
Abstract
Molecular targeted therapy for cancer has been a research hotspot for decades. AXL is a member of the TAM family with the high-affinity ligand growth arrest-specific protein 6 (GAS6). The Gas6/AXL signalling pathway is associated with tumour cell growth, metastasis, invasion, epithelial-mesenchymal transition (EMT), angiogenesis, drug resistance, immune regulation and stem cell maintenance. Different therapeutic agents targeting AXL have been developed, typically including small molecule inhibitors, monoclonal antibodies (mAbs), nucleotide aptamers, soluble receptors, and several natural compounds. In this review, we first provide a comprehensive discussion of the structure, function, regulation, and signalling pathways of AXL. Then, we highlight recent strategies for targeting AXL in the treatment of cancer.AXL-targeted drugs, either as single agents or in combination with conventional chemotherapy or other small molecule inhibitors, are likely to improve the survival of many patients. However, future investigations into AXL molecular signalling networks and robust predictive biomarkers are warranted to select patients who could receive clinical benefit and to avoid potential toxicities.
Similar content being viewed by others

Introduction
Cancer is not a single-cell disease but rather the result of complex interactions of tumour cells with surrounding matrix and immune cells. In recent years, molecular targeted therapy for cancer has been a research hotspot. Tyrosine kinase inhibitors (TKIs) targeting epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGF) and platelet-derived growth factor receptor (PDGFR) have been evaluated in clinical trials with promising results, which prompted the search for additional diagnostic and prognostic biomarkers [1,2,3]. AXL has emerged as a novel biomarker due to its role in biological processes and tumourigenesis [4]. AXL is a member of the TAM family that includes TYRO3, AXL and MER. Growth arrest-specific protein 6 (GAS6) serves as a ligand for AXL with high binding affinity. GAS6/AXL signalling functions as an important pathway driving cancer cell survival, proliferation, migration and invasion, which makes AXL a potential target in cancer treatment [5, 6].
The structure and function of AXL
The gene AXL, located at chromosome 19q13.2, was first identified in patients with chronic myeloid leukaemia (CML) [7]. The word AXL, coming from the Greek word “anexelekto”, means uncontrolled. The protein encoded by the gene AXL, called AXL (UFO, ARK, Tyro7, or JTK11), is a member of the TAM family of receptor tyrosine kinases (RTKs). AXL is composed of an extracellular, transmembrane and intracellular domain [8]. The extracellular structure consists of two immunoglobulin (Ig)-like repeats and two fibronectin type III (Fro III)-like repeats that resemble neural cell adhesion molecules (NCAMs) [9]. The Ig motifs are involved in the binding of AXL with its ligand Gas6 under regulation by Fro III [10]. The intracellular domain is critical for auto-phosphorylation and subsequent kinase activity [11]. The TAM family-specific KW(I/L)A(I/L)ES sequence resides within this intracellular domain, which shares homology with AXL-related RTKs, such as RET [12], and plays an important role in tyrosine kinase activity (Fig. 1a). The TAM family is widely expressed in normal cells and tissues, such as monocytes, platelets, endothelial cells, hippocampus, cerebellum, heart, and liver [13,14,15,16,17,18,19,20], wherein it regulates cell survival, the non-inflammatory clearance of apoptotic cells by phagocytic cells, natural killer cell differentiation, platelet aggregation, etc. [10, 21,22,23].

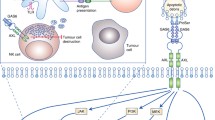
Basic structure, signaling pathways and activation of AXL. (a) Schematic diagram representing the structure of AXL receptor tyrosine kinase. AXL is composed of two immunoglobulin (Ig)-like repeats and two fibronectin type III (Fro III)-like repeats, a transmembrane domain and an intracellular kinase domain. (b) AXL signaling networks upon classical GAS6-mediated activation function in proliferation and survival, migration and invasion, epithelial-to-mesenchymal transition (EMT), angiogenesis, resistance to therapy, immune suppression, and stem cell maintenance. (c) AXL activation patterns: 1) classical GAS6 ligand-dependent dimerization; 2) Gas6 ligand-independent dimerization; 3) heterophilic dimerazation of AXL with a TAM family member like MER or TYRO3; 4) heterophilic dimerazation of AXL with a non-TAM family protein; and 5) ligand-independent activation of AXL through transcellular homophilic binding
The regulation of AXL gene expression
The synthesis of AXL is regulated at many levels. Five transcription factors act on the AXL promotor, including activator protein 1 (AP1) [24], Sp1/Sp3, YAP/TAZ/TEAD, hypoxia inducible factor 1α (HIF1α) [25] and myeloid zinc finger 1 protein (MZF-1) [26]. Activation of TLR signalling upregulates AXL mRNA in dendritic cells and macrophages [27, 28]. The transcription process is also feedback controlled by other RTKs. In non-small-cell lung cancer (NSCLC) and head and neck squamous cell carcinoma (HNSCC), activated EGFR pathways and downstream MEK/ERK signalling promote AXL mRNA expression through the JUN transcription factor [29]. In urothelial carcinoma, AXL mRNA expression is induced after activation of MET and downstream MEK/ERK signalling [30]. AXL mRNA expression is inhibited by two microRNAs (miRNAs), miR-34a and miR-199a/b [31, 32]. In addition, AXL mRNA expression is subject to epigenetic modification, including histone acetylation and histone/DNA methylation [33, 34]. Although full-length AXL contains 894 amino acids and encodes a predicted protein of 98 kDa, the actual observed molecular weight ranges from 100 to 140 kDa due to post-transcriptional regulation of the activated AXL receptor by glycosylation, phosphorylation and multiple sites of monoubiquitination [35,36,37,38,39].
In adults, AXL expression is relatively low [40, 41], but aberrant expression of Gas6/AXL has been shown in a number of human malignancies, including breast cancer, chronic lymphocytic leukaemia (CLL), NSCLC, pancreatic cancer, glioblastoma, melanoma, renal cell carcinoma (RCC), prostate cancer, and oesophageal cancer [4, 23, 42,43,44,45,46,47,48,49,50,51,52,53,54,55], and this altered expression is associated with disease progression and shortened overall survival (OS). In various in vivo breast cancer models, AXL expression was found to be higher in metastatic nodules than in primary tumours, and downregulating AXL with miRNAs inhibited downstream AKT phosphorylation and diminished the motility, metastasis and invasion of tumour cells [47]. Given the role of AXL in cancer development, progression and drug resistance, AXL holds great promise as a prognostic biomarker and therapeutic target.
Gas6/AXL axis and its role in tumour development and progression
Common ligands for the TAM family include Gas6, protein S, Tubby, Tubby-like protein 1 (TULP-1) and Galectin-3 [56, 57]. Studies on Gas6 and protein S are abundant, as these two ligands were the first to be discovered. Gas6 can bind to all three members of the TAM family, whereas protein S binds only to MER and TYRO3. The affinity of Gas6 for AXL is 3–10 times higher than that for the other two members in the family [8, 58]. Gas6 is encoded by growth arrest-specific gene 6 (Gas6) and belongs to the vitamin K-dependent protein family [59]. Upon high-affinity binding to its ligand Gas6, the AXL receptor undergoes homo-dimerization and subsequent trans-autophosphorylation within the intracellular kinase domain, thus recruiting adaptor molecules and effector proteins containing Src homology 2 (SH2) or other phosphotyrosine-binding domains (PTBs) and activating downstream signalling pathways [60, 61]. Six phosphorylation sites have been found in AXL, Tyr698, Tyr702, Tyr703, Tyr779, Tyr821 and Tyr866. Three N-terminal tyrosine residues, 779, 821 and 866, are related to auto-phosphorylation and AXL activation, while the other three C-terminal sites are rather conserved among the TAM receptors and indispensable for complete functions of the kinase [10]. A study on a EGFR-AXL chimeric receptor has revealed that the phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR signalling pathway is induced by binding of Tyr779 and Tyr821 to p85, and the MEK/ERK cascade is activated by binding of phosphorylated Tyr821 to GRB2 [62]. In addition, phosphorylated Tyr821 and Tyr866 can bind to SRC, LYK and PLCα and activate protein kinase C and STAT [4, 10, 15, 61]. Therefore, the PI3K/AKT/mTOR, JAK/STAT, NF-κB, and RAS/RAF/MEK/ERK signalling pathways function as important downstream pathways for the Gas6/AXL axis and play major roles in tumour cell survival, anti-apoptosis signalling, mitogenesis, migration, invasion, drug resistance, angiogenesis and the tumour-host relationship [63,64,65,66] (Fig. 1b).
In addition to Gas6-dependent AXL activation, AXL might also be activated via Gas6-independent mechanisms [62]. Overexpression of AXL leads to aggregation of AXL extracellular domains or ligand-independent homo-dimerization, thereby leading to receptor activation [13, 53]. Moreover, the broad homology AXL shared with other TAM family members, as well as with other non-TAM RTKs such as fibroblast growth factor receptor (FGFR), EGFR, PDGFR and MET [4], encourages the formation of heterodimers and the activation of AXL-dependent signalling [67, 68] (Fig. 1c). In multiple tumour tissues, aberrant AXL expression is related to the extent of malignancy, metastasis and a poor prognosis.
AXL promotes cell proliferation
AXL potentially drives cell proliferation through effector molecules in the PI3K/AKT/mTOR, RAS/RAF/MEK/ERK, JAK/STAT, and NF-κB signalling pathways [10, 69]. AXL fosters cell survival by regulating NF-κB nuclear translocation, increasing the expression of anti-apoptotic markers (survivin, BCL-2, and BCL-XL) and reducing the activity of pro-apoptotic proteins (BAD and caspase-3) [45, 52, 70]. Knockdown of AXL with short hairpin RNA (shRNA) reduces Ki67 expression and increases apoptosis-related protein levels [18, 71]. In CLL, the suppression of AXL promotes apoptosis with reduced levels of the anti-apoptotic protein MCL-1 [72]. Blockade of Gas6/AXL signalling pathway is sufficient to suppress ectopic and orthotopic glioma growth, leading to a marked prolongation of survival [73]. Similar results have been reported in prostate cancer, mesothelioma, lung adenocarcinoma and colorectal cancer [74, 75]. Thus, AXL may be implicated in protecting tumour cells from the apoptotic effects of numerous drugs.
AXL mediates migration and invasion
AXL was shown to be a driving force in the spread of tumours in both in vivo and in vitro studies. AXL activity is critical for cell migration phenotypes, including the increase in the GTP-binding proteins Rho and Rac [76] and the formation of filopodia [77]. The overexpression of AXL in cells with low metastatic colonization potential leads to augmented migratory and invasive abilities [78, 79]. It has been shown that AXL mediates Yes-associated protein (YAP)-dependent oncogenic functions that potentiate migration and invasion in hepatocellular carcinoma (HCC) [80]. In oesophageal adenocarcinoma (EAC) cell lines, AXL is responsible for the peripheral distribution of lysosomes and the secretion of cathepsin B, which promote cell invasion [55]. Matrix metalloproteinase 9 (MMP9) has been identified as a required factor for AXL-mediated invasion in vitro and in vivo [76, 78]. AXL activation promotes the expression of p-AKT and MMP9 through activation of NF-κB and Brg-1 [78, 81, 82]. Depletion of TIG1, which stabilizes AXL and prevents AXL from degradation, leads to a reduction in MMP9 expression in inflammatory breast cancer cell lines, reducing the in vitro migration and invasion of cells [71, 83]. Attenuation of the AXL signalling axis with the anti-AXL monoclonal antibody (mAb) 20G7-D9 dramatically reduces the number of bone metastases after intracardiac injection of breast cancer cells [84]. In HCC, downregulation of AXL by shRNA inhibits cell invasion through the PI3K/AKT-P21-activated kinase-1 (PAK1) signalling pathway. The reduced migration and invasion of cancer cells upon RNA interference (RNAi)-mediated knockdown of AXL or blockade of AXL signalling were also reported in liposarcoma, pancreatic cancer, lung adenocarcinoma, breast cancer and thyroid cancer [69, 76, 85,86,87], thus demonstrating the role of AXL signalling as a good target for conferred migratory and invasive properties.
AXL affects epithelial-mesenchymal transition (EMT)
Evidence supporting a pro-tumourigenic role for AXL in promoting EMT has recently been described in multiple studies. EMT is a reversible event in which cells undergo a transition from an epithelial phenotype to a mesenchymal phenotype through a special programme, and this transition is critical for foetal development and wound healing [88,89,90]. Cell-cell adhesion in normal epithelial cells helps maintain tissue integrity; on the other hand, mesenchymal cells are migratory and invasive [91]. Characteristic protein changes during EMT include a reduction in epithelial markers such as E-cadherin and an increase in mesenchymal markers such as N-cadherin, Snail, Vimentin, Slug, α-catenin and α-SMA [92, 93]. AXL activation drives EMT and enables cells to retain a mesenchymal phenotype [94]. In human breast cancer epithelial cells, transfection of SLUG and SNAIL into MCF10A cells is associated with increased expression of AXL, which induces the loss of epithelial-type morphology and the gain of mesenchymal-related markers [47]. Cells may respond to AXL deprivation by losing the expression of EMT-associated transcription factors (i.e., Slug, Zeb1, snail and Twist) [76, 95, 96], stimulating E-cadherin expression and cell-cell adhesion, and attenuating the activity of TGFâ-R and WNT signalling, thereby reversing to an epithelial-type morphology [44, 97, 98].
AXL in angiogenesis
Angiogenesis is a normal physiological process during foetal development, growth, wound healing, tissue reconstruction and repair [99]. However, it may also provide oxygen, nutrients and essential hormones to tumour cells, contributing to tumour growth, expansion and metastasis [100]. In addition to their oncogenic functions, TAM family members play important roles in vessel integrity and promoting angiogenesis with VEGF, fibroblast growth factor (FGF) and platelet-derived growth factor (PDGF) [10, 28, 101,102,103]. AXL is broadly expressed in endothelial cells and vascular smooth muscle cells; it promotes the stabilization of aggregated platelets, survival of endothelial cells and remodelling of endothelial barriers in wound healing and vessel impairment [104]. The autocrine and paracrine loops of AXL and its ligand Gas6 may enhance the activity and proliferation of endothelial cells, regulate the function of integrin, and promote the migration and survival of endothelial cells and tumour cells through RAC and AKT signalling [8]. AXL catalytic activity induces human umbilical vein endothelial cell (HUVEC) growth, migration and tube formation [105]. Knockdown of either the AXL or Gas6 gene will impair endothelial tube formation and functional circulation [106, 107]. However, Gallicchio et al. [108] implied an anti-angiogenic role for AXL by showing that the Gas6/AXL axis might antagonize VEGFR2-dependent angiogenesis. Studies have demonstrated that AXL expression is associated with antiangiogenic resistance, while the combination of AXL inhibitors with antiangiogenic agents could reduce vessel density in renal cell carcinoma patient-derived xenografts [109,110,111]. The complex role of AXL in angiogenesis under normal or pathological conditions needs to be further discussed.
AXL is associated with drug resistance in cancer treatment
Drug resistance is a thorny problem and a major hindrance during cancer management that often leads to treatment failure or disease recurrence. Numerous examples of the association of AXL expression with treatment resistance have been reported in prostate, breast, ovarian, colorectal and lung cancers [29, 48, 51, 65, 112, 113]. AXL could cause innate or acquired resistance to chemotherapy, immune therapy, molecular targeted therapy or even radiation therapy [98, 114,115,116,117,118]. Molecular targeted therapy leads to an increase in AXL expression, which confers refractoriness or an insufficient response of cells to ERK, BRAF, PI3Kα, ALK, EGFR and VEGFR inhibitors [29, 114, 117, 119, 120]. The suppression of AXL, whether by genetic knockdown or pharmacological inhibition, is effective in circumventing chemoresistance to certain drugs in an otherwise resistant cell line. For example, AXL was identified as a “tyrosine kinase switch”: overexpression of AXL/Gas6 and low KIT expression were found in an imatinib (KIT/PDGF inhibitor)-resistant gastrointestinal stromal tumour (GIST) model [118]. In cisplatin-resistant ovarian cancer cells, AXL mRNA expression was twice as high as that in cisplatin-sensitive cells [121]. Selective silencing or inhibition of AXL resensitized CML cells to imatinib [49] and prostate cancer cells to docetaxel with reduced ATP-binding cassette B1 (ABCB1) levels [98]. In radiation-resistant HNSCC cell lines, marked AXL overexpression was found in cancer cell xenografts and patient-derived xenografts (PDXs), whereas resensitization to chemotherapy and radiation was achieved after AXL knockdown [114, 122].
Over the years, many studies have investigated the mechanisms through which AXL induces drug resistance. These underlying mechanisms might involve crosstalk between AXL and other RTK family members. As mentioned previously, AXL can heterodimerize with non-TAM RTKs such as EGFR, MET and PDGF, which helps avoid the effects of certain RTK inhibitors. For instance, the hetero-interaction between AXL and human epidermal growth factor receptor 2 (HER2) leads to downstream PI3K/AKT and ERK signalling and allows cells to evade the inhibitory effects of lapatinib in HER2-positive breast cancer [123]. Moreover, in PI3K inhibitor-resistant squamous cell carcinoma, AXL binds to EGFR and activates the PLCα/PKC/mTOR signalling pathway to maintain tumour progression [48]. AXL expression also sustains the effects of PI3K/AKT and MEK/ERK, and positive feedback from MEK/ERK induces AXL transcription through JUN. In acute myeloid leukaemia (AML), AXL is associated with upregulated BCL-2 and Twist and participates in the malignant progression of cells, inducing EMT and drug resistance [45]. These findings indicate that AXL is a promising target for the salvage treatment of cancer recurrence.
AXL regulates the immune response
TAM family members are important negative inflammatory mediators inhibiting certain signalling pathways that activate dendritic cells, natural killer cells and macrophages [27, 103, 124, 125], attenuating their ability to eliminate metastases [27, 126, 127]. It was reported that TAM receptors could suppress cytokine production and the TLR-dependent inflammatory response through hijacking pro-inflammatory signals, thereby serving as a feedback mechanism to prevent autoimmune responses [27].
Given the recent growing interest in immune checkpoint blockade, the role of AXL in immune surveillance has garnered much attention. AXL activation is involved in immune evasion through the upregulation of BCL-2 and Twist, the suppression of TLR inflammatory signalling and natural killer cells, and the limited expression of pro-inflammatory cytokines [4, 45], and AXL loss-of-function enhances chronic inflammation and autoimmunity [23, 27, 128,129,130,131]. The combined deletion of AXL and MER has been shown to increase the risk of colitis and colitis-associated cancer [132]. The role of AXL in radioresistant and checkpoint immune-resistant tumours has been described, and the mechanism is thought to involve the ability of AXL to suppress antigen presentation through MHC-I and to enhance myeloid-supporting cytokines and chemokines, resulting in a limited initial immune response [133]. These findings are further supported by the recent genomic and transcriptomic data from metastatic melanoma patients suggesting that AXL overexpression might influence innate sensitivity or cause resistance to anti-PD-1 therapy [115]. Interestingly, the immunomodulatory function of AXL in oncogenesis is paradoxical. Diminishing AXL and MER signalling in normal mouse tissue induces the production of inflammatory cytokines, which favour a pro-malignancy environment [131]. Thus, the mechanisms by which AXL inhibitors modulate the immune response will be important to decipher for additional insight into anticancer therapeutic approaches.
AXL is related to stem cell maintenance
Cancer stem cells (CSCs) are a small subpopulation of cells that reside within the tumour; possess the capabilities of self-renewal, differentiation and tumourigenicity; and exert an immense influence on tumour resistance, recurrence and metastasis [134, 135]. AXL expression is found in human gliomas with high expression of EZH2, which plays a crucial role in stem cell maintenance [33]. Additionally, AXL is correlated with the expression of stem cell marker genes such as Isl1, Cdc2a, Bglap1, CD44 and ALDH1, which increase the tumourigenicity of breast cancer stem cells [96] and enhance the resistance of cutaneous squamous cell carcinoma to chemotherapy [95]. Targeting AXL holds great therapeutic potential to diminish WNT/â-catenin and TGFâR signalling and sphere formation ability, and therefore, to repress cancer resistance and progression [136,137,138].
AXL-targeted therapies
Because of the pleiotropic role of AXL in tumour pathophysiology and drug resistance, AXL represents a promising therapeutic target in the management of cancer. AXL inhibitors have been shown to potentiate tumour cell apoptosis and suppress migration and invasion [43]. Targeting AXL enhances the therapeutic efficacy of chemotherapy and other small-molecule inhibitors, such as VEGF, EGFR, PI3K, PARP, and HER2 inhibitors [54, 123, 139]. Different therapeutic agents have been developed over the decades, including small molecule inhibitors, anti-AXL mAbs, nucleotide aptamers, soluble receptors, and several natural compounds (Fig. 2).

Different therapeutic agents targeting AXL have been developed, including 1) small molecule inhibitors that block AXL auto-phosphorylation and kinase activities; 2) anti-AXL monoclonal antibodies (mAbs); 3) nucleotide aptamers; 4) soluble receptors; and 5) several natural compounds
Small molecule inhibitors
Most small molecules that have inhibitory effects on AXL were not synthesized to act primarily on AXL; thus, the AXL inhibitory activity is not as potent as the inhibitory activity against other kinases [46, 140, 141]. However, BMS-777607, originally designed as a MET-branded inhibitor, was approximately three times more potent against AXL than MET [142]. AXL-specific inhibitors were also developed with high selectivity, including SGI-7079, TP-0903, BGB324, DP3975 and NA80xl. All compounds described here are ATP-competitive inhibitors, either the most commonly observed type I inhibitors that preferentially occupy the ATP-bound pocket of the kinase in the active “aspartate-phenylalanine-glycine (DFG)-in” conformation, or type II inhibitors that prefer the inactive DFG-out conformation [143, 144] and that are reported to be not intrinsically more selective than type I inhibitors [145]. Several AXL inhibitors have been considered promising upon emerging from preclinical studies and are thus advanced into different stages of clinical investigation [146,147,148,149,150,151,152,153].
Type I AXL inhibitors (Table 1)
BGB324 (Bemcentinib, R428; Rigel Pharmaceuticals/BerGenBio)
R428, a specific and highly selective AXL inhibitor, acts on AXL at nanomolar concentrations (IC50 = 14 nM) with 100-fold higher affinity for AXL over ABL in cellular assays [46, 154]. R428 blocks the catalytic activities of AXL and reduces AXL and p-AXL expression [46, 58, 155]. When used alone or in combination with cytotoxic agents to treat AML cells, R428 inhibits the AKT and MAPK pathways through the upregulation of Puma and subsequently suppresses BCL-2 [156]. R428 has been shown to recover drug sensitivity in many models of acquired resistance; for example, R428 inhibits erlotinib-resistant head and neck cancer cell growth and migration [114, 116]. In 2014, R428 was the first AXL-specific TKI to enter clinical trials, and this drug is now in phase I/II clinical trials for AML, myelodysplastic syndromes (MDS), triple-negative breast cancer (TNBC), metastatic melanoma, pancreatic cancer, and NSCLC either alone or in combination with other chemotherapy regimens (NCT02424617, NCT02922777, NCT03184558, NCT02488408, NCT03184571, NCT03649321, NCT03824080, and NCT02872259).
TP-0903 (Tolero Pharmaceuticals)
TP-0903 is a novel selective AXL inhibitor with an in vitro IC50 of 27 nM [157, 158]. TP-0903 disrupts the phosphorylation of AXL; reverses EMT; enhances the depletion of anti-apoptotic proteins such as MCL-1, XIAP, and BCL-2; and induces dose-dependent CLL cell death [72, 159]. Moreover, it also enhances neuroblastoma cell sensitivity to conventional chemotherapy [160]. TP-0903 is currently being evaluated for safety, pharmacokinetics, pharmacodynamics, and antitumour activity in patients with CLL and refractory solid tumours (NCT03572634 and NCT02729298).
Crizotinib (PF-02341066, marketed as Xalkori; Pfizer)
As a multi-target TKI, crizotinib inhibits ALK, MET, RON and AXL [161]. Early preclinical testing of crizotinib revealed potent in vitro and in vivo antitumour effects in a variety of malignancies, including gastric carcinoma, NSCLC, RCC, prostate carcinoma, malignant glioblastoma, anaplastic large cell lymphoma (ALCL), and osteosarcoma [162,163,164,165], which prompted abundant clinical research. In phase I and II studies, crizotinib resulted in significant and rapid improvements in treatment responses in patients with ALK-positive NSCLC and was generally well tolerated [166]; therefore, crizotinib was approved by the FDA to treat patients with ALK-positive metastatic NSCLC. Later, it was further approved for the treatment of metastatic NSCLC patients with ROS1 gene alterations [167]. However, the unavoidable acquired resistance to first-line crizotinib is a major problem to be managed in patients with ALK-positive NSCLC [168]. Recently, clinical trials have been initiated to compare crizotinib and other ALK inhibitors (NCT02737501) and to assess crizotinib in combination with other agents, such as immune checkpoint inhibitors, mTOR inhibitors, and antiangiogenesis drugs (NCT02292550, NCT02321501, NCT02521051, NCT01998126, NCT02511184, and NCT02393625).
Bosutinib (SKI-606, Bosulif®; Pfizer)
Originally developed as an inhibitor of SRC and ABL kinases, the second-generation TKI bosutinib also has potent inhibitory activity against AXL auto-phosphorylation [79], accompanied by the suppression of Slug expression, stabilization of cell-to-cell adhesion and increased membrane localization of â-catenin [141, 169]. Treatment with bosutinib induces the dose- and time-dependent induction of apoptosis in CLL B cells [154] and suppresses the motility and invasiveness of HCC and breast cancer cells [79, 170]. The administration of bosutinib causes the regression of K562 and colorectal xenografts in nude mice [171, 172]. Bosutinib was reported to have significant clinical activity in CML patients who were resistant/intolerant to prior TKIs and was generally well tolerated in these populations in phase I/II clinical trials [173]; thus, it was approved for use in CML at 400 mg q.d. as first-line therapy and at 500 mg q.d. in patients who have failed prior imatinib, nilotinib, or dasatinib [174, 175]. Bosutinib is currently in multiple phase I-IV clinical trials for the treatment of several cancer types, including breast cancer, CML, NSCLC, and glioblastoma.
Gilteritinib (ASP2215; Astellas Pharma/Kotobuki Pharmaceutical)
Gilteritinib is a novel, highly specific, dual FMS-like tyrosine kinase 3 (FLT3)/AXL inhibitor that has demonstrated robust antileukaemic activity in patients with relapsed/refractory AML. AXL is known to promote constitutively active FLT3 and to be responsible for resistance to FLT3 inhibitors [176, 177]. Targeting AXL and FLT3 with gilteritinib translated to tumour regression and reduced proliferation in FLT3 mutation-positive cellular and mouse models of AML [178]. Promising early phase I/II trial data of gilteritinib demonstrated antileukaemic activity and acceptable side effects in relapsed/refractory AML patients from the USA, Germany, Italy and Japan (NCT02181660 and NCT02014558) [179, 180]; thus, it was approved in Japan and the USA for the treatment of relapsed or refractory AML with FLT3 mutation [181]. The clinical development of gilteritinib for advanced solid tumours is also underway in several countries worldwide (NCT02456883, NCT02561455, and NCT02622932).
S49076 (Servier)
S49076 is described as a novel ATP-competitive TKI of MET, AXL, and FGFR1/2/3 [182]. S49076 exerts its cytotoxic activity at low doses on MET- and FGFR2-dependent cells, while it blocks the proliferation of MET-independent cells at higher but clinically relevant doses through targeting Aurora B [183]. In tumour xenograft models, S49076 enhances the antitumour efficacy by synergizing with bevacizumab or radiotherapy [182, 183]. In the first-in-human phase I study (ISRCTN00759419), S49076 demonstrated limited single-agent activity with a tolerable safety profile at a recommended dose of 600 mg once daily in patients with advanced solid tumours [146]. S49076 was recommended for combination therapies; therefore, it is currently in phase I/II clinical trials in combination with gefitinib in MET/AXL-dysregulated NSCLC patients progressing on prior EGFR-TKI treatment [184].
Amuvatinib (MP-470; Astex Pharmaceuticals)
Amuvatinib, a multitargeted RTK inhibitor that targets AXL, KIT, and PDGFRα [140], was shown to disrupt DNA damage repair through the inhibition of Rad51 [185], to suppress AXL expression [186] and to resensitize cells to radio- and chemotherapies in GIST, lung cancer and glioblastoma cells [118, 140, 185,186,187]. MP-470 was recently reported to produce a limited response in a refractory GIST patient with KIT mutations and to show antitumour activity when combined with standard-of-care chemotherapy in neuroendocrine tumours, NSCLC, and small cell lung cancer (SCLC) [147, 188].
Sunitinib (SU11248, Sutent; Pfizer)
Sunitinib is an oral multi-targeted TKI with activity against PDGFR, VEGFR2, FLT3, KIT, and AXL [189,190,191]. It was the first drug jointly approved by the FDA for the treatment of both advanced RCC and imatinib-resistant/intolerant GIST in 2006 and was later approved for pancreatic neuroendocrine tumours (PNETs) [5, 192,193,194,195]. Resistance to sunitinib, whether intrinsic or acquired, still remains a challenge limiting its optimal clinical benefit [196]. Sunitinib is currently in clinical trials for multiple solid tumours, including thymic carcinoma, GIST, cholangiocarcinoma, urothelial carcinoma, NSCLC, soft tissue sarcoma and RCC (NCT01499121, NCT01498835, NCT00794950, NCT01718327, NCT01824615, NCT02623127, NCT03673501, and NCT00372775).
SNS314 (Sunesis Pharmaceuticals)
SNS314 is a pan-selective Aurora kinase inhibitor [197]. Although preclinical studies showed potent anti-tumor activity [198], results of a phase I clinical trial for the treatment of patients with advanced solid tumors were not satisfactory as no responses were observed. Thus, further development of SNS-314 was suspended [5].
Other type I AXL inhibitors in the preclinical stage
2-D08 is a unique inhibitor of protein sumoylation that has strong potency against AXL kinase with an in vitro IC50 of 0.49 nM [199]. 2-D08 is known to inhibit the phosphorylation of AKT and ERK, increase the expression of epithelial surfactant protein, and suppress EMT-mediating transcription factors, including SNAI2, HOXA5 and TBX2/3 [200]. In addition, the dual MER/FLT3 inhibitor UNC2025 [201], the selective AXL inhibitor SGI-7079 [202], and the novel MER and AXL inhibitor UNC569 [203, 204] have also been reported as promising molecules emerging from preclinical studies.
Type II AXL inhibitors (Table 2)
Cabozantinib (XL184, Cometriq®; Exelixis/Ipsen company)
Developed as a non-selective TKI that inhibits multiple RTKs, including MET, AXL, VEGFR2, RET, KIT, and ROS1 [205], and the related angiogenesis and metastasis processes [206, 207], cabozantinib has shown vast in vitro antitumour activity in extensive studies on human umbilical vein endothelial cells (HUVECs) and RCC, HCC, medullary thyroid cancer, and ovarian cancer cells [148, 208,209,210,211,212,213,214]. In vivo studies using xenograft models of breast cancer, lung cancer and glioma also showed the dose-dependent antitumour efficacy of cabozantinib [150, 206]. The capsule form of cabozantinib, Cometriq®, has been approved by the FDA for the treatment of advanced RCC and metastatic medullary thyroid carcinoma [215]. Numerous phase I/II trials have been conducted in patients with melanoma, metastatic breast cancer, NSCLC, AML, castration-resistant prostate cancer and HCC with promising preliminary activities [148,149,150, 216,217,218,219,220]. However, a randomized phase II study of 60 mg p.o. daily cabozantinib versus 80 mg/m2 paclitaxel weekly revealed that cabozantinib at this dose was not recommended over paclitaxel for the treatment of recurrent ovarian cancer [221]. Recently, the final results of the phase III CELESTIAL trial were published, which demonstrated a prolonged median OS for cabozantinib treatment compared with placebo (10.2 months vs. 8.0 months, hazard ratio (HR) = 0.76 (0.63–0.92), p = 0.005) [216]. Other phase III trials are now ongoing in differentiated thyroid cancer (NCT03690388), sorafenib pre-treated HCC (NCT01908426), and carcinoid tumours (NCT03375320).
BMS-777607 (ASLAN002; Bristol-Myers Squibb/Aslan Pharmaceuticals)
BMS-777607 was initially designed to inhibit MET kinase, but in fact, it was found to be a potent AXL kinase inhibitor with an IC50 of 1.1 nM in cell-free assays [222,223,224]. It significantly inhibits MET auto-phosphorylation; the activation of downstream molecules including ERK, AKT, p70S6K and S6; colony formation, migration; invasion; and HGF-induced cell scattering in GTL-16, H1993, U87, PC-3 and DU145 cells [142, 222, 225, 226]. This compound also demonstrates significant in vivo antitumour activity through increased apoptosis and decreased proliferation and migration in the GTL-16 human gastric carcinoma xenograft model [222]. In a KHT sarcoma rodent tumour model, BMS-777607 impaired metastasis [225], led to the regression of intracranial glioma tumour growth and reduced AXL-related tumour angiogenesis [43]. In addition, BMS-777607 is a potent polyploidy inducer that promotes the megakaryocytic differentiation of CHRF-288-11 cells [227]. This compound is now undergoing phase I/II clinical trials in patients with advanced or metastatic tumours (NCT01721148 and NCT00605618).
LY2801653 (Merestinib; Eli Lilly and Company/Dana-Farber Cancer Institute)
LY2801653 is a dual MET/AXL inhibitor that targets RON, MET, and AXL [228, 229]. LY2801653 potently blocks the phosphorylation of MET and AXL and the activation of their downstream signalling molecules. In cholangiocarcinoma, LY2801653 inhibits migration, invasion, colony formation, and concomitant in vivo tumour growth through the suppression of MET and downstream targets [230]. LY2801653 disrupts the activity of mesenchymal glioma stem cells through the inhibition of MAPK-interacting kinases (MNKs) [73] and has a demonstrated antitumour effect in xenograft models of AML, gastric cancer, cholangiocarcinoma and lung cancer cells [229, 231, 232]. LY2801653 is now being investigated in patients with breast cancer, AML, biliary tract cancer and NSCLC (NCT03027284, NCT01285037, NCT03125239, NCT03292536, and NCT02711553).
Foretinib (XL880, EXEL-2880, GSK1363089; GSK)
Foretinib is an oral multi-kinase inhibitor of AXL, MET, VEGFR, ROS, RON, and TIE-2 [161, 233]. Foretinib blocks AXL phosphorylation and is associated with suppressed cell proliferation, dissemination and survival and the inhibition of in vivo tumour growth and peritoneal metastasis in an orthotopic colorectal cancer xenograft model [234]. In HER2-positive breast cancer cells that overexpress AXL, treatment with foretinib in combination with HER2-targeted therapies renders cells more vulnerable to lapatinib [123]. In a phase I clinical trial, the observation of three confirmed partial responses and 22 cases of stable disease in a total of 40 patients confirmed the antitumour activity of foretinib [235]. Data from phase I/II studies showed evidence of tumour regression in patients with advanced papillary renal cell carcinoma (PRCC), TNBC, NSCLC and HCC [151, 153, 236, 237]. However, in a recent phase II study evaluating foretinib in gastric cancer, single-agent foretinib lacked efficacy even in MET-amplified patients with metastatic gastric cancer [152], which demonstrated the requirements for ascertaining the mechanisms of gastric cancer oncogenesis and molecular patient selection. The toxicity profile was relatively manageable, and hypertension and elevated aspartate aminotransferase (AST) were common side effects in patients with cancer [151,152,153, 235,236,237].
MGCD516 (Sitravatinib; Mirati Therapeutics Inc.)
MGCD516 blocks a closely related spectrum of RTKs, including KIT, PDGFRβ, PDGFRα, MET, and AXL [238]. MGCD516 demonstrates better in vitro and in vivo efficacy in sarcoma cell lines than two well-known TKIs, imatinib and crizotinib, and augments immune checkpoint blockade in unresponsive tumours [238, 239]. A phase I trial with MGCD516 is now recruiting patients with solid tumours (NCT02219711).
MGCD265 (Glesatinib; Mirati Therapeutics)
MGCD265 is a small molecule multi-targeted TKI that targets MET, VEGFR1/2/3, RON, TIE-2 and AXL [240], and it has been shown to have a potent clinical response in patients with metastatic NSCLC with AXL amplification [241]. Phase I/II clinical trials in patients with metastatic NSCLC harbouring genetic alterations in MET and advanced malignancies [242] have been completed, but the results are not yet available.
RXDX-106 (Ignyta, Inc.)
RXDX-106 is a selective and potent pan-TAM family inhibitor that exerts antitumour efficacy through regulating immune cells, including M1-polarized intra-tumoural macrophages, NK cells, CD8+ T cells and dendritic cells, and may lead to suppressed tumour growth and progression [243].
Rebastinib (DCC-2036; Deciphera Pharmaceuticals LLC)
Rebastinib, designed as a switch-control inhibitor of the BCR-ABL1 tyrosine kinase [244], also has striking activity against AXL in TNBC cells [245]. It is now in phase I/II clinical trials for the treatment of locally advanced or metastatic solid tumours, CML and breast cancer (NCT03717415, NCT03601897, NCT00827138, and NCT02824575).
Other type II inhibitors at the preclinical stage
NPS-1034 is a newly developed dual AXL/MET inhibitor that exerts efficacy against cancer cells harbouring activated or mutated MET or AXL [246, 247]. LDC1267, a highly selective TAM kinase inhibitor, is able to awaken the innate immune system and enhance NK cell activity to kill cancer metastases in vivo [127].
Anti-AXL mAbs (Table 3)
Current AXL molecular targeted therapeutics exhibit either modest antitumour efficacy, cellular cytotoxicity, or significant off-target effects [46, 229, 248], which prompted the emergence of high affinity anti-AXL mAbs. YW327.6S2 is a blocking antibody that binds to both human and murine AXL, limiting receptor activation and downstream signalling through the ligand Gas6. YW327.6S2 attenuates tumour growth, metastasis, angiogenesis and the secretion of inflammatory cytokines and chemokines from tumour-associated macrophages (TAMs) and potentiates the efficacy of chemotherapy and other small-molecule inhibitors [139].
Two other selected anti-AXL mAbs, D9 and E8, are efficient in inhibiting the proliferation and migration of pancreatic cancer cells through blocking the phosphorylation of AXL and downstream molecules without affecting GAS6 binding [249]. Other anti-AXL mAbs include 20G7-D9 [84], MAb173 [250], 64Cu-labelled anti-AXL antibody [251], antibody-based agents such as the antibody-drug conjugate (ADC) AXL-107-MMAE [252] and AXL-specific CAR and SynNotch receptor [253], which have also shown promising results in preclinical studies.
Nucleotide aptamers (Table 3)
Nucleotide aptamers are immerging alternatives with higher affinity and lower toxicity than current standard therapies [248, 249, 254]. Aptamers are short structured single-stranded RNAs or DNAs that can act as ligands by binding to their targets. Their low cost, convenient generation, low immunogenicity, sufficient stability, and potential as targeted delivery tools for nanoparticles, chemotherapeutics or siRNAs make them promising therapeutics in neoplastic diseases [255, 256].
The RNA aptamer GL21.T was designed to specifically recognize the extracellular domain of AXL. It hampers AXL-dependent downstream ERK and AKT phosphorylation; interferes with cell migration, invasion and colony formation; and inhibits in vivo tumour growth in a mouse xenograft model of human NSCLC cells [255]. The conjugate of miR-34c and the GL21.T aptamer, GL21.T/miR-34c, exhibits dual functional and transcriptional inhibition of AXL in NSCLC cells [257]. Based on the sequence of GL21.T, the corresponding DNA aptamer was synthesized and was more resistant to hydrolysis. This DNA AXL-APTAMER could inhibit AXL phosphorylation and the related cell proliferation in vitro and in vivo and potentiate chemotherapy efficacy in ovarian cancer models [42].
Soluble receptors
Since the TAM family members conventionally undergo alternative splicing or proteolytic cleavage of extracellular domains [10], these soluble extracellular domains produced might act as a ligand sink to downregulate the receptor [258]. The soluble ectodomain of AXL, termed soluble AXL (International Patent application WO2008098139), acts as a ‘decoy receptor’ that binds GAS6, abrogating AXL signalling and Gas6-induced mitogenic effects, and has shown promising results in animal models of metastasis [10, 255, 259, 260]. In addition, soluble AXL has considerable potential as a diagnostic marker in patients at early stage HCC and cirrhosis [261].
Other natural compounds as inhibitors of AXL
Natural compounds could also function as inhibitors of AXL. For example, celastrol exhibits a synergistic effect with gefitinib in suppressing cell proliferation and migration and increases the susceptibility of EGFR-mutant NSCLC cells to gefitinib [262]. Dihydroartemisinin (DHA), the active derivative of the well-known anti-malarial drug artemisinin, blocks AXL expression and the related proliferation, migration, and tumour development of prostate cancer cells via the miR-34a/miR-7/JARID2 pathway [263]. Recently, a new class of quinolone-based compounds has emerged as selective AXL inhibitors that could inhibit TGF-â1-induced MDA-MD-231 breast cancer cell migration and invasion in a dose-dependent manner [264]. In addition, mistletoe extract of Viscum album extract (VAE) was reported to inhibit AXL expression, suppress cell proliferation and overcome cisplatin- and erlotinib-resistance in NSCLC cells [265].
Future perspectives and conclusion
Although AXL-targeted therapies appear promising in treating malignancies, many questions remain unanswered. AXL small molecule inhibitors have pronounced therapeutic effects; nonetheless, their inherent limitations, the off-target effects, might give rise to the inhibition of additional kinases and subsequent unexpected toxicities, limiting their clinical use. A variety of side effects have been reported for molecular targeted therapies, especially for multi-targeted AXL TKIs [148, 151,152,153, 210, 220, 235,236,237].
In addition, many patients do not respond to anti-AXL treatment or acquire resistance to these agents. Therefore, it is beneficial to select the optimal patients who could draw clinical benefits from AXL inhibitors and to avoid potential toxicities. Reliable integrated biomarker design is warranted to guide treatment strategies in predicting response and overcoming therapeutic resistance.
AXL inhibitors, either as single agents or in combination with conventional chemotherapy or other inhibitors such as immune checkpoint inhibitors, angiogenesis inhibitors and other TKIs, are likely to improve the survival of many patients. However, rational combination approaches, the sequence of administration, and the right time of incorporation of anti-AXL agents into treatment regimens should be taken into consideration before clinical use. Dissection of AXL molecular signalling networks and further investigations into the relationships between AXL and other kinases need to be performed to improve antitumour therapies and personalized cancer treatment.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- TKIs:
-
Tyrosine kinase inhibitors
- RTKs:
-
Receptor tyrosine kinases
- Gas6 :
-
Growth arrest specific gene 6
- GAS6:
-
Growth arrest-specific protein 6
- EGFR:
-
Epidermal growth factor receptor
- VEGF:
-
Vascular endothelial growth factor
- PDGF:
-
Platelet-derived growth factor
- PDGFR:
-
Platelet-derived growth factor receptor
- FGF:
-
Fibroblast growth factor
- FGFR:
-
Fibroblast growth factor receptor
- PI3K:
-
PHOSPHATIDYLINOSITOL 3-kinase
- PAK1:
-
PI3K/AKT-P21-activated kinases-1
- HER2:
-
Human epidermal growth factor receptor 2
- EMT:
-
Epithelial mesenchymal transition
- mAbs:
-
Monoclonal antibodies
- FLT3:
-
FMS-like Tyrosine Kinase 3
- MNKs:
-
MAPK-interacting kinases
- Fro III:
-
Fibronectin type III
- Ig:
-
Immunoglobulin
- NCAMs:
-
Neural cell adhesion molecules
- AP1:
-
Activator protein 1
- HIF1α:
-
Hypoxia inducible factor 1α
- MZF-1:
-
Myeloid zinc finger 1 protein
- TULP-1:
-
Tubby-like protein 1
- SH2:
-
Src homology 2
- PTBs:
-
Phosphotyrosine-binding domains
- miRNAs:
-
microRNAs
- shRNA:
-
short hairpin RNA
- siRNA:
-
Small interfering RNA
- RNAi:
-
RNA interference
- HUVECs:
-
Human umbilical vein endothelial cells
- TAMs:
-
Tumor-associated macrophages
- ADC:
-
Antibody-drug conjugate
- DHA:
-
Dihydroartemisinin
- VAE:
-
Visucm album extract
- SCLC:
-
Small cell lung cancer
- NSCLC:
-
Non-small cell lung cancer
- HNSCC:
-
Head and neck squamous cell carcinoma
- AML:
-
Acute myeloid leukemia
- CLL:
-
Chronic lymphocytic leukemia
- CML:
-
Chronic myeloid leukemia
- HCC:
-
Hepatocellular carcinoma
- RCC:
-
Renal cell carcinoma
- PRCC:
-
Papillary renal cell carcinoma
- GIST:
-
Gastrointestinal stromal tumor
- MDS:
-
Myelodysplastic syndromes
- TNBC:
-
Triple-negative breast cancer
- ALCL:
-
Anaplastic large cell lymphoma
- PNETs:
-
Pancreatic neuroendocrine tumors
- OS:
-
Overall survival
- HR:
-
Hazard ratio
- AST:
-
Aspartate aminotransferase
- PDXs:
-
Patient-derived xenografts
References
Ko YJ, Small EJ, Kabbinavar F, Chachoua A, Taneja S, Reese D, DePaoli A, Hannah A, Balk SP, Bubley GJ. A multi-institutional phase ii study of SU101, a platelet-derived growth factor receptor inhibitor, for patients with hormone-refractory prostate cancer. Clin Cancer Res. 2001;7(4):800–5.
Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–99.
Subbiah V, Meyer C, Zinner R, Meric-Bernstam F, Zahurak ML, O'Connor A, Roszik J, Shaw K, Ludwig JA, Kurzrock R, et al. Phase Ib/II Study of the Safety and Efficacy of Combination Therapy with Multikinase VEGF Inhibitor Pazopanib and MEK Inhibitor Trametinib In Advanced Soft Tissue Sarcoma. Clin Cancer Res. 2017;23(15):4027–34.
Gay CM, Balaji K, Byers LA. Giving AXL the axe: targeting AXL in human malignancy. Br J Cancer. 2017;116(4):415–23.
Feneyrolles C, Spenlinhauer A, Guiet L, Fauvel B, Dayde-Cazals B, Warnault P, Cheve G, Yasri A. Axl kinase as a key target for oncology: focus on small molecule inhibitors. Mol Cancer Ther. 2014;13(9):2141–8.
Barata PC, Rini BI. Treatment of renal cell carcinoma: Current status and future directions. CA Cancer J Clin. 2017;67(6):507–24.
Liu E, Hjelle B, Bishop JM. Transforming genes in chronic myelogenous leukemia. Proc Natl Acad Sci U S A. 1988;85(6):1952–6.
Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer. 2014;14(12):769–85.
Yamagata M, Sanes JR, Weiner JA. Synaptic adhesion molecules. Curr Opin Cell Biol. 2003;15(5):621–32.
Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res. 2008;100:35–83.
Kimani SG, Kumar S, Bansal N, Singh K, Kholodovych V, Comollo T, Peng Y, Kotenko SV, Sarafianos SG, Bertino JR, et al. Small molecule inhibitors block Gas6-inducible TAM activation and tumorigenicity. Sci Rep. 2017;7:43908.
Toledo RA, Qin Y, Cheng ZM, Gao Q, Iwata S, Silva GM, Prasad ML, Ocal IT, Rao S, Aronin N, et al. Recurrent Mutations of Chromatin-Remodeling Genes and Kinase Receptors in Pheochromocytomas and Paragangliomas. Clin Cancer Res. 2016;22(9):2301–10.
Bellosta P, Costa M, Lin DA, Basilico C. The receptor tyrosine kinase ARK mediates cell aggregation by homophilic binding. Mol Cell Biol. 1995;15(2):614–25.
Prasad D, Rothlin CV, Burrola P, Burstyn-Cohen T, Lu Q. Garcia de Frutos P, Lemke G: TAM receptor function in the retinal pigment epithelium. Mol Cell Neurosci. 2006;33(1):96–108.
Mark MR, Scadden DT, Wang Z, Gu Q, Goddard A. Godowski PJ: rse, a novel receptor-type tyrosine kinase with homology to Axl/Ufo, is expressed at high levels in the brain. J Biol Chem. 1994;269(14):10720–8.
Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001;293(5528):306–11.
Lai C, Gore M, Lemke G. Structure, expression, and activity of Tyro 3, a neural adhesion-related receptor tyrosine kinase. Oncogene. 1994;9(9):2567–78.
Katagiri M, Hakeda Y, Chikazu D, Ogasawara T, Takato T, Kumegawa M, Nakamura K, Kawaguchi H. Mechanism of stimulation of osteoclastic bone resorption through Gas6/Tyro 3, a receptor tyrosine kinase signaling, in mouse osteoclasts. J Biol Chem. 2001;276(10):7376–82.
Gal-Moscovici A, Scherzer P, Rubinger D, Weiss R, Dranitzki-Elhalel M, Popovtzer MM. Stimulation of osteoclastic bone resorption in a model of glycerol-induced acute renal failure: evidence for a parathyroid hormone-independent mechanism. Bone. 2002;31(4):488–91.
Angelillo-Scherrer A, de Frutos P, Aparicio C, Melis E, Savi P, Lupu F, Arnout J, Dewerchin M, Hoylaerts M, Herbert J, et al. Deficiency or inhibition of Gas6 causes platelet dysfunction and protects mice against thrombosis. Nat Med. 2001;7(2):215–21.
Scaltriti M, Elkabets M, Baselga J. Molecular Pathways: AXL, a Membrane Receptor Mediator of Resistance to Therapy. Clin Cancer Res. 2016;22(6):1313–7.
Meertens L, Labeau A, Dejarnac O, Cipriani S, Sinigaglia L, Bonnet-Madin L, Le Charpentier T, Hafirassou ML, Zamborlini A, Cao-Lormeau VM, et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017;18(2):324–33.
Fujimori T, Grabiec AM, Kaur M, Bell TJ, Fujino N, Cook PC, Svedberg FR, MacDonald AS, Maciewicz RA, Singh D, et al. The Axl receptor tyrosine kinase is a discriminator of macrophage function in the inflamed lung. Mucosal Immunol. 2015;8(5):1021–30.
Mudduluru G, Leupold JH, Stroebel P, Allgayer H. PMA up-regulates the transcription of Axl by AP-1 transcription factor binding to TRE sequences via the MAPK cascade in leukaemia cells. Biol Cell. 2010;103(1):21–33.
Rankin EB, Fuh KC, Castellini L, Viswanathan K, Finger EC, Diep AN, LaGory EL, Kariolis MS, Chan A, Lindgren D, et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci U S A. 2014;111(37):13373–8.
Mudduluru G, Vajkoczy P, Allgayer H. Myeloid zinc finger 1 induces migration, invasion, and in vivo metastasis through Axl gene expression in solid cancer. Mol Cancer Res. 2010;8(2):159–69.
Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131(6):1124–36.
Zagorska A, Traves PG, Lew ED, Dransfield I, Lemke G. Diversification of TAM receptor tyrosine kinase function. Nat Immunol. 2014;15(10):920–8.
Brand TM, Iida M, Stein AP, Corrigan KL, Braverman CM, Luthar N, Toulany M, Gill PS, Salgia R, Kimple RJ, et al. AXL mediates resistance to cetuximab therapy. Cancer Res. 2014;74(18):5152–64.
Yeh CY, Shin SM, Yeh HH, Wu TJ, Shin JW, Chang TY, Raghavaraju G, Lee CT, Chiang JH, Tseng VS, et al. Transcriptional activation of the Axl and PDGFR-alpha by c-Met through a ras- and Src-independent mechanism in human bladder cancer. BMC Cancer. 2011;11:139.
Mudduluru G, Ceppi P, Kumarswamy R, Scagliotti GV, Papotti M, Allgayer H. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene. 2011;30(25):2888–99.
Li R, Shi X, Ling F, Wang C, Liu J, Wang W, Li M. MiR-34a suppresses ovarian cancer proliferation and motility by targeting AXL. Tumour Biol. 2015;36(9):7277–83.
Ott M, Litzenburger UM, Sahm F, Rauschenbach KJ, Tudoran R, Hartmann C, Marquez VE, von Deimling A, Wick W, Platten M. Promotion of glioblastoma cell motility by enhancer of zeste homolog 2 (EZH2) is mediated by AXL receptor kinase. PLoS One. 2012;7(10):e47663.
Mudduluru G, Allgayer H. The human receptor tyrosine kinase Axl gene--promoter characterization and regulation of constitutive expression by Sp1, Sp3 and CpG methylation. Biosci Rep. 2008;28(3):161–76.
Valverde P. Effects of Gas6 and hydrogen peroxide in Axl ubiquitination and downregulation. Biochem Biophys Res Commun. 2005;333(1):180–5.
Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM, Graham DK. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood. 2007;109(3):1026–33.
O'Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B, Prokop C, Espinosa R 3rd, Le Beau MM, Earp HS, Liu ET. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. 1991;11(10):5016–31.
Lu Q, Gore M, Zhang Q, Camenisch T, Boast S, Casagranda F, Lai C, Skinner MK, Klein R, Matsushima GK, et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature. 1999;398(6729):723–8.
Mosesson Y, Shtiegman K, Katz M, Zwang Y, Vereb G, Szollosi J, Yarden Y. Endocytosis of receptor tyrosine kinases is driven by monoubiquitylation, not polyubiquitylation. J Biol Chem. 2003;278(24):21323–6.
Vajkoczy P, Knyazev P, Kunkel A, Capelle HH, Behrndt S, von Tengg-Kobligk H, Kiessling F, Eichelsbacher U, Essig M, Read TA, et al. Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc Natl Acad Sci U S A. 2006;103(15):5799–804.
Funakoshi H, Yonemasu T, Nakano T, Matumoto K, Nakamura T. Identification of Gas6, a putative ligand for Sky and Axl receptor tyrosine kinases, as a novel neurotrophic factor for hippocampal neurons. J Neurosci Res. 2002;68(2):150–60.
Kanlikilicer P, Ozpolat B, Aslan B, Bayraktar R, Gurbuz N, Rodriguez-Aguayo C, Bayraktar E, Denizli M, Gonzalez-Villasana V, Ivan C, et al. Therapeutic Targeting of AXL Receptor Tyrosine Kinase Inhibits Tumor Growth and Intraperitoneal Metastasis in Ovarian Cancer Models. Mol Ther Nucleic Acids. 2017;9:251–62.
Onken J, Torka R, Korsing S, Radke J, Krementeskaia I, Nieminen M, Bai X, Ullrich A, Heppner F, Vajkoczy P. Inhibiting receptor tyrosine kinase AXL with small molecule inhibitor BMS-777607 reduces glioblastoma growth, migration, and invasion in vitro and in vivo. Oncotarget. 2016;7(9):9876–89.
Hsieh MS, Yang PW, Wong LF, Lee JM. The AXL receptor tyrosine kinase is associated with adverse prognosis and distant metastasis in esophageal squamous cell carcinoma. Oncotarget. 2016;7(24):36956–70.
Hong CC, Lay JD, Huang JS, Cheng AL, Tang JL, Lin MT, Lai GM, Chuang SE. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008;268(2):314–24.
Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, Duan M, Torneros A, Yu J, Heckrodt TJ, et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010;70(4):1544–54.
Gjerdrum C, Tiron C, Hoiby T, Stefansson I, Haugen H, Sandal T, Collett K, Li S, McCormack E, Gjertsen BT, et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci U S A. 2010;107(3):1124–9.
Elkabets M, Pazarentzos E, Juric D, Sheng Q, Pelossof RA, Brook S, Benzaken AO, Rodon J, Morse N, Yan JJ, et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell. 2015;27(4):533–46.
Dufies M, Jacquel A, Belhacene N, Robert G, Cluzeau T, Luciano F, Cassuto JP, Raynaud S, Auberger P. Mechanisms of AXL overexpression and function in Imatinib-resistant chronic myeloid leukemia cells. Oncotarget. 2011;2(11):874–85.
Dransfield I, Farnworth S. Axl and Mer Receptor Tyrosine Kinases: Distinct and Nonoverlapping Roles in Inflammation and Cancer? Adv Exp Med Biol. 2016;930:113–32.
Debruyne DN, Bhatnagar N, Sharma B, Luther W, Moore NF, Cheung NK, Gray NS, George RE. ALK inhibitor resistance in ALK(F1174L)-driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene. 2016;35(28):3681–91.
Corno C, Gatti L, Lanzi C, Zaffaroni N, Colombo D, Perego P. Role of the Receptor Tyrosine Kinase Axl and its Targeting in Cancer Cells. Curr Med Chem. 2016;23(15):1496–512.
Burchert A, Attar EC, McCloskey P, Fridell YW, Liu ET. Determinants for transformation induced by the Axl receptor tyrosine kinase. Oncogene. 1998;16(24):3177–87.
Balaji K, Vijayaraghavan S, Diao L, Tong P, Fan Y, Carey JP, Bui TN, Warner S, Heymach JV, Hunt KK, et al. AXL Inhibition Suppresses the DNA Damage Response and Sensitizes Cells to PARP Inhibition in Multiple Cancers. Mol Cancer Res. 2017;15(1):45–58.
Maacha S, Hong J, von Lersner A, Zijlstra A, Belkhiri A. AXL Mediates Esophageal Adenocarcinoma Cell Invasion through Regulation of Extracellular Acidification and Lysosome Trafficking. Neoplasia. 2018;20(10):1008–22.
Caberoy NB, Alvarado G, Bigcas JL, Li W. Galectin-3 is a new MerTK-specific eat-me signal. J Cell Physiol. 2012;227(2):401–7.
Caberoy NB, Zhou Y, Li W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010;29(23):3898–910.
Wu X, Liu X, Koul S, Lee CY, Zhang Z, Halmos B. AXL kinase as a novel target for cancer therapy. Oncotarget. 2014;5(20):9546–63.
Varnum BC, Young C, Elliott G, Garcia A, Bartley TD, Fridell YW, Hunt RW, Trail G, Clogston C, Toso RJ, et al. Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature. 1995;373(6515):623–6.
Sasaki T, Knyazev PG, Clout NJ, Cheburkin Y, Gohring W, Ullrich A, Timpl R, Hohenester E. Structural basis for Gas6-Axl signalling. EMBO J. 2006;25(1):80–7.
Braunger J, Schleithoff L, Schulz AS, Kessler H, Lammers R, Ullrich A, Bartram CR, Janssen JW. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene. 1997;14(22):2619–31.
Fridell YW, Jin Y, Quilliam LA, Burchert A, McCloskey P, Spizz G, Varnum B, Der C, Liu ET. Differential activation of the Ras/extracellular-signal-regulated protein kinase pathway is responsible for the biological consequences induced by the Axl receptor tyrosine kinase. Mol Cell Biol. 1996;16(1):135–45.
Dent P. Crosstalk between ERK, AKT, and cell survival. Cancer Biol Ther. 2014;15(3):245–6.
Huang H, Liu H, Yan R, Hu M. PI3K/Akt and ERK/MAPK Signaling Promote Different Aspects of Neuron Survival and Axonal Regrowth Following Rat Facial Nerve Axotomy. Neurochem Res. 2017;42(12):3515–24.
Sen T, Tong P, Diao L, Li L, Fan Y, Hoff J, Heymach JV, Wang J, Byers LA. Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin Cancer Res. 2017;23(20):6239–53.
Kariolis MS, Miao YR, Diep A, Nash SE, Olcina MM, Jiang D, Jones DS 2nd, Kapur S. Mathews, II, Koong AC et al: Inhibition of the GAS6/AXL pathway augments the efficacy of chemotherapies. J Clin Invest. 2017;127(1):183–98.
Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. 2013;6(287):ra66.
Vouri M, Croucher DR, Kennedy SP, An Q, Pilkington GJ, Hafizi S. Axl-EGFR receptor tyrosine kinase hetero-interaction provides EGFR with access to pro-invasive signalling in cancer cells. Oncogenesis. 2016;5(10):e266.
May CD, Garnett J, Ma X, Landers SM, Ingram DR, Demicco EG, Al Sannaa GA, Vu T, Han L, Zhang Y, et al. AXL is a potential therapeutic target in dedifferentiated and pleomorphic liposarcomas. BMC Cancer. 2015;15:901.
Hasanbasic I, Cuerquis J, Varnum B, Blostein MD. Intracellular signaling pathways involved in Gas6-Axl-mediated survival of endothelial cells. Am J Physiol Heart Circ Physiol. 2004;287(3):H1207–13.
Han J, Tian R, Yong B, Luo C, Tan P, Shen J, Peng T. Gas6/Axl mediates tumor cell apoptosis, migration and invasion and predicts the clinical outcome of osteosarcoma patients. Biochem Biophys Res Commun. 2013;435(3):493–500.
Sinha S, Boysen J, Nelson M, Secreto C, Warner SL, Bearss DJ, Lesnick C, Shanafelt TD, Kay NE, Ghosh AK. Targeted Axl Inhibition Primes Chronic Lymphocytic Leukemia B Cells to Apoptosis and Shows Synergistic/Additive Effects in Combination with BTK Inhibitors. Clin Cancer Res. 2015;21(9):2115–26.
Bell JB, Eckerdt FD, Alley K, Magnusson LP, Hussain H, Bi Y, Arslan AD, Clymer J, Alvarez AA, Goldman S, et al. MNK Inhibition Disrupts Mesenchymal Glioma Stem Cells and Prolongs Survival in a Mouse Model of Glioblastoma. Mol Cancer Res. 2016;14(10):984–93.
Sainaghi PP, Castello L, Bergamasco L, Galletti M, Bellosta P, Avanzi GC. Gas6 induces proliferation in prostate carcinoma cell lines expressing the Axl receptor. J Cell Physiol. 2005;204(1):36–44.
Yuen HF, McCrudden CM, Huang YH, Tham JM, Zhang X, Zeng Q, Zhang SD, Hong W. TAZ expression as a prognostic indicator in colorectal cancer. PLoS One. 2013;8(1):e54211.
Koorstra JB, Karikari CA, Feldmann G, Bisht S, Rojas PL, Offerhaus GJ, Alvarez H, Maitra A. The Axl receptor tyrosine kinase confers an adverse prognostic influence in pancreatic cancer and represents a new therapeutic target. Cancer Biol Ther. 2009;8(7):618–26.
Lay JD, Hong CC, Huang JS, Yang YY, Pao CY, Liu CH, Lai YP, Lai GM, Cheng AL, Su IJ, et al. Sulfasalazine suppresses drug resistance and invasiveness of lung adenocarcinoma cells expressing AXL. Cancer Res. 2007;67(8):3878–87.
Tai KY, Shieh YS, Lee CS, Shiah SG, Wu CW. Axl promotes cell invasion by inducing MMP-9 activity through activation of NF-kappaB and Brg-1. Oncogene. 2008;27(29):4044–55.
Zhang YX, Knyazev PG, Cheburkin YV, Sharma K, Knyazev YP, Orfi L, Szabadkai I, Daub H, Keri G, Ullrich A. AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res. 2008;68(6):1905–15.
Xu MZ, Chan SW, Liu AM, Wong KF, Fan ST, Chen J, Poon RT, Zender L, Lowe SW, Hong W, et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene. 2011;30(10):1229–40.
Rankin EB, Fuh KC, Taylor TE, Krieg AJ, Musser M, Yuan J, Wei K, Kuo CJ, Longacre TA, Giaccia AJ. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010;70(19):7570–9.
Demarchi F, Verardo R, Varnum B, Brancolini C, Schneider C. Gas6 anti-apoptotic signaling requires NF-kappa B activation. J Biol Chem. 2001;276(34):31738–44.
Wang X, Saso H, Iwamoto T, Xia W, Gong Y, Pusztai L, Woodward WA, Reuben JM, Warner SL, Bearss DJ, et al. TIG1 promotes the development and progression of inflammatory breast cancer through activation of Axl kinase. Cancer Res. 2013;73(21):6516–25.
Leconet W, Chentouf M, du Manoir S, Chevalier C, Sirvent A, Ait-Arsa I, Busson M, Jarlier M, Radosevic-Robin N, Theillet C, et al. Therapeutic Activity of Anti-AXL Antibody against Triple-Negative Breast Cancer Patient-Derived Xenografts and Metastasis. Clin Cancer Res. 2017;23(11):2806–16.
Avilla E, Guarino V, Visciano C, Liotti F, Svelto M, Krishnamoorthy G, Franco R, Melillo RM. Activation of TYRO3/AXL tyrosine kinase receptors in thyroid cancer. Cancer Res. 2011;71(5):1792–804.
Li Y, Ye X, Tan C, Hongo JA, Zha J, Liu J, Kallop D, Ludlam MJ, Pei L. Axl as a potential therapeutic target in cancer: role of Axl in tumor growth, metastasis and angiogenesis. Oncogene. 2009;28(39):3442–55.
Shieh YS, Lai CY, Kao YR, Shiah SG, Chu YW, Lee HS, Wu CW. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia. 2005;7(12):1058–64.
Chen T, You Y, Jiang H, Wang ZZ. Epithelial-mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J Cell Physiol. 2017;232(12):3261–72.
Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119(6):1438–49.
Du B, Shim JS. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules. 2016;21(7). https://doi.org/10.3390/molecules21070965.
Nieto MA. Epithelial-Mesenchymal Transitions in development and disease: old views and new perspectives. Int J Dev Biol. 2009;53(8–10):1541–7.
Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166(1):21–45.
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90.
Goyette MA, Duhamel S, Aubert L, Pelletier A, Savage P, Thibault MP, Johnson RM, Carmeliet P, Basik M, Gaboury L, et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep. 2018;23(5):1476–90.
Cichon MA, Szentpetery Z, Caley MP, Papadakis ES, Mackenzie IC, Brennan CH, O'Toole EA. The receptor tyrosine kinase Axl regulates cell-cell adhesion and stemness in cutaneous squamous cell carcinoma. Oncogene. 2014;33(32):4185–92.
Asiedu MK, Beauchamp-Perez FD, Ingle JN, Behrens MD, Radisky DC, Knutson KL. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene. 2014;33(10):1316–24.
Singh M, Yelle N, Venugopal C, Singh SK. EMT: Mechanisms and therapeutic implications. Pharmacol Ther. 2018;182:80–94.
Lin JZ, Wang ZJ, De W, Zheng M, Xu WZ, Wu HF, Armstrong A, Zhu JG. Targeting AXL overcomes resistance to docetaxel therapy in advanced prostate cancer. Oncotarget. 2017;8(25):41064–77.
Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9(6):653–60.
Fridell YW, Villa J Jr, Attar EC, Liu ET. GAS6 induces Axl-mediated chemotaxis of vascular smooth muscle cells. J Biol Chem. 1998;273(12):7123–6.
Ramjiawan RR, Griffioen AW, Duda DG. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis. 2017;20(2):185–204.
Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964–7.
Rothlin CV, Lemke G. TAM receptor signaling and autoimmune disease. Curr Opin Immunol. 2010;22(6):740–6.
Ruan GX, Kazlauskas A. Lactate engages receptor tyrosine kinases Axl, Tie2, and vascular endothelial growth factor receptor 2 to activate phosphoinositide 3-kinase/Akt and promote angiogenesis. J Biol Chem. 2013;288(29):21161–72.
Holland SJ, Powell MJ, Franci C, Chan EW, Friera AM, Atchison RE, McLaughlin J, Swift SE, Pali ES, Yam G, et al. Multiple roles for the receptor tyrosine kinase axl in tumor formation. Cancer Res. 2005;65(20):9294–303.
Pinato DJ, Chowdhury S, Stebbing J. TAMing resistance to multi-targeted kinase inhibitors through Axl and Met inhibition. Oncogene. 2016;35(21):2684–6.
Linger RM, Keating AK, Earp HS, Graham DK. Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin Ther Targets. 2010;14(10):1073–90.
Gallicchio M, Mitola S, Valdembri D, Fantozzi R, Varnum B, Avanzi GC, Bussolino F. Inhibition of vascular endothelial growth factor receptor 2-mediated endothelial cell activation by Axl tyrosine kinase receptor. Blood. 2005;105(5):1970–6.
Xiao Y, Zhao H, Tian L, Nolley R, Diep AN, Ernst A, et al. S100A10 is a critical mediator of GAS6/AXL-induced angiogenesis in renal cell carcinoma. Cancer Res. 2019. https://doi.org/10.1158/0008-5472.CAN-19-1366
Ben-Batalla I, Erdmann R, Jorgensen H, Mitchell R, Ernst T, von Amsberg G, Schafhausen P, Velthaus JL, Rankin S, Clark RE, et al. Axl Blockade by BGB324 Inhibits BCR-ABL Tyrosine Kinase Inhibitor-Sensitive and -Resistant Chronic Myeloid Leukemia. Clin Cancer Res. 2017;23(9):2289–300.
Schoumacher M, Burbridge M. Key Roles of AXL and MER Receptor Tyrosine Kinases in Resistance to Multiple Anticancer Therapies. Curr Oncol Rep. 2017;19(3):19.
Kim KC, Baek SH, Lee C. Curcumin-induced downregulation of Axl receptor tyrosine kinase inhibits cell proliferation and circumvents chemoresistance in non-small lung cancer cells. Int J Oncol. 2015;47(6):2296–303.
Heckmann D, Maier P, Laufs S, Li L, Sleeman JP, Trunk MJ, Leupold JH, Wenz F, Zeller WJ, Fruehauf S, et al. The disparate twins: a comparative study of CXCR4 and CXCR7 in SDF-1alpha-induced gene expression, invasion and chemosensitivity of colon cancer. Clin Cancer Res. 2014;20(3):604–16.
Brand TM, Iida M, Stein AP, Corrigan KL, Braverman CM, Coan JP, Pearson HE, Bahrar H, Fowler TL, Bednarz BP, et al. AXL Is a Logical Molecular Target in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res. 2015;21(11):2601–12.
Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165(1):35–44.
Giles KM, Kalinowski FC, Candy PA, Epis MR, Zhang PM, Redfern AD, Stuart LM, Goodall GJ, Leedman PJ. Axl mediates acquired resistance of head and neck cancer cells to the epidermal growth factor receptor inhibitor erlotinib. Mol Cancer Ther. 2013;12(11):2541–58.
Tian Y, Zhang Z, Miao L, Yang Z, Yang J, Wang Y, Qian D, Cai H, Wang Y. Anexelekto (AXL) Increases Resistance to EGFR-TKI and Activation of AKT and ERK1/2 in Non-Small Cell Lung Cancer Cells. Oncol Res. 2016;24(5):295–303.
Mahadevan D, Cooke L, Riley C, Swart R, Simons B, Della Croce K, Wisner L, Iorio M, Shakalya K, Garewal H, et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene. 2007;26(27):3909–19.
Shen Y, Chen X, He J, Liao D, Zu X. Axl inhibitors as novel cancer therapeutic agents. Life Sci. 2018;198:99–111.
Taniguchi H, Yamada T, Wang R, Tanimura K, Adachi Y, Nishiyama A, Tanimoto A, Takeuchi S, Araujo LH, Boroni M, et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat Commun. 2019;10(1):259.
Macleod K, Mullen P, Sewell J, Rabiasz G, Lawrie S, Miller E, Smyth JF, Langdon SP. Altered ErbB receptor signaling and gene expression in cisplatin-resistant ovarian cancer. Cancer Res. 2005;65(15):6789–800.
Skinner HD, Giri U, Yang LP, Kumar M, Liu Y, Story MD, Pickering CR, Byers LA, Williams MD, Wang J, et al. Integrative Analysis Identifies a Novel AXL-PI3 Kinase-PD-L1 Signaling Axis Associated with Radiation Resistance in Head and Neck Cancer. Clin Cancer Res. 2017;23(11):2713–22.
Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, Halsey W, Sathe GM, Martin AM, Gilmer TM. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 2009;69(17):6871–8.
Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol. 2008;8(5):327–36.
Kurowska-Stolarska M, Alivernini S, Melchor EG, Elmesmari A, Tolusso B, Tange C, Petricca L, Gilchrist DS, Di Sante G, Keijzer C, et al. MicroRNA-34a dependent regulation of AXL controls the activation of dendritic cells in inflammatory arthritis. Nat Commun. 2017;8:15877.
Chan PY, Carrera Silva EA, De Kouchkovsky D, Joannas LD, Hao L, Hu D, Huntsman S, Eng C, Licona-Limon P, Weinstein JS, et al. The TAM family receptor tyrosine kinase TYRO3 is a negative regulator of type 2 immunity. Science. 2016;352(6281):99–103.
Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, Jamieson AM, Langdon WY, Ikeda F, Fededa JP, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. 2014;507(7493):508–12.
Nguyen KQ, Tsou WI, Kotenko S, Birge RB. TAM receptors in apoptotic cell clearance, autoimmunity, and cancer. Autoimmunity. 2013;46(5):294–7.
Weinger JG, Brosnan CF, Loudig O, Goldberg MF, Macian F, Arnett HA, Prieto AL, Tsiperson V, Shafit-Zagardo B. Loss of the receptor tyrosine kinase Axl leads to enhanced inflammation in the CNS and delayed removal of myelin debris during experimental autoimmune encephalomyelitis. J Neuroinflammation. 2011;8:49.
Sharif MN, Sosic D, Rothlin CV, Kelly E, Lemke G, Olson EN, Ivashkiv LB. Twist mediates suppression of inflammation by type I IFNs and Axl. J Exp Med. 2006;203(8):1891–901.
Bosurgi L, Bernink JH, Delgado Cuevas V, Gagliani N, Joannas L, Schmid ET, Booth CJ, Ghosh S, Rothlin CV. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc Natl Acad Sci U S A. 2013;110(32):13091–6.
Uribe DJ, Mandell EK, Watson A, Martinez JD, Leighton JA, Ghosh S, Rothlin CV. The receptor tyrosine kinase AXL promotes migration and invasion in colorectal cancer. PLoS One. 2017;12(7):e0179979.
Aguilera TA, Giaccia AJ. Molecular Pathways: Oncologic Pathways and Their Role in T-cell Exclusion and Immune Evasion-A New Role for the AXL Receptor Tyrosine Kinase. Clin Cancer Res. 2017;23(12):2928–33.
Prieto-Vila M, Takahashi RU, Usuba W, Kohama I, Ochiya T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int J Mol Sci. 2017;18(12). https://doi.org/10.3390/ijms18122574.
Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang CY, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526(7571):131–5.
Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17(5):548–58.
Jin Y, Nie D, Li J, Du X, Lu Y, Li Y, Liu C, Zhou J, Pan J. Gas6/AXL Signaling Regulates Self-Renewal of Chronic Myelogenous Leukemia Stem Cells by Stabilizing beta-Catenin. Clin Cancer Res. 2017;23(11):2842–55.
Yen SY, Chuang HM, Huang MH, Lin SZ, Chiou TW, Harn HJ. n-Butylidenephthalide Regulated Tumor Stem Cell Genes EZH2/AXL and Reduced Its Migration and Invasion in Glioblastoma. Int J Mol Sci. 2017;18(2). https://doi.org/10.3390/ijms18020372.
Ye X, Li Y, Stawicki S, Couto S, Eastham-Anderson J, Kallop D, Weimer R, Wu Y, Pei L. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene. 2010;29(38):5254–64.
Qi W, Cooke LS, Stejskal A, Riley C, Croce KD, Saldanha JW, Bearss D, Mahadevan D. MP470, a novel receptor tyrosine kinase inhibitor, in combination with Erlotinib inhibits the HER family/PI3K/Akt pathway and tumor growth in prostate cancer. BMC Cancer. 2009;9:142.
Vultur A, Buettner R, Kowolik C, Liang W, Smith D, Boschelli F, Jove R. SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells. Mol Cancer Ther. 2008;7(5):1185–94.
Dai Y, Siemann DW. BMS-777607, a small-molecule met kinase inhibitor, suppresses hepatocyte growth factor-stimulated prostate cancer metastatic phenotype in vitro. Mol Cancer Ther. 2010;9(6):1554–61.
Myers SH, Brunton VG, Unciti-Broceta A. AXL Inhibitors in Cancer: A Medicinal Chemistry Perspective. J Med Chem. 2016;59(8):3593–608.
Zhao Z, Wu H, Wang L, Liu Y, Knapp S, Liu Q, Gray NS. Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery? ACS Chem Biol. 2014;9(6):1230–41.
Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29(11):1046–51.
Rodon J, Postel-Vinay S, Hollebecque A, Nuciforo P, Azaro A, Cattan V, Marfai L, Sudey I, Brendel K, Delmas A, et al. First-in-human phase I study of oral S49076, a unique MET/AXL/FGFR inhibitor, in advanced solid tumours. Eur J Cancer. 2017;81:142–50.
Tibes R, Fine G, Choy G, Redkar S, Taverna P, Oganesian A, Sahai A, Azab M, Tolcher AW. A phase I, first-in-human dose-escalation study of amuvatinib, a multi-targeted tyrosine kinase inhibitor, in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71(2):463–71.
Vergote IB, Smith DC, Berger R, Kurzrock R, Vogelzang NJ, Sella A, Wheler J, Lee Y, Foster PG, Weitzman R, et al. A phase 2 randomised discontinuation trial of cabozantinib in patients with ovarian carcinoma. Eur J Cancer. 2017;83:229–36.
Daud A, Kluger HM, Kurzrock R, Schimmoller F, Weitzman AL, Samuel TA, Moussa AH, Gordon MS, Shapiro GI. Phase II randomised discontinuation trial of the MET/VEGF receptor inhibitor cabozantinib in metastatic melanoma. Br J Cancer. 2017;116(4):432–40.
Wakelee HA, Gettinger S, Engelman J, Janne PA, West H, Subramaniam DS, Leach J, Wax M, Yaron Y, Miles DR, et al. A phase Ib/II study of cabozantinib (XL184) with or without erlotinib in patients with non-small cell lung cancer. Cancer Chemother Pharmacol. 2017;79(5):923–32.
Leighl NB, Tsao MS, Liu G, Tu D, Ho C, Shepherd FA, Murray N, Goffin JR, Nicholas G, Sakashita S, et al. A phase I study of foretinib plus erlotinib in patients with previously treated advanced non-small cell lung cancer: Canadian cancer trials group IND.196. Oncotarget. 2017;8(41):69651–62.
Shah MA, Wainberg ZA, Catenacci DV, Hochster HS, Ford J, Kunz P, Lee FC, Kallender H, Cecchi F, Rabe DC, et al. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PLoS One. 2013;8(3):e54014.
Rayson D, Lupichuk S, Potvin K, Dent S, Shenkier T, Dhesy-Thind S, Ellard SL, Prady C, Salim M, Farmer P, et al. Canadian Cancer Trials Group IND197: a phase II study of foretinib in patients with estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2-negative recurrent or metastatic breast cancer. Breast Cancer Res Treat. 2016;157(1):109–16.
Ghosh AK, Secreto C, Boysen J, Sassoon T, Shanafelt TD, Mukhopadhyay D, Kay NE. The novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of nonreceptor kinases: implications for therapy. Blood. 2011;117(6):1928–37.
Chen F, Song Q, Yu Q. Axl inhibitor R428 induces apoptosis of cancer cells by blocking lysosomal acidification and recycling independent of Axl inhibition. Am J Cancer Res. 2018;8(8):1466–82.
Ben-Batalla I, Schultze A, Wroblewski M, Erdmann R, Heuser M, Waizenegger JS, Riecken K, Binder M, Schewe D, Sawall S, et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood. 2013;122(14):2443–52.
Mollard A, Warner SL, Call LT, Wade ML, Bearss JJ, Verma A, Sharma S, Vankayalapati H, Bearss DJ. Design, Synthesis and Biological Evaluation of a Series of Novel Axl Kinase Inhibitors. ACS Med Chem Lett. 2011;2(12):907–12.
Jimenez L, Wang J, Morrison MA, Whatcott C, Soh KK, Warner S, Bearss D, Jette CA, Stewart RA. Phenotypic chemical screening using a zebrafish neural crest EMT reporter identifies retinoic acid as an inhibitor of epithelial morphogenesis. Dis Model Mech. 2016;9(4):389–400.
Patel V, Keating MJ, Wierda WG, Gandhi V. Preclinical combination of TP-0903, an AXL inhibitor and B-PAC-1, a procaspase-activating compound with ibrutinib in chronic lymphocytic leukemia. Leuk Lymphoma. 2016;57(6):1494–7.
Aveic S, Corallo D, Porcu E, Pantile M, Boso D, Zanon C, Viola G, Sidarovich V, Mariotto E, Quattrone A, et al. TP-0903 inhibits neuroblastoma cell growth and enhances the sensitivity to conventional chemotherapy. Eur J Pharmacol. 2018;818:435–48.
Dantas-Barbosa C, Lesluyes T, Loarer FL, Chibon F, Treilleux I, Coindre JM, Meeus P, Brahmi M, Bally O, Ray-Coquard I, et al. Expression and role of TYRO3 and AXL as potential therapeutical targets in leiomyosarcoma. Br J Cancer. 2017;117(12):1787–97.
Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, Yamazaki S, Alton GR, Mroczkowski B, Los G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6(12 Pt 1):3314–22.
Sampson ER, Martin BA, Morris AE, Xie C, Schwarz EM, O'Keefe RJ, Rosier RN. The orally bioavailable met inhibitor PF-2341066 inhibits osteosarcoma growth and osteolysis/matrix production in a xenograft model. J Bone Miner Res. 2011;26(6):1283–94.
Cullinane C, Dorow DS, Jackson S, Solomon B, Bogatyreva E, Binns D, Young R, Arango ME, Christensen JG, McArthur GA, et al. Differential (18)F-FDG and 3′-deoxy-3′-(18)F-fluorothymidine PET responses to pharmacologic inhibition of the c-MET receptor in preclinical tumor models. J Nucl Med. 2011;52(8):1261–7.
Gong HC, Wang S, Mayer G, Chen G, Leesman G, Singh S, Beer DG. Signatures of drug sensitivity in nonsmall cell lung cancer. Int J Proteomics. 2011;2011:215496.
Blackhall F, Cappuzzo F. Crizotinib: from discovery to accelerated development to front-line treatment. Ann Oncol. 2016;27(Suppl 3):iii35–41.
Poon CC, Kelly JJ. Development of crizotinib, a rationally designed tyrosine kinase inhibitor for non-small cell lung cancer. Int J Cancer. 2017;140(9):1945–54.
Tucker ER, Danielson LS, Innocenti P, Chesler L. Tackling Crizotinib Resistance: The Pathway from Drug Discovery to the Pediatric Clinic. Cancer Res. 2015;75(14):2770–4.
Boschelli DH, Ye F, Wang YD, Dutia M, Johnson SL, Wu B, Miller K, Powell DW, Yaczko D, Young M, et al. Optimization of 4-phenylamino-3-quinolinecarbonitriles as potent inhibitors of Src kinase activity. J Med Chem. 2001;44(23):3965–77.
Lee HJ, Jeng YM, Chen YL, Chung L, Yuan RH. Gas6/Axl pathway promotes tumor invasion through the transcriptional activation of Slug in hepatocellular carcinoma. Carcinogenesis. 2014;35(4):769–75.
Golas JM, Arndt K, Etienne C, Lucas J, Nardin D, Gibbons J, Frost P, Ye F, Boschelli DH, Boschelli F. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003;63(2):375–81.
Golas JM, Lucas J, Etienne C, Golas J, Discafani C, Sridharan L, Boghaert E, Arndt K, Ye F, Boschelli DH, et al. SKI-606, a Src/Abl inhibitor with in vivo activity in colon tumor xenograft models. Cancer Res. 2005;65(12):5358–64.
Khoury HJ, Cortes JE, Kantarjian HM, Gambacorti-Passerini C, Baccarani M, Kim DW, Zaritskey A, Countouriotis A, Besson N, Leip E, et al. Bosutinib is active in chronic phase chronic myeloid leukemia after imatinib and dasatinib and/or nilotinib therapy failure. Blood. 2012;119(15):3403–12.
Garcia-Gutierrez V, Milojkovic D, Hernandez-Boluda JC, Claudiani S, Martin Mateos ML, Casado-Montero LF, Gonzalez G, Jimenez-Velasco A, Boque C, Martinez-Trillos A, et al. Safety and efficacy of bosutinib in fourth-line therapy of chronic myeloid leukemia patients. Ann Hematol. 2019;98(2):321–30.
Cortes JE, Gambacorti-Passerini C, Deininger MW, Mauro MJ, Chuah C, Kim DW, Dyagil I, Glushko N, Milojkovic D, le Coutre P, et al. Bosutinib Versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia: Results From the Randomized BFORE Trial. J Clin Oncol. 2018;36(3):231–7.
Park IK, Mishra A, Chandler J, Whitman SP, Marcucci G, Caligiuri MA. Inhibition of the receptor tyrosine kinase Axl impedes activation of the FLT3 internal tandem duplication in human acute myeloid leukemia: implications for Axl as a potential therapeutic target. Blood. 2013;121(11):2064–73.
Park IK, Mundy-Bosse B, Whitman SP, Zhang X, Warner SL, Bearss DJ, Blum W, Marcucci G, Caligiuri MA. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia. 2015;29(12):2382–9.
Mori M, Kaneko N, Ueno Y, Yamada M, Tanaka R, Saito R, Shimada I, Mori K, Kuromitsu S. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Invest New Drugs. 2017;35(5):556–65.
Usuki K, Sakura T, Kobayashi Y, Miyamoto T, Iida H, Morita S, Bahceci E, Kaneko M, Kusano M, Yamada S, et al. Clinical profile of gilteritinib in Japanese patients with relapsed/refractory acute myeloid leukemia: An open-label phase 1 study. Cancer Sci. 2018;109(10):3235–44.
Perl AE, Altman JK, Cortes J, Smith C, Litzow M, Baer MR, Claxton D, Erba HP, Gill S, Goldberg S, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 2017;18(8):1061–75.
Dhillon S. Gilteritinib: First Global Approval. Drugs. 2019;79(3):331–9.
Burbridge MF, Bossard CJ, Saunier C, Fejes I, Bruno A, Leonce S, Ferry G, Da Violante G, Bouzom F, Cattan V, et al. S49076 is a novel kinase inhibitor of MET, AXL, and FGFR with strong preclinical activity alone and in association with bevacizumab. Mol Cancer Ther. 2013;12(9):1749–62.
Clemenson C, Chargari C, Liu W, Mondini M, Ferte C, Burbridge MF, Cattan V, Jacquet-Bescond A, Deutsch E. The MET/AXL/FGFR Inhibitor S49076 Impairs Aurora B Activity and Improves the Antitumor Efficacy of Radiotherapy. Mol Cancer Ther. 2017;16(10):2107–19.
Viteri S, Chang GC, Chiari R, Cho BC, Ciardiello F, Curigliano G, et al. Combination of the S49076 with gefitinib in NSCLC patients progressing on EGFR-TKI and harboring MET/AXL dysregulation. Ann Oncol. 2018;29.
Zhao H, Luoto KR, Meng AX, Bristow RG. The receptor tyrosine kinase inhibitor amuvatinib (MP470) sensitizes tumor cells to radio- and chemo-therapies in part by inhibiting homologous recombination. Radiother Oncol. 2011;101(1):59–65.
Welsh JW, Mahadevan D, Ellsworth R, Cooke L, Bearss D, Stea B. The c-Met receptor tyrosine kinase inhibitor MP470 radiosensitizes glioblastoma cells. Radiat Oncol. 2009;4:69.
Mahadevan D, Theiss N, Morales C, Stejskal AE, Cooke LS, Zhu M, Kurtzman D, Swart R, Ong E, Qi W. Novel receptor tyrosine kinase targeted combination therapies for imatinib-resistant gastrointestinal stromal tumors (GIST). Oncotarget. 2015;6(4):1954–66.
Mita M, Gordon M, Rosen L, Kapoor N, Choy G, Redkar S, Taverna P, Oganesian A, Sahai A, Azab M, et al. Phase 1B study of amuvatinib in combination with five standard cancer therapies in adults with advanced solid tumors. Cancer Chemother Pharmacol. 2014;74(1):195–204.
Patyna S, Arrigoni C, Terron A, Kim TW, Heward JK, Vonderfecht SL, Denlinger R, Turnquist SE, Evering W. Nonclinical safety evaluation of sunitinib: a potent inhibitor of VEGF, PDGF, KIT, FLT3, and RET receptors. Toxicol Pathol. 2008;36(7):905–16.
Sun L, Liang C, Shirazian S, Zhou Y, Miller T, Cui J, Fukuda JY, Chu JY, Nematalla A, Wang X, et al. Discovery of 5-[5-fluoro-2-oxo-1,2- dihydroindol-(3Z)-ylidenemethyl]-2,4- dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J Med Chem. 2003;46(7):1116–9.
Kitagawa D, Yokota K, Gouda M, Narumi Y, Ohmoto H, Nishiwaki E, Akita K, Kirii Y. Activity-based kinase profiling of approved tyrosine kinase inhibitors. Genes Cells. 2013;18(2):110–22.
Nassif E, Thibault C, Vano Y, Fournier L, Mauge L, Verkarre V, Timsit MO, Mejean A, Tartour E, Oudard S. Sunitinib in kidney cancer: 10 years of experience and development. Expert Rev Anticancer Ther. 2017;17(2):129–42.
Braconi C, Bracci R, Cellerino R. Molecular targets in Gastrointestinal Stromal Tumors (GIST) therapy. Curr Cancer Drug Targets. 2008;8(5):359–66.
Gomez-Saez JM. Sunitinib for the treatment of thyroid cancer. Expert Opin Investig Drugs. 2016;25(11):1345–52.
Young K, Iyer R, Morganstein D, Chau I, Cunningham D, Starling N. Pancreatic neuroendocrine tumors: a review. Future Oncol. 2015;11(5):853–64.
Abdel-Aziz AK, Abdel-Naim AB, Shouman S, Minucci S, Elgendy M. From Resistance to Sensitivity: Insights and Implications of Biphasic Modulation of Autophagy by Sunitinib. Front Pharmacol. 2017;8:718.
Oslob JD, Romanowski MJ, Allen DA, Baskaran S, Bui M, Elling RA, Flanagan WM, Fung AD, Hanan EJ, Harris S, et al. Discovery of a potent and selective aurora kinase inhibitor. Bioorg Med Chem Lett. 2008;18(17):4880–4.
Arbitrario JP, Belmont BJ, Evanchik MJ, Flanagan WM, Fucini RV, Hansen SK, Harris SO, Hashash A, Hoch U, Hogan JN, et al. SNS-314, a pan-Aurora kinase inhibitor, shows potent anti-tumor activity and dosing flexibility in vivo. Cancer Chemother Pharmacol. 2010;65(4):707–17.
Kim YS, Keyser SG, Schneekloth JS Jr. Synthesis of 2′,3′,4′-trihydroxyflavone (2-D08), an inhibitor of protein sumoylation. Bioorg Med Chem Lett. 2014;24(4):1094–7.
Fujino N, Kubo H, Maciewicz RA. Phenotypic screening identifies Axl kinase as a negative regulator of an alveolar epithelial cell phenotype. Lab Invest. 2017;97(9):1047–62.
Zhang W, DeRyckere D, Hunter D, Liu J, Stashko MA, Minson KA, Cummings CT, Lee M, Glaros TG, Newton DL, et al. UNC2025, a potent and orally bioavailable MER/FLT3 dual inhibitor. J Med Chem. 2014;57(16):7031–41.
Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19(1):279–90.
Christoph S, Deryckere D, Schlegel J, Frazer JK, Batchelor LA, Trakhimets AY, Sather S, Hunter DM, Cummings CT, Liu J, et al. UNC569, a novel small-molecule mer inhibitor with efficacy against acute lymphoblastic leukemia in vitro and in vivo. Mol Cancer Ther. 2013;12(11):2367–77.
Liu J, Yang C, Simpson C, Deryckere D, Van Deusen A, Miley MJ, Kireev D, Norris-Drouin J, Sather S, Hunter D, et al. Discovery of Novel Small Molecule Mer Kinase Inhibitors for the Treatment of Pediatric Acute Lymphoblastic Leukemia. ACS Med Chem Lett. 2012;3(2):129–34.
Torres KE, Zhu QS, Bill K, Lopez G, Ghadimi MP, Xie X, Young ED, Liu J, Nguyen T, Bolshakov S, et al. Activated MET is a molecular prognosticator and potential therapeutic target for malignant peripheral nerve sheath tumors. Clin Cancer Res. 2011;17(12):3943–55.
Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10(12):2298–308.
You WK, Sennino B, Williamson CW, Falcon B, Hashizume H, Yao LC, Aftab DT, McDonald DM. VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res. 2011;71(14):4758–68.
Viola D, Cappagli V, Elisei R. Cabozantinib (XL184) for the treatment of locally advanced or metastatic progressive medullary thyroid cancer. Future Oncol. 2013;9(8):1083–92.
Elisei R, Schlumberger MJ, Muller SP, Schoffski P, Brose MS, Shah MH, Licitra L, Jarzab B, Medvedev V, Kreissl MC, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31(29):3639–46.
Powles T, Motzer RJ, Escudier B, Pal S, Kollmannsberger C, Pikiel J, Gurney H, Rha SY, Park SH, Geertsen PF, et al. Outcomes based on prior therapy in the phase 3 METEOR trial of cabozantinib versus everolimus in advanced renal cell carcinoma. Br J Cancer. 2018;119(6):663–9.
Motzer RJ, Escudier B, Powles T, Scheffold C, Choueiri TK. Long-term follow-up of overall survival for cabozantinib versus everolimus in advanced renal cell carcinoma. Br J Cancer. 2018;118(9):1176–8.
Choueiri TK, Escudier B, Powles T, Tannir NM, Mainwaring PN, Rini BI, Hammers HJ, Donskov F, Roth BJ, Peltola K, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016;17(7):917–27.
Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F, Hammers H, Hutson TE, Lee JL, Peltola K, et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373(19):1814–23.
Kelley RK, Verslype C, Cohn AL, Yang TS, Su WC, Burris H, Braiteh F, Vogelzang N, Spira A, Foster P, et al. Cabozantinib in hepatocellular carcinoma: results of a phase 2 placebo-controlled randomized discontinuation study. Ann Oncol. 2017;28(3):528–34.
Spigel DR, Ervin TJ, Ramlau RA, Daniel DB, Goldschmidt JH Jr, Blumenschein GR Jr, Krzakowski MJ, Robinet G, Godbert B, Barlesi F, et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2013;31(32):4105–14.
Abou-Alfa GK, Meyer T, Cheng AL, El-Khoueiry AB, Rimassa L, Ryoo BY, Cicin I, Merle P, Chen Y, Park JW, et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N Engl J Med. 2018;379(1):54–63.
Schoffski P, Gordon M, Smith DC, Kurzrock R, Daud A, Vogelzang NJ, Lee Y, Scheffold C, Shapiro GI. Phase II randomised discontinuation trial of cabozantinib in patients with advanced solid tumours. Eur J Cancer. 2017;86:296–304.
Tolaney SM, Nechushtan H, Ron IG, Schoffski P, Awada A, Yasenchak CA, Laird AD, O'Keeffe B, Shapiro GI, Winer EP. Cabozantinib for metastatic breast carcinoma: results of a phase II placebo-controlled randomized discontinuation study. Breast Cancer Res Treat. 2016;160(2):305–12.
Smith DC, Smith MR, Sweeney C, Elfiky AA, Logothetis C, Corn PG, Vogelzang NJ, Small EJ, Harzstark AL, Gordon MS, et al. Cabozantinib in patients with advanced prostate cancer: results of a phase II randomized discontinuation trial. J Clin Oncol. 2013;31(4):412–9.
Hellerstedt BA, Vogelzang NJ, Kluger HM, Yasenchak CA, Aftab DT, Ramies DA, Gordon MS, Lara P Jr. Results of a Phase II Placebo-controlled Randomized Discontinuation Trial of Cabozantinib in Patients with Non-small-cell Lung Carcinoma. Clin Lung Cancer. 2019;20(2):74–81 e71.
Matulonis UA, Sill MW, Makker V, Mutch DG, Carlson JW, Darus CJ, et al. A randomized phase II study of cabozantinib versus weekly paclitaxel in the treatment of persistent or recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol Oncol. 2018. https://doi.org/10.1016/j.ygyno.2018.12.008.
Schroeder GM, An Y, Cai ZW, Chen XT, Clark C, Cornelius LA, Dai J, Gullo-Brown J, Gupta A, Henley B, et al. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl )-2-oxo-1,2-dihydropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the Met kinase superfamily. J Med Chem. 2009;52(5):1251–4.
Sharma S, Zeng JY, Zhuang CM, Zhou YQ, Yao HP, Hu X, Zhang R, Wang MH. Small-molecule inhibitor BMS-777607 induces breast cancer cell polyploidy with increased resistance to cytotoxic chemotherapy agents. Mol Cancer Ther. 2013;12(5):725–36.
Kim KS, Zhang L, Schmidt R, Cai ZW, Wei D, Williams DK, Lombardo LJ, Trainor GL, Xie D, Zhang Y, et al. Discovery of pyrrolopyridine-pyridone based inhibitors of Met kinase: synthesis, X-ray crystallographic analysis, and biological activities. J Med Chem. 2008;51(17):5330–41.
Dai Y, Bae K, Pampo C, Siemann DW. Impact of the small molecule Met inhibitor BMS-777607 on the metastatic process in a rodent tumor model with constitutive c-Met activation. Clin Exp Metastasis. 2012;29(3):253–61.
Cheng CT, Chen YY, Wu RC, Tsai CY, Chiang KC, Yeh TS, Chen MH, Yeh CN. METRON dual inhibitor, BMS777607, suppresses cholangiocarcinoma cell growth, and METRON upregulation indicates worse prognosis for intrahepatic cholangiocarcinoma patients. Oncol Rep. 2018;40(3):1411–21.
Nurhayati RW, Ojima Y, Taya M. BMS-777607 promotes megakaryocytic differentiation and induces polyploidization in the CHRF-288-11 cells. Hum Cell. 2015;28(2):65–72.
Yan SB, Um SL, Peek VL, Stephens JR, Zeng W, Konicek BW, Liu L, Manro JR, Wacheck V, Walgren RA. MET-targeting antibody (emibetuzumab) and kinase inhibitor (merestinib) as single agent or in combination in a cancer model bearing MET exon 14 skipping. Invest New Drugs. 2018;36(4):536–44.
Yan SB, Peek VL, Ajamie R, Buchanan SG, Graff JR, Heidler SA, Hui YH, Huss KL, Konicek BW, Manro JR, et al. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Invest New Drugs. 2013;31(4):833–44.
Barat S, Bozko P, Chen X, Scholta T, Hanert F, Gotze J, Malek NP, Wilkens L, Plentz RR. Targeting c-MET by LY2801653 for treatment of cholangiocarcinoma. Mol Carcinog. 2016;55(12):2037–50.
Wu W, Bi C, Credille KM, Manro JR, Peek VL, Donoho GP, Yan L, Wijsman JA, Yan SB, Walgren RA. Inhibition of tumor growth and metastasis in non-small cell lung cancer by LY2801653, an inhibitor of several oncokinases, including MET. Clin Cancer Res. 2013;19(20):5699–710.
Kawada I, Hasina R, Arif Q, Mueller J, Smithberger E, Husain AN, Vokes EE, Salgia R. Dramatic antitumor effects of the dual MET/RON small-molecule inhibitor LY2801653 in non-small cell lung cancer. Cancer Res. 2014;74(3):884–95.
Qian F, Engst S, Yamaguchi K, Yu P, Won KA, Mock L, Lou T, Tan J, Li C, Tam D, et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009;69(20):8009–16.
Martinelli E, Martini G, Cardone C, Troiani T, Liguori G, Vitagliano D, Napolitano S, Morgillo F, Rinaldi B, Melillo RM, et al. AXL is an oncotarget in human colorectal cancer. Oncotarget. 2015;6(27):23281–96.
Eder JP, Shapiro GI, Appleman LJ, Zhu AX, Miles D, Keer H, Cancilla B, Chu F, Hitchcock-Bryan S, Sherman L, et al. A phase I study of foretinib, a multi-targeted inhibitor of c-Met and vascular endothelial growth factor receptor 2. Clin Cancer Res. 2010;16(13):3507–16.
Choueiri TK, Vaishampayan U, Rosenberg JE, Logan TF, Harzstark AL, Bukowski RM, Rini BI, Srinivas S, Stein MN, Adams LM, et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013;31(2):181–6.
Yau TCC, Lencioni R, Sukeepaisarnjaroen W, Chao Y, Yen CJ, Lausoontornsiri W, Chen PJ, Sanpajit T, Camp A, Cox DS, et al. A Phase I/II Multicenter Study of Single-Agent Foretinib as First-Line Therapy in Patients with Advanced Hepatocellular Carcinoma. Clin Cancer Res. 2017;23(10):2405–13.
Patwardhan PP, Ivy KS, Musi E, de Stanchina E, Schwartz GK. Significant blockade of multiple receptor tyrosine kinases by MGCD516 (Sitravatinib), a novel small molecule inhibitor, shows potent anti-tumor activity in preclinical models of sarcoma. Oncotarget. 2016;7(4):4093–109.
Du W, Huang H, Sorrelle N, Brekken RA. Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI Insight. 2018;3(21). https://doi.org/10.1172/jci.insight.124184
Bonfils C, Beaulieu N, Fournel M, Ste-Croix H, Besterman JM, Maroun CR. The combination of MGCD265, a Met/VEGFR inhibitor in clinical development, and erlotinib potently inhibits tumor growth by altering multiple pathways including glycolysis. Cancer Res. 2012;72. https://doi.org/10.1158/1538-7445.Am2012-1790.
Do KT, Macconaill L, Dubuc A, Chen I, Chao R, Tassell V, Christensen J, Shapiro GI, Sholl LM. Evaluation of the MET/AXL Receptor Tyrosine Kinase (RTK) Inhibitor MGCD265 in a Patient with Metastatic Non-Small Cell Lung Cancer (NSCLC) Harboring AXL Amplification. J Thorac Oncol. 2015;10(9):S797.
Linklater ES, Tovar EA, Essenburg CJ, Turner L, Madaj Z, Winn ME, Melnik MK, Korkaya H, Maroun CR, Christensen JG, et al. Targeting MET and EGFR crosstalk signaling in triple-negative breast cancers. Oncotarget. 2016;7(43):69903–15.
Yokoyama Y, Lew ED, Seelige R, Tindall EA, Walsh C, Fagan PC, et al. Immuno-Oncological Efficacy of RXDX-106, a Novel Small Molecule Inhibitor of the TAM (TYRO3, AXL, MER) Family of Kinases. Cancer Res. 2019. https://doi.org/10.1158/0008-5472.CAN-18-2022
Chan WW, Wise SC, Kaufman MD, Ahn YM, Ensinger CL, Haack T, Hood MM, Jones J, Lord JW, Lu WP, et al. Conformational control inhibition of the BCR-ABL1 tyrosine kinase, including the gatekeeper T315I mutant, by the switch-control inhibitor DCC-2036. Cancer Cell. 2011;19(4):556–68.
Shen Y, Zhang W, Liu J, He J, Cao R, Chen X, Peng X, Xu H, Zhao Q, Zhong J, et al. Therapeutic activity of DCC-2036, a novel tyrosine kinase inhibitor, against triple-negative breast cancer patient-derived xenografts by targeting AXL/MET. Int J Cancer. 2019;144(3):651–64.
Rho JK, Choi YJ, Kim SY, Kim TW, Choi EK, Yoon SJ, Park BM, Park E, Bae JH, Choi CM, et al. MET and AXL inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of MET or AXL activation. Cancer Res. 2014;74(1):253–62.
Shin JS, Hong SW, Moon JH, Kim JS, Jung KA, Kim SM, Lee DH, Kim I, Yoon SJ, Lee CG, et al. NPS-1034, a novel MET inhibitor, inhibits the activated MET receptor and its constitutively active mutants. Invest New Drugs. 2014;32(3):389–99.
Sawyers CL. Opportunities and challenges in the development of kinase inhibitor therapy for cancer. Genes Dev. 2003;17(24):2998–3010.
Leconet W, Larbouret C, Chardes T, Thomas G, Neiveyans M, Busson M, Jarlier M, Radosevic-Robin N, Pugniere M, Bernex F, et al. Preclinical validation of AXL receptor as a target for antibody-based pancreatic cancer immunotherapy. Oncogene. 2014;33(47):5405–14.
Liu R, Gong M, Li X, Zhou Y, Gao W, Tulpule A, Chaudhary PM, Jung J, Gill PS. Induction, regulation, and biologic function of Axl receptor tyrosine kinase in Kaposi sarcoma. Blood. 2010;116(2):297–305.
Wang W, Zhao J, Wen X, Lin CC, Li J, Huang Q, Yu Y, Lin SY, Li C. MicroPET/CT Imaging of AXL Downregulation by HSP90 Inhibition in Triple-Negative Breast Cancer. Contrast Media Mol Imaging. 2017;2017:1686525.
Boshuizen J, Koopman LA, Krijgsman O, Shahrabi A, van den Heuvel EG, Ligtenberg MA, Vredevoogd DW, Kemper K, Kuilman T, Song JY, et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat Med. 2018;24(2):203–12.
Cho JH, Okuma A, Al-Rubaye D, Intisar E, Junghans RP, Wong WW. Engineering Axl specific CAR and SynNotch receptor for cancer therapy. Sci Rep. 2018;8(1):3846.
Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157(2):220–33.
Cerchia L, Esposito CL, Camorani S, Rienzo A, Stasio L, Insabato L, Affuso A, de Franciscis V. Targeting Axl with an high-affinity inhibitory aptamer. Mol Ther. 2012;20(12):2291–303.
Esposito CL, Catuogno S, de Franciscis V, Cerchia L. New insight into clinical development of nucleic acid aptamers. Discov Med. 2011;11(61):487–96.
Russo V, Paciocco A, Affinito A, Roscigno G, Fiore D, Palma F, Galasso M, Volinia S, Fiorelli A, Esposito CL, et al. Aptamer-miR-34c Conjugate Affects Cell Proliferation of Non-Small-Cell Lung Cancer Cells. Mol Ther Nucleic Acids. 2018;13:334–46.
O'Bryan JP, Fridell YW, Koski R, Varnum B, Liu ET. The transforming receptor tyrosine kinase, Axl, is post-translationally regulated by proteolytic cleavage. J Biol Chem. 1995;270(2):551–7.
Costa M, Bellosta P, Basilico C. Cleavage and release of a soluble form of the receptor tyrosine kinase ARK in vitro and in vivo. J Cell Physiol. 1996;168(3):737–44.
Kariolis MS, Miao YR, Jones DS 2nd, Kapur S, Mathews GAJ II, Cochran JR. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat Chem Biol. 2014;10(11):977–83.
Dengler M, Staufer K, Huber H, Stauber R, Bantel H, Weiss KH, Starlinger P, Pock H, Kloters-Plachky P, Gotthardt DN, et al. Soluble Axl is an accurate biomarker of cirrhosis and hepatocellular carcinoma development: results from a large scale multicenter analysis. Oncotarget. 2017;8(28):46234–48.
Lee YJ, Kim SY, Lee C. Axl is a novel target of celastrol that inhibits cell proliferation and migration, and increases the cytotoxicity of gefitinib in EGFR mutant nonsmall cell lung cancer cells. Mol Med Rep. 2019. https://doi.org/10.3892/mmr.2019.9957.
Paccez JD, Duncan K, Sekar D, Correa RG, Wang Y, Gu X, Bashin M, Chibale K, Libermann TA, Zerbini LF. Dihydroartemisinin inhibits prostate cancer via JARID2/miR-7/miR-34a-dependent downregulation of Axl. Oncogenesis. 2019;8(3):14.
Tan L, Zhang Z, Gao D, Chan S, Luo J, Tu ZC, Zhang ZM, Ding K, Ren X, Lu X. Quinolone antibiotic derivatives as new selective Axl kinase inhibitors. Eur J Med Chem. 2019;166:318–27.
Kim S, Kim KC, Lee C. Mistletoe (Viscum album) extract targets Axl to suppress cell proliferation and overcome cisplatin- and erlotinib-resistance in non-small cell lung cancer cells. Phytomedicine. 2017;36:183–93.
Acknowledgements
Not applicable.
Authors’ contributors
WXW provided the idea. ZCJ wrote the article. WYQ helped with the final revision of the article. All authors reviewed the manuscript and approved the final manuscript.
Funding
This work is supported by the National Natural Science Foundation of China (No. 81602492), the National Key Research, Development Program of China (No.2016YFA0201402) and the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (No. 2018ZX09733001) and by the Excellent Youth Foundation of Sichuan Scientific Committee Grant in China (No.2019JDJQ008).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhu, C., Wei, Y. & Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer 18, 153 (2019). https://doi.org/10.1186/s12943-019-1090-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-019-1090-3