
Abstract
Recent insights into the molecular and cellular mechanisms underlying cancer development have revealed the tumor microenvironment (TME) immune cells to functionally affect the development and progression of breast cancer. However, insufficient evidence of TME immune modulators limit the clinical application of immunotherapy for advanced and metastatic breast cancers. Intercellular STAT3 activation of immune cells plays a central role in breast cancer TME immunosuppression and distant metastasis. Accumulating evidence suggests that targeting STAT3 and/or in combination with radiotherapy may enhance anti-cancer immune responses and rescue the systemic immunologic microenvironment in breast cancer. Indeed, apart from its oncogenic role in tumor cells, the functions of STAT3 in TME of breast cancer involve multiple types of immunosuppression and is associated with tumor cell metastasis. In this review, we summarize the available information on the functions of STAT3-related immune cells in TME of breast cancer, as well as the specific upstream and downstream targets. Additionally, we provide insights about the potential immunosuppression mechanisms of each type of evaluated immune cells.

Video abstract
Similar content being viewed by others

Introduction
Breast cancer is one of the most prevalent type of gynecological cancer across the globe. As per estimates 2,088,849 new breast cancer cases and 626,679 breast cancer related deaths were reported in 2018 [1]. Owing to its complex pathology, breast cancer is generally diagnosed at advanced stages when it has already spread to different and distant body parts [2]. As a highly malignant tumor, breast cancer exhibits considerable metastatic potential and often leads to treatment failure and death [3]. Breast cancer can be divided into three types: hormone receptor-positive (estrogen receptor (ER) or progesterone receptor (PR)) constitutes 70% of breast cancer cases, ERBB2-positive constitutes 15–20% of breast cancer cases and triple negative breast cancer (ER−, PR− and ERBB2−) constitutes 15% of breast cancer cases [2]. For management of breast cancer, patients with local disease usually undergo surgery and/or radiation therapy, while the cytotoxic chemotherapy, biologic therapy and endocrine therapy are generally applied to systemic metastasis [4]. Although the breast cancer death rate has decreased by 39% in the last one decade owing to recent advancements in breast cancer diagnostics and therapeutics, almost all patients who are diagnosed with advanced stage and metastatic disease eventually succumb to it [5]. Innovative approaches to reduce frequent relapse of breast cancer and to decrease the death rate is need of the hour. There is concrete evidence in literature that immune system plays an important role in the response of patient reaction to both standard and long-term therapy [6]. The evolving interaction between breast tumor and human immunity was characterized by immunoediting, such as tumor cells death, dendritic cells (DCs) maturation, and effector T cells response [7]. Owing to the heterogeneity of the breast cancer [8], the effect of complex tumor microenvironment (TME) on immunotherapy still lacks sufficient validation.
STAT3 is the family member of signal transduction and transcriptional activators (STAT) proteins, which regulate the gene expression related to cell survival and immune response associated with tumor progression and malignancy [9,10,11]. Generally, STAT3 is localised in the cytoplasm of resting cells in an inactive form [12]. Once activated, STAT3 undergoes phosphorylation, homodimerization, nuclear translocation and DNA binding, subsequently driving the tumor proliferation, differentiation, apoptosis, cell transformation, invasion, angiogenesis, and immune evasion [13] (Fig. 1). Inhibitors of STAT3 have been reported to inhibit cell proliferation and promote the apoptosis of lung cancer, gastric cancer, colorectal cancer, leukemia, melanoma, renal cancer and breast cancer, to name a few [14]. Likewise, immunosuppression and tumor promotion are magically integrated into STAT3 cascade [15,16,17]. Accumulating evidence revealed that STAT3 is an important oncoprotein in an overly complex TME [18,19,20,21]. Breast cancers risk, metastasis, recurrence, and response to treatment is affected by multiple non-malignant cell types in TME, such as macrophages, mast cells, B cells, regulatory T cells (Tregs), DCs, and natural killer cells (NKs) [22, 23]. Recently, STAT3 signaling has been shown to pay a role in immune cells and promotes immunosuppressive function in the TME [24]. It is widely accepted that the immunological insult in TME and the activated immunosuppressive molecules in human cancers are essential in modulating the tumor milieu and tumor progression [18, 25]. STAT3 signaling activation is magically converged in both tumor promotion and immunosuppression, such as the crosstalk between tumor cells and immune cells [18]. Meanwhile, the immune system has a key role in the standard treatment response and long-term survival of breast cancer patients [6]. Recent insights indicate that breast cancer outcomes are determined by the type of elicited immune responses [26,27,28]. Of note, a lot of studies have revealed that STAT3 cascade was associated with breast cancer immune responses [29, 30], which can potentiate signaling in TME immune cells and tumor cells. However, the effects of STAT3 cascade on immune cells in breast cancer tumor immune microenvironment is yet to be elucidated.

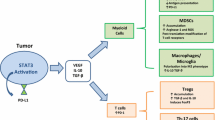
A schematic depiction of constitutive STAT3 activation in immune cells. The phosphorylation of STAT3 is triggered by tumor-derived factors and genetic stress. The activated STAT3 subsequently translocated to the nuclear, where it binds to the DNA at specific site and leads to increased transcription of target genes, thereby contributing to immune cells generation, polarization and immunosuppression properties
In this review, we intend to establish the interaction between STAT3 and immune cells in TME of breast cancer, focusing on the immune cells function and population. Describing above appears to imply an ideal target for breast cancer immunotherapy. STAT3 is not only overactive in different cells of breast cancer milieu, but also simultaneously determines opposite patterns of anti-tumor and pro-tumor immune cells, such as downregulated T cells and upregulated Tregs. The specific inhibition of STAT3 has emerged as a promising strategy to improve the TME, immune surveillance, tumor progression and metastasis of breast cancer. The treatment arms of STAT3 inhibition in combination with radiotherapy exhibits the potential to ameliorate the immunosuppression and favors the systemic immune response.
STAT3-related immune cells in breast cancer tumor microenvironment
The TME plays a crucial role in tumor progression, treatment response and patient prognosis. Myeloid-derived suppressor cells (MDSCs), DCs, Tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), NKs, B cells and T cells are the main immune cells in the TME [31, 32]. As one of many important regulatory factors in TME, STAT3 is a key target that connects the microenvironment with tumor cells [31]. The following is the role of STAT3-related immune cells (MDSCs, macrophages, DCs, and T cells) in the TME of breast cancer (Table 1).
MDSCs
As a heterogeneous population of myeloid progenitor cells in TME, myeloid-derived suppressor cells (MDSCs) are associated with inflammation, tumor progression and metastasis [33, 34]. By suppressing CD4+ T cells, CD8+ T cells and NK cells, recruited MDSCs can inhibit innate and adaptive immune response, leading to the eliminative dysfunction of immune system and suppression of immune surveillance [33, 35]. While markers for the heterogeneous MDSCs have not been defined well because different tumor types have different markers for MDSCs [36]. In breast cancer, phosphorylated-STAT3 directly induced indoleamine 2,3-dioxygenase (IDO) expression in MDSCs by binding to the promotor of IDO which is involved in immunosuppressive effects between breast cancer-derived MDSCs on T cells [37] (Fig. 2a). IDO, a rate-limiting enzyme in tryptophan catabolism, is highly expressed in MDSCs isolated from fresh breast cancer tissues and is associated with tumor-induced immunosuppression by suppressing T cell function [38, 39]. Moreover, MDSCs have been reported to activate the STAT3 mediated inhibition of T cell expansion and Th1 polarization via the IDO manner in breast cancer [39]. Meanwhile, STAT3 cascade blocking has been shown to significantly decrease the IDO expression in MDSCs, tumor development, and metastasis [40]. Noteworthy, intranuclear p52 and RelB (p52/RelB complex) has also been found to be dramatically decreased after STAT3 blocking, which is a dimer involved in NF-κB pathway activation and specific immunological processes [41], implicating that noncanonical NF-κB pathway participated in the STAT3-induced IDO expression and TME immunosuppression [37, 42]. Through the negative association between IDO and T cells CD3ζ-chain/IFN-γ expression, activated IDO from the MDSCs directly abolished the T cells immunity in TME [43]. Furthermore, STAT3 has also been shown to be negatively associated with the downstream interferon regulatory factor–8 (IRF-8) expression via the promoter engagement. This eventually promotes the MDSCs phenotype, which is directly mediated by G-CSF in breast cancer [44, 45]. Downregulation of IRF-8 has been found to facilitate the development and accumulation of MDSCs [44, 46] (Fig. 2d). Therefore, STAT3-related MDSCs generation is considered as a major obstacle to anti-tumor immunotherapy.

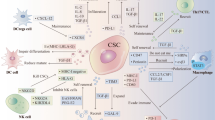
The mechanism of STAT3-related immune cells at in breast cancer TME. Three primer immune cells can be targeted when STAT3 influences the TME of breast cancer. The immune cells population, phenotypes and related gene expression were shaped in tumor milieu. a Node A: MDSCs conditioned by STAT3 cascade in TME induced paralysis of T lymphocytes, activity of CSCs, and carcinogenic factors generation. Meanwhile, the release of ROS might enhance the immunosuppression in various routes. b Node B: Intracellular STAT3 cascade in the macrophages affects the PGE2 and PD-L1 secretion, and induces the Sox2-positive CSCs in TME. Moreover, the HA and A-FABPs induced STAT3 activation is directly associated with TAMs formation and enables the interaction between tumor cells and macrophages, such as promoting TGF-β1 and HIF-1α generation. c Node C: STAT3 cascade suppressed DCs differentiation and deprived the DCs ability to stimulate T cells. Through inhibiting CD86 expression, STAT3 indirectly inhibited the CTLA-4 and promoted IFN-γ expression in TME. Moreover, FLT3L-induced DCs accumulated in immunization site and significantly increased the anti-tumor T cells response and remarkably delayed the tumor growth. The FL3TL/STAT3/Pu.1 cascade promote the differentiation and maturation of DCs, while FL3TL/STAT3 interacts with E2-2/Tcf4 pathway to enhance pDCs-related immune response. d Node D: STAT3 cascade in tumor cells inhibits the MDSCs in TME, which was directly mediated by intercellular G-CSF/IRF-8 function. The co-culture between tumor cells and DCs stimulated STAT3-related HER-2/neu, TGF-β1 and HIF-1α generation. Moreover, the macrophages related PEG2 in TME might stimulate PI3K/Akt pathway via the tumor surface EP4 receptor recognition, which was closely connected to breast cancer cells metastasis
For the upstream, reactive oxygen species (ROS) and IL-6 triggered STAT3 activation regulates MDSCs expansion in the breast cancer, which are both typical characteristics of MDSCs [40, 43, 47]. The IL-6 level in TME has been shown to be positively correlated with infiltration of MDSCs in situ and contributed to worse clinical outcomes [48, 49]. Among which, ADAM proteases (ADAM10)-induced soluble IL-6 was particularly involved in IL-6 trans-signaling and accompanied with the enhanced STAT3 phosphorylation in MDSCs (Fig. 2a). Additionally, infiltrated MDSCs might facilitate the shedding of IL-6 receptor, eventually promoting invasion and metastasis of breast cancer cells via IL-6 trans-signaling [50]. With the IL6-dependent STAT3 stimulation, MDSCs-derived nitric oxide (NO)NO/NOTCH signaling can promote and maintain the persistent phosphorylation of STAT3 [51]. Furthermore, cancer stem cells (CSCs) are associated with tumor progression and treatment resistance [52]. Through STAT3 and NOTCH signaling, MDSCs induced human breast CSCs in TME and exhibited poor survival rate [51]. Among which, IL-6 and MDSCs-derived NO collaboratively stimulated STAT3 and NOTCH signaling. In addition to IL-6, IL-17 appeared to positively regulate the differentiation of MDSCs into macrophages and mDCs, as well as activation of STAT3 [53]. Data also indicated that the levels of IL-17 were considerably downregulated in patients with breast cancer as compared to the healthy blood donors [53].
Recently, Yin et al. reported that the inhibitor of Aurora-A kinase (Alisertib) plays a remarkable role in regulating the immunosuppressive functions of STAT3 and MDSCs in the TME of breast cancer [54]. Aurora-A is a conserved serine/threonine kinase and is associated with poor prognosis and drug resistance [55, 56]. Of note, Alisertib has been used in many preclinical studies and clinical trials, including a recent five-arm phase II study against advanced breast cancer and small cell lung cancer [57]. Alisertib can significantly decrease the MDSCs and enrich T lymphocytes in the TME of breast tumor and peripheral organs, which were essential for the breast cancer regression. Indeed, the administration of alisertib ameliorated immunosuppressive function of MDSCs by inhibiting STAT3-mediated ROS generation in breast cancer (Fig. 2a) [54]. Activated-STAT3 (Tyr705) and ROS levels have been shown to significantly downregulated in MDSCs from alisertib-treated mice. T-cell suppressive activity of MDSCs has been found to be positively associated with ROS generation in a STAT3-dependent manner [58]. Moreover, S100A8/A9 (two modulators of the ROS generation in MDSCs) expression was considerably decreased after alisertib treatment [59, 60], which both have been demonstrated to be regulated by STAT3 [61]. Therefore, constitutive activation of STAT3 regulates MDSCs and anti-cancer T lymphocytes population in breast cancer TME.
Macrophages
Macrophages are essential for host defense and can be divided into two distinct forms, M1-macrophages and M2-macrophages. While the M1-macrophages produce IL-12 to promote Th1 response, and the M2-macrophages sustain Th2-associated effector functions and secrete tumor supportive factors [62, 63]. TAMs are closely similar to M2-polarized macrophages and promote immune evasion of breast tumor cells [64, 65]. Clinical and experimental data demonstrated that a high density of TAMs is associated with both poor prognosis and metastasis in breast cancer patients [66,67,68]. In TME of breast cancer, ERK/STAT3 cascade has emerged as a pivotal regulator to stimulate macrophage M2-like polarization and promote tumor progression and metastasis [64]. Accordingly, STAT3 inhibitor in combination with ERK inhibitor has been found to significantly suppress the M2 macrophage polarization and expression of markers including CD206 and Arg-1. Likewise, the expression of hyaluronan (HA) in TME of breast cancer patients is positively correlated with the amount of M2 macrophages [69] (Fig. 2b). HA is the most important component of extracellular matrix (ECM) and is associated with poor prognosis of breast cancer [70]. With the recognition by HA major receptor CD44 on macrophages [71], TME-derived HA activated the STAT3 cascade and formation of TAMs [72]. In addition, STAT3-inhibitor S3I-201 [72], simultaneously suppressed the STAT3 phosphorylation and macrophages transformation in breast cancer TME as compared to control. Meanwhile, STAT3 blocking in macrophages is positively associated with PTGS2 expression, which triggers the cyclooxygenase-2 (COX-2) to drive PGE2 generation [73]. Intriguingly, increased pro-tumorigenic factor COX-2 in TME might contribute to the attenuated therapeutic responses of breast cancer to ruxolitinib and promote tumor progresses [73, 74]. The PGE2 has been reported to active AKT via PI3K signaling pathway, which is associated with tumor cells proliferation and survival [75, 76]. Consistently, COX-2-mediated PEG2 expression has been shown to be intricately connected to lymph angiogenesis and lymphatic metastasis via PI3K/Akt-dependent receptor EP4 recognition in breast cancer cells [77, 78] (Fig. 2d). Thus, the pros or cons of STAT3 cascade target therapy need to be further illustrated.
Recently, Hao et al. reported the intracellular adipocyte/macrophage fatty acid binding protein (A-FABP) expression to be negatively associated with breast cancer survival via facilitation of the STAT3 cascade in TAM [67] (Fig. 2b). FABP does not only regulate the inflammatory and cellular metabolic pathways, but also affects the macrophages function and phenotype [79,80,81]. A-FABP has been shown to exhibit the ability to promote IL-6 production through NFκB/miR29b pathway in macrophages, which eventually boosted the phosphorylation of STAT3 [67]. In contrary, A-FABP knockdown or anti-IL-6 significantly decreased the STAT3 phosphorylation level in macrophages, subsequently inhibiting the tumor colony formation and metastasis. Moreover, IL-6 activated the STAT3 was specifically enriched in co-culture system between macrophages and breast cancer MCF-7 cells as compared to control, which also showed significant upregulation of transforming growth factor β (TGF-β1) and hypoxia-inducible factor-1α (HIF-1α) mRNA levels [82] (Fig. 2d). Noteworthy, HIF-1α, as a STAT3 downstream target, has been proved to be associated with angiogenesis [83, 84]. STAT3 positively has been shown to positively regulate the expression of TGF-β1 in breast cancer [85], thereby promoting cancer cell proliferation and metastasis [86]. Another co-culture system between macrophages and 4T1 cells revealed that STAT3 was not only activated in macrophages, but also promoted the PD-L1 secretion on macrophages during interaction with breast cancer cells (Fig. 2b) [87]. High PD-L1 expression macrophages have been reported to directly promote apoptosis and inhibit the proliferation via suppressing of activation of autologous T cells [88]. Of note, the interaction between PD-L1 and macrophages surface PD-1 induced an alternative M2-like polarization, resulting in crippling effects on tumor immunity [89]. Furthermore, the crosstalk between TAMs and breast cancer cells showed an ability to induce CSCs phenotype formation with upregulated Sox-2 expression via activation of STAT3 cascade [90]. Accordingly, Sox-2 expression was directly inhibited by STAT3 inhibitor 2-cyano-3,12 dioxooleana-1,9 dien-28-imidazolide (CDDO-Im) treatment. Increased CSCs have been shown to boost the metastasis, chemotherapy resistance, and tumorigenicity in vivo [90, 91]. Although targeting TAMs is a promising clinical tumor immunotherapy strategy, it is difficult to achieve in practice due to the heterogeneous and dynamic nature of macrophages in TME [63, 92, 93]. Against this backdrop, modulating the intercellular STAT3 cascade of macrophages may provide an opportunity to improve therapeutic efficacy of the breast cancer immunotherapy.
Dendritic cells
As primary antigen-presenting cells (APCs), conventional dendritic cells (cDCs) play a main role in adaptive immune response initiation and regulation [94, 95]. The cDCs consist of type 1 (cDC1) and type 2 (cDC2) subsets [96]. In immune system, DCs suppress cancer growth and spread, even when the disease has advanced [97]. The presence of DCs in TME is positively correlated with patient survival. It is now thought that tumor-derived factors (TDFs) IL-6, VEGF, and G-CSF secreted by tumor itself in TME of breast cancer have the ability to stimulate STAT3 cascade in myeloid cells differentiation [34, 98]. As a result, STAT3 activation impaired the generation of DCs as well as DCs function. Mechanically, in both advanced breast cancer patients and breast tumor-bearing mice, STAT3 directly decreased the PKCβII protein and PRKCB2 expression by binding to negative regulatory elements of the PRKCB promoter [98], eventually leading to the suppression of DCs formation and this subtle change can delay the T cells activity, namely CD8+ CTLs (Fig. 2c) [99, 100]. PKCβII has been deemed as splice variant of the PRKCB gene [101], which drives the differentiation of myeloid progenitor cells to DCs [99, 102]. As a negative feedback loop, PKCβII activity inhibited the ability of TDFs to activate STAT3 cascade, via reduction of abundance of the cell surface receptors recognition to TDFs [98]. Knock down of STAT3 in TME of breast tumor results in the downregulation of the surface expression of on tumor cell via the DCs activation [103], and HER-2/neu is a proto-oncogene linked to breast cancer progression and metastasis [103, 104]. Recently, STAT3-deficient cDCs (STAT3− cDCs) was reported to inhibit the breast cancer growth and poor survival as a cell-based vaccine. STAT3− cDCs injection was positively associated with antigen-specific T cells accumulation in breast TME and tumor-related lymph nodes compared to blank control [105]. In addition, upregulated surface CD86 expression was observed rather than CD80 or MHC II due to STAT3 abolishment. DCs-generated co-stimulatory activation (CD86) inhibited the CTLA-4 secretion in breast cancer lymph nodes, promoted intercellular IFN-γ expression, and recognized the CD28 receptor in T cells, therefore promoting T cell accumulation and activity [106,107,108] (Fig. 2c). Furthermore, with the specific receptor IL10RB interaction, IL-10-related signaling plays an important role in STAT3-elicited cDCs immunosuppressive responses of breast TME [105]. Ablating the IL-10/STAT3 cascade dramatically improved the effector T cells amounts. Consistently, increased amount of T cells in TME was linked to efficacy of immunotherapy in breast cancer [109]. The inhibition of STAT3 in migratory cDCs might be a novel immunotherapy strategy for management of metastatic and advanced breast cancer [105].
Intriguingly, another important DCs growth regulator, FMS-related tyrosine kinase 3 ligand (FLT3L) showed the ability to promote DCs proliferation via STAT3-dependent manner [110, 111]. STAT3 activation acts as a checkpoint of FLT3L-regulated DC diversification (Fig. 2c). Moreover, in TME of breast cancer, FLT3L-induced DCs accumulated in immunization site and significantly increased the anti-tumor T cells response and remarkably delayed the tumor growth [112, 113]. While the expression of FLT3L is prevalent in lymphoid (60%–70%) and myeloid (50%–65%) progenitors, absence of STAT3 restricted myeloid differentiated into DCs progeny than T and B lymphocytes [111]. Therefore, STAT3 is a non-redundant regulator of FLT3L-mediated DCs differentiation. One hypothesis is that STAT3-actived Pu.1 expression in lymphocyte and myeloid cells differentiation [114, 115], and high level of FLT3L initiated DCs differentiation via the STAT3/Pu.1 cascade [116]. Nonetheless, the high expression of IL-32 in breast cancer might suppress the STAT3 binding to the Pu.1 promoter, thereby leading to the abrogated maturation of DCs and tumor progression [114, 117].
Furthermore, in plasmacytoid DCs (pDCs), FLT3L used STAT3 to selectively induce the E protein E2-2/Tcf4 interaction, which is essential for the pDC population (Fig. 2c), but not for the other DC lineages in TME [110, 118]. Among which, STAT3 physical interacted with Tcf4 promoters suggesting a direct mechanism of gene induction [110]. Although it is generally assumed that binding of β-catenin to members of the TCF family is cancer-promoting, recent studies have shown that Tcf4 functions instead as a repressor that restricts the breast cancer cell growth [119]. In addition, activation of pDCs contributed to higher killing efficacy of effector lymphocyte in TME of breast cancer model, including FLT3L-induced pDCs [120, 121]. In yet another study, simultaneous inhibition of STAT3 and Tcf4 signaling pathways was reported to suppress the breast cancer cells metastasis both in vitro and in vivo [122]. Despite insufficient evidence, FLT3L/STAT3 cascade directly conducted pDC and anti-tumor immunity via Tcf4 in breast cancer. As such Tcf4 might be used act as a new prognostic biomarker and valuable molecular target for breast cancer immunotherapy.
STAT3-relevant T cells immunosuppression
STAT3 is constitutively activated in all subtypes of breast cancers and particularly plays an essential role in their immunosuppression [123]. Activated-STAT3 in tumor cells not only dampens the generation of immunostimulatory mediator, but also boosts the release of the immunosuppressive factors leading to the stimulation of crosstalk between tumor cells and T cells in TME [124] (Fig. 3). Herein, we discuss the STAT3 related T cells immunosuppression in breast cancer as follows.

The roles of STAT3 in different immune cells immunosuppression. a: For immune activation cells (Left panel), STAT3 directly regulate MDSCs to product IL-10 and ROS in TME, both of which indirectly cause CD4+ T cells to lose their ability to inhibit tumors. The STAT3-induced ROS impaired the CD8+ T cells, while STAT3 also indirectly suppress the CD8+ T cells through inhibiting the INF-α/β generation in breast cancer TME. As a nonredundant regulator of CD8+ T cells, activated-STAT3 was also involved in intercellular glycolysis via promoting the FAO expression. b For immunosuppressive cells, however (Right panel), STAT3 not merely promote the Tregs through triggering IDO (RelB-p52 binding) and IL-10 generation from MDSCs, but also was directly involved in Tregs specific marker Foxp3 expression and immunosuppressive effect. The activated-STAT3 promoted the naive peripheral CD4 + T cells conversion into Tregs, simultaneously inhibiting the IL-2 and IFN-γ production from converted lymphocytes
CD4+ T cells
As previous study has reported that blocking of STAT3 in breast cancer cells induced an antitumor immune response involving CD4+ T cells (Fig. 3a), which may ameliorate TME via secretion of cytokines, such as TNF-α, IFN-γ, IL-6, and IL-5, as well as chemokines CCL5 and CXCL10 [125]. Importantly, TNF-α and IFN-γ synergistically induced signaling in CD4+ T cells that prevents immune evasion, tumor cell proliferation, and multistage carcinogenesis [126, 127]. Moreover, tumor-infiltrating NKs and MDSCs underwent transcriptional reprogramming and enhanced the IL-10 production via STAT3 in breast cancer [58, 128]. As a result, increased IL-10 in TME directly suppressed the generation of CD4+ T cells, thereby impairing tumor immunity. Moreover, IL-10 has pro-tumor activity in TME, and IL-10 facilitated Tregs and M2-like macrophages development [129], as well as counteracting CD4+ T cells, cytotoxic CD8+ T cells and NKs tumoricidal function [34]. Therefore, IL-10 in TME of breast cancer induces immunosuppression and assists the evasion from tumor immune surveillance, promoting tumor cell development and metastasis [130]. Furthermore, much of the suppressive activity of T cells is due to the ROS production, including CD4+ T cells [131,132,133]. The S100A8/A9-mediated ROS suppression was reported to improve the CD4+ T cell accumulation in TME of breast cancer, which has been known to be regulated in STAT3-dependent mechanism in MDSCs [54, 58, 59, 61].
Tregs
Tregs is a subset of CD4+ T cells which sustain the immunosuppressive environment in human cancers. Generally, Tregs trigger immunosuppression by initiating competition for microenvironment factors, thereby leading to cytokine deprivation-induced apoptosis in the target effector T cells [134]. Olkhanud et al. showed that Tregs to be necessary for breast cancer cell metastasis to lungs, which was accompanied with chemokine receptor-mediated chemotaxis and killing function of NKs [135]. In breast cancer animal model with tumor metastasis, activated STAT3 and higher tumor-specific Tregs population showed co-existence and contributed to immunosuppression [136] (Fig. 3b). Moreover, increased intercellular STAT3 in Tregs resulted in enhanced proliferation and suppressive functions of Tregs, further supporting the STAT3-associated Tregs immunosuppression in TME of breast cancer [137].
Furthermore, the expression of Forkhead box P3 (Foxp3), a fundamental mediator of Tregs, is directly regulated by intercellular STAT3 cascade [138]. Activated Foxp3 can bind to a proximal site of the il2 promoter in vivo, leading to the upregulation of several Treg-associated markers expression [135]. The molecular events driving Foxp3 expression required T-cell receptor (TCR)/CD28 to stimulate STAT3 transcription, which in turn promoted the naive peripheral CD4+ T cells conversion into Tregs and acquisition of Tregs suppressive function. Moreover, it also caused, inhibition of IL-2 and IFN-γ production from converted lymphocytes [139, 140]. The positive function of STAT3 is that it can bind to the Foxp3 locus (Exon 2 β sheet region) and promote gene expression, thereby providing an important mechanism by which STAT3 can promote Tregs [141,142,143]. Meanwhile, Tregs lacking STAT3 are devoid of expansion, differentiation and immunosuppressive abilities. Moreover, Treg number has been found to be decreased in the TME of STAT3-deficient mice [139]. Specifically, elevated Foxp3 gene expression and accumulated Foxp3+ Tregs amount were detected in close proximity to lung metastases of breast cancer, as well as higher STAT3 activities [144]. STAT3 cascade may elicit immunosuppressive TME by recruiting Foxp3+ Tregs to the metastatic milieu. Concordantly, some studies have revealed that blocking of STAT3 in breast cancer animals significantly decreased the Tregs proportion in the TME especially the Foxp3+ Tregs [30, 125, 145]. Additionally, STAT3-blocking suppressed the pro-inflammatory and the anti-inflammatory Tregs, thereby adding to T cell cytotoxicity which is conducive for the anti-tumor effects [30].
Previously, Kyung et al. has reported that target TME Tregs regulator indoleamine-2,3-dioxegenase 1 (IDO1) achieved great success in various tumor types (11% achieved partial response or complete response), including breast cancer [146]. IDO was firstly found in 1950s and inhibits the T cell immunity by inducing differentiation and maturation of Treg cells, which is a poor prognosis factor for breast cancer [147,148,149]. Notably, MDSCs-produced IDO1 favors Foxp3 + Tregs and tumor-induced immunosuppression, which in turn leads to advanced breast cancer clinical stage [150, 151]. Furthermore, STAT3-mediated IDO1 expression was found to be upregulated in breast cancer cell-induced MDSCs, which suppressed effector T cells and hyperactivated the infiltration of the Foxp3+ Tregs in TME [37, 151, 152]. In contrary, STAT3 blocking downregulated the IDO1 proteins in MDSCs and stimulated T cell proliferation [37, 152]. Yu et al. showed that STAT3 activation induced IDO through NF-κB activation rather than by its directly binding function, in which, NF-κB subunits (RelB-p52 dimers) directly bind to the IDO promoter thereby triggering IDO expression [40]. Although STAT3 has been reported to be strongly associated with MDSCs and IDO1 in interacting with Foxp3+ Tregs, the STAT3-dependent regulation of IDO1 expression still remains unclear. Therefore, elucidating concrete molecular mechanisms modulating the STAT3/Tregs in breast cancer derived TME may prove essential in the development of novel immunotherapy strategies.
CD8+ T cells
T cells play a central role in human adaptive immune response to cancer, especially the function of CD8+ T-cell to kill malignant cells [25]. Tumor-infiltrating CD8+ T cells in TME are related to distinct clinical outcomes and survival in breast cancer [153]. STAT3 has been demonstrated to be an important checkpoint that blocks anti-tumor immune responses in a variety of immune cells, especially CD8+ T cells [154,155,156,157] (Fig. 3a). Recently, Li et al. reported that inhibition of STAT3 activity in breast cancer metastasis model simultaneously impaired matrix metalloproteinase (MMP)-2/9 and ROS generation and increased the CD8+ T cells in the TME [158]. Noteworthy, MMP-2 and -9 expression has been reported to enhance CD8+ T cells response in liver [159], and MMP-9 is known as a mediator of T cells migration [160, 161]. While, decreased ROS ameliorates the TME-mediated CD8+ T cells suppression [54, 162]. Therefore, STAT3-mediated ROS accretion might play a pivotal role in CD8+ T cell responses of breast cancer than MMPs. Apart from this, STAT3 inhibition can regulate the production of various immunomodulator factors in TME of breast cancer, such as upregulation of INFs, GM-CSF and IL-2, downregulation of TGF-b, IL-6, and IL-10 proteins [163]. These factors regulate inflammatory and antitumor functions of immune cells, including CD8+ T cells [164]. Similarly, it has been reported that blocking of STAT-3 in mice caused significantly higher activation of CD8+ T cells in TME as compared to the control [163]. Consistently, STAT3 blocking in 4T1 syngeneic mouse markedly suppressed the tumor by triggering CD8+ T cells priming to eliminate tumor cells [165], which may be manipulated by STAT3-blocking triggered INF-α/β production [166, 167]. The CD8+ T cells in 4T1 mouse model were also found to decrease the tumor volume, inhibit lung metastasis and prolong the overall survival (OS) [168]. Reports have also revealed that INF-α/β can stimulate the CD8+ T cells [169]. Hence, it is possible that high levels of STAT3-mediated IFN-α/β in the TME favors the function of CD8+ T cells.
Of note, it has been reported that increased 705-tyrosine phosphorylated STAT3 (STAT3-pY705) level was detected in CD8+ T cells of breast tumor tissues [170]. The stimulation of the human breast cancer CD8+ T cells also requires intercellular STAT3 regulation. For example, in the TME of breast cancer, STAT3 activation promotes the expression of fatty acid oxidation (FAO) in CD8+ T cells, which subsequently inhibits the cellular glycolysis and other functions [170]. Wang et al. also showed that FAO pathway in breast cancer stem cells is regulated by JAK/STAT3 signaling pathway [171]. Moreover, inhibition of CD8+ TEFF cells via STAT3 decreased breast tumor burdens and lung metastasis incidence in the Mouse Mammary Tumor Virus-Polyoma Virus Middle T antigen (MMTV-PyMT) transgenic mice. Meanwhile, it has been found that obesity-triggered breast tumor reduced the tumor-infiltrating CD8+ TEFF cells and promoted its progression [170]. In addition, STAT3 activation repressed the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and hexokinase II (HK2), both of which are essential glycolic indicators for T cells [172,173,174]. Conversely, blocking the FAO has been shown to cause significant promotion of CD8+ T cells accumulation and breast cancer regression. On the other hand, genetic instability of SNP allele rs351855-A in fibroblast growth factor receptor 4 (FGFR4) enhances STAT3 cascade and shapes TME in multiple cancer types, attributed to STAT3-pY705 elevation [137]. The suppressed levels of infiltrated CD8+ T cells in TME were observed in rs351855-A transgenic breast cancer mouse model, as a systemic trait, which determined the immune evasion in the TME and accelerated the tumor progression [137]. Thus, host-specific genetic variants might dictate immunosuppressive crosstalk between tumor cells and CD8+ T cells through STAT3 pleiotropic functions in T cells. Similarly, targeting of the STAT3 upstream, downregulating the intercellular pSTAT3-Y705 activity and upregulating glycolysis by leptin or PD-1 intervention markedly ameliorated the CD8+ T effector cells function in TME and prevented the development of breast cancer [170]. Metabolic reprogramming to regulate the function of T cells and upregulate the glycolysis in CD8+ T effector cells can promote their anti-tumor activity and IFN-γ production [175, 176]. Collectively, these findings suggest that targeting STAT3 may lead to a potent anti-tumor T cells immune response in breast cancer with pleiotropic functions.
Targeting STAT3 for breast cancer immunotherapy
The gene-therapy strategies were designed to inhibit the STAT3 signaling and improve the TME in the breast cancer model have proved the potential of STAT3 as a valid target for immunotherapy [177,178,179]. Inhibition of STAT3 activity by ruxolitinib can remarkably inhibit the breast cancer invasion in vivo [180]. Blocking of STAT3 in breast cancer not only suppressed the tumor progression, but also conferred sensitivity to chemotherapeutic drugs [181]. Apart from these advantages, STAT3 inhibition was recently proposed to improve innate and adaptive anti-tumor immunity and immune surveillance [182]. STAT3 inhibition in tumor cells increases the expression of cytokines and chemokines that ameliorate the TME, including DCs and tumor-specific T cells response [182]. Moreover, targeting STAT3 in human breast cancer cells was reported to suppress the tumor progression by regulating the expression of crucial proteins in tumor milieu, such as survivin, chemokines (CCL5 and CXCL10) and proinflammatory cytokines (IL-6, IL-5, TNF-a, and IFN-γ) [183,184,185]. In breast cancer, the STAT3 inhibitors presented a positive feedback in tumor intervention. The direct antitumor effect of STAT3 inhibitors alone has been established in several pre- clinical breast cancer studies (Table 2). As previously reported, preclinical studies of small-molecule STAT3 inhibitor S3I-201 and C188 significantly retarded the breast cancer cell growth in xenografts [186, 187]. Stattic, a nonpeptidic small molecule has been shown to selectively inhibit the phosphorylation, dimerization, and nuclear translocation of STAT3, which consequently promotes the STAT3-dependent breast cancer cell death [188]. Moreover, another low-molecular compound STA-21 has also been identified as a selective STAT3 inhibitor that influences the STAT3 dimerization and DNA binding ability in breast cancer [189]. In addition, an antidiarrheal agent (nifuroxazide) was also identified as a potent inhibitor of STAT3, which markedly inhibited the phosphorylated-Stat3Tyr705 in breast cancer, and induced cancer cell apoptosis in a dose-dependent manner [190]. Additionally, the nifuroxazide also been found to exhibit the potential to inhibit the breast cancer metastasis without detectable toxicity, and the decreased MDSCs in lungs as ascertained in mouse model [190]. Recently, different types of novel inhibitors have been proposed. For instance, Eucannabinolide (Euc) suppressed the STAT3 activation and DNA binding capacity, eventually leading to the inhibition of breast cancer cell viability, proliferation and metastasis [191]. Similarly, 10,11-dehydrocurvularin (DCV), a natural-product macrolide derived from marine fungus, has been shown to selectively suppress the STAT3, to consequently inhibit the proliferation, migration and invasion of breast cancer cells (MDA-MB-231 and MDA-MB-468), via induction of apoptosis [192].
Inactivation of STAT3 contributed to breast cancer immunogenic phenotype, which involved the participation of CD4+ T cells and NKs, and decreased Tregs in the TME [125]. In addition, along with the TME immune response, visible inhibition of the lung metastasis was observed in inhibitor group (STAT3-pY705) as compared to the blank control. Consistently, Mara et al. indicated that ablating STAT3 in both breast cancer and melanoma was associated with the activation of CD4+ T cells and NKs [145]. Meanwhile, combination of the STAT3 inhibitor and anti-PD-1 antibody synergistically improved T cells activation, CCL5 and IFN-γ releasing in the TME as compared toanti-PD-1 alone, and decreased tumor-infiltrating Tregs, therefore inhibiting the breast cancer progression and metastasis [145]. STAT3-blocking induced CCL5 is important for T cell proliferation and migration, which regulates the immune cell based autonomous processes [193,194,195,196]. Thus, STAT3 blocking has been proposed to be a promising adjuvant for the tumor immunotherapy. Pyrimethamine (PYR), an FDA approved anti-microbial drug, has safe inhibition function for STAT3 [197]. Owing to its immunomodulatory effects, PYR treatment significantly inhibited the metastasis and proliferation of breast cancer cells and remarkably attenuated the density of F4/80+ TAMs in breast cancer milieu [30]. Furthermore, PYR-mediated STAT3 inhibition significantly decreased the frequencies of Foxp3+ Tregs, while enhanced the frequency of CD8+ T cell numbers. The inhibitor against the Y239/240-ShcA phosphorylation site has emerged as a novel therapeutic strategy to inhibit STAT3 activation and increase sensitivity breast cancer cells to immunotherapy. This lead to a direct amelioration of immune suppression in breast cancer and increased tumoricidal properties of immune cells, such as NKs and cytotoxic T cells [15]. In addition, CpG-induced STAT3 activation could counteract by the inhibitor AG490 (STAT3i), which shown the ability to promote the Th1 skewing accumulation by counterbalancing CpG-induced Th2/Tregs and the DC maturation through NF-κB activation [198]. The specific T cell reactivity restores the antitumor immunity in patients with breast cancer, which is affected by release of local cytokine and chemokines like CXC10, CXC9, IFNγ, IL-4 and IL-5 [198]. Furthermore, a natural flavonoid compound, pectolinarigenin, inhibited breast cancer lung metastasis, which simultaneously enhanced the CD8+ T cells recruitment and inhibited the STAT3 phosphorylation [158]. The re-activated local immune response is an essential component to sensitize tumors to immunotherapies [199]. Therefore, STAT3 signaling is a most potential therapeutic target for breast cancer systematic immunotherapy.
Previous studies demonstrate that STAT3 activation not merely act as a predictive biomarker for downregulated immune cells response, but also the type of immunomodulator that is strongly associated with programmed death ligand 1 (PD-L1) expression in the TME of breast cancer [15, 29, 200]. The STAT3 translocates to the nucleus where it induces the PD-L1 expression by binding to DNA-response elements in PD-L1 promoter [29, 201]. Disrupting STAT3 cascade could prevent PD-L1 expression [202] and improve the immune surveillance with effector T cells in immune microenvironment [203]. The observation highlighted the therapeutic potentials of targeting STAT3 in TME, especially in breast cancer patients with high PD-L1 expression. Of note, pharmacologic inhibition of STAT3 showed the ability to enhance the efficacy of anti-PD-L1/PD-1 monoclonal antibodies, which was proved effective for patients with metastatic triple negative breast cancer [204]. Ongoing phase I trial (NCT03195699) in advanced-stage breast cancer patients applied the STAT3 SH2-domain binder inhibitor C188-9. Of note, selective inhibition of the SH2-domain by 3-(2,4-dichloro-phenoxymethyl)-5-trichloromethyl-[1,2,4]oxadiazole (ODZ10117) has been reported to significantly inhibit the migration, invasion and distant metastasis of breast cancer [205]. Moreover, a phase II trial combing pSTAT3 inhibitor (napabucasin) with the anti-PD-1 antibody nivolumab in modulating TME is also processing in colorectal cancer (NCT03647839). Therefore, the therapeutic combinations of STAT3 and PD-L1 immune check point inhibition may pave way for the prospective immunotherapy.
Combing immuno/radio-therapy and STAT3 inhibition in breast cancer
Radiotherapy invokes immune-related responses in cancer by several mechanisms. A growing number of studies have revealed that radiation invokes several systemic biological responses, such as adaptive and innate immune-related activities (T cells, macrophages, Tregs, NKs and CTLs) that affect tumor development [206,207,208,209,210,211]. In breast cancer, precise radiotherapy favors the local control of treated lesions and evokes the systematic antitumor immunity of tumor-associated antigens (TAAs). For example, stereotactic body radiotherapy (SBRT) further enhanced major histocompatibility complexes (MHCs) expression and immune cells activity in breast cancer [211,212,213,214]. While Timaner et al. reported that radiotherapy’s antitumor immune effects can be blunted by mechanisms of immune evasion and immune-suppression, such as radiation-mediated PD-L1 upregulation in the TME of breast cancer [215]. The irradiated tumor cells also increase the secretion of immuno-suppressive factors that promote the infiltration of Tregs, MDSCs, and macrophages [216,217,218]. These mechanisms potentially limit the anti-tumor effects of radiotherapy. Of note, a better therapeutic strategy, radiotherapy in combination with immune checkpoint inhibition not merely improves the breast cancer therapeutic effect, but also induces abscopal immune responses outside the radiation field [215, 219]. However, the abscopal responses are rarely observed after radiotherapy alone [220]. To data, heterogeneity or complete lack of abscopal response reports in combing immuno/radio-therapy in breast cancer further hindered the ability to define the populations most likely to benefit [220]. Developing predictive biomarkers of treatment response and efficacy in clinically relevant preclinical models is necessary.
Recently, tumor patients receiving standard radiation therapy were benefited from STAT3 inhibition. The STAT3 inhibition in combination with radiation therapy in tumors statistically reduced the radiation-related Tregs and MDSCs accumulation in the TME and further improved the therapeutic effect [221]. In breast cancer, applying the niclosamide to block the STAT3 overcame the radioresistance and significant increase of radiation-induced ROS, which offers an effective alternative approach for improving the breast cancer radiation therapy [222]. Similarly, S3I-201 has been reported to suppress the radiation-induced STAT3 phosphorylation and increase the radiation-induced cell death in breast cancer [223]. STAT3 is multipotent regulator of both tumor cells and immune cells [30, 137, 224]. In addition, there is an evidence that indicates that T cells are indispensable for radiotherapy and STAT3 inhibition synergistic treatment, but the therapeutic efficacy of radiotherapy and STAT3 inhibition cannot preclude the bridge between T cells and myeloid cells [221]. Generally, Tregs depletion alone is not sufficient to orchestrate an anti-tumor immune response, because of deficient TAAs to attract effector T cells infiltration and killing functions [225, 226]. Radiotherapy is well-suited for improving immunotherapy effect through distinct mechanisms, which exposes TAAs, boosts immune chemokines secretion and enhances the diversity of the TCR repertoire of intratumoral T cells [212, 227, 228]. Moreover, it has been reported that in glioma, DCs antigen presentation and T cell effector functions are also enhanced upon combination of STAT3 inhibition and radiotherapy [229]. The survival time and immunological memory were both improved in the synergistic treatment group. Owing to the heterogeneity of breast cancer [230], single immunotherapy strategies are unlikely to achieve uniform, consistent therapeutic responses among all patients. Immune clearance of a tumor is not determined by a solo immune cell population. Therefore, a rational approach to build upon the STAT3 target immunotherapy strategy such as radiation therapy, which might enhance the systematic antitumor immune responses and therapeutic effects in breast cancers.
Conclusions and future perspectives
Numerous studies support the role of STAT3 in immune cells and dictates the immunomodulatory effects to the TME of breast cancer. Activated STAT3 constitutively suppresses the CD4+/CD8+ T cells and DCs, and favors the MDSCs, Macrophages and Tregs, implying a key role in breast cancer progression, metastasis and immunity. Here, we provided an outline of STAT3 function in immune cells of breast cancer TME, as well as their cascade gene activation and clinical outcomes. The effects are complex and, in some cases, apparently discrepant. Most likely this could be explained by the differential role of STAT3 in various cell types and its participation in different intracellular pathways. Consistently, immune modulators generation has been described on the one hand as a mechanism of intercellular STAT3 induced TME decay, whereas on the other hand the role of immune cells systematic effect on breast cancer metastasis is influenced by STAT3. The resultant outcome reflects the intricate TME between tumor cells and immune cells with STAT3 aberrant functions. Emerging evidence indicates that targeting STAT3 not merely improves the anti-tumor immunity in TME, but also enhances the immunotherapy effect, therefore rendering STAT3 as a promising therapeutic target. Of note, the radiotherapy in combination with STAT3 target immunotherapy might pave way to further improve types of TME immune cell accumulation, systemic immune response and antitumor therapeutic effect. These avenues provide new opportunity for innovations towards advanced and/or metastasis breast cancer efficient immunotherapy.
In general, most of the immune cells intercellular STAT3 induce immunosuppressive effects and are thus tumor promoted. However, STAT3 has also been partly associated with DCs immune activation and IFN-γ secretion and has anti-tumor properties. Nonetheless, which role of STAT3 determines the outcome is still unclear. Future studies also need to explore pros or cons of STAT3 intervention in regulating immunological environment of breast cancer, especially tumor specific immune cells alteration. In this regard, it will be interesting to determine the diverse abilities of STAT3 in the context of TME regulation and systematic eradication of breast cancer.
Availability of data materials
Not applicable.
Abbreviations
- STAT3:
-
Signal transducer and activator of transcription 3
- TME:
-
Tumor microenvironment
- ER:
-
Estrogen receptor
- PR:
-
Progesterone receptor
- DCs:
-
Dendritic cells
- NKs:
-
Natural killer cells
- Tregs:
-
Regulatory T cells
- MDSCs:
-
Myeloid-derived suppressor cells
- IDO:
-
Indoleamine 2,3-dioxygenase
- IRF-8:
-
Interferon regulatory factor–8
- ROS:
-
Reactive oxygen species
- CSCs:
-
Cancer stem cells
- TAMs:
-
Tumor-associated macrophage
- HA:
-
Hyaluronan
- COX-2:
-
Cyclooxygenase-2
- A-FABP:
-
Adipocyte/macrophage fatty acid binding protein
- HIF-1α:
-
Hypoxia-inducible factor-1α
- TGF-β1:
-
Transforming growth factor β
- APCs:
-
Antigen-presenting cells
- TDFs:
-
Tumor-derived factors
- FLT3L:
-
FMS-related tyrosine kinase 3 ligand
- Foxp3:
-
Forkhead box P3
- TCR:
-
T-cell receptor
- FAO:
-
Fatty acid oxidation
- HK2:
-
Hexokinase II
- FGFR4:
-
Fibroblast growth factor receptor 4
- TAAs:
-
Tumor-associated antigens
- SBRT:
-
Stereotactic body radiotherapy
- MHCs:
-
Major histocompatibility complexs
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Waks AG, Winer EP. Breast cancer treatment: a review. JAMA. 2019;321:288–300.
Liu WS, Chan SH, Chang HT, Li GC, Tu YT, Tseng HH, Fu TY, Chang HY, Liou HH, Ger LP, Tsai KW. Isocitrate dehydrogenase 1-snail axis dysfunction significantly correlates with breast cancer prognosis and regulates cell invasion ability. Breast Cancer Res. 2018;20:25.
Gradishar WJ, Anderson BO, Balassanian R, Blair SL, Burstein HJ, Cyr A, Elias AD, Farrar WB, Forero A, Giordano SH, et al. Invasive breast cancer version 1.2016, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2016;14:324–54.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30.
Savas P, Salgado R, Denkert C, Sotiriou C, Darcy PK, Smyth MJ, Loi S. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat Rev Clin Oncol. 2016;13:228–41.
Emens LA. Breast cancer immunotherapy: facts and hopes. Clin Cancer Res. 2018;24:511–20.
Houthuijzen JM, Jonkers J. Cancer-associated fibroblasts as key regulators of the breast cancer tumor microenvironment. Cancer Metastasis Rev. 2018;37:577–97.
Furtek SL, Backos DS, Matheson CJ, Reigan P. Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem Biol. 2016;11:308–18.
Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–56.
Gharibi T, Babaloo Z, Hosseini A, Abdollahpour-Alitappeh M, Hashemi V, Marofi F, Nejati K, Baradaran B. Targeting STAT3 in cancer and autoimmune diseases. Eur J Pharmacol. 2020;878:173107.
Kalantari Khandani N, Ghahremanloo A, Hashemy SI. Role of tumor microenvironment in the regulation of PD-L1: a novel role in resistance to cancer immunotherapy. J Cell Physiol. 2020.
Chen J, Wang J, Lin L, He L, Wu Y, Zhang L, Yi Z, Chen Y, Pang X, Liu M. Inhibition of STAT3 signaling pathway by nitidine chloride suppressed the angiogenesis and growth of human gastric cancer. Mol Cancer Ther. 2012;11:277–87.
Mohassab AM, Hassan HA, Abdelhamid D, Abdel-Aziz M. STAT3 transcription factor as target for anti-cancer therapy. Pharmacol Rep. 2020;72:1101–24.
Ahn R, Sabourin V, Bolt AM, Hébert S, Totten S, De Jay N, Festa MC, Young YK, Im YK, Pawson T, et al. The Shc1 adaptor simultaneously balances Stat1 and Stat3 activity to promote breast cancer immune suppression. Nat Commun. 2017;8:14638.
Owen KL, Brockwell NK, Parker BS. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers (Basel). 2019;11.
Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting STAT3 in cancer immunotherapy. Mol Cancer. 2020;19:145.
Wang Y, Shen Y, Wang S, Shen Q, Zhou X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018;415:117–28.
Bournazou E, Bromberg J. Targeting the tumor microenvironment: JAK-STAT3 signaling. Jakstat. 2013;2:e23828.
Bohrer LR, Chuntova P, Bade LK, Beadnell TC, Leon RP, Brady NJ, Ryu Y, Goldberg JE, Schmechel SC, Koopmeiners JS, et al. Activation of the FGFR-STAT3 pathway in breast cancer cells induces a hyaluronan-rich microenvironment that licenses tumor formation. Cancer Res. 2014;74:374–86.
Sakurai T, Yada N, Hagiwara S, Arizumi T, Minaga K, Kamata K, Takenaka M, Minami Y, Watanabe T, Nishida N, Kudo M. Gankyrin induces STAT3 activation in tumor microenvironment and sorafenib resistance in hepatocellular carcinoma. Cancer Sci. 2017;108:1996–2003.
Flister MJ, Bergom C. Genetic modifiers of the breast tumor microenvironment. Trends Cancer. 2018;4:429–44.
DeNardo DG, Coussens LM. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 2007;9:212.
Liu Q, Yu S, Li A, Xu H, Han X, Wu K. Targeting interlukin-6 to relieve immunosuppression in tumor microenvironment. Tumour Biol. 2017;39:1010428317712445.
Rébé C, Ghiringhelli F. STAT3, a master regulator of anti-tumor immune response. Cancers (Basel). 2019;11.
AlHossiny M, Luo L, Frazier WR, Steiner N, Gusev Y, Kallakury B, Glasgow E, Creswell K, Madhavan S, Kumar R, Upadhyay G. Ly6E/K signaling to TGFβ promotes breast cancer progression, immune escape, and drug resistance. Cancer Res. 2016;76:3376–86.
Hagerling C, Gonzalez H, Salari K, Wang CY, Lin C, Robles I, van Gogh M, Dejmek A, Jirström K, Werb Z. Immune effector monocyte-neutrophil cooperation induced by the primary tumor prevents metastatic progression of breast cancer. Proc Natl Acad Sci U S A. 2019;116:21704–14.
Stakheyeva M, Eidenzon D, Slonimskaya E, Patysheva M, Bogdashin I, Kolegova E, Grigoriev E, Choinzonov E, Cherdyntseva N. Integral characteristic of the immune system state predicts breast cancer outcome. Exp Oncol. 2019;41:32–8.
Sasidharan Nair V, Toor SM, Ali BR, Elkord E. Dual inhibition of STAT1 and STAT3 activation downregulates expression of PD-L1 in human breast cancer cells. Expert Opin Ther Targets. 2018;22:547–57.
Khan MW, Saadalla A, Ewida AH, Al-Katranji K, Al-Saoudi G, Giaccone ZT, Gounari F, Zhang M, Frank DA, Khazaie K. The STAT3 inhibitor pyrimethamine displays anti-cancer and immune stimulatory effects in murine models of breast cancer. Cancer Immunol Immunother. 2018;67:13–23.
Li L, Yu R, Cai T, Chen Z, Lan M, Zou T, Wang B, Wang Q, Zhao Y, Cai Y. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. Int Immunopharmacol. 2020;88:106939.
Katsuta E, Rashid OM, Takabe K. Clinical relevance of tumor microenvironment: immune cells, vessels, and mouse models. Hum Cell. 2020;33:930–7.
Stewart TJ, Abrams SI. How tumours escape mass destruction. Oncogene. 2008;27:5894–903.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68.
Shou D, Wen L, Song Z, Yin J, Sun Q, Gong W. Suppressive role of myeloid-derived suppressor cells (MDSCs) in the microenvironment of breast cancer and targeted immunotherapies. Oncotarget. 2016;7:64505–11.
Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–44.
Yu J, Du W, Yan F, Wang Y, Li H, Cao S, Yu W, Shen C, Liu J, Ren X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190:3783–97.
Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, Extermann M, Mellor AL, Munn DH, Antonia SJ. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int J Cancer. 2002;101:151–5.
Andersen MH. Comment on “Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer.” J Immunol. 2013;190:5341.
Yu J, Wang Y, Yan F, Zhang P, Li H, Zhao H, Yan C, Yan F, Ren X. Noncanonical NF-κB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J Immunol. 2014;193:2574–86.
Sun SC. The noncanonical NF-κB pathway. Immunol Rev. 2012;246:125–40.
Nadiminty N, Lou W, Lee SO, Lin X, Trump DL, Gao AC. Stat3 activation of NF-{kappa}B p100 processing involves CBP/p300-mediated acetylation. Proc Natl Acad Sci U S A. 2006;103:7264–9.
Mougiakakos D, Jitschin R, von Bahr L, Poschke I, Gary R, Sundberg B, Gerbitz A, Ljungman P, Le Blanc K. Immunosuppressive CD14+HLA-DRlow/neg IDO+ myeloid cells in patients following allogeneic hematopoietic stem cell transplantation. Leukemia. 2013;27:377–88.
Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, Bogner PN, Farren MR, Lee KP, Liu K, Abrams SI. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest. 2013;123:4464–78.
Waight JD, Hu Q, Miller A, Liu S, Abrams SI. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS ONE. 2011;6:e27690.
Netherby CS, Messmer MN, Burkard-Mandel L, Colligan S, Miller A, Cortes Gomez E, Wang J, Nemeth MJ, Abrams SI. The granulocyte progenitor stage is a key target of IRF8-mediated regulation of myeloid-derived suppressor cell production. J Immunol. 2017;198:4129–39.
Narayanan PD, Nandabalan SK, Baddireddi LS. Role of STAT3 phosphorylation in ethanol-mediated proliferation of breast cancer cells. J Breast Cancer. 2016;19:122–32.
Jiang M, Chen J, Zhang W, Zhang R, Ye Y, Liu P, Yu W, Wei F, Ren X, Yu J. Interleukin-6 trans-signaling pathway promotes immunosuppressive myeloid-derived suppressor cells via suppression of suppressor of cytokine signaling 3 in breast cancer. Front Immunol. 1840;2017:8.
Beury DW, Parker KH, Nyandjo M, Sinha P, Carter KA, Ostrand-Rosenberg S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J Leukoc Biol. 2014;96:1109–18.
Oh K, Lee OY, Shon SY, Nam O, Ryu PM, Seo MW, Lee DS. A mutual activation loop between breast cancer cells and myeloid-derived suppressor cells facilitates spontaneous metastasis through IL-6 trans-signaling in a murine model. Breast Cancer Res. 2013;15:R79.
Peng D, Tanikawa T, Li W, Zhao L, Vatan L, Szeliga W, Wan S, Wei S, Wang Y, Liu Y, et al. Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH cross-talk signaling. Cancer Res. 2016;76:3156–65.
Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells—an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–9.
Ma M, Huang W, Kong D. IL-17 inhibits the accumulation of myeloid-derived suppressor cells in breast cancer via activating STAT3. Int Immunopharmacol. 2018;59:148–56.
Yin T, Zhao ZB, Guo J, Wang T, Yang JB, Wang C, Long J, Ma S, Huang Q, Zhang K, et al. Aurora A inhibition eliminates myeloid cell-mediated immunosuppression and enhances the efficacy of anti-PD-L1 therapy in breast cancer. Cancer Res. 2019;79:3431–44.
Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL Jr, Golemis EA. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci. 2013;70:661–87.
Damodaran AP, Vaufrey L, Gavard O, Prigent C. Aurora A kinase is a priority pharmaceutical target for the treatment of cancers. Trends Pharmacol Sci. 2017;38:687–700.
Melichar B, Adenis A, Lockhart AC, Bennouna J, Dees EC, Kayaleh O, Obermannova R, DeMichele A, Zatloukal P, Zhang B, et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. Lancet Oncol. 2015;16:395–405.
Sinha P, Ostrand-Rosenberg S. Myeloid-derived suppressor cell function is reduced by Withaferin A, a potent and abundant component of Withania somnifera root extract. Cancer Immunol Immunother. 2013;62:1663–73.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74.
Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182:5693–701.
Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59.
Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166–73.
Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25:315–22.
Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, Yang J, Pan J, Hu S, Zhang C, et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle. 2018;17:428–38.
Niu Z, Shi Q, Zhang W, Shu Y, Yang N, Chen B, Wang Q, Zhao X, Chen J, Cheng N, et al. Caspase-1 cleaves PPARγ for potentiating the pro-tumor action of TAMs. Nat Commun. 2017;8:766.
Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6.
Hao J, Yan F, Zhang Y, Triplett A, Zhang Y, Schultz DA, Sun Y, Zeng J, Silverstein KAT, Zheng Q, et al. Expression of adipocyte/macrophage fatty acid-binding protein in tumor-associated macrophages promotes breast cancer progression. Cancer Res. 2018;78:2343–55.
Su S, Liu Q, Chen J, Chen J, Chen F, He C, Huang D, Wu W, Lin L, Huang W, et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell. 2014;25:605–20.
Zhang G, Guo L, Yang C, Liu Y, He Y, Du Y, Wang W, Gao F. A novel role of breast cancer-derived hyaluronan on inducement of M2-like tumor-associated macrophages formation. Oncoimmunology. 2016;5:e1172154.
Tammi RH, Kultti A, Kosma VM, Pirinen R, Auvinen P, Tammi MI. Hyaluronan in human tumors: pathobiological and prognostic messages from cell-associated and stromal hyaluronan. Semin Cancer Biol. 2008;18:288–95.
Duff MD, Mestre J, Maddali S, Yan ZP, Stapleton P, Daly JM. Analysis of gene expression in the tumor-associated macrophage. J Surg Res. 2007;142:119–28.
Barbieri I, Pensa S, Pannellini T, Quaglino E, Maritano D, Demaria M, Voster A, Turkson J, Cavallo F, Watson CJ, et al. Constitutively active Stat3 enhances neu-mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res. 2010;70:2558–67.
Irey EA, Lassiter CM, Brady NJ, Chuntova P, Wang Y, Knutson TP, Henzler C, Chaffee TS, Vogel RI, Nelson AC, et al. JAK/STAT inhibition in macrophages promotes therapeutic resistance by inducing expression of protumorigenic factors. Proc Natl Acad Sci U S A. 2019;116:12442–51.
Britschgi A, Andraos R, Brinkhaus H, Klebba I, Romanet V, Müller U, Murakami M, Radimerski T, Bentires-Alj M. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell. 2012;22:796–811.
Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30:377–86.
Hill R, Li Y, Tran LM, Dry S, Calvopina JH, Garcia A, Kim C, Wang Y, Donahue TR, Herschman HR, Wu H. Cell intrinsic role of COX-2 in pancreatic cancer development. Mol Cancer Ther. 2012;11:2127–37.
Nandi P, Girish GV, Majumder M, Xin X, Tutunea-Fatan E, Lala PK. PGE2 promotes breast cancer-associated lymphangiogenesis by activation of EP4 receptor on lymphatic endothelial cells. BMC Cancer. 2017;17:11.
Majumder M, Nandi P, Omar A, Ugwuagbo KC, Lala PK. EP4 as a therapeutic target for aggressive human breast cancer. Int J Mol Sci. 2018;19.
Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov. 2008;7:489–503.
Storch J, Corsico B. The emerging functions and mechanisms of mammalian fatty acid-binding proteins. Annu Rev Nutr. 2008;28:73–95.
Zhang Y, Sun Y, Rao E, Yan F, Li Q, Zhang Y, Silverstein KA, Liu S, Sauter E, Cleary MP, Li B. Fatty acid-binding protein E-FABP restricts tumor growth by promoting IFN-β responses in tumor-associated macrophages. Cancer Res. 2014;74:2986–98.
Zhou N, Zhang Y, Zhang X, Lei Z, Hu R, Li H, Mao Y, Wang X, Irwin DM, Niu G, Tan H. Exposure of tumor-associated macrophages to apoptotic MCF-7 cells promotes breast cancer growth and metastasis. Int J Mol Sci. 2015;16:11966–82.
Qiang L, Wu T, Zhang HW, Lu N, Hu R, Wang YJ, Zhao L, Chen FH, Wang XT, You QD, Guo QL. HIF-1α is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012;19:284–94.
Philip B, Ito K, Moreno-Sánchez R, Ralph SJ. HIF expression and the role of hypoxic microenvironments within primary tumours as protective sites driving cancer stem cell renewal and metastatic progression. Carcinogenesis. 2013;34:1699–707.
Hendrayani SF, Al-Khalaf HH, Aboussekhra A. The cytokine IL-6 reactivates breast stromal fibroblasts through transcription factor STAT3-dependent up-regulation of the RNA-binding protein AUF1. J Biol Chem. 2014;289:30962–76.
Muraoka-Cook RS, Kurokawa H, Koh Y, Forbes JT, Roebuck LR, Barcellos-Hoff MH, Moody SE, Chodosh LA, Arteaga CL. Conditional overexpression of active transforming growth factor beta1 in vivo accelerates metastases of transgenic mammary tumors. Cancer Res. 2004;64:9002–11.
Jing W, Guo X, Wang G, Bi Y, Han L, Zhu Q, Qiu C, Tanaka M, Zhao Y. Breast cancer cells promote CD169(+) macrophage-associated immunosuppression through JAK2-mediated PD-L1 upregulation on macrophages. Int Immunopharmacol. 2020;78:106012.
Wang X, Ni S, Chen Q, Ma L, Jiao Z, Wang C, Jia G. Bladder cancer cells induce immunosuppression of T cells by supporting PD-L1 expression in tumour macrophages partially through interleukin 10. Cell Biol Int. 2017;41:177–86.
Diskin B, Adam S, Cassini MF, Sanchez G, Liria M, Aykut B, Buttar C, Li E, Sundberg B, Salas RD, et al. PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat Immunol. 2020;21:442–54.
Yang J, Liao D, Chen C, Liu Y, Chuang TH, Xiang R, Markowitz D, Reisfeld RA, Luo Y. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells. 2013;31:248–58.
Valeta-Magara A, Gadi A, Volta V, Walters B, Arju R, Giashuddin S, Zhong H, Schneider RJ. Inflammatory breast cancer promotes development of M2 tumor-associated macrophages and cancer mesenchymal cells through a complex chemokine network. Cancer Res. 2019;79:3360–71.
Mills CD, Lenz LL, Harris RA. A breakthrough: macrophage-directed cancer immunotherapy. Cancer Res. 2016;76:513–6.
Panni RZ, Linehan DC, DeNardo DG. Targeting tumor-infiltrating macrophages to combat cancer. Immunotherapy. 2013;5:1075–87.
Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811.
Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13:114–9.
Durai V, Murphy KM. Functions of murine dendritic cells. Immunity. 2016;45:719–36.
Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4:941–52.
Farren MR, Carlson LM, Netherby CS, Lindner I, Li PK, Gabrilovich DI, Abrams SI, Lee KP. Tumor-induced STAT3 signaling in myeloid cells impairs dendritic cell generation by decreasing PKCβII abundance. Sci Signal. 2014;7:16.
Cejas PJ, Carlson LM, Zhang J, Padmanabhan S, Kolonias D, Lindner I, Haley S, Boise LH, Lee KP. Protein kinase C betaII plays an essential role in dendritic cell differentiation and autoregulates its own expression. J Biol Chem. 2005;280:28412–23.
Jones LM, Broz ML, Ranger JJ, Ozcelik J, Ahn R, Zuo D, Ursini-Siegel J, Hallett MT, Krummel M, Muller WJ. STAT3 Establishes an immunosuppressive microenvironment during the early stages of breast carcinogenesis to promote tumor growth and metastasis. Cancer Res. 2016;76:1416–28.
Liu WS, Heckman CA. The sevenfold way of PKC regulation. Cell Signal. 1998;10:529–42.
Lin YF, Lee HM, Leu SJ, Tsai YH. The essentiality of PKCalpha and PKCbetaI translocation for CD14+monocyte differentiation towards macrophages and dendritic cells, respectively. J Cell Biochem. 2007;102:429–41.
Hira SK, Manna PP. Down regulation of CD24 and HER-2/neu in breast carcinoma cells by activated human dendritic cell role of STAT3. Cell Immunol. 2012;275:69–79.
Wiedermann U, Davis AB, Zielinski CC. Vaccination for the prevention and treatment of breast cancer with special focus on Her-2/neu peptide vaccines. Breast Cancer Res Treat. 2013;138:1–12.
Chrisikos TT, Zhou Y, Li HS, Babcock RL, Wan X, Patel B, Newton K, Mancuso JJ, Watowich SS. STAT3 inhibits CD103(+) cDC1 vaccine efficacy in murine breast cancer. Cancers (Basel). 2020;12.
da Cunha A, Antoniazi Michelin M, Cândido Murta EF. Phenotypic profile of dendritic and T cells in the lymph node of Balb/C mice with breast cancer submitted to dendritic cells immunotherapy. Immunol Lett. 2016;177:25–37.
Bak SP, Barnkob MS, Bai A, Higham EM, Wittrup KD, Chen J. Differential requirement for CD70 and CD80/CD86 in dendritic cell-mediated activation of tumor-tolerized CD8 T cells. J Immunol. 2012;189:1708–16.
Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355:1423–7.
Vonderheide RH, Domchek SM, Clark AS. Immunotherapy for breast cancer: what are we missing? Clin Cancer Res. 2017;23:2640–6.
Li HS, Yang CY, Nallaparaju KC, Zhang H, Liu YJ, Goldrath AW, Watowich SS. The signal transducers STAT5 and STAT3 control expression of Id2 and E2–2 during dendritic cell development. Blood. 2012;120:4363–73.
Laouar Y, Welte T, Fu XY, Flavell RA. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity. 2003;19:903–12.
Sang H, Pisarev VM, Chavez J, Robinson S, Guo Y, Hatcher L, Munger C, Talmadge CB, Solheim JC, Singh RK, Talmadge JE. Murine mammary adenocarcinoma cells transfected with p53 and/or Flt3L induce antitumor immune responses. Cancer Gene Ther. 2005;12:427–37.
Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 2014;26:638–52.
Kang JW, Park YS, Kim MS, Lee DH, Bak Y, Ham SY, Song YS, Hong JT, Yoon DY. IL-32α down-regulates β2 integrin (CD18) expression by suppressing PU.1 expression in myeloid cells. Cell Signal. 2014;26:1514–22.
Hegde S, Ni S, He S, Yoon D, Feng GS, Watowich SS, Paulson RF, Hankey PA. Stat3 promotes the development of erythroleukemia by inducing Pu.1 expression and inhibiting erythroid differentiation. Oncogene. 2009;28:3349–59.
Onai N, Obata-Onai A, Tussiwand R, Lanzavecchia A, Manz MG. Activation of the Flt3 signal transduction cascade rescues and enhances type I interferon-producing and dendritic cell development. J Exp Med. 2006;203:227–38.
Wang S, Chen F, Tang L. IL-32 promotes breast cancer cell growth and invasiveness. Oncol Lett. 2015;9:305–7.
Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, Holmberg D, Zweier C, den Hollander NS, Kant SG, et al. Transcription factor E2–2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. 2008;135:37–48.
Beildeck ME, Islam M, Shah S, Welsh J, Byers SW. Control of TCF-4 expression by VDR and vitamin D in the mouse mammary gland and colorectal cancer cell lines. PLoS ONE. 2009;4:e7872.
Ghochikyan A, Pichugin A, Bagaev A, Davtyan A, Hovakimyan A, Tukhvatulin A, Davtyan H, Shcheblyakov D, Logunov D, Chulkina M, et al. Targeting TLR-4 with a novel pharmaceutical grade plant derived agonist, Immunomax®, as a therapeutic strategy for metastatic breast cancer. J Transl Med. 2014;12:322.
Chen W, Chan AS, Dawson AJ, Liang X, Blazar BR, Miller JS. FLT3 ligand administration after hematopoietic cell transplantation increases circulating dendritic cell precursors that can be activated by CpG oligodeoxynucleotides to enhance T-cell and natural killer cell function. Biol Blood Marrow Transplant. 2005;11:23–34.
Liu Y, Li H, Liu F, Gao LB, Han R, Chen C, Ding X, Li S, Lu K, Yang L, et al. Heterogeneous nuclear ribonucleoprotein A2/B1 is a negative regulator of human breast cancer metastasis by maintaining the balance of multiple genes and pathways. EBioMedicine. 2020;51:102583.
Lei J, Rudolph A, Moysich KB, Behrens S, Goode EL, Bolla MK, Dennis J, Dunning AM, Easton DF, Wang Q, et al. Genetic variation in the immunosuppression pathway genes and breast cancer susceptibility: a pooled analysis of 42,510 cases and 40,577 controls from the Breast Cancer Association Consortium. Hum Genet. 2016;135:137–54.
Kortylewski M, Yu H. Role of Stat3 in suppressing anti-tumor immunity. Curr Opin Immunol. 2008;20:228–33.
Tkach M, Coria L, Rosemblit C, Rivas MA, Proietti CJ, Díaz Flaqué MC, Beguelin W, Frahm I, Charreau EH, Cassataro J, et al. Targeting Stat3 induces senescence in tumor cells and elicits prophylactic and therapeutic immune responses against breast cancer growth mediated by NK cells and CD4+ T cells. J Immunol. 2012;189:1162–72.
Datta J, Rosemblit C, Berk E, Showalter L, Namjoshi P, Mick R, Lee KP, Brod AM, Yang RL, Kelz RR, et al. Progressive loss of anti-HER2 CD4(+) T-helper type 1 response in breast tumorigenesis and the potential for immune restoration. Oncoimmunology. 2015;4:e1022301.
Müller-Hermelink N, Braumüller H, Pichler B, Wieder T, Mailhammer R, Schaak K, Ghoreschi K, Yazdi A, Haubner R, Sander CA, et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell. 2008;13:507–18.
Armando I, Grossman E, Hoffman A, Goldstein DS. Method for measuring endogenous 3-O-methyldopa in urine and plasma. J Chromatogr. 1991;568:45–54.
Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, Kronenberg M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. 2009;10:1178–84.
Hamidullah CB, Konwar R. Role of interleukin-10 in breast cancer. Breast Cancer Res Treat. 2012;133:11–21.
Kraaij MD, van der Kooij SW, Reinders ME, Koekkoek K, Rabelink TJ, van Kooten C, Gelderman KA. Dexamethasone increases ROS production and T cell suppressive capacity by anti-inflammatory macrophages. Mol Immunol. 2011;49:549–57.
Belikov AV, Schraven B, Simeoni L. T cells and reactive oxygen species. J Biomed Sci. 2015;22:85.
Previte DM, Piganelli JD. Reactive oxygen species and their implications on CD4(+) T cells in type 1 diabetes. Antioxid Redox Signal. 2018;29:1399–414.
Sojka DK, Huang YH, Fowell DJ. Mechanisms of regulatory T-cell suppression—a diverse arsenal for a moving target. Immunology. 2008;124:13–22.
Olkhanud PB, Baatar D, Bodogai M, Hakim F, Gress R, Anderson RL, Deng J, Xu M, Briest S, Biragyn A. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. 2009;69:5996–6004.
Mucha J, Rybicka A, Dolka I, Szymańska J, Manuali E, Parzeniecka-Jaworska M, Kluciński W, Król M. Immunosuppression in dogs during mammary cancer development. Vet Pathol. 2016;53:1147–53.
Kogan D, Grabner A, Yanucil C, Faul C, Ulaganathan VK. STAT3-enhancing germline mutations contribute to tumor-extrinsic immune evasion. J Clin Invest. 2018;128:1867–72.
Hossain DM, Panda AK, Chakrabarty S, Bhattacharjee P, Kajal K, Mohanty S, Sarkar I, Sarkar DK, Kar SK, Sa G. MEK inhibition prevents tumour-shed transforming growth factor-β-induced T-regulatory cell augmentation in tumour milieu. Immunology. 2015;144:561–73.
Pallandre JR, Brillard E, Créhange G, Radlovic A, Remy-Martin JP, Saas P, Rohrlich PS, Pivot X, Ling X, Tiberghien P, Borg C. Role of STAT3 in CD4+CD25+FOXP3+ regulatory lymphocyte generation: implications in graft-versus-host disease and antitumor immunity. J Immunol. 2007;179:7593–604.
Walker MR, Kasprowicz DJ, Gersuk VH, Benard A, Van Landeghen M, Buckner JH, Ziegler SF. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25- T cells. J Clin Invest. 2003;112:1437–43.
Laurence A, Amarnath S, Mariotti J, Kim YC, Foley J, Eckhaus M, O’Shea JJ, Fowler DH. STAT3 transcription factor promotes instability of nTreg cells and limits generation of iTreg cells during acute murine graft-versus-host disease. Immunity. 2012;37:209–22.
Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, O’Shea JJ. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–20.
Hossain DM, Panda AK, Manna A, Mohanty S, Bhattacharjee P, Bhattacharyya S, Saha T, Chakraborty S, Kar RK, Das T, et al. FoxP3 acts as a cotranscription factor with STAT3 in tumor-induced regulatory T cells. Immunity. 2013;39:1057–69.
Kim SJ, Garcia-Recio S, Creighton CJ, Perou CM, Rosen JM. Alterations in Wnt- and/or STAT3 signaling pathways and the immune microenvironment during metastatic progression. Oncogene. 2019;38:5942–58.
De Martino M, Tkach M, Bruni S, Rocha D, Mercogliano MF, Cenciarini ME, Chervo MF, Proietti CJ, Dingli F, Loew D, et al. Blockade of Stat3 oncogene addiction induces cellular senescence and reveals a cell-nonautonomous activity suitable for cancer immunotherapy. Oncoimmunology. 2020;9:1715767.
Jung KH, LoRusso P, Burris H, Gordon M, Bang YJ, Hellmann MD, Cervantes A, de Olza MO, Marabelle A, Hodi FS, et al. Phase I study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) administered with PD-L1 Inhibitor (atezolizumab) in advanced solid tumors. Clin Cancer Res. 2019;25:3220–8.
Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, Muller AJ. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. 2014;63:721–35.
Sun J, Yu J, Li H, Yang L, Wei F, Yu W, Liu J, Ren X. Upregulated expression of indoleamine 2, 3-dioxygenase in CHO cells induces apoptosis of competent T cells and increases proportion of Treg cells. J Exp Clin Cancer Res. 2011;30:82.
Chen JY, Li CF, Kuo CC, Tsai KK, Hou MF, Hung WC. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res. 2014;16:410.
Werfel TA, Elion DL, Rahman B, Hicks DJ, Sanchez V, Gonzales-Ericsson PI, Nixon MJ, James JL, Balko JM, Scherle PA, et al. Treatment-induced tumor cell apoptosis and secondary necrosis drive tumor progression in the residual tumor microenvironment through MerTK and IDO1. Cancer Res. 2019;79:171–82.
Li F, Zhao Y, Wei L, Li S, Liu J. Tumor-infiltrating Treg, MDSC, and IDO expression associated with outcomes of neoadjuvant chemotherapy of breast cancer. Cancer Biol Ther. 2018;19:695–705.
Cheng JT, Deng YN, Yi HM, Wang GY, Fu BS, Chen WJ, Liu W, Tai Y, Peng YW, Zhang Q. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis. 2016;5:e198.
Marrogi AJ, Munshi A, Merogi AJ, Ohadike Y, El-Habashi A, Marrogi OL, Freeman SM. Study of tumor infiltrating lymphocytes and transforming growth factor-beta as prognostic factors in breast carcinoma. Int J Cancer. 1997;74:492–501.
Herrmann A, Kortylewski M, Kujawski M, Zhang C, Reckamp K, Armstrong B, Wang L, Kowolik C, Deng J, Figlin R, Yu H. Targeting Stat3 in the myeloid compartment drastically improves the in vivo antitumor functions of adoptively transferred T cells. Cancer Res. 2010;70:7455–64.
Kujawski M, Zhang C, Herrmann A, Reckamp K, Scuto A, Jensen M, Deng J, Forman S, Figlin R, Yu H. Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res. 2010;70:9599–610.
Moreira D, Adamus T, Zhao X, Su YL, Zhang Z, White SV, Swiderski P, Lu X, DePinho RA, Pal SK, Kortylewski M. STAT3 inhibition combined with CpG immunostimulation activates antitumor immunity to eradicate genetically distinct castration-resistant prostate cancers. Clin Cancer Res. 2018;24:5948–62.
Zhang C, Xin H, Zhang W, Yazaki PJ, Zhang Z, Le K, Li W, Lee H, Kwak L, Forman S, et al. CD5 binds to interleukin-6 and induces a feed-forward loop with the transcription factor STAT3 in B cells to promote cancer. Immunity. 2016;44:913–23.
Li Y, Gan C, Zhang Y, Yu Y, Fan C, Deng Y, Zhang Q, Yu X, Zhang Y, Wang L, et al. Inhibition of Stat3 signaling pathway by natural product pectolinarigenin attenuates breast cancer metastasis. Front Pharmacol. 2019;10:1195.
Yang S, Wang L, Pan W, Bayer W, Thoens C, Heim K, Dittmer U, Timm J, Wang Q, Yu Q, et al. MMP2/MMP9-mediated CD100 shedding is crucial for inducing intrahepatic anti-HBV CD8 T cell responses and HBV clearance. J Hepatol. 2019;71:685–98.
Murooka TT, Rahbar R, Platanias LC, Fish EN. CCL5-mediated T-cell chemotaxis involves the initiation of mRNA translation through mTOR/4E-BP1. Blood. 2008;111:4892–901.
Tsilingaridis G, Yucel-Lindberg T, Concha Quezada H, Modéer T. The relationship between matrix metalloproteinases (MMP-3, -8, -9) in serum and peripheral lymphocytes (CD8+, CD56+ ) in down syndrome children with gingivitis. J Periodontal Res. 2014;49:742–50.
Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989–99.
Liao D, Liu Z, Wrasidlo WJ, Luo Y, Nguyen G, Chen T, Xiang R, Reisfeld RA. Targeted therapeutic remodeling of the tumor microenvironment improves an HER-2 DNA vaccine and prevents recurrence in a murine breast cancer model. Cancer Res. 2011;71:5688–96.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99.
Pei J, Zhang Y, Luo Q, Zheng W, Li W, Zeng X, Li Q, Quan J. STAT3 inhibition enhances CDN-induced STING signaling and antitumor immunity. Cancer Lett. 2019;450:110–22.
Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–42.
Wang WB, Levy DE, Lee CK. STAT3 negatively regulates type I IFN-mediated antiviral response. J Immunol. 2011;187:2578–85.
Liu J, Yang Y, Wang H, Wang B, Zhao K, Jiang W, Bai W, Liu J, Yin J. Syntenin1/MDA-9 (SDCBP) induces immune evasion in triple-negative breast cancer by upregulating PD-L1. Breast Cancer Res Treat. 2018;171:345–57.
Robbins SH, Bessou G, Cornillon A, Zucchini N, Rupp B, Ruzsics Z, Sacher T, Tomasello E, Vivier E, Koszinowski UH, Dalod M. Natural killer cells promote early CD8 T cell responses against cytomegalovirus. PLoS Pathog. 2007;3:e123.
Zhang C, Yue C, Herrmann A, Song J, Egelston C, Wang T, Zhang Z, Li W, Lee H, Aftabizadeh M, et al. STAT3 Activation-induced fatty acid oxidation in CD8(+) T effector cells is critical for obesity-promoted breast tumor growth. Cell Metab. 2020;31:148–61.
Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, Zhang C, Lifshitz V, Song J, Yuan Y, et al. JAK/STAT3-regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 2018;27:136–50.
Li M, Jin R, Wang W, Zhang T, Sang J, Li N, Han Q, Zhao W, Li C, Liu Z. STAT3 regulates glycolysis via targeting hexokinase 2 in hepatocellular carcinoma cells. Oncotarget. 2017;8:24777–84.
He Y, Deng F, Zhao S, Zhong S, Zhao J, Wang D, Chen X, Zhang J, Hou J, Zhang W, et al. Analysis of miRNA-mRNA network reveals miR-140-5p as a suppressor of breast cancer glycolysis via targeting GLUT1. Epigenomics. 2019;11:1021–36.
Douros JD, Baltzegar DA, Reading BJ, Seale AP, Lerner DT, Grau EG, Borski RJ. Leptin stimulates cellular glycolysis through a STAT3 dependent mechanism in Tilapia. Front Endocrinol (Lausanne). 2018;9:465.
Gemta LF, Siska PJ, Nelson ME, Gao X, Liu X, Locasale JW, Yagita H, Slingluff CL, Jr., Hoehn KL, Rathmell JC, Bullock TNJ. Impaired enolase 1 glycolytic activity restrains effector functions of tumor-infiltrating CD8(+) T cells. Sci Immunol. 2019;4.
Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–41.
Piperi C, Papavassiliou KA, Papavassiliou AG. Pivotal role of STAT3 in shaping glioblastoma immune microenvironment. Cells. 2019;8.
Dufait I, Van Valckenborgh E, Menu E, Escors D, De Ridder M, Breckpot K. Signal transducer and activator of transcription 3 in myeloid-derived suppressor cells: an opportunity for cancer therapy. Oncotarget. 2016;7:42698–715.
Zhao X, Zhang Z, Moreira D, Su YL, Won H, Adamus T, Dong Z, Liang Y, Yin HH, Swiderski P, et al. B cell lymphoma immunotherapy using TLR9-targeted oligonucleotide STAT3 inhibitors. Mol Ther. 2018;26:695–707.
Siersbæk R, Scabia V, Nagarajan S, Chernukhin I, Papachristou EK, Broome R, Johnston SJ, Joosten SEP, Green AR, Kumar S, et al. IL6/STAT3 signaling hijacks estrogen receptor α enhancers to drive breast cancer metastasis. Cancer Cell. 2020;38:412–23.
Real PJ, Sierra A, De Juan A, Segovia JC, Lopez-Vega JM, Fernandez-Luna JL. Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene. 2002;21:7611–8.
Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10:48–54.
Yi EH, Lee CS, Lee JK, Lee YJ, Shin MK, Cho CH, Kang KW, Lee JW, Han W, Noh DY, et al. STAT3-RANTES autocrine signaling is essential for tamoxifen resistance in human breast cancer cells. Mol Cancer Res. 2013;11:31–42.
Gritsko T, Williams A, Turkson J, Kaneko S, Bowman T, Huang M, Nam S, Eweis I, Diaz N, Sullivan D, et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin Cancer Res. 2006;12:11–9.
Linher-Melville K, Sharma M, Nakhla P, Kum E, Ungard R, Park J, Rosa D, Gunning P, Singh G. Inhibiting STAT3 in a murine model of human breast cancer-induced bone pain delays the onset of nociception. Mol Pain. 2019;15:1744806918823477.
Li G, Zhao L, Li W, Fan K, Qian W, Hou S, Wang H, Dai J, Wei H, Guo Y. Feedback activation of STAT3 mediates trastuzumab resistance via upregulation of MUC1 and MUC4 expression. Oncotarget. 2014;5:8317–29.
Dave B, Landis MD, Tweardy DJ, Chang JC, Dobrolecki LE, Wu MF, Zhang X, Westbrook TF, Hilsenbeck SG, Liu D, Lewis MT. Selective small molecule Stat3 inhibitor reduces breast cancer tumor-initiating cells and improves recurrence free survival in a human-xenograft model. PLoS ONE. 2012;7:e30207.
Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–42.
Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci U S A. 2005;102:4700–5.
Yang F, Hu M, Lei Q, Xia Y, Zhu Y, Song X, Li Y, Jie H, Liu C, Xiong Y, et al. Nifuroxazide induces apoptosis and impairs pulmonary metastasis in breast cancer model. Cell Death Dis. 2015;6:e1701.
Zhu Z, Yuan J, Xu X, Wei Y, Yang B, Zhao H. Eucannabinolide, a novel sesquiterpene lactone, suppresses the growth, metastasis and BCSCS-like traits of TNBC via inactivation of STAT3. Neoplasia. 2020;23:36–48.
Zhao Q, Bi Y, Zhong J, Li X, Guo J, Liu YX, Pan LR, Tan Y, Deng ZS, Yu XJ. 10,11-dehydrocurvularin exerts antitumor effect against human breast cancer by suppressing STAT3 activation. Acta Pharmacol Sin. 2020.
McLoughlin RM, Jenkins BJ, Grail D, Williams AS, Fielding CA, Parker CR, Ernst M, Topley N, Jones SA. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc Natl Acad Sci U S A. 2005;102:9589–94.
Zhang Y, Liao S, Fan W, Wei W, Wang C, Sun S. Tunicamycin-induced ER stress regulates chemokine CCL5 expression and secretion via STAT3 followed by decreased transmigration of MCF-7 breast cancer cells. Oncol Rep. 2014;32:2769–76.
Dutta P, Sarkissyan M, Paico K, Wu Y, Vadgama JV. MCP-1 is overexpressed in triple-negative breast cancers and drives cancer invasiveness and metastasis. Breast Cancer Res Treat. 2018;170:477–86.
Jin J, Zhao Q. Emerging role of mTOR in tumor immune contexture: impact on chemokine-related immune cells migration. Theranostics. 2020;10:6231–44.
Takakura A, Nelson EA, Haque N, Humphreys BD, Zandi-Nejad K, Frank DA, Zhou J. Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Hum Mol Genet. 2011;20:4143–54.
van Pul KM, Vuylsteke R, de Beijer MTA, van de Ven R, van den Tol MP, Stockmann H, de Gruijl TD. Breast cancer-induced immune suppression in the sentinel lymph node is effectively countered by CpG-B in conjunction with inhibition of the JAK2/STAT3 pathway. J Immunother Cancer. 2020;8.
Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. 2016;44:343–54.
Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, Nie L, Chen Y, Wang YC, Liu C, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22:1264–75.
Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, Wang HY, Wysocka M, Cheng M, Ruggeri BA, Wasik MA. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7–H1). Proc Natl Acad Sci U S A. 2008;105:20852–7.
Wölfle SJ, Strebovsky J, Bartz H, Sähr A, Arnold C, Kaiser C, Dalpke AH, Heeg K. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur J Immunol. 2011;41:413–24.
Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064–74.
Zerdes I, Wallerius M, Sifakis EG, Wallmann T, Betts S, Bartish M, Tsesmetzis N, Tobin NP, Coucoravas C, Bergh J, et al. STAT3 activity promotes programmed-death ligand 1 expression and suppresses immune responses in breast cancer. Cancers (Basel). 2019;11.
Kim BH, Lee H, Song Y, Park JS, Gadhe CG, Choi J, Lee CG, Pae AN, Kim S, Ye SK. Development of oxadiazole-based ODZ10117 as a small-molecule inhibitor of STAT3 for targeted cancer therapy. J Clin Med. 2019;8.
Barcellos-Hoff MH, Park C, Wright EG. Radiation and the microenvironment-tumorigenesis and therapy. Nat Rev Cancer. 2005;5:867–75.
Formenti SC, Demaria S. Systemic effects of local radiotherapy. Lancet Oncol. 2009;10:718–26.
Balázs K, Kis E, Badie C, Bogdándi EN, Candéias S, Garcia LC, Dominczyk I, Frey B, Gaipl U, Jurányi Z, et al. Radiotherapy-induced changes in the systemic immune and inflammation parameters of head and neck cancer patients. Cancers (Basel). 2019;11.
Russell JS, Brown JM. The irradiated tumor microenvironment: role of tumor-associated macrophages in vascular recovery. Front Physiol. 2013;4:157.
Mortezaee K, Parwaie W, Motevaseli E, Mirtavoos-Mahyari H, Musa AE, Shabeeb D, Esmaely F, Najafi M, Farhood B. Targets for improving tumor response to radiotherapy. Int Immunopharmacol. 2019;76:105847.
Muraro E, Furlan C, Avanzo M, Martorelli D, Comaro E, Rizzo A, Fae DA, Berretta M, Militello L, Del Conte A, et al. Local high-dose radiotherapy induces systemic immunomodulating effects of potential therapeutic relevance in oligometastatic breast cancer. Front Immunol. 2017;8:1476.
Bernal-Estévez D, Sánchez R, Tejada RE, Parra-López C. Chemotherapy and radiation therapy elicits tumor specific T cell responses in a breast cancer patient. BMC Cancer. 2016;16:591.
Ho AY, Barker CA, Arnold BB, Powell SN, Hu ZI, Gucalp A, Lebron-Zapata L, Wen HY, Kallman C, D’Agnolo A, et al. A phase 2 clinical trial assessing the efficacy and safety of pembrolizumab and radiotherapy in patients with metastatic triple-negative breast cancer. Cancer. 2020;126:850–60.
Chen HM, Ma G, Gildener-Leapman N, Eisenstein S, Coakley BA, Ozao J, Mandeli J, Divino C, Schwartz M, Sung M, et al. Myeloid-derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clin Cancer Res. 2015;21:4073–85.
Timaner M, Kotsofruk R, Raviv Z, Magidey K, Shechter D, Kan T, Nevelsky A, Daniel S, de Vries EGE, Zhang T, et al. Microparticles from tumors exposed to radiation promote immune evasion in part by PD-L1. Oncogene. 2020;39:187–203.
Liu S, Sun X, Luo J, Zhu H, Yang X, Guo Q, Song Y, Sun X. Effects of radiation on T regulatory cells in normal states and cancer: mechanisms and clinical implications. Am J Cancer Res. 2015;5:3276–85.
Vatner RE, Formenti SC. Myeloid-derived cells in tumors: effects of radiation. Semin Radiat Oncol. 2015;25:18–27.
Seifert L, Werba G, Tiwari S, Giao Ly NN, Nguy S, Alothman S, Alqunaibit D, Avanzi A, Daley D, Barilla R, et al. Radiation therapy induces macrophages to suppress T-cell responses against pancreatic tumors in mice. Gastroenterology. 2016;150:1659–72.
Hu ZI, Ho AY, McArthur HL. Combined radiation therapy and immune checkpoint blockade therapy for breast cancer. Int J Radiat Oncol Biol Phys. 2017;99:153–64.
Abuodeh Y, Venkat P, Kim S. Systematic review of case reports on the abscopal effect. Curr Probl Cancer. 2016;40:25–37.
Oweida AJ, Darragh L, Phan A, Binder D, Bhatia S, Mueller A, Court BV, Milner D, Raben D, Woessner R, et al. STAT3 modulation of regulatory T cells in response to radiation therapy in head and neck cancer. J Natl Cancer Inst. 2019;111:1339–49.
Lu L, Dong J, Wang L, Xia Q, Zhang D, Kim H, Yin T, Fan S, Shen Q. Activation of STAT3 and Bcl-2 and reduction of reactive oxygen species (ROS) promote radioresistance in breast cancer and overcome of radioresistance with niclosamide. Oncogene. 2018;37:5292–304.
Kim JS, Kim HA, Seong MK, Seol H, Oh JS, Kim EK, Chang JW, Hwang SG, Noh WC. STAT3-survivin signaling mediates a poor response to radiotherapy in HER2-positive breast cancers. Oncotarget. 2016;7:7055–65.
Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, Malchinkhuu E, Wersto RP, Biragyn A. Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4+ T cells to T-regulatory cells. Cancer Res. 2011;71:3505–15.
Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307.
Nagorsen D, Scheibenbogen C, Marincola FM, Letsch A, Keilholz U. Natural T cell immunity against cancer. Clin Cancer Res. 2003;9:4296–303.
Oweida A, Lennon S, Calame D, Korpela S, Bhatia S, Sharma J, Graham C, Binder D, Serkova N, Raben D, et al. Ionizing radiation sensitizes tumors to PD-L1 immune checkpoint blockade in orthotopic murine head and neck squamous cell carcinoma. Oncoimmunology. 2017;6:e1356153.
Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–7.
Ott M, Kassab C, Marisetty A, Hashimoto Y, Wei J, Zamler DB, Leu JS, Tomaszowski KH, Sabbagh A, Fang D, et al. Radiation with STAT3 blockade triggers dendritic cell-T cell interactions in the glioma microenvironment and therapeutic efficacy. Clin Cancer Res. 2020.
Ma D, Jiang YZ, Liu XY, Liu YR, Shao ZM. Clinical and molecular relevance of mutant-allele tumor heterogeneity in breast cancer. Breast Cancer Res Treat. 2017;162:39–48.
Acknowledgements
We would like apologize for the omission of any primary citations.
Funding
This study was funded by the Grants from the Union Project of Luzhou City and the Southwest Medical University (Nos. 14JC0144, 2013LZLY-J40).
Author information
Authors and Affiliations
Contributions
J.J. and L.Y. conceived and drafted the manuscript. J.J. drew the Figures. C.Y. and W.J.W. discussed the concepts of the manuscript. Z.Q.J. gave the constructive guidance. W.J.B. and F.S.Z. approved the version to be submitted. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jin, J., Li, Y., Zhao, Q. et al. Coordinated regulation of immune contexture: crosstalk between STAT3 and immune cells during breast cancer progression. Cell Commun Signal 19, 50 (2021). https://doi.org/10.1186/s12964-021-00705-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-021-00705-2