
Abstract
Pancreatic cancer (PC) is one of the most fatal diseases with a very high rate of metastasis and low rate of survival. Despite the advances in understanding this devastating disease, PC still accounts for 3% of all cancers and causes almost 7% of death of cancer patients. Recent studies have demonstrated that the transcription factor nuclear factor-erythroid 2-related factor 2 (Nrf2) and its key negative regulator Kelch-like ECH-associated protein 1 (Keap1) are dysregulated in PC and the Keap1-Nrf2 pathway is an emerging target for PC prevention and therapy. Indeed, Nrf2 plays an either tumor-suppressive or promoting function in PC, which depends on the developmental stages of the disease and the cellular context. Several natural-product Nrf2 activators have been developed to prevent pancreatic carcinogenesis, while the Nrf2 inhibitors have been examined for their efficacy in inhibiting PC growth and metastasis and reversing chemoresistance. However, further preclinical and clinical studies for determining the effectiveness and safety of targeting the Keap1-Nrf2 pathway for PC prevention and therapy are warranted. In this review, we comprehensively discuss the dual roles of the Keap1-Nrf2 signaling pathway in PC as well as the current targeting strategies and known activators and inhibitors of Nrf2. We also propose new strategies that may be used to address the current issues and develop more specific and more effective Nrf2 activator/inhibitors for PC prevention and therapy.
Similar content being viewed by others

Background
Pancreatic cancer (PC) remains the most aggressive and most lethal cancer with the worst survival and prognosis, although significant advances have been made in PC research, especially in its molecular mechanisms, epidemiology, pathophysiology, and treatment in the past decades [1,2,3,4]. Because the initiation of PC is often quite nonspecific and subtle, this notorious disease is very difficult to be diagnosed in its early phases and it is always detected at the advanced stages with metastasis [3]. Since only about 10 to 15% PC are diagnosed at the resectable stage and can be treated with surgery, many patients prolong their lives by relying on radiotherapy and chemotherapy, which may also decrease the quality of their lives due to the side effects [5]. Numerous studies on molecular abnormalities in PC have been performed, leading to the identification of several driver mutations, such as KRAS, TP53, CDKN2A, and SMAD4, which contribute to the initiation and development of PC [6, 7]. Targeted agents that specifically inhibit these molecular changes have been developed, e.g. Erlotinib and Larotrectinib [8]. Although these targeted therapies alone or in combination with chemotherapy, e.g. gemcitabine have shown some efficacy in patients with unresectable PC, only a very limited increase in survival time of patients has been observed [8]. Therefore, identifying novel molecular targets and developing safe and effective targeted therapeutics are still urgent for PC prevention and therapy.
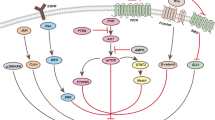
The abnormal expression and activation of the transcription factor nuclear factor-erythroid 2-related factor 2 (Nrf2) and its major negative regulator Kelch-like ECH-associated protein 1 (Keap1) have been observed at different stages of PC and correlated with its initiation, progression, metastasis, and chemoresistance (as shown in Fig. 1) [9, 10]. During the early stage of pancreatic carcinogenesis, Nrf2 exerts a tumor-suppressive role by binding to antioxidant response elements (AREs) and activating its downstream target genes that regulate the cellular antioxidant/detoxification response and immune surveillance [11,12,13]. However, in the PC progression and metastasis phases, Keap1 mutation and silencing are frequently observed and cause the aberrant stabilization of Nrf2 [9]. Consequently, Nrf2 is constitutively activated and promotes PC growth, metastasis, and chemoresistance by regulating the downstream genes that are involved in proliferation, cell cycle progression, apoptosis, ferroptosis, senescence, autophagy, stem cell self-renewal, angiogenesis, metastasis, drug resistance, and metabolic reprogramming [9, 10, 14, 15]. Recent studies have also shown the preventive efficacy of Nrf2 activators in pancreatic tumorigenesis and the suppressive effects of Nrf2 inhibitors on PC growth and metastasis [16,17,18,19]. Because of the opposite roles of Nrf2 at the early and progression stages of PC, a better understanding of the Keap1-Nrf2 pathway and its role in PC may provide a promising strategy for developing novel preventive and therapeutic agents for this dreadful disease.

The dual roles of Keap1-Nrf2 signaling pathway in pancreatic cancer. In normal cells, Nrf2 is temporarily activated when exposed to electrophiles and ROS and activates the transcription of genes that increase the capabilities of detoxification, antioxidant, and immune surveillance, preventing chemical-induced carcinogenesis. In Nrf2-addicted cancer cells, Keap1 is deleted or expressed at a very low level. Nrf2 is overexpressed and constitutively activated and promotes cancer growth and metastasis by regulating its downstream target genes
In the present review, we comprehensively discuss the new findings related to the regulation of the Keap1-Nrf2 pathway, its target genes, and its dual roles in PC initiation, progression, metastasis, and drug resistance. We also review various activators and inhibitors of Nrf2 as well as the current strategies for identifying new Nrf2-targeted agents for PC prevention and therapy.
Structures of Keap1 and Nrf2 proteins
The transcription factor Nrf2 contains 605 amino acids distributed into seven highly conserved Nrf2-ECH homology (Neh) domains, Neh1-Neh7, as depicted in Fig. 2a. Among these domains, Neh1 has a cap ‘n’ collar (CNC) basic-region leucine zipper (bZIP) domain, which is critical for binding to DNA and forming heterodimers with small MAF (sMAF) proteins [20]. In addition, Neh1 contains a nuclear localization signal (NLS) that allows the nuclear translocation of Nrf2 [21]. The Neh2 domain is located in the N-terminal region and contains lysine residues. Neh2 is mainly responsible for the binding with Keap1 homodimers as well as the subsequent ubiquitination and proteasomal degradation of Nrf2 [22]. In addition, Neh6, rich in serine residues, is another negative regulatory domain by binding to β-transducin repeat-containing protein (β-TrCP) and causing Nrf2 ubiquitination and degradation [23]. Conversely, the Neh3 domain endows the Nrf2 protein better stability [24]. In addition, Neh3, Neh4, and Neh5 are transactivation domains that interact with other coactivators [24, 25]. Neh7 is a newly defined domain that is necessary for the binding of Nrf2 to retinoic X receptor α (RXRα), resulting in the inhibition of the Nrf2-ARE signaling pathway [26].

Schematic structures of Keap1 and Nrf2. a Nrf2 comprises seven Nrf2-ECH homology (Neh) domains, Neh1-Neh7. Among these domains, Neh2 and Neh6 are important for binding with the negative regulators Keap1 and β-TrCP, respectively, consequently causing Nrf2 ubiquitination and degradation. Neh1 contains a cap ‘n’ collar (CNC) basic-region leucine zipper (bZIP) domain that is important for interacting with small MAF (sMAF) proteins and DNA. Neh1 also holds a nuclear localization signal (NLS) which is required for the nuclear translocation of Nrf2. Neh3, Neh4, and Neh5 domains are necessary for transactivation. Neh7 is important for binding with an Nrf2 repressor, the retinoic X receptor α (RXRα). b Keap1 comprises an N-terminal region (NTR), a broad complex, Tramtrack and Bric-à-Brac (BTB) domain, an intervening region (IVR), six Kelch repeats, and a C-terminal region (CTR). Among these domains, BTB domain is responsible for the homodimerization of Keap1 and the binding with Cullin3 (Cul3) E3 ligase. BTB also harbors cysteine residues, which are reactive to electrophiles and reactive oxygen species (ROS). Kelch repeats contain binding sites that are important for interacting with Nrf2, p62, and other E/STGE proteins. IVR contains a nuclear export signal (NES), which regulates the cytoplasmic localization of Keap1
Keap1, the key Nrf2 negative regulator, has 624 amino acids that are distributed into five domains (as shown in Fig. 2b), including an N-terminal region (NTR), a broad complex, Tramtrack and Bric-à-Brac (BTB) domain, a cysteine-rich intervening region (IVR), six Kelch repeats, and a C-terminal region (CTR) [27]. Among these domains, the BTB domain is critical for the homodimerization of Keap1 and the binding with Cullin3 (Cul3) E3 ligase [28]. The Kelch repeats are required for the binding of Keap1 with the Neh2 domain of Nrf2 as well as p62 and other E/STGE-containing proteins [29, 30]. IVR is located between BTB and Kelch repeats and has a nuclear export signal (NES) that regulates the cytoplasmic localization of Keap1 [31]. In addition, Keap1 is highly rich in cysteine residues that act as sensors for electrophiles and reactive oxygen species (ROS), which is important for protecting Nrf2 from proteasomal degradation [32].
Regulation of Keap1-Nrf2 signaling pathway in pancreatic cancer
Keap1-Nrf2 signaling pathway
The Keap1-Nrf2 signaling pathway has been extensively reviewed recently [33,34,35]. Here, we present a brief overview of the Keap1-Nrf2 signaling pathway and its key regulators which are depicted in Fig. 3. Under normal physiological conditions, Nrf2 activity and stability are tightly controlled by Keap1 [33]. Keap1 homodimer directly interacts with Cullin3 (Cul3) and forms the Keap1-Cul3-RBX1 (Ring box protein-1) E3 ligase complex, which targets Nrf2 and induces its polyubiquitination and protein degradation by the 26S proteasome [36]. Under oxidative stress, electrophiles and ROS react with the cysteine residues, especially cysteine 151 in Keap1, which leads to the alteration of Keap1 conformation and its inactivation. Consequently, Nrf2 is released from Keap1-Cul3-RBX1 complex and translocates into the nucleus. The nuclear Nrf2 then forms heterodimers with sMAF proteins and binds to the AREs, activating the transcription of ARE-driven genes [33].

The Keap1-Nrf2 signaling pathway. Under normal physiological conditions, the Nrf2 protein level is tightly controlled by Keap1. Keap1 dimerizes through the N-terminal BTB domain and forms E3 ubiquitin ligase complexes with Cullin3 (Cul3) and Ring box protein-1 (RBX1), then promoting Nrf2 ubiquitination and degradation. Nrf2 is also negatively regulated by the E3 ubiquitin ligase complexes, the β-TrCP-SKP1-Cullin1 (Cul1)-RBX1 and HRD1. When cells are exposed to electrophiles or ROS or under endoplasmic reticulum (ER) stress, the Nrf2 protein level is increased. Nrf2 then translocates into the nucleus, forms heterodimers with sMAF proteins, binds to the antioxidant response elements (AREs), and then activates the transcription of ARE-driven genes. p62 also interacts with the Nrf2-binding site on Keap1 and releases Nrf2 from Keap1-mediated protein degradation
Nrf2 is also negatively regulated by the E3 ubiquitin ligase complex β-TrCP in a glycogen synthase kinase 3β (GSK-3β)-dependent but Keap1-independent manner (Fig. 3) [37]. Nrf2 is phosphorylated by GSK3β at Ser342 and Ser347 residues in the Neh6 domain, which is required for the binding of Nrf2 to β-TrCP as well as the subsequent ubiquitination and degradation [23]. In addition, the E3 ubiquitin ligase HRD1 also compromises Nrf2 activation under endoplasmic reticulum (ER) stress and ROS, independent of Keap1 and β-TrCP [38]. The activation of the ER stress pathway upregulates X-box-binding protein 1 (XBP1)-mediated expression of HRD1, which then directly interacts with Nrf2 and promotes its ubiquitination and proteasomal degradation (Fig. 3) [38]. Moreover, the autophagy substrate p62 directly interacts with the Nrf2-binding site on Keap1, competitively inhibits the binding of Nrf2 to Keap1, and protects Nrf2 from Keap1-Cul3-RBX1-mediated protein degradation [30].
Direct regulation of Keap1 in pancreatic cancer
Several Keap1 regulators have recently been found to modulate the expression of Keap1 and Keap1-mediated Nrf2 degradation, consequently affecting PC growth and metastasis (as summarized in Table 1). The epigenetic regulator UHRF1 (ubiquitin-like containing PHD and RING finger domains 1) is overexpressed in PC and correlated to tumor growth [39]. Recent studies have shown that UHRF1 suppresses Keap1 expression by inducing KEAP1 promoter methylation, which causes Nrf2 activation and promotes PC cell proliferation and cell cycle progression [39]. Another epigenetic repressor, MBD1 (methyl-CpG binding domain protein 1) is also highly expressed in PC and negatively regulates the expression of Keap1 by enhancing KEAP1 promoter methylation, in which c-Myc plays a critical role [40].
In addition to p62, two new Keap1-binding proteins, aPKCι (atypical protein kinase Cι) and PALB2 (partner and localizer of BRCA2) have been reported to inhibit ROS and promote tumor growth and drug resistance by inducing Nrf2 accumulation, nuclear translocation, and activation (Table 1) [41, 42]. Mechanistically, aPKCι and PALB2 share with Nrf2 a highly conserved Keap1-binding motif and compete with Nrf2 for Keap1 binding, protecting Nrf2 from Keap1-mediated protein degradation [41, 42]. Moreover, microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) have also been found to regulate the expression of Keap1 in PC [43, 52]. A lncRNA, KRAL (Keap1 regulation-associated lncRNA) directly interacts with miR-141as a competing endogenous RNA (ceRNA) and increases the expression of Keap1, leading to the inactivation of Nrf2 and the enhancement of chemosensitization to cancer cells [43].
Direct regulation of Nrf2 in pancreatic cancer
Except for Keap1-mediated Nrf2 degradation, several regulators have been reported to modulate the expression and transcriptional activity of Nrf2 in PC in a Keap1-independent manner (as summarized in Table 1). The oncogene aldolase A (ALDOA) promotes PC cell proliferation and invasion by increasing the expression of its downstream targets, including Nrf2 [44]. The oncogene Kras also increases the expression of Nrf2, in which ERK (extracellular signal-regulated kinase) and PIN1 (Peptidyl-prolyl cis/trans isomerase NIMA-interacting 1) play a critical role [45,46,47]. The Kras/ERK/Nrf2 signaling pathway has also been found to promote PC cell growth and cause drug resistance [47]. Also, the glucose regulatory protein 78 (GRP78)-mediated unfolded protein response (UPR) increases the Nrf2 activity and the resistance of PC cells to gemcitabine [48]. The transcription factor STAT3 (signal transducer and activator of transcription 3) has also been shown to upregulate the expression of Nrf2 and induce EMT (epithelial-to-mesenchymal transition) in PC [14].
Several negative regulators of Nrf2 have been found and correlated to PC growth, metastasis, and drug resistance (Table 1). Apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/Ref-1) directly interacts with Nrf2 and inhibits its activity in PC cells [49]. Ref-1 also associates with the HMOX-1 (heme oxygenase-1) promoter via the Nrf2-binding site, competitively inhibits the Nrf2-HMOX-1 promoter binding, and down-regulates the expression of HMOX-1 [49]. Decreased deoxycytidine kinase (dCK) has been shown to inhibit PC cell growth and metastasis and sensitize PC cells to gemcitabine treatment by down-regulating the Nrf2 transcriptional activity and decreasing the expression of ARE-driven antioxidant genes [50]. LncRNAs and miRNAs are also involved in the regulation of Nrf2 expression in PC [52]. A lncRNA, termed NRAL (Nrf2 regulation-associated lncRNA) directly binds to miR-340-5p and inhibits the miR-340-5p-mediated repressing activity of the Nrf2–3’UTR, therefore increasing the expression of Nrf2 and inducing drug resistance in PC cells [51].
The dual roles of Keap1-Nrf2 signaling pathway in pancreatic cancer initiation and progression
The Keap1-Nrf2 signaling pathway has been shown to affect PC initiation and progression in a dual manner, which mainly depends on the stages of this disease and the statuses of Keap1 and Nrf2. In the early phase of pancreatic tumorigenesis, Nrf2 activation prevents carcinogen-induced carcinogenesis by activating the transcription of genes (as summarized in Table 2) that regulate detoxification, antioxidation, and immune surveillance whereas Keap1 inhibits Nrf2 stability and activity and promotes pancreatic carcinogenesis [12, 13, 74, 80, 81]. However, at the stage of cancer progression, the loss-of-function mutations in Keap1 and the activating mutations in Nrf2 often happen and lead to the disruption of Keap1-Nrf2 binding and the stabilization and constitutive activation of Nrf2, herein increasing the expression of genes (as summarized in Table 2) that are important for cancer cell proliferation, stem cell self-renewal, cell cycle arrest, apoptosis, ferroptosis, senescence, autophagy, angiogenesis, metastasis, drug resistance, metabolic reprogramming, genome stability and proteotoxic stress [9, 12, 18, 53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73, 75,76,77,78,79, 82]. Of note, somatic mutations in Keap1 (e.g. G333C, G350S, L413R, D236H) and the methylation of the Keap1 promoter are frequently observed in lung cancer whereas Nrf2 mutations (e.g. W24C, E82D, D77V) happen more often in esophageal cancer, which have been reviewed recently [83]. However, there is few study on the accurate detection of mutations in Keap1 and Nrf2 in pancreatic cancer, and the detailed investigations on these mutations are urgently needed.
Nrf2 also promotes mRNA translation and supports efficient protein syntheses, which are partially responsible for Nrf2-regulated redox homeostasis, initiation, and maintenance of PC [10]. More importantly, the high nuclear expression of Nrf2 predicts a worse survival of PC patients, and Nrf2 may be a promising prognostic factor in PC [84]. Recently, Nrf2 has also been found to be elevated in early precursor lesions in the pancreas and contributes to pancreatic carcinogenesis [85,86,87]. These controversial results can at least partially be explained by recent analyses of the Nrf2-responsive genes in PC cells, which have shown that Nrf2 regulates the expression of both oncogenes and tumor suppressor genes [9, 88]. In this section, we present a comprehensive overview of the dual roles of the Keap1-Nrf2 pathway in PC.
The tumor suppressive role of Keap1-Nrf2 pathway in pancreatic cancer
Recent studies have provided new evidence showing the preventive effects of Nrf2 on pancreatic carcinogenesis and pancreatic tumor growth and metastasis. Kim et al have recently shown that Nrf2 activation by oxidative stress protects pancreatic beta cells from damage and apoptotic cell death, which is important for preventing pancreatic carcinogenesis induced by oxidants and carcinogens [89]. Nrf2 activation by a natural product has also been found to inhibit PC cell growth and induce apoptosis by upregulating HO-1, although the direct association of Nrf2 activation and the anticancer efficacy still needs further investigation [90]. Satoh et al have demonstrated a critical role of Nrf2 in preventing lung metastasis using an Nrf2-null (Nrf2−/−) and Keap1-knockdown (Keap1f/f) mouse model [91]. It has been reported that Nrf2 is highly expressed in Keap1f/f mice, decreases the ROS in the immunosuppressive myeloid-derived suppressor cells (MDSCs), and prevents cancer cell metastasis to the lung after implantation of a lung carcinoma cell line 3LL [91]. However, the Nrf2-deficient Keap1f/f mice showed a significantly higher lung metastatic rate after they lost the ability to maintain the redox balance in the immune and hematopoietic systems [91].
The carcinogenic role of Keap1-Nrf2 pathway in pancreatic cancer
Despite the protective effects of Nrf2 on oxidative stress-induced carcinogenesis, a large number of studies have demonstrated a carcinogenic role of the Keap1-Nrf2 pathway in PC. Previous studies have shown that the Kras oncogene induces Nrf2 expression via the Kras/ERK/NRF2 axis [47, 92]. Kras-mediated Nrf2 expression and activation causes low intracellular ROS levels and promotes pancreatic tumorigenesis and metastasis while Nrf2 inhibition blocks Kras-induced cell proliferation, tumorigenesis, and metastasis [46, 85, 87, 93]. Namada et al have recently examined the role of Nrf2 in pancreatic carcinogenesis using a mouse model carrying pancreas-specific Kras and p53 mutations, named KPC mouse model [85]. It has been found that Nrf2 deletion in the KPC mice causes a decrease in the formation of precancerous lesions and slows down the development of invasive pancreatic cancer. Furthermore, two cell lines from Nrf2 deleted KPC tumors (KPCN) have been established and lack the expression of Nrf2 and its downstream target genes, including NQO1. However, the KPCN-derived cell lines still maintain similar features as the cell lines from KPC tumors, except for their increased sensitivity to oxidative stress and gemcitabine [85].
Interestingly, the simultaneous activation of Kras and Nrf2 by Kras mutation and Keap1 deletion, respectively, does not promote PC development as expected but causes pancreatic atrophy [94]. Namada et al have also generated a pancreatic cancer mouse model harboring pancreas-specific K-ras mutation and Keap1 deletion (KC::Keap1) and a KPC mouse model carrying Keap1 deletion (KPC::Keap1) [94]. Both KC::Keap1 and KPC::Keap1 mice contain constitutively activated Nrf2 due to Keap1 deletion, which does not promote the development of pancreatic cancer but causes the progressive atrophy of pancreatic parenchyma. Consequently, these Keap1 deletion mice become weak and even start to die around 40 days after birth [94]. Further studies have shown that Nrf2 deletion can rescue the phenotypic changes in KC::Keap1 mice, which confirms the role of Nrf2 in pancreatic atrophy. Kras also activates Nrf2 by driving the expression of UHRF1, which preserves Keap1 promoter methylation and inhibits Keap1 expression [39]. Deletion of UHRF1 inhibits PC cell growth and induces cell cycle arrest at G2/M phase and apoptosis by reducing the expression of Nrf2 and increasing ROS level. However, concomitant deletion of Keap1 and UHRF1 rescues cells from G2/M phase arrest by restoring Nrf2 expression [39].
Nrf2 exerts its carcinogenic role not only by activating the transcription of its target genes but also by promoting mRNA translation [10, 34, 95]. Kha et al have recently discovered that Nrf2 activation protects premalignant pancreatic ductal epithelial (PDE) cells from apoptosis and accelerates the formation and growth of pancreatic tumors by inducing the expression of a splicing variant of ATF3 (activating transcription factor 3), termed ΔZip2 [96]. Nrf2 also attenuates TGF-β1 (transforming growth factor-β1)-mediated growth inhibition of PDE cells by decreasing the expression levels of p21, phosphor-p38 and phosphor-Smad3 [86]. Moreover, Nrf2 inhibits PC cell apoptosis by upregulating the expression of anti-apoptotic proteins (e.g., Bcl2) and other cytoprotective proteins and enzymes [97].
Pancreatic stellate cells (PSC)-secreted SDF-1α (stromal-derived factor-1α) and IL-6 (interleukin-6) activates Nrf2, which further induces metabolic reprogramming and ROS detoxification and promotes PC cell proliferation [98]. Further studies have shown that PSC-secreted IL-6 also promotes EMT in PC cells by activating the JAK (Janus kinase)/STAT3/Nrf2 pathway. It has been observed that Nrf2 activation upregulates the expression of N-cadherin, fibronectin, Twist2, Snail, and Slug, which contribute to the increased EMT phenotypes [14]. Nrf2 has also been reported to induce EMT by regulating the cancer cells and macrophages interaction [99]. PC cells elevate the intracellular ROS level in macrophages via lactate secretion, which further activates Nrf2, induces macrophage M2 phenotype transformation, and increases VEGF (vascular endothelial growth factor) expression. The cancer cell-educated macrophages then induce Nrf2 activation in PC cells via VEGF secretion and promote EMT [99].
Pancreatitis-induced autophagy substrate p62 has been found to promote PC progression by activating the Keap1/Nrf2/MDM2 (murine double minute 2) signaling pathway [60, 100]. Mechanistically, p62 accumulation stabilizes Nrf2 protein and increases Nrf2-mediated MDM2 expression, which further accelerates the progression of pancreatic intraepithelial neoplasia (PanIN) to PC through both p53-dependent and p53-independent mechanisms [60]. These results also support that directly inhibiting MDM2 may be effective in preventing PanIN to PC progression, regardless of the p53 status [101, 102]. In addition to the regulation by the autophagy adaptor p62, Nrf2 has been reported to promote autophagy by targeting miR-129-3p in PC cells under the treatment of histone deacetylase inhibitors (HDACis) [103]. Further studies have indicated that mammalian target of rapamycin (mTOR) is a target of miR-129-3p and the Nrf2-miR-129-3p-mTOR axis is mainly responsible for HDACis-induced autophagy in PC cells [103]. However, Nrf2 activation has also shown a negative interaction with autophagy under ROS stress [104]. ROS stimulation induces both Nrf2 activation and autophagy in PC cells and inhibiting either Nrf2 or autophagy will lead to the enhancement of another one [104]. Interestingly, the combined inhibition of Nrf2 pathway and autophagy also increases PC cell apoptosis by chemotherapy, which may provide a new treatment approach for PC patients [105].
The Keap1-Nrf2 pathway is also involved in chemoresistance in PC by regulating the expression of drug resistance-associated genes and cytoprotective antioxidant genes (Table 2) [15, 106]. Furthermore, ALDH1A1 (aldehyde dehydrogenase 1 family, member A1) and ALDH3A1 (aldehyde dehydrogenase 3 family, member A1) are up-regulated by Nrf2 and may also contribute to drug resistance in PC [107]. LncRNAs, KRAL and NRAL have been reported to exert an opposite role in mediating the drug resistance by regulating the miR-141/Keap1 axis and the miR-340-5p/Nrf2 axis, respectively [43, 51]. Chemotherapies, e.g. gemcitabine have also been found to increase Nrf2 expression, while pretreatment with Nrf2 inhibitors enhances the sensitivity of PC cells to chemotherapy [108].
Targeting Keap1-Nrf2 signaling pathway for pancreatic cancer prevention and therapy
The Keap1-Nrf2 pathway has been considered as a potential target for PC prevention and therapy. Several Nrf2 activators and inhibitors have been identified and shown efficacy in PC models in vitro and in vivo (as summarized in Table 3). In this section, we focus on the current activators and inhibitors, their efficacy and mechanisms of action. We also review known targeting strategies and propose new targeting strategies (Fig. 4) that may be used to develop more specific and effective Nrf2-targeted agents for PC prevention and therapy.

Targeting Keap1-Nrf2 signaling pathway for pancreatic cancer prevention and therapy. Several strategies have been proposed to target the Nrf2 signaling pathway in human pancreatic cancer: (1) modulating Nrf2 expression at the transcriptional level, (2) modulating the Nrf2 activity by targeting its upstream activators and stabilizers, (3) affecting the nuclear translocation of Nrf2, (4) targeting the Keap1-Nrf2 binding for modulating Nrf2 protein stability, (5) targeting the β-TrCP-Nrf2 binding or the HRD1-Nrf2 binding and modulating Nrf2 ubiquitination and degradation, (6) modulating the binding of Nrf2 with its co-activators in the nucleus, and (7) modulating the binding of Nrf2 with its downstream target genes. Many small-molecule Nrf2 activators and inhibitors have been discovered and shown efficacy in pancreatic cancer cells in vitro and in vivo
Nrf2 activators
Considering the critical role of Keap1 in regulating Nrf2 stability, targeting the Keap1-Nrf2 binding has been demonstrated as a promising strategy for identifying specific activators of Nrf2 (as shown in Fig. 4). A recent study has discovered a dihydroxy coumarin derivative, termed esculetin that directly binds to Keap1 and inhibits its binding to Nrf2 [109]. The released Nrf2 by esculetin has been observed to reduce ROS level, inhibit cell growth, arrest cells at G1 phase, and induce apoptosis and loss of mitochondrial membrane potential in PC cells in vitro [109]. The activation of caspase cascade and inhibition of NF-κB (nuclear factor-κB) also contribute to the anticancer activity of esculetin. However, the selectivity of esculetin to Keap1 and its in vivo efficacy still need further investigation in clinically relevant models of PC. Because Keap1 is often deleted and mutated in pancreatic cancer cells, which also causes the aberrant activation of Nrf2, disrupting the Keap1-Nrf2 binding for reactivating Nrf2 could not be an effective strategy for PC therapy. Therefore, it is possible that the reactivation of Nrf2 by esculetin may not be mainly responsible for its anticancer activity.
Currently, most of the identified Nrf2 activators, including MT477 [16], oleanolic acid (OA) [45], fisetin [110], QD325 [17], resveratrol [61], sulforaphane (SFN) [90], and alphalipoic acid [111] have been found to increase Nrf2 expression and/or enhance Nrf2 activity (Table 3). Among them, OA and SFN have been demonstrated to activate the upstream regulators Nrf2 pathway, ERK and AMPK (AMP-activated protein kinase), respectively, which are important for the increased expression of Nrf2 by these compounds [45, 90]. However, OA-induced Nrf2 activation has been found to compromise its anticancer activity and the suppression of ERK/Nrf2 pathway enhances OA-induced cancer cell apoptosis in vitro and its inhibitory effects on the growth of xenograft tumors in vivo [45]. Although SFN has been shown to inhibit PC cell growth and invasion in vitro and suppress tumor growth in both Panc-1 xenograft model and a transgenic mouse model in vivo [90], the importance of Nrf2 activation in SFN’s anti-PC activity as well as the detailed molecular mechanisms should be further investigated.
All the other known Nrf2 activators have also been shown to inhibit PC cell survival in vitro and/or suppress the growth of PC xenograft tumors in vivo (Table 3). Among them, both MT477 and QD325 have shown in vivo efficacy in MiaPaca-2 xenograft model [16, 17], whereas alphalipoic acid has been found to inhibit the growth of CFPAC-1 xenograft tumor [111]. However, it is still unknown whether Nrf2 activation is responsible for their anti-PC activities. They may activate Nrf2 by inducing cellular ROS accumulation and/or affecting Nrf2 upstream regulators, which need to be investigated for the detailed molecular mechanisms. More importantly, the mutations in Keap1 and constitutive activation of Nrf2 are frequently observed in various cancers, including PC, the safety and feasibility of using Nrf2 activators for treating PC should be carefully examined.
Nrf2 inhibitors
The currently identified Nrf2 inhibitors have been reported to suppress Nrf2 expression, induce Nrf2 protein degradation, or inhibit Nrf2 nuclear translocation (as shown in Fig. 4). A natural product termed brusatol has been reported to reduce Nrf2 protein levels without affecting the expression of Keap1 in PC cells [112]. The down-regulation of Nrf2 by brusatol further caused the repression of its downstream targets that are responsible for drug resistance (MRD1 and MRP5) and antioxidant response (HQO1 and HO-1). Because the upregulation of Nrf2 contributes to gemcitabine resistance, brusatol has also been evaluated in combination with gemcitabine. As expected, brusatol significantly enhanced the anticancer activity of gemcitabine in PC cells in vitro and in vivo [112]. However, brusatol has been reported to be a broad inhibitor of protein translation and could not be used as a specific Nrf2 inhibitor. Another natural-product Nrf2 inhibitor, digoxin has been found to down-regulate Nrf2 expression at the transcription level by inhibiting PI3K (Phosphoinositide 3-kinase)/Akt pathway [19]. Consistently, digoxin-induced Nrf2 inhibition also reverses the resistance of gemcitabine-resistant PC cells to gemcitabine in vitro and in vivo [19].
Nrf2 protein stability is mainly mediated by the ubiquitin-proteasome system. There are at least three E3 ubiquitin ligase complexes, including Keap1-Cul3-RBX1 complex, β-TrCP, and HRD1 that control the ubiquitination of Nrf2 [23, 36, 38]. Therefore, inducing Nrf2 ubiquitination and proteasomal degradation may be an effective approach to inhibit Nrf2. PIK-75, a previously reported PI3K/DNA-PK inhibitor has recently been identified to inhibit Nrf2 by inducing its proteasome-mediated protein degradation [18]. Further studies have also shown its inhibitory effects on Nrf2 transcriptional activity. The anticancer efficacy of PIK-75 alone or in combination with gemcitabine has been demonstrated in PC models in vitro and in vivo [18]. Very recently, a drug-repositioning screening of ~ 4000 clinical compounds has been performed and led to the identification of a potent Nrf2 inhibitor, clobetasol propionate (CP) [113]. CP has been shown to prevent Nrf2 nuclear accumulation and promote its protein degradation in a glucocorticoid receptor-, GSK3-, and β-TrCP-dependent manner. More importantly, CP not only inhibits the growth of tumors with Keap1 and/or LKB1 (liver kinase B1) mutations but also enhances the anticancer activity of rapamycin in vitro and in vivo [113]. These results have suggested that CP could be developed as a therapeutic agent for cancers, especially those with Nrf2 activation and Keap1 and LKB1 mutations.
Directly targeting the active domain of Nrf2 has recently been considered as a potential strategy for developing Nrf2 inhibitors. Singh et al have performed a high-throughput screen of a compound library containing ~ 400,000 small molecules and identified a specific Nrf2 inhibitor, termed ML385, which directly binds to the CNC-bZIP domain of Nrf2 and inhibits the DNA binding activity of the MafG-Nrf2 protein complex [114]. Further studies have shown that ML385 exerts selective cytotoxicity against cancer cells with Keap1 mutations and increases the toxicity of carboplatin in vitro and in vivo [114]. However, it is still unclear whether ML385 is a selective Nrf2 inhibitor or a broad inhibitor of various transcription factors. Another high throughput screening has also led to the identification of a novel Nrf2 inhibitor, named AEM1, which can inhibit Nrf2 transcriptional activity and decrease the expression of its target genes [115]. AEM1 alone and in combination with chemotherapy has been shown to inhibit the growth of cancer cells harboring mutant Keap1 and constitutively activated Nrf2 in vitro and in vivo [115]. However, the cellular target of AEM1, as well as its associated molecular mechanism, is still unknown. In addition, the inhibition of nuclear Nrf2 and its transcription activity has been reported to be involved in the anti-PC activity of pterostilbene in vitro and in vivo, but the detailed molecular mechanisms are still not clear yet [116].
As summarized in Fig. 4 and Table 3, most of the known Nrf2 activators and inhibitors have been developed to (1) modulate Nrf2 expression, (2) target Nrf2 upstream regulators, (3) affect the nuclear translocation of Nrf2, or (4) target Keap1-Nrf2 binding. However, there are still several strategies that need to be further examined for developing novel and specific Nrf2-targeted agents, such as (5) affectingNrf2 ubiquitination and degradation by targeting the β-TrCP-Nrf2 binding and HRD1-Nrf2 binding or developing Nrf2-targeted PROTACs (proteolysis targeting chimeras), (6) modulating the binding of Nrf2 with its co-activators in the nucleus, and (7) modulating the binding of Nrf2 with its downstream target genes.
Conclusions and future direction
During the early stage of pancreatic tumorigenesis, Nrf2 is tightly controlled by Keap1. Nrf2 activation by oxidative stress prevents PC initiation by regulating antioxidant and detoxification response and immune surveillance. In the phases of PC progression and metastasis, Keap1 is mutated or deleted and Nrf2 is constitutively activated. At this stage, the role of Nrf2 is switched from tumor suppression to tumor promotion. Nrf2 can promote pancreatic tumor growth, metastasis, and chemoresistance by activating its downstream target genes and promoting mRNA translation. It has been shown that Nrf2 activators can prevent pancreatic tumorigenesis but may also induce chemoresistance. Conversely, Nrf2 inhibitors can inhibit pancreatic tumor growth and metastasis and sensitize PC cells to chemotherapies, especially gemcitabine. However, the feasibility of the clinical application of Nrf2-targeted agents for PC prevention and therapy still needs further investigation due to the controversial roles of Nrf2 at different stages of PC. Furthermore, the majority of the current Nrf2 activators and inhibitors have been found to have other molecular targets and more specific Nrf2-targeted compounds should be developed. Several unexplored strategies, i.e. targeting the β-TrCP-Nrf2 binding and HRD1-Nrf2 binding or developing Nrf2 PROTACs may be investigated for specific Nrf2-targeted agents for PC prevention and therapy.
Availability of data and materials
Not applicable.
Abbreviations
- ALDH1A1:
-
Aldehyde dehydrogenase 1 family, member A1
- ALDH3A1:
-
Aldehyde dehydrogenase 3 family, member A1
- ALDOA:
-
Aldolase A
- AMPK:
-
AMP-activated protein kinase
- APE1/Ref-1:
-
Apurinic/apyrimidinic endonuclease/redox factor-1
- aPKCι:
-
Atypical protein kinase Cι
- AREs:
-
Antioxidant response elements
- ATF3:
-
Activating transcription factor 3
- BTB domain:
-
Broad complex, Tramtrack andBric-à-Brac domain
- bZIP:
-
Basic-region leucine zipper
- ceRNA:
-
Competing endogenous RNA
- CNC:
-
Cap ‘n’ collar
- CP:
-
Clobetasol propionate
- CTR:
-
C-terminal region
- Cul1:
-
Cullin1
- Cul3:
-
Cullin3
- dCK:
-
Deoxycytidine kinase
- EMT:
-
Epithelial-to-mesenchymal transition
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular signal-regulated kinase
- GRP78:
-
Glucose regulatory protein 78
- GSK-3β:
-
Glycogen synthase kinase 3β
- HDACis:
-
Histone deacetylase inhibitors
- HMOX-1:
-
Heme oxygenase-1
- IL-6:
-
Interleukin-6
- IVR:
-
Intervening region
- JAK:
-
Janus kinase
- KC::Keap1 mice:
-
Mice harboring pancreas-specific K-ras mutation and Keap1 deletion
- Keap1:
-
Kelch-like ECH-associated protein 1
- KPC::Keap1 mice:
-
KPC mice bearing Keap1 deletion
- KPCN mice:
-
KPC mice bearing Nrf2 deletion
- KRAL:
-
Keap1 regulation-associated lncRNA
- LKB1:
-
Liver kinase B1
- LncRNA:
-
Long non-coding RNA
- MBD1:
-
Methyl-CpG binding domain protein 1
- MDM2:
-
Murine double minute 2
- MDSCs:
-
Myeloid-derived suppressor cells
- miRNAs:
-
MicroRNAs
- mTOR:
-
Mammalian target of rapamycin
- Neh:
-
Nrf2-ECH homology
- NES:
-
Nuclear export signal
- NF-κB:
-
Nuclear factor-κB
- NLS:
-
Nuclear localization signal
- NRAL:
-
Nrf2 regulation-associated lncRNA
- Nrf2:
-
Nuclear factor-erythroid 2-related factor 2
- NTR:
-
N-terminal region
- OA:
-
Oleanolic acid
- PALB2:
-
Partner and localizer of BRCA2
- PanIN:
-
Pancreatic intraepithelial neoplasia
- PC:
-
Pancreatic cancer
- PDE:
-
Pancreatic ductal epithelial
- PI3K:
-
Phosphoinositide 3-kinase
- PIN1:
-
Peptidyl-prolyl cis/trans isomerase NIMA-interacting 1
- PROTACs:
-
Proteolysis targeting chimeras
- PSC:
-
Pancreatic stellate cells
- RBX1:
-
Ring box protein-1
- ROS:
-
Reactive oxygen species
- RXRα:
-
Retinoic X receptor α
- SDF-1α:
-
Stromal-derived factor-1α
- SFN:
-
Sulforaphane
- sMAF:
-
Small MAF
- STAT3:
-
Signal transducer and activator of transcription 3
- TGF-β1:
-
Transforming growth factor-β1
- UHRF1:
-
Ubiquitin-like containing PHD and RING finger domains 1
- UPR:
-
Unfolded protein response
- VEGF:
-
Vascular endothelial growth factor
- XBP1:
-
X-box-binding protein 1
- β-TrCP:
-
β-transducin repeat-containing protein
References
Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2019;16(4):207–20.
Murakami T, Hiroshima Y, Matsuyama R, Homma Y, Hoffman RM, Endo I. Role of the tumor microenvironment in pancreatic cancer. Ann Gastroenterol Surg. 2019;3(2):130–7.
Idachaba S, Dada O, Abimbola O, Olayinka O, Uma A, Olunu E, Fakoya AOJ. A review of pancreatic cancer: epidemiology, genetics, screening, and management. Open Access Maced J Med Sci. 2019;7(4):663–71.
Annese T, Tamma R, Ruggieri S, Ribatti D. Angiogenesis in pancreatic cancer: Pre-clinical and clinical studies. Cancers (Basel). 2019;11(3):381.
Heinrich S, Lang H. Neoadjuvant therapy of pancreatic cancer: definitions and benefits. Int J Mol Sci. 2017;18(8):1622.
Strimpakos A, Saif MW, Syrigos KN. Pancreatic cancer: from molecular pathogenesis to targeted therapy. Cancer Metastasis Rev. 2008;27(3):495–522.
Tanaka S. Molecular pathogenesis and targeted therapy of pancreatic cancer. Ann Surg Oncol. 2016;23(Suppl 2):S197–205.
Chandana S, Babiker HM, Mahadevan D. Therapeutic trends in pancreatic ductal adenocarcinoma (PDAC). Expert Opin Inv Drug. 2019;28(2):161–77.
Lister A, Nedjadi T, Kitteringham NR, Campbell F, Costello E, Lloyd B, Copple IM, Williams S, Owen A, Neoptolemos JP, et al. Nrf2 is overexpressed in pancreatic cancer: implications for cell proliferation and therapy. Mol Cancer. 2011;10:37.
Chio IIC, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, Wilson J, Sangar V, Hao Y, Ohlund D, et al. NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell. 2016;166(4):963–76.
Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62(18):5196–203.
Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–60.
Saddawi-Konefka R, O'Sullivan T, Gross ET, Washington A Jr, Bui JD. Tumor-expressed IL-17D recruits NK cells to reject tumors. Oncoimmunology. 2014;3(12):e954853.
Wu YS, Chung I, Wong WF, Masamune A, Sim MS, Looi CY. Paracrine IL-6 signaling mediates the effects of pancreatic stellate cells on epithelial-mesenchymal transition via Stat3/Nrf2 pathway in pancreatic cancer cells. Biochim Biophys Acta Gen Subj. 2017;1861(2):296–306.
Sano M, Homma T, Hayashi E, Noda H, Amano Y, Tsujimura R, Yamada T, Quattrochi B, Nemoto N. Clinicopathological characteristics of anaplastic carcinoma of the pancreas with rhabdoid features. Virchows Arch. 2014;465(5):531–8.
Jasinski P, Zwolak P, Terai K, Vogel RI, Borja-Cacho D, Dudek AZ. MT477 acts in tumor cells as an AURKA inhibitor and strongly induces NRF-2 signaling. Anticancer Res. 2011;31(4):1181–7.
Kuang Y, Sechi M, Nurra S, Ljungman M, Neamati N. Design and synthesis of novel reactive oxygen species inducers for the treatment of pancreatic ductal adenocarcinoma. J Med Chem. 2018;61(4):1576–94.
Duong HQ, Yi YW, Kang HJ, Hong YB, Tang W, Wang A, Seong YS, Bae I. Inhibition of NRF2 by PIK-75 augments sensitivity of pancreatic cancer cells to gemcitabine. Int J Oncol. 2014;44(3):959–69.
Zhou Y, Zhou Y, Yang M, Wang K, Liu Y, Zhang M, Yang Y, Jin C, Wang R, Hu R. Digoxin sensitizes gemcitabine-resistant pancreatic cancer cells to gemcitabine via inhibiting Nrf2 signaling pathway. Redox Biol. 2019;22:101131.
Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol Cell Biol. 2009;29(10):2658–72.
Theodore M, Kawai Y, Yang J, Kleshchenko Y, Reddy SP, Villalta F, Arinze IJ. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J Biol Chem. 2008;283(14):8984–94.
Katoh Y, Iida K, Kang MI, Kobayashi A, Mizukami M, Tong KI, McMahon M, Hayes JD, Itoh K, Yamamoto M. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch Biochem Biophys. 2005;433(2):342–50.
Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, Tobon-Velasco JC, Devijver H, Garcia-Mayoral MF, Van Leuven F, et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol Cell Biol. 2012;32(17):3486–99.
Nioi P, Nguyen T, Sherratt PJ, Pickett CB. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol Cell Biol. 2005;25(24):10895–906.
Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells. 2001;6(10):857–68.
Wang H, Liu K, Geng M, Gao P, Wu X, Hai Y, Li Y, Li Y, Luo L, Hayes JD, et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013;73(10):3097–108.
Li X, Zhang D, Hannink M, Beamer LJ. Crystal structure of the Kelch domain of human Keap1. J Biol Chem. 2004;279(52):54750–8.
Zipper LM, Mulcahy RT. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J Biol Chem. 2002;277(39):36544–52.
Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34(4):176–88.
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–23.
Ogura T, Tong KI, Mio K, Maruyama Y, Kurokawa H, Sato C, Yamamoto M. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc Natl Acad Sci U S A. 2010;107(7):2842–7.
McMahon M, Lamont DJ, Beattie KA, Hayes JD. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc Natl Acad Sci U S A. 2010;107(44):18838–43.
Pandey P, Singh AK, Singh M, Tewari M, Shukla HS, Gambhir IS. The see-saw of Keap1-Nrf2 pathway in cancer. Crit Rev Oncol Hematol. 2017;116:89–98.
Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34(1):21–43.
Cloer EW, Goldfarb D, Schrank TP, Weissman BE, Major MB. NRF2 activation in cancer: from DNA to protein. Cancer Res. 2019;79(5):889–98.
Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24(16):7130–9.
Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31(6):1121–33.
Wu T, Zhao F, Gao B, Tan C, Yagishita N, Nakajima T, Wong PK, Chapman E, Fang D, Zhang DD. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28(7):708–22.
Abu-Alainin W, Gana T, Liloglou T, Olayanju A, Barrera LN, Ferguson R, Campbell F, Andrews T, Goldring C, Kitteringham N, et al. UHRF1 regulation of the Keap1-Nrf2 pathway in pancreatic cancer contributes to oncogenesis. J Pathol. 2016;238(3):423–33.
Zhang B, Xu J, Li C, Shi S, Ji S, Xu W, Liu J, Jin K, Liang D, Liang C, et al. MBD1 is an epigenetic regulator of KEAP1 in pancreatic cancer. Curr Mol Med. 2016;16(4):404–11.
Tian L, Lu Y, Yang T, Deng Z, Xu L, Yao W, Ma C, Li X, Zhang J, Liu Y, et al. aPKCiota promotes gallbladder cancer tumorigenesis and gemcitabine resistance by competing with Nrf2 for binding to Keap1. Redox Biol. 2019;22:101149.
Ma J, Cai H, Wu T, Sobhian B, Huo Y, Alcivar A, Mehta M, Cheung KL, Ganesan S, Kong AN, et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol Cell Biol. 2012;32(8):1506–17.
Wu L, Pan C, Wei X, Shi Y, Zheng J, Lin X, Shi L. lncRNA KRAL reverses 5-fluorouracil resistance in hepatocellular carcinoma cells by acting as a ceRNA against miR-141. Cell Commun Signal. 2018;16(1):47.
Ji S, Zhang B, Liu J, Qin Y, Liang C, Shi S, Jin K, Liang D, Xu W, Xu H, et al. ALDOA functions as an oncogene in the highly metastatic pancreatic cancer. Cancer Lett. 2016;374(1):127–35.
Liu J, Ma L, Chen X, Wang J, Yu T, Gong Y, Ma A, Zheng L, Liang H. ERK inhibition sensitizes cancer cells to oleanolic acid-induced apoptosis through ERK/Nrf2/ROS pathway. Tumour Biol. 2016;37(6):8181–7.
Kong B, Qia C, Erkan M, Kleeff J, Michalski CW. Overview on how oncogenic Kras promotes pancreatic carcinogenesis by inducing low intracellular ROS levels. Front Physiol. 2013;4:246.
Liang C, Shi S, Liu M, Qin Y, Meng Q, Hua J, Ji S, Zhang Y, Yang J, Xu J, et al. PIN1 maintains redox balance via the c-Myc/NRF2 axis to counteract Kras-induced mitochondrial respiratory injury in pancreatic cancer cells. Cancer Res. 2019;79(1):133–45.
Dauer P, Sharma NS, Gupta VK, Nomura A, Dudeja V, Saluja A, Banerjee S. GRP78-mediated antioxidant response and ABC transporter activity confers chemoresistance to pancreatic cancer cells. Mol Oncol. 2018;12(9):1498–512.
Fishel ML, Wu X, Devlin CM, Logsdon DP, Jiang Y, Luo M, He Y, Yu Z, Tong Y, Lipking KP, et al. Apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/Ref-1) redox function negatively regulates NRF2. J Biol Chem. 2015;290(5):3057–68.
Hu Q, Qin Y, Xiang J, Liu W, Xu W, Sun Q, Ji S, Liu J, Zhang Z, Ni Q, et al. dCK negatively regulates the NRF2/ARE axis and ROS production in pancreatic cancer. Cell Prolif. 2018;51(4):e12456.
Wu LL, Cai WP, Lei X, Shi KQ, Lin XY, Shi L. NRAL mediates cisplatin resistance in hepatocellular carcinoma via miR-340-5p/Nrf2 axis. J Cell Commun Signal. 2019;13(1):99–112.
Karihtala P, Porvari K, Soini Y, Eskelinen M, Juvonen P, Haapasaari KM. Expression levels of microRNAs miR-93 and miR-200a in pancreatic adenocarcinoma with special reference to differentiation and relapse-free survival. Oncology. 2019;96(3):164–70.
Wakabayashi N, Shin S, Slocum SL, Agoston ES, Wakabayashi J, Kwak MK, Misra V, Biswal S, Yamamoto M, Kensler TW. Regulation of notch1 signaling by nrf2: implications for tissue regeneration. Sci Signal. 2010;3(130):ra52.
Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW, et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010;38(17):5718–34.
DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47(12):1475–81.
He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, Alam J. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem. 2001;276(24):20858–65.
Yoon DS, Choi Y, Lee JW. Cellular localization of NRF2 determines the self-renewal and osteogenic differentiation potential of human MSCs via the P53-SIRT1 axis. Cell Death Dis. 2016;7:e2093.
Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–29.
Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D, Zhang DD. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009;34(6):663–73.
Todoric J, Antonucci L, Di Caro G, Li N, Wu X, Lytle NK, Dhar D, Banerjee S, Fagman JB, Browne CD, et al. Stress-activated NRF2-MDM2 cascade controls neoplastic progression in pancreas. Cancer Cell. 2017;32(6):824–39 e828.
Cheng L, Yan B, Chen K, Jiang Z, Zhou C, Cao J, Qian W, Li J, Sun L, Ma J, et al. Resveratrol-induced downregulation of NAF-1 enhances the sensitivity of pancreatic cancer cells to gemcitabine via the ROS/Nrf2 signaling pathways. Oxidative Med Cell Longev. 2018;2018:9482018.
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173–84.
Osburn WO, Wakabayashi N, Misra V, Nilles T, Biswal S, Trush MA, Kensler TW. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch Biochem Biophys. 2006;454(1):7–15.
You A, Nam CW, Wakabayashi N, Yamamoto M, Kensler TW, Kwak MK. Transcription factor Nrf2 maintains the basal expression of Mdm2: an implication of the regulation of p53 signaling by Nrf2. Arch Biochem Biophys. 2011;507(2):356–64.
Pajares M, Jimenez-Moreno N, Garcia-Yague AJ, Escoll M, de Ceballos ML, Van Leuven F, Rabano A, Yamamoto M, Rojo AI, Cuadrado A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy. 2016;12(10):1902–16.
Toth RK, Warfel NA. Strange bedfellows: Nuclear factor, erythroid 2-Like 2 (Nrf2) and hypoxia-inducible factor 1 (HIF-1) in tumor hypoxia. Antioxidants. (Basel). 2017;6(2):27.
Oh ET, Kim JW, Kim JM, Kim SJ, Lee JS, Hong SS, Goodwin J, Ruthenborg RJ, Jung MG, Lee HJ, et al. NQO1 inhibits proteasome-mediated degradation of HIF-1alpha. Nat Commun. 2016;7:13593.
Zhao Q, Mao A, Guo R, Zhang L, Yan J, Sun C, Tang J, Ye Y, Zhang Y, Zhang H. Suppression of radiation-induced migration of non-small cell lung cancer through inhibition of Nrf2-notch axis. Oncotarget. 2017;8(22):36603–13.
Wagner PJ, Park HR, Wang Z, Kirchner R, Wei Y, Su L, Stanfield K, Guilarte TR, Wright RO, Christiani DC, et al. In vitro effects of lead on gene expression in neural stem cells and associations between up-regulated genes and cognitive scores in children. Environ Health Perspect. 2017;125(4):721–9.
Hong YB, Kang HJ, Kwon SY, Kim HJ, Kwon KY, Cho CH, Lee JM, Kallakury BV, Bae I. Nuclear factor (erythroid-derived 2)-like 2 regulates drug resistance in pancreatic cancer cells. Pancreas. 2010;39(4):463–72.
Heiss EH, Schachner D, Zimmermann K, Dirsch VM. Glucose availability is a decisive factor for Nrf2-mediated gene expression. Redox Biol. 2013;1:359–65.
Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22(1):66–79.
Wu KC, Cui JY, Klaassen CD. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol Sci. 2011;123(2):590–600.
Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi H, Nakayama K, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. 2016;7:11624.
Singh B, Chatterjee A, Ronghe AM, Bhat NK, Bhat HK. Antioxidant-mediated up-regulation of OGG1 via NRF2 induction is associated with inhibition of oxidative DNA damage in estrogen-induced breast cancer. BMC Cancer. 2013;13:253.
Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014;15(1):7–18.
Dayalan Naidu S, Kostov RV, Dinkova-Kostova AT. Transcription factors Hsf1 and Nrf2 engage in crosstalk for cytoprotection. Trends Pharmacol Sci. 2015;36(1):6–14.
Dayalan Naidu S, Dinkova-Kostova AT. Regulation of the mammalian heat shock factor 1. FEBS J. 2017;284(11):1606–27.
Arlt A, Bauer I, Schafmayer C, Tepel J, Muerkoster SS, Brosch M, Roder C, Kalthoff H, Hampe J, Moyer MP, et al. Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene. 2009;28(45):3983–96.
Isohookana J, Haapasaari KM, Soini Y, Karihtala P. Keap1 expression has independent prognostic value in pancreatic adenocarcinomas. Diagn Pathol. 2015;10:28.
Arfmann-Knubel S, Struck B, Genrich G, Helm O, Sipos B, Sebens S, Schafer H. The crosstalk between Nrf2 and TGF-beta1 in the epithelial-mesenchymal transition of pancreatic duct epithelial cells. PLoS One. 2015;10(7):e0132978.
Li L, Leung PS. Use of herbal medicines and natural products: an alternative approach to overcoming the apoptotic resistance of pancreatic cancer. Int J Biochem Cell Biol. 2014;53:224–36.
Taguchi K, Yamamoto M. The KEAP1-NRF2 system in cancer. Front Oncol. 2017;7:85.
Soini Y, Eskelinen M, Juvonen P, Karja V, Haapasaari KM, Saarela A, Karihtala P. Nuclear Nrf2 expression is related to a poor survival in pancreatic adenocarcinoma. Pathol Res Pract. 2014;210(1):35–9.
Hamada S, Taguchi K, Masamune A, Yamamoto M, Shimosegawa T. Nrf2 promotes mutant K-ras/p53-driven pancreatic carcinogenesis. Carcinogenesis. 2017;38(6):661–70.
Genrich G, Kruppa M, Lenk L, Helm O, Broich A, Freitag-Wolf S, Rocken C, Sipos B, Schafer H, Sebens S. The anti-oxidative transcription factor nuclear factor E2 related factor-2 (Nrf2) counteracts TGF-beta1 mediated growth inhibition of pancreatic ductal epithelial cells -Nrf2 as determinant of pro-tumorigenic functions of TGF-beta1. BMC Cancer. 2016;16:155.
Al Saati T, Clerc P, Hanoun N, Peuget S, Lulka H, Gigoux V, Capilla F, Beluchon B, Couvelard A, Selves J, et al. Oxidative stress induced by inactivation of TP53INP1 cooperates with KrasG12D to initiate and promote pancreatic carcinogenesis in the murine pancreas. Am J Pathol. 2013;182(6):1996–2004.
Yi YW, Oh S. Comparative analysis of NRF2-responsive gene expression in AcPC-1 pancreatic cancer cell line. Genes Genomics. 2015;37:97–109.
Kim MH, Kim EH, Jung HS, Yang D, Park EY, Jun HS. EX4 stabilizes and activates Nrf2 via PKCdelta, contributing to the prevention of oxidative stress-induced pancreatic beta cell damage. Toxicol Appl Pharmacol. 2017;315:60–9.
Chen X, Jiang Z, Zhou C, Chen K, Li X, Wang Z, Wu Z, Ma J, Ma Q, Duan W. Activation of Nrf2 by sulforaphane inhibits high glucose-induced progression of pancreatic cancer via AMPK dependent signaling. Cell Physiol Biochem. 2018;50(3):1201–15.
Satoh H, Moriguchi T, Taguchi K, Takai J, Maher JM, Suzuki T, Winnard PT Jr, Raman V, Ebina M, Nukiwa T, et al. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis. 2010;31(10):1833–43.
Chio II, Tuveson DA. ROS in translation: chink in the armor. Cell Cycle. 2017;16(4):297–8.
DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–9.
Hamada S, Shimosegawa T, Taguchi K, Nabeshima T, Yamamoto M, Masamune A. Simultaneous K-ras activation and Keap1 deletion cause atrophy of pancreatic parenchyma. Am J Physiol Gastrointest Liver Physiol. 2018;314(1):G65–74.
Tummala KS, Kottakis F, Bardeesy N. NRF2: translating the redox code. Trends Mol Med. 2016;22(10):829–31.
Kha ML, Hesse L, Deisinger F, Sipos B, Rocken C, Arlt A, Sebens S, Helm O, Schafer H. The antioxidant transcription factor Nrf2 modulates the stress response and phenotype of malignant as well as premalignant pancreatic ductal epithelial cells by inducing expression of the ATF3 splicing variant DeltaZip2. Oncogene. 2019;38(9):1461–76.
Nahdi A, John A, Raza H. Elucidation of molecular mechanisms of streptozotocin-induced oxidative stress, apoptosis, and mitochondrial dysfunction in Rin-5F pancreatic beta-cells. Oxidative Med Cell Longev. 2017;2017:7054272.
Wu YS, Looi CY, Subramaniam KS, Masamune A, Chung I. Soluble factors from stellate cells induce pancreatic cancer cell proliferation via Nrf2-activated metabolic reprogramming and ROS detoxification. Oncotarget. 2016;7(24):36719–32.
Feng R, Morine Y, Ikemoto T, Imura S, Iwahashi S, Saito Y, Shimada M. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun Signal. 2018;16(1):54.
Torrente L, DeNicola GM. Stressing out PanIN: NRF2 pushes over the edge. Cancer Cell. 2017;32(6):723–5.
Wang W, Qin JJ, Voruganti S, Wang MH, Sharma H, Patil S, Zhou J, Wang H, Mukhopadhyay D, Buolamwini JK, et al. Identification of a new class of MDM2 inhibitor that inhibits growth of orthotopic pancreatic tumors in mice. Gastroenterology. 2014;147(4):893–902 e892.
Wang W, Qin JJ, Voruganti S, Nijampatnam B, Velu SE, Ruan KH, Hu M, Zhou J, Zhang R. Discovery and characterization of dual inhibitors of MDM2 and NFAT1 for pancreatic cancer therapy. Cancer Res. 2018;78(19):5656–67.
Sun W, Yi Y, Xia G, Zhao Y, Yu Y, Li L, Hua C, He B, Yang B, Yu C, et al. Nrf2-miR-129-3p-mTOR axis controls an miRNA regulatory network involved in HDACi-induced autophagy. Mol Ther. 2019;27(5):1039–50.
Zhang L, Li J, Ma J, Chen X, Chen K, Jiang Z, Zong L, Yu S, Li X, Xu Q, et al. The relevance of Nrf2 pathway and autophagy in pancreatic cancer cells upon stimulation of reactive oxygen species. Oxidative Med Cell Longev. 2016;2016:3897250.
Li X, Liang M, Jiang J, He R, Wang M, Guo X, Shen M, Qin R. Combined inhibition of autophagy and Nrf2 signaling augments bortezomib-induced apoptosis by increasing ROS production and ER stress in pancreatic cancer cells. Int J Biol Sci. 2018;14(10):1291–305.
Ju HQ, Gocho T, Aguilar M, Wu M, Zhuang ZN, Fu J, Yanaga K, Huang P, Chiao PJ. Mechanisms of overcoming intrinsic resistance to gemcitabine in pancreatic ductal adenocarcinoma through the redox modulation. Mol Cancer Ther. 2015;14(3):788–98.
Duong HQ, You KS, Oh S, Kwak SJ, Seong YS. Silencing of NRF2 reduces the expression of ALDH1A1 and ALDH3A1 and sensitizes to 5-FU in pancreatic cancer cells. Antioxidants. (Basel). 2017;6(3):52.
Palam LR, Gore J, Craven KE, Wilson JL, Korc M. Integrated stress response is critical for gemcitabine resistance in pancreatic ductal adenocarcinoma. Cell Death Dis. 2015;6:e1913.
Arora R, Sawney S, Saini V, Steffi C, Tiwari M, Saluja D. Esculetin induces antiproliferative and apoptotic response in pancreatic cancer cells by directly binding to KEAP1. Mol Cancer. 2016;15(1):64.
Youns M, Abdel Halim Hegazy W. The natural flavonoid fisetin inhibits cellular proliferation of hepatic, colorectal, and pancreatic cancer cells through modulation of multiple signaling pathways. PLoS One. 2017;12(1):e0169335.
Sun H, Guo X, Wang Z, Wang P, Zhang Z, Dong J, Zhuang R, Zhou Y, Ma G, Cai W. Alphalipoic acid prevents oxidative stress and peripheral neuropathy in nab-paclitaxel-treated rats through the Nrf2 signalling pathway. Oxidative Med Cell Longev. 2019;2019:3142732.
Xiang Y, Ye W, Huang C, Yu D, Chen H, Deng T, Zhang F, Lou B, Zhang J, Shi K, et al. Brusatol enhances the chemotherapy efficacy of gemcitabine in pancreatic cancer via the Nrf2 signalling pathway. Oxidative Med Cell Longev. 2018;2018:2360427.
Choi EJ, Jung BJ, Lee SH, Yoo HS, Shin EA, Ko HJ, Chang S, Kim SY, Jeon SM. A clinical drug library screen identifies clobetasol propionate as an NRF2 inhibitor with potential therapeutic efficacy in KEAP1 mutant lung cancer. Oncogene. 2017;36(37):5285–95.
Singh A, Venkannagari S, Oh KH, Zhang YQ, Rohde JM, Liu L, Nimmagadda S, Sudini K, Brimacombe KR, Gajghate S, et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem Biol. 2016;11(11):3214–25.
Bollong MJ, Yun H, Sherwood L, Woods AK, Lairson LL, Schultz PG. A small molecule inhibits deregulated NRF2 transcriptional activity in cancer. ACS Chem Biol. 2015;10(10):2193–8.
Benlloch M, Obrador E, Valles SL, Rodriguez ML, Sirerol JA, Alcacer J, Pellicer JA, Salvador R, Cerda C, Saez GT, et al. Pterostilbene decreases the antioxidant defenses of aggressive cancer cells in vivo: a physiological glucocorticoids- and Nrf2-dependent mechanism. Antioxid Redox Signal. 2016;24(17):974–90.
Acknowledgements
We thank the current and former members of our laboratories and collaborators for their contributions to the publications cited in this review article. The research field in Keap1 and Nrf2 is rapidly growing, and we apologize for not being able to cite all the recent publications, due to space limitation.
Funding
JJQ was supported by National Natural Science Foundation of China (81903842) and Zhejiang Chinese Medical University Startup Funding (111100E014). WDZ was supported by Professor of Chang Jiang Scholars Program, NSFC (81520108030, 21472238), Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (16DZ2280200), the Scientific Foundation of Shanghai China (13401900103, 13401900101), and the National Key Research and Development Program of China (2017YFC1700200). XDC was supported by the Zhejiang Provincial Science and Technology Projects (2018C37045) and Zhejiang Provincial Medical and Healthy Science and Technology Projects (WKJ-ZJ-1728 and 2016KYB220).
Author information
Authors and Affiliations
Contributions
JJQ and WDZ conceptualized the manuscript. JJQ, XDC, and JZ collected the literature, wrote the manuscript and made the figures. JJQ and WDZ edited and made significant revisions to the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Qin, JJ., Cheng, XD., Zhang, J. et al. Dual roles and therapeutic potential of Keap1-Nrf2 pathway in pancreatic cancer: a systematic review. Cell Commun Signal 17, 121 (2019). https://doi.org/10.1186/s12964-019-0435-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-019-0435-2