette F, Barraza ML, Otero DC, Magana J, Bosenberg MW, Swain SL, Bradley LM. PSGL-1 is an immune checkpoint regulator that promotes T Cell exhaustion. Immunity. 2016;44:1470–1470.
Thakkar D, Paliwal S, Dharmadhikari B, Guan S, Liu L, Kar S, Tulsian NK, Gruber JJ, DiMascio L, Paszkiewicz KH, et al. Rationally targeted anti-VISTA antibody that blockades the C-C ’ loop region can reverse VISTA immune suppression and remodel the immune microenvironment to potently inhibit tumor growth in an Fc independent manner. J Immunother Cancer. 2022;10(e003382):2022.
Mao ZW, Xiao HY, Shen PP, Yang Y, Xue J, Yang YY, Shang YG, Zhang LL, Li X, Zhang YY, et al. KRAS(G12D) can be targeted by potent inhibitors via formation of salt bridge. Cell Discov. 2022;8:5.
Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, Dahlke JR, Fell JB, Fischer JP, Gunn RJ, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRAS(G12D) inhibitor. J Med Chem. 2022;65:3123–33.
Johnston CD, Bullman S. The tumour-associated microbiome. Nat Rev Gastroenterol Hepatol. 2022;19:347–8.
Alam A, Levanduski E, Denz P, Villavicencio HS, Bhatta M, Alhorebi L, Zhang YL, Gomez EC, Morreale B, Senchanthisai S, et al. Fungal mycobiome drives IL-33 secretion and type 2 immunity in pancreatic cancer. Cancer Cell. 2022;40:153–67.
Schuijs MJ, Png S, Richard AC, Tsyben A, Hamm G, Stockis J, Garcia C, Pinaud S, Nicholls A, Ros XR, et al. ILC2-driven innate immune checkpoint mechanism antagonizes NK cell antimetastatic function in the lung. Nat Immunol. 2020;21:998–1009.
Spits H, Mjosberg J. Heterogeneity of type 2 innate lymphoid cells. Nat Rev Immunol. 2022;22:701–12.
Kadosh E, Snir-Alkalay I, Venkatachalam A, May S, Lasry A, Elyada E, Zinger A, Shaham M, Vaalani G, Mernberger M, et al. The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature. 2020;586:133–8.
Ramirez-Labrada AG, Isla D, Artal A, Arias M, Rezusta A, Pardo J, Galvez EM. The influence of lung microbiota on lung carcinogenesis, immunity, and immunotherapy. Trends Cancer. 2020;6:86–97.
Li Y, Fu R, Jiang T, Duan D, Wu Y, Li C, Li Z, Ni R, Li L, Liu Y. Mechanism of lethal skin toxicities induced by epidermal growth factor receptor inhibitors and related treatment strategies. Front Oncol. 2022;12: 804212.
Chin HM, Lai DK, Falchook GS. Extracellular Signal-Regulated Kinase (ERK) Inhibitors in Oncology Clinical Trials. J Immunother Precis Oncol. 2019;2:10–6.
Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, Falchook GS, Price TJ, Sacher A, Denlinger CS, et al. KRAS(G12C) inhibition with Sotorasib in advanced solid tumors. N Engl J Med. 2020;383:1207–17.
Livingstone E, Zimmer L, Vaubel J, Schadendorf D. BRAF, MEK and KIT inhibitors for melanoma: adverse events and their management. Chin Clin Oncol. 2014;3:29.
Gillson J, Ramaswamy Y, Singh G, Gorfe AA, Pavlakis N, Samra J, Mittal A, Sahni S. Small molecule KRAS inhibitors: The future for targeted pancreatic cancer therapy? Cancers (Basel). 2020. https://doi.org/10.3390/cancers12051341.
Parikh K, Banna G, Liu SV, Friedlaender A, Desai A, Subbiah V, Addeo A. Drugging KRAS: current perspectives and state-of-art review. J Hematol Oncol. 2022;15:152.
Plangger A, Rath B, Stickler S, Hochmair M, Lang C, Weigl L, Funovics M, Hamilton G. Cytotoxicity of combinations of the pan-KRAS SOS1 inhibitor BAY-293 against pancreatic cancer cell lines. Discov Oncol. 2022;13:84.
Graham L, Banda K, Torres A, Carver BS, Chen Y, Pisano K, Shelkey G, Curley T, Scher HI, Lotan TL, et al. A phase II study of the dual mTOR inhibitor MLN0128 in patients with metastatic castration resistant prostate cancer. Invest New Drugs. 2018;36:458–67.
Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for cancer therapy. J Hematol Oncol. 2019;12:71.
Ceschi A, Noseda R, Palin K, Verhamme K. Immune checkpoint inhibitor-related cytokine release syndrome: analysis of WHO global pharmacovigilance database. Front Pharmacol. 2020;11:557.
Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, Shabafrouz K, Ribi C, Cairoli A, Guex-Crosier Y, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16:563–80.
Brammer JE, Braunstein Z, Katapadi A, Porter K, Biersmith M, Guha A, Vasu S, Yildiz VO, Smith SA, Buck B, et al. Early toxicity and clinical outcomes after chimeric antigen receptor T-cell (CAR-T) therapy for lymphoma. J Immunother Cancer. 2021. https://doi.org/10.1136/jitc-2020-002303.
Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, Komanduri KV, Lin Y, Jain N, Daver N, et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15:47–62.
Wang H, Zhou F, Zhao C, Cheng L, Zhou C, Qiao M, Li X, Chen X. Interleukin-10 is a promising marker for immune-related adverse events in patients with non-small cell lung cancer receiving immunotherapy. Front Immunol. 2022;13: 840313.
Noel M, O’Reilly EM, Wolpin BM, Ryan DP, Bullock AJ, Britten CD, Linehan DC, Belt BA, Gamelin EC, Ganguly B, et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest New Drugs. 2020;38:800–11.
Ascierto PA, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, Pereira RP, Eigentler T, Rutkowski P, Demidov L, et al. Overall survival with first-line atezolizumab in combination with vemurafenib and cobimetinib in BRAF(V600) mutation-positive advanced melanoma (IMspire150): second interim analysis of a multicentre, randomised, phase 3 study. Lancet Oncol. 2023;24:33–44.
Chen L, Kong X, Fang Y, Paunikar S, Wang X, Brown JAL, Bourke E, Li X, Wang J. Recent advances in the role of discoidin domain receptor tyrosine Kinase 1 and discoidin domain receptor tyrosine Kinase 2 in breast and ovarian cancer. Front Cell Dev Biol. 2021;9: 747314.
Sang YB, Kim JH, Kim CG, Hong MH, Kim HR, Cho BC, Lim SM. The development of AXL Inhibitors in lung cancer: recent progress and challenges. Front Oncol. 2022;12: 811247.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10.
McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–9.
Acknowledgements
We thank Mathew Garnett for comments and discussion, the Translational Cancer Genomics team and the iFuncOnc project team, especially Gabriele Picco for comments on the manuscript. Figures were created with BioRender.com.
Funding
This research was funded in whole, or in part, by the Wellcome Trust Grant 206194 and 220540/Z/20/A. For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission. AW and MAC are funded by Open Targets (OTAR2061).
Author information
Authors and Affiliations
Contributions
AW and MAC designed figures and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors have no competing interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Watterson, A., Coelho, M.A. Cancer immune evasion through KRAS and PD-L1 and potential therapeutic interventions. Cell Commun Signal 21, 45 (2023). https://doi.org/10.1186/s12964-023-01063-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01063-x