
Abstract
Immune evasion contributes to cancer growth and progression. Cancer cells have the ability to activate different immune checkpoint pathways that harbor immunosuppressive functions. The programmed death protein 1 (PD-1) and programmed cell death ligands (PD-Ls) are considered to be the major immune checkpoint molecules. The interaction of PD-1 and PD-L1 negatively regulates adaptive immune response mainly by inhibiting the activity of effector T cells while enhancing the function of immunosuppressive regulatory T cells (Tregs), largely contributing to the maintenance of immune homeostasis that prevents dysregulated immunity and harmful immune responses. However, cancer cells exploit the PD-1/PD-L1 axis to cause immune escape in cancer development and progression. Blockade of PD-1/PD-L1 by neutralizing antibodies restores T cells activity and enhances anti-tumor immunity, achieving remarkable success in cancer therapy. Therefore, the regulatory mechanisms of PD-1/PD-L1 in cancers have attracted an increasing attention. This article aims to provide a comprehensive review of the roles of the PD-1/PD-L1 signaling in human autoimmune diseases and cancers. We summarize all aspects of regulatory mechanisms underlying the expression and activity of PD-1 and PD-L1 in cancers, including genetic, epigenetic, post-transcriptional and post-translational regulatory mechanisms. In addition, we further summarize the progress in clinical research on the antitumor effects of targeting PD-1/PD-L1 antibodies alone and in combination with other therapeutic approaches, providing new strategies for finding new tumor markers and developing combined therapeutic approaches.
Similar content being viewed by others

Introduction
Immune suppression largely contributes to cancer occurrence and progression. The programmed cell death protein 1 (PD-1, also known as PDCD1 and CD279) was originally identified by Ishida et al. in apoptotic mouse T-cell tumors [1]. PD-1 is a transmembrane protein belonging to the CD28/CTLA-4 superfamily. It is widely expressed at the surface of activated T cells, B cells, monocytes, and other immune cells, and negatively regulates human immune response through binding with its two ligands, namely programmed cell death 1 ligands (PD-L1 or PD-L2). PD-L1 (B7-H1; CD274) and PD-L2 (B7-DC; CD273) belong to the B7 family of T cell co-inhibitory molecules. PD-L1 is widely expressed in antigen-presenting cells and tissues, such as heart and lung [2, 3]. The interaction of PD-1 with PD-L1 or PD-L2 provides inhibitory signals responsible for inhibiting T cell signaling, mediating the mechanisms of tolerance, and providing immune homeostasis. Therefore, PD-1 suppresses autoimmunity and prevents the occurrence of autoimmune diseases. In addition, PD-L1 or PD-L2 expressed by cancer cells binds to PD-1 on the surface of T cells, thereby inhibiting T cell activation and leading to cancer immune escape [4]. Numerous studies revealed that PD-L1 expression is very high in lung cancer, melanoma, glioma, breast cancer and other malignant tumor cells, forming an immunosuppressive tumor microenvironment [5].
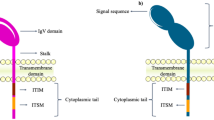
PD-1 mainly consists of extracellular IgV-like domain region, hydrophobic transmembrane region and cytoplasmic region, and the tail of the cytoplasmic region has immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM) [6, 7], which is an important structural basis for PD-1 to transmit inhibitory signals and perform immunosuppressive functions. PD-L1 is structurally similar to PD-1 and is more conserved and widely expressed than PD-L2 [8], so it plays the leading effect in tumor cells immune evasion. In recent years, antagonistic antibodies against PD-1 or PD-L1 have been approved by the FDA to treat cancer, opening a new chapter in tumor immunotherapy across the era [9].
Anti-PD-1/PD-L1 inhibitors have become effective immune checkpoint inhibitors (ICIs) and are rapidly becoming the standard therapy for various cancers. Tumor immunotherapy aims to block the activity of inhibitory immune checkpoint proteins and promote T cell activation to achieve anti-tumor immune effects [10]. Owing to their safety and precision, these inhibitors hold significant promise in tumor immunotherapy. Research indicates that the PD-1/PD-L1 pathway plays a crucial role in regulating autoimmunity responses and peripheral tolerance. Notably, anti-PD-1/PD-L1 immunotherapy can effectively block the PD-1/PD-L1 signaling pathway, restore T cell activity, enhance anti-tumor immunity, and then eliminate tumor cells [11, 12]. Therefore, the discovery of multiple immunotherapies, such as PD-1 and PD-L1 inhibitors, has significant clinical implications for tumor-specific immunotherapy. This paper aims to provide a relatively comprehensive review of the mechanisms of PD-1/PD-L1 signaling in autoimmunity and tumor immunity, as well as its clinical efficacy in different tumors. It serves as a reference for exploring the internal regulatory mechanisms and resistance mechanisms of cancer immunotherapy targeting PD-1/PD-L1. We aim to offer new strategies for identifying novel tumor markers and developing combined drug treatments, thereby achieving early diagnosis and personalized treatment for patients.
The physiological function and immune regulation mechanism of the PD-1/PD-L1 axis as an immune checkpoint
Under normal physiological conditions, PD-1 and PD-L1 molecules are among the primary participants in maintaining immune homeostasis. Their binding reduces autoimmune cell attacking on self-tissues, maintains immune balance by inhibiting the activation of T cells. T cell activation primarily relies on a “dual-signal” system. Antigen-presenting cells (APCs) capture and process bacterial antigens, subsequently presenting them to the T-cell receptor (TCR) through the major histocompatibility complex (MHC) molecule. Upon recognition and binding to form the TCR-MHC complex, a series of co-stimulatory signals are required to further induce the immune response of effector T cells [13, 14]. Co-stimulatory molecules expressed on APCs, such as CD80 and CD86, bind to CD28 on T-cells to provide co-stimulatory signals, thoroughly activating T-cells to specifically target and kill cells infected by pathogens. Negative co-stimulatory molecules like CTLA4 and PD-1/PD-L1 play an essential role in preventing tissue inflammation and autoimmune diseases under normal physiological conditions by avoiding excessive activation of T cells. When cells become cancerous, they will exploit this inhibitory pathway to escape the siege of the immune system [15, 16]. Furthermore, the interaction between TCR and MHC is highly specific and sensitive, enabling T cells to detect rare antigenic epitopes on APCs. Due to central and peripheral immune tolerance, autoreactive T cells that recognize specific antigens are usually in an unresponsive state [17]. This might explain why reactive T cells detected in patients typically fail to control or eradicate advanced diseases.
In the human immune system, the binding of PD-L1 to PD-1 primarily exerts an immunosuppressive regulatory effect through Src homology region 2 domain-containing phosphatase-2 (SHP2), thereby attenuating the immune response of T cells [18]. SHP2 is a tyrosine phosphatase composed of N-SH2, the C-SH2 structural domain, the PTP structural domain, and the C-terminal tail, among which N-SH2 plays a core function in the activation process of SHP2. In the inactive state, an intramolecular interaction occurs between the N-SH2 and PTP domains of SHP2, effectively obstructing the entry of SHP2 substrates into the catalytic site. The activation of SHP2 requires the removal of the PTP domain from the N-SH2 domain while concurrently binding to a specific phosphotyrosine motif, thereby destroying its self-inhibition state and exerting its dephosphorylation function [19]. When PD-1 binds to PD-L1, it can induce phosphorylation of tyrosine residues in the ITSM and ITIM structural domains in the cytoplasmic region of PD-1. The phosphorylated ITSM then binds C-SH2 with high affinity, recruiting SHP2 to PD-1. The phosphorylated ITMM binds to N-SH2 and activates SHP2 [20, 21]. Subsequently, activated SHP-2 dephosphorylates TCR-associated CD3 and ZAP70 signalosomes, further attenuating TCR downstream signaling intensity and cytokine secretion such as IL-2 [7]. In bone marrow myelocytes, the PD-1-SHP-2 axis also restrains myeloid cell differentiation by impeding HOXA10 and IRF8 phosphorylation. Targeted deletion of PD-1 or SHP-2 in mice induces the differentiation of myeloid cells into monocytes with strengthened antigen presentation and T cell co-stimulation capabilities, thereby boosting immunity [22]. However, research also shows that, under chronic viral infections, SHP-2 is dispensable for T cell exhaustion and PD-1 signaling [23]. Additionally, the inhibitory target of SHP2 is likely the T-cell co-stimulatory receptor CD28. PD-1 can inhibit T cell function through SHP2-mediated dephosphorylation of CD28 rather than directly inhibiting TCR signaling [24]. This also indicates that, besides TCR, the major targets of PD-1 signaling need to extensively consider the role of CD28 or other co-stimulatory molecules. Therefore, based on the aforementioned molecular regulatory mechanisms of the PD-1/PD-L1 signaling pathway, it is not difficult to see that pathogen-infected cells utilize the PD-1/PD-L1 axis to promote the occurrence of immune inflammation in the local tumor microenvironment, breaking the immune balance of the organism and evading the attack of the host immune system. Blocking the PD-1/PD-L1 pathway can enhance the efficacy of T cells, making pathogen-infected cells more sensitive to immune checkpoint blockade therapy (Fig. 1).
The PD-1 molecule plays a crucial role in cellular adhesion and migration. Activated Tregs highly expressed PD-1, PD-1/PD-L1 signaling regulates T cell migration across lymphatic endothelial cells through PI3K/Akt in vivo [25]. The binding of PD-1 to PD-L1 also regulates the formation of memory T cells, affecting the memory and persistence of immune responses. Tissue-Resident Memory T Cells mediate protective immune responses and control tissue immune homeostasis in the human pancreas through the PD-1/PD-L1 inhibitory pathway [26]. Additionally, PD-1 can influence glycolysis and other metabolic pathways by inhibiting the downstream signal transduction of T cell signaling molecules, thereby obstructing cellular bioenergetics [27]. Moreover, PD-1 plays a key physiological role in both the central and peripheral nervous systems. Junli Zhao et al. find that PD-1/PD-L1 signaling in hippocampal neurons regulates synaptic transmission and cognitive behaviors. Inhibiting PD-1 alleviates the decline in learning and memory following traumatic brain injury [28]. Under physiological conditions, PD-L1 is widely expressed across numerous tissues, though its exact role in tissue and organ development and regeneration remains unclear. Recent research has uncovered that PD-L1 is abundantly present in breast stem cells and promotes the development and regeneration of mammary glands, offering new insights into the physiological functions of PD-L1 [29]. During pregnancy, the expression of PD-L1 in the placenta contributes to the maternal immune tolerance to fetal antigens and prevents immune attacks on the fetus [30]. A study has shown that PD-L1+ senescent cells, which are resistant to T cell surveillance, gradually accumulate with ageing. Blocking the PD-L1 signaling by PD-1 antibody activates CD8+ T cells to clear p16+ senescent cells as well as PD-L1+ senescent cell population in vivo, ameliorating ageing-related phenotypes in mice [31].

The immune regulation mechanism of the PD-1/PD-L1 axis. Antigen-presenting cells (APCs) deliver tumor antigens to the T-cell receptor (TCR) via the major histocompatibility complex(MHC), and when the MHC-antigen complex specifically binds to the TCR, it triggers a series of signal transductions, including the phosphatidylinositol signaling and mitogen- activated protein kinases signaling pathways, thus activating the immune responses of effector T cells. Upon PD-L1 binding to PD-1, phosphorylation of tyrosine residues in the ITSM and ITIM domains of the PD-1 cytoplasmic region occurs, recruiting and activating SHP2. Subsequently, recruited SHP-2 mediates dephosphorylation of TCR-associated CD3 and ZAP70 signalosomes, while inhibiting CD28 co-stimulatory signals. This further attenuates downstream TCR signaling strength and cytokine secretion, such as IL-2, ultimately inhibiting the function of T cells. Figure created with BioRender
Roles of PD-1/PD-L1 in transplantation and autoimmune diseases
The PD-1/PD-L1 signaling pathway is critical in autoimmune diseases, viral infections, transplantation and tumor immunity [32, 33]. Under a normal physiologic state, PD-1/PD-L1 signaling can effectively inhibit the excessive activation of immune cells, thereby avoiding severe persistent tissue damage and participating in maintaining immune tolerance to self-antigens. Disruption of the balance between PD-1 and PD-L1 can lead to the occurrence of various autoimmune diseases, including type 1 diabetes mellitus (T1DM), multiple sclerosis (MS), systemic lupus erythematosus (SLE), and rheumatoid arthritis (RA) [34].
Transplantation
After transplantation, due to mutual intolerance between the graft and the host, it often leads to reciprocal attack and rejection of immune cells between the recipient’s immune system and the donor graft. The most common reaction is the host versus graft reaction (HVGR) during organ transplantation, while individual patients also experience a graft versus host reaction (GVHR) where T/B immune cells in the graft attack the host [35]. The negative regulatory signals mediated by PD-1/PD-L1 can inhibit the activation and proliferation of T cells, induce T cell dysfunction, and effectively reduce immune rejection between the host and the donor [36]. For example, the PD-1/PD-L1 signaling pathway participates in the rejection of human allografts. Shi’s team performs biopsies on transplanted livers of recipients experiencing acute rejection induced by inflammation. They note that PD-L1 expression is increased in the portal veins and lobular regions of the transplanted liver, as well as high expression of PD-1 on infiltrating T cells within the graft, with critical interactions between the donor PD-L1 and recipient PD-1. Blocking PD-1/PD-L1 enhances the proliferation of infiltrating T cells within the graft, exacerbating the immune rejection after transplantation [37]. Similar experimental results are observed in the mouse model of renal allograft rejection. PD-1 is widely expressed on most T cells within the transplanted kidney, and the upregulation of PD-L1 expression on renal tubular epithelial cells may inhibit T cell activation and proliferation, reducing T cell-mediated excessive immune damage [38]. These results suggest that upregulation of PD-1/PD-L1 expression may be a negative feedback protective mechanism in the immune response to parenchymatous organ transplantation, reducing postoperative immune reactions (Fig. 2).

Role of PD-1/PD-L1 in transplantation and autoimmune diseases. During organ transplantation, PD-1 is highly expressed on the surface of infiltrating T cells in grafts. The negative regulatory signal mediated by PD-1/PD-L1 can inhibit the excessive activation of T cells, induce immune tolerance, and effectively reduce the immune rejection between the host and the donor after surgery. Blocking PD-1/PD-L1 promotes proliferation of infiltrating T cells in grafts, exacerbating post-transplant immune rejection reactions and inducing severe and persistent tissue damage. Similarly, the disruption of the balance between PD-1 and PD-L1 signals can lead to the occurrence of many autoimmune diseases, such as type 1 diabetes mellitus (T1DM), multiple sclerosis (MS), systemic lupus erythematosus (SLE), and rheumatoid arthritis (RA). Figure created with BioRender
Clinical data has shown that recipients with recurrent and metastatic liver cancer after liver transplantation may experience immune-related adverse reactions after undergoing immunotherapy, leading to a poor prognosis for the patients. For example, Friend et al. describe two patients with recurrent and refractory hepatocellular carcinoma (HCC) after orthotopic liver transplantation, who rapidly develop irreversible acute liver rejection and eventually die shortly after receiving the PD-1 inhibitor nivolumab [39]. Likewise, Fisher et al. also evaluated the safety and efficacy of immunotherapies such as PD-1 inhibitors in 57 metastatic cancer patients with a history of solid organ transplantation (liver, kidney, and heart). The results demonstrate that 37% of the patients experience organ rejection reactions, and 14% die due to graft rejection. Among them, the rejection rate is highest after using nivolumab, and patients who use Ipilimumab have the highest rate of malignant tumor progression. Kidney transplant recipients exhibit the highest rejection rate, followed by liver transplant recipients [40]. In a clinically relevant case, a 63-year-old female patient with type 1 diabetes develops pulmonary metastatic melanoma 10 years after kidney transplantation. Within one week of initiating first-line treatment with nivolumab for melanoma, the patient experiences acute renal allograft rejection, renal failure, and concurrent diabetic ketoacidosis [41], suggesting a potential association between the use of anti-PD-1 antibodies and allograft rejection. Therefore, the PD-1/PD-L1 pathway is critical for inducing and maintaining transplant tolerance.
So how can we apply immunotherapy to exert anti-tumor effects in the body without triggering immune rejection? I believe that the first step is to identify biomarkers that can predict the risk of transplant rejections and tumor responses. For example, recipients experiencing acute rejection show high expression of PD-L1 in biopsy samples. Based on this, we can characterize the expression of PD-L1 on grafts before using anti-PD-1 drugs and select the appropriate immune checkpoint inhibitor for blockade therapy according to the patient’s individual condition and the possibility of rejection. Secondly, when administering immune checkpoint blockade therapy to patients with a history of organ transplantation, we must be cautious and consider the risk of immune rejection in the transplant rather than purely the anti-tumor treatment effects. Finally, it is important to develop novel immunosuppressive therapies to minimize the use of ICI therapy and devise personalized immunosuppression plans for patients. For example, targeting PD-1 with appropriate agonists (such as PD-L1 Ig fusion protein) can reduce rejection reactions and optimize therapeutic approaches. Thus, PD-1/PD-L1 signaling may have a major role in graft tolerance and the prevention of late rejection after transplantation.
Autoimmune diseases
PD-1/PD-L1 also has significant clinical relevance to autoimmune diseases (Fig. 2).
Type 1 diabetes mellitus (T1DM) is a severe auto-immune disease caused by severe destruction of insulin-producing β-cells in the pancreas, resulting in hyperglycemia [42]. Recently, it has been discovered that IFN-α and IFN-γ can induce PD-L1 expression in human islet cells, which can limit the immunological killing of islet cells after the activation of autoreactive T cells, thereby preventing autoimmune damage. Blocking JAKs or IRF1 (the recommended strategies for T1DM treatment) can prevent PD-L1 induction [43]. During the progression of T1DM in NOD (non-obese diabetic) mice, T cells attack pancreatic islet cells, causing the loss of beta cells. PD-L1 expression is significantly increased in beta cells that can withstand autoimmune attacks and survive for a long time, thus preventing type 1 diabetes [44]. Similarly, platelets genetically engineered to overexpress PD-L1 accumulate in pancreas and inhibit the activity of islet-specific autoreactive T cells, reversing new-onset T1D [45]. PD-1 also regulates autoimmunity to limit the development of diabetes by inhibiting the proliferation and infiltration of pancreatic T cells. Single-cell RNA-seq and TCR-seq of spontaneous T1D vs. anti-PD-1 induced T1D show that PD-1-induced T1D significantly increases the proliferation of exhausted/effector-like T cells [46]. When using PD-1/PD-L1 inhibitors, the PD-L1 molecule on β cells can’t bind to PD-1 on autoreactive T cells, while the inflammation-stimulated autoreactive T cells are over-activated, greatly promoting the progression of diabetes. This means that PD-1 or PD-L1 antibodies used in cancer immunotherapies may induce autoimmune insulin-dependent diabetes mellitus. For instance, a case-review analysis by Stamatouli et al. [47] found that among 2960 patients treated with immune checkpoint inhibitors, 27 cases (an incidence of 0.9%) experienced acute episodes of insulin-deficient diabetes. Of these 27 diabetic patients, 59% also have diabetic ketoacidosis, 42% have pancreatitis, 85% exhibit rapid loss of beta-cell function, manifested by rapid progression of hyperglycemia. Moreover, this type of immune-associated diabetes occurs more frequently in patients receiving anti-PD-1/PD-L1 than in those treated solely with anti-CTLA-4, indicating that T1DM is primarily associated with anti-PD-1/PD-L1 immunosuppressive therapy. Increasing clinical data suggests that in a majority of cancer patients, the use of PD-1 or PD-L1 antibody treatments can induce diabetic ketoacidosis (DKA) or trigger acute onset of type 1 diabetes shortly after initiation, significantly reducing the clinical efficacy of antitumor therapies [48, 49] Therefore, based on the above research and case analysis, we know that tumors associated with autoimmune diabetes have no advantage during immunotherapy. For patients with a previous history of diabetes, the risk of anti-PD-1/PD-L1-induced autoimmune diabetes should be evaluated before choosing immunotherapy. If biomarkers related to the progression of diabetes can be identified for personalized drug management, it would contribute to reducing immune-related adverse effects in patients and even preventing diabetes onset. Meanwhile, patients should be closely followed up during first-line immunotherapy, with routine blood glucose monitoring before each administration and periodic testing for islet autoantibodies to prevent DKA.
Systemic lupus erythematosus (SLE) is one of the most common systemic auto-immune diseases, which is characterized by the destruction of self-antigen immune tolerance and excessive accumulation of self-antibodies, resulting in multi-organ damage [50]. Among them, type I interferons and Toll-like receptors regulate PD-1 and its ligand PD-L1 expression by activating NF-κB or STAT1, playing a crucial role in SLE pathogenesis [51]. PD-1 is significantly upregulated on CD4 T cells and in a pathogenic activation state during SLE progression, the mitochondrial membrane potential of PD-1+CD4 T cells shows abnormal activation, thus promoting the progression of SLE. When using cell membrane biomimetic membrane nanoparticles with high PD-L1 expression (IM-MNPs/DXM, a functional-driven, disease-relevant CD4 T cell-targeted drug carrier), it kills pathogenic CD4 T cells and inhibits the expression of pro-inflammatory factors, reshaping the balance between the effector T cells and Tregs in the microenvironment, thereby alleviating SLE [52]. Similarly, in SLE mouse models, PD-1 blockade can activate suppressive CD8+ T cells, thereby delaying the onset of renal disease and the progression of SLE, ultimately leading to an increase in the survival rates of the mice [53]. Recent studies have found that anti-PD-1 autoantibodies are elevated in the serum of newly diagnosed SLE patients, promoting T cell proliferation and positively correlating with the disease activity of SLE [54]. This indicates that PD-1 antibodies may accelerate the severity of lupus by blocking the biological functions of PD-1 signaling, providing new insights into the regulatory pathways of PD-1 in SLE. Furthermore, the frequency of PD-L1 expressing neutrophils in SLE patients is significantly increased, it is positively correlated with disease activity and severity, serving as a novel indicator for evaluating SLE [55]. We hypothesize that neutrophils expressing PD-L1 may inhibit T cell-mediated immune responses through multiple mechanisms, it may be a negative feedback mechanism to prevent potential tissue damage caused by excessive autoimmune responses in SLE patients. In conclusion, the regulatory function of the PD-1/PD-L1 pathway in SLE patients is complex and may be related to PD-1 polymorphisms or the quantity and immunosuppressive function of Tregs in SLE. It may also be context-dependent, requiring further preclinical studies to elucidate its contributions to immune regulatory functions.
Multiple sclerosis (MS) is an inflammatory demyelinating disease of the central nervous system mediated by immune dysregulation, mainly characterized by motor and sensory disorders, ataxia, etc. PD-1/PD-L1 signaling plays a key role in the pathogenesis of MS by downregulating the immune responses of autoreactive T cells in the central nervous system [56]. Previous studies have shown increased expression of PD-L1 and PD-1 in the central nervous system of animal models, such as experimental autoimmune encephalomyelitis (EAE). Blocking PD-1 in EAE leads to antigen-specific T cell expansion and increased cytokine production, thereby enhancing the autoimmune response and accelerating the progression of EAE symptoms [57]. Antje Kroner et al. find that the PD-1 intron 7146G/A polymorphism is associated with disease progression in MS. In patients carrying the PD-1 polymorphism, mutations in the regulatory region of the PD-1 gene may affect PD-1 signal transduction, thereby impairing the function of inhibiting T cell proliferation mediated by PD-1 and accelerating disease progression in MS patients [58]. Koto et al. observe that PD-1+ CD8 T cells were reduced in the peripheral blood of MS patients during the MS disease remission state. Conversely, during MS relapses, IFN-β stimulation induces the enrichment of PD-1+CD8 T cells in the cerebrospinal fluid, which contributes to self-immune regulation and is correlated with a positive response to subsequent treatment [59]. In summary, these findings suggest that PD-1/PD-L1 plays a protective role in EAE and can be used as a new target for therapeutic intervention.
Rheumatoid arthritis (RA) is an autoimmune systemic inflammatory disease with a complex pathogenesis, primarily characterized by the production of autoantibodies such as immunoglobulin G (IgG) and anti-citrullinated protein antibodies (ACPA). Overexpression of PD-1/PD-L1 in macrophages and synovial T cells in RA patients has been significantly associated with disease progression and a lower overall survival rate [60, 61]. Wan et al. find that IFN-γ and TNF-α selectively induce overexpression of negative co-stimulatory molecules such as PD-1 and CTLA-4 in synovial T cells and APCs of RA patients. However, the overexpression of these negative co-stimulatory molecules, which downregulates T cell responses, presents a clear contradiction with the persistent activation of autoreactive T cells in RA. Further research reveals that this functional abnormality of negative co-stimulatory molecules appears to be mediated by soluble PD-1 (sPD-1), which is secreted abundantly in the serum and synovial fluid of RA patients. Specifically, the PD-1 alternative splice variant (PD-1Δex3) derived from RA T cells produces sPD-1 and antagonizes the inhibitory efficacy of membrane PD-1 on T cells, which is specific to RA and unrelated to the activation state of T cells. The use of a human recombinant fusion protein of soluble PD-1 (hPD-1-Ig) can effectively block the immunosuppressive function mediated by membrane PD-1 and PD-L1 [62]. This study provides new evidence for the role of PD-1 soluble factors in the pathogenesis of RA and other autoimmune diseases, contributing to the development of new therapeutic strategies. RA-related inflammatory factors are also involved in PD-L1 regulation, leading to the high expression of PD-L1 in RA. For example, Haiyan Zhou et al. evaluated circulating T cell subsets and cytokine expression profiles in RA patients. The results exhibited a remarkable increase in pro-inflammatory cytokines such as MCP-1, IL-6, and IL-1β in RA. PD-1+ TEM (effector memory T cell), PD-1+ TCM (central memory T cell), and PD-1+ Tfh (follicular helper T cells) subsets were positively correlated with RA disease activity DAS28, suggesting a potential as a new indicator for RA treatment [63]. Additionally, Kostine et al. conducted a retrospective assessment of the incidence of rheumatic immune-related adverse events (irAEs) and the tumor response rates in 524 patients receiving ICIs. The results show that 35 patients (6.6%) are referred to rheumatology, nearly all rheumatic irAEs occur in the context of anti-PD-1/PD-L1 antibody treatments, and patients with rheumatic irAEs show a higher tumor response rate [64].
These above findings provide preliminary evidence for the regulatory functions of PD-1/PD-L1 signaling in autoimmune diseases, the exact mechanisms still need further elucidation in future studies. Traditional approaches involving gene knockout or inhibition of PD-1/PD-L1 may cause activated immune cells to become uncontrolled by immune checkpoints, eventually exacerbating autoimmunity mediated by T cells and B cells in autoimmune diseases. So, is there a therapy that can preserve normal immune cells while targeting and eliminating dysfunctional immune cells? In recent years, in addition to the research targeting PD-1/PD-L1 checkpoint alone, researchers have found that depletion of PD-1+ T lymphocytes also significantly improved the progression of autoimmune diseases. For example, targeted immunotoxins against PD-1 (composed of the anti-PD-1 single-chain variable fragment, the albumin binding domain and the Pseudomonas exotoxin) selectively bind to and eliminate PD-1 expressing cells, as well as reduce the proliferation of autoreactive T cells in inflamed organs [65]. Further studies found that in the T1D mouse models, immunotoxin-treated PD-1 cell depletion significantly inhibited the onset of T1D and prolonged the mice’s survival. In the EAE mouse models, treated mice even regained walking abilities with no signs of relapse. Importantly, each mouse retains normal immune responses and their immune functions are not compromised. The efficacy of immunotoxin-induced PD-1 cell depletion can be further improved through pharmacokinetic-pharmacodynamic optimization, thereby preventing autoimmune diseases. In the future, targeted depletion of PD-1+ cells may be an efficient and widely applicable treatment to alleviate autoimmunity.
Roles of PD-1/PD-L1 axis in tumors
Tumor invasion, metastasis, and recurrence are complex processes with multi-gene involvement and multi-step development, which are formed by the interaction of intrinsic factors and external factors such as viral infection, and represent a dynamic interplay of clearance, balance, and evasion between the immune system and the tumor [66]. The interaction between PD-1 and PD-L1 can transmit inhibitory signals within cells, suppressing lymphocyte proliferation and activation, thereby diminishing immune capacity. Consequently, tumor cells often promote their own occurrence and growth by ‘hijacking’ this axis [67, 68]. When tumor-infiltrating lymphocytes (TILs) recognize tumor antigens, they initiate “adaptive resistance” mediated by the PD-1/PD-L1 pathway to assist tumor immune evasion and distant metastasis [69]. Specifically, this is manifested as follows, (I) PD-1/PD-L1 interaction suppresses the activation and proliferation of T cells, promoting T cell dysfunction and apoptosis. (II) PD-1/PD-L1 interaction enhances the function of regulatory T cells (Tregs) and induces immune tolerance. (III;) PD-1/PD-L1 interaction promotes the polarization of TAM and other immune cells into tumor-promoting phenotypes, facilitating immune escape and cancer progression. (III) Signaling of PD-L1 within cancer cells may prevent the apoptosis of tumor cells themselves, and the interaction of PD-L1 with CD80 can suppress the immune response, etc [70].
PD-1/PD-L1 axis promotes tumor occurrence and development
PD-1/PD-L1 interaction promotes effector T cell exhaustion and apoptosis
T cells play a major role in tumor immunity, as well as being an important mediator of the activation or inhibitory signals transmitted by T cell co-inhibitory receptor molecules. PD-1, a checkpoint receptor expressed on T cells, causes T cell exhaustion and prevents excessive immune stimulation when it receives signals from its ligand PD-L1 [71]. Under normal conditions, PD-L1 is lowly expressed on most cells, but it is highly expressed on the surface of human cancer cells. Prolonged exposure to viral antigen stimulation may result in T cell subset dysregulation or dysfunction, being in a state of diminished responsiveness, known as “T cell exhaustion” (Tex cells) [72]. Tex cells have impaired killing effects and undergo different patterns of differentiation. They are characterized by increased co-expression of inhibitory receptors (PD-1, LAG3, and TIGIT, etc.), reduced production of cytokines (IL-2, TNF, and IFN-γ, etc.), metabolic changes, and impaired proliferation capacity and survival rate [73]. Finally, T cells are completely exhausted and undergo apoptosis, many mechanisms can explain this process (Fig. 3a).

The regulation of PD-1/PD-L1 signaling on immune cells. a. The PD-1/PD-L1 pathway promotes the exhaustion and apoptosis of effector T cells. Tex cells are characterized by increased expression of highly inhibitory receptors, including PD-1, LAG3, and TIGIT, decreased cytokine production such as TNF, IL-2, and IFN-γ, metabolic alterations, and impaired proliferative capacity and survival. b. PD-1/PD-L1 promotes the generation and development of induced Tregs (iTregs) by reducing the phosphorylation of PI3K/Akt/mTOR and S6, while enhancing PTEN, thus enhancing the immune suppression functions of Treg cells and inducing immune tolerance. c. PD-1/PD-L1 can promote the polarization of tumor-associated macrophages (TAM) toward the M2 phenotype, releasing large amounts of fibroblast growth factor, VEGF, TNF-α, and other cytokines to promote angiogenesis and support immune suppression, invasion, and metastasis of cancer cells, accelerating cancer progression. d. PD-1 on NK cells binds to PD-L1 on cancer cells, inhibiting the degranulation and cytotoxic function of NK cells, decreasing their ability to kill tumor cells, and promoting tumor immune escape. The use of PD-1 and PD-L1 inhibitors may reactivate the anti-tumor immune response of the above immune cells. Figure created with BioRender
Upon the binding of PD-1 on tumor-infiltrating lymphocytes to PD-L1 on tumor cells, an inhibitory signal is transmitted to T cells, which can block the TCR signal cascade responses and its co-stimulatory signal transduction, inducing dysfunction and even exhaustion of effector T cells, thus causing tumor cell immune escape [74]. Previous studies mostly considered that PD-1 is preferentially expressed on chronic virus-specific CD8 T cells and is a major regulator of T cell exhaustion and apoptosis [75]. PD-1 blocks the anti-apoptotic gene Bcl-xL expression by inhibiting Akt activation, which in turn reduces T cell survival rates [76]. Blocking the PD-1/PD-L1 interaction with mAb restores T cell proliferation, cytokine secretion, and cytotoxicity, to a certain extent reversing T cell exhaustion [77], thus identifying PD-1 as a promising therapeutic approach for restoring HIV-specific exhausted T cells. In tumors or chronic viral infections, PD-1 is overexpressed on functionally exhausted T cells. Persistent activation and PD-1 signaling can inhibit glycolysis and mitochondrial metabolism, this biological energy deficiency exacerbates early T cell dysfunction and exhaustion [78], indicating that the intensity of PD-1 signaling also determines the severity of T cell exhaustion. PD-1 signaling also prevents the conversion of CD8+ central memory T cells (TCM) into CD8+ effector memory T cells (TEM) [79], reducing the long-term immune memory that may prevent future metastatic diseases. Additionally, PD-L1 on tumor cells also interacts with CD80 on activated T cells, decreasing the activation of effector T cells and even inducing apoptosis of activated T cells [80]. Diskin et al. found that, under the stimulation of tumor antigens and inflammatory cytokines, the expression of PD-L1 in tumor-infiltrating T cells can be activated. At this time, PD-L1 acts as both ligand and receptor to transmit forward and backward signals to regulate the immune responses in tumor tissues. Specifically, PD-L1 in T cells induces intracellular signaling, its inhibitory effect is the same as that of PD-1. PD-L1+ T cells also promote the differentiation of STAT6-dependent M2 type macrophages and inhibit the function of adjacent effector T cells through the typical PD-L1/PD-1 axis [81]. Thus, blocking PD-1/PD-L1 can reduce T cell apoptosis and promote anti-tumor immunity.
PD-1/PD-L1 blockade therapy is the foundation of current tumor immunotherapy, but the specific anti-tumor immune mechanisms are still unclear. It was previously generally believed that the anti-tumor immune effect was exerted by restoring the activity of exhausted T cells or inhibiting the activity of Treg cells within the tumors [82]. In recent years, there has been ongoing controversy regarding whether exhausted T cells can be reverted back to an active state, as increasing evidence suggests that blockade of the PD-1/PD-L1 axis may not be able to “rescue” terminally exhausted T cells. So, what specific type of T cell can actually respond to PD-1/PD-L1 inhibitor therapy? Margaret et al. found that new T cell clones appeared after anti-PD-1 therapy, recruiting tumor-infiltrating lymphocytes to the tumor microenvironment [83]. Miller et al. found that differences among exhausted CD8+ T cell subsets affected their anti-tumor functions and the efficacy of ICI therapy. Compared to terminally exhausted T cells, progenitor exhausted CD8+ TILs are better at controlling tumor growth, melanoma patients with a higher proportion of progenitor exhausted CD8+ TILs have a longer response to PD-1 blockade therapy [84]. Subsequently, Chen et al. discovered that combining Alarmin HMGN1 peptide with PD-L1 blockade therapy promotes the proliferation and activates the anti-tumor activity of stem-like/progenitor-exhausted CD8+ T (Tstem/Tpex) cells [85]. Therefore, a detailed understanding of T cell exhaustion, especially the subset of stem-like CD8 T cells, is crucial for developing effective combination immunotherapies. According to the latest research by Lilin Ye et al., there exists a group of TCF-1+ TOX− tumor-specific memory CD8+ T cells (TTSM) in tumor-draining lymph nodes (TdLN), which have a typical memory T cell phenotype and potent anti-tumor capabilities with less exhaustion, representing the real CD8+T cell subpopulation that responds to PD-1/PD-L1 inhibitors [86]. Treatment with PD-L1 inhibitors promoted substantial expansion of TdLN-TTSM cells, resulting in the accumulation of TPEX (TCF-1+TOX+, exhausted precursor T cells) and TEX (TCF-1− TOX+) cells in TME. Injecting PD-L1 inhibitors into TdLNs effectively inhibits tumor growth, the anti-tumor effects of PD-L1 inhibitors disappear after the surgical excision of TdLNs, providing mechanistic support for preoperative immune neoadjuvant therapy. A study by Zhu Bo’s team also redefined the unique hematopoietic development pattern in the tumor context. Erythroid precursor cells CD45+EPC, already on the erythroid development trajectory, shifted towards differentiation into tumor-promoting erythroid-derived myeloid cells (EDMCs) under the “coercion” of tumors [87]. The expansion of EDMCs was positively associated with intra-tumor PD-L1 expression, T cell exhaustion and immune tolerance in TME, which significantly inhibited antitumor immune responses of CD8+ T cells and greatly reduced the efficacy of immune checkpoint inhibitors, thus providing valuable prognostic biomarkers and a new therapeutic goal for clinical ICI treatment. Therefore, the above findings indicate that although the extent of T cell exhaustion in the TME of patients with effective ICI therapy is reduced, this does not necessarily result from exhausted T cells regaining vitality. Further exploration into how ICI therapy improves and restores the function of exhausted T cells is needed.
PD-1/PD-L1 interaction promotes the generation and function of Treg cells
Regulatory T cells (Tregs) are a special subset of CD4+ T cells. Tregs can produce certain pro-inflammatory cytokines in the inflammatory microenvironment and suppress autoimmunity by secreting cytokines including adenosine, TGF-β1, and IDO [88], thereby mediating tumor cell immune escape and promoting tumor growth. As many tumors are highly infiltrated by immunosuppressive Treg cells, this may further trigger strong immunosuppression and hinder the functions of effector T cells, which is associated with poor prognosis in various cancers [89].
Recent research has revealed that the PD-1 or PD-L1 pathway can promote the generation of Treg cells and enhance their function in TME, which is closely correlated with the immune escape of tumor cells and tumor development. For instance, Francisco et al. discovered that PD-L1 itself can promote the generation and development of induced Tregs (iTregs) by reducing the phosphorylation of Akt–mTOR, S6, and ERK2, while enhancing PTEN, thereby strengthening the immune suppressive functions of Tregs and suppressing the responses of autoreactive T cells [90]. High levels of PD-L1 expression usually lead to an increase in the number of Foxp3+ Treg cells [91]. Patients with tumors infiltrated by a large number of Foxp3+Treg cells often have a poor prognosis [92, 93]. Removing Foxp3+Treg cells from tumor tissues can enhance anti-tumor immunity, hence a combined strategy of blocking PD-L1/PD-1 while depleting Tregs might show promise in improving treatment outcomes for these patients. Similar to effector T cells, Treg cells also express PD-1, and their survival and functions in the TME depend on TCR and CD28 signals [94]. The regulation of these signaling molecules promotes the induction and maintenance of Treg cells, inhibits T cell responses, and participates in the distant metastasis and recurrence of tumors. PD-1 expression on Treg cells can also promote the transformation of naive CD4+T cells into Treg cells by inhibiting the phosphorylation of Smad3 by cyclin-dependent kinase (Cdk2), thereby suppressing normal immune responses [95]. (Fig. 3b).
During the process of tumor immunotherapy, the application of PD-1 or PD-L1 antibody blockade therapy affects the proliferation and function of Tregs, and may even trigger drug resistance and hyper-progressive disease (HPD). On the one hand, PD-1/PD-L1 antibodies inhibit the differentiation, proliferation, and function of Tregs induced by PD-1/PD-L1. For example, PD-L1, which is upregulated in glioblastoma (GBM), induces and maintains Treg proliferation by interacting with PD-1 on Tregs, thereby exerting its immunosuppressive function. Using nivolumab (a PD-1 inhibitor) suppresses PD-1/PD-L1-induced Treg expansion, significantly prolonging the survival of glioma patients [96]. Similarly, Gambichler et al. evaluate the effects of PD-1 antibody blockade treatment on Treg subsets in the blood of melanoma patients, showing that the use of nivolumab or pembrolizumab leads to a rapid decrease in PD-1+ Tregs in peripheral blood, reducing the risk of melanoma-specific death [97]. Conversely, in some patients with poor innate immunity, T cells remain at a low level. When PD-L1 expression is upregulated on tumor cells, immune therapy mediated by PD-1/PD-L1 antibodies may enhance the TCR signaling and CD28 signaling of Tregs, thereby activating Tregs and enhancing their inhibitory functions, resisting tumor immunity and ultimately leading to the occurrence of hyper-progressive disease (HPD). For example, PD-L1/PD-1 interactions restrict effector regulatory T cells (eTregs) during homeostasis and infection. Under steady-state conditions, activated T cell receptors promote the production of PD-1 in eTregs, and blockade of PD-1 signaling enhances the activity of eTregs. During Toxoplasma infection, IFN-γ promotes PD-L1 upregulation in bone marrow cells through STAT1, associated with a reduction in the Treg population. Blockade of PD-L1 prevents the loss of eTregs and mitigates the development of immunopathology [98]. Anti-PD-1 mAbs treatments cause proliferation of PD-1+ Treg cells in some gastric cancer patients, thereby increasing Treg cell-mediated immunosuppressive activity and promoting disease hyper-progression during PD-1 blockade therapy [99]. Studies have shown that Tregs efficiently uptake lactic acid from the tumor microenvironment through MCT1, convert lactic acid into malic acid and citric acid, and then transfer it to mitochondria to participate in the tricarboxylic acid cycle, enhancing PD-1 expression and Treg immunosuppressive function, and reducing the effectiveness of immunotherapy [100]. We speculate that this may also be a reason for the occurrence of disease hyperprogression. Lactate can also regulate the generation of Tregs by lactylation at Lys72 of MOESIN, thereby improving the interaction between MOESIN and TGF-β/SMAD3 signaling. Combined treatment with anti-PD-1 and lactate dehydrogenase inhibitors has a stronger anti-tumor effect [101], thereby preventing hyper-progression disease. This suggests that in immunotherapy, the exhaustion of PD-1+ Tregs or weakening their immunosuppressive function combined with ICI therapy can be potential interventions for HPD, thus improving the treatment efficacy. In conclusion, these studies indicate that Tregs serve as biomarkers to predict the occurrence of HPD in anti-PD-1/PD-L1 immunotherapies. More research is still needed to clarify the dual effects of PD-1/PD-L1 antibody blockade therapy on Tregs, which may depend on the regulation of multiple factors, such as differences in Treg subgroups, the effect of Tregs on different target cells (such as APCs) in chronic viral infections or cancer, and different downstream molecular mechanisms of regulation, etc. In the future, we should deeply explore the complex relationships between PD-1/PD-L1 and Tregs in tumor immunity and combination therapy to provide new strategies for improving the efficacy of immunotherapies for solid tumors.
PD-1/PD-L1 interaction promotes cancer progression by polarizing tumor-associated macrophages (TAMs)
Macrophages are mainly divided into M1 and M2 types. M1 type macrophages exert immune surveillance functions by recruiting chemokines (CXCL10) and activating other immune effector cells to target cancer cells indirectly. In contrast, M2 type macrophages promote immunosuppression, cancer cell invasion and metastasis, and angiogenesis by releasing various cytokines, playing a key role in tumor progression [102]. Tumor-associated macrophages (TAMs) refer to a specific type of macrophage that infiltrates into tumors, typically characterized by two polarization states but whose functional phenotype is closer to that of M2 macrophages [103]. The autophagosome TRAP released by tumor cells induces macrophages to polarize to the M2 phenotype and upregulates PD-L1 and IL-10 expression in TAMs through the TLR4-MyD88-p38-STAT3 signaling pathway, suppressing T cell proliferation and promoting tumor growth [104]. TAM not only secretes basic vascular endothelial growth factor A, fibroblast growth factor and adrenomedullin to stimulate angiogenesis but also degrades the extracellular matrix, providing nutrition to tumor cells and supporting distant metastasis of tumors [105].
TAMs secrete various cytokines (such as IFN-γ, TNF) to upregulate the PD-L1 expression in tumor cells. With the growth of tumors, PD-1 expression in TAMs increases exponentially [106]. PD-1+ TAMs expressed more CD206, CD11c, and fewer MHC class II molecules [107], suggesting that PD-1 promotes macrophage polarization towards the M2 phenotype. When PD-1 is deficient, it drives macrophage polarization to the M1 phenotype by increasing the phosphorylation of STAT1/NF-κB p65 and downregulating p-STAT6, ultimately leading to richer M1 macrophage infiltration and higher levels of pro-inflammatory cytokine secretion [108]. However, due to long-term interaction with macrophages, CD8+ T cells may mediate lymphocyte trapping, making it difficult for lymphocytes to migrate and infiltrate into tumor islets [109]. Thus, macrophage exhaustion may reactivate CD8+ T cell migration into tumor islets, enhancing the efficacy of anti-PD-1 therapy. Moreover, in patients with various cancers, PD-L1 induces an immune-suppressive phenotype in macrophages through negative signaling, usually associated with poor prognosis. Anti-PD-L1 treatment can reverse this phenotype and induce macrophage-mediated anti-tumor activity [110, 111]. Studies in nearly 500 cases of NSCLC have found that 80% of CD68 macrophages highly expressed PD-L1, these patients had longer overall survival (OS) when treated with anti-PD-1/PD-L1 axis therapy [112]. In hepatocellular carcinoma cells, tumor-specific SLFN11 inhibits hepatocellular carcinoma proliferation and metastasis, knockdown of SLFN11 in HCC cells promotes macrophage migration and polarization towards the M2 phenotype in a CCL2 cytokine-dependent manner, thereby activating the NF-κB pathway, upregulating the expression of PD-L1 and accelerating tumor progression. Pharmacological antagonism of the CCL2/CCR2 signals can enhance the antitumor effect of anti-PD-1 monoclonal antibodies [106]. The Hippo signaling pathway reduces T cell infiltration by directly regulating IL-34 transcription and PD-L1 expression in TNBC, thereby promoting TAM recruitment and reshaping the tumor immune microenvironment. Targeting TAMs with IL-34/CSF-1R inhibitors enhances the antitumor efficacy of immune checkpoint inhibitors [113]. IFN-γ, secreted by inflammatory cells such as macrophages and NK cells, upregulates PD-L1 expression on tumor cells [114]. With the upregulation of PD-L1 expression on tumor cells, it will produce adaptive resistance to IFN-γ released by cytotoxic T lymphocytes, thus forming a vicious cycle that aggravates disease progression and produces primary resistance to ICIs treatment.
PD-1/PD-L1 axis inhibits NK cell killing function to promote immune escape
Natural killer cells (NK) originate from pluripotent hematopoietic stem cells in the bone marrow, which can recognize and kill tumor or virus-infected cells. They also coordinate immune responses by releasing immunomodulatory cytokines such as PGE2, IDO, and TGF-β, serving as the first line of defense against viral infections and preventing malignant transformation of cells, having a crucial role in immune surveillance [115]. Therefore, enhancing the quantity and killing capacity of NK cells in cancer patients contributes to improving the effects of anti-tumor immunotherapy.
In recent years, there has been an increasing exploration of NK cell- based tumor immunotherapy. However, it is still unclear regarding the mechanisms of PD-1/PD-L1 molecule expression on NK cells, their impact on NK cell functions, and their role in regulating anti-tumor immunity. Most research has demonstrated that, in addition to T cells, the PD-1/PD-L1 axis also inhibits NK cell-induced antitumor immunity in vivo. Joy Hsu et al. find that PD-1 is highly expressed on activated NK cells, and its binding to PD-L1 on cancer cells inhibits NK cell degranulation and cytotoxic functions, thereby reducing its antitumor immune responses, promoting tumor immune evasion and progression. Using PD-1 and PD-L1 inhibitors reactivates NK cell anti-tumor responses [116]. Huang et al. find that PD-1 signal transduction on the NK cell inhibits lytic granule polarization and disrupts the “ inside-out” signal of integrin, suppressing the cytotoxic effects of NK cells, such as granzyme secretion, and reducing their ability to kill tumor cells [117] (Fig. 3d). However, Judge et al. find that under various heterogeneous conditions that stimulate high NK cell activation in vitro and in vivo, there is no significant expression of PD-1 on NK cells of humans and mice [118]. This contradictory phenomenon has prompted us to wonder, if NK cells do not express PD-1 themselves, how does the PD-1/PD-L1 pathway inhibit NK cell activity? Recent studies have revealed that in leukemia mouse models, PD-1 on the surface of NK cells is not endogenously expressed but entirely derived from leukemic cells in a SLAM receptor-dependent manner. Further research has found that frequent trogocytosis (cell nibbling) between lymphocytes and tumor cells leads to NK cells acquiring PD-1 from leukemic cells, thus inhibiting NK cell-mediated tumor immune surveillance functions [119]. Trogocytosis refers to lymphocytes “gnawing off” part of the cell membrane from the antigen-presenting cells through the immune synapses, this part of the cell membrane usually carries the cell membrane surface molecules of donor cells, such as PD-1 [120]. This study provides a new perspective on the regulatory mechanisms of PD-1 expression in NK cells, we believe more similar discoveries will be made in the future.
In terms of clinical treatment, attention has begun to focus on NK cells. For example, Lin et al. evaluate the efficiency and safety of NK cell re-infusion combined with PD-1 antibody therapy in patients suffering from advanced non-small cell lung cancer. The results show that, compared to patients treated with Pembrolizumab monotherapy, patients receiving multiple NK cell re-infusions combined with Pembrolizumab have a prolonged overall survival of 18.5 months and an objective response rate (ORR) of up to 36.5%. Furthermore, tumor markers such as circulating tumor cells (CTC) significantly decrease after treatment, preventing cancer recurrence and metastasis [121]. Dong et al. find that PD-L1 expression on NK cells can be induced via the PI3K/AKT/NF-κB signaling pathway in human leukemia, increasing the cytotoxic function of NK cells. The use of anti-PD-L1 mAbs directly targets PD-L1+ NK (PD-L1-expressing NK) cells through p38 signaling to counteract PD-L1-tumors [122]. Therefore, the combined therapy of anti-PD-L1 mAb and PD-L1+ NK cells significantly improves the efficacy of cancer immunotherapies. This anti-tumor mechanism, independent of PD-1, provides new insights into the activation of NK cells and for the first time provides a potential explanation for why some patients lacking PD-L1 expression on tumor cells still respond to anti-PD-L1 monoclonal therapies. Additionally, most clinical trials indicate that NK cell immunotherapy does not significantly shrink tumors in advanced cancer patients but can serve as a postoperative adjuvant therapy to inhibit recurrence. In the future, we aim to truly achieve the precise killing of tumor cells through various immune combination therapies.
PD-1/PD-L1 signaling affects cellular energy synthesis and metabolism
As tumor cells grow rapidly, some may die due to insufficient oxygen and nutrients. Under conditions where cell growth is rapid and surrounding nutrients are lacking, the anaerobic metabolism of glucose increases, leading to the accumulation of lactic acid. Lactic acid derived from tumors causes upregulation of PD-L1 in lung cancer cells and inhibits the function of T cells by disrupting aerobic glycolysis [123], thus creating an optimal environment for PD-1/PD-L1 interactions and tumor immune escape. Besides the supply of biological energy and biosynthesis, metabolic pathways such as glycolysis activation, increased lipid metabolism, and enhanced mitochondrial biosynthesis in TME also play a role in PD-1/PD-L1 signaling, reshaping the local tumor microenvironment and altering the metabolic adaptability of immune cells (Fig. 4). For example, PD-1 signaling stimulates the overexpression of the rate-limiting enzyme CPT1A in fatty acid oxidation (FAO), leading T cells to rely on FAO over glycolysis as their main source of energy [124]. This transition facilitates the transformation of effector T cells into memory T cells, FAO stimulates the production of more Treg cells, thus shaping an immunosuppressive microenvironment that promotes tumor escape. T cells require significant energy to combat cancer. Creatine is an important metabolic regulator controlling T cell anti-tumor immunity, it serves as a “molecular battery” to store bioenergy and provides power for T cell activity [125]. Insufficient intake of creatine severely impairs the anti-tumor immune responses of T cells, supplementing mice with creatine can significantly inhibit tumor growth. Moreover, the combination of creatine supplementation with PD-1/PD-L1 blockade therapy provides a continuous supply of energy to T cells, effectively preventing tumor progression for a long time and significantly improving T cell-based cancer immunotherapies.

PD-1/PD-L1 promotes tumorigenesis and development. PD-1/PD-L1 signaling can alter cellular energy synthesis and metabolic pathways by disrupting aerobic glycolysis, thereby promoting fatty acid oxidation (FAO) as the main source of energy in T cells. Additionally, NAMPT, a component of NAD+ metabolism, can enhance PD-L1 expression in tumor cells through Stat1-dependent IFN-γ signaling, thereby inhibiting T cell function and remodeling the local tumor microenvironment, ultimately exerting significant effects on tumor metastasis, recurrence, and prognosis. Furthermore, the gut microbiota can also improve tumor progression and enhance T cell immune responses, suggesting its potential use in improving the efficacy of PD-1 blockade therapy. Figure created with BioRender
In terms of immunometabolism, NAMPT (nicotinamide phosphoribosyltransferase) in NAD+ (nicotinamide adenine dinucleotide) metabolism increases PD-L1 expression in tumor cells through the Stat1-dependent IFN-γ signaling pathway, inducing CD8+ T cell exhaustion, thus resulting in tumor immune escape and poor prognosis for patients [126]. Further studies have shown that tumors with high expression of NAMPT are more sensitive to PD-1/PD-L1 blockade therapy. Supplementing with NAD+ precursors (β-nicotinamide mononucleotide, NMN) significantly increases sensitivity to immune therapy in tumors resistant to such treatments. This strategy of combining NAD+ supplementation with anti-PD-1/PD-L1 antibodies provides a novel therapeutic strategy for immune therapy-resistant tumors (Fig. 4). Additionally, Chao Zhong’s team [127] found that during DSS-induced colitis, the activation of intestinal lymphoid tissue inducer cells (LTi) is accompanied by increased PD-1 expression. Blocking the PD-1 signaling pathway leads to metabolic reprogramming of LTi cells, mainly manifested by reduced glycolysis, increased FAO, and decreased production of IL-22, eventually resulting in intestinal inflammation. Since excessive fatty acids promote FAO in LTi cells, obese patients may be more prone to intestinal complications caused by anti-PD-1 antibodies. Inhibiting FAO may have significant implications for intervening in intestinal inflammation. Qorraj et al. [128] find that the metabolic phenotype of monocytes is altered in chronic lymphocytic leukemia (CLL). Glucose uptake, glycolysis molecules, and glucose transporter expression decrease. Further research finds that the interaction between the PD-1 expressed on monocytes and the PD-L1 on CLL cells impairs glycolysis, phagocytosis, and BTK signaling, contributing to tumor immune evasion and weakening immune-based therapy. Conversely, disrupting PD-1/PD-L1 signaling reverses these immune metabolic dysfunctions and restores the anti-tumor activity of monocytes in CLL.
The gut microbiota environment is also an essential factor in regulating the immunity and sensitivity of PD-1 blockade therapies (Fig. 4). For example, Andrew’s team uses melanoma models and anti-PD-1/CTLA-4 therapy to demonstrate that immune checkpoint therapy induces the translocation of specific intestinal bacteria to secondary lymphoid organs and tumors, thus activating Dendritic Cells (DCs) and triggering anti-tumor responses of T cells. The use of antibiotics reduces the translocation of the gut microbiome to the mesenteric lymph nodes (MLN) and tumor-draining lymph nodes (TDLN), decreasing the responses of DCs and effector CD8 T cells [129]. This study suggests that the gut microbiome might also be a key mechanism in synergistically promoting extra-intestinal anti-cancer immunity and is critical for ICT treatment. Matson et al. find that the symbiotic microbiome improves tumor control, enhances T cell responses, and increases the effectiveness of anti-PD-L1 therapy in patients with metastatic melanoma, so maintaining a healthy gut microbiota helps patients fight against cancer [130]. Gopalakrishnan et al. investigate the oral and intestinal microbiota of 112 patients receiving anti-PD-1 immunotherapies, where 30 “responsive” patients contain a higher abundance of Ruminococcaceae family bacteria in their feces and a more abundant synthetic metabolic pathway in their bodies, resulting in stronger systemic immunity and anti-tumor immunity [131]. Zhao’s team selected 65 patients with advanced hepatobiliary cancer undergoing anti-PD-1 therapies and divided them into the clinical benefit response (CBR) group and the non-clinical benefit (NCB) group. Bioinformatics analysis discovers that gut microorganisms might influence immune therapy responses by mediating metabolic pathways. The enriched microbiome in the CBR group is related to energy metabolism, significantly prolonging the OS of patients undergoing immunotherapy. Whereas the enriched microbiome in the NCB group is related to amino acid metabolism, with patients having a shorter OS [132]. In conclusion, these findings have tremendous implications for cancer immunotherapy, suggesting that modulating the gut microbiota and metabolism improves the effectiveness of PD-1 blockade therapy. Metabolic pathways involving different gut microbes may represent potential important mechanisms affecting the effects and survival benefits of anti-PD-1 immunotherapy in hepatobiliary tumors, serving as efficacious biomarkers for predicting immunotherapy. Perhaps one day, supplementing the body with specific gut bacteria beneficial for the action of PD-1 antibodies or undergoing specific fecal transplantation, can increase the effectiveness of immunotherapy.
Effects of PD-1/PD-L1 signaling on tumor metastasis, recurrence, and prognosis
PD-1/PD-L1 is involved not only in the regulation of tumor immunity but also plays a certain role in tumor metastasis and recurrence. For example, PD-1 or PD-L1 promotes the malignant growth and autophagy of tumors such as ovarian cancer and melanoma by activating the mTORC signaling pathway [133, 134]. PD-L1 is lowly expressed in poorly differentiated and metastatic GC cells, knocking down PD-L1 reduces the expression of E-cadherin, promotes cell migration and wound healing capabilities, and prolongs the survival time of patients [135]. PD-L1 also promotes the growth and metastasis of cervical cancer by activating the ITGB4/SNAI1/SIRT3 signaling pathway. Specifically, PD-L1 activates the AKT/GSK3β signaling pathway by directly binding to integrin β4 (ITGB4), thereby inducing the expression of the transcriptional repressor SNAI1. SNAI1, in turn, affects the gene expression involved in EMT and regulates glucose metabolism by inhibiting the activity of the SIRT3 promoter [136]. Therefore, overexpression of PD-L1 significantly increases tumor glucose uptake, promotes lymph node metastasis, and is significantly associated with poor prognosis in patients, targeting PD-L1 and its downstream effector molecules as a potential approach to interfere with cervical cancer growth and metastasis. In addition, CD8+ T cells induce PD-L1 expression on hepatocellular carcinoma cells in an IFN-γ-dependent manner, which in turn promotes the apoptosis of CD8+ T cells, leading to disease progression and postoperative recurrence, blocking PD-L1 reverses this phenomenon [137].
High PD-L1 expression makes tumor cells more sensitive to PD-1/PD-L1 inhibitors, using PD-1 or PD-L1 antibody therapy can stimulate immune cells to “recognize” cancer cells again, thus enhancing their own immunity to attack cancer cells and significantly prolonging patient survival. For example, results from first-line clinical trials in advanced NSCLC patients show that compared to platinum-based chemotherapy, treatment with Pembrolizumab provides PD-L1+ patients with longer progression-free survival and overall survival, as well as fewer immune-related adverse events [138]. However, some patients with advanced malignancies may be at risk of experiencing hyper-progressive disease after receiving ICI therapy. Kim et al. evaluated 263 patients with recurrent and metastatic NSCLC receiving PD-1/PD-L1 inhibitors. The results show that PD-1/PD-L1 blockade leads to the occurrence of HPD in many NSCLC patients. The frequency of severely exhausted CD8 T cells (TIGIT+ PD-1+ CD8 T cells) in the peripheral blood of HPD patients is higher than that of non-HPD patients, and the frequency of effector/memory subsets (CCR7− CD45RA−) is lower, with worse PFS and OS [139]. Li et al. find that specific carcinogenic pathways promote the development of HPD following PD-1/PD-L1 blockade therapy. ICI-activated cytotoxic CD8+ T cells released IFN-γ, promoting tumor FGF2 signaling, thereby inhibiting PKM2 activity and reducing NAD levels, enhancing SIRT1-mediated acetylation of β-catenin, ultimately activating the Wnt-β-catenin pathway and leading to HPD [140] (Fig. 4).
The above studies suggest that the overexpression of PD-1/PD-L1 in cancer can promote the occurrence of malignant tumor behavior through multiple signaling pathways. Although targeting PD-L1-mediated extracellular signaling pathway brings effective anti-cancer therapies, some patients have experienced rapid cancer progression during ICI immunotherapy, making it difficult for patients to benefit from clinical treatment. It can be seen that the regulatory roles of the PD-1/PD-L1 pathway in tumor initiation and progression are complex and varied, possibly related to specific genetic backgrounds, tumor biology behavior, or post-translational modifications specific to PD-L1 or PD-1. In the future, exploring and identifying biomarkers that can predict the risk of HPD in response to ICI treatment, preventing the occurrence of HPD by exhausting CD8+ T cells, and overcoming tumor immune resistance to prevent tumor metastasis and recurrence, holds significant clinical significance.
The effect of PD-1/PD-L1 signal on the tumor itself
Traditionally, most studies have emphasized the molecular interactions and modifications between PD-1 and PD-L1 to seek novel tumor immunotherapy approaches. Recent research has revealed that PD-L1 exhibits extensive intracellular and extracellular distribution. It can be expressed in various forms in tumor cells, such as cytoplasmic PD-L1 (cPD-L1), membrane PD-L1 (mPD-L1), soluble PD-L1 (sPD-L1), and extracellular vesicular PD-L1 (EV PD-L1) [141]. Currently, only the function of mPD-L1 has been widely recognized and confirmed. mPD-L1 inhibits T cell function by binding to PD-1 on T cells, and its high expression is often related to tumor progression and poor prognosis in patients [142]. The distribution and immune regulatory function of non-cellular membrane PD-L1 have not been fully demonstrated. Recent studies have suggested that besides its role in promoting tumor cell escape from immune surveillance, PD-L1 is also considered a crucial effector molecule that transduces intrinsic signals in tumor cells to prevent their own apoptosis or to facilitate tumor progression in an immune-independent manner.
As early as 2010, Ghebeh et al. reported that chemotherapy induces nuclear translocation of PD-L1 in breast cancer cells, suggesting that PD-L1 has functions beyond inhibiting T cells [143]. Subsequently, the Shulin Li team found that circulating tumor cells (CTCs) from patients with prostate and colorectal cancers have higher expressions of nPD-L1, indicating that nuclear localization of PD-L1 may be involved in cancer progression and metastasis, holding a significant prognostic value [144]. In TNBC, nuclear PD-L1 is important for regulating the cohesion of sister chromatids and the genomic stability of cancer cells. PD-L1 deficiency leads to the formation of multinucleated cells and the separation of sister chromatids, thereby promoting tumor growth [145]. Du et al. find that in non-small cell lung cancer, highly expressed PD-L1 enters the nucleus by binding to the nucleoprotein KPNB1, then activates the Gas6/MerTK and its downstream AKT and Erk signaling pathways, thereby promoting tumor cell proliferation [146], offering a novel perspective for the optimization of lung cancer immunotherapy. Gao et al. find that under hypoxic conditions, treatment with TNFα and CHX promotes the nuclear translocation of PD-L1, where it interacts with phosphorylated (p)-Stat3-Y705. Subsequently, p-Stat3-Y705 binds to the gasdermin C (GSDMC) promoter region, upregulating GSDMC gene expression, and then caspase-8 cleavage and activation of GSDMC, inducing pyroptosis in cells and resulting in necrosis in the hypoxic region of tumors [147]. Moreover, nuclear PD-L1 also induces tumor cells to express other immune checkpoint molecules, including PD-L2 and VISTA, thus enhancing the anti-tumor responses to PD-1 blockade [148]. Since these molecules are not the targets of the PD-1/PD-L1 blockade, they may contribute to acquired resistance to immune therapy. In bladder cancer, intrinsic PD-L1 can also activate intrinsic cancer cell signals in a PD-1 independent manner and enhance cancer cell proliferation and survival by activating mTOR and inhibiting autophagy [149]. Therefore, the non-immune-dependent functions of nuclear PD-L1, such as participating in tumor pro-inflammatory pathways and reprogramming the expression of genes related to immune responses, may be significant for immunotherapy (Fig. 5).
Not only that, the inherent PD-1 in tumor cells can act as a potential tumor suppressor to inhibit tumor growth. For instance, Wang’s team found that not only PD-L1 is highly expressed in various tumor cells, including lung and liver cancer, but PD-1 is also widely expressed. PD-1 reduces tumor cell proliferation and colony formation by inhibiting the classical AKT and ERK1/2 signaling pathways. Similarly, Shisuo Du et al. also find high expression of inherent PD-1 in tumor cells of NSCLC patients. Treatment with PD-1 targeted antibodies enhances the survival ability of lung cancer cells in vitro, and even potentially develops resistance to PD-1 inhibitor therapy [150]. This also provides some evidence for why some patients do not respond effectively to PD-1 antibody treatment. In contrast, Kleffel et al. detect PD-1 expression on the surface of melanoma cells. The inherent PD-1 binds to its ligand PD-L1 and activates the downstream mTOR signaling through PI3K/AKT-independent pathway, thereby promoting tumor cell self-proliferation, even in mice lacking adaptive immunity. Additionally, PD-L1 expressed in melanoma also promotes tumor growth through paracrine or autocrine interaction with PD-1 [134]. Gupta et al. discover that tumor-initiating cells (TICs) highly expressed PD-L1 in ovarian cancer and melanoma, the intrinsic cell signals produced by PD-L1 promote TIC proliferation and mTORC1 activation, and PD-L1 expression also increases the sensitivity of TIC to IFN-γ and rapamycin, thereby resisting tumor immunotherapy and promoting tumor recurrence [151]. So does PD-1 expressed in tumors play a role in promoting or inhibiting cancer? We speculate this may be context-dependent, as different types of tumor cells or selective signaling pathways may mediate different regulatory functions in tumor cells. More studies are still needed to explain the distinct roles of PD-1 signaling in different cancer types, as well as the specific molecular and cellular mechanisms involved in its regulation. In conclusion, these studies suggest that atypical local PD-1 and PD-L1 may also serve as new targets for cancer therapy, offering new insights for devising optimal ICI treatment strategies to benefit cancer patients.

The effects of nuclear PD-L1 on the tumor itself. PD-L1 not only promotes tumor cells to evade immune surveillance but also facilitates tumor progression in an immune-independent manner. PD-L1, which is expressed at high levels in tumor cells, can enter the nucleus by binding to the nuclear entry protein KPNB1 and exert pro-cancer effects. Nuclear PD-L1 can also trigger the upregulation of immune checkpoint genes, including PD-L2 and VISTA, thereby enhancing the anti-tumor responses of PD-1 inhibition. Under hypoxic conditions, treatment with TNFα and CHX can promote the nuclear translocation of PD-L1, which then interacts with p-Stat3-Y705. Subsequently, p-Stat3-Y705 combines with the GSDMC promoter region, leading to the upregulation of GSDMC gene expression. Furthermore, GSDMC is cleaved and activated by caspase-8, triggering pyroptosis in cells and necrosis in the hypoxic areas of tumors. Figure created with BioRender
The regulatory mechanisms of PD-1/PD-L1 expression in cancers
The activation of PD-1/PD-L1 signaling is not only associated with maintaining autoimmune homeostasis and establishing peripheral immune tolerance, but also involved in tumor progression and immune evasion, serving as a crucial inhibitor of both innate immune and adaptive immune systems [152]. However, the underlying mechanisms regulating the expression of the PD1/PD-L1 pathway are not fully understood. We speculate that specific signals transmitted by cells or inflammatory factors in the tumor may play a dominant role in the regulation of PD-1/PD-L1 expression. Therefore, we summarize the previous regulatory mechanisms of PD-1/PD-L1 expression in tumors, and on this basis, look forward to the application prospects of tumor immunotherapy targeting the PD-1/PD-L1 axis, which could contribute to exploring new therapies for tumor immunity.
Regulation of PD-1 expression in tumor immunity
PD-1 is a major immune checkpoint that regulates T cell activation and ensures peripheral tolerance. It is typically highly expressed in activated T and B lymphocytes, NK cells, and tumor-infiltrating lymphocytes. PD-1 expression is a complex and dynamic regulatory process influenced by various factors, including antigen signaling stimulation, inflammatory factors, and cell types within TME.
During the initial immune phase, T cells, under the stimulation of the MHC-TCR signaling pathway, enhance the enhancer activity within the major transcription regulatory regions (CR-B and CR-C) of PD-1 through reversible demethylation actions [153], and activate the initial transcription of PD-1 under the synergistic actions of various transcription activators such as NF-κB [154], NFATc1 [155], STAT3/4 [156]. When acute virus infection occurs, TCR signaling gradually diminishes as the antigen is cleared, the methylation level of PD-1 enhancers gradually recovers, weakening their transcriptional activation and strengthening their inhibitory effects [157,158,159]. Thus, PD-1 is not expressed on resting memory T cells during acute infection. Conversely, under chronic stimulation and persistent antigen infection, cytotoxic T lymphocytes may become dysfunctional, with impaired secretion of cytokines including TNF-α, IFN-γ, and IL-2, leading to a significant increase in PD-1 expression on T cells. Continuously high expression of PD-1 will transmit inhibitory signals, inducing immune tolerance. For example, upregulation of PD-1 expression in melanoma promotes tumor evasion from immune surveillance, protecting cancer cell growth [134]. At this time, the regulation of PD-1 expression occurs as follows, sustained TCR signaling leads to irreversible demethylation of PD-1 enhancers, relieving the restriction of TCR signaling and driving PD-1 into a state of “uncontrolled” transcriptional activation [160, 161], which further exacerbates T cell functional exhaustion and results in severe immune suppression.
Moreover, other signaling pathways and factors are still involved in the subsequent expression regulation of PD-1 due to the influence of the tumor microenvironment and abnormal cytokine secretion. For example, the transcriptional activator FoxO1 enhances PD-1 expression by binding to the corresponding element within CR-C of PD-1, thereby activating inhibitory signaling pathways, leading to impaired downstream PI3K/AKT/mTOR signaling, so that FoxO1 accumulates in the nucleus and forms a negative feedback regulatory mechanism to enhance the expression of PD-1 [162]. IFN-α activates the JAK/STATs pathway, forming an ISGF3 complex composed of STAT1-STAT2 heterodimers and interferon response factor 9 (IRF9), the complex binds to the interferon-sensitive response element within CR-C to enhance PD-1 expression [163]. Type I IFN also induces the secretion of IL-7 to suppress PD-1 expression in acute viral hepatitis, maintaining antiviral CD8+ T cell response and internal balance in vivo [164]. The inflammatory factor TGF-β1 upregulates PD-1 and CTLA-4 expression in T lymphocytes through the calcium-regulated phosphatase nuclear factor of activated T cells 1 (CaN/NFATc1) pathway, thereby inducing immune escape [165]. More signaling pathways associated with PD-1 expression regulation await further research.
Increasing research has discovered that tumors maintain a state of T cell exhaustion through PD1 signaling, blocking the PD-1 signaling pathway restores or reverses the activity and immune function of exhausted T cells to some extent, enhancing the effectiveness of cancer immunotherapy [77, 166, 167]. Currently, PD-1 blockade therapy has been applied in the immunotherapy of various advanced cancers including melanoma [168], non-small cell lung cancer [169], and colorectal cancer [170].
Regulatory mechanisms of PD-L1 expression in cancers
The impact of PD-L1 in tumors is not fully understood and represents a dynamic and variable process. Numerous reports have indicated that PD-L1 expression is regulated by different intrinsic and extrinsic mechanisms, including genomic alterations, post-transcriptional regulations, post-translational modifications of proteins, carcinogenic signals, inflammatory cytokines, etc. Increased PD-L1 expression in TME restricts host immune responses and induces the development of an immunosuppressive microenvironment in cancer [171, 172]. Therefore, PD-L1 is widely regarded as a crucial immune checkpoint associated with cancer progression and poor patient prognosis, and it holds broad clinical application prospects as a predictive biomarker and therapeutic target in the field of oncology.
Regulatory mechanisms of PD-L1 expression at the gene level
The PD-L1 gene is located on chromosome 9p24.1. Research has shown that PD-L1 gene amplification and structural variations (SVs), chromosomal aberrations, and chromosomal diversity can all cause the upregulation of PD-L1 expression in tumors, making it a promising biomarker for tumor treatment. Copy number variations (CNVs) of the PD-L1 gene affect PD-L1 gene expression levels. In nodular sclerosing Hodgkin’s lymphoma, amplification of the 9p24.1 chromosome is directly related to increased PD-L1 expression on Reed-Sternberg cells [173]. Tumors with an amplified PD-L1 gene often have a higher mutational burden, closely associated with increased PD-L1 expression in malignant cells and poor patient outcomes [174, 175]. However, PD-L1 gene amplification is not always associated with high PD-L1 positivity [176]. PD-L1 amplification occurs only in a small subset of malignancies, such as oral squamous cell carcinoma [177], non-small cell lung cancer [178], cervical squamous cell carcinoma [179], and triple-negative breast cancer [180]. Additionally, in the classical Hodgkin lymphoma and the primary mediastinal large B-cell lymphoma, the amplified region of the 9p24.1 chromosome also includes the Janus kinase 2 (JAK2) locus. JAK2 amplification can enhance PD-L1 expression by activating the IFN-γ pathway and increase sensitivity to JAK2 inhibition [181, 182]. Using the JAK2 inhibitor TG-101348 significantly reduces PD-L1 protein expression levels. The amplification of PD-L1 is caused by genomic rearrangements, ultimately resulting in high PD-L1 expression. Chong LC et al. comprehensively characterized 36 novel PD-L1 structural rearrangements (SRs) in B-cell non-Hodgkin’s lymphoma, including intrachromosomal inversions, duplications, deletions, and translocations of locus fragments, and found that PD-L1 SRs are significantly correlated with PD-L1 protein expression [183]. PD-L1 chromosomal translocations also lead to PD-L1 overexpression to promote tumorigenesis in some diffuse large B-cell lymphomas [184]. In addition, some non-CNV changes in the PD-L1 gene, such as deletion of PD-L1 exons, SNP site mutation [185], DNA double strand break [186], and PD-L1 3’ UTR structural variation [187], are emerging as new genetic mechanisms regulating PD-L1 gene expression.
Regulatory mechanisms of PD-L1 expression at the epigenomic level
Apart from the gene level, epigenetic modifications (DNA methylation, histone methylation, histone acetylation, etc.) in the PD-L1 promoter are also involved in the regulation of PD-L1 expression. Aberrant promoter methylation and histone modifications of PD-L1 may be potential mechanisms leading to its upregulation.
PD-L1 promoter methylation relates to PD-L1 expression, methylated DNA downregulates PD-L1 expression by suppressing its gene transcription. Methylation of the PD-L1 promoter has been found to be negatively correlated with its mRNA and protein expression in various cancers such as gastric cancer [188], diffuse low-grade glioma [189], and non-small cell lung cancer [190]. Hypomethylation of the PD-L1 promoter typically results in upregulated PD-L1 expression, thus exerting immune inhibitory effects [191]. DNMT (DNA methyltransferase) inhibitors induce PD-L1 protein expression and improve the response to PD-L1 blockade therapy in tumors [192]. In the EMT pathway of NSCLC, TNF-α/TGFβ1 induces demethylation of the PD-L1 promoter by reducing DNMT1 levels, which leads to upregulation of PD-L1 expression [193]. Low-dose decitabine (a DNA demethylation drug) combined with PD-1 inhibitors can promote the expansion and effector function of CD8+ exhausted precursor T cells by maintaining the activity of the AP-1/JunD pathway, thus achieving more potent and durable anti-tumor activity. The combination synergistically improves immune surveillance efficacy against tumors [194]. Data from a phase II clinical trial in relapsed or refractory Hodgkin lymphoma shows that, compared to monotherapy with camrelizumab, patients treated with Decitabine and camrelizumab (anti-PD-1 monoclonal antibody) combination therapy have an increased percentage of circulating peripheral central memory T cells, an overall remission rate (ORR) of up to 95%, and significantly prolong progression-free survival (PFS) [195]. Therefore, combining demethylation inhibitors with anti-PD-L1 antibodies may be a more effective oncology treatment strategy.
Acetylation of the histones in the PD-L1 gene promoter region can induce PD-L1 expression. HDAC primarily participates in regulating acetylation by removing acetyl groups from the N-acetylated lysine residues of histones [196]. In breast cancer, HDAC1/2 is recruited to the PD-L1 promoter by the TET2 protein, leading to deacetylation of H3K27ac, thus inhibiting PD-L1 transcription [197]. Treatment with HDAC inhibitors mainly upregulates histone acetylation on the upstream bases of the PD-L1 exons, causing chromatin relaxation and increasing the activity of the PD-L1 promoter fragment, thus enhancing PD-L1 gene expression [198]. In non-small cell lung cancer, hepatocellular carcinoma, and breast cancer, the reduction of COP1 (E3 ligase constitutive photomorphogenesis protein 1) increases c-Jun accumulation and subsequently inhibits the expression of histone deacetylase 3 (HDAC3), thereby enhancing histone H3 acetylation and promoting PD-L1 transcription [199]. Furthermore, BRD4 (bromodomain-containing protein 4), a member of the BET (bromodomain and extra-terminal domain) family, plays a crucial role in chromatin remodeling and transcriptional activation. BRD4 effectively enhances the PD-L1 gene transcription by RNA polymerase II through binding to acetylated histone H3K27Ac in the PD-L1 gene promoter region and distal enhancers [200]. In pancreatic cancer, histone acetyltransferase 1 promotes PD-L1 gene transcription by facilitating the binding of a complex containing BRD4 to acetylated histone H4 [201]. Romidepsin (selective inhibitors of HDAC1 and 2) mainly enhances PD-L1 expression in colorectal cancer by modulating histone H3 and H4 acetylation [202]. HDAC3 inhibitors quickly increase the recruitment of BRD4 to the PD-L1 gene promoter region, thus activating PD-L1 transcription [203].
In pancreatic cancer, the histone methyltransferase MLL1 can enrich H3K4me3 on the PD-L1 promoter, promoting the expression of PD-L1. Inhibiting MLL1 significantly reduces H3K4me3 enrichment in the PD-L1 promoter region [189]. Conversely, in liver cancer, the histone methyltransferase EZH2 acts as an intrinsic modifier to negatively regulate IFN-γ-induced PD-L1 expression. EZH2 suppresses PD-L1 expression by directly upregulating H3K27me3 in the promoters of PD-L1 and IRF1, without affecting the activation of the IFNγ-STAT1 signaling pathway [204]. In breast cancer, Histone Lysine-specific Demethylase 1 (LSD1) inhibitors increase the enrichment of H3K4me2 at the proximal element or core regions of the transcription start site on the PD-L1 promoter, increasing PD-L1 expression in TNBC cells. The combination of an LSD1 inhibitor and PD-1 mAb synergistically enhances tumor immunogenicity and the anti-tumor efficacy of immunotherapy [205]. In some HCC cells, Lysine-specific demethylase 1 A decreases MEF2D methylation by interacting with it, demethylated MEF2D binds to the PD-L1 promoter and activates PD-L1 expression, thus promoting immune inhibitory activity [206].
Mechanisms of PD-L1 expression regulation in transcriptional and post-transcriptional
Transcriptional regulation
Transcription factors involved in PD-L1 regulation
The regulation of PD-L1 expression mainly involves the participation of transcriptional regulators, upstream and downstream signaling molecules and receptors. At present, multiple transcriptional regulators involved in regulating PD-L1 expression have been identified.
MYC
Overexpression of MYC is observed in approximately 70% of tumorigenesis. The transcription factor MYC can directly regulate PD-L1 transcriptional initiation through binding to the PD-L1 promoter and enhance PD-L1 expression. Inactivation of MYC has been found to downregulate PD-L1 mRNA and protein expression, enhancing the anti-tumor immune responses in tumor tissues such as hepatocellular carcinoma, acute T-lymphocytic leukemia, and melanoma [207]. In esophageal squamous cell carcinoma, ChIP sequencing results show that C-Myc upregulates the expression of PD-L1 through binding to the PD-L1 promoter, thereby promoting tumor immune escape and resulting in poorer overall survival in patients [208]. Treatment with c-Myc inhibitors significantly inhibits PD-L1 transcription, such as in lung cancer, where Bafetinib (a tyrosine kinase inhibitor) can reduce PD-L1 transcription by inhibiting the transcription of c-Myc [209].
CDK
The cyclin-dependent kinase (CDK) family also plays a major role in PD-L1 regulation. For instance, the transcription factor TFIIH is important in transcription and DNA repair, its CDK7 subunit can regulate the transcription of PD-L1 in a MYC-dependent manner and promote tumor progression. In NSCLC cells, the CDK7 inhibitor THZ1 inhibits MYC transcriptional activity by downregulating p38α, thereby suppressing PD-L1 expression and improving the anti-tumor immunity against PD-1 therapy in vivo by recruiting infiltrating CD8+ T cells [210]. Similarly, the expression levels of cyclin-dependent kinase 5 (CDK5) and PD-L1 are positively correlated in a variety of tumors. IFN-γ-induced upregulation of PD-L1 requires Cdk5. The absence of Cdk5 induces the expression of PD-L1 transcriptional inhibitors (IRF2 and IRF2BP2), maintaining transcriptional repression of PD-L1 after IFN-γ stimulation, resulting in increased infiltration of CD4 T cells and promoting anti-tumor immune responses [211].
STAT
After binding to its receptor, IFN-γ activates the downstream JAK-STAT signaling pathway, mainly through STAT1 to stimulate the expression of downstream transcriptional regulators. The induction of IRF1 by STAT acts as the major transcriptional regulator of PD-L1 expression induced by IFN-γ. Garcia-Diaz et al. have identified two IRF1 binding sites in the PD-L1 promoter region, the abolition of the IRF1 site leads to a reduction in PD-L1 levels [212]. TET2 is an important factor in the IFN-γ/JAK/STAT1 pathway, it can bind to STAT1 activated by IFN-γ, catalyze the hydroxylation of 5mC, and activate the expression of Th1-type chemokines and PD-L1 [213]. HDAC2 (histone deacetylase 2) enhances PD-L1 expression by increasing the phosphorylation of JAKs/STAT1 and recruiting STAT1 to the PD-L1 promoter. Simultaneously, HDAC2 is also recruited to the PD-L1 promoter by STAT1. Knockout of HDAC2 impairs the IFN-γ-induced H3K27 upregulation, H3K9 acetylation, and recruitment of BRD4 to the PD-L1 promoter region, inhibiting the immune escape and metastasis of triple-negative breast cancer (TNBC) [214]. Chidamide (a class I HDAC inhibitor) upregulates the expression of PD-L1 by activating the transcription factor STAT1 in chondrosarcoma [215]. STAT3 is another important oncogenic transcription factor that directly binds to the PD-L1 promoter to regulate PD-L1 transcription [216]. It also inhibits the elevated PD-L1 protein expression caused by NPM/ALK (nucleophosmin/anaplastic lymphoma kinase) mutations by silencing STAT3 [217]. Additionally, HDAC3 upregulates PD-L1 expression through the STAT3 pathway in pancreatic cancer. Using an HDAC3 inhibitor or knocking down HDAC3 weakens the binding of STAT3 to the PD-L1 promoter, thereby destroying the HDAC3/STAT3/PD-L1 signal transduction and improving the immunotherapy efficacy in pancreatic cancer [218]. Similarly, in melanoma, HDAC8 participates in the transcriptional regulation of PD-L1 by interacting with the transcriptional complex of HOXA5 and STAT3, thereby inhibiting PD-L1 expression [219].
NF-κB
PD-L1 expression in cancer cells also depends on the transcription factor NF-κB. Both the promoter region of the PD-L1 gene and the enhancer region 140 Kb downstream of the PD-L1 gene contain NF-κB binding sites, the NF-κB subunit RelA can bind to the PD-L1 promoter and directly participate in the regulation of PD-L1 expression [220]. NF-κB is activated by various oncogene mutations and inflammatory signaling pathways. Among which, TNF-α and INF-γ induce PD-L1 expression through the NF-κB pathway in breast cancer, prostate cancer, and colon cancer. In particular, TNF-α-dependent activation of IKKε induces the recruitment of p65 to the PD-L1 promoter region, thereby upregulating PD-L1 expression [193]. Ultraviolet radiation (UVR) promotes the immune escape of cancerous melanocytes and the development of melanoma by upregulating NF-κB and IRF3-dependent PD-L1 transcription [221]. There are reports that NF-κB inhibitors can significantly suppress the expression of PD-L1 [222].
Other oncogenic transcription factors
AP-1, a transcriptional regulator composed of FOS, c-Jun, MAF, and ATF subunits, binds to the enhancer region of the PD-L1 gene located about 5 kb downstream of the transcription start site to participate in the transcriptional regulation of PD-L1, promoting its expression in Hodgkin’s lymphoma [223]. Hypoxic conditions induce the activation of hypoxia-inducible factor-1α (HIF-1α) and the accumulation of lactic acid, with both HIF-1α and HIF-2α being able to directly bind to the hypoxia response element in the PD-L1 promoter region, participating in the transcriptional regulation of PD-L1 [224]. The transcriptional activator NRF2, upon stimulation by IFN-γ, directly binds to the PD-L1 promoter and induces the transcription of PD-L1, mediating its upregulation [225]. The endogenous transcriptional regulator nucleophosmin 1 specifically binds to the PD-L1 promoter and activates the PD-L1 transcription in triple-negative breast cancer cells, thereby increasing PD-L1 mRNA and protein expression [226].
The carcinogenic pathway participates in the transcriptional regulation of PD-L1
Several oncogenic signaling pathways, such as Akt/mTOR, JAK/STAT3, and EGFR/MAPK, or the loss or silencing of PTEN, ALK, and EGFR, are involved in the regulation of PD-L1 transcription, potentially leading to the upregulation of PD-L1 expression in tumor cells [227]. IFN-γ released by T cells, is a crucial regulator inducing PD-L1 expression in tumor cells, where the IFN-γ/JAKs/STATs/IRF1 axis is the main pathway to induce and maintain PD-L1 expression in malignancies. The binding of IFN-γ to its receptor leads to the phosphorylation of tyrosine on JAK1 and JAK2, initiating the phosphorylation of STAT1/STAT3 proteins [228]. STAT1 then translocates to the nucleus, binds as a transcription factor to genes that possess gamma interferon activation sites (GAS) to induce the transcription of IFN-stimulated genes (ISG), including IRF1 and IRF9. IRF1 binds to the PD-L1 promoter, significantly enhances PD-L1 expression [229, 230]. IFN-γ also directly upregulates the expression of PD-L1 in tumor and immune cells in TME by activating NF-κB and PKD2 signaling pathways [231]. Phosphorylation of JAK has also been shown to activate other important pathways, including PI3K, RAS, AKT, and MAPK [232]. Loss-of-function mutations in the JAK1/JAK2 pathway result in the absence of PD-L1, which triggers primary and acquired resistance to PD-1 blockade therapy. PD-L1 transcription is also regulated by PTEN/PI3K/AKT/mTOR signaling, where the loss of PTEN or the activation of mTOR can promote PD-L1 expression, high PD-L1 expression also in turn promotes the loss of PTEN expression in cells, suggesting a reciprocal interaction between the two [233, 234]. Some lymphocyte chemokines, such as CXCR2 and CXCL5, increase PD-L1 expression by activating the PI3K/Akt/GSK-3β/Snail signaling pathway, leading to epithelial-mesenchymal transition and promoting cancer invasion and metastasis [235].
In melanoma cells with BRAF mutations, PD-L1 expression is promoted through the activation of STAT3 and the downstream MAPK signal c-Jun, using MEK inhibitors significantly downregulates MAPK signaling and suppresses the production of PD-L1 [236]. When the Anaplastic Lymphoma Kinase (ALK) is mutated and abnormally activated, it often alters its own kinase activity, thereby activating downstream signaling pathways and leading to tumor development. For example, in ALK+ anaplastic large cell lymphoma, NPM-ALK activates downstream MEK-ERK and STAT3 pathways to promote PD-L1 expression [237]. KRAS mutations activate the downstream MEK/ERK signaling pathway, which promotes PD-L1 expression [238]. EGFR mutations, upon stimulation by its ligand EGF, activate downstream signaling pathways such as ERK, mTOR, NF-κBp65, STAT3, and JAK2-STAT1 to promote PD-L1 expression. Inhibiting the MAPK or WNT signaling pathways prevents the upregulation of PD-L1 protein induced by EGF (epidermal growth factor) and IFN-γ, making cancer cells resistant to chemotherapy and conventional cancer treatments [239,240,241] (Fig. 6).

Genetic and transcriptional regulatory mechanisms of PD-L1 expression. At the gene level, tumors with PD-L1 gene amplification in chromosome 9p24.1 have a higher mutation load and a close correlation with increased PD-L1 expression. Epigenetic modifications of the PD-L1 promoter region, including DNA methylation, histone methylation, and acetylation, are also important in the regulation of PD-L1 expression. For example, TNF-α/TGFβ1 induces demethylation of the PD-L1 promoter by decreasing DNMT1 (DNA methyltransferase) levels, resulting in PD-L1 up-regulation and thus exerting immunosuppressive effects. At the transcriptional level, PD-L1 expression is primarily regulated by transcription factors, including STAT, MYC, NF-κB, IRF1, AP-1, and HIF-1α, as well as signaling pathway effector molecules such as MAPK/PI3K/Akt, JAK/STAT3, and EGFR/MAPK. Figure created with BioRender
Post-transcriptional regulation
Non-coding RNAs involved in post-transcriptional regulation of PD-L1
Non-coding RNAs such as miRNA, lncRNA, and circRNA bind the 3’ untranslated region (3’-UTR) of mRNA or act as an upstream regulator to block PD-L1 expression. They are crucial to the development of different types of cancer, their aberrant expression has become an important diagnostic marker and therapeutic candidate.
MicroRNAs (miRNAs)
are small non-coding RNAs (18–25 nucleotides) that regulate PD-L1 gene expression by directly binding to the 3’-UTR (direct action) or by targeting other genes involved in the regulation of PD-L1 expression (indirect action) [242].
MicroRNAs
directly suppress PD-L1 mRNA post-transcriptional expression. miRNAs bind to the 3′-UTR of mRNA, resulting in mRNA cleavage or translation inhibition, thus negatively regulating the expression of PD-L1 at the post-transcriptional level. In NSCLC, wild-type P53-induced miR-34 directly binds to the 3′-UTR of PD-L1 to inhibit PD-L1 mRNA expression, the therapeutic delivery of miR-34a combined with radiotherapy can increase T cell infiltration and inhibit tumor growth [243]. Probes targeting the PD-L1 3’-UTR in lung cancers also enrich Let-7b miRNA, directly inhibiting PD-L1 expression. Inhalation nebulization treatment with let-7b significantly increases the anti-tumor function of CD8+TIL and reduces the expression of PD-L1 in lung cancer cells, showing remarkable tumor inhibitory effects with lower side effects [244]. Daniel et al. find that treating HDLEC (human dermal lymphatic vascular endothelial cells) with the pro-inflammatory cytokines IFN-γ and TNF-α induces miR-155 expression. Further studies reveal that the PD-L1 3’-UTR contains two functional miR-155 binding sites, overexpression of miR-155 inhibits PD-L1 expression, while inhibition of miR-155 results in increased PD-L1 expression following IFN-γ and TNF-α stimulation [245]. However, in diffuse large B-cell lymphoma, miR155 upregulates PD-L1 expression by directly binding to PD-L1 3′-UTR and recruits CD8+ T cells in a PD-1/PD-L1-dependent manner [246]. For these contradictory results, we speculate that there may be multiple cytokines or pathways to regulate PD-L1 expression, and miR-155 is just one of them. More mechanisms need to be further investigated (Fig. 7).
MicroRNAs
also indirectly regulate PD-1/PD-L1 through upstream and downstream pathways. For example, miR-3127-5p activates the phosphorylation of STAT3 by inhibiting autophagy, thereby inducing the upregulation of PD-L1 expression, inhibiting T cell proliferation and promoting immune evasion and drug resistance in lung cancer cells [247]. After treating breast cancer cells with exosomes derived from cancer-associated fibroblasts, the expression of miR-92 increases. The target gene of miR-92, LATS2, interacts with YAP1 and binds to the enhancer region of PD-L1, thereby enhancing the transcriptional activity of PD-L1 [248]. miR-23a-3p, derived from exosomes released by HCC cells under endoplasmic reticulum stress, upregulates PD-L1 expression in macrophages by the PTEN/AKT pathway and inhibits the function of T cells [249]. In gastric cancer, miR-675-3p (derived from GC-EV) inhibits the expression of the target gene CXXC4 and promotes PD-L1 expression by activating the MAPK pathway, thereby stimulating immune evasion of GC cells [250].
Long non-coding RNAs (lncRNAs)
are a highly conserved class of non-coding RNAs longer than 200 nucleotides that have become important regulators of gene expression in various diseases. lncRNA mainly acts as an upstream regulator of PD-1/PD-L1, affecting anti-tumor immunity [251]. Recent research has shown that lncRNAs interact with miRNAs to regulate physiological and pathological processes by inhibiting the transport of target mRNAs or promoting mRNA degradation, thus promoting PD-L1 expression [252]. For example, LncRNA MALAT1 promotes tumor genesis and immune escape in diffuse large B-cell lymphoma by sponging miR-195 to directly target PD-L1 mRNA, while treatment with shMALAT1 increases miR-195 levels and decreases the expression of PD-L1 [253]. LINC01140 directly inhibits miR-377-3p and miR-155-5p expression levels by sponging, and both miR-377-3p and miR-155-5p directly bind to PD-L1 3′-UTR, which leads to upregulation of the downstream target PD-L1 and promotes proliferation, migration and invasion of lung cancer cells [254]. LncRNA HOTAIR also induces PD-L1 overexpression by abnormally activating the TNFα/NF-κB signaling, promoting the immune escape of glioma cells [255]. The latest research shows that the lncRNA IFITM4P upregulates the expression of PD-L1 in oral cancer through a dual mechanism. Firstly, IFITM4P reduces PTEN transcription by increasing the binding of KDM5A to the PTEN promoter in the nucleus, upregulating PD-L1 expression. Secondly, IFITM4P acts as a scaffold in the cytoplasm, recruiting SASH1 to bind and phosphorylate TAK1 (Thr187), thereby promoting phosphorylation of NF-κB (Ser536) and concomitantly inducing PD-L1 expression, inhibiting tumor immune responses [256]. Apart from PD-L1 mRNA, the PD-L1 gene in human LUAD also produces a long non-coding RNA (PD-L1-lnc) by alternative splicing, whose expression is significantly upregulated by IFN-γ. PD-L1-lnc promotes the progression of lung adenocarcinoma by directly binding to c-Myc and enhancing c-Myc transcriptional activity [257].
Circular RNAs (circRNAs)
are a kind of functional non-coding circular RNAs that regulate biological processes by mediating RNA alternative splicing (AS), cis-regulation of transcription, and acting as competitive endogenous RNAs [258]. Many CircRNA sequences contain nucleotide binding sites corresponding to miRNAs, functioning as miRNA sponges to participate in post-transcriptional gene expression and regulation, which are of significant importance in the progression, metastasis, treatment resistance, and drug resistance of tumors [259]. The regulation of the PD-1/PD-L1 pathway by circRNAs is based on the binding of miRNAs to immune checkpoint mRNA. In melanoma, circ_0020710 acts as a sponge for miR-370-3p and directly binds to it, which in turn upregulates CXCL12 expression. CXCL12 recruits suppressive immune cells to form an immune-inhibitory microenvironment, ultimately promoting the proliferation, migration and invasion of melanoma cells [260]. In HNSC, miR-382-3p downregulates the expression of PD-L1 through binding to PD-L1 3′-UTR, while circ_0000052 binds to miR-382-3p through a competitive endogenous RNA mechanism, thereby relieving its inhibitory effect on PD-L1 and inducing excessive PD-L1 expression [261]. similarly, circ-CPA4 targets and weakens the expression of let-7 miRNA, then upregulates PD-L1 expression to promote tumorigenesis and EMT in NSCLC cells. Exosomes derived from NSCLC cells, containing PD-L1, also promote stemness and increase the resistance of NSCLC cells to cisplatin [262].
Post-transcriptional m6A modification of PD-L1
RNA methylation is an important post-transcriptional modification, where the N6-methyladenosine (m6A) modification serves as the most common type of mRNA methylation in mammals. The m6A modification maintains mRNA stability, pre-mRNA splicing, enhances translation efficiency, and promotes polyadenylation and other biological processes. This post-transcriptional m6A modification of RNA is dynamically reversible, mainly involving three types of regulatory factors, methyltransferases (writers), demethylases (erasers), and m6A-binding proteins (readers) [263].
M6A RNA modifications are critical regulators of PD-L1 expression, stability, and T cell-mediated tumor killing. Reader proteins, including the YTHDF family, IGF2PBs, and HNRNPs, recognize the base information modified by m6A methylation and further interact with RNA to exert major regulatory effects, such as participating in downstream translation and mRNA degradation processes [264]. During the process of PD-L1 transcription from DNA to RNA, adenosine is methylated at the N6 position under the action of methyltransferases including METTL3, METTL14, WTAP, and RBM15, these types of methyltransferases are referred to as writers. For example, in breast cancer, METTL3-mediated activation of PD-L1 mRNA is IGF2BP3-dependent, the METTL3/IGF2BP3 axis upregulates the m6A modification of PD-L1 mRNA, further increasing the stability of PD-L1 mRNA and inhibiting tumor immune surveillance [265]. In colorectal cancer and melanoma, defects in METTL3 or METTL14 promote IFN-γ-Stat1-Irf1 signaling by stabilizing STAT1 and IRF1 mRNA expression in an m6A-YTHDF2-dependent manner, thus upregulating PD-L1 expression and making tumors more sensitive to PD-L1 [266]. In HCC, lipopolysaccharide (LPS) promotes the m6A methylation of the lncRNA MIR155HG by upregulating METTL14, thereby maintaining the stability of MIR155HG expression in the reader protein ELAVL1-dependent pathway. Subsequently, MIR155 HG acts as a competitive endogenous RNA through the miR-223/STAT1 axis to upregulate PD-L1 expression, leading to immune escape in HCC cells [267]. Erasers refer to a class of demethylases that mediate the demethylation of m6A, mainly including FTO and ALKBH enzymes, making RNA methylation a reversible response. For example, in colorectal cancer, fat mass and obesity-associated protein (FTO) removes the m6A modifications of PD-L1 mRNA, enhancing its stability and promoting high expression of PD-L1 in an IFN-γ-independent way. Depleting FTO downregulates PD-L1 expression in colon cancer cells [268]. Xinyao Qiu et al. confirm through N6-methyladenosine sequencing (m6A-seq) that PD-L1 mRNA is a direct target of m6A modification [269]. The demethylase ALKBH5 maintains the stability of PD-L1 mRNA in intrahepatic cholangiocarcinoma (ICC) cells by interacting with PD-L1 mRNA and inhibiting T cell anti-tumor immunity in a PD-L1-dependent manner. Lack of ALKBH5 enhances m6A modification on the PD-L1 3′UTR region and accelerates its degradation in a YTHDF2-dependent manner, thereby reducing PD-L1 protein levels (Fig. 7).
In the above studies, it was observed that m6A modification mediated by methyltransferases and demethylases has a dual regulatory role in different types of cancer. So does m6A modification play a role in maintaining or reducing the stability of PD-L1? We speculate that this may not be related to the m6A modification itself but depends on the regulatory function of the bound reader protein, it may even be associated with immune cell infiltration in TME. For instance, in a Meta-analysis on the expression and prognosis of m6A regulators and PD-L1 in HCC, they find that the transcription of regulators is upregulated in HCC patients and demonstrate a significant association with PD-L1 expression, the expression of the YTHDF family is also significantly related to immune infiltration in the HCC microenvironment. Furthermore, overexpression of YTHDF1/2, YTHDC1, RBM15, and METTL3 is significantly correlated with the clinical stage of hepatocellular carcinoma, while downregulation of ZC3H13 and METTL14 is associated with poor prognosis [270]. In recent years, despite the fact that the study of m6A modification has made gratifying progress, its underlying mechanisms in early-stage malignancies remain unknown. Therefore, targeting m6A modification holds promise as an unknown field in future research on tumor immunotherapy.
In addition to m6A modifications, RNA also undergoes other modifications such as N1-methyladenosine (m1A), 5-methylcytosine (m5C), and N7-methylguanosine (m7G). For example, Yun et al. [271] identify through bioinformatics analysis that m5C methylation regulators are related to poor prognosis in patients with pancreatic ductal adenocarcinoma, tumors with high m5C scores exhibit lower CD8 T cell infiltration and higher PD-L1 expression levels. In conclusion, the current evidence for the impact of these modifications on the regulation of immune checkpoint molecules remains unclear and requires further investigation.
Regulatory mechanisms of PD-L1 expression in post-translational modification
In general, the eIF4F complex, a eukaryotic translation initiation complex, regulates the translation of mRNA encoding STAT1 (one of the main transcriptional regulators of PD-L1) by binding to the RNA sequence in the 5’-UTR of STAT1, thereby modulating the expression of IFN-γ-induced PD-L1 on cancer cells [272].Subsequently, post-translational modifications, including phosphorylation, ubiquitination, glycosylation, and palmitoylation regulate the expression of PD-L1 protein by affecting activity, stability, and membrane expression in cancer cells [273, 274] (Fig. 7).
N-glycosylation modification of PD-L1 protein
Glycosylation is a major post-translational modification that is important for protein folding, localization, intracellular transport, and immunogenicity [275]. N-glycosylation is the primary form of glycosylation modification for PD-L1, which is crucial in maintaining the stability of PD-L1 itself.
There are multiple regulatory mechanisms for PD-L1 glycosylation in cancer cells. Studies have shown that PD-L1 is N-glycosylated only at N35, N192, N200, and N219, which stabilizes the protein and prevents its degradation by the 26 S proteasome [276]. N-glycosylation modifications at these sites produce a steric hindrance effect, obstructing the interaction between glycogen synthase kinase 3β (GSK3β) and PD-L1, thus stabilizing PD-L1 and suppressing cytotoxic T cell activity. Non-glycosylated PD-L1 is less stable and, when bound to GSK3β, is phosphorylated and degraded through the ubiquitin/proteasome system [277]. For example, in breast cancer, EGF-induced inactivation of GSK3β induces an increase in PD-L1 glycosylation, maintaining stable PD-L1 expression. However, gefitinib destabilizes PD-L1 by inhibiting EGF signaling, enhancing anti-tumor T cell immunity [277]. EGF/EGFR signaling also promotes PD-L1 glycosylation at positions N192 and N200 by upregulating the expression of the glycosyltransferase B3GNT3, ensuring PD-1/PD-L1 binding. The specific recognition of the poly-LacNAc part by tomato lectin blocks the interaction between PD-L1 and PD-1, thereby promoting PD-L1 internalization and degradation [278]. Therefore, constructing monoclonal antibody conjugate complexes (STM108-ADC) targeting the N192 and N200 glycosylation sites of PD-L1 can serve as a promising target for post-translational modifications of immune checkpoints. Research also finds that the N-glycosylation modification of PD-L1 and its binding with PD-1 are closely related to immunosuppression, blocking the glycosylation of PD-L1 greatly improves the anti-tumor effects. For instance, EMT induces N-glycosyltransferase STT3 transcription by β-catenin, which in turn maintains the stability of STT3-dependent PD-L1 N-glycosylation and induces the upregulation of PD-L1 expression in tumor stem cells, enabling immune evasion. This situation can be reversed when using etoposide [279]. In hepatocellular carcinoma, IL-6 induces phosphorylation of the Y112 site of PD-L1 by JAK1, this phosphorylation facilitates STT3A-catalyzed glycosylation of PD-L1. Blocking the IL-6/JAK1 signaling axis inhibits the binding of STT3A to PD-L1, leading to PD-L1 degradation [280]. N-glycosylation of the PD-L1 protein is detrimental to the detection of PD-L1, making it difficult to bind with diagnostic antibodies for PD-L1. After the glycan chains of PD-L1 are hydrolyzed using N-glycosidase (PNGase F), the detection rate of the PD-L1 protein is significantly improved, increasing the affinity for binding to PD-L1 antibodies [281]. Therefore, deglycosylated PD-L1 protein is expected to become a biomarker for guiding cancer immunotherapy.
Phosphorylation modification of PD-L1 protein
PD-L1 phosphorylation sites are predominantly located in the extracellular domain, different kinases induce PD-L1 phosphorylation at different sites, producing diverse effects. For example, when using EGFR inhibitors activates GSK3α by inhibiting AKT activity, subsequently GSK3α promotes the phosphorylation of Ser279 and Ser283 residues of PD-L1 [282]. Meanwhile, GSK3β, an isomer of GSK3α, induces non-glycosylated PD-L1 to phosphorylate, resulting in polyubiquitination and degradation of PD-L1 in the cytoplasm [277]. PARP1 inhibitors olaparib [283] and c-MET inhibitors [284] can downregulate PD-L1 expression by modulating GSK3β activity. Furthermore, AMPK activated by metformin directly binds to PD-L1 in the endoplasmic reticulum (ER), causing PD-L1 to be phosphorylated at the S195 site, leading to abnormal glycosylation of PD-L1 and endoplasmic reticulum associated degradation (ERAD) [285]. The energy state changes induced by the ketogenic diet activate AMPK, which then phosphorylates the Ser283 site on PD-L1, thereby destroying its interaction with CMTM4, subsequently triggering PD-L1 degradation through lysosomes [286].
Ubiquitination modification of PD-L1 protein
Ubiquitination modification refers to the processes where a ubiquitin molecule covalently binds to a target protein molecule under the action of E1, E2, and E3 ligases and specifically modifies it. When the target protein binds to a single ubiquitin molecule, it is called monoubiquitination. When multiple lysines of the target protein are simultaneously tagged by a single ubiquitin molecule, it is referred to as polyubiquitination [287]. PD-L1 expression is highly regulated by ubiquitin-proteasome systems (UPS), such as STUB1 and SPOP, promoting PD-L1 polyubiquitination and degradation, while CSN5 and the deubiquitinating enzyme DUB antagonize its degradation [288].
Ubiquitin ligase
NEDD4 is an E3 ubiquitin ligase of the HECT domain family that acts as a regulator of PD-L1 ubiquitination. In bladder cancer, NEDD4 is phosphorylated by FGFR3, followed by interacting with PD-L1 and catalyzing its polyubiquitination via Lys48 (K48) linkage [289]. Within the ER, the E3 ubiquitin ligase HRD1 (HMG-CoA reductase degradation 1) initiates the polyubiquitination of abnormally glycosylated PD-L1, resulting in endoplasmic reticulum associated degradation [285]. CyclinD-CDK4 and the Cullin3SPOP E3 ligase regulate PD-L1 expression through ubiquitin-proteasome system-mediated degradation. During the S and G2 phases, CyclinD-CDK4 phosphorylates SPOP (speckle type BTB/POZ protein) and dissociates it from Cullin 3, thereby enhancing the stability of Cullin 3, inducing PD-L1 polyubiquitination and reducing PD-L1 expression levels. Inhibition of CDK4 and CDK6 in vivo reduces SPOP phosphorylation, promoting APC/Cdh1-mediated SPOP degradation, resulting in high PD-L1 expression [290]. Additionally, inhibition of CDK5 increases ubiquitin E3 ligase FBXO22 levels, which degrades PD-L1 and makes tumor cells more sensitive to DNA damage [291]. In breast cancer, the transmembrane and ubiquitin-like structural domain-containing protein 1 (TMUB1) competes with HUWE1 (an E3 ubiquitin ligase) for binding to PD-L1 and inhibiting its ubiquitination at K281 in the ER [292]. The E3 ubiquitin ligase STUB1 (STIP1 homology and U-box containing protein 1) also polyubiquitinates PD-L1 in the cytoplasmic domain at lysine residues and downregulates PD-L1 expression, whereas CMTM6 maintains PD-L1 stability by blocking ubiquitination in tumor cells [293]. Wu et al. identify a novel E3 ubiquitin ligase ARIH1 that targets the PD-L1 protein. In breast cancer cells, ES-072 (an EGFR inhibitor) increases the phosphorylation levels at S279 and S283 sites of the PD-L1 protein intracellular segment through the EGFR-GSK3α pathway, promoting the interaction between PD-L1 and ARIH1, thereby regulating PD-L1 degradation through the proteasome and promoting anti-tumor immunity [282].
Deubiquitinating enzymes(DUB)
Ubiquitination is a dynamic and reversible modification in which ubiquitin molecules and polyubiquitin chains on target proteins can be dissociated by deubiquitinating enzymes, thus stabilizing PD-L1 protein levels in cancer cells. For example, in gastric cancer, PD-L1 expression is positively related to the expression of ubiquitin-specific protease 7 (USP7), which directly interacts with PD-L1 and promotes its deubiquitination and stabilization. Targeting USP7 enhances CD8 T cell infiltration and cytotoxicity in TME and reduces PD-L1 expression in tumor cells [294]. In pancreatic cancer, ubiquitin-specific protease 8 (USP8) catalyzes the deubiquitination of PD-L1 by inhibiting ubiquitin-regulated proteasome degradation, the combined therapy of USP8 inhibitors with anti-PD-L1 effectively inhibits pancreatic tumor growth by activating cytotoxic T cells [295]. In hepatocellular carcinoma, the deubiquitinating enzyme USP22 directly interacts with the C-terminus of PD-L1, catalyzing its deubiquitination and stabilization. USP22 is highly expressed in HCC, closely correlated with poor prognosis in patients [296]. Furthermore, pro-inflammatory cytokines enhance the deubiquitination of PD-L1, thereby increasing its stability. For example, TNF-α released by M2 macrophages induces the binding of NF-κB p65 to the CSN5 promoter, promoting CSN5 transcription and increasing CSN5 expression. Subsequently, TNF-α also inhibits the ubiquitination and degradation of PD-L1 protein through COP9 signalosome subunit 5 (CSN5), thereby enhancing the stability of PD-L1 and sensitizing cancer cells to immunotherapy [297].
Palmitoylation modification of PD-L1
Palmitoylation modification refers to the attachment of fatty acyl groups to the cysteines of the target proteins through thioester bonds, which in turn regulates the membrane localization and function of the protein. Recent studies have indicated that palmitoylation of PD-L1 has become a novel post-translational modification, which is critical for maintaining PD-L1 stability and improving the efficacy of tumor immunotherapy. However, it is still in the research stage [298].
In different types of tumors, the palmitoyltransferases mediating the palmitoylation modification of PD-L1 may vary. In the study conducted by the Xu lab using a mouse model of colon cancer, the cysteine residue at position 272 (Cys272) of the PD-L1 protein is palmitoylated by the palmitoyltransferase DHHC3 (aspartate-histidine-histidine-cysteine 3), thereby inhibiting the ubiquitination and degradation of PD-L1 and increasing PD-L1 expression levels. Treatment with 2-bromopalmitate or silencing DHHC3 activates anti-tumor immunity by inhibiting PD-L1 palmitoylation. They also designed a competitive inhibitor of PD-L1 palmitoylation according to the amino acid sequences near the Cys272 site, which specifically inhibits PD-L1 palmitoylation, reduces PD-L1 expression in cancer cells and enhances the anti-tumor immune responses mediated by T cells [274]. In the same year, Yang et al. also reported that in breast cancer, the palmitoyl transferase DHHC9 (aspartate-histidine-histidine-cysteine 9) mediates palmitoylation modification of PD-L1. DHHC9 covalently attaches palmitate to cysteine residues(C272) of PD-L1, thereby blocking the ubiquitination of PD-L1 to increase its stability. Disrupting PD-L1 palmitoylation through specific point mutations at the Cys272 site or inhibiting ZDHHC9 expression makes breast cancer cells sensitive to T-cell killing, thereby inhibiting tumor growth [299]. PD-L1 is highly palmitoylated in cisplatin-resistant bladder cancer cells, inhibition of fatty acid synthase (FASN) suppresses PD-L1 expression and palmitoylation, suggesting that the FASN-PD-L1 targeting therapy may be used for bladder cancer patients [300].
Lysosomal-mediated degradation
In addition to the above proteasome degradation pathways, lysosomes in cells also participate in the degradation of various endocytic proteins. PD-1/PD-L1, as a cell membrane protein, can be imported into lysosomes for degradation. For example, the HIP1R protein carries a lysosomal targeting signal and delivers PD-L1 to lysosomes for degradation by binding to PD-L1, thereby enhancing the killing effect of T cells on cancer cells. Moreover, a peptide (PD-LYSO) designed based on the lysosomal sorting signals and the binding sequences of HIP1R and PD-L1can successfully deplete PD-L1 expression in tumor cells [301]. Furthermore, cancer cells have also evolved mechanisms to enhance PD-L1 stability and inhibit lysosomal degradation. The transmembrane proteins CMTM4 and CMTM6 (CKLF-like MARVEL transmembrane domain containing 4/6) are key regulators of PD-L1, primarily stabilizing PD-L1 protein expression through the lysosomal pathway [293]. The binding of CMTM6 to PD-L1 inhibits the lysosomal degradation of PD-L1, after knocking out CMTM6, the E3 ubiquitin ligase STUB1 induces PD-L1 polyubiquitination in the circulating endosomes, thereby degrading it via the lysosomal pathway [302].

Mechanisms of PD-L1 expression regulation at post-transcriptional and post-translational. Non-coding RNAs such as miR-34, miR-200, and miR-197 can suppress PD-L1 mRNA expression by directly binding to PD-L1 3′UTR. The m6A modification of PD-L1 mRNA is critical for regulating the expression and stability of PD-L1 and mediating tumor immune escape. For instance, demethylases such as FTO and ALKBH can remove the m6A modification on PD-L1 mRNA, increasing its stability and promoting high PD-L1 expression. The METTL3/IGF2BP3 axis also enhances the stability of PD-L1 mRNA by upregulating its m6A modification, further promoting tumor immune evasion. Additionally, post-translational modifications, including phosphorylation, ubiquitination, glycosylation, and palmitoylation can regulate PD-L1 protein expression by influencing its activity, stability, and membrane expression in cancer cells. Figure created with BioRender
Regulation of PD-L1 by tumor microenvironment
Previous research has concentrated on the intrinsic mechanisms regulating PD-L1 expression in tumor cells, but the expression of PD-L1 is also influenced by a pro-inflammatory tumor microenvironment (TME) created by exposure to infiltrating immune cells, which includes pro-inflammatory cytokines, exosomes, metabolites, etc [303]. Beyond the aforementioned interferon signaling pathways, there are many specific cytokines within the TME that significantly modulate the expression of PD-1/PD-L1. For example, IL-17 A increases PD-L1 expression in colorectal cancer through the p65/NRF1/miR-15b-5p pathway and promotes resistance to anti-PD-1 therapies [304]. IL-6 derived from glioblastoma induces PD-L1 expression on bone marrow cells via a STAT3-dependent mechanism, therapeutic blockade of IL-6 signaling inhibits tumor growth and improves survival rates [305]. Tumor-derived TNF-α induces PD-L1 expression on mast cells through activating the NF-κB pathway, thus inhibiting T cell immunity and gastric cancer progression [306]. TGF-β from TAM inhibits T cell activity by reducing the expression levels of IFN-γ and granzyme B through the inhibition of Smad2/3 phosphorylation and mitochondrial respiration [307]. Nutrients in the tumor microenvironment, especially glucose, induce cancer cells to upregulate PD-L1 expression. Specifically, in human glioblastoma cells, high glucose levels promote the dissociation of hexokinase 2 (HK2) from mitochondria, subsequently mediating phosphorylation at the IκBα T291 site and promoting IκBα degradation, thus activating the NF-κB pathway and upregulating PD-L1 expression at the transcriptional level. Combined treatment with an HK2 inhibitor and anti-PD-1 antibody significantly inhibits tumor growth in mice and prolongs survival [308]. Exosomes secreted by cells contain abundant miRNAs, mRNAs, and functional proteins, which mediate intercellular signaling in the TME [309]. Bo Gong et al. find two secretory PD-L1 splicing variants lacking transmembrane domains in aPD-L1-resistant NSCLC patients, which can mediate resistance to PD-L1 antibody blockade treatment by inhibiting T cell activity [310]. Exosomes derived from glioblastoma stem cells further enhance immunosuppression by upregulating phosphorylated STAT3, thus promoting macrophage polarization to M2 macrophages and increasing PD-L1 expression [311]. Exosomes released by metastatic melanoma carry PD-L1 on their surface, the levels of circulating exosomal PD-L1 are positively correlated with IFN-γ levels. IFN-γ stimulation increases the amount of PD-L1 on these vesicles, thereby inhibiting CD8 T cell function and promoting tumor growth [312]. These findings collectively highlight the crucial roles of various molecules in PD-L1 regulation in different cancer cells, indicating their potential as targets for immunotherapy.
In summary, PD-1/PD-L1 expression is subject to multiple regulatory mechanisms and varies greatly at different stages of tumor development. Given the unique microenvironment and intricate signaling pathways in tumors, it is necessary for future research to delve into which PD-1/PD-L1 regulatory mechanisms play a key role in certain types of cancer. These studies will advance our understanding of tumor immune evasion mechanisms and provide new insights for tumor immunotherapy. We believe that in the near future, as the veil is progressively lifted on the various regulatory mechanisms governing the expression of immune checkpoint molecules, the prospects for inhibiting tumor immunity via these checkpoints will be more promising.
Blockade of PD-1/PD-L1 signaling in tumor immunotherapy
Nowadays, tumor immunotherapy, compared to traditional therapy methods such as surgery and chemotherapy, focuses more on utilizing own immune system to eliminate tumors. Due to its fewer adverse side effects and broader applicability, it has made significant progress in treating various types of cancer. Currently, the therapeutic approaches targeting PD-1/PD-L1 include immune checkpoint inhibitors, adoptive T cell therapy (ACT), gene editing therapies, and tumor vaccines, which have been widely applied clinically in advanced cancer patients, particularly in melanoma and lung cancer, achieving remarkable early success and greatly improving the overall survival and progression-free survival of cancer patients [313]. Unfortunately, there are still limitations such as low response rates and resistance in some cancer patients, resulting in only a minority of patients benefiting clinically. Therefore, identifying suitable candidates for immune checkpoint inhibitor therapy, clarifying patient resistance mechanisms, and exploring novel strategies for combined drug immunotherapy hold significant research significance.
With the discovery of immune escape mechanisms, drug development has been greatly advanced. At present, ICIs based on the PD-1/PD-L1 axis have received clinical approval in many countries. These ICIs reactivate T cell immune responses against tumors by inhibiting immune checkpoints, thereby enhancing immune system surveillance and exerting anti-tumor effects [314]. Antibodies targeting the PD-1/PD-L1 pathway are already approved to treat various human cancers, such as head and neck cancer (pembrolizumab and nivolumab), melanoma (pembrolizumab and nivolumab), urothelial carcinoma (nivolumab, pembrolizumab, durvalumab, atezolizumab, and avelumab), non-small cell lung cancer (pembrolizumab, nivolumab, and atezolizumab), renal cell carcinoma (nivolumab), Hodgkin lymphoma (pembrolizumab and nivolumab), skin squamous cell carcinoma (cemiplimab), and so on [315, 316]. However, these therapies need to be used cautiously, as most patients will develop drug resistance. Excessive blockade of PD-1/PD-L1 may lead to autoimmune diseases and produce side effects.
Targeting PD-1/PD-L1 by antibodies
PD-1 or PD-L1 antibodies can specifically block the binding between PD-1 and PD-L1, thereby reactivating T cells and restoring anti-tumor immune function, making them potential anti-tumor drugs. The drug development teams utilize the molecular targeting principle of monoclonal antibodies and the theory of tumor immunology as the basis for developing PD-1 and PD-L1 monoclonal antibodies. PD-1 monoclonal antibodies work by reducing the stimulatory signals produced by the binding of PD-1/PD-L1, restoring the T cell immune responses and enhancing T cell attack against cancer cells. PD-L1 monoclonal antibodies follow the principle that drug molecules preferentially target PD-L1 on cancer cells, reducing the negative regulatory factors released by cancer cells to the immune system [317]. Moreover, PD-L1 inhibitors are particularly effective against cancers with a high mutation burden, such as colon cancer with microsatellite instability. As heightened mutagenicity triggers responses from pre-existing T cells, using PD-L1 inhibitors reduces the immunosuppressive effect on T cells [318]. So far, six immune checkpoint inhibitors targeting PD-1 or PD-L1 have already been approved for clinical therapy in cancer patients, including PD-1 inhibitors like pembrolizumab, nivolumab, cemiplimab, and PD-L1 inhibitors like atezolizumab, avelumab, and durvalumab [319] (Table 1).
Anti-PD-1 antibody
Pembrolizumab
Pembrolizumab (trade name Keytruda) is a high-affinity humanized IgG4 anti-PD-1 antibody, it was the first treatment globally approved by the FDA for patients with metastatic melanoma [320] and the first therapy approved to treat all high-mutated solid tumors [321]. Phase I clinical trials have proven the safety and efficacy of pembrolizumab in treating melanoma, and phase II clinical trials have shown that pembrolizumab’s efficacy in treating advanced melanoma is significantly superior to that of ipilimumab (anti-CTLA-4 antibody) [322]. Due to its significant anti-tumor effects observed in clinical trials, pembrolizumab has been approved for first-line therapy in various advanced melanomas [323]. Subsequently, its indications have extended to recurrent head and neck squamous cell carcinoma [324], advanced or metastatic NSCLC with PD-L1 positivity [325], metastatic urothelial carcinoma [326], cervical cancer [327], classical Hodgkin lymphoma [328], microsatellite-unstable or mismatch repair-deficient cancer [329], gastric cancer [330] and primary mediastinal large B-cell lymphoma [331], among others. In conclusion, pembrolizumab treatment has greatly improved the overall survival of cancer patients.
Nivolumab
Nivolumab (trade name Opdivo) is a humanized IgG4 anti-PD-1 antibody that exhibits high affinity for PD-1, effectively blocking the binding between PD-1 and PD-L1 [332]. Nivolumab can be used to treat unresectable or metastatic melanoma following Ipilimumab (anti-CTLA-4), with a standalone objective response rate (ORR) of 31.7% [333]. After the success of the phase I-III clinical trial, Nivolumab is approved as a first-line treatment for metastatic melanoma [334], second-line therapy for NSCLC [335] and renal cell carcinoma (RCC) [336]. In NSCLC patients, nivolumab’s objective response rate is 23.7%, significantly prolonging the overall survival, response rate, and progression-free survival (PFS) in patients in a manner independent of PD-L1 expression [337]. Additionally, Nivolumab has been demonstrated to be effective against many other malignancies, including classical Hodgkins lymphoma [173], recurrent head and neck squamous cell carcinoma [338], locally advanced or metastatic urothelial carcinoma [339], and microsatellite instability or mismatch repair-deficient colorectal cancer [340].
Cemiplimab
Cemiplimab (trade name Libtayo) is an anti-PD-1 antibody with high affinity and is the first immune checkpoint therapeutic drug designed for the specific treatment of metastatic cutaneous squamous cell carcinoma (CSCC) [341]. Clinical phase 1 trials in advanced CSCC patients demonstrated that after Cemiplimab treatment, there were no signs of disease recurrence for over 16 months, and the response was persistent [342]. The phase II trial of Cemiplimab treatment showed an objective response rate of up to 47% [343]. Subsequently, Cemiplimab has also been approved for the first-line clinical therapy of advanced NSCLC with high PD-L1 expression [344] and metastatic basal cell carcinoma [345].
In addition, four other PD-1 antibodies (Sintilima, Toripalimab, Penpilimab, and Zzimberelimab) have been approved in China for the therapy of resectable or metastatic melanoma, nasopharyngeal carcinoma, urothelial carcinoma, as well as relapsed or refractory classical Hodgkin lymphoma [346].
Anti-PD-L1 antibody
Atezolizumab
Atezolizumab (trade name Tecentriq) is a phage-derived human IgG1 antibody with a genetically engineered modified Fc fragment. Consequently, Atezolizumab can block PD-L1 on the tumor surface while avoiding antibody-dependent cell-mediated cytotoxicity effects [347]. As a PD-L1 immune checkpoint inhibitor, Atezolizumab has demonstrated significant efficacy in various cancers, including breast cancer, renal cell carcinoma, and bladder transitional cell carcinoma. It significantly improves the survival rate of NSCLC patients, and this response is associated with PD-L1 expression in tumor-infiltrating immune cells [348]. In advanced or metastatic urothelial carcinoma, atezolizumab is also approved as a first-line treatment, where increased PD-L1 expression on immune cells can enhance the responses to Atezolizumab [349]. Atezolizumab may also be applied to patients with EGFR-sensitive mutations who are receiving targeted therapy [350].
Avelumab
Avelumab (trade name Bavencio) is a human IgG1 monoclonal antibody that has been approved for the therapy of refractory metastatic urothelial carcinoma [351] and Merkel cell carcinoma [352]. The therapeutic action of Avelumab involves the blockade of PD-L1 to reinvigorate T cells and induce the occurrence of antibody-dependent cell-mediated cytotoxicity through its natural Fc region [353]. In the therapy of metastatic Merkel cell carcinoma, Avelumab has shown an objective response rate of 62.1% [354], and many other studies on Avelumab as a monotherapy or in combination with other therapies are underway.
Duravulumab
Duravulumab (trade name Imfinzi) is a humanized antibody that can directly target PD-L1, blocking the interaction between PD-L1 and PD-1, thereby preventing tumor immune evasion and enhancing the immune responses [355]. Durvalumab is mainly approved for the therapy of platinum-resistant primary advanced or metastatic urothelial carcinoma in PD-L1-positive patients [356] and small cell lung cancer [357]. According to reports, Durvalumab’s overall response rate (ORR) in the treatment of HNSCC is 9.2% [358], with progression-free survival rates of 20% for all HNSCC patients, reaching 25% in PD-L1-positive patients [359]. In NSCLC, the ORR of Durvalumab treatment can reach 66.3%, significantly extending the overall survival of patients with phase III and unresectable NSCLC [360]. The drug has tolerable toxicity characteristics and appears to be more effective in patients with high PDL-1 expression. However, further research is needed to determine its efficacy at different disease stages and its applicability to other tumor types.
Targeting PD-1/PD-L1 by small molecule inhibitors
Due to the fact that large molecule monoclonal antibodies such as PD-1 and PD-L1 monoclonal antibodies only recognize receptors on the surface of cells and cannot act directly in the cells, resulting in low drug utilization, long-term use may easily cause acquired drug resistance. Consequently, combination therapy with other treatments is necessary to improve therapeutic efficacy. In recent years, researchers have expanded the inhibition of PD-1/PD-L1 pathways into the realm of traditional small-molecule drugs. Non-monoclonal antibodies that block PD-1/PD-L1 binding, such as small molecule inhibitors, cyclic peptide inhibitors, and other macrocyclic compounds, have gained more and more attention [361, 362]. In the following discussion, we will focus on PD-1/PD-L1 small-molecule inhibitors.
Small-molecule inhibitors are divided into sulfonamides, biphenyls, oxadiazoles, and other heterocyclic compounds [363]. In the molecular structure, the transmembrane protein PD-L1 participates in PD-1/PD-L1 binding in a dimeric form. While the small-molecule compounds BMS202 and BMS-8 act on the PD-L1 protein, causing PD-L1 to form dimers. Subsequently, BMS202 and BMS-8 bind to the cylindrical hydrophobic cavities of the two dimerized PD-L1s respectively, preventing normal interaction between PD-1 and PD-L1 [364], reducing the negative regulation of immune function by PD-1/PD-L1. Additionally, the BMS family’s BMS-1001 and BMS-1166 alleviate PD-1/PD-L1-mediated Jurkat T lymphocyte exhaustion by inhibiting PD-L1, activating T lymphocyte immune activity [365].
The research findings on small-molecule drugs for tumor immunotherapy currently require extensive clinical trials for validation, and many compounds with potential biological activities are still in the preclinical research stage. With in-depth research on the interaction information between PD-1 and PD-L1 and the surface binding of the formed dimers, it will provide an essential structural basis for the design of small-molecule drugs. Furthermore, there are many tumor-associated regulatory factors and signaling pathways, which also offer abundant targets for small-molecule compounds.
Combination therapy
Owing to the heterogeneity of cancers and genetic differences between individuals, the efficacy of targeting the PD-1/PD-L1 pathway alone is unsatisfactory [366]. Therefore, there is an urgent need for combination therapies that can be tailored to different patients in order to increase the response rates to PD-1/PD-L1 inhibitors and overcome resistance to anti-PD-1/PD-L1 therapy. Recent studies have shown that combination therapies such as radiotherapy and chemotherapy, targeted therapies such as VEGF/VEGFR, cellular therapy, tumor vaccines, and oncolytic viruses can regulate PD-L1 in certain cancers [10, 12], making them ideal candidates for combination therapy with anti-PD-1/PD-L1. These approaches have achieved initial success in treating various types of cancer.
PD-1/PD-L1 inhibitors combined with other immune checkpoint inhibitors (ICIs)
PD-1/PD-L1 inhibitor combined with CTLA-4 inhibitor treatment
In T cells, CTLA-4 generates potent inhibitory signaling to prevent T cell activation and growth. Blocking CTLA-4 inhibits Tregs, reactivates T cells suppressed within the TME, and accompanies an increase in IFN-γ production, thereby inducing PD-L1 expression in the TME and subsequently inhibiting T cell anti-tumor responses [367]. For certain patients with low PD-L1 expression, CTLA-4 reduces the risk of resistance to PD-1 and demonstrates synergistic anti-tumor effects. Mounting evidence suggests that the dual blockade of PD-1/PD-L1 and CTLA-4 exhibits stronger anti-tumor activity in certain cancer types [368]. It has become the most widely used and deeply explored immunotherapy approach, approved for the therapy of metastatic melanoma, advanced renal cell carcinoma, and mismatch repair deficient/highly microsatellite instable colon cancer. When the PD-1 inhibitor Nivolumab is combined with the CTLA-4 inhibitor Ipilimumab, it significantly increases the progression-free survival rate and median overall survival in advanced NSCLC patients, greatly improving the patient prognosis with more durable therapeutic effects [369]. In B16 melanoma, the combined blockade of CTLA-4 and PD-1 negative co-stimulatory receptors significantly increases the Teff infiltration and production of inflammatory cytokines in tumors, and reduces Tregs and myeloid cells within the tumor, thus counteracting tumor-induced immune suppression and promoting tumor rejection [370]. In advanced melanoma, the five-year survival rates of combined therapy with Nivolumab and Ipilimumab reach 52%, with a positive response rate of 63.8% in clinical phase II and III trials [371, 372]. In clinical phase III trials of patients with metastatic renal cell carcinoma (mRCC) [373] or unresectable malignant pleural mesothelioma (CheckMate 743) [374], similar efficacy is observed in the combination of anti-PD-1 (nivolumab) and anti-CTLA-4 (ipilimumab) treatment, significantly improving patient overall survival. Moreover, a meta-analysis by Kongju et al. [368] reveals that the combination therapy of PD-1 with CTLA-4 is a feasible strategy, offering enhanced efficacy and acceptable adverse events.
PD-1/PD-L1 inhibitors combined with other ICIs
In addition to targeting CTLA-4, other dual immune checkpoint blockade strategies, such as PD-1/PD-L1 inhibitors combined with immune checkpoint inhibitors like TIM-3, TIGIT, LAG-3, and PVRIG, are still under clinical trials [375]. LAG-3 and PD-1 synergistically regulate T cell functions, thereby attenuating anti-tumor immune responses. In untreated metastatic or inoperable melanoma patients, combination treatment of Relatlimab (an anti-LAG-3 monoclonal antibody) with nivolumab demonstrates anti-tumor activity, significantly improving patients’ progression-free survival [376]. TIM-3 inhibits anti-cancer immunity and mediates resistance to PD-1 and PD-L1 inhibitors. Clinical Phase Ia/b results of LY3321367 (a novel TIM-3 monoclonal antibody) show that the ORR and DCR (disease control rate) of PD-1 or PD-L1 inhibitors combined with TIM-3 inhibitors are 4% and 42%, respectively, with favorable pharmacokinetics/pharmacodynamics [377]. In the CITYSCAPE trial, PD-1 or PD-L1 inhibitors combined with a novel anti-TIGIT inhibitory immune checkpoint drug (Tiragolumab) showed clinically meaningful improvements in objective remission rates and progression-free survival rates, serving as a potential first-line therapy for locally advanced unresectable or metastatic NSCLC [378].
Furthermore, PD-1/PD-L1 inhibitors also regulate T cell function by combining with immune checkpoint agonists like CD137 (4-1BB), ICOS, CD134 (OX40), GITR, and CD27. For example, the results of a phase Ib clinical trial (NCT02179918) find that in advanced solid tumor patients treated with Utomilumab (a human monoclonal antibody agonist targeting the T cell co-stimulatory receptor CD137) combined with pembrolizumab, higher levels of activated memory/effector peripheral blood CD8 T cells are observed, leading to profound and durable anti-tumor activity and high safety [379]. Several ongoing clinical studies are exploring the combination of other co-stimulatory molecule agonists with PD-1/PD-L1 antibodies. Initial clinical data indicate that this therapy has superior anti-tumor efficacy, thus providing a solid scientific basis for subsequent cancer immunotherapy.
PD-1/PD-L1 inhibitors combined with targeted drugs
Targeted therapy can precisely target transcription regulatory factors to activate or inhibit key signaling pathways in cellular metabolism, it activates T cells to exert immune functions and inhibits tumor cell proliferation, thus inhibiting tumor disease progression at the level of cellular metabolism. Since PD-1/PD-L1 antibodies take longer to take effect, typically 2 to 3 months, combining PD-L1/PD1 immunotherapy with targeted therapy significantly improves therapeutic effects.
PD-1/PD-L1 inhibitors combined with anti-angiogenic drugs
Tumor growth depends on the formation of new blood vessels to ensure a continuous supply of oxygen and nutrients. Targeting the inhibition of tumor vasculature is primarily achieved by blocking the pathways of tumor angiogenesis, cutting off the nutrient supply to tumor cells and achieving a treatment for inhibiting tumor growth [380]. However, studies have found that inhibiting tumor angiogenesis may further aggravate vascular damage, making it difficult for drugs that kill cancer cells to reach the interior of the tumor. With the proposal and clinical application of the “vascular normalization” theory, new progress has been made in tumor treatment. The theory aims to induce structural and functional changes in tumor blood vessels by using anti-angiogenic drugs, rendering them more similar to normal blood vessels, thereby increasing blood flow and allowing cytotoxic drugs to more easily penetrate the tumors [381, 382]. Although vessel normalization delays tumor progression, the underlying regulatory mechanisms are poorly understood. Zhang’s team discovered that gene expression related to vascular normalization is linked to the activation of T-lymphocytes’ immune signaling pathways, helper T (Th) cells are crucial in vascular normalization and immune programming. In mice with defective vascular normalization, Th cell infiltration in tumors is reduced. When Th cells are activated using immune checkpoint blockade methods, the ability for vascular normalization is strengthened [383]. This suggests that there is mutual regulation between vascular normalization and the immune system. Huang et al. also discover that in mouse breast cancer models, using a low dose of vascular normalization agent, anti-VEGFR2 antibodies, can increase CD4+ and CD8+ T cell infiltration, reprogram the tumor microenvironment, and enhance the efficacy of immunotherapy [384]. Therefore, anti-angiogenesis therapy can not only suppress angiogenesis but also obstruct the accumulation of immunosuppressive cells through vascular normalization and promote the influx of effector T cells into tumors.
Currently, vascular endothelial growth factor (VEGF) is the major target of anti-angiogenic agents, which mainly sends signals through VEGFR2 and is critical for regulating the biological function of endothelial cells, vascular permeability, and angiogenesis [385]. Extensive clinical data indicate that the combination of PD-1/PD-L1 blockade with anti-angiogenic drugs significantly enhances the efficacy of PD-1/PD-L1 inhibitors and improves patient survival. Studies have found that anti-VEGF or VEGFR2 upregulates PD-L1 expression levels, related to the extent of CTL activity in tumors. Therefore, VEGF/VEGFR monoclonal antibodies or tyrosine kinase inhibitors make tumor cells more sensitive to PD-L1 inhibitors, combining them with PD-L1 inhibitors enhances the sensitivity and effectiveness of anti-angiogenic therapy [386]. Clinically, anti-angiogenic drugs are mainly divided into monoclonal antibodies targeting VEGF and VEGFR, and tyrosine kinase inhibitors (TKI) blocking the intracellular domain of VEGFR [387]. For example, patients with surgically unresectable HCC who received combination therapy with Bevacizumab (a VEGF antibody) and Atezolizumab (a PD-L1 antibody) have an objective response rate of 36% and a disease control rate of 71%, with a reduced risk of death [388]. This indicates that the combination therapy scheme can provide longer progression-free survival (PFS) than targeting PD-L1 alone. Subsequently, to compare this combination treatment with the current standard first-line therapy for HCC (sorafenib monotherapy), the Phase 3 IMbrave150 clinical trial (NCT03434379) is conducted. The data indicate that the overall survival rate with ATZ and BVZ combination therapy is 67.2% at 12 months, with a 42% reduction in the risk of death and significant improvements in OS and PFS compared to sorafenib treatment [389]. Therefore, combining atezolizumab and bevacizumab may improve the anti-tumor immune responses, providing more lasting clinical benefits for patients with hepatocellular carcinoma.
Moreover, Hiroto Kikuchi et al. find that when hepatocellular carcinoma is first treated with PD-1 inhibitors, subsequent administration of the anti-angiogenic agent sorafenib (a multi-kinase inhibitor with anti-VEGFR1-3 activity) can enhance the response of hepatocellular carcinoma to sorafenib. This enhancement is accompanied by an increase in CD8 T cell infiltration and the promotion of tumor vessel normalization, thereby inducing an effective anti-tumor immune response and increasing the survival rate of mice [390]. In a clinical phase 1b trial for advanced RCC patients, the combination of the VEGF receptor tyrosine kinase inhibitor Axitinib and the PD-1 monoclonal antibody Pembrolizumab yielded encouraging results. Out of the patients treated, 73% achieve objective responses, with 8% achieving complete remission, and more than 90% of patients exhibit tumor shrinkage, the median progression-free survival exceeds 20 months [391]. This indicates that the combined therapy of Axitinib and Pembrolizumab is safe and tolerable in the initial treatment of patients with advanced RCC, offering unprecedented anti-tumor activity. Subsequently, Rini et al. performed a phase III clinical trial for advanced RCC, enrolling a total of 861 patients. The findings reveal that compared to the standard targeted therapy drug Sunitinib for renal cancer, the combination therapy of Axitinib and Pembrolizumab achieves an objective remission rate of nearly 60%, with a maximum reduction of 47% in the risk of death [392]. This suggests that the combination therapy significantly prolongs OS and PFS, and achieves higher objective response rates, making it a potential first-line therapy for advanced RCC. In the same year, Motzer et al. conducted a comparison between PD-L1 antibody avelumab combined with Axitinib and Sunitinib monotherapy in 886 patients with advanced renal cancer. The results show that in 560 PD-L1 positive patients, the combination therapy significantly increased the objective response rates, there was little difference in side effects between the two groups during the treatment period [393]. Moreover, Ciccarese et al. conducted a meta-analysis and revealed that VEGFR-TKIs in combination with ICIs significantly improved the PFS of patients with mRCC [394].
In summary, these studies suggest that the development of combination therapies involving ICIs and anti-angiogenic drugs holds promising potential as a novel approach, enhancing the anti-tumor immune response in cancer patients. In the future, further analysis of its efficacy and safety should be conducted, with the goal of translating these findings into clinical practice to improve the immunotherapy outcomes of cancer patients.
PD-1/PD-L1 inhibitors combined with EGFR-TKIs therapy
The Epidermal Growth Factor Receptor (EGFR) on the cell surface is phosphorylated to form a dimer after binding to its ligand, activating downstream pathways like RAS/RAF/MEK/MAPK and PI3K/AKT, subsequently causing tumor cell proliferation, invasion, and apoptosis [395]. EGFR tyrosine kinase inhibitors (EGFR-TKIs) are effective targeted drugs for NSCLC patients with EGFR mutations, acting as analogs of adenosine triphosphate (ATP) that competitively bind to the intracellular EGFR tyrosine kinase site, thereby blocking the activation of downstream pathways and inhibiting tumor growth. However, TKI-targeted therapy also promotes the occurrence of resistance in patients, primarily related to EGFR gene amplification or secondary mutations, and may even lead to hyper-progressive disease [396].
Recent research has shown that in NSCLC with EGFR mutations, EGFR-TKIs regulate PD-1 and PD-L1 expression by inhibiting downstream signaling. In NSCLC patients, activated EGFR signaling upregulates PD-L1 expression by phosphorylating ERK1/2 and c-Jun. Simultaneously, PD-L1 induces the apoptosis of T lymphocytes in the TME through the PD-L1/PD-1 axis, thereby mediating tumor immune evasion. If using EGFR-TKI to inhibit EGFR, it can suppress PD-L1 expression and reactivate anti-tumor immunity in T cells [397]. In a mouse model of EGFR-mutated lung adenocarcinoma, which has a non-inflammatory immunosuppressive tumor microenvironment, EGFR signaling recruits CD4+ regulatory T cells and CD8+ T cell infiltration by activating JNK/cJun and inhibiting IRF1. Treatment with erlotinib (EGFR-TKIs) in combination with anti-PD-1 mAb has better antitumor efficacy than monotherapy [398]. However, existing clinical research indicates that NSCLC patients with EGFR mutations have a poor response to EGFR-TKIs combined with PD-1/PD-L1 inhibitor immunotherapy, the toxicity reaction significantly increases. For instance, in a phase I clinical trial of 21 advanced EGFR-mutated NSCLC patients, the ORR for Nivolumab combined with Erlotinib (the first generation of EGFR-TKIs) is 15%, and the 24-week PFS rate is 48%. Although this combined therapy shows some preliminary efficacy, the incidence of AEs is also higher [399]. Similarly, in a phase Ib clinical trial of Osimertinib (a third-generation EGFR TKI) combined with the PD-L1 monoclonal antibody durvalumab for the therapy of NSCLC patients with EGFR mutations, the objective remission rate in the Osimertinib monotherapy group is 80%, but only 64% in the combination therapy group. The incidence of interstitial lung disease increases in patients receiving combination therapy, leading to the early termination of the clinical trial [400]. In addition, Su et al. find that EGFR-TKI primary-resistant patients have higher PD-L1 expression compared to those with acquired resistance, resulting in a significant reduction in the objective remission rate and shorting the progression-free survival [401]. This indicates that EGFR-mutated NSCLC patients may have a limited response to EGFR-TKI treatment. Therefore, the safety of the combination therapy of PD-1/PD-L1 inhibitors and EGFR-TKI therapy in NSCLC patients remains unclear, possibly related to PD-L1 expression, tumor mutation burden (TMB), and potential differences in different tumors. Hence, a cautious approach is required when selecting treatment options.
At present, the combination therapy of TKIs and immune checkpoint inhibitors is being studied in various clinical trials, but the results of these trials are not yet mature. Future research should focus on exploring the mechanisms of combination therapy, optimizing drug sequence and dosage, enhancing the research on overcoming TKI treatment resistance, and minimizing the adverse reactions related to tumor immunotherapy, aiming to maximize the benefits of TKIs combined with ICI in clinical treatment.
PD-1/PD-L1 inhibitors combined with PARP kinase inhibitors
PARPs are a family of proteins involved in DNA damage repair, maintenance of genomic stability, and promotion of apoptosis in tumor cells. Inhibition of PARP may result in the accumulation of DNA damage and double-stranded DNA breaks, thereby increasing tumor mutation and neoantigen load. PARP plays an important role in tumor immunity, DNA damage caused by PARP inhibitors (PARPi) upregulates PD-L1 expression through the cGAS/STING pathway, inhibiting the functions of effector T cells. Alternatively, it promotes inflammatory responses by altering the extrinsic immunogenicity of the tumor. When combined with immune checkpoint inhibitors, PARPi regulates T cell activity in the immune microenvironment, thus activating anti-tumor immune responses [402].
Recently, researchers discovered that PARPi acts as immune modulators in tumor therapy, enhancing the efficacy of immune checkpoint inhibitors. In BRCA1 gene-deficient breast cancer, PARPi also upregulates PD-L1 expression on tumor cell surfaces by inactivating GSK3β, thereby inhibiting the activation of tumor-infiltrating T lymphocytes. Combining PARPi and PD-L1 inhibitors can reactivate anti-tumor immunity [283]. In BRCA1 gene-deficient ovarian cancer, PARPi upregulates PD-L1 expression by promoting the phosphorylation of CHK1 (a cell cycle checkpoint kinase), which is associated with reduced infiltration of CTL. The use of PD-L1 blockers reverses the inhibitory efficacy of PARPi on CD8 T cells, thereby enhancing the antitumor effects of PARPi [403], which also provides a theoretical basis for the combination therapy of PARPi and PD-L1 antibodies in ovarian cancer. Since the accumulation of DNA damage caused by PARPi, there has been an enhancement of the antitumor activity of the PD-L1 antibody by increased expression of new antigens and immune recognition of the tumor. Therefore, it is not difficult to find that cancer patients carrying BRCA gene mutations have specific DNA repair deficiencies and are more sensitive to PARP inhibitors that impede DNA repair. When combined with ICI inhibitors, the anti-tumor effect is better. More signaling regulatory mechanisms need to be further studied.
In the TOPACIO/KEYNOTE-162 trial of combined PARPi and ICI, researchers analyzed the efficacy of Niraparib combined with Pembrolizumab in recurrent ovarian cancer and metastatic TNBC [404]. The results show that in 60 patients with stage 1 and stage 2 ovarian cancer, the ORR is 18% and the DCR is 65%. Among them, 3 cases achieve a complete remission, 8 cases have a partial remission, and 28 cases are stable. Subsequent MEDIOLA clinical trial analysis shows that PARP inhibitor Olaparib combined with PD-L1 inhibitor durvalumab has good efficacy in metastatic breast cancer with BRCA1/2 mutations, with a response rate of 52% and a disease control rate of 80% [405]. Therefore, this combined therapy has shown promising anti-tumor activity in tumor therapy and also lays the foundation for further large-sample randomized controlled trials. Currently, the clinical application of PARPi in combination with ICIs for tumor treatment is still in its infancy, and the development of PARP inhibitors is becoming a hotspot in the field of tumor immunity.
PD-1/PD-L1 inhibitors combined with targeted receptor agonists
IL-2, a type I cytokine, is the earliest approved for cancer therapy because of its significant and long-lasting efficacy, but the high toxicity of IL-2 at high-dose administration limits its use. NKTR-214 is a selective agonist targeting the IL-2R dimer CD122, it exhibits favorable pharmacological effects with relatively low toxicity [406]. Therefore, it has been used in combination with ICIs in recent years. Studies have shown that after patients receive NKTR-214 treatment, the number of CD8+ T cells and NK cells in tumors increases linearly, and the expression of PD-1 and PD-L1 on the cell surface increases [407], suggesting a potential synergistic mechanism between NKTR-214 and PD-1 inhibitors. Currently, results from phase I/II clinical trials combining nKTR-214 with nivolumab have shown a total ORR of 59.5% in 38 patients with specific immunotherapy-naive advanced solid tumors, including 7 complete remissions (18.9%), with good tolerability [408]. Therefore, targeting IL-2 receptor agonists combined with ICI therapy serves as a dual immunotherapy approach for a range of advanced solid tumors.
Toll-like receptors (TLRs) are pattern recognition receptors of the innate immune system, recently been considered key factors in the pathogenesis of tumors. TLR signaling not only affects immune cell activation, maturation and function, but also influences tumor metabolism and metastasis [409]. TLR agonists have been proven to enhance the efficacy of ICIs by increasing the proportion of tumor-specific cytotoxic T cells in preclinical models. For example, in a phase Ib clinical trial for advanced melanoma patients who are unresponsive or have failed PD-1 monotherapy, the TLR9 agonist Vidutolimod induces and attracts anti-tumor T cells by triggering a strong IFN response, thereby reversing PD-1 blockade resistance. When Vidutolimod is combined with pembrolizumab, durable responses and tumor regression are observed in 25% of patients, with controllable safety [410]. Other TLR agonists have also carried out phase I clinical trials combined with PD-1/PD-L1 inhibitors, but specific clinical data have not yet been reported.
Besides the targeted TLR and IL-2 receptor agonist therapies mentioned above, there are ongoing developments in new combinations and targets to enhance the efficacy of ICIs in patients. For example, inhibitors of the adenosine pathway, anti-HER2 agents in gastric cancer, BTK inhibitors, histone deacetylase (HDAC) inhibitors, and demethylating agents are being explored. These targeted therapeutic approaches provide a new way for ICI treatment, but clinical application still needs further study.
PD-1/PD-L1 inhibitors combined with radiotherapy and chemotherapy
Radiation therapy directly interferes with a patient’s primary tumor, transforming it into situ vaccine. This process recruits more T lymphocytes to infiltrate the tumor lesion and releases immunogenic factors (such as TNF, IL-1, HMGB1, etc.) to indirectly stimulate the immune system, exerting an immunological killing effect [411]. Besides the tumor lesions irradiated by radiation therapy, distant non-irradiated metastatic lesions also shrink, this radiation-induced abscopal effect is likely related to the activation of systemic immune responses. It appears with a relatively higher probability in radiation therapy combined with immunotherapy, significantly increasing patients’ survival [412]. For instance, Yan Lan et al. find that a bifunctional fusion protein, bintrafusp alfa (BA), that simultaneously inhibits TGF-β and PD-L1, effectively synergizes with radiation therapy. The combination of BA+ and radiotherapy (BART) increases tumor-infiltrating leukocytes, overcoming tumor immune escape. It also reduces radiation-induced pulmonary fibrosis, ultimately eradicating cancer lesions while preserving normal tissues [413]. In malignant pleural effusion models, radiation tumor cell-released microparticles (RT-MPs) can precisely target M2-TAMs in the TME and convert them into M1-TAMs by activating the JAK-STAT and MAPK pathways, facilitating an anti-tumor immune response. The internalization of RT-MP also significantly enhances PD-L1 expression in TAMs, RT-MP combined with anti-PD-1 therapy shows good biocompatibility and elicits memory immune responses [414]. Combining nanoparticle-mediated immune radiotherapy with dual blockade of LAG3 and TIGIT can enhance the treatment effect of anti-PD-1 resistant lung cancer [415].
In clinical research, radiation therapy has demonstrated synergistic effects with various immunotherapies. Combining radiation therapy with PD-1/PD-L1 blockade therapy can activate tumor-specific T cells in the TME and reduce the accumulation of MDSCs and Tregs, thereby enhancing anti-tumor immunity [416]. A phase II trial reports that radiation therapy improves the response of MSS CRC and PDAC patients to ipilimumab and nivolumab immunotherapy [417]. Theelen et al.’s retrospective analysis found that patients with metastatic NSCLC benefit from the abscopal effect of radiotherapy in combination with Pembrolizumab immunotherapy, with a favorable clinical prognosis. The combination of PD-1 monoclonal antibody and radiation therapy (iRT) shows a significantly higher abscopal response rate (ARR) than the PD-1 monoclonal antibody treatment group (41.7% vs. 19.7%). For non-irradiated lesions, radiation therapy at least doubles the efficacy of PD-1 monoclonal antibody, significantly improving the treatment outcome and extending the patients’ survival [418]. These studies have also propelled the conduct of a randomized open-phase III trial of Nivolumab combined with radiation therapy for treating glioblastoma [419].
Chemotherapy mainly kills cancer cells by targeting their DNA synthesis and replication processes. Preclinical studies have shown that chemotherapy promotes the infiltration of T cells into tumor tissues and decreases the number of immunosuppressive cells, thus enhancing the sensitivity of tumor cells to immune checkpoint blockade therapy [420]. Therefore, the appropriate combination of chemotherapy drugs with PD-1/PD-L1 inhibitors enhances the effects of anti-PD-1/PD-L1 therapy and significantly improves overall survival in patients. For example, In clinical randomized trials, the combination of Pembrolizumab with chemotherapy can significantly improve the OS for patients with metastatic triple-negative breast cancer [421] and metastatic squamous NSCLC [422], and also prolong PFS. Similarly, in patients with advanced NSCLC and PD-L1 expression ≥ 50%, the combination of first-line cemiplimab (a PD-1 inhibitor) and chemotherapy significantly improves OS and PFS [423]. These findings indicate that adding PD-1/PD-L1 inhibitors to standard chemotherapy is important in the first-line treatment of various solid tumors. However, chemotherapy drugs not only kill tumor cells but often have serious side effects. Common side effects include bone marrow suppression, which leads to decreased white blood cells, red blood cells, and platelets. Severe bone marrow suppression results in extremely low immunity and coagulation disorders, early symptoms may not be apparent. If not promptly detected and corrected in time, it can lead to severe infection and vital organ bleeding, and other fatal complications [424]. Therefore, when choosing chemotherapy, patients need an experienced physician to develop reasonable plans, closely observe adverse reactions during chemotherapy, timely monitor routine blood tests and pay attention to other symptoms related to chemotherapy. If discomfort is found after chemotherapy, communicate with the attending physician in time and take relevant treatment measures.
In summary, more and more clinical trials suggest that when combining PD-1/PD-L1 inhibitors with different radiotherapy/chemotherapy protocols, the impact of different dosage regimens in terms of efficacy and prognosis is quite different. Therefore, the exploration of larger sample sizes is required to extensively investigate the underlying mechanisms behind the differential treatment outcomes among different groups, and to validate the feasibility of combined therapeutic approaches involving chemotherapy and PD-1 antibodies.
Photodynamic therapy (PDT) combined with PD-1/PD-L1 inhibitors for anti-tumor therapy
Photodynamic therapy (PDT) is a novel adjuvant tumor therapy based on photosensitizers, oxygen molecules and light sources. When the photosensitizer is absorbed and metabolized by the target tissue, cell-toxic singlet oxygen (1O2) and reactive oxygen species (ROS) are generated through a series of photochemical reactions under the stimulation of an appropriate light source in the presence of oxygen. These reactive species kill the tumor tissue either by directly causing cell death through triggering signal cascade reactions, thereby achieving therapeutic purposes [425].
In recent years, researchers have combined immune checkpoint inhibitors (ICI) with PDT for adjuvant anti-tumor therapy, achieving preliminary success in animal models of breast cancer, colon cancer, renal cancer, lung cancer, and skin cancer [426]. For example, small-molecule nano-inhibitor targeting the PD-1/PD-L1 axis (BMS202) mediates ICI and PDT synergistically in the therapy of breast cancer, leading to primary tumor regression and reducing the occurrence of lung metastasis [427]. Hwang et al. use a combination of PDT and flagellin-assisted tumor-specific peptide vaccine (FlaB-Vax) in treating B16-F10 melanoma mouse models that had undergone PD-1 blockade therapy. This combination effectively induces systemic anti-tumor immune responses, elevating CD8+ T cell infiltration and systemic IFN-γ secretion, enhancing the accumulation of effector memory CD8+ T lymphocytes, further strengthening the efficacy of PD-1 blockade therapy [428]. Yuan et al. find that the multifunctional mTHPC@VeC/T-RGD NPs platform (a multifunctional nanoparticle loaded with photosensitive mTHPC) mediated PDT treatment enhances the anti-tumor efficacy of PD-L1 inhibitors in colorectal cancer therapy. Under 660 nm near-infrared (NIR) laser irradiation, mTHPC@VeC/T-RGD NPs can kill tumor cells by inducing apoptosis and stimulating a systemic immune response, while PD-L1 blockade further suppresses tumor growth, metastasis, and forms long-term host immune memory to prevent tumor recurrence [429]. Peng Huang’s team developed a biomimetic nanoemulsion expressing PD-1 on its cell membrane, which is used for synergetic photodynamic immunotherapy against hypoxic breast cancer, the treatments inhibit tumor growth by promoting dendritic cell maturation and tumor infiltration by cytotoxic T lymphocytes. Additionally, the co-delivering photosensitizer with PD-1 protein using this nanoemulsion achieved synergistic effects of PDT and immunotherapy [430].
Therefore, PDT possesses advantages such as precise targeting, repeatable treatment, and low toxic side effects, making it a good clinical application prospect for combination therapy with ICI. However, due to the limited penetration depth of traditional PDT light sources, it is more suitable for treating superficial tumors and less effective for deep-seated tumors. At present, there are few clinical studies combining ICI and PDT, with only a few case reports and one ongoing clinical trial. In the future, how to further improve the selectivity and specificity of photosensitizers to target tumor therapy, enhance the penetration depth of PDT, and develop new technologies and methods that combine nanophotonic technology with biomedical applications for diagnosis and treatment will be the new directions for PDT development. With the advancement of technology, PDT technology will make greater contributions to human health.
Aptamer drug conjugates (ApDC) combined with PD-1/PD-L1 inhibitor for anti-tumor therapy
Aptamer drug conjugates(ApDC)are highly promising novel immunotherapies. Studies have shown that ApDC provides immune modulators to restrict immune co-stimulation to the tumor area and also induces new antigens within the tumor, thereby blocking immune checkpoints and activating functional immune cells, prolonging anti-tumor immunity [431]. With the development of cancer immunotherapy, therapeutic aptamers have been reported for targeting immune checkpoints, including CTLA-4, PD-1, and PD-L1. For example, self-assembling nanoparticles of multivalent aptamer-drug conjugates (ApMDC) enhance the anti-tumor responses of anti-PD-1 immunotherapy [432]. Engineered multifunctional aptamers (P1/C4-bi-apt) improve anti-tumor immune responses by blocking the dual immune checkpoints of CTLA-4 and PD-L1 [433]. Additionally, DNA aptamers targeting PD-1/PD-L1 have been developed to disrupt the immunosuppressive interaction between the immune-inhibitory PD-1 and PD-L1 molecules. Prodeus et al. discover that MP7 (a short synthetic anti-PD-1 DNA aptamer) specifically binds to the extracellular domain of PD-1, competitively antagonizes the direct binding between PD-1 and PD-L1 molecules, functionally blocking PD-L1-mediated immune suppression signals and ultimately inhibiting the growth of colon cancer [434]. LAI et al. discover that aptPD-L1 (a PD-L1 antagonistic DNA aptamer) specifically binds to PD-L1, thereby blocking the interaction between PD-1 and PD-L1. In vitro experiments show that aptPD-L1 promotes lymphocyte proliferation and inhibits tumor growth in vivo, it helps in restoring T cell function and altering the TME without causing hepatorenal toxicity [435]. Li Wenzhe et al. develop a metal-organic framework nanoparticle (M@O-A) based on the PD-L1 nucleic acid aptamer (aptPD-L1) to achieve efficient and low-toxic tumor photo-immunotherapy [436]. It first uses aptPD-L1 to block the immune suppressive function of PD-L1 protein on the surface of tumor cells, then under near-infrared light irradiation, the nanosystem can perform PDT simultaneously, thus inducing immunogenic cell death (ICD) in tumor cells, synergistically activating the immune system with potent tumor cell killing capability and lower immune-related toxicity.
These aptamers, due to their similar tumor inhibitory efficacy to antibodies and easier synthesis compared to antibodies, have advantages such as low immunogenicity, low risk of inducing autoimmune toxicity, high targeting specificity, and low toxicity. Therefore, they serve as alternative therapeutic agents for cancer immunotherapy. Even though nucleic acid aptamers have made numerous advances in the field of tumor immunotherapy research, applying them to clinical drug treatments still faces many challenges. For example, the screening process of nucleic acid aptamers is often long and nucleic acid aptamers usually require appropriate chemical modification to exert their anti-tumor effects. Further preclinical studies are needed to address issues such as the stability of nucleic acid aptamers in the bloodstream, the targeting efficiency of drug delivery systems, and the metabolism and clearance pathways of drugs in the body.
PD-1/PD-L1 inhibitors combined with immune cell therapy
Adoptive cell therapy (ACT) involves the infusion of T cells derived from TILs, or T cells that have been genetically engineered to express tumor-recognizing receptors into the body, allowing them to directly exert anti-tumor effects and promote the host’s anti-tumor immune responses [437]. Preclinical studies on ACT using genetically engineered T cell receptors (TCRs) and chimeric antigen receptors (CARs) have achieved promising results, some T cell therapies have received regulatory approval for the therapy of patients with B cell malignancies.
Research has found that combining PD-1/PD-L1 blockade antibodies with ACT increases the speed of tumor therapy responses. For example, in NSCLC, combining anti-PD-1/PD-L1 with CAR-T cell therapy promotes the restoration of normal immune recognition function and maintains immune system homeostasis [438]. Phase 1 clinical trial results of TIL adoptive cell therapy combined with Nivolumab (NCT03215810) show that among 13 patients, 3 confirmed responses, 11 have reduced tumor burden, including 2 who achieved complete remission after 1.5 years, effectively preventing subsequent relapse associated with PD-L1 expression [439]. Therefore, TIL mediates an effective response in tumors that are insensitive to traditional ICIs. With the advancement of ACT cell immunotherapy technology, researchers are considering whether, in addition to using PD-1 or PD-L1 inhibitors, CAR-T cells secreting PD-1 antibodies can be used to treat recurrent solid tumors. Fang et al. first report that patients with advanced refractory ovarian cancer who received autologous PD-1-meso CAR-T cells (secreting full-length PD-1 antibodies) treatment have expanded CAR-T cell numbers in the bloodstream, and their own CD8+ T cell activity is also significantly enhanced, indicating that CAR-T cells infused back into the human body can mobilize immune cells to target tumor cells efficiently and precisely, rapidly improving the tumor local microenvironment’s immune function to kill cancer cells without causing significant adverse reactions [440]. In addition, Lu’s team conducted the first phase I clinical trial to inject CRISPR/Cas9 edited PD-1 knockout T cells into patients with advanced NSCLC. The results show that edited T cells are detected in peripheral blood, with a median PFS of 7.7 weeks and a median OS of 42.6 weeks, with 2 patients experiencing stable disease. This research demonstrates that the clinical application of CRISPR/Cas9 knocked-out PD-1 T cells is safe and feasible, but lacks significant efficacy in NSCLC patients [441]. Similarly, the Stadtmauer team uses CRISPR technology to genetically modify T cells in 3 patients with advanced cancer, they delete three genes, TCRα, TCRβ, and PD-1, to enhance the functions of T cells. They also introduce a synthetic cancer-specific TCR transgenic (NY-ESO-1) to recognize tumor cells, and finally the multiple-engineered T cells are reinfused into the patients. Studies show that all three patients tolerated the treatment well, these genetically edited T cells survive for up to 9 months in the patients, exhibiting the ability to consistently attack and kill tumors with few treatment-related serious adverse reactions [442]. Therefore, cell therapy that uses CRISPR-Cas9 technology to specifically knockout PD-1 in CART-T cells or T cells to improve the anti-tumor activity of effector T cells and overcome drug resistance in advanced metastatic tumors has been clinically proven to be safe and effective. In the future, new strategies should be developed to avoid the off-target risk of CRISPR/Cas9 gene editing. Meanwhile, the CRISPR/Cas9 system, as an emerging technology in cell therapy, is expected to achieve greater breakthroughs in combination therapy with PD-1/PD-L1 in the field of tumor immunotherapy.
PD-1/PD-L1 inhibitors combined with oncolytic virus therapy
Oncolytic virus therapy is a series of immune cascade reactions that directly kill tumor cells by injecting oncolytic viruses (OVs) into tumors and causing systemic anti-tumor immune responses, which promote the antitumor responses of tumor-specific T cells [443]. Currently, various natural and transgenic viruses have been developed as oncolytic agents. OVs improve the therapeutic responses to PD-1 therapy by reversing immune suppressive factors, increasing the number and diversity of infiltrating lymphocytes and promoting PD-L1 expression in tumors [444]. For example, Hamdan et al. find that the oncolytic adenovirus Ad-Cab secretes a cross-hybrid Fc fusion peptide targeting PD-L1, thereby activating the Fc effector mechanisms of IgA1 (neutrophil activation) and IgG1 (natural killer and complement activation). Compared to the conventional PD-L1 inhibitor Atezolizumab, Ad-Cab demonstrates higher anti-tumor activity and activates multiple immune effector cell populations, thereby enhancing tumor killing capacity in vivo or in vitro, and in patient-derived tumor organoids [445]. Kiyokawa et al. find that treating mice with GBM using ICOVIR17 (an oncolytic adenovirus expressing hyaluronidase) increases CD8+ T-cell and macrophage infiltration and upregulates PD-L1 expression. Combining ICOVIR17 with PD-1 blockade antibodies induces tumor-associated pro-inflammatory macrophages and tumor-specific T cell cytotoxicity locally and systemically, enhancing tumor killing and further extending the survival of GBM mice [446]. Hence, OVs combined with PD-1/PD-L1 inhibitors may be beneficial to patients who have a poor response to immune checkpoint inhibition.
PD-1/PD-L1 inhibitors combined with cancer vaccines
Cancer vaccines work by enhancing immune recognition by using genetically modified genes or new antigens, enabling stronger immune responses to kill cancer cells and preventing their growth and recurrence. Compared to chemotherapy and radiotherapy, cancer vaccines usually have minimal side effects [447]. In recent years, there has been extensive research on personalized cancer vaccines for cancer treatment. For example, FixVac (BNT111), a nanoparticulate liposomal RNA (RNA-LPX) vaccine for intravenous administration developed by Ugur Sahin et al., has shown that it induces PD-1+ effector memory T cells in Phase I clinical trials for patients with advanced melanoma. Some patients who received FixVac after failing anti-PD-1 therapy exhibit tumor regression. The combination of FixVac and anti-PD-1 treatment is particularly effective for patients with a lower mutation burden, with a tumor regression rate exceeding 35% [448]. Additionally, a clinical phase I/II trial shows that Nivolumab combined with IO102/IO103 (an immune regulatory vaccine targeting IDO and PD-L1) achieves a high ORR of 80% in metastatic melanoma patients, with 43% achieving complete remission, this combination significantly decreases the tumor burden and extends PFS to 26 months [449]. Therefore, tumor vaccination in conjunction with checkpoint inhibition may be an effective immunotherapy regimen.
In summary, combination therapy offers more advantages, can also reduce the toxicity of anti-PD-1/PD-L1 immunotherapy and the possibility of acquired drug resistance. However, the long-term impact of chemotherapy on long-term immune cells is still unknown due to the insufficient number of clinical patient samples. As clinical studies evaluating combined immunotherapies rapidly increase, it is crucial to comprehensively understand the molecular mechanisms of anti-PD-1/PD-L1 therapies and identify more valuable targets to enhance the effectiveness of immunotherapy, broaden the beneficiary population, and further improve immune efficacy.
PD-1/PD-L1-mediated immune resistance and side effects
In recent years, although immune checkpoint blockade therapy targeting the PD-1/PD-L1 axis has shown significant therapeutic effects in the treatment of multiple malignancies, the response rate of anti-PD-1/PD-L1 mAb in the overall patient population is far from satisfactory. A large portion of patients do not respond, one of the major obstacles to the ICI therapy found in clinical use is that most patients develop resistance and relapse in the later stages of treatment [450]. Due to the high incidence of primary and acquired drug resistance, most patients cannot benefit from these therapies. Moreover, the complex resistance mechanisms of tumor cells to immunotherapy impact the clinical prognosis of patients [451]. Studies have found that the drug resistance to the PD-1/PD-L1 monoclonal antibody may be associated with the following factors, the intrinsic immune evasion genetic pathways in tumor cells, tumor antigen mutations and presentation processes, interactions among multiple immune checkpoints, suppressive tumor microenvironment (tumor intrinsic factors, inflammatory environment), oncogenic pathways in tumors, epigenetic changes in key tumor proteins, accumulation of metabolites, etc [452]. Therefore, it is urgent to understand the mechanisms of resistance to PD-1/PD-L1 inhibitors in immunotherapy, to discover universal biomarkers across multiple tumors, to develop combination therapies to sensitize resistant patients, and to find new strategies to enhance the efficacy of ICIs.
Among various types of cancer, although PD-1/PD-L1 blockade has clinical benefits, most patients do not experience durable or even curative remission after treatment and eventually relapse. The reasons for insufficient tumor immunotherapy response may be due to the heterogeneity of PD-1/PD-L1 expression levels in different tumor patients, low tumor mutation burden, and low tumor immunogenicity. Although the expression of PD-1/PD-L1 is crucial during ICI treatment, targeting PD-1 with antibodies disrupts PD-1 signaling on all cells, not just tumor-specific T cells, which may induce an imbalance in the immune system and cause collateral damage. Furthermore, studies have detected the same TCR sequences in both tumor and normal tissues [453], suggesting that during ICI treatment, T cells may infiltrate both sites, leading to indiscriminate immune attacks on systemic tissues during ICI treatment, thereby producing immune-related side effects and ultimately leading to treatment failure in cancer patients.
PD-1/PD-L1 immunotherapy is often accompanied by life-threatening immune-related adverse events (irAEs), irAEs induced by immunotherapy are usually insidious in onset, lack specificity, and have a broad toxic spectrum [454]. Physicians need to enhance the management of irAEs in terms of prevention, examination, and treatment to effectively control the diseases. Compared to conventional treatment, the main side effects caused by anti-PD-1/PD-L1 treatment include Atezolizumab causing hyperthyroidism, nausea, etc. Nivolumab causes endocrine toxicity and cardiotoxicity, etc. Pembrolizumab causes arthralgia, fatigue, hepatotoxicity, etc. Ipilimumab causes skin and gastrointestinal toxicity, etc. Tremelimumab causes rash, diarrhea, fatigue, etc [455]. Taking cardiotoxicity and myositis in irAEs as examples, Johnson et al. report fatal myocarditis in two melanoma patients after combination treatment with nivolumab and ipilimumab. Both patients developed myocarditis with rhabdomyolysis, cardiac electrical instability, and massive infiltration of T cells and macrophages. The severity of myocarditis is also more severe than monotherapy [456]. Subsequently, Wei et al. identify a preclinical mouse model of ICI-related myocarditis (Ctla4+/−Pdcd1−/−mice) induced by this combination therapy, and prove that intervention treatment with Abatacept (a recombinant soluble fusion protein of CTLA-4) inhibits T cell co-stimulation and improves disease progression [457]. In terms of early intervention, the latest study by the Joe-Elie SALEM team found that early management measures for ICI myocarditis patients, such as mechanical ventilation, high-dose Abatacept and Ruxolitinib (a JAK1/JAK2 inhibitor) treatment combined with CD86 RO (CD86 receptor occupancy on circulating monocytes) monitoring, hold promise for reducing the high mortality rate caused by ICI-induced myocarditis, but further research is still needed to evaluate the optimal drug combination and dosage [458]. Therefore, further research might be required to investigate the mechanisms of these checkpoint therapies to improve these toxicities, effective and accurate diagnosis and therapeutic approaches should be sought to reduce the severity of complications.
Conclusion
The clinical application of immune checkpoint inhibitors (ICI) targeting PD-1/PD-L1 has completely transformed traditional cancer treatment. With the successful clinical trials of novel ICB drugs and combination therapies, as well as innovative breakthroughs in mRNA vaccines and nanomaterial delivery system technologies, ICIs show great promise in cancer therapy. Despite the significant effects of PD-1/PD-L1 tumor immunotherapy in prolonging patient survival and improving the prognosis of advanced cancer patients, the development of PD-1/PD-L antibodies in tumor immunity will also face more challenges, such as resistance developed from long-term use of PD-1 or PD-L1 inhibitors, how to improve drug utilization, and how to select the best combination therapy. In addition, PD-1/PD-L1 blockade is limited by the low response rates in certain cancers, the lack of known biomarkers, immune-related toxicity, and inherent and acquired resistance. Therefore, in basic mechanism research, PD-1/PD-L1 expression levels, IFN-γ, CD8+ T cell infiltration, and tumor mutational burden (TMB) are the most widely used biomarkers. In the future, we will explore new mechanisms of targeting PD-1 or PD-L1 and new directions in combination therapy, jointly targeting the adaptive stress pathways related to metabolism, oxidation, and DNA damage repair in the tumor, increasing the abundance of tumor immune cells, to provide options for improving response rates and preventing drug resistance. We also attempt to change the local tumor microenvironment, neutralize immune suppressive factors within the TME, block immune suppressive responses, restore the immune system’s homeostasis, and reactivate T cells’ immunity and the patient’s autoimmunity against cancer.
Beyond the conclusions discussed in this review, the regulatory differences in immune systems among species, the selection of preclinical animal models, the conduct of clinical trials, the clinical use of approved ICB drugs, the optimization of combination therapy regimens, and the individualization of immunotherapy based on the specificity of patients and the high heterogeneity of tumors are all important directions to consider. In clinical applications, when different clinical factors (such as age, gender, and obesity) are involved, patients’ responses to drug treatment may slightly vary. There is still a lack of optimal in vitro and in vivo experimental models for assessing the efficacy, immune mechanisms, and drug toxicity of new immunotherapies. Therefore, developing 3D co-culture models of tumor cells and immune cells or patient-derived xenografts (PDXs) and organoid model technologies represents the future of precision treatment, which is important for accelerating the development, screening, clinical translation, and evaluation of targeted therapies for the next generation of ICB drugs. Due to the complex dynamic equilibrium in the immune microenvironment and the heterogeneity of tumors in each patient, personalized immunotherapies need to be investigated in the future. The emergence of single-cell multi-omics studies and innovative high-throughput sequencing technologies opens new avenues towards personalized patient treatment. For example, in heterogeneous diseases such as bladder cancer, multi-omics sequencing and analysis may help provide stronger biomarkers for predicting chemosensitivity [459]. Gene expression models established based on multi-omics sequencing data can be used to screen specific patient groups that may respond well to cytotoxic drugs, achieving precise targeted therapy. We believe that in the future, the latest advancements in artificial intelligence-based algorithms and models will soon be able to predict responses to immunotherapy and combination therapy at a personalized level. Undeniably, since PD-1/PD-L1 inhibitors are still expensive drugs in many countries, adopting combination therapy regimens will impose a significant financial burden on patients. Hence, developing effective predictive biomarkers to select patients who may benefit from ICI treatment, optimizing combination therapy regimens (including drug dosages and usage sequences, etc.), optimizing pharmaceutical processes, reducing drug prices, and preventing patients from incurring huge healthcare costs due to over-treatment, is a key issue for improving people’s livelihoods.
In summary, the regulation of the PD-1/PD-L1 pathway in tumor immunity is complex and fascinating. A comprehensive understanding of the metabolic mechanisms behind immune escape, the accurate and reliable identification of predictive biomarkers, and therapeutic targets for the response of different cancers to immune checkpoint inhibitors are necessary to improve immunotherapy for tumors. Notably, during PD-1/PD-L1 therapy, the immune system may be in an over-activated state, damaging organ systems and even causing immune-related adverse events such as myocarditis, making it imperative to comprehensively monitor the immune system and prevent systemic diseases in a timely manner. It is believed that with continued research, the role and resistance mechanisms of the PD-1/PD-L1 pathway in the regulation of the body’s immunity and tumor immunotherapy will be thoroughly elucidated. This will provide new strategies for combined immunotherapy in the clinical treatment of solid tumors, increase the personalization of cancer treatment, and benefit more patients.
Abbreviations
- APC:
-
Antigen-presenting cells
- ACT:
-
Adoptive cell therapy
- ApDC:
-
Aptamer drug conjugates
- ALK:
-
Anaplastic Lymphoma Kinase
- BRD4:
-
Bromodomain-containing protein 4
- BET:
-
Bromodomain and extra-terminal domain
- CTC:
-
Circulating tumor cell
- CLL:
-
Chronic lymphocytic leukemia
- CBR:
-
Clinical benefit response
- CNV:
-
Copy number variation
- ceRNAs:
-
Competitive endogenous RNAs
- circRNAs:
-
Circular RNAs
- CMTM6:
-
CKLF-like MARVEL transmembrane domain containing 6
- CSCC:
-
Cutaneous squamous cell carcinoma
- CDK:
-
Cyclin-dependent kinase
- CAR:
-
Chimeric antigen receptor
- DLBCL:
-
Diffuse large B-cell lymphoma
- DC:
-
Dendritic cell
- DHHC3:
-
Aspartate-histidine-histidine-cysteine 3
- DHHC9:
-
Aspartate-histidine-histidine-cysteine 9
- DCR:
-
Disease control rate
- EAE:
-
Experimental autoimmune encephalomyelitis
- EMT:
-
Epithelial-mesenchymal transition
- EDMC:
-
Erythroid-Derived Myeloid Cells
- EGF:
-
Epidermal growth factor
- ER:
-
Endoplasmic reticulum
- ERAD:
-
Endoplasmic reticulum associated degradation
- FAO:
-
Fatty acid oxidation
- FTO:
-
Obesity-associated protein
- GVHR:
-
Graft-versus-host reaction
- GADA:
-
Glutamic acid decarboxylase antibody
- GC:
-
Gastric cancer
- GSDMC:
-
Gasdermin C
- GSK3β:
-
Glycogen synthase kinase 3β
- HLA/APM:
-
Antigen-presenting machinery elements
- HVGR:
-
Host versus graft reaction
- HCC:
-
Hepatocellular carcinoma
- HPD:
-
Hyperprogressive disease
- HIF-1α:
-
Hypoxia-inducible factor-1α
- HDAC3:
-
Histone deacetylase 3
- ITIM:
-
Immunoreceptor tyrosine-based inhibitory motif
- ITSM:
-
Immunoreceptor tyrosine-based switch motif
- ICIs:
-
Immune checkpoint inhibitors
- IgG:
-
Immunoglobulin G
- irAEs:
-
Immune-related adverse events
- ICT:
-
Immune checkpoint therapy
- JAK2:
-
Janus kinase 2
- LTi:
-
Lymphoid tissue inducer cells
- lncRNAs:
-
Long non-coding RNAs
- LSD1:
-
Lysine-specific Demethylase 1
- MHC:
-
Major histocompatibility complex
- MS:
-
Multiple sclerosis
- mAbs:
-
Monoclonal antibodies
- miRNAs:
-
MicroRNAs
- m6A:
-
N6-methyladenosine
- mRCC:
-
Metastatic renal cell carcinoma
- NOD:
-
Non-obese diabetic
- NK:
-
Natural killer cell
- NAMPT:
-
Nicotinamide phosphoribosyltransferase
- NAD+ :
-
Nicotinamide adenine dinucleotide
- NSCLC:
-
Non-small cell lung cancer
- OS:
-
Overall survival
- ORR:
-
Objective response rate
- OVs:
-
Oncolytic viruses
- PD-1:
-
Programmed cell death protein 1
- PD-Ls:
-
Programmed cell death ligands
- PFS:
-
Progression-free survival
- PDT:
-
Photodynamic therapy
- RA:
-
Rheumatoid arthritis
- SHP2:
-
Src homology region 2 domain-containing phosphatase-2
- SLE:
-
Systemic lupus erythematosus
- sPD-1:
-
Soluble PD-1
- STS:
-
Soft tissue sarcoma
- SR:
-
Structural rearrangement
- SPOP:
-
Speckle type BTB/POZ protein
- STUB1:
-
STIP1 homology and U-box containing protein 1
- Tregs:
-
Regulatory T cells
- TCR:
-
T-cell receptor
- T1DM:
-
Type 1 diabetes mellitus
- TILs:
-
Tumor-infiltrating lymphocytes
- Tex cells:
-
T cell exhaustion
- TME:
-
Tumor microenvironment
- TdLN:
-
Tumor-draining lymph node
- TAM:
-
Tumor-associated macrophage
- TIC:
-
Tumor-initiating cell
- TNBC:
-
Triple-negative breast cancer
- TKI:
-
Tyrosine kinase inhibitor
- TLR:
-
Toll-like receptor
- VEGF:
-
Vascular endothelial growth factor
- 3'-UTR:
-
3’ untranslated region
References
Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887–95.
Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol Baltim Md. 1950. 2002;169:5538–45.
Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33:2706–16.
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the Pd-1 Immunoinhibitory receptor by a Novel B7 Family Member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800.
Zhang X, Schwartz J-CD, Guo X, Bhatia S, Cao E, Lorenz M, et al. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity. 2004;20:337–47.
Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209:1201–17.
Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–8.
Singh S, Hassan D, Aldawsari HM, Molugulu N, Shukla R, Kesharwani P. Immune checkpoint inhibitors: a promising anticancer therapy. Drug Discov Today. 2020;25:223–9.
Kruger S, Ilmer M, Kobold S, Cadilha BL, Endres S, Ormanns S, et al. Advances in cancer immunotherapy 2019 – latest trends. J Exp Clin Cancer Res CR. 2019;38:268.
Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118:9–16.
Jiang Y, Chen M, Nie H, Yuan Y. PD-1 and PD-L1 in cancer immunotherapy: clinical implications and future considerations. Hum Vaccines Immunother. 2019;15:1111–22.
Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–58.
Liechtenstein T, Dufait I, Lanna A, Breckpot K, Escors D, MODULATING CO-STIMULATION DURING, ANTIGEN PRESENTATION TO ENHANCE CANCER IMMUNOTHERAPY. Immunol Endocr Metab Agents Med Chem. 2012;12:224–35.
Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–42.
Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009;229:12–26.
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70.
Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293–7.
Song Z, Wang M, Ge Y, Chen X-P, Xu Z, Sun Y, et al. Tyrosine phosphatase SHP2 inhibitors in tumor-targeted therapies. Acta Pharm Sin B. 2021;11:13–29.
Marasco M, Berteotti A, Weyershaeuser J, Thorausch N, Sikorska J, Krausze J, et al. Molecular mechanism of SHP2 activation by PD-1 stimulation. Sci Adv. 2020;6:eaay4458.
Patsoukis N, Duke-Cohan JS, Chaudhri A, Aksoylar H-I, Wang Q, Council A, et al. Interaction of SHP-2 SH2 domains with PD-1 ITSM induces PD-1 dimerization and SHP-2 activation. Commun Biol. 2020;3:128.
Christofides A, Katopodi X-L, Cao C, Karagkouni D, Aliazis K, Yenyuwadee S, et al. SHP-2 and PD-1-SHP-2 signaling regulate myeloid cell differentiation and antitumor responses. Nat Immunol. 2023;24:55–68.
Rota G, Niogret C, Dang AT, Barros CR, Fonta NP, Alfei F, et al. Shp-2 is dispensable for establishing T cell exhaustion and for PD-1 signaling in vivo. Cell Rep. 2018;23:39–49.
Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science. 2017;355:1428–33.
Piao W, Li L, Saxena V, Iyyathurai J, Lakhan R, Zhang Y, et al. PD-L1 signaling selectively regulates T cell lymphatic transendothelial migration. Nat Commun. 2022;13:2176.
Weisberg SP, Carpenter DJ, Chait M, Dogra P, Gartrell-Corrado RD, Chen AX, et al. Tissue-Resident memory T cells mediate Immune Homeostasis in the human pancreas through the PD-1/PD-L1 pathway. Cell Rep. 2019;29:3916–e39325.
Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the Tumor Microenvironment is a driver of Cancer Progression. Cell. 2015;162:1229–41.
Zhao J, Bang S, Furutani K, McGinnis A, Jiang C, Roberts A, et al. PD-L1/PD-1 checkpoint pathway regulates hippocampal neuronal excitability and learning and memory behavior. Neuron. 2023;111:2709–e27269.
Wang R, Huang F, Wei W, Zhou Y, Ye Z, Yu L, et al. Programmed cell death Ligand 1 is enriched in mammary stem cells and promotes Mammary Development and Regeneration. Front Cell Dev Biol. 2021;9:772669.
Moore AR, Vivanco Gonzalez N, Plummer KA, Mitchel OR, Kaur H, Rivera M, et al. Gestationally dependent immune organization at the maternal-fetal interface. Cell Rep. 2022;41:111651.
Wang T-W, Johmura Y, Suzuki N, Omori S, Migita T, Yamaguchi K, et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature. 2022;611:358–64.
Chikuma S. Basics of PD-1 in self-tolerance, infection, and cancer immunity. Int J Clin Oncol. 2016;21:448–55.
Chamoto K, Al-Habsi M, Honjo T. Role of PD-1 in Immunity and Diseases. In: Yoshimura A, editor. Emerg Concepts Target Immune Checkp Cancer Autoimmun [Internet]. Cham: Springer International Publishing; 2017 [cited 2023 Feb 13]. pp. 75–97. https://doi.org/10.1007/82_2017_67.
Gianchecchi E, Delfino DV, Fierabracci A. Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmun Rev. 2013;12:1091–100.
Langewisch E, Mannon RB. Chronic allograft Injury. Clin J Am Soc Nephrol CJASN. 2021;16:1723–9.
Righi I, Vaira V, Morlacchi LC, Croci GA, Rossetti V, Blasi F, et al. Immune checkpoints expression in chronic lung allograft rejection. Front Immunol. 2021;12:714132.
Shi X-L, Mancham S, Hansen BE, de Knegt RJ, de Jonge J, van der Laan LJW, et al. Counter-regulation of rejection activity against human liver grafts by donor PD-L1 and recipient PD-1 interaction. J Hepatol. 2016;64:1274–82.
Shim YJ, Khedraki R, Dhar J, Fan R, Dvorina N, Valujskikh A, et al. Early T cell infiltration is modulated by programed cell death-1 protein and its ligand (PD-1/PD-L1) interactions in murine kidney transplants. Kidney Int. 2020;98:897.
Friend BD, Venick RS, McDiarmid SV, Zhou X, Naini B, Wang H, et al. Fatal orthotopic liver transplant organ rejection induced by a checkpoint inhibitor in two patients with refractory, metastatic hepatocellular carcinoma. Pediatr Blood Cancer. 2017;64:e26682.
Fisher J, Zeitouni N, Fan W, Samie FH. Immune checkpoint inhibitor therapy in solid organ transplant recipients: a patient-centered systematic review. J Am Acad Dermatol. 2020;82:1490–500.
Ong M, Ibrahim AM, Bourassa-Blanchette S, Canil C, Fairhead T, Knoll G. Antitumor activity of nivolumab on hemodialysis after renal allograft rejection. J Immunother Cancer. 2016;4:64.
Katsarou A, Gudbjörnsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Primer. 2017;3:17016.
Colli ML, Hill JLE, Marroquí L, Chaffey J, Dos Santos RS, Leete P, et al. PDL1 is expressed in the islets of people with type 1 diabetes and is up-regulated by interferons-α and-γ via IRF1 induction. EBioMedicine. 2018;36:367–75.
Rui J, Deng S, Arazi A, Perdigoto AL, Liu Z, Herold KC. β cells that resist immunological attack develop during progression of Autoimmune Diabetes in NOD mice. Cell Metab. 2017;25:727–38.
Zhang X, Kang Y, Wang J, Yan J, Chen Q, Cheng H, et al. Engineered PD-L1-Expressing platelets Reverse New-Onset type 1 diabetes. Adv Mater. 2020;32:1907692.
Collier JL, Pauken KE, Lee CAA, Patterson DG, Markson SC, Conway TS, et al. Single-cell profiling reveals unique features of diabetogenic T cells in anti-PD-1-induced type 1 diabetes mice. J Exp Med. 2023;220:e20221920.
Stamatouli AM, Quandt Z, Perdigoto AL, Clark PL, Kluger H, Weiss SA, et al. Collateral damage: insulin-dependent diabetes Induced with checkpoint inhibitors. Diabetes. 2018;67:1471–80.
Godwin JL, Jaggi S, Sirisena I, Sharda P, Rao AD, Mehra R, et al. Nivolumab-induced autoimmune diabetes mellitus presenting as diabetic ketoacidosis in a patient with metastatic lung cancer. J Immunother Cancer. 2017;5:40.
Marsiglio J, McPherson JP, Kovacsovics-Bankowski M, Jeter J, Vaklavas C, Swami U, et al. A single center case series of immune checkpoint inhibitor-induced type 1 diabetes mellitus, patterns of disease onset and long-term clinical outcome. Front Immunol. 2023;14:1229823.
Almaani S, Meara A, Rovin BH. Update on Lupus Nephritis. Clin J Am Soc Nephrol CJASN. 2017;12:825–35.
Curran CS, Gupta S, Sanz I, Sharon E. PD-1 immunobiology in systemic lupus erythematosus. J Autoimmun. 2019;97:1–9.
Guo Q, Chen C, Wu Z, Zhang W, Wang L, Yu J, et al. Engineered PD-1/TIGIT dual-activating cell-membrane nanoparticles with dexamethasone act synergistically to shape the effector T cell/Treg balance and alleviate systemic lupus erythematosus. Biomaterials. 2022;285:121517.
Wong M, La Cava A, Singh RP, Hahn BH. Blockade of programmed Death-1 in Young (New Zealand Black × New Zealand White)F1 mice promotes the activity of suppressive CD8 + T cells that protect from Lupus-Like Disease. J Immunol. 2010;185:6563–71.
Shi H, Ye J, Teng J, Yin Y, Hu Q, Wu X, et al. Elevated serum autoantibodies against co-inhibitory PD-1 facilitate T cell proliferation and correlate with disease activity in new-onset systemic lupus erythematosus patients. Arthritis Res Ther. 2017;19:52.
Luo Q, Huang Z, Ye J, Deng Y, Fang L, Li X, et al. PD-L1-expressing neutrophils as a novel indicator to assess disease activity and severity of systemic lupus erythematosus. Arthritis Res Ther. 2016;18:47.
Ibañez-Vega J, Vilchez C, Jimenez K, Guevara C, Burgos PI, Naves R. Cellular and molecular regulation of the programmed death-1/programmed death ligand system and its role in multiple sclerosis and other autoimmune diseases. J Autoimmun. 2021;123:102702.
Salama AD, Chitnis T, Imitola J, Ansari MJI, Akiba H, Tushima F, et al. Critical role of the programmed Death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med. 2003;198:71–8.
Kroner A, Mehling M, Hemmer B, Rieckmann P, Toyka KV, Mäurer M, et al. A PD-1 polymorphism is associated with disease progression in multiple sclerosis. Ann Neurol. 2005;58:50–7.
Koto S, Chihara N, Akatani R, Nakano H, Hara A, Sekiguchi K, et al. Transcription factor c-Maf promotes immunoregulation of programmed cell death 1–Expressed CD8 + T cells in multiple sclerosis. Neurol Neuroimmunol Neuroinflammation. 2022;9:e1166.
Belkhir R, Burel SL, Dunogeant L, Marabelle A, Hollebecque A, Besse B, et al. Rheumatoid arthritis and polymyalgia rheumatica occurring after immune checkpoint inhibitor treatment. Ann Rheum Dis. 2017;76:1747–50.
Bartosińska J, Zakrzewska E, Król A, Raczkiewicz D, Purkot J, Majdan M, et al. Differential expression of programmed death 1 (PD-1) on CD4 + and CD8 + T cells in rheumatoid arthritis and psoriatic arthritis. Pol Arch Intern Med. 2017;127:815–22.
Wan B, Nie H, Liu A, Feng G, He D, Xu R et al. Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J Immunol Baltim Md. 1950. 2006;177:8844–50.
Zhou H, Hu B, Zhaopeng Z, Liu J, Zhong Q, Fan Y, et al. Elevated circulating T cell subsets and cytokines expression in patients with rheumatoid arthritis. Clin Rheumatol. 2019;38:1831–9.
Kostine M, Rouxel L, Barnetche T, Veillon R, Martin F, Dutriaux C, et al. Rheumatic disorders associated with immune checkpoint inhibitors in patients with cancer-clinical aspects and relationship with tumour response: a single-centre prospective cohort study. Ann Rheum Dis. 2018;77:393–8.
Zhao P, Wang P, Dong S, Zhou Z, Cao Y, Yagita H, et al. Depletion of PD-1-positive cells ameliorates autoimmune disease. Nat Biomed Eng. 2019;3:292–305.
Efremova M, Rieder D, Klepsch V, Charoentong P, Finotello F, Hackl H, et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat Commun. 2018;9:32.
Kuol N, Stojanovska L, Nurgali K, Apostolopoulos V. PD-1/PD-L1 in disease. Immunotherapy. 2018;10:149–60.
Li X, Shao C, Shi Y, Han W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J Hematol OncolJ Hematol Oncol. 2018;11:31.
Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of Immune escape. Sci Transl Med. 2012;4:127ra37.
Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in Tolerance and Autoimmunity. Immunol Rev. 2010;236:219–42.
Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4.
Zarour HM. Reversing T-cell dysfunction and exhaustion in Cancer. Clin Cancer Res off J Am Assoc Cancer Res. 2016;22:1856–64.
Konen JM, Rodriguez BL, Fradette JJ, Gibson L, Davis D, Minelli R, et al. Ntrk1 promotes resistance to PD-1 checkpoint blockade in Mesenchymal Kras/p53 mutant Lung Cancer. Cancers. 2019;11:462.
Bardhan K, Anagnostou T, Boussiotis VA. The PD1:PD-L1/2 pathway from Discovery to clinical implementation. Front Immunol. 2016;7:550.
Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, et al. PD-1 is a regulator of virus-specific CD8 + T cell survival in HIV infection. J Exp Med. 2006;203:2281–92.
Guo X, Jiang H, Shi B, Zhou M, Zhang H, Shi Z, et al. Disruption of PD-1 enhanced the anti-tumor activity of Chimeric Antigen Receptor T Cells against Hepatocellular Carcinoma. Front Pharmacol. 2018;9:1118.
Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7.
Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by PD-1 are an early driver of CD8 + T cell exhaustion. Immunity. 2016;45:358–73.
Charlton JJ, Chatzidakis I, Tsoukatou D, Boumpas DT, Garinis GA, Mamalaki C. Programmed Death-1 shapes memory phenotype CD8 T cell subsets in a cell-intrinsic manner. J Immunol. 2013;190:6104–14.
Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell–mediated antitumor immunity. Nat Med. 2003;9:562–7.
Diskin B, Adam S, Cassini MF, Sanchez G, Liria M, Aykut B, et al. PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat Immunol. 2020;21:442–54.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Callahan MK, Wolchok JD. Recruit or Reboot? How does Anti-PD-1 therapy change tumor-infiltrating lymphocytes? Cancer Cell. 2019;36:215–7.
Miller BC, Sen DR, Abosy RA, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8 + T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20:326–36.
Chen C-Y, Ueha S, Ishiwata Y, Shichino S, Yokochi S, Yang D, et al. Combining an Alarmin HMGN1 peptide with PD-L1 blockade results in Robust Antitumor effects with a concomitant increase of Stem-Like/Progenitor exhausted CD8 + T cells. Cancer Immunol Res. 2021;9:1214–28.
Huang Q, Wu X, Wang Z, Chen X, Wang L, Lu Y, et al. The primordial differentiation of tumor-specific memory CD8 + T cells as bona fide responders to PD-1/PD-L1 blockade in draining lymph nodes. Cell. 2022;185:4049–e406625.
Long H, Jia Q, Wang L, Fang W, Wang Z, Jiang T, et al. Tumor-induced erythroid precursor-differentiated myeloid cells mediate immunosuppression and curtail anti-PD-1/PD-L1 treatment efficacy. Cancer Cell. 2022;40:674–e6937.
Turner JA, Stephen-Victor E, Wang S, Rivas MN, Abdel-Gadir A, Harb H, et al. Regulatory T cell-derived TGF-β1 controls multiple checkpoints governing Allergy and Autoimmunity. Immunity. 2020;53:1202–e12146.
Wing JB, Tanaka A, Sakaguchi S. Human FOXP3 + Regulatory T cell heterogeneity and function in autoimmunity and Cancer. Immunity. 2019;50:302–16.
Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–29.
Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, akt, and mTOR. Proc Natl Acad Sci U S A. 2008;105:7797–802.
Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. 2016;22:679–84.
Que Y, Xiao W, Guan Y, Liang Y, Yan S, Chen H, et al. PD-L1 expression is Associated with FOXP3 + Regulatory T-Cell infiltration of soft tissue sarcoma and poor patient prognosis. J Cancer. 2017;8:2018–25.
Gianchecchi E, Fierabracci A. Inhibitory receptors and pathways of lymphocytes: the role of PD-1 in Treg Development and their involvement in Autoimmunity Onset and Cancer Progression. Front Immunol. 2018;9:2374.
Patsoukis N, Sari D, Boussiotis VA. PD-1 inhibits T cell proliferation by upregulating p27 and p15 and suppressing Cdc25A. Cell Cycle. 2012;11:4305–9.
DiDomenico J, Lamano JB, Oyon D, Li Y, Veliceasa D, Kaur G, et al. The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology. 2018;7:e1448329.
Gambichler T, Schröter U, Höxtermann S, Susok L, Stockfleth E, Becker JC. Decline of programmed death-1-positive circulating T regulatory cells predicts more favourable clinical outcome of patients with melanoma under immune checkpoint blockade. Br J Dermatol. 2020;182:1214–20.
Perry JA, Shallberg L, Clark JT, Gullicksrud J, DeLong JH, Douglas BB, et al. PD-L1 – PD-1 interactions limit effector regulatory T cell populations at homeostasis and during infection. Nat Immunol. 2022;23:743–56.
Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, et al. PD-1 + regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U S A. 2019;116:9999–10008.
Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin Y-T, Togashi Y, et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. 2022;40:201–e2189.
Gu J, Zhou J, Chen Q, Xu X, Gao J, Li X, et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-β signaling in regulatory T cells. Cell Rep. 2022;39:110986.
Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–37.
Qian B, Pollard JW. Macrophage diversity enhances Tumor Progression and Metastasis. Cell. 2010;141:39–51.
Zf W, M HLRG et al. Z, J M, Y Z,. Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. J Immunother Cancer [Internet]. 2018 [cited 2023 Feb 21];6. https://pubmed.ncbi.nlm.nih.gov/30563569/.
Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17:887–904.
Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumor-associated macrophages inhibits phagocytosis and tumor immunity. Nature. 2017;545:495–9.
Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495–9.
Chen W, Wang J, Jia L, Liu J, Tian Y. Attenuation of the programmed cell death-1 pathway increases the M1 polarization of macrophages induced by zymosan. Cell Death Dis. 2016;7:e2115–2115.
Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc Natl Acad Sci. 2018;115:E4041–50.
Jeong H, Kim S, Hong B-J, Lee C-J, Kim Y-E, Bok S, et al. Tumor-Associated macrophages enhance Tumor Hypoxia and Aerobic Glycolysis. Cancer Res. 2019;79:795–806.
Hartley GP, Chow L, Ammons DT, Wheat WH, Dow SW. Programmed cell death Ligand 1 (PD-L1) signaling regulates macrophage proliferation and activation. Cancer Immunol Res. 2018;6:1260–73.
Liu Y, Zugazagoitia J, Ahmed FS, Henick BS, Gettinger SN, Herbst RS, et al. Immune cell PD-L1 co-localizes with macrophages and is associated with outcome in PD-1 pathway blockade therapy. Clin Cancer Res off J Am Assoc Cancer Res. 2020;26:970–7.
Wang Z, Wang F, Ding X-Y, Li T-E, Wang H-Y, Gao Y-H, et al. Hippo/YAP signaling choreographs the tumor immune microenvironment to promote triple negative breast cancer progression via TAZ/IL-34 axis. Cancer Lett. 2022;527:174–90.
Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K, et al. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br J Cancer. 2015;112:1501–9.
Franks SE, Wolfson B, Hodge JW. Natural Born killers: NK cells in Cancer Therapy. Cancers. 2020;12:2131.
Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault M-C, Trevino TN et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest 128:4654–68.
Huang Y, Chen Z, Jang JH, Baig MS, Bertolet G, Schroeder C, et al. PD-1 blocks lytic granule polarization with concomitant impairment of integrin outside-in signaling in the natural killer cell immunological synapse. J Allergy Clin Immunol. 2018;142:1311–e13218.
Judge SJ, Dunai C, Aguilar EG, Vick SC, Sturgill IR, Khuat LT et al. Minimal PD-1 expression in mouse and human NK cells under diverse conditions. J Clin Invest 130:3051–68.
Hasim MS, Marotel M, Hodgins JJ, Vulpis E, Makinson OJ, Asif S et al. When killers become thieves: trogocytosed PD-1 inhibits NK cells in cancer. Sci Adv 8:eabj3286.
Bettadapur A, Miller HW, Ralston KS. Biting off what can be chewed: trogocytosis in Health, infection, and Disease. Infect Immun. 2020;88:e00930–19.
Lin M, Luo H, Liang S, Chen J, Liu A, Niu L et al. Pembrolizumab plus allogeneic NK cells in advanced non–small cell lung cancer patients. J Clin Invest 130:2560–9.
Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z, et al. The mechanism of anti-PD-L1 antibody efficacy against PD-L1 negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discov. 2019;9:1422–37.
Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and Effector functions in CD8 + T cells. Eur J Immunol. 2008;38:2438–50.
Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692.
Di Biase S, Ma X, Wang X, Yu J, Wang Y-C, Smith DJ, et al. Creatine uptake regulates CD8 T cell antitumor immunity. J Exp Med. 2019;216:2869–82.
Lv H, Lv G, Chen C, Zong Q, Jiang G, Ye D, et al. NAD + metabolism maintains inducible PD-L1 expression to Drive Tumor Immune Evasion. Cell Metab. 2021;33:110–e1275.
Wu D, Hu L, Han M, Deng Y, Zhang Y, Ren G, et al. PD-1 signaling facilitates activation of lymphoid tissue inducer cells by restraining fatty acid oxidation. Nat Metab. 2022;4:867–82.
Qorraj M, Bruns H, Böttcher M, Weigand L, Saul D, Mackensen A, et al. The PD-1/PD-L1 axis contributes to immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia. Leukemia. 2017;31:470–8.
Choi Y, Lichterman JN, Coughlin LA, Poulides N, Li W, Del Valle P, et al. Immune checkpoint blockade induces gut microbiota translocation that augments extraintestinal antitumor immunity. Sci Immunol. 2023;8:eabo2003.
Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre M-L, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104–8.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103.
Mao J, Wang D, Long J, Yang X, Lin J, Song Y, et al. Gut microbiome is associated with the clinical response to anti-PD-1 based immunotherapy in hepatobiliary cancers. J Immunother Cancer. 2021;9:e003334.
Gao H, Zhang J, Ren X. PD-L1 regulates tumorigenesis and autophagy of ovarian cancer by activating mTORC signaling. Biosci Rep. 2019;39:BSR20191041.
Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015;162:1242–56.
He P-X, Ma Z-L, Han H, Zhang X-Y, Niu S-H, Du L-N, et al. Expression of programmed death ligand 1 (PD-L1) is associated with metastasis and differentiation in gastric cancer. Life Sci. 2020;242:117247.
Wang S, Li J, Xie J, Liu F, Duan Y, Wu Y, et al. Programmed death ligand 1 promotes lymph node metastasis and glucose metabolism in cervical cancer by activating integrin β4/SNAI1/SIRT3 signaling pathway. Oncogene. 2018;37:4164–80.
Shi F, Shi M, Zeng Z, Qi R-Z, Liu Z-W, Zhang J-Y, et al. PD-1 and PD-L1 upregulation promotes CD8(+) T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int J Cancer. 2011;128:887–96.
Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive non-small-cell Lung Cancer. N Engl J Med. 2016;375:1823–33.
Cg K, Kh K, Kh P, Cf X, Mh H, Bc A et al. Hyperprogressive disease during PD-1/PD-L1 blockade in patients with non-small-cell lung cancer. Ann Oncol Off J Eur Soc Med Oncol [Internet]. 2019 [cited 2023 Mar 19];30. https://pubmed.ncbi.nlm.nih.gov/30977778/.
Li G, Choi JE, Kryczek I, Sun Y, Liao P, Li S, et al. Intersection of immune and oncometabolic pathways drives cancer hyperprogression during immunotherapy. Cancer Cell. 2023;41:304–e3227.
Ying H, Zhang X, Duan Y, Lao M, Xu J, Yang H, et al. Non-cytomembrane PD-L1: an atypical target for cancer. Pharmacol Res. 2021;170:105741.
Thompson RH, Kuntz SM, Leibovich BC, Dong H, Lohse CM, Webster WS, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res. 2006;66:3381–5.
Ghebeh H, Lehe C, Barhoush E, Al-Romaih K, Tulbah A, Al-Alwan M, et al. Doxorubicin downregulates cell surface B7-H1 expression and upregulates its nuclear expression in breast cancer cells: role of B7-H1 as an anti-apoptotic molecule. Breast Cancer Res BCR. 2010;12:R48.
Satelli A, Batth IS, Brownlee Z, Rojas C, Meng QH, Kopetz S, et al. Potential role of nuclear PD-L1 expression in cell-surface vimentin positive circulating tumor cells as a prognostic marker in cancer patients. Sci Rep. 2016;6:28910.
Yu J, Qin B, Moyer AM, Nowsheen S, Tu X, Dong H, et al. Regulation of sister chromatid cohesion by nuclear PD-L1. Cell Res. 2020;30:590.
Du W, Zhu J, Zeng Y, Liu T, Zhang Y, Cai T, et al. KPNB1-mediated nuclear translocation of PD-L1 promotes non-small cell lung cancer cell proliferation via the Gas6/MerTK signaling pathway. Cell Death Differ. 2021;28:1284–300.
Hou J, Zhao R, Xia W, Chang C-W, You Y, Hsu J-M, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22:1264–75.
Gao Y, Nihira NT, Bu X, Chu C, Zhang J, Kolodziejczyk A, et al. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat Cell Biol. 2020;22:1064–75.
Zhang D, Reyes RM, Osta E, Kari S, Gupta HB, Padron AS, et al. Bladder cancer cell-intrinsic PD‐L1 signals promote mTOR and autophagy activation that can be inhibited to improve cytotoxic chemotherapy. Cancer Med. 2021;10:2137–52.
Du S, McCall N, Park K, Guan Q, Fontina P, Ertel A, et al. Blockade of Tumor-expressed PD-1 promotes lung cancer growth. Oncoimmunology. 2018;7:e1408747.
Gupta HB, Clark CA, Yuan B, Sareddy G, Pandeswara S, Padron AS, et al. Tumor cell-intrinsic PD-L1 promotes tumor-initiating cell generation and functions in melanoma and ovarian cancer. Signal Transduct Target Ther. 2016;1:16030.
Liu J, Chen Z, Li Y, Zhao W, Wu J, Zhang Z. PD-1/PD-L1 Checkpoint Inhibitors in Tumor Immunotherapy. Front Pharmacol [Internet]. 2021 [cited 2023 Jan 30];12. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8440961/.
Bally APR, Tang Y, Lee JT, Barwick BG, Martinez R, Evavold BD et al. Conserved region C functions to regulate PD-1 expression and subsequent CD8 T cell memory. J Immunol Baltim Md. 1950. 2017;198:205–17.
Bally APR, Lu P, Tang Y, Austin JW, Scharer CD, Ahmed R et al. NF-κB regulates PD-1 expression in macrophages. J Immunol Baltim Md. 1950. 2015;194:4545–54.
Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 Regulates Programmed Death-1 Expression Upon T Cell Activation. J Immunol Baltim Md. 1950. 2008;181:4832–9.
Austin JW, Lu P, Majumder P, Ahmed R, Boss JM. STAT3, STAT4, NFATc1, and CTCF regulate PD-1 through multiple novel regulatory regions in murine T cells. J Immunol Baltim Md 1950. 2014;192:4876–86.
Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8:765–72.
Bally APR, Neeld DK, Lu P, Majumder P, Tang Y, Barwick BG et al. PD-1 expression during acute infection is repressed through a LSD1- Blimp-1 axis. J Immunol Baltim Md. 1950. 2020;204:449–58.
Brown KE, Freeman GJ, Wherry EJ, Sharpe AH. Role of PD-1 in regulating acute infections. Curr Opin Immunol. 2010;22:397–401.
Youngblood B, Oestreich KJ, Ha S-J, Duraiswamy J, Akondy RS, West EE, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8 + T cells. Immunity. 2011;35:400–12.
Ahn E, Youngblood B, Lee J, Lee J, Sarkar S, Ahmed R. Demethylation of the PD-1 promoter is imprinted during the Effector phase of CD8 T cell exhaustion. J Virol. 2016;90:8934.
Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8 + T cells during chronic infection. Immunity. 2014;41:802–14.
Nakayamada S, Poholek AC, Lu KT, Takahashi H, Kato M, Iwata S et al. Type I Interferon induces binding of STAT1 to Bcl6: Divergent Roles of STAT-family transcription factors in the TFH cell genetic program. J Immunol Baltim Md. 1950. 2014;192:2156.
Hou L, Jie Z, Liang Y, Desai M, Soong L, Sun J. Type 1 interferon-induced IL-7 maintains CD8 + T-cell responses and homeostasis by suppressing PD-1 expression in viral hepatitis. Cell Mol Immunol. 2015;12:213–23.
Bao S, Jiang X, Jin S, Tu P, Lu J. TGF-β1 induces Immune escape by enhancing PD-1 and CTLA-4 expression on T lymphocytes in Hepatocellular Carcinoma. Front Oncol. 2021;11:694145.
Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477–86.
Pan J, Li X, Shao B, Xu F, Huang X, Guo X, et al. Self-blockade of PD-L1 with Bacteria-derived outer-membrane vesicle for enhanced Cancer Immunotherapy. Adv Mater Deerfield Beach Fla. 2022;34:e2106307.
Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med. 2019;25:454–61.
Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, et al. Neoadjuvant PD-1 blockade in Resectable Lung Cancer. N Engl J Med. 2018;378:1976–86.
Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola-Essel M, et al. PD-1 blockade in Mismatch Repair–Deficient, locally advanced rectal Cancer. N Engl J Med. 2022;386:2363–76.
Yi M, Niu M, Xu L, Luo S, Wu K. Regulation of PD-L1 expression in the tumor microenvironment. J Hematol OncolJ Hematol Oncol. 2021;14:10.
Shen X, Zhang L, Li J, Li Y, Wang Y, Xu Z-X. Recent findings in the regulation of programmed death Ligand 1 expression. Front Immunol. 2019;10:1337.
Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with Nivolumab in Relapsed or Refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372:311–9.
Huang RSP, Murugesan K, Montesion M, Pavlick DC, Mata DA, Hiemenz MC, et al. Pan-cancer landscape of CD274 (PD-L1) copy number changes in 244 584 patient samples and the correlation with PD-L1 protein expression. J Immunother Cancer. 2021;9:e002680.
Budczies J, Mechtersheimer G, Denkert C, Klauschen F, Mughal SS, Chudasama P, et al. PD-L1 (CD274) copy number gain, expression, and immune cell infiltration as candidate predictors for response to immune checkpoint inhibitors in soft-tissue sarcoma. Oncoimmunology. 2017;6:e1279777.
Goodman AM, Piccioni D, Kato S, Boichard A, Wang H-Y, Frampton G, et al. Prevalence of PDL1 amplification and preliminary response to Immune Checkpoint Blockade in Solid tumors. JAMA Oncol. 2018;4:1237.
Straub M, Drecoll E, Pfarr N, Weichert W, Langer R, Hapfelmeier A, et al. CD274/PD-L1 gene amplification and PD-L1 protein expression are common events in squamous cell carcinoma of the oral cavity. Oncotarget. 2016;7:12024–34.
Ikeda S, Okamoto T, Okano S, Umemoto Y, Tagawa T, Morodomi Y, et al. PD-L1 is upregulated by simultaneous amplification of the PD-L1 and JAK2 genes in Non-small Cell Lung Cancer. J Thorac Oncol off Publ Int Assoc Study Lung Cancer. 2016;11:62–71.
Howitt BE, Sun HH, Roemer MGM, Kelley A, Chapuy B, Aviki E, et al. Genetic basis for PD-L1 expression in squamous cell carcinomas of the Cervix and Vulva. JAMA Oncol. 2016;2:518–22.
Guo L, Li W, Zhu X, Ling Y, Qiu T, Dong L, et al. PD-L1 expression and CD274 gene alteration in triple-negative breast cancer: implication for prognostic biomarker. SpringerPlus. 2016;5:805.
Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–77.
Prestipino A, Emhardt AJ, Aumann K, O’Sullivan D, Gorantla SP, Duquesne S, et al. Oncogenic JAK2V617F causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci Transl Med. 2018;10:eaam7729.
Comprehensive characterization of. Programmed death ligand structural rearrangements in B-cell non-hodgkin lymphomas. Blood. 2016;128:1206–13.
Georgiou K, Chen L, Berglund M, Ren W, de Miranda NFCC, Lisboa S, et al. Genetic basis of PD-L1 overexpression in diffuse large B-cell lymphomas. Blood. 2016;127:3026–34.
Wagner M, Tupikowski K, Jasek M, Tomkiewicz A, Witkowicz A, Ptaszkowski K, et al. SNP-SNP Interaction in genes encoding PD-1/PD-L1 Axis as a potential risk factor for Clear Cell Renal Cell Carcinoma. Cancers. 2020;12:3521.
Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun. 2017;8:1751.
Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature. 2016;534:402–6.
Lv D, Xing C, Cao L, Zhuo Y, Wu T, Gao N. PD-L1 gene promoter methylation represents a potential diagnostic marker in advanced gastric cancer. Oncol Lett. 2020;19:1223–34.
Röver LK, Gevensleben H, Dietrich J, Bootz F, Landsberg J, Goltz D, et al. PD-1 (PDCD1) promoter methylation is a prognostic factor in patients with diffuse Lower-Grade Gliomas Harboring Isocitrate dehydrogenase (IDH) mutations. EBioMedicine. 2018;28:97–104.
Zhang Y, Xiang C, Wang Y, Duan Y, Liu C, Zhang Y. PD-L1 promoter methylation mediates the resistance response to anti-PD-1 therapy in NSCLC patients with EGFR-TKI resistance. Oncotarget. 2017;8:101535–44.
Chatterjee A, Rodger EJ, Ahn A, Stockwell PA, Parry M, Motwani J, et al. Marked global DNA hypomethylation is Associated with constitutive PD-L1 expression in Melanoma. iScience. 2018;4:312–25.
Sheikh TN, Chen X, Xu X, McGuire JT, Ingham M, Lu C et al. Growth Inhibition and Induction of Innate Immune Signaling of Chondrosarcomas with Epigenetic Inhibitors. Mol Cancer Ther. 2021;10.1158/1535–7163.MCT-21–0066.
Asgarova A, Asgarov K, Godet Y, Peixoto P, Nadaradjane A, Boyer-Guittaut M, et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology. 2018;7:e1423170.
Li X, Li Y, Dong L, Chang Y, Zhang X, Wang C et al. Decitabine-priming increases anti-PD-1 antitumor efficacy by promoting CD8 + progenitor exhausted T-cell expansion in tumor models. J Clin Invest. 2023;e165673.
Liu Y, Wang C, Li X, Dong L, Yang Q, Chen M, et al. Improved clinical outcome in a randomized phase II study of anti-PD-1 camrelizumab plus decitabine in relapsed/refractory Hodgkin lymphoma. J Immunother Cancer. 2021;9:e002347.
Hesham HM, Lasheen DS, Abouzid KAM. Chimeric HDAC inhibitors: Comprehensive review on the HDAC-based strategies developed to combat cancer. Med Res Rev. 2018;38:2058–109.
Shen Y, Liu L, Wang M, Xu B, Lyu R, Shi YG, et al. TET2 inhibits PD-L1 gene expression in breast Cancer cells through Histone Deacetylation. Cancers. 2021;13:2207.
Woods DM, Sodré AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC inhibition upregulates PD-1 ligands in Melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res. 2015;3:1375–85.
Wang H, Fu C, Du J, Wang H, He R, Yin X, et al. Enhanced histone H3 acetylation of the PD-L1 promoter via the COP1/c-Jun/HDAC3 axis is required for PD-L1 expression in drug-resistant cancer cells. J Exp Clin Cancer Res CR. 2020;39:29.
Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, et al. BET-Bromodomain inhibitors engage the host Immune System and regulate expression of the Immune checkpoint ligand PD-L1. Cell Rep. 2017;18:2162–74.
Fan P, Zhao J, Meng Z, Wu H, Wang B, Wu H, et al. Overexpressed histone acetyltransferase 1 regulates cancer immunity by increasing programmed death-ligand 1 expression in pancreatic cancer. J Exp Clin Cancer Res CR. 2019;38:47.
Shi Y, Fu Y, Zhang X, Zhao G, Yao Y, Guo Y, et al. Romidepsin (FK228) regulates the expression of the immune checkpoint ligand PD-L1 and suppresses cellular immune functions in colon cancer. Cancer Immunol Immunother. 2021;70:61–73.
Deng S, Hu Q, Zhang H, Yang F, Peng C, Huang C. HDAC3 inhibition upregulates PD-L1 expression in B-Cell lymphomas and augments the efficacy of Anti-PD-L1 therapy. Mol Cancer Ther. 2019;18:900–8.
Xiao G, Jin L-L, Liu C-Q, Wang Y-C, Meng Y-M, Zhou Z-G, et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J Immunother Cancer. 2019;7:300.
Qin Y, Vasilatos SN, Chen L, Wu H, Cao Z, Fu Y, et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances Antitumor Efficacy of Immune Checkpoint Blockade. Oncogene. 2019;38:390–405.
Wang Y, Cao K. KDM1A promotes immunosuppression in Hepatocellular Carcinoma by regulating PD-L1 through demethylating MEF2D. J Immunol Res. 2021;2021:9965099.
Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the Anti-tumor Immune response through CD47 and PD-L1. Science. 2016;352:227–31.
Liang M-Q, Yu F-Q, Chen C. C-Myc regulates PD-L1 expression in esophageal squamous cell carcinoma. Am J Transl Res. 2020;12:379–88.
Chen X, Du Q, Guo H, He Q, Yang B, Ding L. Bafetinib suppresses the transcription of PD-L1 through c-Myc in Lung Cancer. Front Pharmacol. 2022;13:897747.
Wang J, Zhang R, Lin Z, Zhang S, Chen Y, Tang J, et al. CDK7 inhibitor THZ1 enhances antiPD-1 therapy efficacy via the p38α/MYC/PD-L1 signaling in non-small cell lung cancer. J Hematol OncolJ Hematol Oncol. 2020;13:99.
Dorand RD, Nthale J, Myers JT, Barkauskas DS, Avril S, Chirieleison SM, et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science. 2016;353:399–403.
Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–201.
Xu Y, Lv L, Liu Y, Smith MD, Li W-C, Tan X et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J Clin Invest 129:4316–31.
Xu P, Xiong W, Lin Y, Fan L, Pan H, Li Y. Histone deacetylase 2 knockout suppresses immune escape of triple-negative breast cancer cells via downregulating PD-L1 expression. Cell Death Dis. 2021;12:779.
Que Y, Zhang X-L, Liu Z-X, Zhao J-J, Pan Q-Z, Wen X-Z, et al. Frequent amplification of HDAC genes and efficacy of HDAC inhibitor chidamide and PD-1 blockade combination in soft tissue sarcoma. J Immunother Cancer. 2021;9:e001696.
Song TL, Nairismägi M-L, Laurensia Y, Lim J-Q, Tan J, Li Z-M, et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood. 2018;132:1146–58.
Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A. 2008;105:20852–7.
Hu G, He N, Cai C, Cai F, Fan P, Zheng Z, et al. HDAC3 modulates cancer immunity via increasing PD-L1 expression in pancreatic cancer. Pancreatol off J Int Assoc Pancreatol IAP Al. 2019;19:383–9.
Wang YF, Liu F, Sherwin S, Farrelly M, Yan XG, Croft A, et al. Cooperativity of HOXA5 and STAT3 is critical for HDAC8 inhibition-mediated transcriptional activation of PD-L1 in human melanoma cells. J Invest Dermatol. 2018;138:922–32.
Antonangeli F, Natalini A, Garassino MC, Sica A, Santoni A, Di Rosa F. Regulation of PD-L1 expression by NF-κB in Cancer. Front Immunol. 2020;11:584626.
Wang W, Chapman NM, Zhang B, Li M, Fan M, Laribee RN, et al. Upregulation of PD-L1 via HMGB1-activated IRF3 and NF-κB contributes to UV radiation-induced immune suppression. Cancer Res. 2019;79:2909–22.
Fang W, Zhang J, Hong S, Zhan J, Chen N, Qin T, et al. EBV-driven LMP1 and IFN-γ up-regulate PD-L1 in nasopharyngeal carcinoma: implications for oncotargeted therapy. Oncotarget. 2014;5:12189–202.
Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O’Donnell E, et al. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin Lymphomas and Post-transplant Lymphoproliferative disorders: implications for targeted therapy. Clin Cancer Res. 2012;18:1611–8.
Bailey CM, Liu Y, Liu M, Du X, Devenport M, Zheng P et al. Targeting HIF-1α abrogates PD-L1–mediated immune evasion in tumor microenvironment but promotes tolerance in normal tissues. J Clin Invest 132:e150846.
Papalexi E, Mimitou EP, Butler AW, Foster S, Bracken B, Mauck WM, et al. Characterizing the molecular regulation of inhibitory immune checkpoints with multi-modal single-cell screens. Nat Genet. 2021;53:322–31.
Qin G, Wang X, Ye S, Li Y, Chen M, Wang S, et al. NPM1 upregulates the transcription of PD-L1 and suppresses T cell activity in triple-negative breast cancer. Nat Commun. 2020;11:1669.
Zerdes I, Matikas A, Bergh J, Rassidakis GZ, Foukakis T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: biology and clinical correlations. Oncogene. 2018;37:4639–61.
Mimura K, Teh JL, Okayama H, Shiraishi K, Kua L, Koh V, et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK‐STAT pathway in gastric cancer. Cancer Sci. 2018;109:43–53.
Bhat MY, Solanki HS, Advani J, Khan AA, Keshava Prasad TS, Gowda H, et al. Comprehensive network map of interferon gamma signaling. J Cell Commun Signal. 2018;12:745–51.
Wu B, Song M, Dong Q, Xiang G, Li J, Ma X, et al. UBR5 promotes tumor immune evasion through enhancing IFN-γ-induced PDL1 transcription in triple negative breast cancer. Theranostics. 2022;12:5086–102.
Chen J, Feng Y, Lu L, Wang H, Dai L, Li Y, et al. Interferon-γ-induced PD-L1 surface expression on human oral squamous carcinoma via PKD2 signal pathway. Immunobiology. 2012;217:385–93.
Chen E, Staudt LM, Green AR. Janus Kinase Deregulation in Leukemia and Lymphoma. Immunity. 2012;36:529–41.
Gao Y, Yang J, Cai Y, Fu S, Zhang N, Fu X, et al. IFN-γ-mediated inhibition of lung cancer correlates with PD-L1 expression and is regulated by PI3K-AKT signaling. Int J Cancer. 2018;143:931–43.
Haddadi N, Lin Y, Travis G, Simpson AM, McGowan EM, Nassif NT. PTEN/PTENP1: ‘Regulating the regulator of RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy. Mol Cancer. 2018;17:37.
Zhou S-L, Zhou Z-J, Hu Z-Q, Li X, Huang X-W, Wang Z, et al. CXCR2/CXCL5 axis contributes to epithelial-mesenchymal transition of HCC cells through activating PI3K/Akt/GSK-3β/Snail signaling. Cancer Lett. 2015;358:124–35.
Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res off J Am Assoc Cancer Res. 2013;19:598–609.
Zhang J-P, Song Z, Wang H-B, Lang L, Yang Y-Z, Xiao W, et al. A novel model of controlling PD-L1 expression in ALK + anaplastic large cell lymphoma revealed by CRISPR screening. Blood. 2019;134:171–85.
Turner E, Chen L, Foulke JG, Gu Z, Tian F. CRISPR/Cas9 edited RAS & MEK Mutant cells acquire BRAF and MEK inhibitor resistance with MEK1 Q56P restoring sensitivity to MEK/BRAF inhibitor combo and KRAS G13D gaining sensitivity to Immunotherapy. Cancers. 2022;14:5449.
Perry JM, Tao F, Roy A, Lin T, He XC, Chen S, et al. Overcoming Wnt-β-catenin dependent anticancer therapy resistance in leukaemia stem cells. Nat Cell Biol. 2020;22:689–700.
Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity. 2016;44:609–21.
Napolitano S, Matrone N, Muddassir AL, Martini G, Sorokin A, De Falco V, et al. Triple blockade of EGFR, MEK and PD-L1 has antitumor activity in colorectal cancer models with constitutive activation of MAPK signaling and PD-L1 overexpression. J Exp Clin Cancer Res CR. 2019;38:492.
Wang Q, Lin W, Tang X, Li S, Guo L, Lin Y, et al. The roles of microRNAs in regulating the expression of PD-1/PD-L1 Immune Checkpoint. Int J Mol Sci. 2017;18:2540.
Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, et al. PDL1 regulation by p53 via miR-34. J Natl Cancer Inst. 2016;108:djv303.
Zhang Q, Pan J, Xiong D, Wang Y, Miller MS, Sei S, et al. Pulmonary aerosol delivery of Let-7b microRNA confers a striking inhibitory effect on lung carcinogenesis through targeting the Tumor Immune Microenvironment. Adv Sci. 2021;8:2100629.
Yee D, Shah KM, Coles MC, Sharp TV, Lagos D. MicroRNA-155 induction via TNF-α and IFN-γ suppresses expression of programmed death ligand-1 (PD-L1) in human primary cells. J Biol Chem. 2017;292:20683–93.
Zheng Z, Sun R, Zhao H-J, Fu D, Zhong H-J, Weng X-Q, et al. MiR155 sensitized B-lymphoma cells to anti-PD-L1 antibody via PD-1/PD-L1-mediated lymphoma cell interaction with CD8 + T cells. Mol Cancer. 2019;18:54.
Tang D, Zhao D, Wu Y, Yao R, Zhou L, Lu L, et al. The miR-3127‐5p/p‐STAT3 axis up‐regulates PD‐L1 inducing chemoresistance in non‐small‐cell lung cancer. J Cell Mol Med. 2018;22:3847–56.
Dou D, Ren X, Han M, Xu X, Ge X, Gu Y, et al. Cancer-Associated fibroblasts-derived Exosomes suppress Immune cell function in breast Cancer via the miR-92/PD-L1 pathway. Front Immunol. 2020;11:2026.
Liu J, Fan L, Yu H, Zhang J, He Y, Feng D, et al. Endoplasmic reticulum stress causes Liver Cancer cells to Release Exosomal miR-23a-3p and Up-regulate programmed death Ligand 1 expression in macrophages. Hepatol Baltim Md. 2019;70:241–58.
Li P, Luo X, Xie Y, Li P, Hu F, Chu J, et al. GC-Derived EVs enriched with MicroRNA-675-3p contribute to the MAPK/PD-L1-Mediated Tumor Immune escape by targeting CXXC4. Mol Ther Nucleic Acids. 2020;22:615–26.
Jiang W, Pan S, Chen X, Wang Z, Zhu X. The role of lncRNAs and circRNAs in the PD-1/PD-L1 pathway in cancer immunotherapy. Mol Cancer. 2021;20:116.
Venkatesh J, Wasson M-CD, Brown JM, Fernando W, Marcato P. LncRNA-miRNA axes in breast cancer: novel points of interaction for strategic attack. Cancer Lett. 2021;509:81–8.
Wang Q-M, Lian G-Y, Song Y, Huang Y-F, Gong Y. LncRNA MALAT1 promotes tumorigenesis and immune escape of diffuse large B cell lymphoma by sponging miR-195. Life Sci. 2019;231:116335.
Xia R, Geng G, Yu X, Xu Z, Guo J, Liu H, et al. LINC01140 promotes the progression and tumor immune escape in lung cancer by sponging multiple microRNAs. J Immunother Cancer. 2021;9:e002746.
Wang Y, Yi K, Liu X, Tan Y, Jin W, Li Y, et al. HOTAIR Up-Regulation activates NF-κB to Induce Immunoescape in Gliomas. Front Immunol. 2021;12:785463.
Shi L, Yang Y, Li M, Li C, Zhou Z, Tang G, et al. LncRNA IFITM4P promotes immune escape by up-regulating PD-L1 via dual mechanism in oral carcinogenesis. Mol Ther J Am Soc Gene Ther. 2022;30:1564–77.
Qu S, Jiao Z, Lu G, Yao B, Wang T, Rong W, et al. PD-L1 lncRNA splice isoform promotes lung adenocarcinoma progression via enhancing c-Myc activity. Genome Biol. 2021;22:104.
Verduci L, Tarcitano E, Strano S, Yarden Y, Blandino G. CircRNAs: role in human diseases and potential use as biomarkers. Cell Death Dis. 2021;12:468.
Chen L-L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat Rev Mol Cell Biol. 2020;21:475–90.
Wei C-Y, Zhu M-X, Lu N-H, Liu J-Q, Yang Y-W, Zhang Y, et al. Circular RNA circ_0020710 drives tumor progression and immune evasion by regulating the miR-370-3p/CXCL12 axis in melanoma. Mol Cancer. 2020;19:84.
Zhang D-J, Fu Z-M, Guo Y-Y, Guo F, Wan Y-N, Guan G-F. Circ_0000052/miR-382-3p axis induces PD-L1 expression and regulates cell proliferation and immune evasion in head and neck squamous cell carcinoma. J Cell Mol Med. 2023;27:113–26.
Hong W, Xue M, Jiang J, Zhang Y, Gao X. Circular RNA circ-CPA4/ let-7 miRNA/PD-L1 axis regulates cell growth, stemness, drug resistance and immune evasion in non-small cell lung cancer (NSCLC). J Exp Clin Cancer Res CR. 2020;39:149.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20:608–24.
Huang J, Chen Z, Chen X, Chen J, Cheng Z, Wang Z. The role of RNA N6-methyladenosine methyltransferase in cancers. Mol Ther Nucleic Acids. 2021;23:887–96.
Wan W, Ao X, Chen Q, Yu Y, Ao L, Xing W, et al. METTL3/IGF2BP3 axis inhibits tumor immune surveillance by upregulating N6-methyladenosine modification of PD-L1 mRNA in breast cancer. Mol Cancer. 2022;21:60.
Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R, et al. m6A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD‐1 therapy. EMBO J. 2020;39:e104514.
Peng L, Pan B, Zhang X, Wang Z, Qiu J, Wang X, et al. Lipopolysaccharide facilitates immune escape of hepatocellular carcinoma cells via m6A modification of lncRNA MIR155HG to upregulate PD-L1 expression. Cell Biol Toxicol. 2022;38:1159–73.
Tsuruta N, Tsuchihashi K, Ohmura H, Yamaguchi K, Ito M, Ariyama H, et al. RNA N6-methyladenosine demethylase FTO regulates PD-L1 expression in colon cancer cells. Biochem Biophys Res Commun. 2020;530:235–9.
Qiu X, Yang S, Wang S, Wu J, Zheng B, Wang K, et al. M6A demethylase ALKBH5 regulates PD-L1 expression and Tumor Immunoenvironment in Intrahepatic Cholangiocarcinoma. Cancer Res. 2021;81:4778–93.
Kong F, Wang K, Wang L. Systematic analysis of the expression profile and prognostic significance of m6A regulators and PD-L1 in hepatocellular carcinoma. Discov Oncol. 2022;13:131.
Yun D, Yang Z, Zhang S, Yang H, Liu D, Grützmann R, et al. An m5C methylation regulator-associated signature predicts prognosis and therapy response in pancreatic cancer. Front Cell Dev Biol. 2022;10:975684.
Cerezo M, Guemiri R, Druillennec S, Girault I, Malka-Mahieu H, Shen S, et al. Translational control of tumor immune escape via the eIF4F–STAT1–PD-L1 axis in melanoma. Nat Med. 2018;24:1877–86.
Hsu J-M, Li C-W, Lai Y-J, Hung M-C. Posttranslational modifications of PD-L1 and their applications in Cancer Therapy. Cancer Res. 2018;78:6349–53.
Yao H, Lan J, Li C, Shi H, Brosseau J-P, Wang H, et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat Biomed Eng. 2019;3:306–17.
Breitling J, Aebi M. N-Linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harb Perspect Biol. 2013;5:a013359.
Benicky J, Sanda M, Brnakova Kennedy Z, Grant OC, Woods RJ, Zwart A, et al. PD-L1 glycosylation and its impact on binding to clinical antibodies. J Proteome Res. 2021;20:485–97.
Li C-W, Lim S-O, Xia W, Lee H-H, Chan L-C, Kuo C-W, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632.
Li C-W, Lim S-O, Chung EM, Kim Y-S, Park AH, Yao J, et al. Eradication of triple negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell. 2018;33:187–e20110.
Hsu J-M, Xia W, Hsu Y-H, Chan L-C, Yu W-H, Cha J-H, et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun. 2018;9:1908.
Chan L-C, Li C-W, Xia W, Hsu J-M, Lee H-H, Cha J-H et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J Clin Invest 129:3324–38.
Lee H-H, Wang Y-N, Xia W, Chen C-H, Rau K-M, Ye L, et al. Removal of N-Linked glycosylation enhances PD-L1 detection and predicts Anti-PD-1/PD-L1 therapeutic efficacy. Cancer Cell. 2019;36:168–e1784.
Wu Y, Zhang C, Liu X, He Z, Shan B, Zeng Q, et al. ARIH1 signaling promotes anti-tumor immunity by targeting PD-L1 for proteasomal degradation. Nat Commun. 2021;12:2346.
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen M-K, Hsu J-M, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res off J Am Assoc Cancer Res. 2017;23:3711–20.
Li H, Li C-W, Li X, Ding Q, Guo L, Liu S, et al. MET inhibitors promote Liver Tumor Evasion of the Immune response by stabilizing PDL1. Gastroenterology. 2019;156:1849–e186113.
Cha J-H, Yang W-H, Xia W, Wei Y, Chan L-C, Lim S-O, et al. Metformin promotes antitumor immunity via endoplasmic reticulum-associated degradation of PD-L1. Mol Cell. 2018;71:606–e6207.
Dai X, Bu X, Gao Y, Guo J, Hu J, Jiang C, et al. Energy status dictates PD-L1 protein abundance and anti-tumor immunity to enable checkpoint blockade. Mol Cell. 2021;81:2317–e23316.
Swatek KN, Komander D. Ubiquitin modifications. Cell Res. 2016;26:399–422.
Hou B, Chen T, Zhang H, Li J, Wang P, Shang G. The E3 ubiquitin ligases regulate PD-1/PD-L1 protein levels in tumor microenvironment to improve immunotherapy. Front Immunol. 2023;14:1123244.
Jing W, Wang G, Cui Z, Xiong G, Jiang X, Li Y, et al. FGFR3 destabilizes PD-L1 via NEDD4 to control T-cell-mediated bladder Cancer Immune Surveillance. Cancer Res. 2022;82:114–29.
Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via Cul3SPOP to control cancer immune surveillance. Nature. 2018;553:91–5.
De S, Holvey-Bates EG, Mahen K, Willard B, Stark GR. The ubiquitin E3 ligase FBXO22 degrades PD-L1 and sensitizes cancer cells to DNA damage. Proc Natl Acad Sci U S A. 2021;118:e2112674118.
Shi C, Wang Y, Wu M, Chen Y, Liu F, Shen Z, et al. Promoting anti-tumor immunity by targeting TMUB1 to modulate PD-L1 polyubiquitination and glycosylation. Nat Commun. 2022;13:6951.
Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature. 2017;549:106–10.
Wang Z, Kang W, Li O, Qi F, Wang J, You Y, et al. Abrogation of USP7 is an alternative strategy to downregulate PD-L1 and sensitize gastric cancer cells to T cells killing. Acta Pharm Sin B. 2021;11:694–707.
Yang H, Zhang X, Lao M, Sun K, He L, Xu J, et al. Targeting ubiquitin-specific protease 8 sensitizes anti-programmed death-ligand 1 immunotherapy of pancreatic cancer. Cell Death Differ. 2023;30:560–75.
Huang X, Zhang Q, Lou Y, Wang J, Zhao X, Wang L, et al. USP22 Deubiquitinates CD274 to suppress anticancer immunity. Cancer Immunol Res. 2019;7:1580–90.
Lim S-O, Li C-W, Xia W, Cha J-H, Chan L-C, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30:925–39.
Liu Z, Xiao M, Mo Y, Wang H, Han Y, Zhao X, et al. Emerging roles of protein palmitoylation and its modifying enzymes in cancer cell signal transduction and cancer therapy. Int J Biol Sci. 2022;18:3447–57.
Yang Y, Hsu J-M, Sun L, Chan L-C, Li C-W, Hsu JL, et al. Palmitoylation stabilizes PD-L1 to promote breast tumor growth. Cell Res. 2019;29:83–6.
Shahid M, Kim M, Jin P, Zhou B, Wang Y, Yang W, et al. S-Palmitoylation as a Functional Regulator of Proteins Associated with Cisplatin Resistance in bladder Cancer. Int J Biol Sci. 2020;16:2490–505.
Wang H, Yao H, Li C, Shi H, Lan J, Li Z, et al. HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nat Chem Biol. 2019;15:42–50.
Burr ML, Sparbier CE, Chan Y-C, Williamson JC, Woods K, Beavis PA, et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549:101–5.
Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18:10.
Liu C, Liu R, Wang B, Lian J, Yao Y, Sun H, et al. Blocking IL-17A enhances tumor response to anti-PD-1 immunotherapy in microsatellite stable colorectal cancer. J Immunother Cancer. 2021;9:e001895.
Lamano JB, Lamano JB, Li YD, DiDomenico JD, Choy W, Veliceasa D, et al. Glioblastoma-derived IL-6 induces immunosuppressive peripheral myeloid cell PD-L1 and promotes Tumor Growth. Clin Cancer Res off J Am Assoc Cancer Res. 2019;25:3643–57.
Lv Y, Zhao Y, Wang X, Chen N, Mao F, Teng Y, et al. Increased intratumoral mast cells foster immune suppression and gastric cancer progression through TNF-α-PD-L1 pathway. J Immunother Cancer. 2019;7:54.
Dimeloe S, Gubser P, Loeliger J, Frick C, Develioglu L, Fischer M, et al. Tumor-derived TGF-β inhibits mitochondrial respiration to suppress IFN-γ production by human CD4 + T cells. Sci Signal. 2019;12:eaav3334.
Guo D, Tong Y, Jiang X, Meng Y, Jiang H, Du L, et al. Aerobic glycolysis promotes tumor immune evasion by hexokinase2-mediated phosphorylation of IκBα. Cell Metab. 2022;34:1312–e13246.
Lo Cicero A, Stahl PD, Raposo G. Extracellular vesicles shuffling intercellular messages: for good or for bad. Curr Opin Cell Biol. 2015;35:69–77.
Gong B, Kiyotani K, Sakata S, Nagano S, Kumehara S, Baba S, et al. Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non–small cell lung cancer. J Exp Med. 2019;216:982–1000.
Gabrusiewicz K, Li X, Wei J, Hashimoto Y, Marisetty AL, Ott M, et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology. 2018;7:e1412909.
Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–6.
Guan J, Lim KS, Mekhail T, Chang C-C. Programmed death Ligand-1 (PD-L1) expression in the programmed death Receptor-1 (PD-1)/PD-L1 blockade: a key player against various cancers. Arch Pathol Lab Med. 2017;141:851–61.
Rao M, Valentini D, Dodoo E, Zumla A, Maeurer M. Anti-PD-1/PD-L1 therapy for infectious diseases: learning from the cancer paradigm. Int J Infect Dis IJID off Publ Int Soc Infect Dis. 2017;56:221–8.
Gong J, Chehrazi-Raffle A, Reddi S, Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8.
Rotte A, Jin JY, Lemaire V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann Oncol off J Eur Soc Med Oncol. 2018;29:71–83.
Tan S, Chen D, Liu K, He M, Song H, Shi Y, et al. Crystal clear: visualizing the intervention mechanism of the PD-1/PD-L1 interaction by two cancer therapeutic monoclonal antibodies. Protein Cell. 2016;7:866–77.
Lin KX, Istl AC, Quan D, Skaro A, Tang E, Zheng X. PD-1 and PD-L1 inhibitors in cold colorectal cancer: challenges and strategies. Cancer Immunol Immunother. 2023;72:3875–93.
Iwai Y, Hamanishi J, Chamoto K, Honjo T. Cancer immunotherapies targeting the PD-1 signaling pathway. J Biomed Sci. 2017;24:26.
Kwok G, Yau TCC, Chiu JW, Tse E, Kwong Y-L. Pembrolizumab (Keytruda). Hum Vaccines Immunother. 2016;12:2777–89.
Patnaik A, Kang SP, Rasco D, Papadopoulos KP, Elassaiss-Schaap J, Beeram M, et al. Phase I study of Pembrolizumab (MK-3475; Anti-PD-1 monoclonal antibody) in patients with Advanced Solid tumors. Clin Cancer Res off J Am Assoc Cancer Res. 2015;21:4286–93.
Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, et al. Safety and tumor responses with Lambrolizumab (Anti–PD-1) in Melanoma. N Engl J Med. 2013;369:134–44.
Schachter J, Ribas A, Long GV, Arance A, Grob J-J, Mortier L, et al. Pembrolizumab versus Ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet. 2017;390:1853–62.
Fuereder T. Immunotherapy for head and neck squamous cell carcinoma. Memo. 2016;9:66–9.
Peters S, Kerr KM, Stahel R. PD-1 blockade in advanced NSCLC: a focus on pembrolizumab. Cancer Treat Rev. 2018;62:39–49.
Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee J-L, Fong L, et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N Engl J Med. 2017;376:1015–26.
Chung HC, Ros W, Delord J-P, Perets R, Italiano A, Shapira-Frommer R, et al. Efficacy and safety of Pembrolizumab in previously treated Advanced Cervical Cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol off J Am Soc Clin Oncol. 2019;37:1470–8.
Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P, et al. Phase II study of the efficacy and safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J Clin Oncol. 2017;35:2125–32.
Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA approval Summary: Pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res. 2019;25:3753–8.
Fashoyin-Aje L, Donoghue M, Chen H, He K, Veeraraghavan J, Goldberg KB, et al. FDA approval Summary: Pembrolizumab for recurrent locally Advanced or metastatic gastric or gastroesophageal Junction Adenocarcinoma expressing PD-L1. Oncologist. 2019;24:103–9.
Zinzani PL, Ribrag V, Moskowitz CH, Michot J-M, Kuruvilla J, Balakumaran A, et al. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood. 2017;130:267–70.
Brahmer JR, Hammers H, Lipson EJ. Nivolumab: targeting PD-1 to bolster antitumor immunity. Future Oncol Lond Engl. 2015;11:1307–26.
Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–84.
Hazarika M, Chuk MK, Theoret MR, Mushti S, He K, Weis SL, et al. U.S. FDA approval Summary: Nivolumab for treatment of unresectable or metastatic melanoma following progression on Ipilimumab. Clin Cancer Res. 2017;23:3484–8.
Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced squamous-cell non–small-cell Lung Cancer. N Engl J Med. 2015;373:123–35.
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373:1803–13.
Kazandjian D, Suzman DL, Blumenthal G, Mushti S, He K, Libeg M, et al. FDA approval Summary: Nivolumab for the treatment of metastatic non-small cell Lung Cancer with Progression on or after platinum-based chemotherapy. Oncologist. 2016;21:634–42.
Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375:1856–67.
Zschäbitz S, Niegisch G. Zweitlinientherapie Des metastasierten urothelkarzinoms. Urol. 2020;59:804–9.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz H-J, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91.
Markham A, Duggan S. Cemiplimab: First Global approval. Drugs. 2018;78:1841–6.
Falchook GS, Leidner R, Stankevich E, Piening B, Bifulco C, Lowy I, et al. Responses of metastatic basal cell and cutaneous squamous cell carcinomas to anti-PD1 monoclonal antibody REGN2810. J Immunother Cancer. 2016;4:70.
Migden MR, Rischin D, Schmults CD, Guminski A, Hauschild A, Lewis KD, et al. PD-1 blockade with Cemiplimab in Advanced Cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379:341–51.
Wang L, Peng Y, Zeng X, Peng L, Li S, Qin S, et al. Cost-effectiveness analysis of Cemiplimab Versus Chemotherapy as First-Line treatment in Advanced NSCLC with PD-L1 expression levels of at least 50%. Adv Ther. 2021;38:4354–65.
Romero D. Cemiplimab is a new option in BCC. Nat Rev Clin Oncol. 2021;18:400.
Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer. 2022;21:28.
Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–62.
Felip E, Altorki N, Zhou C, Csőszi T, Vynnychenko I, Goloborodko O, et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB-IIIA non-small-cell lung cancer (IMpower010): a randomised, multicentre, open-label, phase 3 trial. Lancet Lond Engl. 2021;398:1344–57.
Balar AV, Galsky MD, Rosenberg JE, Powles T, Petrylak DP, Bellmunt J, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet Lond Engl. 2017;389:67–76.
Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Atezolizumab plus Bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med. 2019;7:387–401.
Apolo AB, Ellerton JA, Infante JR, Agrawal M, Gordon MS, Aljumaily R, et al. Avelumab as second-line therapy for metastatic, platinum-treated urothelial carcinoma in the phase ib JAVELIN solid tumor study: 2-year updated efficacy and safety analysis. J Immunother Cancer. 2020;8:e001246.
Shirley M, Avelumab. A review in metastatic Merkel Cell Carcinoma. Target Oncol. 2018;13:409–16.
Boyerinas B, Jochems C, Fantini M, Heery CR, Gulley JL, Tsang KY, et al. Antibody-dependent cellular cytotoxicity (ADCC) activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol Res. 2015;3:1148–57.
D’Angelo SP, Russell J, Lebbé C, Chmielowski B, Gambichler T, Grob J-J, et al. Efficacy and safety of first-line Avelumab Treatment in patients with Stage IV Metastatic Merkel Cell Carcinoma. JAMA Oncol. 2018;4:e180077.
Alvarez-Argote J, Dasanu CA. Durvalumab in cancer medicine: a comprehensive review. Expert Opin Biol Ther. 2019;19:927–35.
Faiena I, Cummings AL, Crosetti AM, Pantuck AJ, Chamie K, Drakaki A. Durvalumab: an investigational anti-PD-L1 monoclonal antibody for the treatment of urothelial carcinoma. Drug Des Devel Ther. 2018;12:209–15.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after Chemoradiotherapy in Stage III Non-small-cell Lung Cancer. N Engl J Med. 2017;377:1919–29.
Amin N, Maroun CA, El Asmar M, Alkhatib HH, Guller M, Herberg ME, et al. Neoadjuvant immunotherapy prior to surgery for mucosal head and neck squamous cell carcinoma: systematic review. Head Neck. 2022;44:562–71.
Siu LL, Even C, Mesía R, Remenar E, Daste A, Delord J-P, et al. Safety and Efficacy of Durvalumab with or without Tremelimumab in patients with PD-L1–Low/Negative recurrent or metastatic HNSCC. JAMA Oncol. 2019;5:195–203.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N Engl J Med. 2018;379:2342–50.
Sasikumar PG, Ramachandra M. Small-molecule Immune checkpoint inhibitors targeting PD-1/PD-L1 and other Emerging Checkpoint pathways. BioDrugs Clin Immunother Biopharm Gene Ther. 2018;32:481–97.
Geng Q, Jiao P, Jin P, Su G, Dong J, Yan B. PD-1/PD-L1 inhibitors for Immuno-oncology: from antibodies to small molecules. Curr Pharm Des. 2018;23:6033–41.
Liu J, Chen Z, Li Y, Zhao W, Wu J, Zhang Z. PD-1/PD-L1 checkpoint inhibitors in Tumor Immunotherapy. Front Pharmacol. 2021;12:731798.
Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B et al. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Struct Lond Engl. 1993. 2015;23:2341–8.
Skalniak L, Zak KM, Guzik K, Magiera K, Musielak B, Pachota M, et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget. 2017;8:72167–81.
Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–61.
Marangoni F, Zhakyp A, Corsini M, Geels SN, Carrizosa E, Thelen M, et al. Expansion of tumor-associated Treg cells upon disruption of a CTLA-4-dependent feedback loop. Cell. 2021;184:3998–e401519.
Wu K, Yi M, Qin S, Chu Q, Zheng X, Wu K. The efficacy and safety of combination of PD-1 and CTLA-4 inhibitors: a meta-analysis. Exp Hematol Oncol. 2019;8:26.
Rijavec E, Indini A, Ghidini M, Tomasello G, Cattaneo M, Barbin F, et al. Nivolumab plus Ipilimumab for the first-line treatment of metastatic NSCLC. Expert Rev Anticancer Ther. 2021;21:705–13.
Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–80.
Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF, et al. Two-year overall survival rates from a randomised phase 2 trial evaluating the combination of nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma. Lancet Oncol. 2016;17:1558–68.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Lao CD, et al. Five-year survival with combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med. 2019;381:1535–46.
Motzer RJ, Rini BI, McDermott DF, Frontera OA, Hammers HJ, Carducci MA, et al. Nivolumab plus Ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: extended follow-up of efficacy and safety results from a randomised phase 3 trial. Lancet Oncol. 2019;20:1370–85.
Baas P, Scherpereel A, Nowak AK, Fujimoto N, Peters S, Tsao AS, et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): a multicentre, randomised, open-label, phase 3 trial. Lancet Lond Engl. 2021;397:375–86.
Wang D-R, Wu X-L, Sun Y-L. Therapeutic targets and biomarkers of tumor immunotherapy: response versus non-response. Signal Transduct Target Ther. 2022;7:331.
Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Gutiérrez EC, et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N Engl J Med. 2022;386:24–34.
Harding JJ, Moreno V, Bang Y-J, Hong MH, Patnaik A, Trigo J, et al. Blocking TIM-3 in treatment-refractory Advanced Solid tumors: a phase Ia/b study of LY3321367 with or without an Anti-PD-L1 antibody. Clin Cancer Res off J Am Assoc Cancer Res. 2021;27:2168–78.
Cho BC, Abreu DR, Hussein M, Cobo M, Patel AJ, Secen N, et al. Tiragolumab plus Atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. 2022;23:781–92.
Tolcher AW, Sznol M, Hu-Lieskovan S, Papadopoulos KP, Patnaik A, Rasco DW, et al. Phase ib study of Utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in combination with Pembrolizumab (MK-3475) in patients with Advanced Solid tumors. Clin Cancer Res. 2017;23:5349–57.
Farnsworth RH, Lackmann M, Achen MG, Stacker SA. Vascular remodeling in cancer. Oncogene. 2014;33:3496–505.
Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7:987–9.
Wang K, Chen Q, Liu N, Zhang J, Pan X. Recent advances in, and challenges of, anti-angiogenesis agents for tumor chemotherapy based on vascular normalization. Drug Discov Today. 2021;26:2743–53.
Tian L, Goldstein A, Wang H, Lo HC, Kim IS, Welte T, et al. Mutual regulation of Tumour Vessel normalization and Immunostimulatory Reprogramming. Nature. 2017;544:250–4.
Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. 2012;109:17561–6.
Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol. 2016;17:611–25.
Hack SP, Zhu AX, Wang Y. Augmenting anticancer immunity through combined targeting of angiogenic and PD-1/PD-L1 pathways: challenges and opportunities. Front Immunol. 2020;11:598877.
Niu G, Chen X. Vascular endothelial growth factor as an anti-angiogenic target for Cancer Therapy. Curr Drug Targets. 2010;11:1000–17.
Lee MS, Ryoo B-Y, Hsu C-H, Numata K, Stein S, Verret W, et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): an open-label, multicentre, phase 1b study. Lancet Oncol. 2020;21:808–20.
Galle PR, Finn RS, Qin S, Ikeda M, Zhu AX, Kim T-Y, et al. Patient-reported outcomes with atezolizumab plus bevacizumab versus sorafenib in patients with unresectable hepatocellular carcinoma (IMbrave150): an open-label, randomised, phase 3 trial. Lancet Oncol. 2021;22:991–1001.
Kikuchi H, Matsui A, Morita S, Amoozgar Z, Inoue K, Ruan Z, et al. Increased CD8 + T-cell infiltration and efficacy for multikinase inhibitors after PD-1 blockade in Hepatocellular Carcinoma. JNCI J Natl Cancer Inst. 2022;114:1301–5.
Atkins MB, Plimack ER, Puzanov I, Fishman MN, McDermott DF, Cho DC, et al. Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: a non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. Lancet Oncol. 2018;19:405–15.
Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. 2019;380:1116–27.
Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. 2019;380:1103–15.
Ciccarese C, Iacovelli R, Porta C, Procopio G, Bria E, Astore S, et al. Efficacy of VEGFR-TKIs plus immune checkpoint inhibitors in metastatic renal cell carcinoma patients with favorable IMDC prognosis. Cancer Treat Rev. 2021;100:102295.
Levantini E, Maroni G, Del Re M, Tenen DG. EGFR signaling pathway as therapeutic target in human cancers. Semin Cancer Biol. 2022;85:253–75.
Tan C-S, Gilligan D, Pacey S. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol. 2015;16:e447–59.
Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, et al. Upregulation of PD-L1 by EGFR activation mediates the Immune escape in EGFR-Driven NSCLC: implication for Optional Immune targeted therapy for NSCLC patients with EGFR Mutation. J Thorac Oncol. 2015;10:910–23.
Sugiyama E, Togashi Y, Takeuchi Y, Shinya S, Tada Y, et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non–small cell lung cancer. Sci Immunol. 2020;5:eaav3937.
Gettinger S, Hellmann MD, Chow LQM, Borghaei H, Antonia S, Brahmer JR, et al. Nivolumab Plus Erlotinib in patients with EGFR-Mutant Advanced NSCLC. J Thorac Oncol off Publ Int Assoc Study Lung Cancer. 2018;13:1363–72.
Yang JC-H, Shepherd FA, Kim D-W, Lee G-W, Lee JS, Chang G-C, et al. Osimertinib Plus Durvalumab versus Osimertinib Monotherapy in EGFR T790M-Positive NSCLC following previous EGFR TKI therapy: CAURAL brief report. J Thorac Oncol off Publ Int Assoc Study Lung Cancer. 2019;14:933–9.
Su S, Dong Z-Y, Xie Z, Yan L-X, Li Y-F, Su J, et al. Strong programmed death Ligand 1 expression predicts poor response and De Novo Resistance to EGFR tyrosine kinase inhibitors among NSCLC patients with EGFR Mutation. J Thorac Oncol off Publ Int Assoc Study Lung Cancer. 2018;13:1668–75.
Pham MM, Ngoi N, Peng G, Tan DS, Yap TA. Development of poly(ADP-Ribose) polymerase (PARP) inhibitor and immunotherapy combinations: Progress, Pitfalls and promises. Trends Cancer. 2021;7:958–70.
Xue C, Xu Y, Ye W, Xie Q, Gao H, Xu B, et al. Expression of PD-L1 in ovarian cancer and its synergistic antitumor effect with PARP inhibitor. Gynecol Oncol. 2020;157:222–33.
Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-arm phases 1 and 2 trial of Niraparib in Combination with Pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol. 2019;5:1141–9.
Domchek SM, Postel-Vinay S, Im S-A, Park YH, Delord J-P, Italiano A, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020;21:1155–64.
Sullivan RJ. Back to the future: rethinking and retooling IL2 in the Immune checkpoint inhibitor era. Cancer Discov. 2019;9:694–5.
Doberstein SK. Bempegaldesleukin (NKTR-214): a CD-122-biased IL-2 receptor agonist for cancer immunotherapy. Expert Opin Biol Ther. 2019;19:1223–8.
Diab A, Tannir NM, Bentebibel S-E, Hwu P, Papadimitrakopoulou V, Haymaker C, et al. Bempegaldesleukin (NKTR-214) plus Nivolumab in patients with Advanced Solid tumors: phase I dose-escalation study of Safety, Efficacy, and Immune activation (PIVOT-02). Cancer Discov. 2020;10:1158–73.
Huang L, Xu H, Peng G. TLR-mediated metabolic reprogramming in the tumor microenvironment: potential novel strategies for cancer immunotherapy. Cell Mol Immunol. 2018;15:428–37.
Ribas A, Medina T, Kirkwood JM, Zakharia Y, Gonzalez R, Davar D, et al. Overcoming PD-1 blockade resistance with CpG-A toll-like receptor 9 Agonist Vidutolimod in patients with metastatic melanoma. Cancer Discov. 2021;11:2998–3007.
Formenti SC, Demaria S. Systemic effects of local radiotherapy. Lancet Oncol. 2009;10:718–26.
Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer. 2018;18:313–22.
Lan Y, Moustafa M, Knoll M, Xu C, Furkel J, Lazorchak A, et al. Simultaneous targeting of TGF-β/PD-L1 synergizes with radiotherapy by reprogramming the tumor microenvironment to overcome immune evasion. Cancer Cell. 2021;39:1388–e140310.
Wan C, Sun Y, Tian Y, Lu L, Dai X, Meng J, et al. Irradiated tumor cell–derived microparticles mediate tumor eradication via cell killing and immune reprogramming. Sci Adv. 2020;6:eaay9789.
Hu Y, Paris S, Bertolet G, Barsoumian HB, He K, Sezen D, et al. Combining a nanoparticle-mediated immunoradiotherapy with dual blockade of LAG3 and TIGIT improves the treatment efficacy in anti-PD1 resistant lung cancer. J Nanobiotechnol. 2022;20:417.
Gong X, Li X, Jiang T, Xie H, Zhu Z, Zhou F, et al. Combined Radiotherapy and Anti-PD-L1 antibody synergistically enhances Antitumor Effect in Non-small Cell Lung Cancer. J Thorac Oncol off Publ Int Assoc Study Lung Cancer. 2017;12:1085–97.
Parikh AR, Szabolcs A, Allen JN, Clark JW, Wo JY, Raabe M, et al. Radiation Therapy enhances Immunotherapy response in microsatellite-stable colorectal and pancreatic adenocarcinoma in a phase II trial. Nat Cancer. 2021;2:1124–35.
Chowdhury PS, Chamoto K, Honjo T. Combination therapy strategies for improving PD-1 blockade efficacy: a new era in cancer immunotherapy. J Intern Med. 2018;283:110–20.
Omuro A, Brandes AA, Carpentier AF, Idbaih A, Reardon DA, Cloughesy T, et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: an international randomized phase III trial. Neuro-Oncol. 2023;25:123–34.
Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014;21:15–25.
Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im S-A, Yusof MM, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet Lond Engl. 2020;396:1817–28.
Paz-Ares L, Vicente D, Tafreshi A, Robinson A, Soto Parra H, Mazières J, et al. A randomized, placebo-controlled trial of Pembrolizumab Plus Chemotherapy in patients with metastatic squamous NSCLC: protocol-specified final analysis of KEYNOTE-407. J Thorac Oncol off Publ Int Assoc Study Lung Cancer. 2020;15:1657–69.
Gogishvili M, Melkadze T, Makharadze T, Giorgadze D, Dvorkin M, Penkov K, et al. Cemiplimab plus chemotherapy versus chemotherapy alone in non-small cell lung cancer: a randomized, controlled, double-blind phase 3 trial. Nat Med. 2022;28:2374–80.
Le Rhun E, Oppong FB, Vanlancker M, Stupp R, Nabors B, Chinot O, et al. Prognostic significance of therapy-induced myelosuppression in newly diagnosed glioblastoma. Neuro-Oncol. 2022;24:1533–45.
Li X, Lovell JF, Yoon J, Chen X. Clinical development and potential of photothermal and photodynamic therapies for cancer. Nat Rev Clin Oncol. 2020;17:657–74.
Anand S, Chan TA, Hasan T, Maytin EV. Current prospects for Treatment of Solid Tumors via Photodynamic, Photothermal, or Ionizing Radiation therapies combined with Immune Checkpoint Inhibition (A Review). Pharm Basel Switz. 2021;14:447.
Zhang R, Zhu Z, Lv H, Li F, Sun S, Li J, et al. Immune Checkpoint Blockade mediated by a small-molecule Nanoinhibitor Targeting the PD-1/PD-L1 pathway synergizes with photodynamic therapy to Elicit Antitumor Immunity and Antimetastatic effects on breast Cancer. Small Weinh Bergstr Ger. 2019;15:e1903881.
Hwang HS, Cherukula K, Bang YJ, Vijayan V, Moon MJ, Thiruppathi J, et al. Combination of photodynamic therapy and a Flagellin-Adjuvanted Cancer Vaccine Potentiated the Anti-PD-1-Mediated melanoma suppression. Cells. 2020;9:2432.
Yuan Z, Fan G, Wu H, Liu C, Zhan Y, Qiu Y, et al. Photodynamic therapy synergizes with PD-L1 checkpoint blockade for immunotherapy of CRC by multifunctional nanoparticles. Mol Ther. 2021;29:2931–48.
Zhang Y, Liao Y, Tang Q, Lin J, Huang P. Biomimetic nanoemulsion for synergistic photodynamic-immunotherapy against hypoxic breast tumor. Angew Chem Int Ed. 2021;60:10647–53.
Zhu G, Chen X. Aptamer-based targeted therapy. Adv Drug Deliv Rev. 2018;134:65–78.
Geng Z, Wang L, Liu K, Liu J, Tan W. Enhancing anti-PD-1 immunotherapy by Nanomicelles Self-assembled from multivalent Aptamer Drug Conjugates. Angew Chem Int Ed. 2021;60:15459–65.
Du Y, Zhang D, Wang Y, Wu M, Zhang C, Zheng Y, et al. A highly stable multifunctional aptamer for enhancing antitumor immunity against hepatocellular carcinoma by blocking dual immune checkpoints. Biomater Sci. 2021;9:4159–68.
Prodeus A, Abdul-Wahid A, Fischer NW, Huang EH-B, Cydzik M, Gariépy J. Targeting the PD-1/PD-L1 Immune Evasion Axis with DNA aptamers as a Novel Therapeutic Strategy for the treatment of disseminated cancers. Mol Ther Nucleic Acids. 2015;4:e237.
Lai W-Y, Huang B-T, Wang J-W, Lin P-Y, Yang P-C. A novel PD-L1-targeting antagonistic DNA aptamer with Antitumor effects. Mol Ther Nucleic Acids. 2016;5:e397.
Zhang J, Li W, Qi Y, Wang G, Li L, Jin Z, et al. PD-L1 aptamer-functionalized metal–Organic Framework nanoparticles for Robust Photo-Immunotherapy against Cancer with enhanced safety. Angew Chem Int Ed. 2023;62:e202214750.
Met Ö, Jensen KM, Chamberlain CA, Donia M, Svane IM. Principles of adoptive T cell therapy in cancer. Semin Immunopathol. 2019;41:49–58.
Sui H, Ma N, Wang Y, Li H, Liu X, Su Y, et al. Anti-PD-1/PD-L1 therapy for non-small-cell lung Cancer: toward Personalized Medicine and Combination strategies. J Immunol Res. 2018;2018:6984948.
Creelan BC, Wang C, Teer JK, Toloza EM, Yao J, Kim S, et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1 resistant metastatic lung cancer: a phase I trial. Nat Med. 2021;27:1410–8.
Fang J, Ding N, Guo X, Sun Y, Zhang Z, Xie B, et al. αPD-1-mesoCAR-T cells partially inhibit the growth of advanced/refractory ovarian cancer in a patient along with daily apatinib. J Immunother Cancer. 2021;9:e001162.
Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat Med. 2020;26:732–40.
Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367:eaba7365.
Shalhout SZ, Miller DM, Emerick KS, Kaufman HL. Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol. 2023;20:160–77.
Nguyen H-M, Bommareddy PK, Silk AW, Saha D. Optimal timing of PD-1 blockade in combination with oncolytic virus therapy. Semin Cancer Biol. 2022;86:971–80.
Hamdan F, Ylösmäki E, Chiaro J, Giannoula Y, Long M, Fusciello M, et al. Novel oncolytic adenovirus expressing enhanced cross-hybrid IgGA fc PD-L1 inhibitor activates multiple immune effector populations leading to enhanced tumor killing in vitro, in vivo and with patient-derived tumor organoids. J Immunother Cancer. 2021;9:e003000.
Kiyokawa J, Kawamura Y, Ghouse SM, Acar S, Barcin E, Martínez-Quintanilla J, et al. Modification of extracellular matrix enhances oncolytic adenovirus immunotherapy in glioblastoma. Clin Cancer Res off J Am Assoc Cancer Res. 2021;27:889–902.
Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018;359:1355–60.
Sahin U, Oehm P, Derhovanessian E, Jabulowsky RA, Vormehr M, Gold M, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. 2020;585:107–12.
Kjeldsen JW, Lorentzen CL, Martinenaite E, Ellebaek E, Donia M, Holmstroem RB, et al. A phase 1/2 trial of an immune-modulatory vaccine against IDO/PD-L1 in combination with nivolumab in metastatic melanoma. Nat Med. 2021;27:2212–23.
O’Donnell JS, Long GV, Scolyer RA, Teng MWL, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. 2017;52:71–81.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707–23.
Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, et al. Resistance mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-intrinsic and -extrinsic factors. Immunity. 2016;44:1255–69.
Berner F, Bomze D, Diem S, Ali OH, Fässler M, Ring S, et al. Association of checkpoint inhibitor-Induced toxic effects with Shared Cancer and tissue antigens in Non-small Cell Lung Cancer. JAMA Oncol. 2019;5:1043–7.
Ramos-Casals M, Brahmer JR, Callahan MK, Flores-Chávez A, Keegan N, Khamashta MA, et al. Immune-related adverse events of checkpoint inhibitors. Nat Rev Dis Primer. 2020;6:38.
Bhardwaj M, Chiu MN, Pilkhwal Sah S. Adverse cutaneous toxicities by PD-1/PD-L1 immune checkpoint inhibitors: pathogenesis, treatment, and surveillance. Cutan Ocul Toxicol. 2022;41:73–90.
Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant myocarditis with combination Immune Checkpoint Blockade. N Engl J Med. 2016;375:1749–55.
Wei SC, Meijers WC, Axelrod ML, Anang N-AAS, Screever EM, Wescott EC, et al. A genetic mouse model recapitulates immune checkpoint inhibitor-associated myocarditis and supports a mechanism-based therapeutic intervention. Cancer Discov. 2021;11:614–25.
Salem J-E, Bretagne M, Abbar B, Leonard-Louis S, Ederhy S, Redheuil A et al. Abatacept/Ruxolitinib and screening for concomitant respiratory muscle failure to Mitigate Fatality of Immune-Checkpoint inhibitor myocarditis. Cancer Discov. 2023;CD-22-1180.
Baysoy A, Bai Z, Satija R, Fan R. The technological landscape and applications of single-cell multi-omics. Nat Rev Mol Cell Biol. 2023;1–19.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from The National Natural Science Foundation of China (No.U23A20456, 82272631, 82072596, 82173339, China), the National “111” Project (No. Project #111-2-12, China), the Hunan Provincial Key Research and Development Program (No.2022SK2026, China), the scientific research program of FuRong laboratory (No.2023SK2094, China), the Natural Science Foundation of Hunan Province, China (No.2024JJ3036, China), the Beijing Xisike Clinical Oncology Research Foundation (No.Y-HR2020ZD-0052, China), the Independent Exploration and Innovation Program of Central South University (No.1053320221155, China).
Author information
Authors and Affiliations
Contributions
X.L. conceived and wrote the manuscript; B.X. conceived the structure of manuscript; K.K., P.C., Z.Z., G.L., W.X., and M.Y. collected materials. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lin, X., Kang, K., Chen, P. et al. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol Cancer 23, 108 (2024). https://doi.org/10.1186/s12943-024-02023-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-024-02023-w