
Abstract
The ASXL1 gene is one of the most frequently mutated genes in malignant myeloid diseases. The ASXL1 protein belongs to protein complexes involved in the epigenetic regulation of gene expression. ASXL1 mutations are found in myeloproliferative neoplasms (MPN), myelodysplastic syndromes (MDS), chronic myelomonocytic leukemia (CMML) and acute myeloid leukemia (AML). They are generally associated with signs of aggressiveness and poor clinical outcome. Because of this, a systematic determination of ASXL1 mutational status in myeloid malignancies should help in prognosis assessment.
Similar content being viewed by others


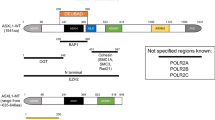
Mutations in the ASXL1 (additional sex combs like 1) gene were first reported in 2009 in myelodysplastic syndromes [1]. ASXL1 maps to chromosome region 20q11, close to the DNMT3B gene, and belongs to a family of three paralogs. ASXL1 comprises 12 exons and is expressed in most hematopoietic cell types.
Function of the ASXL1 protein
ASXL1 codes for a nuclear protein of 1084 residues characterized by an N-terminal helix-turn-helix domain, HARE-HTH [2], and an unusual C-terminal plant homeodomain (PHD), which may bind methylated lysines (Figure 1). The central part of ASXL1 contains an ASXH globular domain that may interact with a polycomb-associated deubiquitinase (DUB) [2, 3]. ASXL1 regulates epigenetic marks and transcription through interaction with polycomb complex proteins and various transcription activators and repressors [3–5]. In Drosophila, ASX forms a complex with the ubiquitin carboxy-terminal hydrolase calypso to constitute the recently identified polycomb repressive deubiquitinase (PR-DUB) complex [3, 6]. Human wild-type ASXL1 associates with the calypso ortholog BAP1 [7]. The calypso/BAP1 DUB deubiquitylates K119ub on histone H2A, leading to gene repression. However, the role of ASXL1 in leukemogenesis does not seem to be mediated by the DUB complex [7]. Recent data have shown that ASXL1 interacts with components of the polycomb complex PRC2, namely EZH2 and SUZ12, two proteins involved in the deposition of H3K27me3 histone repressive marks. These two PRC2 components are also mutated in myeloid malignant diseases [8–11]. Inhibition of ASXL1 function leads to loss of H3K27me3 histone marks. ASXL1 role could be to recruit the PRC2 complex to known leukemogenic loci such as HOXA genes [7]. ASXL1 also associates with HP1α/CBX5, a component of the heterochromatin repressive complex [6, 12]. HP1α binds to histone H3. JAK2 phosphorylates histone H3 and excludes HP1α from chromatin [13]. Thus, a potential functional link may exist between ASXL1 and JAK2 mutations but this remains to be demonstrated.

Distribution of ASXL1 mutations along the protein. From top to bottom are shown the localization of the ASXL1 gene on chromosome region 20q11, the exon structure of ASXL1, and the ASXL1 protein with its conserved motifs and binding regions: HARE helix-turn-helix at the N-terminus, HP1/CBX5 binding region, ASXH, an α-helical domain that contains LXXLL (nuclear receptor boxes), and the C-terminal plant homeodomain (PHD) finger. Below reported mutations (see Table 1) are shown along the protein: circles and triangles indicate frameshift and nonsense mutations, respectively, and the colors correspond to the exon location.
The functions of the other ASXL proteins are poorly defined. ASXL2 has been shown to regulate heart [14] and bone development, as well as adipogenesis. Mouse ASXL2 has been identified as a regulator of bone mineral density and osteoclastogenesis [15] and whereas ASXL1 represses, ASXL2 increases the expression of adipogenic genes [16]. ASXL3 expression and functions remain to be determined [17].
ASXL1 and concomitant mutations in myeloid malignancies
The vast majority of the ASXL1 mutations found in myeloid malignancies affect the twelfth exon of the gene although rare mutations in other exons have been detected [18]. ASXL1 mutations are frameshift and nonsense mutations that are supposed to result in C-terminal truncation of the protein upstream of the PHD finger (Figure 1). The functional relevance of some reported missense mutations is not clear. The most frequent mutation, which accounts for more than 50% of all ASXL1 mutations, is a duplication of a guanine nucleotide (c.1934dupG); it leads to a frameshift (p.Gly646TrpfsX12). One study has described this mutation as a PCR artefact [19], but because it is not found in germ-line DNAs, control DNAs or other studied types of cancers such as breast cancer, it is now generally considered to be a bona fide mutation.
ASXL1 mutations are usually heterozygous, suggesting that haplo-insufficiency is the key pathological factor; however, the truncated ASXL1 protein could also have a dominant negative role in titrating out an interacting protein. Actually, recent data have demonstrated a loss of ASXL1 protein in leukemia samples with ASXL1 mutation, indicating that these mutations are loss-of-function disease alleles [7].
ASXL1 is mutated in all types of malignant myeloid diseases, including myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPN), chronic myelomonocytic leukemia (CMML) and acute myeloid leukemia (AML). According to the series studied, ASXL1 mutation frequency varies from a few percent to more than 50% of cases (Table 1). ASXL1 mutations are most frequent in CMML (~ 45%). In MPNs, they are frequent in primary myelofibrosis (PMF)(34.5%) and rare in polycythemia vera (PV) or essential thrombocythemia (ET). In AML, they are found in secondary (30%) rather than in de novo cases (6.5%), and in AML with normal karyotype ASXL1 mutations are mutually exclusive with NPM1 mutations [20]. ASXL1 is the second most frequently mutated gene in MDSs after TET2[21]. In MDSs, ASXL1 mutations are more frequent in refractory anemia with excess of blasts (RAEB) than in the other forms such as refractory anemia with ring sideroblasts (RARS) [1, 5, 22]. ASXL1 mutations are further detected in rare cases of juvenile myelomonocytic leukemia (JMML) [23] and in RARS-T [24].
With the exception of NPM1 and FLT3, it seems that ASXL1 mutations coincide with mutations in many known genes including EZH2[18], IDH1/2, RUNX1 and TET2[21, 22]. Although ASXL1 functions are related to the PRC2 complex, which includes EZH2, ASXL1 and EZH2 mutations are not mutually exclusive [18, 38]. ASXL1 mutations can also cooperate with mutations in genes encoding signaling (CBL, JAK2, NF1, RAS) and splicing proteins (SF3B1, SRSF2, U2AF35). For example, in MDSs, ASXL1 mutations are more frequent in U2AF35-mutated patients than in U2AF35 wild-type patients [39]. In MPNs, ASXL1 mutations are found with the same frequency in JAK2V617F and JAK2 wild-type cases [26, 36]. In MDSs, ASXL1 mutations are often associated with RUNX1 mutations, and, in AMLs, with RUNX1 and CEBPA. [29, 33, 40].
Other alterations in ASXL1, ASXL2 and ASXL3
Few deletions of the gene have been reported and ASXL1 is generally not included in the more telomeric 20q13 deletion that is often observed in myeloid diseases. The ASXL1 gene can be translocated and fused to the PAX5 gene in acute lymphoblastoid leukemia [41] and altered by germ-line mutations in the Bohring-Opitz syndrome; this severe syndrome leads to death at an early age preventing to know whether susceptibility to hematopoietic diseases might result from ASXL1 germ-line mutations [42]. In recent genome sequencing studies rare mutations in ASXL1 and ASXL3 have also been found in chronic lymphocytic leukemia [43] but not in T-cell acute leukemia [44]. Mutations in ASXL2 and ASXL3 have not been found in myeloid diseases so far, but ASXL2-MYST3 and EPC1-ASXL2 fusions have been identified in myelodysplastic syndrome and T-cell acute leukemia, respectively [45, 46]. Both MYST3 and EPC1 are epigenetic regulators and these fusion proteins probably disrupt epigenetic protein complexes.
Animal models of ASXL1 loss
In a first model of Asxl1 gene knock-out in the mouse ASXL1 loss mildly perturbed myelopoiesis but did not trigger an actual hematological malignancy [47]. However, the effect of the absence of ASXL1 protein may have been masked by partially penetrant perinatal lethality. In another, more recent model of conditional Asxl1 gene knock-out, the animals developed a strong hematopoietic phenotype consistent with an MDS with myeloproliferative features. In cooperation with NRAS oncogenic mutation the absence of ASXL1 triggered an MDS/MPN. These observations were confirmed by experiments in hematopoietic cells using shRNA directed against ASXL1, which were highly coherent with the expected role of ASXL1 in leukemogenesis [7].
ASXL1 mutations in disease evolution
Like TET2 mutations, ASXL1 mutations are found in chronic and acute stages of myeloid malignancies. In a study of MPNs, with the exception of a single patient who acquired both ASXL1 and TET2 mutations, all patients with ASXL1 mutation at leukemic transformation already had ASXL1 mutation at the chronic stage [25]. In a series of secondary AML with multilineage dysplasia we found that in cases resulting from a transformation of a known MDS the same ASXL1 mutation was present at both the chronic and acute stages (Devillier et al., submitted). These observations suggest that ASXL1 mutations may constitute early hits in leukemogenesis and precede other alterations such as JAK2 and TET2 mutations [24, 25, 28]. However, there is also evidence to suggest that the opposite is true in some cases. In MPNs, for example, the proportion of ASXL1 mutations is higher in post-PV myelofibrosis (MF) and post-ET MF than in PV and ET. This suggests that the ASXL1 mutation may follow a JAK2 mutation and could therefore help predict the risk of evolution from PV and ET to MF [26, 36, 48]. As such, ASXL1 mutations may play a crucial role in the pathogenesis of PMF, as well as in the molecular progression from the chronic phase of a previous PV or ET to MF. Finally, in MDSs and CMML, ASXL1 mutations seem to be present in chronic phases and precede transformation and in rare cases, ASXL1 mutations can be lost or acquired during relapse of de novo AML [29].
ASXL1 mutations in disease outcome
A number of studies have linked ASXL1 mutations to the outcome of malignant myeloid diseases. In a study of MPNs based on the DIPSS-plus score [49] (Dynamic International Prognostic Scoring System for primary myelofibrosis), ASXL1 mutation tended to be associated with an aggressive disease and a poor overall survival [26]. In a large study of PMF patients ASXL1 mutations were associated with shorter overall survival [50]. In CMML, the presence of an ASXL1 mutation could help predict transformation to AML [30]. In MDSs, ASXL1 mutations are associated with a reduced time to progression in AML and constitute an independent prognostic marker [37]. Finally, a study of 18 genes in a large cohort of MDSs showed that mutations in 5 genes had prognostic impact: TP53, EZH2, ETV6, RUNX1 and ASXL1[21]. Coupled with the standardized international prognostic scoring system (IPSS), mutations in these five genes could help refine the prognosis evaluation of MDSs.
By contrast, a study of a large cohort of 605 AML cases without cytogenetic prognostic markers other than 11q23 abnormalities, reported that ASXL1 mutations were not associated with outcome [35]. However, they were associated with shorter overall survival in patients with intermediate-risk AML [29, 33]. A recent study of 476 cases with intermediate-risk de novo AML showed that ASXL1 mutations have a major impact on outcome [51]. According to the current European LeukemiaNet (ELN) guidelines for the diagnosis and management of AML, AMLs with normal karyotype are classified into two genetic categories based on their NPM1, FLT3-ITD and CEBPA mutation status: the ELN Favorable category is defined as mutated CEBPA and/or mutated NPM1 without FLT3-IT; all remaining cases (ie, those with wild-type CEBPA, and wild-type NPM1 with or without FLT3-ITD or mutated NPM1 with FLT3-ITD) form the ELN Intermediate-I category [52, 53]. ASXL1 mutations have been associated with inferior survival among ELN Favorable, but not among ELN Intermediate-I patients [40]. Taken together, these data show that ASXL1 mutations have prognostic value in certain subgroups of AML patients.
Conclusion
In almost all studies, and whatever the type of myeloid malignancy, ASXL1 mutations are associated with adverse features including, but not limited to myelodysplasia, myelofibrosis or progression to AML. Systematic detection of ASXL1 mutations could thus help in the assessment of disease and should perhaps be implemented in routine practice, whether associated with already systematically-surveyed mutations (CEBPA, JAK2, FLT3, NPM1) or in upcoming systematic genome analyses.
References
Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, Carbuccia N, Lagarde A, Prebet T, Nezri M, Sainty D, Olschwang S, Xerri L, Chaffanet M, Mozziconacci MJ, Vey N, Birnbaum D: Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009, 145: 788-800. 10.1111/j.1365-2141.2009.07697.x.
Aravind L, Iyer LM: The HARE-HTH and associated domains: Novel modules in the coordination of epigenetic DNA and protein modifications. Cell Cycle. 2012, 11 (1): 119-131. 10.4161/cc.11.1.18475.
Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, Wilm M, Muir TW, Müller J: Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010, 465: 243-247. 10.1038/nature08966.
Cho YS, Kim EJ, Park UH, Sin HS, Um SJ: Additional sex comb-like 1 (ASXL1), in cooperation with SRC-1, acts as a ligand-dependent coactivator for retinoic acid receptor. J Biol Chem. 2006, 281: 17588-17598. 10.1074/jbc.M512616200.
Boultwood J, Perry J, Pellagatti A, Fernandez-Mercado M, Fernandez-Santamaria C, Calasanz MJ, Larrayoz MJ, Garcia-Delgado M, Giagounidis A, Malcovati L, Della Porta MG, Jädersten M, Killick S, Hellström-Lindberg E, Cazzola M, Wainscoat JS: Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia. 2010, 24: 1062-1065. 10.1038/leu.2010.20.
Beisel C, Paro R: Silencing chromatin: comparing modes and mechanisms. Nat Rev Genet. 2011, 12: 123-135.
Abdel-Wahab O, Adli M, Saunders L, Gao J, Shih AH, Pandey S, Jaffe J, Zhao X, Perna F, Carroll M, Melnick A, Nimer SD, Aifantis I, Bernstein B, Levine RL: ASXL1 Mutations Promote Myeloid Transformation Through Inhibition of PRC2-Mediated Gene Repression. Abstract 405, ASH December 2011. Session: 603 Oncogenes and Tumor Suppressors: From Genomic-based Discovery to Functional Validation.
Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG, Duncombe A, Cervantes F, Oscier D, Boultwood J, Grand FH, Cross NC: Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010, 42: 722-726. 10.1038/ng.621.
Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tönnissen ER, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, van der Reijden BA, Jansen JH: Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010, 42: 665-667. 10.1038/ng.620.
Brecqueville M, Cervera N, Adelaide J, Rey J, Carbucia N, Chaffanet M, Vey N, Murati A: Mutations and deletions of the SUZ12 polycomb gene in myeloproliferative neoplasms. Blood Cancer Journal. 2011, 1: 33-10.1038/bcj.2011.31.
Score J, Hidalgo-Curtis C, Jones AV, Winkelmann N, Skinner A, Ward D, Zoi K, Ernst T, Stegelmann F, Döhner K, Chase A, Cross NC: Inactivation of polycomb repressive complex 2 components in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Blood. 2012, 119: 1208-1213. 10.1182/blood-2011-07-367243.
Lee SW, Cho YS, Na JM, Park UH, Kang M, Kim EJ, Um SJ: ASXL1 represses retinoic acid receptor-mediated transcription through associating with HP1 and LSD1. J Biol Chem. 2010, 285: 18-29. 10.1074/jbc.M109.065862.
Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, Green AR, Kouzarides T: JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009, 461: 819-822. 10.1038/nature08448.
Baskind HA, Na L, Ma Q, Patel MP, Geenen DL, Wang QT: Functional conservation of Asxl2, a murine homolog for the Drosophila enhancer of trithorax and polycomb group gene Asx. PLoS One. 2009, 4: e4750-10.1371/journal.pone.0004750.
Farber CR, Bennett BJ, Orozco L, Zou W, Lira A, Kostem E, Kang HM, Furlotte N, Berberyan A, Ghazalpour A, Suwanwela J, Drake TA, Eskin E, Wang QT, Teitelbaum SL, Lusis AJ: Mouse genome-wide association and systems genetics identify Asxl2 as a regulator of bone mineral density and osteoclastogenesis. PLoS Genet. 2011, 7: e1002038-10.1371/journal.pgen.1002038.
Park UH, Yoon SK, Park T, Kim EJ, Um SJ: Additional sex comb-like (ASXL) proteins 1 and 2 play opposite roles in adipogenesis via reciprocal regulation of peroxisome proliferator-activated receptor {gamma}. J Biol Chem. 2011, 286: 1354-1363. 10.1074/jbc.M110.177816.
Katoh M, Katoh M: Identification and characterization of human CXXC10 gene in silico. Int J Oncol. 2004, 25: 1193-1199.
Abdel-Wahab O, Pardanani A, Patel J, Wadleigh M, Lasho T, Heguy A, Beran M, Gilliland DG, Levine RL, Tefferi A: Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia. 2011, 25: 1200-1202. 10.1038/leu.2011.58.
Abdel-Wahab O, Kilpivaara O, Patel J, Busque L, Levine RL: The most commonly reported variant in ASXL1 (c.1934dupG;p.Gly646TrpfsX12) is not a somatic alteration. Leukemia. 2010, 24: 1656-1657. 10.1038/leu.2010.144.
Carbuccia N, Trouplin V, Gelsi-Boyer V, Murati A, Rocquain J, Adélaïde J, Olschwang S, Xerri L, Vey N, Chaffanet M, Birnbaum D, Mozziconacci MJ: Mutual exclusion of ASXL1 and NPM1 mutations in a series of acute myeloid leukemias. Leukemia. 2010, 24: 469-473. 10.1038/leu.2009.218.
Bejar R, Stevenson K, Abdel-Wahab O, Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, Ebert BL: Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011, 364: 2496-2506. 10.1056/NEJMoa1013343.
Rocquain J, Carbuccia N, Trouplin V, Raynaud S, Murati A, Nezri M, Tadrist Z, Olschwang S, Vey N, Birnbaum D, Gelsi-Boyer V, Mozziconacci MJ: Combined mutations of ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 genes in myelodysplastic syndromes and acute myeloid leukemias. BMC Cancer. 2010, 10: 401-407. 10.1186/1471-2407-10-401.
Sugimoto Y, Muramatsu H, Makishima H, Prince C, Jankowska AM, Yoshida N, Xu Y, Nishio N, Hama A, Yagasaki H, Takahashi Y, Kato K, Manabe A, Kojima S, Maciejewski : Spectrum of molecular defects in juvenile myelomonocytic leukaemia includes ASXL1 mutations. Br J Haematol. 2010, 150: 83-87.
Szpurka H, Jankowska AM, Makishima H, Bodo J, Bejanyan N, Hsi ED, Sekeres MA, Maciejewski JP: Spectrum of mutations in RARS-T patients includes TET2 and ASXL1 mutations. Leuk Res. 2010, 34: 969-973. 10.1016/j.leukres.2010.02.033.
Abdel-Wahab O, Manshouri T, Patel J, Harris K, Yao J, Hedvat C, Heguy A, Bueso-Ramos C, Kantarjian H, Levine RL, Verstovsek S: Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010, 70: 447-452. 10.1158/0008-5472.CAN-09-3783.
Brecqueville M, Rey J, Bertucci F, Coppin E, Finetti P, Carbuccia N, Cervera N, Gelsi-Boyer V, Arnoulet C, Gisserot O, Verrot D, Slama B, Vey N, Mozziconacci MJ, Birnbaum D, Murati A: Mutation analysis of ASXL1, CBL, DNMT3A, IDH1, IDH2, JAK2, MPL, NF1, SF3B1, SUZ12 and TET2 in myeloproliferative neoplasms. Genes Chromosome Cancer. 2012,
Boultwood J, Perry J, Zaman R, Fernandez-Santamaria C, Littlewood T, Kusec R, Pellagatti A, Wang L, Clark RE, Wainscoat JS: High-density single nucleotide polymorphism array analysis and ASXL1 gene mutation screening in chronic myeloid leukemia during disease progression. Leukemia. 2010, 24: 1139-1145. 10.1038/leu.2010.65.
Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adélaïde J, Rey J, Vainchenker W, Bernard OA, Chaffanet M, Vey N, Birnbaum D, Mozziconacci MJ: Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia. 2009, 23: 2183-2186. 10.1038/leu.2009.141.
Chou WC, Huang HH, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Ko BS, Wu SJ, Huang SY, Hsu SC, Chen YC, Huang YN, Chang YC, Lee FY, Liu MC, Liu CW, Tseng MH, Huang CF, Tien HF: Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2011, 116: 4086-4094.
Gelsi-Boyer V, Trouplin V, Roquain J, Adélaïde J, Carbuccia N, Esterni B, Finetti P, Murati A, Arnoulet C, Zerazhi H, Fezoui H, Tadrist Z, Nezri M, Chaffanet M, Mozziconacci MJ, Vey N, Birnbaum D: ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br J Haematol. 2010, 151: 365-375. 10.1111/j.1365-2141.2010.08381.x.
Grossmann V, Kohlmann A, Eder C, Haferlach C, Kern W, Cross NC, Haferlach T, Schnittger S: Molecular profiling of chronic myelomonocytic leukemia reveals diverse mutations in > 80% of patients with TET2 and EZH2 being of high prognostic relevance. Leukemia. 2012, 25: 877-879.
Jankowska AM, Makishima H, Tiu RV, Szpurka H, Huang Y, Traina F, Visconte V, Sugimoto Y, Prince C, O'Keefe C, Hsi ED, List A, Sekeres MA, Rao A, McDevitt MA, Maciejewski JP: Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood. 2011, 118: 3932-3941. 10.1182/blood-2010-10-311019.
Pratcorona M, Abbas S, Sanders M, Koenders J, Kavelaars F, Erpelinck-Verschueren C, Zeilemaker A, Lowenberg B, Valk P: Acquired mutations in ASXL1 in acute myeloid leukemia: prevalence and prognostic value. Haematologica. 2012, 97: 388-392. 10.3324/haematol.2011.051532. doi:10.3324
Ricci C, Fermo E, Corti S, Molteni M, Faricciotti A, Cortelezzi A, Lambertenghi Deliliers G, Beran M, Onida F: RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clin Cancer Res. 2010, 16: 2246-2256. 10.1158/1078-0432.CCR-09-2112.
Shen Y, Zhu YM, Fan X, Shi JY, Wang QR, Yan XJ, Gu ZH, Wang YY, Chen B, Jiang CL, Yan H, Chen FF, Chen HM, Chen Z, Jin J, Chen SJ: Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood. 2011, 118: 5593-5603. 10.1182/blood-2011-03-343988.
Stein BL, Williams DM, O'Keefe C, Rogers O, Ingersoll RG, Spivak JL, Verma A, Maciejewski JP, McDevitt MA, Moliterno AR: Disruption of the ASXL1 gene is frequent in primary, post-essential thrombocytosis and post-polycythemia vera myelofibrosis, but not essential thrombocytosis or polycythemia vera: analysis of molecular genetics and clinical phenotypes. Haematologica. 2011, 96: 1462-1469. 10.3324/haematol.2011.045591.
Thol F, Friesen I, Damm F, Yun H, Weissinger EM, Krauter J, Wagner K, Chaturvedi A, Sharma A, Wichmann M, Göhring G, Schumann C, Bug G, Ottmann O, Hofmann WK, Schlegelberger B, Heuser M, Ganser A: Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol. 2011, 29: 2499-2506. 10.1200/JCO.2010.33.4938.
Guglielmelli P, Biamonte F, Score J, Hidalgo-Curtis C, Cervantes F, Maffioli M, Fanelli T, Ernst T, Winkelman N, Jones AV, Zoi K, Reiter A, Duncombe A, Villani L, Bosi A, Barosi G, Cross NC, Vannucchi AM: EZH2 mutational status predicts poor survival in myelofibrosis. Blood. 2011, 118: 5227-5234. 10.1182/blood-2011-06-363424.
Damm F, Kosmider O, Gelsi-Boyer V, Renneville A, Carbuccia N, Hidalgo-Curtis C, Della-Valle V, Couronné L, Scourzic L, Chesnais V, Guerci-Bresler A, Slama B, Beyne-Rauzy O, Schmidt-Tanguy A, Stamatoullas-Bastard A, Dreyfus F, Prebet T, Debotton S, VEY N, Morgan MA, Cross NCP, Preudhomme C, Birnbaum D, Bernard OA, Fontenay M: Mutations affecting m RNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood. 2012,
Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrózek K, Nicolet D, Whitman SP, Wu YZ, Schwind S, Powell BL, Carter TH, Wetzler M, Moore JO, Kolitz JE, Baer MR, Carroll AJ, Larson RA, Caligiuri MA, Marcucci G, Bloomfield CD: ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood. 2011, 118: 6920-6929. 10.1182/blood-2011-08-368225.
An Q, Wright SL, Moorman AV, Parker H, Griffiths M, Ross FM, Davies T, Harrison CJ, Strefford JC: Heterogeneous breakpoints in patients with acute lymphoblastic leukemia and the dic(9;20)(p11-13;q11) show recurrent involvement of genes at 20q11.21. Haematologica. 2009, 94: 1164-1169. 10.3324/haematol.2008.002808.
Hoischen A, van Bon BW, Rodriguez-Santiago B, Gilissen C, Vissers LE, de Vries P, Janssen I, van Lier B, Hastings R, Smithson SF, Newbury-Ecob R, Kjaergaard S, Goodship J, McGowan R, Bartholdi D, Rauch A, Peippo M, Cobben JM, Wieczorek D, Gillessen-Kaesbach G, Veltman JA, Brunner HG, de Vries BB: De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat Genet. 2011, 43: 729-731. 10.1038/ng.868.
Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L, Ramsay AJ, Beà S, Pinyol M, Martínez-Trillos A, López-Guerra M, Colomer D, Navarro A, Baumann T, Aymerich M, Rozman M, Delgado J, Giné E, Hernández JM, González-Díaz M, Puente DA, Velasco G, Freije JM, Tubío JM, Royo R, Gelpí JL, Orozco M, Pisano DG, Zamora J, Vázquez M: Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011, 44: 47-52. 10.1038/ng.1032.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen SC, Wei L, Collins-Underwood JR, Ma J, Roberts KG, Pounds SB, Ulyanov A, Becksfort J, Gupta P, Huether R, Kriwacki RW, Parker M, McGoldrick DJ, Zhao D, Alford D, Espy S: The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012, 481: 157-163. 10.1038/nature10725.
Imamura T, Kakazu N, Hibi S, Morimoto A, Fukushima Y, Ijuin I, Hada S, Kitabayashi I, Abe T, Imashuku S: Rearrangement of the MOZ gene in pediatric therapy-related myelodysplastic syndrome with a novel chromosomal translocation t(2;8)(p23;p11). Genes Chromosomes Cancer. 2003, 36: 413-419. 10.1002/gcc.10172.
Nakahata S, Saito Y, Hamasaki M, Hidaka T, Arai Y, Taki T, Taniwaki M, Morishita K: Alteration of enhancer of polycomb 1 at 10p11.2 is one of the genetic events leading to development of adult T-cell leukemia/lymphoma. Genes Chromosomes Cancer. 2009, 48: 768-776. 10.1002/gcc.20681.
Fisher CL, Pineault N, Brookes C, Helgason CD, Ohta H, Bodner C, Hess JL, Humphries RK, Brock HW: Loss-of-function Additional sex combs like 1 mutations disrupt hematopoiesis but do not cause severe myelodysplasia or leukemia. Blood. 2010, 115: 38-46. 10.1182/blood-2009-07-230698.
Ricci C, Spinelli O, Salmoiraghi S, Finazzi G, Carobbio A, Rambaldi A: ASXL1 mutations in primary and secondary myelofibrosis. Br J Haematol. 2012, 156: 404-407. 10.1111/j.1365-2141.2011.08865.x.
Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S, Van Dyke D, Hanson C, Wu W, Pardanani A, Cervantes F, Passamonti F, Tefferi A: DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011, 29: 392-397. 10.1200/JCO.2010.32.2446.
Guglielmelli P, Biamonte F, Score J, Hidalgo-Curtis C, Cervantes F, Maffioli M, Fanelli T, Ernst T, Winkelman N, Jones AV, Zoi K, Reiter A, Duncombe A, Villani L, Paoli C, Bosi A, Barosi G, Cross NCP, Vannucchi AM: Prognostic Impact of EZH2 and ASXL1 Mutation in Myelofibrosis. Abstract 2811 ASH, December 2011. Session: 634. Myeloproliferative Syndromes: Poster II.
Schnittger S, Eder C, Alpermann T, Fasan A, Grossmann V, Alexander Kohlmann A, Kern W, Haferlach C, Haferlach T: ASXL1 exon 12 Mutations Are Frequent in AML with Intermediate Risk Karyotype and Are Independently Associated with An Extremely Poor Outcome. Abstract 416 ASH, December 2011. Session: 611. Leukemias - Biology, Cytogenetics and Molecular Markers in Diagnosis and Prognosis: Prognostic Biomarkers in Adult AML.
Rockova V, Abbas S, Wouters BJ, Erpelinck CA, Beverloo HB, Delwel R, van Putten WL, Löwenberg B, Valk PJ: Risk stratification of intermediate-risk acute myeloid leukemia: integrative analysis of a multitude of gene mutation and gene expression markers. Blood. 2011, 118: 1069-1076. 10.1182/blood-2011-02-334748.
Dohner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz MA, Sierra J, Tallman MS, Löwenberg B, Bloomfield CD: Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010, 115: 453-474. 10.1182/blood-2009-07-235358.
Acknowledgements
Our work in this field is supported by Inserm, Institut Paoli-Calmettes and grants from the Association pour la Recherche sur le Cancer (DB) and Association Laurette Fugain (MJM 2010).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors have contributed ideas, discussions, and have participated in the writing of the manuscript. All authors read and approved the final manuscript
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gelsi-Boyer, V., Brecqueville, M., Devillier, R. et al. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol 5, 12 (2012). https://doi.org/10.1186/1756-8722-5-12
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-8722-5-12