
Abstract
Divergent N6-methyladenosine (m6A) modifications are dynamic and reversible posttranscriptional RNA modifications that are mediated by m6A regulators or m6A RNA methylation regulators, i.e., methyltransferases (“writers”), demethylases (“erasers”), and m6A-binding proteins (“readers”). Aberrant m6A modifications are associated with cancer occurrence, development, progression, and prognosis. Numerous studies have established that aberrant m6A regulators function as either tumor suppressors or oncogenes in multiple tumor types. However, the functions and mechanisms of m6A regulators in cancer remain largely elusive and should be explored. Emerging studies suggest that m6A regulators can be modulated by epigenetic modifications, namely, ubiquitination, SUMOylation, acetylation, methylation, phosphorylation, O-GlcNAcylation, ISGylation, and lactylation or via noncoding RNA action, in cancer. This review summarizes the current roles of m6A regulators in cancer. The roles and mechanisms for epigenetic modification of m6A regulators in cancer genesis are segregated. The review will improve the understanding of the epigenetic regulatory mechanisms of m6A regulators.
Similar content being viewed by others

Background
Similar to DNA and proteins, RNA can undergo more than 170 post-transcriptional modifications [1]. In the 1970s, adenosine, an RNA building block, was demonstrated to be methylated at N6 nitrogen atom (i.e., N6-methyladenosine (m6A) formation) [2, 3]. Consequently, m6A modification has been identified as the most abundant cellular modification in mammalian mRNA. A pioneer study demonstrated, for the first time, role of m6A in mRNA stability [4], followed by cloning and discovery (in 1997) of methyltransferase-like protein 3 (METTL3), which synthesizes nearly all m6A in the mRNA transcriptome (Fig. 1) [5]. In addition, other studies have shown that m6A is essential for regulation of many developmental processes [6, 7]. This has resulted in rapid development of detection and transcriptome-wide mapping technologies for m6A-containing transcripts, enabling detection in nearly all types of RNAs, including mRNAs, small nuclear RNAs (snRNAs), ribosomal RNAs (rRNAs), and several species of regulatory RNAs [8]. Previous studies have largely focused on delineating the role of m6A methylation in mRNA metabolism and tumor progression; however, emerging evidence has revealed that m6A is involved in almost all RNA metabolic processes, such as mRNA maturation, transcription, translation, degradation, and stability. Dysregulation of m6A results in pathogenesis of multiple human diseases, including cancer. Growing evidence suggests m6A alteration is involved in tumorigenesis through many regulatory mechanisms in programmed cell death [9], metabolism [10], drug resistance [11], oncogene and/or tumor suppressor expression [12], immunotherapy [13], and targeted therapy [14]. The m6A RNA modification is dynamically and reversibly regulated by three enzymes, namely, m6A methyltransferases (“writers”), m6A demethylases (“erasers”), and m6A binding proteins (“readers”), that establish a complex interplay between m6A incorporation, degradation, and recognition [15, 16]. Enzymes mediating m6A effects are defined as m6A regulators or m6A RNA methylation regulators (Fig. 2) [14, 16]. Methyltransferases install m6A, demethylases remove m6A, and m6A-binding proteins recognize and act upon m6A-modified RNA. While writers and erasers determine the distribution and prevalence of m6A, readers mediate m6A-dependent functions [16]. Accumulating evidence has revealed that writers, erasers, and readers are frequently disordered and are involved in cancer pathogenesis by regulating the expression of oncogenes and/or tumor suppressors, promoting cancer proliferation, development, metastasis, and tumorigenesis [10,11,12, 14, 17, 18]. While previous studies mostly focused on the role of m6A RNA methylation in tumorigenesis, recent studies have explored m6A regulators in cancer genesis. Nevertheless, the functions and mechanisms of m6A regulators are unknown and need to be elucidated in cancer. Since 2015 [19], studies have revealed that m6A regulatory proteins are regulated by epigenetic modifications, such as ubiquitination, SUMOylation, acetylation, methylation, phosphorylation, and lactylation, or via noncoding RNA action, in cancer. In this review, a concise overview of the current understanding of the role of m6A regulators in cancer is provided. Additionally, the roles and mechanisms of epigenetic modifications of m6A regulators in cancer genesis are delineated. This review will enhance the understanding of the epigenetic regulatory mechanisms of m6A regulators.

Timeline diagram depicting essential discoveries in the field of m6A research

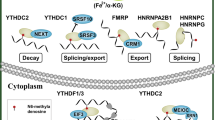
m6A regulator proteins and the underlying mechanisms of m6A modification. The m6A modification of mRNA is mainly catalyzed by the core methylase complex METTL3-WTAP-METTL14. RBM15/15B, VIRMA/KIAA1429, and ZC3H13 are newly identified mRNA modification writers; METTL4, and METTL16 are snRNA modification writhers; and METTL5 and ZCCHC4 are rRNA m6A writers. The m6A modification is removed by FTO, ALKBH5, and ALKBH3. Readers that include members of the YTH domain-containing family, the IGF2BP family, the HNRNP family, eIF3, PRRC2A, and FMRP, recognize modification and affect various functions of RNAs
m6A regulator proteins: m6A writers, erasers, and readers
The m6A writers, erasers, and readers constitute the molecular composition of m6A RNA methylation regulator proteins [14]. These are proteins that insert (writers), remove (erasers), and recognize (readers) m6A on mRNAs or noncoding RNAs. Proteins that mediate the effects of m6A establish a complex interplay between the above three m6A functions [15]. The effects of m6A on mRNA expression are mediated by an expanding list of m6A readers and m6A writer-complex components, as well as potential erasers. The mechanisms and effects of m6A-modifying regulatory proteins on RNA metabolism are summarized in Table 1.
Writers
The currently known m6A methyltransferases, or “m6A writers”, include methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), wilms tumor 1-associated protein (WTAP), RNA binding motif protein 15/15B (RBM15/RBM15B), vir-like m6A methyltransferase associated (VIRMA or KIAA1429), zinc finger CCCH-type containing 13 (ZC3H13), methyltransferase-like 16 (METTL16), methyltransferase-like 4 (METTL4), methyltransferase-like 5 (METTL5), and zinc finger CCHC-type containing 4 (ZCCHC4) (Table 1). The m6A, first reported om 1994, is a multicomponent methyltransferase complex [56]. Subsequently, METTL3, an S-adenosyl-methionine-binding protein with methyltransferase activity, was identified [5]. Recent studies have identified additional components of the m6A methyltransferase complex in mammals, namely, METTL14 [22, 57] and WTAP [22, 23], which are known to form a complex with METTL3 and are anchored to the nucleus to catalyze m6A methyltransferases [22, 23]. While METTL3 functions as a key catalytic component of the m6A methyltransferase complex [5], METTL14 is the core subunit of m6A methyltransferase for m6A installation [22] and WTAP is the regulatory subunit of m6A methyltransferase facilitating m6A modification [22, 23]. RBM15/15B is a subunit of the writer complex and facilitates the recruitment of the m6A writer complex to RNA by interacting with METTL3 in a WTAP-dependent manner [26, 58]. VIRMA (originally known as KIAA1429) is a regulatory subunit of m6A methyltransferase that facilitates m6A installation and functions as a WTAP interactor to associate with the METTL3/METTL14/WTAP complex, coordinatively modulating m6A modification [25, 58]. The ablation of VIRMA leads to a substantial loss of m6A in D. melanogaster [59] and mammalian cells [24]. VIRMA recruits the m6A complex to specific RNA sites and interacts with the polyadenylation cleavage factors CPSF5 and CPSF6, resulting in prolonged 3ʹUTR selection [25]. ZC3H13 interacts with WTAP and anchors it in the nucleus to promote m6A modification [31, 58], facilitating m6A addition and stem cell renewal [31]. Deletion of ZC3H13 resulted in the loss of m6A in D. melanogaster [30, 60] and approximately 80% loss of cellular m6A in mammalian cells [30], suggesting that some m6A sites are formed independent of ZC3H13. Similar to WTAP, ZC3H13 is important for the nuclear localization of the writer complex [31] and is assumed to promote RBM15/15B interaction with WTAP to facilitate methylation [30]. METTL16 mediates the insertion of m6A in small nuclear RNA (snRNAs) (e.g., the spliceosome component U6 snRNA) [27, 29]. METTL16 also functions as a methyltransferase and catalyzes m6A addition in U6-like sequences of MAT2A mRNA, the enzyme required for the biosynthesis of S-adenosylmethionine (SAM) [27, 61]. In addition, METTL16 catalyzes the addition of m6A in a small number of noncoding RNAs and mRNAs [29]. ZCCHC4 is a ribosomal RNA (rRNA)-adenosine-methyltransferase responsible for the formation of a single m6A residue in the 28 S ribosomal RNA (rRNA) [32, 62]. The addition of m6A on unique, highly conserved sites in the 18 S rRNA of eukaryotes is mediated by METTL5-TRMT112 complex, in which METTL5 functions as the catalytic subunit and TRMT112 as an allosteric adaptor [32]. METTL4 mediates m6A methylation of U2 snRNAs to regulate pre-mRNA splicing [36, 63].
Erasers
The m6A incorporation and removal in mRNA is a dynamic and reversible process, confirmed in 2011 with the discovery of the fat mass and obesity-associated protein (FTO), which is the first m6A demethylase that removes the methyl group to restore the methylated base to the adenine base [37]. FTO displays m6A demethylase activity and demethylates m6A residues in mRNA indicating the reversibility of this modification [37]. Mauer et al. characterized FTO as a m6A demethylase that regulates mRNA stability and suggested that m6A is a dynamic reversible modification, rekindling interest in the biological relevance of m6A [64]. Furthermore, Zheng et al. discovered the second mammalian m6A demethylase, namely, alkB homologue 5 (ALKBH5), that affects mouse spermatogenesis and demonstrated that m6A is a dynamic reversible modification of mRNA [38]. FTO and ALKBH5 facilitate the removal of m6A and potentially affect different subsets of target mRNAs because of their distinct subcellular and tissue distributions [37, 38]. The first evidence of reversible post-transcriptional modification was given when FTO and ALKBH5 removed addition of m6A in mRNA and certain noncoding RNAs transcribed by RNA polymerase II [37, 38]. By definition, ALKBH3 is an eraser responsible for the removal of the m6A modification on the tRNA [39].
Readers
m6A can recruit m6A-binding proteins or m6A readers that mediate m6A-dependent functions to regulate the fate of mRNAs [16, 65]. The m6A readers regulate mRNA nuclear export, splicing, degradation, translation, and stability. The first discovered m6A reader family, providing a mechanistic basis for understanding the effects of m6A on mRNA, was the YT521-B homology (YTH) domain family of proteins [66]. The YTH domain family includes YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2. The nuclear m6A readers are YTHDC1, HNRNPC11, HNRNPA2B1, and HNRNPG, whereas m6A readers in the cytosol are YTHDC2, YTHDF1/2/3, and IGF2BP1/2/3. Different readers have different m6A positioning functions [67]. YTHDF2, the first discovered m6A-binding protein, regulates mRNA degradation by mediating the lifetime of target transcripts [41, 66]. Similarly, YTHDF1 promotes translation of m6A-modified mRNAs in the cytosol [42], while YTHDF3 cooperates with YTHDF1 and YTHDF2 to modulate the translation and degradation of m6A-labelled mRNA and inversely regulates their functions [50]. The insulin-like growth factor 2 mRNA-binding proteins (IGF2BP1/2/3) promote mRNA stability and translation [52]. FMRP enhances the nuclear export and stability of m6A-decorated RNAs [53, 54]. Furthermore, YTHDC1 modulates nuclear export and splicing of m6A-modified RNAs [47, 48], while YTHDC2 regulates the translation and abundance of target genes [51]. As a multiprotein complex that recruits small ribosomal subunits to mRNAs, Eukaryotic initiation factor 3 (eIF3) preferentially binds to m6A-decorated mRNA and is involved in mRNA translation [42, 68]. YTHDF1 recruits eIF3 to the 5’ end of the transcripts, resulting in YTHDF1 looping that modulates initiation of translation [42]. The heterogeneous nuclear ribonucleoprotein (HNRNP) proteins include HNRNPA2B1, HNRNPC, and HNRNPG. HNRNPA2B1 [44] and HNRNPC[45] are active splicing regulators that can selectively bind m6A-decorated mRNAs [45, 69, 70]. HNRNPA2B1 recognizes m6A-labelled primary miRNAs (pri-miRNAs) and regulates alternative splicing events [44] and miRNA biogenesis [44, 71]. HNRNPC recognizes m6A-induced changes in secondary mRNA structures [45], and HNRNPG is an RNA-binding protein involved in the splicing of m6A-labelled mRNA[72]. Proline-rich coiled-coil 2 A (PRRC2A) was later identified as a novel m6A reader that binds to a consensus GGACU motif in the Olig2 coding sequence to stabilize Olig2 mRNA [46].
m6A regulator proteins and cancer
Previous studies have shown that m6A is associated with numerous human diseases, including cancer. Pioneering studies have provided molecular evidence of the direct regulatory roles of m6A in cancer [73, 74]. The ablation of METTL3 caused apoptosis and reduced the invasiveness of lung adenocarcinoma cells [73], whereas hypoxia-activated m6A demethylase ALKBH5 induces the accumulation of breast cancer stem cells through HIF-dependent and ALKBH5-mediated m6A demethylation of NANOG mRNA [74]. Recent evidence has indicated that m6A regulatory proteins, i.e., writers, erasers, and readers, play a role in various types of human cancers by contributing to malignancy. This includes cancer cell proliferation, self-renewal of cancer stem cells, and resistance to radiotherapy or chemotherapy. Comprehensive reviews for detailed discussions on the role of m6A regulatory proteins in cancer are already available in literature [11, 12, 14, 17, 18, 67, 75,76,77,78,79]. However, the functions and mechanisms of m6A regulators in cancer remain largely unestablished and need future investigations.
Epigenetic modification of m6A regulators and tumorigenesis
Epigenetics is a reversible and dynamic process that regulates gene expression without altering DNA. There are four major mechanisms of epigenetic regulation: DNA methylation, histone modification, chromatin structure regulation, and noncoding RNA regulation [80, 81]. All mechanisms, except chromatin structure regulation, have been studied extensively [82]. The histone subunit in the nucleosome possesses a characteristic tail containing specific amino acids for covalent posttranslational modifications (PTMs), such as acetylation, methylation, ubiquitylation, phosphorylation, glycosylation, sumoylation, acylation, glycation, hydroxylation, serotonylation, and ADP-ribosylation [83,84,85,86]. Recent studies have suggested that m6A regulators in cancer can be modulated by epigenetic modifications, including ubiquitination, SUMOylation, acetylation, lactylation, O-GlcNAcylation, methylation, phosphorylation, ISGylation, and noncoding RNA. Hence, this section focuses on the roles and mechanisms of the epigenetic modification of m6A regulators in cancer genesis. The effects and mechanisms of epigenetic modification of m6A regulatory proteins in tumorigenesis are summarized in Table 2.
Ubiquitination/deubiquitination
Ubiquitination, a highly conserved and key protein PTM, plays an important role in controlling substrate degradation of various proteins [121, 122]. The deubiquitinases (DUBs) can reverse ubiquitination by removing ubiquitin chains, resulting in the termination of ubiquitination and preservation of substrate protein expression levels [122]. The interaction between ubiquitination and deubiquitination plays an essential role in controlling all aspects of biological activity, including cancer. Recent studies have shown that ubiquitination/deubiquitination is involved in the regulation of m6A regulatory proteins in cancer (Fig. 3).

Epigenetic modification of m6A regulator proteins by ubiquitination and SUMOylation in cancer. BC, Breast cancer; CRC, colorectal cancer; GBC, gallbladder cancer; GBM, glioblastoma; HCC, hepatocellular carcinoma; NSCLC, non-small cell lung carcinoma; PC, Pancreatic cancer
Ubiquitination/deubiquitination of writers
USP38 mediates METTL14 protein deubiquitination; therefore, METTL14 overexpression inhibits bladder cancer cell (BCa) malignancy. METTL14 stabilizes USP38 mRNA through m6A modification in a YTHDF2-dependent manner, demonstrating that METTL14 suppresses BCa progression and forms a feedback loop with USP38 [96]. Similarly, USP29 upregulation mediates KIAA1429 deubiquitination, thereby stabilizing SOX8 mRNA and protein levels through m6A modification to facilitate malignant proliferation in colorectal carcinoma (CRC) [95]. In addition, METTL3 expression has been shown to significantly increase with tumor progression and positively correlate with peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (PIN1) expression in breast cancer tissues. PIN1 interacts with and stabilizes METTL3 by preventing its ubiquitin-dependent proteasomal and lysosomal degradation, thereby increasing the m6A modification of transcriptional coactivator with PDZ-binding motif (TAZ) and epidermal growth factor receptor (EGFR) mRNA, resulting in their efficient translation [97]. This suggests that PIN1 regulates METTL3 through ubiquitination in breast cancer [97].
Ubiquitination/deubiquitination of erasers
Downregulation of GSK3β inhibits the ubiquitination of FTO, in turn, stabilizing FTO levels. In succession, FTO increases MZF1 expression by mediating the FTO-regulated m6A modification of MZF1 and consequently, promotes c-Myc expression and cell proliferation [87]. The former study suggests that GSK3β acts as a suppressor in CRC. This observation was later confirmed by other studies, wherein FTO was shown to act as a tumor suppressor in CRC by reducing the expression of metastasis-associated protein 1 (MTA1) in an m6A-dependent manner using IGF2BP2 [88]. The hypoxic tumor microenvironment reduces FTO protein expression by increasing serine/threonine kinase receptor-associated protein (STRAP)-mediated ubiquitination and facilitates CRC metastasis [88]. Ubiquitin-specific peptidase 18 (USP18) upregulates FTO levels through post-translational deubiquitination while decreasing m6A levels in PYCR1, thereby stabilizing the PYCR1 transcript and promoting bladder cancer initiation and progression [89]. Collectively, the above findings define the crucial role played by ubiquitination/deubiquitination in the modulation of FTO in cancer and reveal a novel epigenetic modification of FTO. In addition, USP36 deubiquitinates and stabilizes ALKBH5. The depletion of USP36 drastically decreases glioma tumorigenesis, impairs cell proliferation, deteriorates the self-renewal of GSCs, and increases the sensitivity of GSCs to temozolomide (TMZ) [90].
Ubiquitination/deubiquitination of readers
The IGF2BP family of m6A regulatory proteins is also modified by ubiquitination or deubiquitination in cancer. The elevation of E3 ubiquitin ligase F-box/SPRY domain-containing protein 1 (FBXO45) promotes hepatocellular carcinoma (HCC) tumorigenesis through IGF2BP1 ubiquitination and activation, resulting in the upregulation of polo-like kinase (PLK1) expression, suggesting possibility of a new therapeutic regimen for HCC that targets the FBXO45/IGF2BP1/PLK1 axis [91]. TEA domain family member 4 (TEAD4)-transcriptionally activated lncRNA MNX1-AS1 suppresses IGF2BP3 degradation by recruiting USP16. The MNX1-AS1/IGF2BP3 axis inhibits the Hippo signaling pathway, thereby activating TEAD4. Consequently, MNX1-AS1 promotes tumorigenesis, progression, and metastasis of gallbladder cancer (GBC) through an MNX1-AS1/IGF2BP3/Hippo pathway positive feedback mechanism [92]. Similarly, USP11 upregulation protects IGF2BP3 from degradation via deubiquitination and promotes CRC tumorigenesis [93]. Another study has shown that upregulated Linc01232 suppresses the ubiquitin-induced degradation of HNRNPA2B1 and activates A-Raf-induced MAPK/ERK, in turn, promoting the metastasis of pancreatic cancer (PC) [94].
SUMOylation
SUMOylation is defined as a post-translational protein modification by conjugation of small ubiquitin-like modifier (SUMO) proteins to substrate proteins. As it is a dynamic as well as reversible process, it has been associated with various cellular processes and is a vital mechanism in cellular stress responses [123]. SUMOylation occurs via an enzymatic cascade involving a dimeric SUMO-activating enzyme E1 (SAE1 and SAE2/UBA2), a single E2 (ubiquitin-conjugating enzyme 9, UBC9), and a limited set of E3 ligases [124]. SUMO-specific proteases (SENPs) cooperate with SUMO molecules to regulate the SUMOylation state of substrate proteins by specifically de-SUMOylating them. SUMOylation is aberrantly upregulated in many cancer stages, including tumorigenesis, epithelial-mesenchymal transition (EMT), metastasis, drug resistance, and antitumor immunity [123, 125].
SUMOylation of writers
SUMO1-mediated SUMOylation of METTL3 promotes tumor progression by regulating Snail mRNA homeostasis in an m6A methyltransferase activity-dependent manner in HCC (Fig. 3) [98]. The upregulated expression of METTL3, circ_0000677, and ABCC1 has been observed in CRC. SUMO1-mediated METTL3 SUMOylation facilitates CRC progression and drug resistance by stabilizing circ_0000677 in an m6A-dependent manner, thereby upregulating ABCC1 expression [99]. SUMOylation of METTL3 by SUMO1 promotes tumorigenesis in human non-small cell lung carcinoma (NSCLC). SUMOylation of METTL3, usually reversed by SENP1, significantly inhibits its m6A methyltransferase activity, leading to decreased m6A mRNA levels [100].
SUMOylation of erasers
A recent study demonstrated that SIRT1 functions as an oncogene by downregulating FTO via RANBP2-mediated FTO SUMOylation and degradation. SIRT1 activates RANBP2, a critical component of the E3 ligase SUMOs and essential for SUMOylation of FTO at the lysine (K-216) site that promotes FTO degradation. As a tumor suppressor in HCC, the guanine nucleotide-binding protein G(o) subunit alpha (GNAO1) is a m6A downstream target of FTO, and SIRT1-mediated ablation of FTO downregulates GNAO1 mRNA expression through increasing m6A modification [101]. This study suggests that SIRT1 destabilizes FTO, steering GNAO1 as an m6A-modified downstream molecule in HCC tumorigenesis.
SUMOylation of readers
HNRNPA2B1 expression is an independent predictor of good prognosis in patients with breast cancer. SUMOylation of HNRNPA2 mediated by a protein inhibitor of activated STAT 2 (PIAS2) functions as an endogenous inhibitor of replication protein A1 (RPA1). HNRNPA2B1 hinders homologous recombination (HR) repair by limiting RPA availability and increasing sensitivity to PARP inhibitors [102]. A recent study demonstrated that hypoxia upregulates UBC9 expression and increases SUMOylation of hnRNP A2/B1, promoting its nuclear export to eliminate miR-204-3p in glioma. As exosomal miR-204-3p is known to promote tube formation in vascular endothelial cells via the ATXN1/STAT3 pathway, TAK-981, a SUMOylation inhibitor, can inhibit miR-204-3p sorting into exosomes and inhibits tumor growth and angiogenesis. This suggests that TAK-981 could be a potential therapeutic target for gliomas [103]. SUMOylation of IGF2BP2 by SUMO1 increases IGF2BP2 expression by blocking its ubiquitin-proteasome pathway-dependent degradation. This upregulation stabilizes lncRNA OIP5-AS1, which in turn, binds to miR-495-3p and decreases the association of miR-495-3p, hypoxia-inducible factor 1 alpha (HIF1A), and matrix metalloproteinase 14 (MMP14) mRNA, ultimately promoting the formation of vasculogenic mimicry in glioma [104]. SUMOylation of YTHDF2 at the major site, K571, can be increased by hypoxia and reduced by oxidative stress and SUMOylation inhibitors. The binding affinity of SUMOylated YTHDF2 to m6A-labelled mRNA is significantly increased and resultant deregulated gene expression causes cancer progression in NSCLC [105]. The above study uncovered a new regulatory mechanism for YTHDF2 recognition by m6A-RNA, highlighting the important role of YTHDF2 SUMOylation in the post-transcriptional regulation of gene expression in NSCLC progression [105].
Acetylation
Protein acylation plays a vital role in key cellular processes involved in physiology and disease, such as enzyme activity, protein stability, subcellular localization, protein-protein interactions, transcriptional activity, and protein-DNA interactions [126]. Histone acetylation was first identified as a mechanism of gene transcription regulation in the early 1960s [127]. After the first finding, acetylation of the non-histone protein, p53, was discovered in the 1980s, followed by identification of multiple non-histone proteins as targets for acylation [126]. A recent study demonstrated that acetylation plays a role in regulating METTL3 localization and tumorigenic progression in breast cancer (Fig. 4) [108]. METTL3 acetylation is a key PTM for determining its cellular translocation. Li et al. demonstrated that METTL3 acetylation by EP300/CBP hinders the migration and invasion potential of breast cancer cells. It is known that physiological stimuli modulate METTL3 nuclear entry. IL-6-induced deacetylation promotes the nuclear shift of METTL3 via the AMPK/SIRT1 axis, whereas ASP/NAM-mediated acetylation decreases its nucleus import [108]. The METTL3-mediated m6A modification of IL-6 mRNA enhances METTL3 deacetylation and nuclear translocation, whereas SIRT1 inhibition counterbalances this deacetylation-mediated nuclear shift of METTL3. Intriguingly, reconstitution of acetylation-mimetic METTL3 mutant resulted in enhanced translation and compromised metastatic potential, revealing an acetylation-mediated regulatory mechanism that determines the subcellular localization of METTL3 [108]. Additionally, lysine acetyltransferase 2 A (KAT2A)-mediated H3K27 acetylation activates METTL3, promoting cancer metastasis by activating early growth response-1 (EGR1)/Snail signaling in a YTHDF3-dependent manner and revealing a susceptibility to METTL3 blockade in esophageal squamous cell carcinoma. The anti-HIV drug elvitegravir inhibited metastasis by directly targeting METTL3 and enhancing stress-inducible phosphoprotein 1 homology and U-box containing protein 1 (STUB1)-mediated proteasomal degradation in esophageal squamous cell carcinoma (ESCC) [107]. METTL3 acetylation mediated reduced N6-Methyladenosine to promote the expression of metal regulatory transcription factor 1(MTF1) and HCC progression [109]. EP300/CBP-mediated histone 3 acetylation upregulates RBM15 and promotes clear cell renal cell carcinoma (ccRCC) progression by stabilizing CXCL11 mRNA in an m6A-dependent manner [106].

Epigenetic modification of m6A regulator proteins by acetylation, methylation, O-GlcNAcylation, ISGylation, phosphorylation, and lactylation, or noncoding RNA in cancer. ccRCC, clear cell renal cell carcinoma; CRC, colorectal cancer; ESCC, esophageal squamous cell carcinoma; GBC, gallbladder cancer; GBM, glioblastoma; HCC, hepatocellular carcinoma; NSCLC, non-small cell lung carcinoma; OC, ovarian cancer; OM, ocular melanoma; OSCC, oral squamous cell carcinoma
Phosphorylation
Phosphorylation is an important epigenetic PTM that strongly correlates with the occurrence and development of multiple diseases, including cancer [128]. Sun et al. demonstrated that activated ERK phosphorylates METTL3 and WTAP. This phosphorylation of METTL3 facilitates its interaction with USP5, thereby stabilizing the m6A METTL3-METTL14-WTAP methyltransferase complex by deubiquitination as shown in Fig. 4 [113]. The loss of METTL3/WTAP phosphorylation reduces the degradation of m6A-labelled pluripotent factor transcripts and traps mouse embryonic stem cells (mESC) in a pluripotent state. METTL3 phosphorylation in ERK-activated tumor cells contributes to CRC tumorigenesis, suggesting that a new function of ERK in regulating m6A methylation exists and that the activation of the ERK-METTL3/WTAP axis promotes tumorigenesis [113].
Lactylation
Lactylation is a novel PTM that was initially reported by Zhao et al. (2019) as an indicator of lactate levels and glycolysis [129]. Lactylation has intrinsic connections with cell lactate metabolism which is linked to metabolic rewiring and epigenetic remodeling. Therefore, it represents a novel epigenetic code that affects cellular dysfunction and carcinogenesis [130]. Recent studies have identified lactate-derived lactylation of lysine (Kla) residues on histones as an epigenetic modification that directly stimulates gene transcription from chromatin [129]. Increasing experimental evidence suggests that lactylation plays a role in tumorigenesis. A recent study provides insight into the lactylome profile of hepatitis B virus (HBV)-related HCC, demonstrating an important role for non-histone Kla in HCC progression, preferentially affecting metabolic proteins as shown in Fig. 4 [131]. Hypoxia-induced glycolysis promotes lactylation, thereby stabilizing catenin and aggravating the malignant behavior of CRC cells [132]. Proprotein convertase subtilisin/kexin type 9 (PCSK9) is involved in the progression and metastasis of CRC by regulating EMT and PI3K/AKT signaling and polarization of macrophages. It acts by mediating migration inhibitory factor (MIF), lactate levels, and protein lactylation [133]. In addition, lactate acts as an essential molecule that boosts regulatory T cells (Treg cells) in the tumor microenvironment by lactylating MOESIN at Lys72. This results in enhanced interaction of MOESIN with transforming growth factor β (TGF-β) receptor I and downstream SMAD3 signaling [134]. Another study showed that HIF1α lactylation enhances transcription of hyaluronic acid (HA) binding protein, KIAA1199, to promote angiogenesis and vasculogenic mimicry in prostate cancer [135]. Therefore, the inhibition of lactylation is a therapeutic target for cancer [136]. Novel studies suggest that lactylation regulates m6A regulator proteins in cancer [110, 111]. Lactylation of METTL3 by acetyltransferase p300 induces Mettl3 expression via H3K18la. Lactylation of the METTL3-JAK1-STAT3 regulatory axis induces immunosuppressive functions in tumor-infiltrating myeloid cells in CRC [110]. Additionally, lactylation drives oncogenesis by facilitating YTHDF2 expression in ocular melanomas [111]. Here, lactylation of YTHDF2 was mediated by EP300 at H3K18la. As YTHDF2 recognizes m6A-labelled PER1 and TP53 mRNAs and promotes their degradation, it accelerates tumorigenesis in ocular melanoma [111].
O-GlcNAcylation
The attachment of O-linked N-acetylglucosamine (O-GlcNAc) moieties to serine or threonine residues of nuclear, cytoplasmic, and mitochondrial proteins is an important PTM that links nutrient flux to gene transcription during virus replication and tumorigenesis [137, 138]. O-GlcNAcylation is dynamically regulated by O-GlcNAc Transferase (OGT) and O-GlcNAcase (OGA). Recently, aberrant O-GlcNAcylation is emerging as a common feature of cancer, owing to deregulated cellular nutrient flux [139, 140]. A recent study, for the first time, showed that O-GlcNAcylation plays a role in the regulation of m6A regulatory proteins in HCC. O-GlcNAcylation of YTHDF2 promotes HBV-associated HCC progression in an m6A-dependent manner, as shown in Fig. 4 [112]. OGT-mediated O-GlcNAcylation of YTHDF2 promotes protein stability and oncogenic activity by inhibiting ubiquitination. YTHDF2 stabilizes minichromosome maintenance protein 2 (MCM2) and MCM5 transcripts in an m6A-dependent manner, promoting cell cycle progression and HBV-related HCC tumorigenesis. OSMI-1, an OGT inhibitor, significantly suppresses HCC progression by targeting YTHDF2 O-GlcNAcylation [112]. Collectively, these findings demonstrate a new regulatory mechanism for YTHDF2 through O-GlcNAcylation and highlight the vital role of YTHDF2 O-GlcNAcylation in m6A RNA methylation and HCC progression.
Methylation
Protein methylation, first discovered in 1959 [141], is a crucial PTM that regulates the functions of both histone and non-histone proteins [142]. Since the discovery of histone methylation in 1964 [143], numerous studies have unveiled the biology behind protein methylation [144]. Protein methylation occurs mainly at the side chains of lysine (Lys) and arginine (Arg) residues [145]. While lysine residues can be mono-, di-, or trimethylated (me1, me2, and me3, respectively) in a SAM-dependent manner [146], arginine residues can be mono- or demethylated at the respective side-chain by protein arginine methyltransferases (PRMTs) with SAM as the methyl donor [145, 147]. Ample evidence exists that shows involvement of dysregulation of protein methylation in the cancer development and progression [148, 149]. A recent study, for the first time, showed that the arginine methylation plays a role in regulating m6A regulatory proteins in leukemia (Fig. 4) [19]. The RNA-binding protein, RBM15, is methylated at residue R578 by PRMT1, leading to its degradation via E3 ligase (CNOT4)-mediated ubiquitylation. RBM15 binds to the pre-messenger RNA intronic regions of RUNX1, GATA1, TAL1, and c-MPL, a mechanism considered important for megakaryopoiesis. Furthermore, PRMT1 regulates alternative RNA splicing by reducing RBM15 protein concentration [19].
ISGylation
Ubiquitin, covalently conjugated to other protein substrates, was first discovered in 1975 [150]. This discovery prompted the finding of ubiquitin-like proteins (UBLs) that are structurally and evolutionarily related to ubiquitin [e.g., interferon-stimulated gene 15 (ISG15), small ubiquitin-like modifier (SUMO), and NEDD8] [151]. The first UBL, ISG15, was discovered in 1979 and can mediate ISGylation or ubiquitin-like covalent modification of other proteins [152]. Two studies suggest a role for ISG15 and ISGylation in cancer progression [151, 153]. A recent study showed that ISG15 suppresses the translation of multidrug resistance-associated protein 2 (MRP2/ABCC2) via ISGylation of hnRNPA2B1 and enhances drug sensitivity in cisplatin-resistant ovarian cancer cells (Fig. 4) [114]. While ISG15 expression is downregulated in cisplatin-resistant ovarian cancer cells, overexpression of wild-type ISG15 increases cisplatin-sensitivity of ovarian cancer cells through ISGylated hnRNPA2B1 blockage of its recruitment, and consequently, decreases MRP2/ABCC2 translation and expression [114].
Noncoding RNA
Noncoding RNA, or ncRNAs, are functional RNA with limited or no protein-coding abilities but are one of the most common epigenetic regulation mechanisms [154, 155]. NcRNAs interact with target molecules and participate in the regulation of disease development, including cancer [156]. Recent evidence indicates a regulatory role for ncRNAs in the control of m6A regulatory proteins in cancer (Fig. 4). It has been shown that upregulated circNEIL3 stabilizes IGF2BP3 by preventing HECTD4-mediated ubiquitination, in turn, promoting tumorigenesis and progression of gliomas [120]. Another study has demonstrated that circEZH2 works as a sponge for miR-133b to upregulate IGF2BP2 and blocks its ubiquitination-dependent degradation, thereby facilitating the proliferation and migration of CRC cells [115]. Hsa_circ_0026134 promotes TRIM25- and IGF2BP3-mediated proliferation and invasion by sponging miR-127-5p [117]. Upregulated lncRNA CYTOR promotes migration, invasion, and EMT. CYTOR inhibits HNRNPC ubiquitination and stabilizes ZEB1 mRNA [119]. Similarly, upregulated LINRIS is demonstrated to promote malignancy. Knockdown of LINRIS decreases IGF2BP2 levels through IGF2BP2 ubiquitination and attenuates MYC-mediated glycolysis in CRC cells [116]. Another study has shown that decreased miR503HG is present in HCC. Enhanced expression of miR503HG significantly inhibits the invasion and metastasis of HCC. miR503HG interacts with HNRNPA2B1 and promotes its degradation via the ubiquitin-proteasome pathway, resulting in decreased stability of p52 and p65 mRNA while suppressing NF-κB signaling in HCC cells [118].
Conclusion and perspectives
While previous studies mainly focused on the role of m6A RNA methylation in tumorigenesis, recent studies provide insight into m6A regulators in cancer genesis. Nevertheless, the functions and mechanisms of m6A regulators are not completely understood and need to be elucidated in cancer. Emerging evidence since 2015 has shown that m6A can be regulated by epigenetic modifications in cancers [19]. In this review, we have discussed the roles and mechanisms of the epigenetic modifications of m6A regulators in cancer genesis and highlighted the crucial role of the epigenetic modification of m6A regulators in tumorigenesis, explaining the regulatory interaction between the epigenetic modification of m6A regulators and m6A modification of RNA in cancer pathogenesis. However, the understanding of epigenetic modification of m6A regulators in cancer is still in its infancy.
Crosstalk between histone modifications occurs when one or more histone modifications modulate the recognition, addition, or removal of another modification, or synergistically function to repress or promote the gene transcription [157, 158]. There is exists an interplay between m6A RNA methylation and other epigenetic regulators [159]. The listed epigenetic modifications on m6A regulators are complete, however most of these studies maybe have some disadvantages for their focus on one epigenetic modifications mechanism on m6A regulators. Nevertheless, continuous progress in this field is taking place, and whether these epigenetic regulatory mechanisms are specific to other types of cancer remains to be explored. Little is known about the interplay between two different epigenetic modifications on the same m6A regulators. In addition to ubiquitination, SUMOylation, acetylation, methylation, phosphorylation, O-GlcNAcylation, ISGylation, and lactylation or via noncoding RNA action, whether other epigenetic modification including malonylation, succinylation, and glutarylation, et al. are involved in regulating m6A regulatory proteins remains unclear. Thus, additional studies of the roles of other potential epigenetic modification on m6A regulatory proteins are warranted.
Growing evidence suggests targeting m6A regulatory proteins maybe work as a novel therapeutic opportunities for immunotherapy or drug resistance in cancer, and m6A regulatory proteins can be feasibly targeted by small-molecules targeting m6A regulators [160]. Revealing epigenetic regulation mechanism of m6A regulatory proteins in cancer will accelerate the development of promising combination therapeutic regimes containing epigenetic agents and targeting m6A regulatory proteins to overcome chemotherapy resistance, and highlights some promising therapeutic avenues that may be used to surmount chemotherapy drug resistance. Whether the epigenetic modification affect multiple m6A regulatory proteins and how these different epigenetic modification corporate with diverse signaling pathways to determine the role of epigenetic modification in cancer. A profound study on the epigenetic modification network of m6A regulatory proteins process requires extensive investigation. We believe that identifying the effects of epigenetic regulation on m6A regulatory proteins will lead to a better understanding of cancer genesis and provide better therapeutic targets.
As concluded, studies about epigenetic modification of m6A regulator proteins is an emerging research field in cancer, and bring a new frontier to cancer research. This implies an additional layer of complexity for the interpretation of m6A modification. The role of epigenetic regulation on m6A regulatory proteins in cancer remains an open conundrum for future investigate on.
Data Availability
All data generated or analyzed during this study are included in this published article.
Abbreviations
- ALKBH3:
-
AlkB homologue 3
- ALKBH5:
-
AlkB homologue 5
- BC:
-
Breast cancer
- BCa:
-
Bladder cancer
- ccRCC:
-
Clear cell renal cell carcinoma
- CRC:
-
Colorectal cancer
- DUBs:
-
Deubiquitinases
- eIF3:
-
Eukaryotic translation initiation factor 3 subunit A
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial-mesenchymal transition
- ESCC:
-
Esophageal squamous cell carcinoma
- FMRP:
-
Fragile X mental retardation protein
- FTO:
-
Fat mass and obesity-associated protein
- GBC:
-
Gallbladder cancer
- GBM:
-
Glioblastoma
- HCC:
-
Hepatocellular carcinoma
- HNRNPC:
-
Heterogeneous nuclear ribonucleoprotein C
- HNRNPA2B1:
-
Heterogeneous nuclear ribonucleoprotein A2/B1
- HNRNPG:
-
Heterogeneous nuclear ribonucleoprotein G
- HNRNPA2B1:
-
Heterogeneous nuclear ribonucleoprotein A2/B1
- IGF2BP1:
-
Insulin-like growth factor 2 mRNA binding protein 1
- IGF2BP2:
-
Insulin-like growth factor 2 mRNA binding protein 2
- IGF2BP3:
-
Insulin-like growth factor 2 mRNA binding protein 3
- ISG15:
-
Ubiquitin-like protein interferon-stimulated gene 15
- m6A:
-
N6-methyladenosine
- MCM2:
-
Minichromosome maintenance protein 2
- MTA1:
-
Metastasis-associated protein 1
- METTL3:
-
Methyltransferase-like protein 3
- METTL4:
-
Methyltransferase-like 4
- METTL14:
-
Methyltransferase-like 14
- METTL5:
-
Methyltransferase-like 5
- METTL16:
-
Methyltransferase-like 16
- NSCLC:
-
Non-small cell lung carcinoma
- OC:
-
Ovarian cancer
- OM:
-
Ocular melanoma
- OSCC:
-
Oral squamous cell carcinoma
- PC:
-
Pancreatic cancer
- PIN1:
-
Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1
- PTM:
-
Posttranslational modification
- RANBP2:
-
RAN-binding protein 2, a small ubiquitin-related modifiers (SUMOs) E3 ligase
- RBM15:
-
RNA binding motif protein 15
- RBM15B:
-
RNA binding motif protein 15B
- PRMT1:
-
Protein arginine methyltransferase 1
- PRRC2A:
-
Proline rich coiled-coil 2 A
- STRAP:
-
Serine/threonine kinase receptor associated protein
- TAZ:
-
Transcriptional coactivator with PDZ-binding motif
- TEAD4:
-
TEA domain family member 4
- USP:
-
Ubiquitin specific peptidase
- VIRMA (KIAA1429):
-
Vir-like m6A methyltransferase associated
- WTAP:
-
Wilms tumor 1- associated protein
- YTHDC1:
-
YTH domain containing 1
- YTHDF1:
-
YTH N6-methyladenosine RNA binding protein 1
- YTHDF2:
-
YTH N6-methyladenosine RNA binding protein 2
- YTHDF3:
-
YTH N6-methyladenosine RNA binding protein 3
- YTHDC2:
-
YTH domain containing 2
- ZC3H13:
-
Zinc finger CCCH-type containing 13
- ZCCHC4:
-
Zinc finger CCHC-type containing 4
References
Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, et al. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res. 2013;41(Database issue):D262–267. https://doi.org/10.1093/nar/gks1007.
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71(10):3971–5.
Perry RP, Kelley DE. Existence of methylated messenger RNA in mouse L cells. Cell. 1974;1(1):37–42. https://doi.org/10.1016/0092-8674(74)90153-6.
Sommer S, Lavi U, Darnell JE Jr. The absolute frequency of labeled N-6-methyladenosine in HeLa cell messenger RNA decreases with label time. J Mol Biol. 1978;124(3):487–99. https://doi.org/10.1016/0022-2836(78)90183-3.
Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997;3(11):1233–47.
Clancy MJ, Shambaugh ME, Timpte CS, Bokar JA. Induction of sporulation in Saccharomyces cerevisiae leads to the formation of N6-methyladenosine in mRNA: a potential mechanism for the activity of the IME4 gene. Nucleic Acids Res. 2002;30(20):4509–18. https://doi.org/10.1093/nar/gkf573.
Zhong S, Li H, Bodi Z, Button J, Vespa L, Herzog M, et al. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell. 2008;20(5):1278–88. https://doi.org/10.1105/tpc.108.058883.
Boulias K, Greer EL. Biological roles of adenine methylation in RNA. Nat Rev Genet. 2023;24(3):143–60. https://doi.org/10.1038/s41576-022-00534-0.
Liu L, Li H, Hu D, Wang Y, Shao W, Zhong J, et al. Insights into N6-methyladenosine and programmed cell death in cancer. Mol Cancer. 2022;21(1):32. https://doi.org/10.1186/s12943-022-01508-w.
An Y, Duan H. The role of m6A RNA methylation in cancer metabolism. Mol Cancer. 2022;21(1):14. https://doi.org/10.1186/s12943-022-01500-4.
Liu Z, Zou H, Dang Q, Xu H, Liu L, Zhang Y, et al. Biological and pharmacological roles of m(6)a modifications in cancer drug resistance. Mol Cancer. 2022;21(1):220. https://doi.org/10.1186/s12943-022-01680-z.
He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-methyladenosine and its role in cancer. Mol Cancer. 2019;18(1):176. https://doi.org/10.1186/s12943-019-1109-9.
Li X, Ma S, Deng Y, Yi P, Yu J. Targeting the RNA m(6)a modification for cancer immunotherapy. Mol Cancer. 2022;21(1):76. https://doi.org/10.1186/s12943-022-01558-0.
Deng LJ, Deng WQ, Fan SR, Chen MF, Qi M, Lyu WY, et al. m6A modification: recent advances, anticancer targeted drug discovery and beyond. Mol Cancer. 2022;21(1):52. https://doi.org/10.1186/s12943-022-01510-2.
Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet. 2014;15:293–306. https://doi.org/10.1038/nrg3724.
Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18(1):31–42. https://doi.org/10.1038/nrm.2016.132.
Wang T, Kong S, Tao M, Ju S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. 2020;19(1):88. https://doi.org/10.1186/s12943-020-01204-7.
Zhou Z, Lv J, Yu H, Han J, Yang X, Feng D, et al. Mechanism of RNA modification N6-methyladenosine in human cancer. Mol Cancer. 2020;19(1):104. https://doi.org/10.1186/s12943-020-01216-3.
Zhang L, Tran NT, Su H, Wang R, Lu Y, Tang H, Aoyagi S, Guo A, Khodadadi-Jamayran A, Zhou D et al. (2015). Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. Elife 4. https://doi.org/10.7554/eLife.07938.
Wang P, Doxtader KA, Nam Y. Structural basis for Cooperative function of Mettl3 and Mettl14 methyltransferases [J]. Mol Cell 2016;63(2):306–17. https://doi.org/10.1016/j.molcel.2016.05.041.
Wang X, et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534:575–8.
Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–5.
Ping XL, Sun BF, Wang L et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase [J]. Cell Res 2014;24(2):177–89. https://doi.org/10.1038/cr.2014.3.
Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 2014;8(1):284–96. https://doi.org/10.1016/j.celrep.2014.05.048.
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. VIRMA mediates preferential m6A mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;410. https://doi.org/10.1038/s41421-018-0019-0.
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. M(6)a RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537(7620):369–73. https://doi.org/10.1038/nature19342.
Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, Conrad NK. The U6 snRNA m(6)a methyltransferase METTL16 regulates SAM synthetase Intron Retention. Cell. 2017;169:824–835e14. https://doi.org/10.1016/j.cell.2017.05.003.
Satterwhite ER, Mansfield KD. RNA methyltransferase METTL16: targets and function [J]. Wiley Interdiscip Rev RNA 2022;13(2):e1681. https://doi.org/10.1002/wrna.1681.
Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Höbartner C, Sloan KE, Bohnsack MT. Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017;18:2004–14. https://doi.org/10.15252/embr.201744940.
Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)a machinery component Wtap/Fl(2)d. Genes Dev. 2018;32(5–6):415–29. https://doi.org/10.1101/gad.309146.117.
Wen J, Lv R, Ma H, Shen H, He C, Wang J, et al. Zc3h13 regulates nuclear RNA m(6)a methylation and mouse embryonic stem cell Self-Renewal. Mol Cell. 2018;69(6):1028–1038e6. https://doi.org/10.1016/j.molcel.2018.02.015.
van Tran N, Ernst F, Hawley BR, Zorbas C, Ulryck N, Hackert P, Bohnsack KE, Bohnsack MT, Jaffrey SR, Graille M, et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019;47:7719–33. https://doi.org/10.1093/nar/gkz619.
Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R, Chen K, Lu Z, Chen H, Shi YG, et al. N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol. 2019;15:88–94. https://doi.org/10.1038/s41589-018-0184-3.
Pinto R, Vågbø CB, Jakobsson ME, Kim Y, Baltissen MP, O’Donohue MF, Guzmán UH, Małecki JM, Wu J, Kirpekar F, et al. The human methyltransferase ZCCHC4 catalyses N6-methyladenosine modification of 28S ribosomal RNA. Nucleic Acids Res. 2020;48:830–46. https://doi.org/10.1093/nar/gkz1147.
Ren W, Lu J, Huang M, Gao L, Li D, Wang GG, Song J. Structure and regulation of ZCCHC4 in m(6)A-methylation of 28S rRNA. Nat Commun. 2019;10:5042. https://doi.org/10.1038/s41467-019-12923-x.
Chen H, Gu L, Orellana EA, Wang Y, Guo J, Liu Q, Wang L, Shen Z, Wu H, Gregory RI, Xing Y, Shi Y. METTL4 is an snRNA m(6)am methyltransferase that regulates RNA splicing. Cell Res. 2020;30:544–7. https://doi.org/10.1038/s41422-019-0270-4.
Jia G, Fu Y, Zhao X, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7. https://doi.org/10.1038/nchembio.687.
Zheng G, Dahl JA, Niu Y, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29. https://doi.org/10.1016/j.molcel.2012.10.015.
Ueda Y, Ooshio I, Fusamae Y, Kitae K, Kawaguchi M, Jingushi K, Hase H, Harada K, Hirata K, Tsujikawa K. AlkB homolog 3-mediated tRNA demethylation promotes protein synthesis in cancer cells. Sci Rep. 2017;7:42271. https://doi.org/10.1038/srep42271.
Du H, Zhao Y, He J et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex [J]. Nat Commun 2016;7:12626. https://doi.org/10.1038/ncomms12626.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20. https://doi.org/10.1038/nature12730.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. N(6)-methyladenosine modulates Messenger RNA translation efficiency. Cell. 2015;161:1388–99. https://doi.org/10.1016/j.cell.2015.05.014.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 5’ UTR m(6)a promotes Cap-Independent translation. Cell. 2015;163:999–1010. https://doi.org/10.1016/j.cell.2015.10.012.
Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m(6)A-Dependent Nuclear RNA Processing events. Cell. 2015;162:1299–308. https://doi.org/10.1016/j.cell.2015.08.011.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–4. https://doi.org/10.1038/nature14234.
Wu R, Li A, Sun B, Sun JG, Zhang J, Zhang T, Chen Y, Xiao Y, Gao Y, Zhang Q, et al. A novel m(6)a reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. 2019;29:23–41. https://doi.org/10.1038/s41422-018-0113-8.
Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, Sha J, Huang X, Guerrero L, Xie P, He E, Shen B, He C. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife 2017;6:e31311. https://doi.org/10.7554/eLife.31311.
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, Wang X, Ma HL, Huang CM, Yang Y, Huang N, Jiang GB, Wang HL, Zhou Q, Wang XJ, Zhao YL, Yang YG. Nuclear m(6)a reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61:507–19. https://doi.org/10.1016/j.molcel.2016.01.012.
Li A, Chen YS, Ping XL et al. Cytoplasmic m(6)a reader YTHDF3 promotes mRNA translation [J]. Cell Res 2017;27(3):444–7. https://doi.org/10.1038/cr.2017.10.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, Liu C, He C. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27:315–28. https://doi.org/10.1038/cr.2017.15.
Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, Qi M, Lu Z, Shi H, Wang J, Cheng Y, Luo G, Dai Q, Liu M, Guo X, Sha J, Shen B, He C. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27:1115–27. https://doi.org/10.1038/cr.2017.99.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, Hu YC, Hüttelmaier S, Skibbe JR, Su R, Deng X, Dong L, Sun M, Li C, Nachtergaele S, Wang Y, Hu C, Ferchen K, Greis KD, Jiang X, Wei M, Qu L, Guan JL, He C, Yang J, Chen J. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95. https://doi.org/10.1038/s41556-018-0045-z.
Edens BM, Vissers C, Su J, Arumugam S, Xu Z, Shi H, Miller N, Ringeling R, Ming F, He GL, Song C, Ma H. Y.C.FMRP modulates neural differentiation through m(6)A-Dependent mRNA Nuclear Export. Cell Rep. 2019;28:845–854e5. https://doi.org/10.1016/j.celrep.2019.06.072.
Zhang F, Kang Y, Wang M, Li Y, Xu T, Yang W, Song H, Wu H, Shu Q, Jin P. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum Mol Genet. 2018;27:3936–50. https://doi.org/10.1093/hmg/ddy292.
Yu F, Zhu AC, Liu S, Gao B, Wang Y, Khudaverdyan N et al. RBM33 is a unique m(6)a RNA-binding protein that regulates ALKBH5 demethylase activity and substrate selectivity. Mol Cell. 2023;83(12):2003-2019. https://doi.org/10.1016/j.molcel.2023.05.010.
Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, Rottman F. Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J Biol Chem. 1994;269(26):17697–704. https://doi.org/10.1016/S0021-9258(17)32497-3.
Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16(2):191–8. https://doi.org/10.1038/ncb2902.
Horiuchi K, Kawamura T, Iwanari H, Ohashi R, Naito M, Kodama T, et al. Identification of Wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J Biol Chem. 2013;288(46):33292–302. https://doi.org/10.1074/jbc.M113.500397.
Lence T, Akhtar J, Bayer M, Schmid K, Spindler L, Ho CH, et al. M(6)a modulates neuronal functions and sex determination in Drosophila. Nature. 2016;540(7632):242–7. https://doi.org/10.1038/nature20568.
Guo J, Tang HW, Li J, Perrimon N, Yan D. (2018). Xio is a component of the Drosophila sex determination pathway and RNA N(6)-methyladenosine methyltransferase complex. Proc. Natl. Acad. Sci. U.S.A. 115(14):3674–3679. https://doi.org/10.1073/pnas.1720945115.
Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y, Kumagai S, Ochiai K, Suzuki T, Igarashi K. S-Adenosylmethionine synthesis is regulated by selective N(6)-Adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep. 2017;21:3354–63. https://doi.org/10.1016/j.celrep.2017.11.092.
Sepich-Poore C, Zheng Z, Schmitt E, Wen K, Zhang ZS, Cui XL, Dai Q, Zhu AC, Zhang L, Sanchez Castillo A, et al. The METTL5-TRMT112 N(6)-methyladenosine methyltransferase complex regulates mRNA translation via 18S rRNA methylation. J Biol Chem. 2022;298:101590. https://doi.org/10.1016/j.jbc.2022.101590.
Goh YT, Koh C, Sim DY, Roca X, Goh W. METTL4 catalyzes m6Am methylation in U2 snRNA to regulate pre-mRNA splicing. Nucleic Acids Res. 2020;48:9250–61. https://doi.org/10.1093/nar/gkaa684.
Mauer J, Luo X, Blanjoie A, et al. Reversible methylation of m6Am in the 5’ cap controls mRNA stability. Nature. 2017;541(7637):371–5. https://doi.org/10.1038/nature21022.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20:608–24. https://doi.org/10.1038/s41580-019-0168-5.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6. https://doi.org/10.1038/nature11112.
Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, Yang C, Chen Y. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6:74. https://doi.org/10.1038/s41392-020-00450-x.
Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, Du P, Kim W, Tang S, Sliz P, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561:556–60. https://doi.org/10.1038/s41586-018-0538-8.
David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–8. https://doi.org/10.1038/nature08697.
König J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17:909–15. https://doi.org/10.1038/nsmb.1838.
Alarcón CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519:482–5. https://doi.org/10.1038/nature14281.
Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45:6051–63. https://doi.org/10.1093/nar/gkx141.
Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)a methyltransferase METTL3 promotes translation in Human Cancer cells. Mol Cell. 2016;62:335–45. https://doi.org/10.1016/j.molcel.2016.03.021.
Zhang C, Samanta D, Lu H, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113(14):E2047–56. https://doi.org/10.1073/pnas.160288311.
Kumar S, Nagpal R, Kumar A, Ashraf MU, Bae YS. Immunotherapeutic potential of m6A-Modifiers and MicroRNAs in Controlling Acute myeloid leukaemia. Biomedicines. 2021;9(6):690. https://doi.org/10.3390/biomedicines9060690.
Kumar S, Sarthi P, Mani I et al. Epitranscriptomic Approach: to improve the efficacy of ICB Therapy by Co-Targeting Intracellular Checkpoint CISH. Cells. 2021;10(9):2250. https://doi.org/10.3390/cells10092250.
Liu Y, Yang D, Liu T, Chen J, Yu J, Yi P. N6-methyladenosine-mediated gene regulation and therapeutic implications. Trends Mol Med. 2023;29(6):454-467. https://doi.org/10.1016/j.molmed.2023.03.005.
Liu Z, Gao L, Cheng L, et al. The roles of N6-methyladenosine and its target regulatory noncoding RNAs in tumors: classification, mechanisms, and potential therapeutic implications. Exp Mol Med. 2023;55(3):487–501. https://doi.org/10.1038/s12276-023-00944-y.
Shen LT, Che LR, He Z, et al. Aberrant RNA m(6)a modification in gastrointestinal malignancies: versatile regulators of cancer hallmarks and novel therapeutic opportunities. Cell Death Dis. 2023;14(4):236. https://doi.org/10.1038/s41419-023-05736-w.
Cao J, Yan Q, Cancer Epigenetics. Tumor immunity, and Immunotherapy. Trends Cancer. 2020;6(7):580–92. https://doi.org/10.1016/j.trecan.2020.02.003.
Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. 2019;571(7766):489–99. https://doi.org/10.1038/s41586-019-1411-0.
Wang N, Ma T, Yu B. Targeting epigenetic regulators to overcome drug resistance in cancers. Signal Transduct Target Ther. 2023;8(1):69. https://doi.org/10.1038/s41392-023-01341-7.
Dai Z, Ramesh V, Locasale JW. The evolving metabolic landscape of chromatin biology and epigenetics. Nat Rev Genet. 2020;21(12):737–53. https://doi.org/10.1038/s41576-020-0270-8.
Chan JC, Maze I. Nothing is yet set in (hi)stone: novel post-translational modifications regulating chromatin function. Trends Biochem Sci. 2020;45(10):829–44. https://doi.org/10.1016/j.tibs.2020.05.009.
Li X, Egervari G, Wang Y, Berger SL, Lu Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat Rev Mol Cell Biol. 2018;19(9):563–78. https://doi.org/10.1038/s41580-018-0029-7.
Park JW, Han JW. Targeting epigenetics for cancer therapy. Arch Pharm Res. 2019;42(2):159–70. https://doi.org/10.1007/s12272-019-01126-z.
Zhang Z, Gao Q, Wang S. Kinase GSK3β functions as a suppressor in colorectal carcinoma through the FTO-mediated MZF1/c-Myc axis. J Cell Mol Med. 2021;25(5):2655–65. https://doi.org/10.1111/jcmm.16291.
Ruan DY, Li T, Wang YN, Meng Q, Li Y, Yu K, et al. FTO downregulation mediated by hypoxia facilitates colorectal cancer metastasis. Oncogene. 2021;40(33):5168–81. https://doi.org/10.1038/s41388-021-01916-0.
Song W, Yang K, Luo J, Gao Z, Gao Y. Dysregulation of USP18/FTO/PYCR1 signaling network promotes bladder cancer development and progression. Aging. 2021;13(3):3909–25. https://doi.org/10.18632/aging.202359.
Chang G, Xie GS, Ma L, Li P, Li L, Richard HT. (2022). USP36 promotes tumorigenesis and drug sensitivity of glioblastoma by deubiquitinating and stabilizing ALKBH5. Neuro-oncology. https://doi.org/10.1093/neuonc/noac238.
Lin XT, Yu HQ, Fang L, Tan Y, Liu ZY, Wu D et al. (2021). Elevated FBXO45 promotes liver tumorigenesis through enhancing IGF2BP1 ubiquitination and subsequent PLK1 upregulation. Elife 10. https://doi.org/10.7554/eLife.70715.
Liu S, Li H, Zhu Y, Ma X, Shao Z, Yang Z, et al. LncRNA MNX1-AS1 sustains inactivation of Hippo pathway through a positive feedback loop with USP16/IGF2BP3 axis in gallbladder cancer. Cancer Lett. 2022;547215862. https://doi.org/10.1016/j.canlet.2022.215862.
Huang YY, Zhang CM, Dai YB, Lin JG, Lin N, Huang ZX, et al. USP11 facilitates colorectal cancer proliferation and metastasis by regulating IGF2BP3 stability. Am J Translational Res. 2021;13(2):480–96.
Meng LD, Shi GD, Ge WL, Huang XM, Chen Q, Yuan H, et al. Linc01232 promotes the metastasis of pancreatic cancer by suppressing the ubiquitin-mediated degradation of HNRNPA2B1 and activating the a-raf-induced MAPK/ERK signaling pathway. Cancer Lett. 2020;494107–120. https://doi.org/10.1016/j.canlet.2020.08.001.
Li J, Yang J, Chen Z, Liu L, Wang H, Deng Q, et al. Promotive role of USP29-mediated deubiquitination in malignant proliferation of colorectal cancer cells via the KIAA1429/SOX8 axis. Bosn J Basic Med Sci. 2022. https://doi.org/10.17305/bjbms.2022.7930.
Huang J, Zhou W, Hao C, He Q, Tu X. The feedback loop of METTL14 and USP38 regulates cell migration, invasion and EMT as well as metastasis in bladder cancer. PLoS Genet. 2022;18(10):e1010366. https://doi.org/10.1371/journal.pgen.1010366.
Bhattarai PY, et al. METTL3 stabilization by PIN1 promotes breast tumorigenesis via enhanced m(6)A-dependent translation. Oncogene. 2023;42:1010–23. https://doi.org/10.1038/s41388-023-02617-6.
Xu H, Wang H, Zhao W, Fu S, Li Y, Ni W, et al. SUMO1 modification of methyltransferase-like 3 promotes tumor progression via regulating snail mRNA homeostasis in hepatocellular carcinoma. Theranostics. 2020;10(13):5671–86. https://doi.org/10.7150/thno.42539.
Liu Q, Huang Q, Liu H, He FJ, Liu JH, Zhou YY, et al. SUMOylation of methyltransferase-like 3 facilitates colorectal cancer progression by promoting circ_0000677 in an m(6) A-dependent manner. J Gastroenterol Hepatol. 2022;37(4):700–13. https://doi.org/10.1111/jgh.15775.
Du Y, Hou G, Zhang H, Dou J, He J, Guo Y, et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 2018;46(10):5195–208. https://doi.org/10.1093/nar/gky156.
Liu X, Liu J, Xiao W, Zeng Q, Bo H, Zhu Y, et al. SIRT1 regulates N(6) -Methyladenosine RNA modification in Hepatocarcinogenesis by Inducing RANBP2-Dependent FTO SUMOylation. Hepatology. 2020;72(6):2029–50. https://doi.org/10.1002/hep.31222.
Zhu S, Hou J, Gao H, Hu Q, Kloeber JA, Huang J, et al. SUMOylation of HNRNPA2B1 modulates RPA dynamics during unperturbed replication and genotoxic stress responses. Mol Cell. 2023. https://doi.org/10.1016/j.molcel.2023.01.003.
Guo Q, et al. Glioblastoma upregulates SUMOylation of hnRNP A2/B1 to eliminate the tumor suppressor miR-204-3p, accelerating angiogenesis under hypoxia. Cell Death Dis. 2023;14:147. https://doi.org/10.1038/s41419-023-05663-w.
Li H, Wang D, Yi B, Cai H, Wang Y, Lou X, et al. SUMOylation of IGF2BP2 promotes vasculogenic mimicry of glioma via regulating OIP5-AS1/miR-495-3p axis. Int J Biol Sci. 2021;17(11):2912–30. https://doi.org/10.7150/ijbs.58035.
Hou G, Zhao X, Li L, Yang Q, Liu X, Huang C, et al. SUMOylation of YTHDF2 promotes mRNA degradation and cancer progression by increasing its binding affinity with m6A-modified mRNAs. Nucleic Acids Res. 2021;49(5):2859–77. https://doi.org/10.1093/nar/gkab065.
Zeng X, Chen K, Li L, Tian J, Ruan W, Hu Z, et al. Epigenetic activation of RBM15 promotes clear cell renal cell carcinoma growth, metastasis and macrophage infiltration by regulating the m6A modification of CXCL11. Free Radic. Biol Med. 2022;184135–147. https://doi.org/10.1016/j.freeradbiomed.2022.03.031.
Liao L, He Y, Li SJ, Zhang GG, Yu W, Yang J, et al. Anti-HIV drug Elvitegravir suppresses Cancer Metastasis via increased proteasomal degradation of m6A methyltransferase METTL3. Cancer Res. 2022;82(13):2444–57. https://doi.org/10.1158/0008-5472.CAN-21-4124.
Li Y, He X, Lu X, Gong Z, Li Q, Zhang L, et al. METTL3 acetylation impedes cancer metastasis via fine-tuning its nuclear and cytosolic functions. Nat Commun. 2022;13(1):6350. https://doi.org/10.1038/s41467-022-34209-5.
Yang Y, Cai Q, Fu QSheng, Wei Dong L, Fan Y, F., and, Wu Z, X. Reduced N6-Methyladenosine mediated by METTL3 Acetylation promotes MTF1 expression and Hepatocellular Carcinoma. Cell Growth Chem Biodivers. 2022;19(11):e202200333. https://doi.org/10.1002/cbdv.202200333.
Xiong J, He J, Zhu J et al. Lactylation-driven METTL3-mediated RNA m(6)a modification promotes immunosuppression of tumor-infiltrating myeloid cells [J]. Mol Cell 2022;82(9):1660–1677e10. https://doi.org/10.1016/j.molcel.2022.02.033.
Yu J, Chai P, Xie M, et al. Histone lactylation drives oncogenesis by facilitating m(6)a reader protein YTHDF2 expression in ocular melanoma [J]. Genome Biol. 2021;22(1):85. https://doi.org/10.1186/s13059-021-02308-z.
Yang Y, Yan Y, Yin J, Tang N, Wang K, Huang L, et al. O-GlcNAcylation of YTHDF2 promotes HBV-related hepatocellular carcinoma progression in an N(6)-methyladenosine-dependent manner. Signal Transduct Target Ther. 2023;8(1):63. https://doi.org/10.1038/s41392-023-01316-8.
Tai H, Wang X, Zhou J, Han X, Fang T, Gong H, et al. Protein kinase Cβ activates fat mass and obesity-associated protein by influencing its ubiquitin/proteasome degradation. FASEB J. 2017;31(10):4396–406. https://doi.org/10.1096/fj.201601159RR.
Wang JM, Liu BQ, Zhang Q, Hao L, Li C, Yan J, et al. ISG15 suppresses translation of ABCC2 via ISGylation of hnRNPA2B1 and enhances drug sensitivity in cisplatin resistant ovarian cancer cells. Biochim Biophys Acta Mol Cell Res. 2020;1867(4):118647. https://doi.org/10.1016/j.bbamcr.2020.118647.
Yao B, Zhang Q, Yang Z, An F, Nie H, Wang H, et al. CircEZH2/miR-133b/IGF2BP2 aggravates colorectal cancer progression via enhancing the stability of m(6)A-modified CREB1 mRNA. Mol Cancer. 2022;21(1):140. https://doi.org/10.1186/s12943-022-01608-7.
Wang Y, Lu JH, Wu QN, Jin Y, Wang DS, Chen YX, et al. LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol Cancer. 2019;18(1):174. https://doi.org/10.1186/s12943-019-1105-0.
Zhang W, Zhu L, Yang G, Zhou B, Wang J, Qu X, et al. Hsa_circ_0026134 expression promoted TRIM25- and IGF2BP3-mediated hepatocellular carcinoma cell proliferation and invasion via sponging miR-127-5p. Biosci Rep. 2020;40(7). https://doi.org/10.1042/BSR20191418.
Wang H, Liang L, Dong Q, Huan L, He J, Li B, et al. Long noncoding RNA miR503HG, a prognostic indicator, inhibits tumor metastasis by regulating the HNRNPA2B1/NF-κB pathway in hepatocellular carcinoma. Theranostics. 2018;8(10):2814–29. https://doi.org/10.7150/thno.23012.
Zhu W, Wang J, Liu X, Xu Y, Zhai R, Zhang J, et al. lncRNA CYTOR promotes aberrant glycolysis and mitochondrial respiration via HNRNPC-mediated ZEB1 stabilization in oral squamous cell carcinoma. Cell Death Dis. 2022;13(8):703. https://doi.org/10.1038/s41419-022-05157-1.
Pan Z, Zhao R, Li B, Qi Y, Qiu W, Guo Q, et al. EWSR1-induced circNEIL3 promotes glioma progression and exosome-mediated macrophage immunosuppressive polarization via stabilizing IGF2BP3. Mol Cancer. 2022;21(1):16. https://doi.org/10.1186/s12943-021-01485-6.
Cockram PE, Kist M, Prakash S, Chen SH, Wertz IE, Vucic D. Ubiquitination in the regulation of inflammatory cell death and cancer. Cell Death Differ. 2021;28:591–605. https://doi.org/10.1038/s41418-020-00708-5.
Liu J, Cheng Y, Zheng M, Yuan B, Wang Z, Li X, Yin J, Ye M, Song Y. Targeting the ubiquitination/deubiquitination process to regulate immune checkpoint pathways. Signal Transduct Target Ther. 2021;6:28. https://doi.org/10.1038/s41392-020-00418-x.
Seeler JS, Dejean A. SUMO and the robustness of cancer. Nat Rev Cancer. 2017;17:184–97. https://doi.org/10.1038/nrc.2016.143.
Kroonen JS, Vertegaal A. Targeting SUMO signaling to Wrestle Cancer. Trends Cancer. 2021;7:496–510. https://doi.org/10.1016/j.trecan.2020.11.009.
Du L, Liu W, Rosen ST. Targeting SUMOylation in cancer. Curr Opin Oncol. 2021;33:520–5. https://doi.org/10.1097/CCO.0000000000000765.
Shang S, Liu J, Hua F. Protein acylation: mechanisms, biological functions and therapeutic targets. Signal Transduct Target Ther. 2022;7:396. https://doi.org/10.1038/s41392-022-01245-y.
Phillips DM. The presence of acetyl groups of histones. Biochem J. 1963;87:258–63. https://doi.org/10.1042/bj0870258.
Pang K et al. Role of protein phosphorylation in cell signaling, disease, and the intervention therapy. MedComm. 2022;3(4):e175. https://doi.org/10.1002/mco2.175.
Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575–80. https://doi.org/10.1038/s41586-019-1678-1.
Li X, Yang Y, Zhang B, Lin X, Fu X, An Y, Zou Y, Wang JX, Wang Z, Yu T. Lactate metabolism in human health and disease. Signal Transduct Target Ther. 2022;7:305. https://doi.org/10.1038/s41392-022-01151-3.
Yang Z, Yan C, Ma J, Peng P, Ren X, Cai S, Shen X, Wu Y, Zhang S, Wang X, et al. Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nat Metab. 2023;5:61–79. https://doi.org/10.1038/s42255-022-00710-w.
Miao Z, Zhao X, Liu X. Hypoxia induced β-catenin lactylation promotes the cell proliferation and stemness of colorectal cancer through the wnt signaling pathway. Exp Cell Res. 2023;422:113439. https://doi.org/10.1016/j.yexcr.2022.113439.
Wang L, Li S, Luo H, Lu Q, Yu S. PCSK9 promotes the progression and metastasis of colon cancer cells through regulation of EMT and PI3K/AKT signaling in tumor cells and phenotypic polarization of macrophages. J Exp Clin Cancer Res. 2022;41:303. https://doi.org/10.1186/s13046-022-02477-0.
Gu J, Zhou J, Chen Q, Xu X, Gao J, Li X, Shao Q, Zhou B, Zhou H, Wei S, et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-β signaling in regulatory T cells. Cell Rep. 2022;39:110986. https://doi.org/10.1016/j.celrep.2022.110986.
Luo Y, Yang Z, Yu Y, Zhang P. HIF1α lactylation enhances KIAA1199 transcription to promote angiogenesis and vasculogenic mimicry in prostate cancer. Int J Biol Macromol. 2022;222:2225–43. https://doi.org/10.1016/j.ijbiomac.2022.10.014.
Pan L, Feng F, Wu J, Fan S, Han J, Wang S, Yang L, Liu W, Wang C, Xu K. Demethylzeylasteral targets lactate by inhibiting histone lactylation to suppress the tumorigenicity of liver cancer stem cells. Pharmacol Res. 2022;181:106270. https://doi.org/10.1016/j.phrs.2022.106270.
Fehl C, Hanover JA. Tools, tactics and objectives to interrogate cellular roles of O-GlcNAc in disease. Nat Chem Biol. 2022;18:8–17. https://doi.org/10.1038/s41589-021-00903-6.
Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. 2017;18:452–65. https://doi.org/10.1038/nrm.2017.22.
Ferreira JA, Peixoto A, Neves M, Gaiteiro C, Reis CA, Assaraf YG, Santos LL. Mechanisms of cisplatin resistance and targeting of cancer stem cells: adding glycosylation to the equation. Drug Resist Updat. 2016;24:34–54. https://doi.org/10.1016/j.drup.2015.11.003.
Slawson C, Hart GW. O-GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer. 2011;11:678–84. https://doi.org/10.1038/nrc3114.
Ambler RP, Rees MW. Epsilon-N-Methyl-lysine in bacterial flagellar protein. Nature. 1959;184:56–7. https://doi.org/10.1038/184056b0.
Bhat KP, Ümit Kaniskan H, Jin J, Gozani O. Epigenetics and beyond: targeting writers of protein lysine methylation to treat disease. Nat Rev Drug Discov. 2021;20:265–86. https://doi.org/10.1038/s41573-020-00108-x.
Murray K, Epsilon-N-methyl lysine in. Histones Biochem 3:10–5. https://doi.org/10.1021/bi00889a003.
Murn J, Shi Y. The winding path of protein methylation research: milestones and new frontiers. Nat Rev Mol Cell Biol. 2017;18:517–27. https://doi.org/10.1038/nrm.2017.35.
Michalak EM, Burr ML, Bannister AJ, Dawson MA. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat Rev Mol Cell Biol. 2019;20:573–89. https://doi.org/10.1038/s41580-019-0143-1.
Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications. Biochim Biophys Acta. 2009;1789:45–57. https://doi.org/10.1016/j.bbagrm.2008.06.005.
Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function. Mol Cell. 2005;18:263–72. https://doi.org/10.1016/j.molcel.2005.04.003.
Hamamoto R, Saloura V, Nakamura Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat Rev Cancer. 2015;15:110–24. https://doi.org/10.1038/nrc3884.
Rodríguez-Paredes M, Lyko F. The importance of non-histone protein methylation in cancer therapy. Nat Rev Mol Cell Biol. 2019;20:569–70. https://doi.org/10.1038/s41580-019-0147-x.
Goldstein G, Scheid M, Hammerling U, Schlesinger DH, Niall HD, Boyse EA. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc Natl Acad Sci U S A. 1975;72:11–5. https://doi.org/10.1073/pnas.72.1.11.
Han HG, Moon HW, Jeon YJ. ISG15 in cancer: beyond ubiquitin-like protein. Cancer Lett. 2018;438:52–62. https://doi.org/10.1016/j.canlet.2018.09.007.
Farrell PJ, Broeze RJ, Lengyel P. Accumulation of an mRNA and protein in interferon-treated Ehrlich ascites tumour cells. Nature. 1979;279:523–5. https://doi.org/10.1038/279523a0.
Yuan Y, Qin H, Li H, Shi W, Bao L, Xu S, Yin J, Zheng L. The functional roles of ISG15/ISGylation in Cancer. Molecules. 2023;28. https://doi.org/10.3390/molecules28031337.
Lin X, Wu Z, Hu H, Luo ML, Song E. Non-coding RNAs rewire cancer metabolism networks. Semin Cancer Biol. 2021;75:116–26. https://doi.org/10.1016/j.semcancer.2020.12.019.
Yin X et al. LncRNAs and CircRNAs in cancer. MedComm. 2022;3(2):e141. https://doi.org/10.1002/mco2.141.
Zhou L, et al. Revisiting cancer hallmarks: insights from the interplay between oxidative stress and non-coding RNAs. Mol Biomed. 2020;1:4. https://doi.org/10.1186/s43556-020-00004-1.
Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Annu Rev Biochem. 2011;80:473–99. https://doi.org/10.1146/annurev-biochem-061809-175347.
Nagarajan S, Johnsen SA. Crosstalk between histone modifications integrates various signaling inputs to Fine-Tune Transcriptional output - ScienceDirect. Chromatin Signal Dis. 2016:217–39. https://doi.org/10.1016/B978-0-12-802389-1.00012-5.
Zhao Y, Chen Y, Jin M, Wang J. The crosstalk between m(6)a RNA methylation and other epigenetic regulators: a novel perspective in epigenetic remodeling. Theranostics. 2021;11:4549–66. https://doi.org/10.7150/thno.54967.
Zhou X, Li C, Chen T, Li W, Wang X, Yang Q. Targeting RNA N6-methyladenosine to synergize with immune checkpoint therapy. Mol Cancer. 2023;22(1):36. https://doi.org/10.1186/s12943-023-01746-6.
Acknowledgements
Not applicable.
Funding
This work was supported in part by the Science Foundation of AMHT (2022YK01), the Science Foundation of CASIC (2020-LCYL-009), the Science Foundation of ASCH (YN202104), and the Hygiene and Health Development Scientific Research Fostering Plan of Haidian District Beijing (HP2021-19-50701).
Author information
Authors and Affiliations
Contributions
YuW, HP and HW researched data for the article and contributed substantially to discussion of the content. HW, yaW, HP and YuW wrote the article. JW and JC reviewed and/or edited the manuscript before submission. YuW and HW conceived of and designed the study. HW, ZC and JW provided administrative support. All authors analysed and interpreted the data. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All of the authors are aware of and agree to the content of the paper and their being listed as a co-author of the paper.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Y., Wang, Y., Patel, H. et al. Epigenetic modification of m6A regulator proteins in cancer. Mol Cancer 22, 102 (2023). https://doi.org/10.1186/s12943-023-01810-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-023-01810-1