
Abstract
Background
The epigenetic regulator additional sex combs-like 1 (ASXL1) is an adverse prognostic factor in acute myeloid leukemia (AML). However, the mutational spectrum and prognostic factors of ASXL1-mutated (ASXL1+) AML are largely unknown. We aim to evaluate the risk factors influencing the prognosis of ASXL1+ AML.
Methods
We performed next-generation sequencing (NGS) in 1047 cases of de novo AML and discovered 91 ASXL1+ AML (8.7%). The Log-Rank test and Kaplan-Meier were used to evaluate survival rate, and the Cox regression model was used to analyze multivariate analysis.
Results
In a total of 91 ASXL1+ AML, 86% had one or more co-mutations. The factors that had adverse impact on overall survival (OS) and event-free survival (EFS) are defined as high risk factors, including age ≥ 60 years, WBC count ≥ 50 × 109/L, FLT3-ITD mutations, RUNX1 mutations, and absence of AML1-ETO fusion gene. ASXL1 mutations without any risk factor were classified as single-hit ASXL1+ AML; ASXL1 mutations accompanied with one of the risk factors was referred to as double-hit ASXL1+ AML; ASXL1 mutations with two or more of the risk factors were designated as triple-hit ASXL1+ AML. The combination of these risk factors had a negative influence on the prognosis of ASXL1+ AML. The median OS was not attained in single-hit ASXL1+ AML, 29.53 months in double-hit ASXL1+ AML, and 6.67 months in triple-hit ASXL1+ AML (P = 0.003). The median EFS was not attained in single-hit ASXL1+ AML, 29.53 months in double-hit ASXL1+ AML, and 5.47 months in triple-hit ASXL1+ AML (P = 0.002). Allogenic hematopoietic stem cell transplantation (allo-HSCT) improved the prognosis of double/triple-hit ASXL1+ AML patients.
Conclusions
Our study provided new insights into the mutational spectrum and prognostic factors of ASXL1+ AML patients. Our primary data suggest that the risk factors in ASXL1+ AML contribute to the poor outcome of these patients. The management of ASXL1+ AML patients should be based on the risk factors and allo-HSCT is highly recommended for consolidation.
Similar content being viewed by others

Background
Acute myeloid leukemia (AML) is a group of hematological malignancies with high heterogeneity [1, 2]. Advances in individualized induction regimens with targeted agents and hematopoietic stem cell transplantation (HSCT) for consolidation have significantly improved the results of AML patients. However, the prognosis in some AML cases remains unsatisfactory. Recurrent chromosomal abnormalities and gene mutations have been implicated in leukemogenesis and are employed in the clinic for risk-adopted AML therapy [3]. The favorable risk factors are t(8;21), inv(16)/t(16;16), t(15;17), and CEBPA double mutations and NPM1 mutations, but the adverse risk factors are t(9;22) and mutations in FLT3-ITD, RUNX1, and AXSL1 [4, 5]. It has been established that not all molecular alterations have prognostic and therapeutic implications in AML. The mutations of CEBPA showed a favorable prognostic impact on AML only when the mutations occurred at both alleles [6]. The FLT3-ITD mutations had a negative prognostic impact when the ratio of the mutant alleles to wild alleles was more than 0.5 [7, 8]. The beneficial prognostic effects of t(8;21) and inv(16)/t(16;16) can be reversed by co-occurring with c-KIT mutations [9], and the same goes for NPM1 co-occurring with FLT3-ITD mutations [10]. Agents that target mutations, such as midostaurin on FLT3-ITD, can rescue patients from unfavorable outcomes [11, 12]. Based on the understanding of gene mutations in the prognosis of AML, hematologists have used innovative and targeted agents in chemotherapy to improve the outcome of these patients [13]. However, some patients may have multiple gene mutations or risk factors simultaneously. The interaction between mutated genes and other risk factors may affect the prognosis of AML patients. For instance, coexistence of ASXL1 and SRSF2 mutations may increase the risk of death in AML patients [14]. Therefore, it is crucial to make precise risk stratification to guide the managements of AML patients. Further research is needed to determine the interaction of co-mutated genes and clinical risk factors in patients carrying certain mutations, such as AXSL1, on the prognosis and treatment options.
ASXL1 is the human homologue of the Drosophila Additional sex combs (Asx) [2]. The ASXL family consists of three members (ASXL1, ASXL2, and ASXL3) with conserved domain structures consisting of ASXN, ASXH, ASXM1, ASXM2, and a PHD finger [15]. ASXL1 encodes a chromatin binding protein of the polycomb group and trithorax complex family [16, 17], which may be involved in epigenetic regulation. ASXL1 is located on chromosome 20q11. ASXL1 acts as a coactivator for the retinoid receptors including retinoic acid receptor (RAR) and retinoid X receptor through binding with steroid receptor coactivator-1 [18]. Moreover, ASXL1 also cooperates with heterochromatin protein-1 and histone H3 demethylase LSD1 to regulate histone methylation and repress retinoic acid-receptor activity [19]. Germline mutations of ASXL1 and ASXL3 can be seen in individuals with congenital abnormalities, such as Bainbridge–Ropers syndrome, while somatic truncation mutants of all three ASXL family members are found in human cancer [15]. ASXL1 is frequently mutated in patients with different types of myeloid malignancies, including myelodysplastic syndromes (MDS), myeloproliferative neoplasms, chronic myelomonocytic leukemia, and AML with MDS-related alterations [20]. ASXL1 mutations are commonly associated with aggressive behaviors and a poor clinical prognosis across the spectrum of malignant myeloid diseases [21]. In mouse model experiments, ASXL1 silencing together with oncogenic NRasG12D generates hepatosplenomegaly and progressive anemia, emphasizing ASXL1’s function in myeloid malignancies [2]. Although the adverse prognostic value of ASXL1 mutations in AML is obvious, previous studies often focused on comparing the difference between mutated and wild-type ASXL1 patients. However, the impact of other factors such as variant allele frequency (VAF) and companion gene mutations (co-mutations) on the prognosis of ASXL1+ AML needs to be evaluated.
In this study, we comprehensively investigated the mutational spectrum and prognostic factors of ASXL1+ AML. We also analyzed the interaction of molecular profiles of gene mutation and clinical risk factors on the survival of ASXL1+ AML patients. Our data demonstrated that the addition of risk factors to ASXL1 mutations were associated with the adverse outcome of AML patients. Meanwhile, Allo-HSCT and AML/ETO fusion gene improved the survival of ASXL1+ AML patients. In conclusion, our data provide new evidence for precise risk stratification and optimal treatments of ASXL1+ AML.
Subjects and methods
Patients
Between May 2016 and January 2020, 1047 cases of de novo AML were examined with next-generation sequencing (NGS) at the First Affiliated Hospital of Zhengzhou University. A total of 91 cases with ASXL1+ AML were identified and included in the research. The WHO 2016 edition of classification of myeloid neoplasms and acute leukemia [22] was used to make the diagnosis and classification of AML. According to the 2017 ELN guideline for adult acute myeloid leukemia [4], patients were categorized into three risk groups: favorable-risk, intermediate-risk, and adverse-risk. This study was approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University. Following the Declaration of Helsinki, all patients or their legal guardians gave their informed permission.
Treatment protocols
All-trans retinoic acid and arsenic trioxide-based chemotherapy was used for induction and consolidation therapy in individuals with acute promyelocytic leukemia (APL). Induction chemotherapy regimens for non-APL patients included the DA, IA, and MA regimens, which consisted of a standard dose of cytarabine (Ara-C; 100 mg/m2/day for 7 days) combined with daunorubicin (60 mg/m2/day for 3 days) or idarubicin (12 mg/m2/day for 3 days) or mitoxantrone (10 mg/m2/day for 3 days). Patients were given cytarabine (2–3 g/m2, once every 12 h for 3 days)-based chemotherapy after remission. The chemotherapy consolidation for older patients was chosen on an individual basis by the specialists. As part of the consolidation process, 12 patients received allo-HSCT. The actual therapy was chosen based on both the doctor’s suggestion and the patient’s desire. The final follow-up for surviving patients occurred in January 2021.
Fusion genes detection
Fresh bone marrow samples were collected using an Ethylene Diamine Tetraacetic Acid (EDTA) anticoagulant tube. Mononuclear cells were extracted by density gradient centrifugation. RNA was extracted using the standard TRIzol technique (Invitrogen, Carlsbad, CA, USA) and reverse-transcribed into cDNA using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Fusion genes were detected by real-time quantitative PCR using Multiplex RT-PCR Fusion Gene Kits (Rightongene, Shanghai, China). A panel of forty-three fusion genes was screened, including MLL-(AF4, AF6, AF9, AF10, AF17, AF1q, AF1p, AFX, ELL, SEPT6, ENL), NUP98-(HoxA11, HoxA13, PMX1), (NPM, F1P1L1, PML, PRKAR1A, STAT5b, NUMA1, PLZF)-RARα, (ETV6, FIPIL1)-PDGFRA, AML1-(ETO, MTG16, MDS1/EV11), TEL-(JAK2, AML1, ABL), NPM-(ALK, MLF), (DEK, SET)-CAN, SIL-TAL1, E2A-HLF, TEL-PDGFRB, TLS-ERG, CBFβ-MYH11, BCR-ABL, E2A-PBX1.
Next-generation sequencing
The mutational hotspots or whole coding regions of 22 genes were assessed by next-generation sequencing, including FLT3, NPM1, KIT, CEBPA, DNMT3A, IDH1, IDH2, TET2, EZH2, RUNX1, ASXL1, PHF6, TP53, SF3B1, SRSF2, U2AF1, ZRSR2, NRAS, CBL, SETBP1, ETV6, and JAK2. The detection was performed utilizing a Rightongene AML/MDS/MPN Sequencing Panel (Rightongene, Shanghai, China) on an Illumina MiSeq System (Illumina, San Diego, CA) high-throughput sequencing platform. The original data after sequencing was analyzed by bioinformatics using NCBI, CCDS, dbSNP (v138), COSMIC, human genome database (HG19) and other databases to determine the pathogenic mutation site. The average depth of the sequencing was 4837.978Kb, detection sensitivity was ~ 5%. Details on variant calling, filtering, and annotation are detailed in our recently published reports [23].
Statistical analysis
SPSS software version 26.0 (Chicago, IL, USA) and GraphPad PrismTM 8.01(San Diego, California, USA) were used for the analysis. Continuous variables were presented as mean values ± standard deviation, or median (range) considering whether the data fit a normal distribution or not; categoric measures were summarized with frequency counts and percentages. Overall survival (OS) is defined as the time from diagnosis to death or the time of the last follow-up. Event-free survival (EFS) is defined as the time from diagnosis to relapse, death, or the time of the last follow-up. The Kaplan-Meier method was used for survival analysis, and the Log-rank test was utilized to assess differences between groups. Univariate analysis and multivariate analysis were performed using the Cox proportional hazard regression model. Multivariable analysis including variables with P<0.05 in univariate analysis were performed for OS and EFS. A two-sided P < 0.05 was regarded as statistically significant.
Results
Clinical features of ASXL1+ AML patients
ASXL1 mutations were found in 8.7% (91 of 1047) of the patients in the whole cohort. The median age of the patients was 50 (33–58) years, with 20 cases older than 60 years and 49 cases being male, as indicated in Table 1. The median white blood cell (WBC) count was 7.5 (2.4-33.3) × 109/L, with 18 cases (19.78%) having a value of ≥ 50 × 109/L. Bone marrow blast percentage of more than 80% was seen in 15 cases (16%). According to the 2017 ELN risk criteria, 27 cases (20%) were favorable-risk AML (including 4 cases of APL), 1 case (1%) was intermediate-risk AML, and 63 cases (69%) were adverse-risk AML. Allo-HSCT was applied in 12 patients (13%). Three cases died within 30 days after induction therapy, and 50 cases (63%) died at the end of the follow-up.
The molecular mutations of ASXL1 were detected in 30 different nucleotide sites, all of which were located in exon 12, including G652S (41.76%), G642fs (12.09%), H630fs (8.79%), S1231F and R693X (5.49%), N986S (4.40%), T1139K (3.33%), G643fs and Y591X (2.20%). The distribution of all nucleotide sites was shown in Additional file 1. Most of the patients carried a single-point mutation, 7 (7.69%) patients carried two-point mutations, and one patient carried three-point mutations (G642fs, G643fs and G645fs). The median VAF value of ASXL1 mutation was 49.17% (1.02–79.28%).
Companion gene mutations and fusion genes in ASXL1+ AML patients
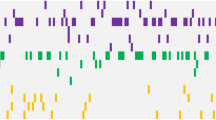
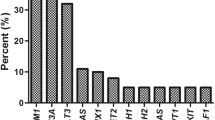
One or more co-mutation of genes was observed in 83 patients (86.46%) of ASXL1+ AML (Fig. 1). TET2 had the highest mutation frequency (48.35%), followed by U2AF1 (16.48%), CEBPA (15.38%), NRAS (14.29%), FLT3-ITD (13.19%), DNMT3A (10.99%), IDH2 (8.79%), RUNX1 (7.69%), KIT (6.59%), and SRSF2 (5.49%). Other mutant genes (including FLT3-TKD, ETV6, IDH1, CBL, SETBP1, NPM1, TP53, EZH2, SF3B1, JAK) are found in fewer than 5% of ASXL1+ AML patients; PHF6 and ZRSR2 mutations are not seen in ASXL1+ AML patients.

The mutational landscape of 91 ASXL1+ AML patients. The landscape displayed all genetic anomalies for each subject. A single patient instance was represented by the boxes in one column. Mutations were color coded according to mutation type. The frequency distribution of all aberrations was depicted by the histogram on the right
The fusion genes were screened in 83 of 91 ASXL1+AML cases. There were 31 cases (37.35%) with fusion gene mutations, including AML1-ETO in 17 cases (20.48%), PML-RARα in 4 cases (4.82%), BCR-ABL, MLL-AF9 and CBFβ-MYH11 in 3 cases (3.61%), MLL-ELL in 1 case (1.20%). The remaining 52 cases (62.65%) were with negative fusion genes.
Risk factors on the prognosis of ASXL1+ AML
In order to understand the prognostic impacts of clinical features and molecular profiles on the outcomes of ASXL1+ AML patients, we analyzed the risk factors on OS and EFS including gender (female vs. male), age (≥ 60 vs. < 60 years), ASXL1 nucleotide sites, ASXL1 VAF (≥ 49.17% vs. < 49.17%), WBC counts (≥ 50 vs. < 50 × 109/L), HGB (≥ 110 vs. < 110 g/L), PLT counts (≥ 100 vs. < 100 × 109/L), bone marrow blasts (≥ 80% vs. < 80%), peripheral blood blasts (≥ 20% vs. < 20%), allo-HSCT (yes vs. no), risk stratification (adverse vs. inter/favorable -risk), AML1-ETO fusion gene (positive vs. negative), CBFβ-MYH11 fusion gene (positive vs. negative), and the mutation status of other common AML co-mutation genes. The median follow-up time was 12.93 (0.37–53.53) months. Table 2 and Additional file 2A revealed that older patients (age ≥ 60 years) had a shorter OS (P = 0.034). Higher WBC counts (≥ 50 × 109/L) were associated with a shorter OS (P = 0.035, Additional file 2C) and EFS (P = 0.006, Additional file 2D). Cases who accepted allo-HSCT had a longer OS (P = 0.024, Additional file 2E) and a better EFS (P = 0.013, Additional file 2F). The adverse risk group had a lower OS (P = 0.005) and EFS (P = 0.004). AML1-ETO coexistence was related to a prolonged OS (P = 0.010, Additional file 3A) and EFS (P = 0.013, Additional file 3B). FLT3-ITD co-mutation was related to a shorter OS (P < 0.001, Additional file 3C) and EFS (P < 0.001, Additional file 3D). However, neither the ASXL1 mutation sites nor the ASXL1 VAF had impacts on EFS or OS.
The factors with P < 0.05 in univariate analyses were included in the multivariate analysis. FLT3-ITD co-mutation had an independent predictive impact on poor OS (Table 3). Allo-HSCT was an independent protective factor for the OS and EFS of ASXL1+ AML patients (Table 3).
Then, we assessed the prognosis effect of the aforementioned factors in the adverse-risk group. The survival study revealed that decreased HGB levels (< 110 g/L), FLT3-ITD mutations, and RUNX1 mutations had a negative influence on the OS of ASXL1+ AML patients (P = 0.045, P = 0.047, and P = 0.027, respectively; Additional file 3E, F). These variables had no impact on EFS. Allo-HSCT recipients had a longer OS and EFS (P = 0.024 and P = 0.013, respectively). HGB levels < 110 g/L and the FLT3-ITD mutations were found to have an independent predictive influence on poor OS in the multivariate analysis.
Increased number of risk factors may shorten the OS and EFS of ASXL1+ AML patients
The aforementioned factors that had adverse impact on OS and EFS are defined as high risk factors, including age ≥ 60 years, WBC count ≥ 50 × 109/L, FLT3-ITD mutations, RUNX1 mutations, and the absence of AML1-ETO fusion gene. ASXL1 mutations without any risk factor were referred to as single-hit ASXL1+ AML. ASXL1 mutations with one risk factor was referred to as double-hit ASXL1+ AML. ASXL1 mutations with two or more risk factors were referred to as triple-hit ASXL1+ AML. The combination of these risk factors had a negative influence on the prognosis of ASXL1+ AML (Fig. 2). The median OS was not attained in single-hit ASXL1+ AML, 29.53 months in double-hit ASXL1+ AML, and 6.67 months in triple-hit ASXL1+ AML (P = 0.003, Fig. 2A). The median EFS in single-hit ASXL1+ AML was not attained in single-hit ASXL1+ AML, 29.53 months in double-hit ASXL1+ AML, and 5.47 months in triple-hit ASXL1+ AML (P = 0.003, Fig. 2B).

Comparison of OS (A) and EFS (B) in single-hit, double-hit and triple-hit ASXL1+ AML. The factors that had adverse impact on OS and EFS are defined as high risk factors, including age ≥ 60 years, WBC count ≥ 50 × 109/L, FLT3-ITD mutations, RUNX1 mutations, and absence of AML1-ETO fusion gene. ASXL1 mutations without any risk factor were classified as single-hit ASXL1+ AML. ASXL1 mutations together with any of the risk factors was referred to as double-hit ASXL1+ AML. ASXL1 mutations along with any two or more of the risk factors were designated as triple-hit ASXL1+ AML
Allo-HSCT improved the survival of double/triple-hit ASXL1+ AML patients
In our study, 12 patients received allo-HSCT as the consolidation management. Eleven of them carried one or more risk factors in addition to ASXL1 mutations. As shown in Fig. 3, allo-HSCT significantly improved the OS (median 29.53 months vs. 11.33 months, P = 0.008, Fig. 3A) and EFS (median 29.53 months vs. 8.53 months, P = 0.007, Fig. 3B) in double or triple-hit ASXL1+ AML patients.

Transplantation can improve OS (A) and EFS (B) of ASXL1+ AML patients. The factors that had adverse impact on OS and EFS are defined as high risk factors, including age ≥ 60 years, WBC count ≥ 50 × 109/L, FLT3-ITD mutations, RUNX1 mutations, and absence of AML1-ETO fusion gene. ASXL1 mutations without any risk factor were classified as single-hit ASXL1+ AML. ASXL1 mutations together with any of the risk factors was referred to as double-hit ASXL1+ AML. ASXL1 mutations along with any two or more of the risk factors were designated as triple-hit ASXL1+ AML
Discussion
Previous researches found that ASXL1 mutations were recurrent in 5–20% of AML patients[24,25,26,27,28,29]. These mutations are heterozygous and result in ASXL1 mutants with a C-terminal truncation[21]. This ASXL1 mutation pattern is characterized by dominant-negative or gain-of-function mutations [30]. ASXL1 gain-of-function mutations have been linked to poor outcomes in AML patients [27, 29, 31, 32]. Based on the adverse outcome of ASXL1+ AML patients, ASXL1 mutations were recognized as a stratification criterion for AML in the 2017 ELN guideline [4]. However, given the prevalence and adverse outcome of ASXL1 mutations in AML, it is critical to identify the molecular landscape of ASXL1+ AML patients for establishing precise risk stratification in this subgroup of AML. This study investigated several key issues related to ASXL1+ AML, and discovered that the addition of other risk factors to ASXL1 mutations worsens the adverse outcome of ASXL1+ AML patients.
The majority of ASXL1 mutations in our study were found on codon 12, which is consistent with earlier reports [25, 33]. The most prevalent mutation was a guanine duplication (c.1934dupG) that results in a frameshift (p.Gly646TrpfsX12) [29, 34, 35]. These ASXL1 mutations in AML patients are regarded as gain-of-function with a negative prognosis [30]. In our study, the distribution of mutation sites was quite diverse, with G652S, G642fs, and H630fs having the highest occurrence. This diversity of nucleotide mutations might be attributed to differences in the selected population and races. There was no statistical difference in OS and EFS across different mutated nucleotides, consistent with a recent study [35]. Furthermore, we found that ASXL1 VAF did not correlate with survival, consistent with Richardson and colleagues’ findings that VAFs of ASXL1 mutations were not significantly associated with OS [14]. Moreover, the current chemotherapy regimen and allo-HSCT may partly overcome the poor prognosis of high VAF and different mutation sites. The functional relevancies of ASXL1 mutation in different nucleotides or frequencies are needed to further study.
Age ≥ 60 years and WBC counts ≥ 50 × 109/L are typically linked with unfavorable risk and poor outcome in AML patients [4]. In our cohort of ASXL1+ AML, age ≥ 60 years also had a detrimental influence on OS. WBC account ≥ 50 × 109/L had a negative impact on OS and EFS. Allo-HSCT, which was formerly thought to be the cure for AML, showed a survival advantage, particularly in individuals with double or triple-hit ASXL1+ AML.
Most primary MDS patients with ASXL1 mutations (85%) have concurrent mutations of genes at the time of diagnosis [36]. The mutational profiles of ASXL1+ AML are complicated and multiple molecular interactions may exist. We observed that 86.46% of ASXL1+AML patients had additional gene mutations. Detailed investigation in the roles of co-occurred mutations is necessary for ASXL1+ AML patients. A previous study showed that RUNX1 mutation promotes leukemogenesis of myeloid malignancies in ASXL1+ leukemia [37] and is associated with adverse prognoses of patients with de novo AML [38]. In our study, RUNX1 did not have effects on OS and EFS in the overall prognostic analysis, but it was associated with shorter OS in high-risk ASXL1+ AML patients. This suggests that RUNX1 mutation does not have prognostic significance in ASXL1+ AML and may be involved in the leukemogenesis of this subtype of AML. In addition, FLT3-ITD mutation is recognized as a poor prognostic factor that is associated with short OS, EFS and DFS [38,39,40]. The ASXL1, FLT3-ITD, and RUNX1 mutations have been identified as major risk factors in AML patients by the ELN guidelines [4]. In this study, FLT3-ITD mutations were also associated with a shorter OS and EFS in ASXL1+ AML patients and correlated with a shorter survival time in high-risk ASXL1+ AML patients. This finding suggests that FLT3-ITD mutations exacerbate the poor prognosis of ASXL1+ AML. The AML1-ETO fusion gene results from the chromosomal translocation t(8;21), and is usually related to good response to induction therapy, as well as high complete remission rates in AML patients [41]. Our findings showed that the AML1-ETO fusion gene was similarly associated with a prolonged OS and EFS in ASXL1+ AML patients. These data demonstrate that the complex molecular interactions may affect the prognosis of ASXL1+ AML patients. Our study further identified the factors associated with prognostic heterogeneity in ASXL1+ AML patients. The application of multiple-hit theory may improve the prognostic stratification schemes, making the prognosis in ASXL1+ AML more precise. Future studies can also formulate a potential scoring system with these prognostic factors after validated on large cohort of ASXL1+ AML cases. As a result, clinicians can develop individualized precision treatment options for each patient.
Currently, clinical diagnoses and risk assessments for AML are mostly based on cytogenetic and genomic changes [4]. The prognosis for AML patients varies substantially, particularly for those with normal karyotype [31]. With the application of NGS in the clinical practice, we can better understand the complex roles and prognostic impacts of molecular mutations of genes in AML. According to the multiple-hit theory of genetic alterations in lymphoma and multiple myeloma, we further analyzed the additional risk factors for the survival of ASXL1+ AML patients. The results showed that the more risk factors, the shorter the OS and EFS for ASXL1+ AML patients. The application of allo-HSCT significantly improved the prognosis of ASXL1+ AML patients [35]. This was also applicable to the double-hit/triple-hit patients defined in our study, further confirming the importance of allo-HSCT in the treatment of AML patients.
Our research had several limitations. First, our study was retrospective and prone to selection biases. Second, owing to technical limitations, certain gene mutations may go undetected. Prognostic implications of some gene mutations may be overlooked. Third, the small sample sizes of several subgroups resulted in relatively low statistical power. Because of these constraints, our findings require confirmation in a larger and prospective population.
Conclusions
This study provides new insights into the mutational spectrum and prognostic factors of ASXL1+ AML patients. The results demonstrate that increasing risk factors are associated with adversary prognosis of ASXL1+ AML patients. Our research further emphasizes the necessity of having the precise risk stratification for ASXL1+ AML patients.
Availability of data and materials
The data that support the findings of our research are available from The First Affiliated Hospital of Zhengzhou University, but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available.
Abbreviations
- ASXL1:
-
Additional sex combs-like 1
- Asx:
-
Additional sex combs
- AML:
-
Acute myeloid leukemia
- APL:
-
Acute promyelocytic leukemia
- MDS:
-
Myelodysplastic syndromes
- allo-HSCT:
-
Allogenic hematopoietic stem cell transplantation
- HSCT:
-
Hematopoietic stem cell transplantation
- RAR:
-
Retinoic acid receptor
- NGS:
-
Next-generation sequencing
- EDTA:
-
Ethylene diamine tetraacetic acid
- VAF:
-
Variant allele frequency
- WBC:
-
White blood cell
- HGB:
-
Hemoglobin
- PLT:
-
Platelet
- OS:
-
Overall survival
- EFS:
-
Event-free survival
References
Chen W-L, Wang J-H, Zhao A-H, Xu X, Wang Y-H, Chen T-L, Li J-M, Mi J-Q, Zhu Y-M, Liu Y-F, et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood. 2014;124(10):1645–54.
Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, Pandey S, Patel JP, Chung YR, Koche R, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22(2):180–93.
Papaioannou D, Petri A, Dovey OM, Terreri S, Wang E, Collins FA, Woodward LA, Walker AE, Nicolet D, Pepe F, et al. The long non-coding RNA HOXB-AS3 regulates ribosomal RNA transcription in NPM1-mutated acute myeloid leukemia. Nat Commun. 2019;10(1):5351.
Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–47.
Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, Rees J, Hann I, Stevens R, Burnett A, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood. 1998;92(7):2322–33.
Fasan A, Haferlach C, Alpermann T, Jeromin S, Grossmann V, Eder C, Weissmann S, Dicker F, Kohlmann A, Schindela S, et al. The role of different genetic subtypes of CEBPA mutated AML. Leukemia. 2014;28(4):794–803.
Schlenk RF, Kayser S, Bullinger L, Kobbe G, Casper J, Ringhoffer M, Held G, Brossart P, Lübbert M, Salih HR, et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood. 2014;124(23):3441–9.
Gale RE, Green C, Allen C, Mead AJ, Burnett AK, Hills RK, Linch DC. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111(5):2776–84.
Paschka P, Marcucci G, Ruppert AS, Mrózek K, Chen H, Kittles RA, Vukosavljevic T, Perrotti D, Vardiman JW, Carroll AJ, et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol. 2006;24(24):3904–11.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–21.
Larson RA, Mandrekar SJ, Huebner LJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, Thiede C, Prior TW, Döhner K, et al. Midostaurin reduces relapse in FLT3-mutant acute myeloid leukemia: the Alliance CALGB 10603/RATIFY trial. Leukemia. 2021. https://doi.org/10.1038/s41375-021-01179-4.
Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, Thiede C, Prior TW, Döhner K, Marcucci G, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3mutation. N Engl J Med. 2017;377(5):454–64.
Upadhyay Banskota S, Khanal N, Bhatt VR. A precision medicine approach to management of acute myeloid leukemia in older adults. Curr Opin Oncol. 2020;32(6):650–5.
Richardson DR, Swoboda DM, Moore DT, Johnson SM, Chan O, Galeotti J, Esparza S, Hussaini MO, Van Deventer H, Foster MC, et al. Genomic characteristics and prognostic significance of co-mutated ASXL1/SRSF2 acute myeloid leukemia. Am J Hematol. 2021;96(4):462–70.
Srivastava A, Ritesh KC, Tsan Y-C, Liao R, Su F, Cao X, Hannibal MC, Keegan CE, Chinnaiyan AM, Martin DM, et al. De novo dominant ASXL3 mutations alter H2A deubiquitination and transcription in Bainbridge-Ropers syndrome. Hum Mol Genet. 2016;25(3):597–608.
Kakosaiou K, Panitsas F, Daraki A, Pagoni M, Apostolou P, Ioannidou A, Vlachadami I, Marinakis T, Giatra C, Vasilatou D, et al. ASXL1 mutations in AML are associated with specific clinical and cytogenetic characteristics. Leukemia Lymphoma. 2018;59(10):2439–46.
Fisher CL, Randazzo F, Humphries RK, Brock HW. Characterization of Asxl1, a murine homolog of Additional sex combs, and analysis of the Asx-like gene family. Gene. 2006;369:109–18.
Cho Y-S, Kim E-J, Park U-H, Sin H-S, Um S-J. Additional sex comb-like 1 (ASXL1), in cooperation with SRC-1, acts as a ligand-dependent coactivator for retinoic acid receptor. J Biol Chem. 2006;281(26):17588–98.
ASXL1 represses. retinoic acid receptor-mediated transcription through associating with HP1 and LSD1. J Biol Chem. 2015;290(10):6008.
Nagase R, Inoue D, Pastore A, Fujino T, Hou H-A, Yamasaki N, Goyama S, Saika M, Kanai A, Sera Y, et al. Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J Exp Med. 2018;215(6):1729–47.
Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci M-J, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol. 2012;5:12.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405.
Yu J, Li Y, Li T, Li Y, Xing H, Sun H, Sun L, Wan D, Liu Y, Xie X, et al. Gene mutational analysis by NGS and its clinical significance in patients with myelodysplastic syndrome and acute myeloid leukemia. Exp Hematol Oncol. 2020;9:2.
Abdel-Wahab O, Manshouri T, Patel J, Harris K, Yao J, Hedvat C, Heguy A, Bueso-Ramos C, Kantarjian H, Levine RL, et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010;70(2):447–52.
Boultwood J, Perry J, Pellagatti A, Fernandez-Mercado M, Fernandez-Santamaria C, Calasanz MJ, Larrayoz MJ, Garcia-Delgado M, Giagounidis A, Malcovati L, et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia. 2010;24(5):1062–5.
Carbuccia N, Trouplin V, Gelsi-Boyer V, Murati A, Rocquain J, Adélaïde J, Olschwang S, Xerri L, Vey N, Chaffanet M, et al. Mutual exclusion of ASXL1 and NPM1 mutations in a series of acute myeloid leukemias. Leukemia. 2010;24(2):469–73.
Chou W-C, Huang H-H, Hou H-A, Chen C-Y, Tang J-L, Yao M, Tsay W, Ko B-S, Wu S-J, Huang S-Y, et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2010;116(20):4086–94.
Rocquain J, Carbuccia N, Trouplin V, Raynaud S, Murati A, Nezri M, Tadrist Z, Olschwang S, Vey N, Birnbaum D, et al. Combined mutations of ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 genes in myelodysplastic syndromes and acute myeloid leukemias. BMC Cancer. 2010;10:401.
Paschka P, Schlenk RF, Gaidzik VI, Herzig JK, Aulitzky T, Bullinger L, Späth D, Teleanu V, Kündgen A, Köhne C-H, et al. ASXL1 mutations in younger adult patients with acute myeloid leukemia: a study by the German-Austrian Acute Myeloid Leukemia Study Group. Haematologica. 2015;100(3):324–30.
Yang H, Kurtenbach S, Guo Y, Lohse I, Durante MA, Li J, Li Z, Al-Ali H, Li L, Chen Z, et al. Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood. 2018;131(3):328–41.
Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrózek K, Nicolet D, Whitman SP, Wu Y-Z, Schwind S, et al. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood. 2011;118(26):6920–9.
Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–89.
Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adélaïde J, Rey J, Vainchenker W, Bernard OA, Chaffanet M, Vey N, et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia. 2009;23(11):2183–6.
Schnittger S, Eder C, Jeromin S, Alpermann T, Fasan A, Grossmann V, Kohlmann A, Illig T, Klopp N, Wichmann HE, et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia. 2013;27(1):82–91.
Zhou L, An J, Hou C, Ding Z, Qiu H, Tang X, Sun A, Chen S, Xu Y, Liu T, et al. Allogeneic hematopoietic stem cell transplantation could improve the survival of acute myeloid leukemia patients with ASXL1 mutations. Hematology. 2021;26(1):340–7.
Chen TC, Hou HA, Chou WC, Tang JL, Kuo YY, Chen CY, Tseng MH, Huang CF, Lai YJ, Chiang YC, et al. Dynamics of ASXL1 mutation and other associated genetic alterations during disease progression in patients with primary myelodysplastic syndrome. Blood Cancer J. 2014;4:e177.
Bera R, Chiu M-C, Huang Y-J, Lin T-H, Kuo M-C, Shih L-Y. RUNX1 mutations promote leukemogenesis of myeloid malignancies in ASXL1-mutated leukemia. J Hematol Oncol. 2019;12(1):104.
Bachas C, Schuurhuis GJ, Reinhardt D, Creutzig U, Kwidama ZJ, Zwaan CM, van den Heuvel-Eibrink MM, De Bont ESJM, Elitzur S, Rizzari C, et al. Clinical relevance of molecular aberrations in paediatric acute myeloid leukaemia at first relapse. Br J Haematol. 2014;166(6):902–10.
Santos FPS, Jones D, Qiao W, Cortes JE, Ravandi F, Estey EE, Verma D, Kantarjian H, Borthakur G. Prognostic value of FLT3 mutations among different cytogenetic subgroups in acute myeloid leukemia. Cancer. 2011;117(10):2145–55.
Whitman SP, Maharry K, Radmacher MD, Becker H, Mrózek K, Margeson D, Holland KB, Wu Y-Z, Schwind S, Metzeler KH, et al. FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood. 2010;116(18):3622–6.
Lagunas-Rangel FA, Chávez-Valencia V, Gómez-Guijosa M, Cortes-Penagos C. Acute myeloid leukemia-genetic alterations and their clinical prognosis. Int J Hematol Oncol Stem Cell Res. 2017;11(4):328–39.
Acknowledgements
We thank all the treating physicians for allowing us to enroll their patients and the patients for allowing us to analyze their data.
Funding
This work was supported by the National Natural Science Foundation of China [Grant Number 81800137 and U1804191] and Henan Medical Science and Technology Research Project (Grant Number 2018020068).
Author information
Authors and Affiliations
Contributions
YFL and SJW designed the study and revised the manuscript. LXL collected and analyzed the data. YF analyzed the data and drafted the manuscript. YFL, YJL and SJW revised the manuscript. ZZW partially collected the data. CW and ZXJ critically viewed and supervised the study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The project has been approved by the ethics committee of The First Affiliated Hospital of Zhengzhou University and each participant has signed written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
ASXL1 mutations at codon 12 of 91 de novo AML patients. Distribution and frequencies are given for ASXL1 mutations at codon 12. The boxes in one column represent single patient case. Mutations were color coded by mutation type. The histogram on the right showed the frequency distribution of all aberrations.
Additional file 2.
Comparison of OS and EFS between different clinical characteristic groups in ASXL1+ AML. OS and EFS were compared in (A-B) patients older than 60 years and patients younger than 60 years; (C-D) patients with WBC≥50 × 109/L vs. <50 × 109/L; (E-F) patients who accepted allo-HSCT or not.
Additional file 3.
Comparison of OS and EFS between different clinical characteristic groups in ASXL1+ AML. OS and EFS were compared in (A-B) patients with AML1-ETO fusion gene or not; (C-D) patients with FLT3-ITD mutations or not; (E-F) adverse-risk patients with RUNX1 mutations or not.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fan, Y., Liao, L., Liu, Y. et al. Risk factors affect accurate prognosis in ASXL1-mutated acute myeloid leukemia. Cancer Cell Int 21, 526 (2021). https://doi.org/10.1186/s12935-021-02233-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-021-02233-y