
Abstract
Lung cancer continues to be the leading cause of cancer-related death worldwide. In the last decade, significant advancements in the diagnosis and treatment of lung cancer, particularly NSCLC, have been achieved with the help of molecular translational research. Among the hopeful breakthroughs in therapeutic approaches, advances in targeted therapy have brought the most successful outcomes in NSCLC treatment. In targeted therapy, antagonists target the specific genes, proteins, or the microenvironment of tumors supporting cancer growth and survival. Indeed, cancer can be managed by blocking the target genes related to tumor cell progression without causing noticeable damage to normal cells. Currently, efforts have been focused on improving the targeted therapy aspects regarding the encouraging outcomes in cancer treatment and the quality of life of patients. Treatment with targeted therapy for NSCLC is changing rapidly due to the pace of scientific research. Accordingly, this updated study aimed to discuss the tumor target antigens comprehensively and targeted therapy-related agents in NSCLC. The current study also summarized the available clinical trial studies for NSCLC patients.
Similar content being viewed by others

Introduction
Lung cancer accounts for about 13% of all cancers and is the number one cause of cancer-related death worldwide which leads to more deaths than colorectal, breast, brain, and prostate cancers [1, 2]. In 2022, the American Cancer Society estimated 236,740 new cases and 130,180 deaths from lung cancer in the United States. The prognosis of those with advanced disease, such as stage IIIB or stage IV, is poor and less than 5% [3]. Only about 15% of patients are diagnosed with early-stage disease, and most (84%) are in an advanced stage at the time of diagnosis. Altogether, Non-small cell lung cancer (NSCLC) patients have a poor prognosis and low 5-year overall survival (OS), approximately 17.4% [4,5,6].
NSCLC is the most common type of lung cancer, accounting for approximately 85% of the cases. The three most common forms of NSCLC are adenocarcinoma, squamous cell carcinoma as well as large cell carcinoma [7,8,9]. Common cancer treatment approaches include surgery, chemotherapy, and radiation therapy. Recent advances in science have led to the emergence of newer and more efficient methods, such as immunotherapy and target therapy, in treating various diseases, including infections, cancers, autoimmunities, and other disorders [10,11,12]. Over the past decade, immunotherapy and targeted therapy, along with other treatments, indicated successful outcomes in treating advanced lung cancer, especially NSCLC. They improved the overall survival (OS) of patients. This is predominantly due to the availability of biomarkers to select patients for targeted and immunotherapy-based treatments. In other words, treatments are shifting toward newer targeted and small molecule therapies to improve outcomes among NSCLC patients. Indeed, investigation of the human genome has permitted more efficient identification of gene alterations that are potential "targets" for therapy [13,14,15]. However, targeted therapy is indicated for those with distant metastases and stage IV disease.
Our main goal in the present study is to investigate categorized targeted therapy strategies in treating NSCLC. In the beginning, we give a general explanation of the NSCLC common treatments and then continue our study by focusing on the targeted therapy of NSCLC, which be discussed separately in the VEGF, KRAS, EGFR, ALK, ROS1, BRAF, RET, MET, NTRK, HER2, HER3, PI3K/AKT/mTORC, PD1, and CTLA-4 related sections.
Treatment approaches for NSCLC
The treatment choices for NSCLC are based mainly on factors such as the tumor grade, size, and location, lymph node status, the patient’s overall health, and lung function. Surgery, chemotherapy, radiation therapy, and targeted therapy are the approved treatment modalities for NSCLC [16]. As a standard therapy approach, Stage 0 NSCLC is generally curable by surgery with no chemotherapy or radiation therapy. In addition, laser therapy, photodynamic therapy, or brachytherapy may be substituted for surgery. For stage I NSCLC, surgery is the main choice, and radiation therapy or adjuvant chemotherapy after surgery may lower the cancer recurrence. In stage II NSCLC, cancer removal may be followed by adjuvant chemotherapy, radiation, and immunotherapy. The initial treatment for stage IIIA NSCLC may comprise some combination of chemotherapy, radiation therapy, and/or surgery accompanied by immunotherapy. For people whose cancer cells have specific mutations in the EGFR gene, adjuvant therapy with the targeted drug osimertinib might be an option at some point additionally. Stage IIIB can’t be entirely removed by surgery, but chemoradiotherapy and immunotherapy are helpful. Stage IVA or IVB can be tough to cure, and treatments such as surgery, photodynamic therapy, laser therapy, radiation therapy, chemotherapy, immunotherapy, and targeted therapy may help by relieving symptoms. For stage IVB cancers that have metastasized all over the body, before any treatments, the cancer cells will be tested for certain gene mutations, including the VEGF, KRAS, EGFR, ALK, ROS1, BRAF, RET, MET, NTRK genes. If one of these genes is mutated, your first treatment will likely be a targeted therapy drug. Identifying gene mutations or some protein expressions in lung cancer has led to the development of molecularly targeted therapy to progress the survival of patients with advanced disease [17].
Targeted drug therapy
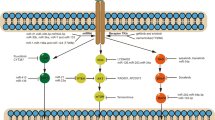
Cancer cells have modifications in their genes or proteins that make them different from normal cells. Consequently, they can develop faster and sometimes could spread. Targeted cancer therapy works by those differences and targets cancer's specific genes and proteins that contribute to cancer growth and survival. So, it chunks the growth and spread of cancer cells, which confines the damage to healthy cells. Targeted drugs are often used for advanced lung cancers when conventional drugs don't affect them, mainly because they have different side effects. Many drugs targeting these pathways have been developed, and there are numerous FDA-approved targeted agents [5]. Today, inhibitors of EGFR, ALK, PI3K/AKT/mTOR, RAS-MAPK, RET, MET, BRAF, and NTRK/ROS1, as well as PD1 and CTLA4 molecules, are available for NSCLC, and many of these are now typical of care for selected patients (Fig. 1). In other words, to find the most effective treatment, personalized cancer therapy should be done by identifying the status of target genes and proteins in the patient by standard tests [5].

Target genes and drugs in NSCLC. Various targeted therapy drugs have been summarized that target different key related genes and their signaling pathways in NSCLC
Despite these new therapeutic options for patients with advanced NSCLC, there continue to be significant challenges as resistance development and disease progression occur in most of these patients [18]. Nevertheless, while target therapy in NSCLC has provided disease control, the tumors inevitably develop drug resistance. Understanding resistance mechanisms and developing combinational therapies are essential for improving treatment outcomes.
Vascular endothelial growth factor (VEGF)
Numerous studies demonstrated the efficient effect of small-molecule inhibitors of the Vascular Endothelial Growth Factor and its receptors (VEGF/VEGFR) in treating various malignancies, including NSCLC. There is two FDA-approved anti-VEGF pathway, Bevacizumab (Avastin/anti-VEGF-antibody) and Ramucirumab (Cyramza/anti-VEGFR antibody), for advanced NSCLC, which are used alone or in combination with chemotherapy. The VEGF pathway has been extensively studied and shown to play a critical role in angiogenesis [19]. Interestingly emerging evidence has demonstrated that VEGF may play a more comprehensive role in the pathogenesis of cancer than was previously thought, as it causes tumor-induced immunosuppression [20]. VEGFs can disrupt the maturation of dendritic cells (DCs) and other hematopoietic lineages and consequently down-regulate the antigen presentation process. Besides, it could enrich the infiltration of regulatory T (Treg) cells, tumor-associated macrophages (M2 macrophages), and Myeloid-Derived Suppressor Cells (MDSC) into the NSCLC niche [20,21,22,23]. In other words, blockade of the VEGF signaling leads to reversing these immune suppressive mechanisms, reducing the recruitment of suppressor cells into the tumor but increasing the infiltration of effector T cells.
Some studies revealed combination therapy of the other anti-angiogenic inhibitor or chemotherapy drug by anti-VEGF exerts a mostly synergistic anti-tumor effect. One study constructed a novel bispecific decoy receptor VEGFR-EGFR/Fc targeting the VEGF and EGF-like ligands in human NSCLC. The constructed dual-specific antibody inhibited tumor invasion, relocation, proliferation, and angiogenesis [24]. Interestingly, "Zhao" et al. revealed EGFR/VEGF inhibition therapy was transcendent to single EGFR inhibition, but not VEGF inhibition, for advanced NSCLC [25]. In addition, the combination of bevacizumab plus gemcitabine/cisplatin chemotherapy for advanced NSCLC had clinical efficacy and prolonged the long-term survival of patients [26]. Furthermore, it has been shown the combination of bevacizumab and erlotinib extended Progression-free survival (PFS) but increased the incidence of adverse events compared to their monotherapy in NSCLC patients [27].
Recently, some studies investigated the modulatory role of miRNA on the VEGF pathway. Wang et al. showed a decreased level of messenger RNA expressions of miR-199a and increased VEGF in NSCLC rat models. They declared that miR-199a prevents the proliferation of NSCLC cells via the targeted down-regulation of the Hypoxia-inducible factor 1-alpha /VEGF signaling pathway [28]. Another study showed that miR-214 targeted the inhibitor of Growth Family Member 4 in lung cancer cells and upregulated the HIF-1α pathway, leading to Matrix Metallopeptidase 2 and VEGF upregulation [29]. These studies indicate that targeted inhibition of the VEGF pathway by another mechanism could be investigated.
KRAS
Kirsten Rat Sarcoma viral oncogene homolog (KRAS) gene is a gene that produces a protein that functions in the cell signaling systems that regulate cell division, maturation, and growth. It is the most mutated oncogene in human cancers, which hinders the development of effective drugs against KRAS [30, 31]. On that account, despite colossal endeavors that contributed to the development of drugs pointed at hindering KRAS or its signaling pathways, KRAS was historically considered undruggable. Nevertheless, several recent research that shows encouraging results initiating a new exciting era [32].
We now have a better understanding of the intricate interactions involving the RAS family of signaling proteins thanks to Ostrem, Shokat, and colleagues' discovery of the switch II pocket on the surface of the active and inactive forms of KRAS [33, 34]. According to all this research, two direct KRAS G12C (OFF) inhibitors, Sotorasib and Adagrasib, earned the breakthrough designation by the U.S. Food and Drug Administration (FDA) for the treatment of patients with KRAS G12C metastatic lung cancer and have shown promising results in phase I and II clinical trials (NCT04625647, NCT04933695, NCT04303780, NCT03785249) [35, 36].
Sotorasib (AMG510), developed by Amgen, is the first FDA-approved drug that irreversibly and selectively inactivates KRAS G12C using the interaction with a surface groove of the histidine 95 next to the cysteine 12 switch II pocket and keeps it in the inactive GDP-bound state. The compound AMG 510 has shown promise in phase I/II study (CodeBreak 100: NCT03600883) across solid tumors. In addition to CodeBreak 100, aiming for KRAS in NSCLC is a field of study. Recently, the phase 3 multicenter CodeBreak 200 clinical trial (NCT04303780) of a type of combined treatment using sotorasib and docetaxel has been used for patients with locally progressive and unresectable or metastatic NSCLC with KRAS G12C mutations [37,38,39,40]. Adagrasib (MRTX849) is another KRAS G12C (OFF) inhibitor that acts the same as sotorasib and binds KRAS G12C irreversibly and preferentially in its inactive GDP-bound state and also secures the switch II pocket. Adagrasib has been shown in clinical studies to have an anticancer effect against brain metastases and can reach the brain and cerebrospinal fluid. Phase I/II research evaluating the efficacy of adagrasib included participants with previously treated advanced or metastatic cancers, including NSCLC that carried the KRASG12C mutation. Adagrasib and docetaxel are also being compared in a phase III trial (KRYSTAL-12) for patients with recently treated KRASG12C-mutated NSCLC (NCT04685135), and numerous combination therapies with adagrasib are being developed [41,42,43,44,45].
EGFR
The epidermal growth factor receptor (also known as ErbB1, HER1, or EGFR) is a member of a family of receptor tyrosine kinases that can activate a wide range of signaling pathways resulting in cell growth, differentiation, proliferation, and survival [46, 47]. After the ligand has been bound, the EGFR tyrosine kinase activates the receptor by homo- or heterodimerizing it and auto-phosphorylating tyrosine-rich cytoplasmic regions. This initiates the PIK3CA/AKT1/mTOR pathway and the RAS/RAF1/MAP2K1/MAPK1 kinases6, two major downstream intermediate pathways [48].
The EGFR mutation is one of the most significant genetic abnormalities in NSCLC patients among the rising driving oncogenes. Enhanced EGFR expression on cancerous cells (which is about 40–80% of NSCLCs), increased ligand synthesis by cancerous cells, and activating mutations of EGFR within cancerous cells are three primary factors that activate EGFR [49, 50]. Over 50% of adenocarcinomas and tumors from East Asians, never smokers, and women have these sorts of mutations, as do 10 to 20% of individuals with advanced NSCLC [51]. Complementary research demonstrated that activating mutations, not EGFR overexpression, were the main therapeutic target.
Around 20% of individuals with lung adenocarcinoma have EGFR mutations, including Exon 19 deletions (60%) and exon 21-point mutations (L858R missense replacements) at position 858 (35%), where leucine is changed to arginine, which cause fundamental activation of the receptor without ligand interaction [52, 53]. The first two drugs to target the tyrosine kinase domain of the EGFR were gefitinib and erlotinib. In both the phase I series and the subsequent phase II studies, these drugs showed encouraging action in NSCLC patients who had previously received chemotherapy. As a result, they have decided to approve the therapy for advanced NSCLC [54, 55]. Afatinib is another EGFR-TKI that is quite effective in treating advanced NSCLC patients with activating EGFR mutations in several clinical studies [56, 57].
In the first-line therapy approach, EGFR-TKIs are advised for NSCLC patients who have to activate EGFR mutations. Notably, chemotherapy is not necessarily the only treatment choice for individuals with wild-type EGFR NSCLC if there is no activating EGFR mutation. It is noteworthy that a sizable fraction of patients may still benefit clinically from EGFR-TKI therapy, even in those with wild-type EGFR NSCLC. There is ample proof that patients with wild-type EGFR NSCLC should not get EGFR-TKIs as their first-line therapy [58, 59]. A different approach to preventing EGFR activation and signaling is represented by monoclonal antibodies. In addition to forming antibody-receptor complexes that are endocytosed and destroyed, they can also completely prevent ligands from binding to the extracellular domain. The anti-EGFR mAbs cetuximab, necitumumab, panitumumab, and matuzumab are now accessible. Cetuximab and platinum doublet chemotherapy were used in two phase III trials, FLEX and BMS099, to treat advanced NSCLC [60, 61]. EGFR-TKIs considerably increase objective response rate (ORR), progression-free survival (PFS), and quality of life (QoL) compared to traditional chemotherapeutic regimens while exhibiting minor toxicity. The use of EGFR-TKIs has made significant advancements in the treatment of NSCLC, ushering in a new age of focused therapy and precision medication [62, 63].
ALK
Anaplastic lymphoma kinase (ALK) is a different tyrosine kinase that has been thoroughly researched as a subject for TKI therapies. This 1620 amino acid transmembrane tyrosine kinase receptor is generated by the proto-oncogene ALK mostly expressed in the nervous system. ALK dimerizes and then auto-phosphorylates the intracellular kinase domain in response to ligand binding to its extracellular domain. This protein aids in regulating cellular development. Anaplastic large cell lymphoma, neuroblastoma, and non-small cell lung cancer are a few examples of cancers in which the anaplastic lymphoma kinase (ALK) gene may be altered. When activated in cancer, the cancer cells may develop and proliferate due to these ALK gene alterations. Gene fusions, chromosomal translocations, gene amplification or deregulation, and activating point mutations are the three main ways by which ALK signaling is triggered in tumor cells [64,65,66,67].
In 60 percent of anaplastic large cell lymphomas, a rare subtype of non-Hodgkin lymphomas, alk rearrangements were first discovered as a fusion to a section of the nucleophosmin (NPM) gene [68]. However, echinoderm microtubule-associated protein-like 4 (EML4) is the most common fusion component, and many EML4 breakpoints have been identified. Other uncommon fusion partners have also been identified; examples include TFG and KIF5B [69, 70]. About 2-7% of stage III or IV NSCLC patients and, more frequently, younger individuals, never or light smokers, which are more likely to develop brain metastases, have been shown to contain oncogenic fusion genes, including EML4 and ALK [71, 72]. Specific inhibitors, such as crizotinib, ceritinib, alectinib, etc., are highly beneficial in treating ALK-positive individuals, particularly those with non-small cell lung cancer [73].
The tyrosine kinases ALK and c-MET are specifically and powerfully inhibited by the tiny drug crizotinib. It is a first-generation c-MET inhibitor called PHA-66752 that was converted into an inhibitor of the 3-benzyloxy-2-aminopyridine series by leveraging the cocrystal structure of the inhibitor and c-MET to enhance active-site binding. In both first-line and second-line settings, crizotinib has been shown to increase progression-free survival (PFS) compared to chemotherapy [74, 75]. A second-generation ALK inhibitor, ceritinib, has been demonstrated to successfully treat advanced or metastatic ALK-positive NSCLC, including in patients who have already received crizotinib [76, 77]. The efficacy of these two ALK-targeted treatments may vary, according to recent research. Preclinical findings show that ceritinib is 20 times more effective against ALK than crizotinib. Additionally, it has been demonstrated that ceritinib is effective in individuals who have gained resistance to crizotinib, with remarkable tumor responses being seen in brain metastases [78, 79]. The evidence for the effectiveness of ceritinib combined with crizotinib in patients with advanced or metastatic NSCLC who have not taken crizotinib comes from five clinical trials: NCT01283516, NCT01685138, NCT00585195, NCT00932451, NCT00932893 [80, 81]. Expanding the therapy options and improving survival are effective targeted medications created against specific molecular subtypes of NSCLC, such as EGFR and ALK.
C-ros oncogene 1 (ROS1)
ROS proto-oncogene 1 receptor tyrosine kinase (ROS1) rearrangement in NSCLC cell lines was first discovered in 2007 [82]. ROS1-rearranged NSCLC includes 1–2% of total NSCLC cases, all detected/diagnosed as adenocarcinomas. Although ROS1-positive prevalence was significantly higher in never-smoker young patients, the overall survival rate of ROS1-positive NSCLC indicated no significant variation compared with ROS1–negative [83]. There are FDA-approved ROS1 tyrosine kinase inhibitors (TKIs), such as crizotinib and entrectinib. Indeed, several other targeted TKIs are being developed or assessed in clinical trials.
Crizotinib (1st generation)
ALK and ROS1 share a 77% amino acid profile in their kinase domains [84]. Crizotinib efficacy in ALK-positive NSCLCs and ALK-RAS structural similarity suggested that ROS1 may be a potent therapeutic target for crizotinib. In 2016, crizotinib (Xalkori, PF-02341066) received approval from European Medicines Agency (EMA) and the United States Food and Drug Administration (FDA) for ROS1-rearranged NSCLC therapy [81]. crizotinib is the standard first-line treatment for ROS1-positive NSCLCs. Despite the significant efficacy of crizotinib against ROS1-rearranged NSCLCs, resistance to crizotinib leads to tumor relapse. The resistance to crizotinib is developed due to the acquired secondary point mutations, mainly attributed to the ROS1 G2032R mutation [85, 86]. This highlighted the pressing need to discover new potent ROS1-rearranged NSCLC-targeted therapies.
Entrectinib (2nd generation)
Entrectinib was first introduced in 2016 as a potent oral multi-target TKI against ROS1, ALK, and TRK kinases and gained FDA approval in ROS1-positive NSCLCs treatment [87]. A study disclosed in vitro and in vivo activities of entrectinib in NSCLC models [88]. Entrectinib showed substantial activity in ROS1-positive models. In addition, it has been reported that entrectinib was well penetrated through the blood–brain barrier (BBB) and induced intracranial activity, which interprets entrectinib potential efficacy in brain metastasis [88]. Multiple phase I/II cohort trials (STARTRK-1, STARTRK-2, and ALKA-372-001) of entrectinib have reported the efficient activity and manageable safety of entrectinib in ROS1 + NSCLC patients. The overall objective response rate was 77%, and the median duration of response was 24·6 months. Serious adverse events were observed in 11% of patients, and high-grade (grade 3 or 4) adverse events were reported in 34% of patients [89]. A recent study has compared the outcomes of two FDA-approved ROS1 inhibitors TKI, entrectinib, and crizotinib. The time-to-treatment discontinuation (TTD), PFS, and OS were variables of this comparison. These reports indicate further prolonged TTD using entrectinib (12.9 months) compared to crizotinib (8.8 months) [90].
Lorlatinib (3rd generation)
Lorlatinib (PF-06463922) is a selective third-generation tyrosine kinase inhibitor (TKI). It has been introduced as a ROS1/ALK inhibitor in crizotinib-resistant ROS1-positive NSCLC and glioblastoma. Lorlatinib showed high potency in NSCLCs with ROS1 G2032R mutation [91], which developed resistance to crizotinib [85, 86]. Oral lorlatinib showed a significantly higher ROS1 inhibition compared with crizotinib, alectinib, and ceritinib, which represents an encouraging alternative treatment in crizotinib-resistant ROS1-positive NSCLCs. In addition, lorlatinib improved CNS penetrance and reached a high level in cerebrospinal fluid (CSF), indicating that lorlatinib is a potential candidate for brain metastasis [91]. Phase I and II clinical trials in 2017–2019 investigated lorlatinib efficacy in ROS1- and ALK-rearranged NSCLC patients via addressing objective response rate (ORR) and median progression-free survival (PFS) [92, 93]. The clinical trials showed substantial activity in advanced ROS1-positive NSCLC patients. Lorlatinib showed a promising function in either crizotinib resistance or CNS metastasis. Additionally, there was no significant toxicity, and lorlatinib was well tolerated [92, 93]. Of note, lorlatinib showed in vitro anti-tumor activity against other crizotinib-resistant mutations such as L2026M, 66, 83 S1986Y/F, 66, and D2033N. Thus, Lorlatinib could be a potential alternative against advanced crizotinib resistance and metastasized NSCLCs.
V-Raf murine sarcoma viral oncogene homolog B1 (BRAF)
BRAF is an oncogenic driver which accelerates the RAS–RAF–MEK–ERK pathway and induces cellular growth and hyper-proliferation [16]. BRAF gene mutations are present in 2–4% of NSCLC patients, of which approximately 50% of BRAF mutations have been detected as V600E [94, 95]. Non-V600E mutations, including G469A and D594G, account for 50% of total BRAF mutations [94,95,96]. Interestingly, in contrast with other rearrangements/drivers, non-V600E BRAF-driven cases are more associated with current/former smoking history [97, 98], while V600E BRAF mutant cases were commonly female or never smokers [98].
There are combined targeted therapies against V600 mutant melanoma that have achieved FDA approval. In 2014 the combined treatment of trametinib and dabrafenib, and later in 2020, the combination of atezolizumab with cobimetinib and vemurafenib was approved by FDA for patients with BRAF V600 mutation-positive melanoma [99]. The similarities in mutations and underlying mechanisms lead to several studies and clinical trials investigating these therapies for BRAF-positive NSCLC. BRAF mutation leads to the hyperactivity of the RAS–RAF–MEK–ERK axis, which exerts oncogenic features and drives hyper-proliferation [100]. Hence, blocking the members of the abovementioned pathway individually or together has shown encouraging effects against tumors.
Single treatment
Vemurafenib (PLX4032) was the first drug approved against BRAF-mutant cancer by the United States in 2011 [101]. Vemurafenib was first introduced 2008 as an oral selective inhibitor of oncogenic B-Raf kinase against melanoma harboring BRAF V600E mutation. Vemurafenib inhibits the activity of V600E BRAF mutation by blocking RAF/MEK/ERK pathway [102]. In 2015, vemurafenib administration by BRAF-positive NSCLC patients indicated a PFS of 7.3 months and an ORR of 42% [103]. In line with this study, in 2017, an open-label phase 2 study indicated that vemurafenib enhanced the PFS in previously untreated patients with V600 BRAF-driven NSCLC. Regarding toxicity, vemurafenib administration has been described as safe in these patients [104]. Dabrafenib selectively inhibits BRAF kinase and functions as an adenosine-triphosphate(ATP)-competitive inhibitor [105]. In 2016, an open-label phase 2 clinical trial study investigated the effect of dabrafenib in V600E BRAF-positive NSCLC cases. It has been reported that most patients had a rapid response to the treatment. Adenocarcinoma was controlled in 58% of patients, whereas the overall response was 33% [106]. This investigation was the first study (cohort A) of a series of three enrolled cohorts [106,107,108]. Cohorts B and C investigated the trametinib and dabrafenib combination, which will be mentioned below.
Adjuvant treatment/Co-administration
Dabrafenib + Trametinib (combined BRAF/MEK inhibition): Several clinical trials have investigated the combination of BRAF/MEK inhibition to develop a synergistic response. Planchard et al., in cohort B study, investigated the therapeutic outcome and toxicity of trametinib and dabrafenib combination for the first time in a phase 2 clinical trial [107]. This combined strategy was investigated in BRAF V600E-mutant metastatic NSCLC patients who were previously treated with platinum-based chemotherapy. The reported median PFS was longer than 9 months, and the overall response rate was more than 50%, which presents this combination as a promising candidate. Of note, in melanoma, intense adverse effects such as pyrexia, anemia, confusional state, decreased appetite, etc., were reported in more than half of the patients (56%), although the safety profile was manageable, and no treatment-related deaths were reported [107]. Later in 2017, in a cohort C study [108], Planchard et al. investigated the abovementioned combination therapy (trametinib plus dabrafenib) in patients with treatment-naïve BRAF V600E-mutant metastatic NSCLC. Thus, the response rate and toxicity are comparable in previously treated vs untreated patients. Due to the promising results of trametinib and dabrafenib combination therapy, in 2017, the FDA approved this treatment option for patients harboring BRAF V600E-positive metastatic NSCLC [109].
Trametinib + Vemurafenib: In 2015, a study compared the outcome in patients treated with trametinib (a MEK inhibitor) with or without vemurafenib. A single administration of vemurafenib affected V600E-BRAF, whereas trametinib affected both V600E-BRAF and non-V600E-BRAF. It has been reported that vemurafenib and trametinib co-administration enhanced BIM (pro-apoptotic protein) and apoptosis compared to single trametinib administration. Thus the combination of a BRAF inhibitor and a MEK inhibitor showed encouraging efficiency. Of note, this study also investigated the vemurafenib and erlotinib combination in BRAF-V600E cells, and they observed no synergistic effect [110].
Rearranged during transfection (RET)
RET fusions have been reported in approximately 1% of NSCLC cases. Similar to the ROS1 fusions, most of the RET-positive population never included smokers and young patients [111]. The autophosphorylation of RET TKI domains drives Proto-Oncogene pathways. It activates downstream pathways, such as phosphatidylinositol 3-kinase (PI3K)/AKT, RAS/MAPK, c-Jun N-terminal kinase (JNK, and RAS/extracellular signal-regulated kinase (ERK), which are involved in the cellular growth, differentiation, and proliferation [112]. Additionally, it has been reported that RET rearrangements in advanced NSCLC patients are independently associated with an increased risk of brain metastases [113].
Both selective and non-selective RET inhibitors have been tested in clinical trials. While the reported outcomes were controversial, FDA has recently approved two selective RET inhibitors (pralsetinib and selpercatinib) as therapeutic options for metastatic NSCLC patients harboring RET fusions [114, 115].
Selective RET inhibitors
Pralsetinib (BLU-667): Pralsetinib is a selective tyrosine TKI, which potently targets and inhibits RET fusions. A multi-cohort, open-label, phase 1/2 study (ARROW) study (NCT03037385) tested the potential activity and safety of the pralsetinib in both previously treated and untreated patients with RET-altered metastatic NSCLC [116]. The maximum tolerated dose was determined once-daily 400 mg, based on a phase 1 study [117]. The response rate was reported based on the ORR. The ORR of previously-treated patients with platinum-based chemotherapy was 61%, of which 6% showed a complete response. While the ORR of treatment-naïve patients was 70%, and the complete response rate was 11%. These data suggested a better ORR in treatment-naïve; however, previously treated patients showed linger median duration of response in the periods of 6 and 12 months [116]. Although no treatment-related deaths were reported, several adverse effects were observed, including neutropenia, hypertension, and anemia.
Altogether, these findings introduce oral pralsetinib as a potent, well-tolerated treatment for RET-altered metastatic NSCLC patients [116]. The response to pralsetinib in RET fusion-positive NSCLC patients was also assessed based on the alterations/clearance of the level of the RET circulating tumor DNA (ctDNA) in blood samples. Interestingly RET ctDNA was rapidly decreased in almost all patients with every range of administered pralsetinib doses. After 8 weeks, RET ctDNA was not detectable in 81% of NSCLC patients, which is a rapid response [118]. A recruiting, randomized, open-label phase III trial compares pralsetinib and conventional first-line treatments based on the PFS of RET-altered NSCLC patients. The experimental arm will randomly receive pralsetinib. The squamous cell lung carcinoma patients of the control arm will receive platinum + gemcitabine, while non-squamous cell lung cancer patients of the control arm will receive platinum + pemetrexed ± pembrolizumab. This study aims to determine the appropriate choice for the first-line treatment [119].
Selpercatinib (LOXO-292): Selpercatinib (LOXO-292) is an oral, highly selective TKI inhibitor. It is an ATP-competitive, small-molecule RET inhibitor which has been developed to inhibit resistant or activating RET mutations. Compared to the multi-kinase inhibitors (MKIs), selpercatinib has shown remarkable activity in RET-inhibition and meaningful less toxicity in vitro and in vivo [120]. Clinically, selpercatinib presented potent activity in a patient with KIF5B-RET fusion-positive lung cancer. The alectinib-resistant patient had previously received treatments, including whole-brain radiotherapy, immunotherapy, and chemotherapy. It has been reported that selpercatinib dramatically reduced neurological [120]. A recent phase I/II study (LIBRETTO-001, NCT03157128) investigated the efficacy and toxicity of selpercatinib separately in two different cohort studies in previously treated and treatment-naïve patients with advanced RET fusion-positive NSCLC [121]. The objective response (partial or complete) was considered the primary endpoint. The PFS, duration of response, and safety were secondary endpoints in this study. Treatment-naïve showed an 85% accurate response, while the objective response of previously treated patients was 64%. In line with prior observations, selpercatinib significantly exerted intracranial activities against brain metastasis. The median CNS duration of response was 10.1 months, and the objective intracranial response was reported to be 91%. Selpercatinib showed intracranial responses, significantly rapid response, and persistent activity. Due to the insignificant/absent off-target activities, selpercatinib induced low-grade toxicity, and most of the drug-induced grade 3 toxicities were manageable via dose modifications. Thus, selpercatinib could be employed as a long-term RET-targeted therapy in patients with NSCLC [121].
Due to the promising reports of selective RET inhibitors, several phase 3 studies investigated novel agents to introduce the best treatment as the first-line treatment option. A phase III study (LIBRETTO-431, NCT04194944) is comparing standard first-line treatments with selpercatinib in RET-fusion-positive NSCLC patients, and this trial is in recruiting stage. In this study, the PFS of the control group who receive platinum-based chemotherapy (carboplatin or cisplatin) with or without pembrolizumab will be those who received selpercatinib [122]. In addition, this recruiting-stage trial aims to investigate the efficacy and safety of selpercatinib in combination with chemotherapy ± pembrolizumab.
Non-selective RET inhibitors
Non-selective RET inhibitors are MKIs, which target multiple kinases, have shown limited efficacy, and induced off-target toxicity. Non-selective RET inhibitors, including vandetanib, cabozantinib, and lenvatinib, were the first MKIs tested in RET-altered NSCLC patients. In a study in 2017, the highest total response rate (37%) and median PFS (3.6 months) were attributed to cabozantinib. The response rates of sunitinib and vandetanib were reported at 22% and 18%, respectively, although no complete response was observed. The median PFS of sunitinib and vandetanib were reported at 2.2 and 2.9 months, respectively [123]. Due to the limited efficacy and potentially high off-target toxicity of the multi-kinase inhibitors (MKIs), selective RET inhibitors appear to be more appropriate candidates. Resistance to both FDA-approved RET inhibitors is Inevitable, and G810C has been reported as the most resistant RET mutant [82].
Thus, further investigations are required to develop novel selective RET inhibitors and combined therapies. A phase I/II clinical trial (NCT04161391) aims to investigate TPX-0046 efficacy, safety, and tolerability in drug-resistant and naïve RET-altered tumor models with Solvent Front Mutations (SMF). TPX-0046 has shown potent anti-tumor activity in vitro and in vivo against various RET alterations, particularly against SFM-mediated resistance. In phase II, this trial will test TPX-0046 in treatment-naive or pretreated stage IV NSCLC patients to investigate the ORR [124].
mesenchymal-epithelial transition (MET)
Mesenchymal-Epithelial Transition (MET) is a proto-oncogene belonging to the tyrosine kinase receptors family that was activated following the binding to its ligand, hepatocyte growth factor (HGF) [125]. Besides the physiological function of MET in cell proliferation [126], this proto-oncogene undergoes various alterations like mutations, amplifications, and overexpression, which could lead to malignant cells [127]. For instance, it has been reported that MET is overexpressed in 20% of NSCLC and amplified in 1–5% of NSCLC [128, 129]. On the other hand, it has been introduced as one of the critical processes in metastasis, which is needed to revert the mesenchymal phenotype to the epithelial phenotype for attaching the cancer cells to other tissues [130]. In this regard, it has been shown that this aberrant MET expression is correlated with a poor clinical prognosis of NSCLC [131]. Thus, targeting MET as a therapeutic option has been investigated in clinical trials.
Small molecule inhibitors of MET are divided into three main groups according to the binding site on MET structure [132]. type I and II prevent ATP binding and activating of the receptor (with the distinct binding site), while type III binds allosteric sites rather than the ATP-binding site [133]. In addition, type I of MET inhibitors include two subtypes; Ia (i.e., Crizotinib) and Ib (i.e., Capmatinib, Tepotinib, and Savolitinib), which Ib group is more specific to MET and has less off-target effects [134]. Of note, Crizotinib was first approved for the treatment of ALK-rearranged NSCLC and ROS1-rearranged NSCLC, but then it has been demonstrated that it could be recruited in NSCLC with MET amplification [135]. After Crizotinib FDA approval, Capmatinib and Tepotinib were approved for metastatic NSCLC in patients with MET exon 14 (METex14) skipping mutation [136], as one of the main mutations, which contributes to NSCLC independently [137]. Type II inhibitors, including Cabozantinib, Glesatinib, and Merestinib, not only bind to a hydrophobic pocket beside the ATP binding site of MET but also can block different kinases such as RON, AXL, VEGFR2, etc. Although Cabozantinib was approved for the treatment of advanced medullary thyroid carcinoma and advanced clear-cell renal-cell carcinoma, the studies on the efficacy and safety of Cabozantinib in MET-mutated NSCLC have shown promising results [138]. Moreover, Tivantinib, as a type-III inhibitor, was used in combination with Erlotinib, as an RGFR inhibitor, which revealed its beneficial effects on patients with NSCLC [139].
Moreover, there are other various strategies in the targeting of MET, which were included as immunotherapy. Regardless the expression of PD-L1 is remarkable in MET exon 14 NSCLC [140], and the immune checkpoint blockage strategy seems ineffective as monotherapy in patients with MET exon 14 NSCLC [141, 142]. Conversely, the combination of Cabozantinib with other anti-PD-L1 monoclonal antibodies in clinical trials revealed the safety and efficacy of this combination approach [143]. In addition to combination therapy, recruiting monoclonal antibodies as a tool for targeting the MET has been developed to disrupt the HGF/MET interaction [144, 145]. In this regard, different MET-specific monoclonal antibodies have been designed and studied, such as Onartuzumab, Telisotuzumab, and Amivantamab [138]. Amivantamab is a bi-specific monoclonal antibody targeting either EGFR or MET approved for patients with advanced metastatic NSCLC [146].Regardless of the progressions in MET targeting strategies and promising results, the tumor heterogeneity and selecting an appropriate population of patients benefiting from targeted therapy is challenging [147]. In addition, acquired resistance is another issue not only in MET targeting but also in all TKI approaches [148].
Neurotrophic tropomyosin receptor kinase (NTRK)
The Neurotrophic tropomyosin receptor kinase (NTRK) involves three transmembrane receptor tyrosine kinases, TRKA, TRKB, and TRKC are responsible for the development and maturation of the central nervous system physiologically [1]. These tyrosine kinase receptors mediate three main signaling pathways; Ras/Raf/MAPK pathway, PI3K/Akt/mTOR pathway, and PLCc/PKC pathway are critical for cell proliferation and plasticity [2, 3]. Physiologically, the activation of these receptors depends on three ligands, including nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and neurotrophin 3 (NT-3) [3, 4]. Otherwise, some genetic alterations in NTRK genes have been shown in malignancies, such as mutations, amplifications, splice variants, and deletions, which could lead to ligand-independent activation of downstream signaling pathways [5, 6]. The most prevalent alteration reported in numerous cancers is NTRK fusions (less than 1% of patients with NSCLC) which leads to the production of a chimeric protein with oncogenic activation [7]. Although it seems that NTRK fusions are just a minority cause of NSCLC, the high prevalence of NSCLC in the world made NTRK an interesting target for developing inhibitory drugs [8].
Regardless of NTRK inhibiting by unselective tyrosine kinase inhibitors (TKIs) such as crizotinib, cabozantinib, etc., which inhibit a range of targets, other more selective drugs have been developed [9]. Consequently, larotrectinib and entrectinib are two drugs that could get FDA approve after showing promising results in clinical trials. Entrectinib is an oral TKI with more selective activity against TRK, ROS1, and ALK. Although this drug is not just particular for TRK, according to the result of clinical trials, the ORR in patients with NTRK fusion-positive NSCLC was 70% with minimal tolerable adverse effects [10, 11]. Finally, FDA granted accelerated approval to entrectinib for adults with NTRK fusion-positive solid tumors. In addition, larotrectinib is a selective pan-TRK inhibitor whose efficacy and safety in NSCLC, and other cancers have been demonstrated [12, 13]. Accordingly, in a short time after clinical trials started, FDA approved it for the treatment of adult and pediatric patients with NTRK fusion-positive tumors. Interestingly, it has been reported that patients enrolled in clinical trials with brain metastases efficiently respond to the treatment with entrectinib and relatively with larotrectinib [10, 14].
Due to the acquired resistance mutations in tumor cells treated with the first generation of NTRK inhibitors which affect the durability of the drug, the second generation of inhibitors has been developed [15]. This new generation of inhibitors has a specific macrocyclic structure which could circumvent the on-target mutation of NTRKs [16]. In this regard, three drugs are under investigation; selitrectinib, taletrectinib, and repotrectinib. Although FDA just granted repotrectinib for Fast-Track designation in patients with advanced solid tumors harboring NTRK gene fusion [17], the results of selitrectinib case reports and phase I/II trials of taletrectinib suggest the efficacy and safety of these medications in the treatment of tumors [18, 19]. furthermore, to clarify the direct effect of these medications on patients with NSCLC, more studies are needed.
Neurotrophic tropomyosin receptor kinase (NTRK)
The Neurotrophic tropomyosin receptor kinase (NTRK) involves three transmembrane receptor tyrosine kinases, TRKA, TRKB, and TRKC, which are responsible for the development and maturation of the central nervous system physiologically [149]. These tyrosine kinase receptors mediate three main signaling pathways; Ras/Raf/MAPK pathway, PI3K/Akt/mTOR pathway, and PLCc/PKC pathway, which are critical for cell proliferation and plasticity [150, 151]. Physiologically, the activation of these receptors depends on three ligands, including nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and neurotrophin 3 (NT-3) [151, 152]. Otherwise, some genetic alterations in NTRK genes have been shown in malignancies, such as mutations, amplifications, splice variants, and deletions, which could lead to ligand-independent activation of downstream signaling pathways [153, 154]. The most prevalent alteration reported in numerous cancers is NTRK fusions (less than 1% of patients with NSCLC), which leads to the production of a chimeric protein with oncogenic activation [155]. Although it seems that NTRK fusions are just a minority cause of NSCLC, the high prevalence of NSCLC in the world made NTRK an interesting target for developing inhibitory drugs [156].
Regardless of NTRK inhibiting by unselective tyrosine kinase inhibitors (TKIs) such as crizotinib, cabozantinib, etc., which inhibit a range of targets, other more selective drugs have been developed [157]. Consequently, larotrectinib and entrectinib are two drugs that could get FDA approve after showing promising results in clinical trials. Entrectinib is an oral TKI with more selective activity against TRK, ROS1, and ALK. Although this drug is not just selective for TRK, according to the result of clinical trials, the ORR in patients with NTRK fusion-positive NSCLC was 70% with minimal tolerable adverse effects [158, 159]. Finally, FDA granted accelerated approval of entrectinib for adults with NTRK fusion-positive solid tumors. In addition, larotrectinib is a selective pan-TRK inhibitor whose efficacy and safety in NSCLC, and other cancers have been demonstrated [160, 161]. Accordingly, in a short time after clinical trials started, FDA approved it for the treatment of adult and pediatric patients with NTRK fusion-positive tumors. Interestingly, it has been reported that patients enrolled in clinical trials with brain metastases efficiently respond to the treatment with entrectinib and relatively with larotrectinib [158, 162].
Due to the acquired resistance mutations in tumor cells treated with the first generation of NTRK inhibitors, which affect the durability of the drug, the second generation of inhibitors has been developed [163]. This new generation of inhibitors has a specific macrocyclic structure, which could circumvent the on-target mutation of NTRKs [164]. In this regard, three drugs are under investigation, selitrectinib, taletrectinib, and repotrectinib. Although FDA just granted repotrectinib for Fast-Track designation in patients with advanced solid tumors harboring NTRK gene fusion [165], the results of selitrectinib case reports and phase I/II trials of taletrectinib suggest the efficacy and safety of these medications in the treatment of tumors [166, 167]. Furthermore, to clarify the direct effect of these medications on patients with NSCLC, more studies are needed.
Human epidermal growth factor receptor 2 (HER2)
The human epidermal growth factor receptors are a series of receptor tyrosine kinase (RTK) which consist of 4 members; HER1-4 encoding by ErbB1-4 genes respectively [168]. HER2 is a transmembrane glycoprotein, and no exact ligand has been demonstrated for it [169]. Although activation of HER2 leads to several proliferative or apoptotic signaling pathways, including MAPK, PI3K/AKT, and JAK/STAT, its oncogenic and tumorigenic roles have been clearly illustrated in different cancers [170]. In this regard, three main genetic alterations have been demonstrated that could lead to NSCLC; mutation, overexpression, and amplification. Their frequency has been reported to be about 2–4%, 11–32%, and 2–23% respectively, in various studies [171, 172]. Nevertheless, the exact definition of HER2-positive lung cancer has not been addressed.
Targeting HER2 by various inhibitors such as pyrotinib, poziotinib, lapatinib afatinib, dacomitinib, and neratinib has been studied as a therapeutic option in patients with NSCLC, but unhopefully, none of them could yet get approved [173,174,175,176]. Despite the unsatisfactory results of various TKIs in treating NSCLC, some clinical trials showed promising results. For instance, in the treatment of NSCLC patients harboring HER2 exon 20 mutations with pyrotinib (as an oral pan-HER TKI), an ORR of 53.3% and a median PFS of 6.4 months with no extreme adverse events have been reported [177]. Moreover, a new small molecule TKI, mobocertinib, which selectively inhibits EGFR insertion and HER2 mutation, is under investigation with primary acceptable results [178]. In immunotherapy, monoclonal antibodies targeting HER2 have been investigated for the treatment of NSCLC. In this regard, two drugs, pertuzumab and trastuzumab, which have been approved for HER2 + breast cancer, have been used in clinical trials, but more studies are needed to confirm their therapeutic effect on NSCLC [179,180,181]. In addition, antibody–drug conjugations have been developed as an effective strategy for targeting HER2. T-DMI and T-DXd are two antibody–drug conjugates in which trastuzumab is attached to emantisine and deruxtecan, respectively. Both of these drugs have been implicated in clinical trials and showed promising results, which caused to T-DXd drug to get FDA grant for HER2-mutant NSCLC and gastric cancer [182, 183]. The main issue in recruiting HER2-targeting therapeutic approaches in treating NSCLC is detecting the proper population of patients sensitive to these medications. In other words, HER2 + NSCLC should be defined clearly for researchers and clinicians. Moreover, the lack of pharmacodynamic and pharmacokinetic information is another limitation for recruiting these medications on larger scales. Accordingly, more clinical and preclinical studies are needed to design the optimal guideline.
PI3K/AKT/mTORC
The PI3K/Akt/mTOR signaling pathway has been considered one of the most commonly altered molecular pathways in NSCLC. EGFR and KRAS, the types of oncogenic drivers in NSCLC, can activate the PI3K/AKT/mTOR pathway, which enhances cancer cell proliferation, metabolism, and survival [184]. Besides, the RTKs are known as an entry point to PI3K/AKT/mTOR pathway activation. RTK mutation and amplification lead to ligand-independent signaling in NSCLC. Accordingly, targeting the PI3K/Akt/mTOR signaling pathway could suppress these signals [185].
PI3K is a family of intracellular lipid kinases classified into three classes based on structure and function. Each class showed distinct roles in signal transduction that regulated oncogenic transformation and tumor maintenance. However, somatic mutation and amplification of PI3K classes have been found in patients with NSCLC [186]. Activation mutations in PIK3CA (encoding the catalytic subunit PI3Kα) and alterations of the tumor suppressor phosphatase and tensin homolog (PTEN) are reported in squamous and non-squamous NSCLC. Preclinical and clinical studies suggested that targeting the PI3K pathway cloud regresses PIK3CA-mutant lung cancer. The growth of PI3K-dependent NSCLC cell lines could block through simultaneous inhibition of multiple PI3K pathway components, reducing the NSCLC progression [187, 188]. PI3K inhibitors are classified as pan-PI3K and selective PI3K inhibitors [184]. Several pan-PI3K inhibitors, including, Pilaralisib (XL-147), Buparlisib (BKM120), PX-866, and Pictilisib (GDC-0941), have been tested in clinical trial phases. The data associated with partial responses did not indicate significant improvement in PFS and OS [184]. Hence, the pan-class I PI3K inhibitor therapies have not shown enough efficacy. Several clinical trials are performed based on isoform-specific class I PI3K inhibitors. Of these, Alpelisib (BYL719) and Taselisib (GDC-0032) are potent PI3K inhibitors, which target the p110α isoform and the p110α, p110γ, and p110δ isoforms, respectively. They have been under evaluation in a phase II study of patients with advanced NSCLC [189, 190]. Other Selective PI3K inhibitors, including INK1117, GSK2636771, AZD8186, and SAR260301, have been investigated in clinical phase I trials [184].
On the other hand, upregulation of the Akt pathway is significant in NSCLC patients associated with increased mTOR [191]. Preclinical studies in NSCLC cell lines demonstrated that Akt activation leads to PTEN, EGFR or PIK3CA mutation, or HER2 amplification [192]. Therefore, targeted inhibition of Akt is an efficient therapeutic strategy, as many studies have characterized the extensive list of Akt pathway inhibitors in development. Perifosine is an alkyl phospholipid (APL) that blocks Akt translocation to the membrane, preventing Akt phosphorylation and activation, the best-tolerated treatment against NSCLC [193]. The evidence illustrated that using perifosine debilitates the translocation of GSK3β to the cell membrane and the consequent phosphorylation by kinases to Akt and ERK.
Moreover, perifosine therapy can inhibit rapamycin and significantly reduce the level of p-GSK3β and GSK3β in A549-RR cells. These results suggested that perifosine can counteract several survival pathways activated by mTORC1 inhibition, including GSK3β. However, recent studies found that a combination of perifosine and rapamycin can be more efficient than monotherapy, with every single reagent inhibiting cell growth by regulating the activity of GSK3β [194]. MK-2206 is another potent and selective inhibitor of AKT with anti-proliferative activity. Based on in vitro and in vivo results, the combined effects of MK-2206 with RTK inhibitors such as lapatinib and erlotinib can significantly exert tumor inhibitory activities [195]. Also phase II trial indicated that the combination of MK2206 and erlotinib met its primary endpoint in patients pretreated with erlotinib [196].
Meanwhile, significant proportions of NSCLC demonstrated upregulation of the mTOR pathway. mTOR complexes (mTORC1 and mTORC2) play an essential role in maintaining cellular homeostasis and growth [197]. Phosphorylation of p70-ribosomal protein S6 kinase 1 (p70-S6K1) and elongation initiation factor (EIF)-4E binding protein 1 (4E-BP1), are two significant substrates of mTORC1, regulate numerous processes such as cell growth, proliferation, migration, and invasion in NSCLC patients [198]. mTORC2 directly phosphorylates and activates Akt and Protein Kinase Cα (PKCα) [199]. Accordingly, mTOR inhibitors can be an attractive strategy to prevent the progression of advanced NSCLC. Ongoing clinical trials have been developed and investigated in preclinical and clinical studies, including Everolimus, Sirolimus, Temsirolimus, and Ridaforolimus. Everolimus is an mTOR inhibitor that explicitly targets mTORC1. Conservative evaluation indicated that side effects were of mild severity in most patients treated with Everolimus [200]. On the other hand, Combination therapy of Everolimus and EGFR inhibitors showed limited antitumor activity in NSCLC patients with a mutation in the PI3K-AKT-mTOR pathway [201]. Based on the phase I/II study, the combination of Sirolimus with Pemetrexed demonstrated potential activity and appeared to have no severe safety concerns [202]. Using Temsirolimus as a single targeted agent failed to meet its efficacy endpoint [203]. Although, the combination of Temsirolimus and Neratinib (HER2 inhibitor) showed a 19% response in patients with HER2-mutant lung cancers [204]. Ridaforolimus treatment showed improvement in PSF and trending for better OS in NSCLC patients with KRAS-mutant [205]. NSCLC involves a multistep process and is associated with several intracellular pathways and several genetic alterations. Therefore, using a single targeted agent may not be optimal [203]. Dual targeting has been developed by blocking both PI3K and mTOR. Cyclin D and FOXO forkhead transcription factors are important downstream targets of PI3K/Akt signaling. BEZ235 downregulated cyclin D1 and cyclin D3 expression in NSCLC through transcriptional repression and proteasome-mediated degradation, ultimately arresting the cell cycle at the G1 phase and inhibiting PI3K/Akt activity [203]. BEZ235 also inhibited mTOR signaling mediated by reduced cyclin D expression [206]. Additionally, XL765 and GDC-0980 were tested in a phase Ib trial that demonstrated an acceptable safety profile in NSCLC patients [207, 208].
Targeting programmed death receptor 1 (PD-1)/ PD-L1 pathway
PD-1/PD-L1 signaling axis is prominent in regulating immune responses and maintaining self-tolerance or tissue integrity [209]. PD-1 is a negative costimulatory receptor expressed primarily on the surface of activated T cells, B cells, natural killer T cells, activated monocytes, and dendritic cells (DCs) [210]. PD-1 binds with its ligands, PD-L1 and PD-L2, which belong to the B7 family and can be expressed by tumor cells, normal cells, and immune cells [211]. However, PD-1-expressed T cells mediated to inhibit effective anti-tumor immune responses. PD-1/PD-L1 interaction inhibits the expression of T cell transcription factors such as GATA-3 and T-bet. It also inhibits CD8+ cytotoxic T lymphocyte (CTL) function, survival, and proliferation and induces apoptosis of tumor-infiltrating T cells. Besides, regulatory T cell (Tregs) differentiation and maintaining their suppressive function can mediate PD-L1 expression. Thereby, inhibition of PD-1/PD-L1 pathways can activate the anti-tumor activity mediated by both effector T cell activation and Treg inhibition [212].
Whereas the migration of immune cells to the tumor exhibit anti-tumor activity, over time, the tumor microenvironment favoring to becomes immunosuppressive, intending the emergence of tumor-promoting cells such as M2 macrophages, T regs, and myeloid-derived suppressor cells (MDSCs). Cancer cells escape from immune recognition and use immune-inhibitory mechanisms to evade the immune system's defenses. Immune editing is a dynamic process used to modulate the immune microenvironment and improve immune cell function so it can induce the alternative mechanisms of immune evasion [213,214,215]. Targeting the PD-1/PD-L1 checkpoint could regulate immune responses to eliminate NSCLC. Modulating the PD-1/PD-L1 pathway is currently under development as potential immunotherapies for patients with NSCLC. Several PD-1/PD-L1 antibodies are approved for the first- and second-line setting that improved efficacy and longer duration of response compared to other standard treatments and demonstrated manageable toxicity profiles [216]. Nivolumab is a fully human monoclonal antibody against PD-1 that was approved for treating patients with NSCLC. The patient treated with nivolumab in first-line monotherapy did not show more PFS than those who received platinum-based chemotherapy in a broad population with a PD-L1 expression level of 5% or more. Nivolumab showed a favorable safety profile as compared with chemotherapy [217]. Besides, based on the phase 3 study, nivolumab was correlated with a significant improvement in overall survival and response rate versus docetaxel in advanced non-squamous NSCLC [218]. Pembrolizumab, MK-3475, a humanized monoclonal IgG4 anti-PD-1 antibody, is another anti-PD1 showing robust anti-tumor activity and significantly improved PFS and OS than chemotherapy as first-line therapy for metastatic NSCLC with PD-L1 tumor proportion score of at least 50%. There was a lower incidence of treatment-related AEs than platinum-based chemotherapy, and did not show any evidence of increased toxicity during long-term follow-up [219]. The analyses demonstrated that pembrolizumab had a better ORR than nivolumab, but the difference in PFS was not significant between pembrolizumab and nivolumab in patients with recurrent or advanced NSCLC [220]. The evidence indicated that NSCLC patients with EGFR- or HER2-mutated and ALK-rearranged do not benefit from immunotherapy. Also, BRAF- and MET-mutated NSCLC are supposed to be as sensitive to anti-PD1/PD-L1 immunotherapy [221]. Although anti-PD1 monotherapy such as nivolumab and pembrolizumab had similar efficacy in older and younger patients with NSCLC, survival was significantly worse in patients with poor performance status (PS). However, an immune checkpoint inhibitor may be considered for NSCLC patients with poor PS harboring positive PD-L1 expression [222]. Other immune checkpoints were potential therapeutic targets, including programmed death receptor ligand 1 (PD-L1). Atezolizumab is a humanized IgG1 antagonist antibody to PD-L1 that blocks the interaction between the PD-L1 and PD-1 receptors activation complex. It is designed to impede inhibitory signals in T cells, with resultant tumor recognition by cytotoxic T cells. atezolizumab therapy improved the OS and ORR in patients with NSCLC expressing PD-L1 [223]. Cemiplimab monotherapy significantly enhanced OS and PFS versus chemotherapy in patients with advanced non-small-cell lung cancer with PD-L1 of at least 50% [224]. Moreover, durvalumab is a selective, high-affinity, engineered, human IgG1 monoclonal antibody that blocks the interaction between PD-L1 and PD-1, allowing T cells to recognize and kill tumor cells. Based on the real-world prospective study, durvalumab therapy had a safety profile and improved progression-free survival and OS among patients with stage III NSCLC [225, 226]. According to meta-analysis data, anti-PD-1 and anti-PD-L1 therapy had significantly longer OS and illustrated a lower ORR, higher 3–4 Grade AEs rate, and higher drug-related death than chemotherapy [227]. However, the Phase 3 trial demonstrated that Avelumab did not significantly improve OS versus docetaxel in patients with NSCLC with PD-L1 + tumors [228].
Additionally, Epigenetic mechanisms were used to modulate PD-L1 expression in cancer cells. miR-200c is one miRNA that directly binds to the 3′UTR of PD-L1 in NSCLC to inhibit PD-L1 expression [229]. miR-135 and miR-3127-5p positively regulate PD-L1 expression in NSCLC that indirectly induce PD-L1 expression by activating the PI3K-AKT-mTOR pathway [230, 231]. On the other hand, the methylation status of the PD-L1 promoter can use to predict the outcome of PD-1/PD-L1-targeted therapy. Anti-PD1 therapy may enhance drug resistance to anti-PD-1 immunotherapy Nivolumab in NSCLC patients through increasing PD-L1 promoter methylation and decreased PD-L1 expression [232].
Cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4)
CTLA-4 (Cytotoxic T-lymphocyte-associated antigen-4, CD152) is a type of immune checkpoint pathway that mediates negative regulation of T cell activation and preservation of self-tolerance. However, CTLA-4 expression is a potential prognostic and predictive biomarker in NSCLC patients. Evidence showed that the expression of CTLA-4 has a different prognostic effect in metastatic NSCLC lymph nodes versus primary tumors, which is mediated by phenotypical differences between the tumor microenvironments of lymph nodes and primary tumors [233]. CTAL-4 is considered a leader of the immune checkpoint inhibitors, which potentially suppressing autoreactive T cells at the initial stage of naive T-cell activation, typically in lymph nodes [234]. The inhibitory function of CTLA-4 was administered through several mechanisms, such as competition with CD28-positive costimulation for binding to their shared B7 ligands (CD80/CD86) [235]. CTLA-4 is considered the homolog of CD28 that shows a higher binding affinity for B7 than CD28. CTLA-4:B7 binding can produce an inhibitory signal through contraction of the stimulatory signals from CD28:B7 and TCR: MHC binding. However, CTLA-4:B7 inhibitory signals include direct inhibition at the TCR immune synapse and enhanced mobility of T cells, causing the reduced ability to interact with APCs [234].
Besides, the independent prognostic effect of CTLA-4 overexpression was indicated in NSCLC, a favorable impact of CTLA-4 overexpression on clinical outcomes. CTLA-4 overexpression can cause a worse prognosis due to an enhanced downregulation of T-cell activation. Indeed, it showed that CTLA-4 mediated negative signals into cancer cells, compared with the ones currently observed in T cells [236]. However, the permanent expression of CTLA-4 on cancer cells showed a critical role in cancer cell progression by producing inhibitory signals to weaken the immune response. Hence, targeting CTLA-4 is an attractive strategy for increasing immune efficacy against malignancies and improving the prognosis of tumor patients. Multiple preclinical and clinical trials were performed to test the antibodies targeting CTLA-4, including ipilimumab and tremelimumab [235]. Ipilimumab is a fully humanized IgG1κ mAb targeting CTLA4. Treatment with ipilimumab improved irPFS and mWHO-PFS and safety and tolerability without the severe impact of toxicities in patients with stage IV NSCLC [237]. Tremelimumab is another monoclonal immunoglobulin G2 antibody against CTLA-4 that enhances immune function via preventing normal downregulation of T cells and prolonging T-cell action [238]. Tremelimumab indicated a safety profile and improved the durability of objective responses than standard chemotherapy in patients with advanced cancer such as NSCLC [239]. Clinical trial results illustrated that anti-CTLA4 mAbs lead to acute immune activation, which increases CD8+ CTL infiltrates in the tumor and may produce transient autoimmune manifestations. Corticosteroid therapy has been planned with CTLA-4 blockage to prevent the adverse event. Besides, a combination of CTLA-4 with GM-CSF-expressing tumor cell vaccine can be therapeutically effective; it targets prominent regulatory pathways of the immune system and modifies the immune response [239].
However, CTLA-4 blockade was suggested as a regulatory checkpoint for therapeutic development. Dual checkpoint therapy may be more efficient and reduce the adverts event of monotherapy with anti-CTLA-4 therapy. Also, PD-L1 expression and tumor mutational burden are potential biomarkers to respond to the combination approaches in these treatments. Based on First-line treatment, a combination of nivolumab plus ipilimumab improved PFS compared with chemotherapy in NSCLC patients with high tumor mutational burden. In comparison, patients with a low tumor mutational burden showed similar PFS in the nivolumab-plus-ipilimumab and chemotherapy groups. Dual therapy exhibited better efficacy than monotherapy in patients with a high tumor mutational burden [240]. Combination therapy tremelimumab with durvalumab has demonstrated clinical activity in advance-NSCLC patients. Based on a phase 3 randomized clinical trial, durvalumab plus tremelimumab did not statistically significantly enhance OS or PFS versus chemotherapy in patients with PD-L1 tumor proportion score ≥ 25%, while there was improved OS or PFS in durvalumab plus tremelimumab than chemotherapy in patients with 25% of tumor cells expressing PD-L1. Also, the dual checkpoint inhibition showed optimal benefit in OS and enhanced PFS. In addition, dual immunotherapy showed a high rate of TRAEs that was causing to discontinuation than durvalumab or chemotherapy [241].
FDA approved Clinical trials for NSCLC-targeted therapy
Clinical and preclinical studies indicated the efficacy and safety of multiple therapies for NSCLC patients with different aberrations, including EGFR exon 19 deletions or exon 21 (L858R) mutations, RET fusion-positive metastatic, ALK genomic tumor aberrations, EGFR exon 20 insertions. However, based on clinical trial results, several targeted therapies were approved by U.S. Food and Drug Administration (FDA) for NSCLC (Table 1). The receptor tyrosine kinase inhibitors, including erlotinib [242], gefitinib [243], osimertinib [244], crizotinib [245], lorlatinib [246], tepotinib [247, 248] received first approval for treating patients with EGFR mutation, acquired EGFR TKI resistance, ALK-positive, METex14 skipping alterations, respectively. In addition, immune checkpoint inhibitors such as atezolizumab [249], cemiplimab [250], and nivolumab plus ipilimumab [251] received FDA approval for treating patients with high PD-L1 expression and no EGFR or ALK genomic tumor aberrations. Moreover, Amivantamab, an intravenously administered bispecific antibody targeting EGFR and c-MET, received its first FDA approval for treating NSCLC with EGFR Exon 20 insertion mutations [146]. Based on findings from the CHRYSALIS clinical trial (NCT02609776), Amivantamab showed robust and durable responses with tolerable safety, which an ORR rate of 40%, a median PFS of 8.3 months, and a median OS of 22.8 months [252]. Unfortunately, according to recent evidence, the disease's progression and no treatment response have been shown in patients [253]. Recently, preclinical investigation supported the clinical development of mobocertinib for treating EGFRex20ins-mutated NSCLC [254].
Combination therapy
In order to overcome the limitations of target therapy and increase its efficiency, using this treatment in combination with other cancer treatment methods can be more effective. There are multiple examples of combined treatment of target therapy with other common treatments used for NSCLC patients, especially chemotherapy. Various studies have been conducted in this field, which can help to increase the efficiency of target therapy and overcome the limitations of this treatment method, especially drug resistance [255]. Extensive research has been conducted on the comparison of targeted therapy drugs with chemotherapy agents, including cetuximab, an anti-EGF-R monoclonal antibody, with cisplatin and vinorelbine [61], Bevacizumab, a mAb targeting the VEGF in combination with carboplatin and paclitaxel [256], and Figitumumab, a mAb targeting insulin-like growth factor type 1 receptor (IGF-1R), along with paclitaxel and carboplatin or gemcitabine/cisplatinum regimen [257]. On average, the results of all studies were similar. They showed that the combination therapy was effective and safe, and the group receiving these treatments had higher response rates, median progression-free survival, and overall survival. The combination of a c-MET inhibitor and the EGF-R inhibitor erlotinib was used compared to using either inhibitor alone in the NSCLC tumor xenograft mice model and indicated a significant anti-tumor function [258]. Vandetanib is a multi-target TKI that selectively targets VEGF-R, EGF-R, and RET tyrosine kinase activity. Similar to mentioned combination therapies, Vandetanib and docetaxel, exhibited promising outcomes with increased progression-free survival (PFS). It also showed that Vandetanib controlled lung cancer symptoms longer than chemotherapy alone [259, 260]. Promising results have also been reported from the conducted and ongoing trials on the effect of combination therapy sunitinib and erlotinib, as well as sunitinib along with platinum-based chemotherapy. Sunitinib is a multitargeted TKI with antiangiogenic and antitumor activities that inhibits VEGFR, PDGFR, KIT, RET, and Flt-3 [261]. Similar studies have reported promising results of combination therapy of everolimus, the mammalian target of rapamycin (mTOR) inhibitor with chemotherapy or combination with chemotherapy/gefitinib regimen, including higher survival, good response to therapy, and improvement in disease symptoms [200, 262]. Also, target therapy in combination with radiotherapy or combined chemotherapy/radiotherapy has resulted in promising results of improved survival and response to treatment. Examples are cetuximab, an EGFR inhibitor, along with radiotherapy and chemotherapy in advanced lung cancer [263, 264], as well as Gefitinib and erlotinib integrated into chemoradiation [265, 266].
Limitations of NSCLC targeted therapy
The widespread application of targeted therapies for advanced NSCLC to potentially cure disease, is hindered by several significant challenges. These include drug resistance, toxicity, and the high cost of these agents, which can limit their accessibility to all NSCLC patients.
Tumor heterogeneity and Lack of validated biomarkers
Another limitation of targeted therapy in NSCLC is the lack of validated biomarkers to predict response to treatment due to tumor heterogeneity [255]. While molecular testing is used to identify patients who may benefit from targeted therapy, not all patients with a particular molecular abnormality will respond to treatment. Additionally, some molecular abnormalities may be present in only a subset of cancer cells within a tumor, leading to incomplete targeting and potential resistance [267, 268]. Most targeted therapies have been developed for advanced-stage NSCLC patients who have already developed molecular abnormalities that can be targeted. However, early-stage NSCLC patients may not have these molecular abnormalities, and there may not be targeted therapies available for their specific subtype of NSCLC [269, 270]. This can make it challenging to identify which patients should receive targeted therapy and which should receive other types of treatment [255, 269, 271]. Additionally, even when molecular abnormalities are identified, there is often a lack of evidence on the optimal treatment approach, including which targeted therapy to use and the best sequencing of therapies.
Drug resistance
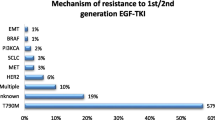
Resistance is a major limitation of targeted therapy in NSCLC, where cancer cells that were initially sensitive to treatment eventually become resistant and continue to grow and spread. Resistance can occur due to various mechanisms, including genetic mutations, alterations in gene expression, and changes in the tumor microenvironment. Sometimes, changes like mutations and amplifications occur in the target gene that allows cells to continue growing even when an inhibitor is present [255]. Targeted therapies can create selective pressure, which causes abnormalities that activate the driver oncogene. Malignant tumors with specific abnormalities initially respond well to selective inhibitors. However, resistance develops over time, often due to genetic aberrations in the target gene. For example, secondary mutations like EGFR C797S have been seen after osimertinib therapy, while EGFR T790M mutations can lead to resistance with erlotinib or gefitinib [272, 273]. Additionally, crizotinib has been shown to lead to on-target resistance in patients with ALK and ROS-1 mutations (L1196M and L2026M, respectively) [274]. These genetic changes and subsequent resistance are significant limitations to the effectiveness of targeted therapies in NSCLC. In NSCLC cells, acquired resistance to gefitinib or osimertinib has shown EMT characteristics, such as a decrease in E-cadherin and an increase in vimentin and stemness, without any secondary EGFR mutations [275]. AXL is implicated in the pathogenesis of EMT, and AXL inhibitors may restore sensitivity to erlotinib in mesenchymal EGFR-mutant cells [276]. While these inhibitors have shown significant efficacy in some patients with NSCLC, resistance can develop due to a variety of mechanisms, including downregulation of antigen presentation, upregulation of alternative immune checkpoints, and alterations in the tumor microenvironment.
Toxicity
Although short-term exposure to targeted therapy drugs is generally manageable, their prolonged use for chronic administration often results in significant toxic side effects. Targeted therapies were initially considered less harmful because they specifically target cancer cells. However, both "on-target" and "off-target" effects of these therapies can lead to toxicity. Also, some targeted therapies can cause serious adverse events such as liver or kidney toxicity, blood clots, and gastrointestinal problems [255]. These side effects can limit the dose and duration of therapy, reducing the effectiveness of the treatment. The on-target toxicity occurs when a drug designed to inhibit cancer-specific targets also inhibits a fixed set of proteins in normal cells, leading to toxicity by inhibiting the signaling pathway. This can result in a range of toxic side effects, including hyperglycemia with PI3K inhibition, hypertension with vascular endothelial growth factor inhibition, and skin rash with EGFR inhibition [277, 278]. Off-target toxicity occurs when a drug blocks a non-target protein, leading to harmful side effects. Unlike on-target toxicity, off-target toxicity is specific to a particular drug and does not exhibit a class effect. For example, osimertinib is associated with cardiac toxicity, such as heart failure, left ventricular dysfunction, conduction abnormalities, and myocardial injury, while gefitinib is not [279, 280].
Conclusions and future perspective
Over the last decade, the progress of molecular pathology has improved our knowledge of the pathophysiology and heterogeneity of NSCLC, resulting in significant evolution in treating patients with more advanced and effective methods. Target therapy and immunotherapy have opened a new and promising perspective in treating lung cancer by achieving successful outcomes. Several signaling pathways and specific oncogenic driver mutations have been identified that cause malignant alterations. New targets, such as microRNAs, VEGF, ALK, MET, HER2, and immune checkpoint inhibitors (PD-1 and CTLA4), as well as signaling pathways like PI3K/AKT/mTOR, RAS/MAPK, and NTRK/ROS1 pathways, are constantly discovered in addition to the epidermal growth factor receptor (EGFR), driving the emergence of new treatments. These factors and pathways have been the target of numerous medicines that have demonstrated clinical efficacy. Currently, some of these drugs, including EGFR inhibitors (gefitinib and erlotinib), PI3K/AKT/mTOR inhibitors (everolimus), and NTRK/ROS1 inhibitors (entrectinib) are being used as first-line therapies instead of chemotherapy. Interestingly, many NSCLC patients hopefully respond to checkpoint inhibitors, such as the anti-PD1 antibodies (nivolumab and pembrolizumab). Additionally, some research has indicated that certain targeted treatments combined with immunotherapies are effective in NSCLC. Drug resistance in the tumors is inevitable even though target therapy for NSCLC has controlled the disease. For improving treatment effectiveness, combination drug development and understanding resistance mechanisms are essential. Up to now, numerous clinical trials evaluating targeted therapy and immunotherapy agents are now being conducted, and the outcomes thus far are encouraging. The findings of trials will shed light on defining targeted therapy involvement in the treatment of NSCLC and clarify the roles of immune-based monotherapies, combined immunotherapies, and targeted therapy-immunotherapy combinations. Targeted therapy may ultimately change the way that lung cancer is treated for patients with few therapeutic options. Clinical research is still being done to discover the predictive factors responding to targeted therapies. Improved molecular biomarker knowledge and developed combined therapies might provide the most effective cure. Proper patient selection using predictive biomarkers will be crucial for theranostics and truly customized oncological treatment in the future to maximize the limited resources available and decrease vulnerability. Future gains in patient survival are anticipated if resistance is addressed, the suitable inhibitor or combination of inhibitors is used, and side effects minimal.
Availability of data and materials
Not applicable.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. 2018;68(6):394–424.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA. 2020;70(1):7–30.
Blumenschein G Jr, Kabbinavar F, Menon H, Mok T, Stephenson J, Beck J, et al. A phase II, multicenter, open-label randomized study of motesanib or bevacizumab in combination with paclitaxel and carboplatin for advanced nonsquamous non-small-cell lung cancer. Ann Oncol. 2011;22(9):2057–67.
García-Campelo R, Bernabé R, Cobo M, Corral J, Coves J, Dómine M, et al. SEOM clinical guidelines for the treatment of non-small cell lung cancer (NSCLC) 2015. Clin Transl Oncol. 2015;17(12):1020–9.
Majeed U, Manochakian R, Zhao Y, Lou Y. Targeted therapy in advanced non-small cell lung cancer: current advances and future trends. J Hematol Oncol. 2021;14(1):1–20.
Polverino A, Coxon A, Starnes C, Diaz Z, DeMelfi T, Wang L, et al. AMG 706, an oral, multikinase inhibitor that selectively targets vascular endothelial growth factor, platelet-derived growth factor, and kit receptors, potently inhibits angiogenesis and induces regression in tumor xenografts. Can Res. 2006;66(17):8715–21.
Lu T, Yang X, Huang Y, Zhao M, Li M, Ma K, et al. Trends in the incidence, treatment, and survival of patients with lung cancer in the last four decades. Cancer Manag Res. 2019;11:943.
Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JH, Beasley MB, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10(9):1243–60.
Clark SB, Alsubait S. Non small cell lung cancer. Petersburg: StatPearls Publishing Treasure Island (FL); 2021.
Al-Haideri M, Tondok SB, Safa SH, Maleki AH, Rostami S, Jalil AT, et al. CAR-T cell combination therapy: the next revolution in cancer treatment. Cancer Cell Int. 2022;22(1):365.
Esmaeilzadeh A, Jafari D, Tahmasebi S, Elahi R, Khosh E. Immune-based therapy for COVID-19. In: Rezaei N, editor. Coronavirus disease—COVID-19. Cham: Springer; 2021.
Sadeghi S, Tehrani FR, Tahmasebi S, Shafiee A, Hashemi SM. Exosome engineering in cell therapy and drug delivery. Inflammopharmacology. 2023;31(1):145–69.
Daniels MG, Bowman RV, Yang IA, Govindan R, Fong KM. An emerging place for lung cancer genomics in 2013. J Thorac Dis. 2013;5(Suppl 5):S491.
Dearden S, Stevens J, Wu Y-L, Blowers D. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Ann Oncol. 2013;24(9):2371–6.
Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12(2):175–80.
Upadhya A, Yadav KS, Misra A. Targeted drug therapy in non-small cell lung cancer: clinical significance and possible solutions—Part I. Expert Opin Drug Deliv. 2021;18(1):73–102.
Alexander M, Kim SY, Cheng H. Update 2020: management of non-small cell lung cancer. Lung. 2020;198(6):897–907.
Lin JJ, Shaw AT. Resisting resistance: targeted therapies in lung cancer. Trends Cancer. 2016;2(7):350–64.
Tian W, Cao C, Shu L, Wu F. Anti-angiogenic therapy in the treatment of non-small cell lung cancer. Onco Targets Ther. 2020;13:12113.
Zhao Y, Guo S, Deng J, Shen J, Du F, Wu X, et al. VEGF/VEGFR-targeted therapy and immunotherapy in non-small cell lung cancer: targeting the tumor microenvironment. Int J Biol Sci. 2022;18(9):3845–58.
Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo: presented in part at the keystone symposium “cellular and molecular biology of dendritic cells”, Santa Fe, NM, March 3–9, 1998, and at the annual meeting of the American association for cancer research, March 28-April 1, 1998. Blood, J Am Soc Hematol. 1998;92(11):4150–66.
Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2(10):1096–103.
Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet A-L, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212(2):139–48.
Guo X-F, Zhang Y-Y, Kang J, Dou Q-H, Zhu X-F. A bispecific decoy receptor VEGFR-EGFR/Fc binding EGF-like ligands and VEGF shows potent antitumor efficacy. J Drug Target. 2022;30(3):302–12.
Zhao Y, Wang H, Shi Y, Cai S, Wu T, Yan G, et al. Comparative effectiveness of combined therapy inhibiting EGFR and VEGF pathways in patients with advanced non-small-cell lung cancer: a meta-analysis of 16 phase II/III randomized trials. Oncotarget. 2017;8(4):7014.
Duan J, Yang Z, Liu D, Shi Y. Clinical efficacy of bevacizumab combined with gemcitabine and cisplatin combination chemotherapy in the treatment of advanced non-small cell lung cancer. J BUON. 2018;23(5):1402–6.
Zhou K, Zhao S, Guo W, Ding L. Efficacy and safety of erlotinib combined with bevacizumab in the treatment of non-small cell lung cancer: a systematic review and meta-analysis. Medicine. 2020;99(3):e18771.
Wang L, Zhang L, Wang L, Zhu L, Ma X. Influence of miR-199a on rats with non-small cell lung cancer via regulating the HIF-1alpha/VEGF signaling pathway. Eur Rev Med Pharmacol Sci. 2019;23(23):10363–9.
Zhang Y, Liu Y, Liu H, Tang WH. Exosomes: biogenesis, biologic function and clinical potential. Cell Biosci. 2019;9(1):19.
Mustachio LM, Chelariu-Raicu A, Szekvolgyi L, Roszik J. Targeting KRAS in cancer: promising therapeutic strategies. Cancers. 2021;13(6):1204.
Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170(1):17–33.
Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discovery. 2020;19(8):533–52.
Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–51.
Ostrem JM, Shokat KM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov. 2016;15(11):771–85.
Jacobs F, Cani M, Malapelle U, Novello S, Napoli VM, Bironzo P. Targeting KRAS in NSCLC: old failures and new options for “non-G12c” patients. Cancers. 2021;13(24):6332.
Koga T, Suda K, Fujino T, Ohara S, Hamada A, Nishino M, et al. KRAS secondary mutations that confer acquired resistance to KRAS G12C inhibitors, sotorasib and adagrasib, and overcoming strategies: insights from in vitro experiments. J Thorac Oncol. 2021;16(8):1321–32.
Hong D, Bang Y, Barlesi F, Durm G, Falchook G, Govindan R, et al. 1257O Durability of clinical benefit and biomarkers in patients (pts) with advanced non-small cell lung cancer (NSCLC) treated with AMG 510 (sotorasib). Ann Oncol. 2020;31:S812.
Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) Inhibition with sotorasib in advanced Solid tumors. N Engl J Med. 2020;383(13):1207–17.
Reck M, Spira A, Besse B, Wolf J, Skoulidis F, Borghaei H, et al. 1416TiP CodeBreak 200: a phase III multicenter study of sotorasib (AMG 510), a KRAS (G12C) inhibitor, versus docetaxel in patients with previously treated advanced non-small cell lung cancer (NSCLC) harboring KRAS p. G12C mutation. Ann Oncol. 2020;31:S894–5.
Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS pG12C mutation. New Engl J Med. 2021;384(25):2371–81.
Fell JB, Fischer JP, Baer BR, Blake JF, Bouhana K, Briere DM, et al. Identification of the clinical development candidate MRTX849, a covalent KRASG12C inhibitor for the treatment of cancer. J Med Chem. 2020;63(13):6679–93.
Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patientstherapeutic insight from the KRASG12C inhibitor MRTX849. Cancer Discov. 2020;10(1):54–71.
Janne P, Rybkin II, Spira A, Riely GJ, Papadopoulos KP, Sabari J, editors. KRYSTAL-1: updated safety and efficacy data with adagrasib (MRTX849) in NSCLC with KRASG12C mutation from a phase 1/2 study. presented at the 32nd EORTC-NCI-AACR symposium; 2020.
Riely GJ, Ou SI, Rybkin I, Spira A, Papadopoulos K, Sabari J, et al. 99O_PR KRYSTAL-1: activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (Pts) with advanced non–small cell lung cancer (NSCLC) harboring KRASG12C mutation. J Thorac Oncol. 2021;16(4):S751–2.
Weiss J, Yaeger R, Johnson M, Spira A, Klempner S, Barve M, et al. LBA6 KRYSTAL-1: adagrasib (MRTX849) as monotherapy or combined with cetuximab (Cetux) in patients (Pts) with colorectal cancer (CRC) harboring a KRASG12C mutation. Ann Oncol. 2021;32:S1294.
Arteaga CL. The epidermal growth factor receptor: from mutant oncogene in nonhuman cancers to therapeutic target in human neoplasia. J Clin Oncol. 2001;19(18 Suppl):32s–40s.
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–37.
Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341–54.
Jackman DM, Yeap BY, Sequist LV, Lindeman N, Holmes AJ, Joshi VA, et al. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12(13):3908–14.
Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361(10):958–67.
Soria JC, Mok TS, Cappuzzo F, Jänne PA. EGFR-mutated oncogene-addicted non-small cell lung cancer: current trends and future prospects. Cancer Treat Rev. 2012;38(5):416–30.
Kawahara A, Azuma K, Sumi A, Taira T, Nakashima K, Aikawa E, et al. Identification of non-small-cell lung cancer with activating EGFR mutations in malignant effusion and cerebrospinal fluid: rapid and sensitive detection of exon 19 deletion E746–A750 and exon 21 L858R mutation by immunocytochemistry. Lung Cancer. 2011;74(1):35–40.
Seo AN, Park TI, Jin Y, Sun PL, Kim H, Chang H, et al. Novel EGFR mutation-specific antibodies for lung adenocarcinoma: highly specific but not sensitive detection of an E746_A750 deletion in exon 19 and an L858R mutation in exon 21 by immunohistochemistry. Lung Cancer. 2014;83(3):316–23.
Kris MG, Natale RB, Herbst RS, Lynch TJ Jr, Prager D, Belani CP, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA. 2003;290(16):2149–58.
Pérez-Soler R, Chachoua A, Hammond LA, Rowinsky EK, Huberman M, Karp D, et al. Determinants of tumor response and survival with erlotinib in patients with non–small-cell lung cancer. J Clin Oncol. 2004;22(16):3238–47.
Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–34.
Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15(2):213–22.
Gridelli C, Ciardiello F, Gallo C, Feld R, Butts C, Gebbia V, et al. First-line erlotinib followed by second-line cisplatin-gemcitabine chemotherapy in advanced non-small-cell lung cancer: the TORCH randomized trial. J Clin Oncol. 2012;30(24):3002–11.
Zhou F, Zhou CC. Targeted therapies for patients with advanced NSCLC harboring wild-type EGFR: what’s new and what’s enough. Chin J Cancer. 2015;34(7):310–9.
Khambata-Ford S, Harbison CT, Hart LL, Awad M, Xu LA, Horak CE, et al. Analysis of potential predictive markers of cetuximab benefit in BMS099, a phase III study of cetuximab and first-line taxane/carboplatin in advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(6):918–27.
Pirker R, Pereira JR, Szczesna A, von Pawel J, Krzakowski M, Ramlau R, et al. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet. 2009;373(9674):1525–31.
Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–46.
Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–42.
Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14(4):439–49.
Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–34.
Palmer RH, Vernersson E, Grabbe C, Hallberg B. Anaplastic lymphoma kinase: signalling in development and disease. Biochem J. 2009;420(3):345–61.
Tan DS, Araújo A, Zhang J, Signorovitch J, Zhou ZY, Cai X, et al. Comparative efficacy of ceritinib and crizotinib as initial ALK-targeted therapies in previously treated ADVANCED NSCLC: an adjusted comparison with external controls. J Thorac Oncol. 2016;11(9):1550–7.
Gandhi L, Jänne PA. Crizotinib for ALK-rearranged non-small cell lung cancer: a new targeted therapy for a new target. Clin Cancer Res. 2012;18(14):3737–42.
Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–9.
Kang J, Zhang XC, Chen HJ, Zhong WZ, Xu Y, Su J, et al. Complex ALK fusions are associated with better prognosis in advanced non-small cell lung cancer. Front Oncol. 2020;10:596937.
Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2008;14(13):4275–83.
Rodig SJ, Mino-Kenudson M, Dacic S, Yeap BY, Shaw A, Barletta JA, et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res. 2009;15(16):5216–23.
Shaw AT, Felip E, Bauer TM, Besse B, Navarro A, Postel-Vinay S, et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017;18(12):1590–9.
Cui JJ, Tran-Dubé M, Shen H, Nambu M, Kung PP, Pairish M, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem. 2011;54(18):6342–63.
Huang Z, Xiong Q, Cui Z, Tao H, Zhang S, Wang L, et al. Efficacy and safety of crizotinib plus bevacizumab in ALK/ROS-1/c-MET positive non-small cell lung cancer: an open-label, single-arm, prospective observational study. Am J Transl Res. 2021;13(3):1526–34.
Felip E, Kim D, Mehra R, Tan DS, Chow L, Camidge DR, et al. Efficacy and safety of ceritinib in patients (pts) with advanced anaplastic lymphoma kinase (ALK)-rearranged (ALK+) non-small cell lung cancer (NSCLC): an update of ASCEND-1. Ann Oncol. 2014;25:iv456.
Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–77.
Gainor JF, Tan DS, De Pas T, Solomon BJ, Ahmad A, Lazzari C, et al. Progression-free and overall survival in ALK-positive NSCLC patients treated with sequential crizotinib and ceritinib. Clin Cancer Res. 2015;21(12):2745–52.
Thomas RK. Overcoming drug resistance in ALK-rearranged lung cancer. N Engl J Med. 2014;370(13):1250–1.
Kim D-W, Ahn M-J, Shi Y, De Pas TM, Yang P-C, Riely GJ, et al. Results of a global phase II study with crizotinib in advanced ALK-positive non-small-cell lung cancer (NSCLC). Ann Oncol. 2012;23:xi32–3.
Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371(21):1963–71.
Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–203.
Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30(8):863–70.
Huber KV, Salah E, Radic B, Gridling M, Elkins JM, Stukalov A, et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature. 2014;508(7495):222–7.
Awad MM, Katayama R, McTigue M, Liu W, Deng YL, Brooun A, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013;368(25):2395–401.
Gainor JF, Tseng D, Yoda S, Dagogo-Jack I, Friboulet L, Lin JJ, et al. Patterns of metastatic spread and mechanisms of resistance to crizotinib in ROS1-positive non-small-cell lung cancer. JCO Precis Oncol. 2017. https://doi.org/10.1200/PO.17.00063.
Menichincheri M, Ardini E, Magnaghi P, Avanzi N, Banfi P, Bossi R, et al. Discovery of entrectinib: a new 3-aminoindazole as a potent anaplastic lymphoma kinase (ALK), c-ros oncogene 1 kinase (ROS1), and pan-tropomyosin receptor kinases (Pan-TRKs) inhibitor. J Med Chem. 2016;59(7):3392–408.
Ardini E, Menichincheri M, Banfi P, Bosotti R, De Ponti C, Pulci R, et al. Entrectinib, a Pan-TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol Cancer Ther. 2016;15(4):628–39.
Drilon A, Siena S, Dziadziuszko R, Barlesi F, Krebs MG, Shaw AT, et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21(2):261–70.
Doebele RC, Perez L, Trinh H, Martinec M, Martina R, Riehl T, et al. Comparative effectiveness analysis between entrectinib clinical trial and crizotinib real-world data in ROS1+ NSCLC. J Comp Eff Res. 2021;10(17):1271–82.
Zou HY, Li Q, Engstrom LD, West M, Appleman V, Wong KA, et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc Natl Acad Sci USA. 2015;112(11):3493–8.
Felip E, Bauer T, Solomon B, Besse B, James L, Clancy J, et al. MA07. 11 safety and efficacy of lorlatinib (PF-06463922) in patients with advanced ALK+ or ROS1+ non-small-cell lung cancer (NSCLC). J Thorac Oncol. 2017;12(1):S383–4.
Shaw AT, Solomon BJ, Chiari R, Riely GJ, Besse B, Soo RA, et al. Lorlatinib in advanced ROS1-positive non-small-cell lung cancer: a multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2019;20(12):1691–701.
Paik PK, Arcila ME, Fara M, Sima CS, Miller VA, Kris MG, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. 2011;29(15):2046–51.
Villaruz LC, Socinski MA, Abberbock S, Berry LD, Johnson BE, Kwiatkowski DJ, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the lung cancer mutation consortium. Cancer. 2015;121(3):448–56.
Tissot C, Couraud S, Tanguy R, Bringuier PP, Girard N, Souquet PJ. Clinical characteristics and outcome of patients with lung cancer harboring BRAF mutations. Lung Cancer. 2016;91:23–8.
Cardarella S, Ogino A, Nishino M, Butaney M, Shen J, Lydon C, et al. Clinical, pathologic, and biologic features associated with BRAF mutations in non-small cell lung cancer. Clin Cancer Res. 2013;19(16):4532–40.
Marchetti A, Felicioni L, Malatesta S, Grazia Sciarrotta M, Guetti L, Chella A, et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol. 2011;29(26):3574–9.
Abdulkarim LS, Motley RJ. First-line advanced cutaneous melanoma treatments: where do we stand? JMIR Cancer. 2021;7(4):e29912.
Amaral T, Sinnberg T, Meier F, Krepler C, Levesque M, Niessner H, et al. The mitogen-activated protein kinase pathway in melanoma part I–activation and primary resistance mechanisms to BRAF inhibition. Eur J Cancer. 2017;73:85–92.
Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, et al. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov. 2012;11(11):873–86.
Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci. 2008;105(8):3041–6.
Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay J-Y, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–36.
Subbiah V, Gervais R, Riely GJ, Hollebecque A, Blay J-Y, Felip E, et al. Efficacy of vemurafenib in patients (pts) with non-small cell lung cancer (NSCLC) with BRAF V600 mutation. Am Soc Clin Oncol. 2017. https://doi.org/10.1200/JCO.2017.35.15_suppl.9074.
King AJ, Arnone MR, Bleam MR, Moss KG, Yang J, Fedorowicz KE, et al. Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions. PLoS ONE. 2013;8(7):e67583.
Planchard D, Kim TM, Mazieres J, Quoix E, Riely G, Barlesi F, et al. Dabrafenib in patients with BRAFV600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17(5):642–50.
Planchard D, Besse B, Groen HJ, Souquet P-J, Quoix E, Baik CS, et al. Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol. 2016;17(7):984–93.
Planchard D, Smit EF, Groen HJ, Mazieres J, Besse B, Helland Å, et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18(10):1307–16.
Food D Administration. FDA grants regular approval to dabrafenib and trametinib combination for metastatic NSCLC with BRAF V600E mutation. 2017.
Joshi M, Rice SJ, Liu X, Miller B, Belani CP. Trametinib with or without vemurafenib in BRAF mutated non-small cell lung cancer. PLoS ONE. 2015;10(2):e0118210.
Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18(3):378–81.
Li AY, McCusker MG, Russo A, Scilla KA, Gittens A, Arensmeyer K, et al. RET fusions in solid tumors. Cancer Treat Rev. 2019;81:101911.
Wang H, Wang Z, Zhang G, Zhang M, Zhang X, Li H, et al. Driver genes as predictive indicators of brain metastasis in patients with advanced NSCLC: EGFR, ALK, and RET gene mutations. Cancer Med. 2020;9(2):487–95.
Food U, D Administration. FDA approves pralsetinib for lung cancer with RET gene fusions. Disponibile su. 2020. https://www.bitly/3eT3VfA. Accessed 17 May 2021.
Zheng H, Chen Z-S, Li J. Selpercatinib for lung and thyroid cancers with RET gene mutations or fusions. Drugs Today (Barcelona, Spain: 1998). 2021;57(10):621–9.
Gainor JF, Curigliano G, Kim D-W, Lee DH, Besse B, Baik CS, et al. Pralsetinib for RET fusion-positive non-small-cell lung cancer (ARROW): a multi-cohort, open-label, phase 1/2 study. Lancet Oncol. 2021;22(7):959–69.
Subbiah V, Taylor M, Lin J, Hu M, Ou S-HI, Brose MS, et al. Abstract CT043: highly potent and selective RET inhibitor, BLU-667, achieves proof of concept in a phase I study of advanced RET-altered solid tumors. Cancer Res. 2018;78(13_Supplement):CT043-CT.
Curigliano G, Subbiah V, Gainor J, Lee D, Taylor M, Zhu V, et al. Treatment with BLU-667, a potent and selective RET inhibitor, provides rapid clearance of ctDNA in patients with RET-altered non-small cell lung cancer (NSCLC) and thyroid cancer. Ann Oncol. 2019;30:v790.
Griesinger F, Curigliano G, Thomas M, Subbiah V, Baik CS, Tan DSW, et al. Safety and efficacy of pralsetinib in RET fusion-positive non-small cell lung cancer including as first-line therapy: update from the ARROW trial. Ann Oncol. 2022. https://doi.org/10.1016/j.annonc.2022.08.002.
Subbiah V, Velcheti V, Tuch B, Ebata K, Busaidy N, Cabanillas M, et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol. 2018;29(8):1869–76.
Drilon A, Oxnard GR, Tan DSW, Loong HHF, Johnson M, Gainor J, et al. Efficacy of selpercatinib in RET fusion-positive non-small-cell lung cancer. N Engl J Med. 2020;383(9):813–24.
Solomon BJ, Zhou CC, Drilon A, Park K, Wolf J, Elamin Y, et al. Phase III study of selpercatinib versus chemotherapy ± pembrolizumab in untreated RET positive non-small-cell lung cancer. Future Oncol. 2021;17(7):763–73.
Gautschi O, Milia J, Filleron T, Wolf J, Carbone DP, Owen D, et al. Targeting RET in patients with RET-rearranged lung cancers: results from the global, multicenter RET registry. J Clin Oncol. 2017;35(13):1403–10.
Drilon AE, Zhai D, Rogers E, Deng W, Zhang X, Ung J, et al. The next-generation RET inhibitor TPX-0046 is active in drug-resistant and naïve RET-driven cancer models. Am Soc Clin Oncol. 2020. https://doi.org/10.1200/JCO.2020.38.15_suppl.3616.
Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011;3(1 Suppl):S7-s19.
Uchikawa E, Chen Z, Xiao GY, Zhang X, Bai XC. Structural basis of the activation of c-MET receptor. Nat Commun. 2021;12(1):4074.
Comoglio PM, Trusolino L, Boccaccio C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat Rev Cancer. 2018;18(6):341–58.
Bubendorf L, Dafni U, Schöbel M, Finn SP, Tischler V, Sejda A, et al. Prevalence and clinical association of MET gene overexpression and amplification in patients with NSCLC: results from the European thoracic oncology platform (ETOP) lungscape project. Lung Cancer. 2017;111:143–9.
Drilon A, Cappuzzo F, Ou SI, Camidge DR. Targeting MET in lung cancer: will expectations finally be met? J Thorac Oncol. 2017;12(1):15–26.
Hamilton G, Rath B. Mesenchymal-epithelial transition and circulating tumor cells in small cell lung cancer. Adv Exp Med Biol. 2017;994:229–45.
Soltermann A, Tischler V, Arbogast S, Braun J, Probst-Hensch N, Weder W, et al. Prognostic significance of epithelial-mesenchymal and mesenchymal–epithelial transition protein expression in non–small cell lung cancer. Clin Cancer Res. 2008;14(22):7430–7.
Hardy-Werbin M, Del Rey-Vergara R, Galindo-Campos MA, Moliner L, Arriola E. MET inhibitors in small cell lung cancer: from the bench to the bedside. Cancers. 2019;11(10):1404.
Backes A, Zech B, Felber B, Klebl B, Müller G. Small-molecule inhibitors binding to protein kinase. Part II: the novel pharmacophore approach of type II and type III inhibition. Expert Opin Drug Discov. 2008;3(12):1427–49.
Gherardi E, Birchmeier W, Birchmeier C, Vande WG. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12(2):89–103.
Reungwetwattana T, Liang Y, Zhu V, Ou SI. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: the why, the how, the who, the unknown, and the inevitable. Lung Cancer. 2017;103:27–37.
Drusbosky LM, Dawar R, Rodriguez E, Ikpeazu CV. Therapeutic strategies in METex14 skipping mutated non-small cell lung cancer. J Hematol Oncol. 2021;14(1):129.
Awad MM, Oxnard GR, Jackman DM, Savukoski DO, Hall D, Shivdasani P, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-Met overexpression. J Clin Oncol. 2016;34(7):721–30.
Garcia-Robledo JE, Rosell R, Ruíz-Patiño A, Sotelo C, Arrieta O, Zatarain-Barrón L, et al. KRAS and MET in non-small-cell lung cancer: two of the new kids on the “drivers” block. Ther Adv Respir Dis. 2022;16:17534666211066064.
Agwa ES, Ma PC. Targeting the MET receptor tyrosine kinase in non-small cell lung cancer: emerging role of tivantinib. Cancer Manag Res. 2014;6:397–404.
Xu Z, Li H, Dong Y, Cheng P, Luo F, Fu S, et al. Incidence and PD-L1 expression of MET 14 skipping in Chinese population: a non-selective NSCLC cohort study using RNA-based sequencing. Onco Targets Ther. 2020;13:6245–53.
Baba K, Tanaka H, Sakamoto H, Shiratori T, Tsuchiya J, Ishioka Y, et al. Efficacy of pembrolizumab for patients with both high PD-L1 expression and an MET exon 14 skipping mutation: a case report. Thoracic Cancer. 2019;10(2):369–72.
Reis H, Metzenmacher M, Goetz M, Savvidou N, Darwiche K, Aigner C, et al. MET expression in advanced non-small-cell lung cancer: effect on clinical outcomes of chemotherapy, targeted therapy, and immunotherapy. Clin Lung Cancer. 2018;19(4):e441–63.
Agarwal N, Vaishampayan U, Green M, Di Nucci F, Chang P-Y, Scheffold C, et al. Phase Ib study (COSMIC-021) of cabozantinib in combination with atezolizumab: results of the dose escalation stage in patients (pts) with treatment-naïve advanced renal cell carcinoma (RCC). Ann Oncol. 2018;29:308.
Huang S, van Duijnhoven SMJ, Sijts A, van Elsas A. Bispecific antibodies targeting dual tumor-associated antigens in cancer therapy. J Cancer Res Clin Oncol. 2020;146(12):3111–22.
Wang J, Anderson MG, Oleksijew A, Vaidya KS, Boghaert ER, Tucker L, et al. ABBV-399, a c-Met antibody-drug conjugate that targets both MET-amplified and c-Met-overexpressing tumors, irrespective of MET pathway dependencea tumor-targeting c-Met ADC. Clin Cancer Res. 2017;23(4):992–1000.
Syed YY. Amivantamab: first approval. Drugs. 2021;81(11):1349–53.
De Mello RA, Neves NM, Amaral GA, Lippo EG, Castelo-Branco P, Pozza DH, et al. The role of MET inhibitor therapies in the treatment of advanced non-small cell lung cancer. J Clin Med. 2020;9(6):1918.
Recondo G, Bahcall M, Spurr LF, Che J, Ricciuti B, Leonardi GC, et al. Molecular mechanisms of acquired resistance to MET tyrosine kinase inhibitors in patients with MET Exon 14-mutant NSCLC. Clin Cancer Res. 2020;26(11):2615–25.
Nakagawara A. Trk receptor tyrosine kinases: a bridge between cancer and neural development. Cancer Lett. 2001;169(2):107–14.
Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10(3):381–91.
Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361(1473):1545–64.
Klein R, Jing SQ, Nanduri V, O’Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65(1):189–97.
Edel MJ, Shvarts A, Medema JP, Bernards R. An in vivo functional genetic screen reveals a role for the TRK-T3 oncogene in tumor progression. Oncogene. 2004;23(29):4959–65.
Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015;5(1):25–34.
Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open. 2016;1(2):e000023.
Farago AF, Taylor MS, Doebele RC, Zhu VW, Kummar S, Spira AI, et al. Clinicopathologic features of non-small-cell lung cancer harboring an NTRK gene fusion. JCO Precis Oncol. 2018. https://doi.org/10.1200/PO.18.00037.
Rohrberg KS, Lassen U. Detecting and targeting NTRK fusions in cancer in the era of tumor agnostic oncology. Drugs. 2021;81(4):445–52.
Doebele RC, Drilon A, Paz-Ares L, Siena S, Shaw AT, Farago AF, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21(2):271–82.
Lee J, Park S, Jung HA, Sun JM, Lee SH, Ahn JS, et al. Evaluating entrectinib as a treatment option for non-small cell lung cancer. Expert Opin Pharmacother. 2020;21(16):1935–42.
Hong DS, Shen L, van Tilburg CM, Tan DS-W, Kummar S, Lin JJ, et al. Long-term efficacy and safety of larotrectinib in an integrated dataset of patients with TRK fusion cancer. Philadelphia: Wolters Kluwer Health; 2021.
Lin JJ, Kummar S, Tan DS-W, Lassen UN, Leyvraz S, Liu Y, et al. Long-term efficacy and safety of larotrectinib in patients with TRK fusion-positive lung cancer. Philadelphia: Wolters Kluwer Health; 2021.
Rosen EY, Schram AM, Young RJ, Schreyer MW, Hechtman JF, Shu CA, et al. Larotrectinib demonstrates CNS efficacy in TRK fusion-positive solid tumors. JCO Precis Oncol. 2019;3:1.
Qin H, Patel MR. The challenge and opportunity of NTRK inhibitors in non-small cell lung cancer. Int J Mol Sci. 2022;23(6):2916.
Murray BW, Rogers E, Zhai D, Deng W, Chen X, Sprengeler PA, et al. Molecular characteristics of repotrectinib that enable potent inhibition of TRK fusion proteins and resistant mutations. Mol Cancer Ther. 2021;20(12):2446–56.
Drilon A, Ou SI, Cho BC, Kim DW, Lee J, Lin JJ, et al. Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/ALK inhibitor that potently inhibits ROS1/TRK/ALK solvent-front mutations. Cancer Discov. 2018;8(10):1227–36.
Florou V, Nevala-Plagemann C, Whisenant J, Maeda P, Gilcrease GW, Garrido-Laguna I. Clinical activity of selitrectinib in a patient with mammary analogue secretory carcinoma of the parotid gland with secondary resistance to entrectinib. J Natl Compr Cancer Netw JNCCN. 2021;19(5):478–82.
Katayama R, Gong B, Togashi N, Miyamoto M, Kiga M, Iwasaki S, et al. The new-generation selective ROS1/NTRK inhibitor DS-6051b overcomes crizotinib resistant ROS1-G2032R mutation in preclinical models. Nat Commun. 2019;10(1):3604.
Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–50.
Wei XW, Gao X, Zhang XC, Yang JJ, Chen ZH, Wu YL, et al. Mutational landscape and characteristics of ERBB2 in non-small cell lung cancer. Thoracic cancer. 2020;11(6):1512–21.
Lv Q, Meng Z, Yu Y, Jiang F, Guan D, Liang C, et al. Molecular mechanisms and translational therapies for human epidermal receptor 2 POSITIVE breast cancer. Int J Molr Sci. 2016;17(12):2095.
Pellegrini C, Falleni M, Marchetti A, Cassani B, Miozzo M, Buttitta F, et al. HER-2/Neu alterations in non-small cell lung cancer: a comprehensive evaluation by real time reverse transcription-PCR, fluorescence in situ hybridization, and immunohistochemistry. Clin Cancer Res. 2003;9(10 Pt 1):3645–52.
Tan D, Deeb G, Wang J, Slocum HK, Winston J, Wiseman S, et al. HER-2/neu protein expression and gene alteration in stage I-IIIA non-small-cell lung cancer: a study of 140 cases using a combination of high throughput tissue microarray, immunohistochemistry, and fluorescent in situ hybridization. Diagn Mol Pathol. 2003;12(4):201–11.
Dziadziuszko R, Smit EF, Dafni U, Wolf J, Wasąg B, Biernat W, et al. Afatinib in NSCLC With HER2 Mutations: results of the prospective, open-label phase II NICHE trial of European thoracic oncology platform (ETOP). J Thorac Oncol. 2019;14(6):1086–94.
Ekman S. HER2: defining a Neu target in non-small-cell lung cancer. Ann Oncol. 2019;30(3):353–5.
Prelaj A, Bottiglieri A, Proto C, Russo GL, Signorelli D, Ferrara R, et al. 1388P Poziotinib in advanced NSCLC with EGFR or HER2 exon 20 insertion mutation: Initial results from a single site expanded access program. Ann Oncol. 2020;31:S882.
Ross HJ, Blumenschein GR Jr, Aisner J, Damjanov N, Dowlati A, Garst J, et al. Randomized phase II multicenter trial of two schedules of lapatinib as first- or second-line monotherapy in patients with advanced or metastatic non-small cell lung cancer. Clin Cancer Res. 2010;16(6):1938–49.
Wang Y, Jiang T, Qin Z, Jiang J, Wang Q, Yang S, et al. HER2 exon 20 insertions in non-small-cell lung cancer are sensitive to the irreversible pan-HER receptor tyrosine kinase inhibitor pyrotinib. Ann Oncol. 2019;30(3):447–55.
Jänne PA, Neal JW, Camidge DR, Spira A, Piotrowska Z, Horn L, et al. Antitumor activity of TAK-788 in NSCLC with EGFR exon 20 insertions. Ann Oncol. 2019;30:108.
Barthélémy P, Leblanc J, Goldbarg V, Wendling F, Kurtz JE. Pertuzumab: development beyond breast cancer. Anticancer Res. 2014;34(4):1483–91.
Mazières J, Barlesi F, Filleron T, Besse B, Monnet I, Beau-Faller M, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: results from the European EUHER2 cohort. Ann Oncol. 2016;27(2):281–6.
Swanton C, Futreal A, Eisen T. Her2-targeted therapies in non-small cell lung cancer. Clin Cancer Res. 2006;12(14 Pt 2):4377s-s4383.
Network CGAR. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543.
Peters S, Stahel R, Bubendorf L, Bonomi P, Villegas A, Kowalski DM, et al. Trastuzumab emtansine (T-DM1) in patients with previously treated HER2-overexpressing metastatic non-Small cell lung cancer: efficacy, safety, and biomarkers. Clin Cancer Res. 2019;25(1):64–72.
Tan AC. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac Cancer. 2020;11(3):511–8.
Papadimitrakopoulou V. Development of PI3K/AKT/mTOR pathway inhibitors and their application in personalized therapy for non-small-cell lung cancer. J Thorac Oncol. 2012;7(8):1315–26.
Scheffler M, Bos M, Gardizi M, König K, Michels S, Fassunke J, et al. PIK3CA mutations in non-small cell lung cancer (NSCLC): genetic heterogeneity, prognostic impact and incidence of prior malignancies. Oncotarget. 2015;6(2):1315–26.
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–6.
Vansteenkiste JF, Canon J-L, De Braud F, Grossi F, De Pas T, Gray JE, et al. Safety and efficacy of buparlisib (BKM120) in patients with PI3K pathway-activated non-small cell lung cancer: results from the phase II BASALT-1 study. J Thorac Oncol. 2015;10(9):1319–27.
Langer CJ, Redman MW, Wade JL 3rd, Aggarwal C, Bradley JD, Crawford J, et al. SWOG S1400B (NCT02785913), a phase II study of GDC-0032 (Taselisib) for previously treated PI3K-positive patients with stage IV squamous cell lung cancer (Lung-MAP Sub-Study). J Thorac Oncol. 2019;14(10):1839–46.
Zhou Q, Zhang X-C, Peng B, Yu X, Akimov M, Weber BL, et al. A phase II cluster study of single agent AUY922, BYL719, INC280, LDK378, and MEK162 in Chinese patients with advanced non-small cell lung cancer (NSCLC). Am Soc Clin Oncol. 2014. https://doi.org/10.1200/jco.2014.32.15_suppl.tps8122.
Balsara BR, Pei J, Mitsuuchi Y, Page R, Klein-Szanto A, Wang H, et al. Frequent activation of AKT in non-small cell lung carcinomas and preneoplastic bronchial lesions. Carcinogenesis. 2004;25(11):2053–9.
Guo Y, Du J, Kwiatkowski DJ. Molecular dissection of AKT activation in lung cancer cell lines. Mol Cancer Res. 2013;11(3):282–93.
Le Grand M, Berges R, Pasquier E, Montero M-P, Borge L, Carrier A, et al. Akt targeting as a strategy to boost chemotherapy efficacy in non-small cell lung cancer through metabolism suppression. Sci Rep. 2017;7(1):45136.
Ma Z, Zhu L, Luo X, Zhai S, Li P, Wang X. Perifosine enhances mTORC1-targeted cancer therapy by activation of GSK3β in NSCLC cells. Cancer Biol Ther. 2012;13(11):1009–17.
Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an Allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivoMK-2206 sensitizes tumors to chemotherapy. Mol Cancer Ther. 2010;9(7):1956–67.
Lara PN Jr, Longmate J, Mack PC, Kelly K, Socinski MA, Salgia R, et al. Phase II study of the AKT inhibitor MK-2206 plus erlotinib in patients with advanced non-small cell lung cancer who previously progressed on erlotinib. Clin Cancer Res. 2015;21(19):4321–6.
Ekman S, Wynes MW, Hirsch FR. The mTOR Pathway in lung cancer and implications for therapy and biomarker analysis. J Thorac Oncol. 2012;7(6):947–53.
Kim J-O, Kim K-H, Song IS, Cheon K-S, Kim O-H, Lee SC, et al. Potentiation of the anticancer effects of everolimus using a dual mTORC1/2 inhibitor in hepatocellular carcinoma cells. Oncotarget. 2017;8(2):2936–48.
Sparks CA, Guertin DA. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene. 2010;29(26):3733–44.
Soria JC, Shepherd FA, Douillard JY, Wolf J, Giaccone G, Crino L, et al. Efficacy of everolimus (RAD001) in patients with advanced NSCLC previously treated with chemotherapy alone or with chemotherapy and EGFR inhibitors. Ann Oncol. 2009;20(10):1674–81.
Fang W, Huang Y, Gu W, Gan J, Wang W, Zhang S, et al. PI3K-AKT-mTOR pathway alterations in advanced NSCLC patients after progression on EGFR-TKI and clinical response to EGFR-TKI plus everolimus combination therapy. Transl Lung Cancer Res. 2020;9(4):1258–67.
Komiya T, Memmott RM, Blumenthal GM, Bernstein W, Ballas MS, De Chowdhury R, et al. A phase I/II study of pemetrexed with sirolimus in advanced, previously treated non-small cell lung cancer. Transl Lung Cancer Res. 2019;8(3):247–57.
Reungwetwattana T, Molina JR, Mandrekar SJ, Allen-Ziegler K, Rowland KM, Reuter NF, et al. Brief report: a phase II “window-of-opportunity” frontline study of the MTOR inhibitor, temsirolimus given as a single agent in patients with advanced NSCLC, an NCCTG study. J Thorac Oncol. 2012;7(5):919–22.
Gandhi L, Besse B, Mazieres J, Waqar S, Cortot A, Barlesi F, et al. MA04. 02 neratinib±temsirolimus in HER2-mutant lung cancers: an international, randomized phase II study. J Thorac Oncol. 2017;12(1):S358–9.
Riely GJ, Brahmer JR, Planchard D, Crinò L, Doebele RC, Mas Lopez LA, et al. A randomized discontinuation phase II trial of ridaforolimus in non-small cell lung cancer (NSCLC) patients with KRAS mutations. Am Soc Clin Oncol. 2012. https://doi.org/10.1200/jco.2012.30.15_suppl.7531.
Qin X, Liu M, Wu Y, Wang S, Lian S, Jia H, et al. Dual blocking of PI3K and mTOR signaling by DHW-221, a novel benzimidazole derivative, exerts antitumor activity in human non-small cell lung cancer. Clin Transl Med. 2021;11(9):e514.
Dolly SO, Wagner AJ, Bendell JC, Kindler HL, Krug LM, Seiwert TY, et al. Phase I Study of apitolisib (GDC-0980), dual phosphatidylinositol-3-kinase and mammalian target of rapamycin kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2016;22(12):2874–84.
Jänne PA, Cohen RB, Laird AD, Macé S, Engelman JA, Ruiz-Soto R, et al. Phase I safety and pharmacokinetic study of the PI3K/mTOR inhibitor SAR245409 (XL765) in combination with erlotinib in patients with advanced solid tumors. J Thorac Oncol. 2014;9(3):316–23.
Lastwika KJ, Wilson W 3rd, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Can Res. 2016;76(2):227–38.
Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–28.
Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26(1):677–704.
Ji M, Liu Y, Li Q, Li X-D, Zhao W-Q, Zhang H, et al. PD-1/PD-L1 pathway in non-small-cell lung cancer and its relation with EGFR mutation. J Transl Med. 2015;13:5.
Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5(200):200ra116.
Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21(4):687–92.
Hudson K, Cross N, Jordan-Mahy N, Leyland R. The extrinsic and intrinsic roles of PD-L1 and Its receptor PD-1: implications for immunotherapy treatment. Front immunol. 2020. https://doi.org/10.3389/fimmu.2020.568931.
Moya-Horno I, Viteri S, Karachaliou N, Rosell R. Combination of immunotherapy with targeted therapies in advanced non-small cell lung cancer (NSCLC). Ther Adv Med Oncol. 2018;10:1758834017745012.
Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med. 2017;376(25):2415–26.
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–39.
Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Five-Year outcomes with pembrolizumab versus chemotherapy for metastatic non-small-cell lung cancer With PD-L1 tumor proportion score ≥ 50. J Clin Oncol. 2021;39(21):2339–49.
Cui P, Li R, Huang Z, Wu Z, Tao H, Zhang S, et al. Comparative effectiveness of pembrolizumab vs nivolumab in patients with recurrent or advanced NSCLC. Sci Rep. 2020;10(1):13160.
Dantoing E, Piton N, Salaün M, Thiberville L, Guisier F. Anti-PD1/PD-L1 immunotherapy for non-small cell lung cancer with actionable oncogenic driver mutations. Int J Mol Sci. 2021;22(12):6288.
Matsubara T, Seto T, Takamori S, Fujishita T, Toyozawa R, Ito K, et al. Anti-PD-1 monotherapy for advanced NSCLC patients with older age or those with poor performance status. Onco Targets Ther. 2021;14:1961–8.
Ilie M, Hofman P. Atezolizumab in advanced non-small cell lung cancer. J Thorac Dis. 2017;9(10):3603–6.
Sezer A, Kilickap S, Gümüş M, Bondarenko I, Özgüroğlu M, Gogishvili M, et al. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: a multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet. 2021;397(10274):592–604.
Yamamoto T, Tsukita Y, Katagiri Y, Matsushita H, Umezawa R, Ishikawa Y, et al. Durvalumab after chemoradiotherapy for locally advanced non-small cell lung cancer prolonged distant metastasis-free survival, progression-free survival and overall survival in clinical practice. BMC Cancer. 2022;22(1):364.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. New Engl J Med. 2018;379(24):2342–50.
Huang G, Sun X, Liu D, Zhang Y, Zhang B, Xiao G, et al. The efficacy and safety of anti-PD-1/PD-L1 antibody therapy versus docetaxel for pretreated advanced NSCLC: a meta-analysis. Oncotarget. 2018;9(3):4239–48.
Park K, Özgüroğlu M, Vansteenkiste J, Spigel D, Yang JCH, Ishii H, et al. Avelumab versus docetaxel in patients with platinum-treated advanced NSCLC: 2-year follow-up from the JAVELIN lung 200 phase 3 trial. J Thorac Oncol. 2021;16(8):1369–78.
Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241.
Wang N, Zhang T. Downregulation of MicroRNA-135 promotes sensitivity of non-small cell lung cancer to gefitinib by targeting TRIM16. Oncol Res. 2018;26(7):1005–14.
Tang D, Zhao D, Wu Y, Yao R, Zhou L, Lu L, et al. The miR-3127-5p/p-STAT3 axis up-regulates PD-L1 inducing chemoresistance in non-small-cell lung cancer. J Cell Mol Med. 2018;22(8):3847–56.
Zhang Y, Xiang C, Wang Y, Duan Y, Liu C, Zhang Y. PD-L1 promoter methylation mediates the resistance response to anti-PD-1 therapy in NSCLC patients with EGFR-TKI resistance. Oncotarget. 2017;8(60):101535–44.
Paulsen EE, Kilvaer TK, Rakaee M, Richardsen E, Hald SM, Andersen S, et al. CTLA-4 expression in the non-small cell lung cancer patient tumor microenvironment: diverging prognostic impact in primary tumors and lymph node metastases. Cancer Immunol Immunother. 2017;66(11):1449–61.
Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39(1):98–106.
Zhao Y, Yang W, Huang Y, Cui R, Li X, Li B. Evolving roles for targeting CTLA-4 in cancer immunotherapy. Cell Physiol Biochem. 2018;47(2):721–34.
Salvi S, Fontana V, Boccardo S, Merlo DF, Margallo E, Laurent S, et al. Evaluation of CTLA-4 expression and relevance as a novel prognostic factor in patients with non-small cell lung cancer. Cancer Immunol Immunother. 2012;61(9):1463–72.
Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30(17):2046–54.
Tarhini AA, Kirkwood JM. Tremelimumab (CP-675,206): a fully human anticytotoxic T lymphocyte-associated antigen 4 monoclonal antibody for treatment of patients with advanced cancers. Expert Opin Biol Ther. 2008;8(10):1583–93.
Tarhini AA, Iqbal F. CTLA-4 blockade: therapeutic potential in cancer treatments. Onco Targets Ther. 2010;3:15–25.
Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378(22):2093–104.
Rizvi NA, Cho BC, Reinmuth N, Lee KH, Luft A, Ahn MJ, et al. Durvalumab with or without tremelimumab vs standard chemotherapy in first-line treatment of metastatic non-small cell lung cancer: the MYSTIC phase 3 randomized clinical trial. JAMA Oncol. 2020;6(5):661–74.
Khozin S, Blumenthal GM, Jiang X, He K, Boyd K, Murgo A, et al. U.S. food and drug administration approval summary: erlotinib for the first-line treatment of metastatic non-small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 (L858R) substitution mutations. Oncologist. 2014;19(7):774–9.
Kazandjian D, Blumenthal GM, Yuan W, He K, Keegan P, Pazdur R. FDA approval of gefitinib for the treatment of patients with metastatic EGFR mutation-positive non-small cell lung cancer. Clin Cancer Res. 2016;22(6):1307–12.
Greig SL. Osimertinib: first global approval. Drugs. 2016;76(2):263–73.
Kazandjian D, Blumenthal GM, Chen HY, He K, Patel M, Justice R, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist. 2014;19(10):e5-11.
Syed YY. Lorlatinib: first global approval. Drugs. 2019;79(1):93–8.
Markham A. Tepotinib: first approval. Drugs. 2020;80(8):829–33.
Sakai H, Morise M, Kato T, Matsumoto S, Sakamoto T, Kumagai T, et al. Tepotinib in patients with NSCLC harbouring MET exon 14 skipping: Japanese subset analysis from the phase II VISION study. Jpn J Clin Oncol. 2021;51(8):1261–8.
FDA approves atezolizumab for first-line treatment of metastatic NSCLC with high PD-L1 expression U.S. food and drug administration. 2020. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-first-line-treatment-metastatic-nsclc-high-pd-l1-expression
FDA approves cemiplimab-rwlc for non-small cell lung cancer with high PD-L1 expression U.S. food and drug administration. 2021. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-cemiplimab-rwlc-non-small-cell-lung-cancer-high-pd-l1-expression
FDA approves nivolumab plus ipilimumab and chemotherapy for first-line treatment of metastatic NSCLC U.S. food and drug administration. 2020. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-nivolumab-plus-ipilimumab-and-chemotherapy-first-line-treatment-metastatic-nsclc
Park K, Haura EB, Leighl NB, Mitchell P, Shu CA, Girard N, et al. Amivantamab in EGFR exon 20 insertion-mutated non-small-cell lung cancer progressing on platinum chemotherapy: initial results from the CHRYSALIS phase I study. J Clinl Oncol. 2021;39(30):3391–402.
Pacheco JM. Mobocertinib: a potential treatment for NSCLC with EGFR exon 20 insertions. Cancer Discov. 2021;11(7):1617–9.
Gonzalvez F, Vincent S, Baker TE, Gould AE, Li S, Wardwell SD, et al. Mobocertinib (TAK-788): a targeted inhibitor of EGFR exon 20 insertion mutants in non-small cell lung cancer. Cancer Discov. 2021;11(7):1672–87.
Rivera-Concepcion J, Uprety D, Adjei AA. Challenges in the use of targeted therapies in non-small cell lung cancer. Cancer Res Treat. 2022;54(2):315–29.
Johnson DH, Fehrenbacher L, Novotny WF, Herbst RS, Nemunaitis JJ, Jablons DM, et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol. 2004;22(11):2184–91.
Haluska P, Shaw HM, Batzel GN, Yin D, Molina JR, Molife LR, et al. Phase I dose escalation study of the anti insulin-like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors. Clin Cancer Res. 2007;13(19):5834–40.
Kim ES, Salgia R. MET pathway as a therapeutic target. J Thorac Oncol. 2009;4(4):444–7.
Herbst RS, Sun Y, Eberhardt WE, Germonpré P, Saijo N, Zhou C, et al. Vandetanib plus docetaxel versus docetaxel as second-line treatment for patients with advanced non-small-cell lung cancer (ZODIAC): a double-blind, randomised, phase 3 trial. Lancet Oncol. 2010;11(7):619–26.
Heymach JV, Johnson BE, Prager D, Csada E, Roubec J, Pesek M, et al. Randomized, placebo-controlled phase II study of vandetanib plus docetaxel in previously treated non small-cell lung cancer. J Clin Oncol. 2007;25(27):4270–7.
Socinski MA, Novello S, Brahmer JR, Rosell R, Sanchez JM, Belani CP, et al. Multicenter, phase II trial of sunitinib in previously treated, advanced non-small-cell lung cancer. J Clin Oncol. 2008;26(4):650–6.
Khuri F, Harvey R, Saba N, Owonikoko T, Kauh J, Shin D, et al. Everolimus, an mTOR inhibitor, in combination with docetaxel for recurrent/refractory NSCLC: a phase I study. J Clin Oncol. 2009;27(15_suppl):8060.
Blumenschein GR Jr, Paulus R, Curran WJ, Robert F, Fossella F, Werner-Wasik M, et al. Phase II study of cetuximab in combination with chemoradiation in patients with stage IIIA/B non-small-cell lung cancer: RTOG 0324. J Clin Oncol. 2011;29(17):2312–8.
Govindan R, Bogart J, Stinchcombe T, Wang X, Hodgson L, Kratzke R, et al. Randomized phase II study of pemetrexed, carboplatin, and thoracic radiation with or without cetuximab in patients with locally advanced unresectable non-small-cell lung cancer: cancer and leukemia group B trial 30407. J Clin Oncol. 2011;29(23):3120–5.
Gatzemeier U, Pluzanska A, Szczesna A, Kaukel E, Roubec J, De Rosa F, et al. Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer: the tarceva lung cancer investigation trial. J Clin Oncol. 2007;25(12):1545–52.
Ready N, Jänne PA, Bogart J, Dipetrillo T, Garst J, Graziano S, et al. Chemoradiotherapy and gefitinib in stage III non-small cell lung cancer with epidermal growth factor receptor and KRAS mutation analysis: cancer and leukemia group B (CALEB) 30106, a CALGB-stratified phase II trial. J Thorac Oncol. 2010;5(9):1382–90.
Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92.
Lovly CM, Salama AK, Salgia R. Tumor Heterogeneity and Therapeutic Resistance. American Society of Clinical Oncology educational book American Society of Clinical Oncology Annual Meeting. 2016;35:e585-93
Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14(8):535–46.
Piotrowska Z, Niederst MJ, Karlovich CA, Wakelee HA, Neal JW, Mino-Kenudson M, et al. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov. 2015;5(7):713–22.
Zito Marino F, Bianco R, Accardo M, Ronchi A, Cozzolino I, Morgillo F, et al. Molecular heterogeneity in lung cancer: from mechanisms of origin to clinical implications. Int J Med Sci. 2019;16(7):981–9.
Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121(9):725–37.
Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. 2017;376(7):629–40.
Pan Y, Deng C, Qiu Z, Cao C, Wu F. The resistance mechanisms and treatment strategies for ALK-rearranged non-small cell lung cancer. Front Oncol. 2021;11:713530.
Weng CH, Chen LY, Lin YC, Shih JY, Lin YC, Tseng RY, et al. Epithelial–mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene. 2019;38(4):455–68.
Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer. 2019;18(1):153.
Perez-Soler R. Rash as a surrogate marker for efficacy of epidermal growth factor receptor inhibitors in lung cancer. Clin Lung Cancer. 2006;8(Suppl 1):S7-14.
Robinson ES, Khankin EV, Karumanchi SA, Humphreys BD. Hypertension induced by vascular endothelial growth factor signaling pathway inhibition: mechanisms and potential use as a biomarker. Semin Nephrol. 2010;30(6):591–601.
Chu CY, Choi J, Eaby-Sandy B, Langer CJ, Lacouture ME. Osimertinib: a novel dermatologic adverse event profile in patients with lung cancer. Oncologist. 2018;23(8):891–9.
Kunimasa K, Kamada R, Oka T, Oboshi M, Kimura M, Inoue T, et al. Cardiac adverse events in EGFR-mutated non-small cell lung cancer treated with osimertinib. JACC CardioOncol. 2020;2(1):1–10.
Acknowledgements
Our sincere gratitude goes to authors who contributed their time and expertise to accomplish this article.
Funding
This research received no grant from any funding agency, commercial or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
M.A., R.M., A.H.M, S.H., and S.R. contributed to data gathering, writing the manuscript, and designing the tables and figures. F.F., S.Kh., and M.A. reviewed the last manuscript, engineered and edited the collection data, and completed the defective subject. A.H., R.A.S. and M.A. contributed to the revision and edition of the manuscript based on reviewers’ comments. S.T. and R.M. contributed to the hypothesis, correspondence, and revision of the final manuscript, scientific and structural editing, and conforming of the manuscript before submission.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Araghi, M., Mannani, R., Heidarnejad maleki, A. et al. Recent advances in non-small cell lung cancer targeted therapy; an update review. Cancer Cell Int 23, 162 (2023). https://doi.org/10.1186/s12935-023-02990-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-023-02990-y