
Abstract
It has been intensively reported that the immunosuppressive tumor microenvironment (TME) results in tumor resistance to immunotherapy, especially immune checkpoint blockade and chimeric T cell antigen therapy. As an emerging therapeutic agent, oncolytic viruses (OVs) can specifically kill malignant cells and modify immune and non-immune TME components through their intrinsic properties or genetically incorporated with TME regulators. Strategies of manipulating OVs against the immunosuppressive TME include serving as a cancer vaccine, expressing proinflammatory factors and immune checkpoint inhibitors, and regulating nonimmune stromal constituents. In this review, we summarized the mechanisms and applications of OVs against the immunosuppressive TME, and strategies of OVs in combination with immunotherapy. We also introduced future directions to achieve efficient clinical translation including optimization of preclinical models that simulate the human TME and achieving systemic delivery of OVs.
Similar content being viewed by others

Background
Increasing studies have been focused on the role of tumor microenvironment (TME) in immunosuppression. Hypoxia, acidosis, low immunogenicity and suppressed immune cells in the TME pose a great challenge to cancer immunotherapy [1]. Although tremendous progress has been achieved in immune checkpoint blockade (ICB) and chimeric antigen T (CAR-T) cell therapies, considering the heterogeneity and immunosuppression of the TME in many tumors, these two leading immunotherapies that require a pre-existing inflammatory microenvironment for optimal efficacy are not a panacea. The limited response rate in ICB-treated patients and modest efficacy of CAR-T cell therapy for solid tumors, especially for those tumors with immunosuppressive TME remain as intractable problems. The current predicament of immunotherapy raises an imperious demand for a proinflammatory shift of the TME [2, 3].
Oncolytic viruses (OVs) are a type of replicative-competent agents that selectively infect and lyse tumor cells and reverse immunosuppression by targeting the TME including both immune and non-immune stromal constituents [4]. As a versatile therapeutic agent, an OV can intrinsically trigger tumor-specific immune responses or be genetically inserted with exogenous therapeutic genes to modulate the TME, bringing potent therapeutic efficacy and relatively low toxicity [5].
In this review, we summarized the modulatory effects of OVs against the immunosuppressive TME as well as the preclinical and clinical applications of OVs in combination with immunotherapy. Developing humanized animal model to simulate human TME and optimizing administration methods of OVs were also discussed.
Immunosuppression in the TME
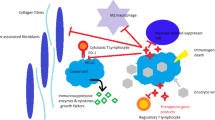
The TME consists of cellular and non-cellular components. The cellular components include neoplastic cells, cancer-associated fibroblasts (CAFs), endothelial cells (ECs), innate immune cells [e.g., neutrophils, dendritic cells (DCs), and natural killer (NK) cells], adaptive immune cells (e.g., T and B cells), and immunosuppressive cells [e.g. myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T cells (Tregs)]. The non-cellular components include the extracellular matrix (ECM), tumor vasculature, and secretory molecules (e.g. cytokines, chemokines, growth factors, and proteases) [6]. Notably, the majority of TME components contribute to the immunosuppressive microenvironment in various manners, as is shown in Fig. 1.

Components of tumor microenvironment (TME) contribute to the immunosuppression in various manners. Tumor cells downregulate expression of major histocompatibility complex-I (MHC-I) and antigens to avoid antigen presentation and T cell recognition, and express immune checkpoint proteins such as programmed cell-death ligand 1 (PD-L1) and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) to inactivate infiltrated T cells. Additionally, tumor cells recruit various immunosuppressive cells [e.g. myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory-T cells (Tregs)] by expressing immunosuppressive molecules [e.g. interleukin (IL)-10, chemokine ligand (CCL)-5, granulocyte–macrophage colony-stimulating factor (GM-CSF), indoleamine-2,3-dioxygenase (IDO) and tumor growth factor-β (TGF-β)]. Tumor cells, immunosuppressive cells and various immunoregulatory molecules [e.g. reactive oxygen species (ROS), arginase-1 (Arg-1), CCL-22, IL-10, and PD-L1] construct an immunosuppressive network in the TME. The activities of dendritic cells (DCs), T cells, natural killer (NK) cells, and other immune cells are therefore repressed severely. Moreover, classical stromal components contribute to immunosuppression. Continuous release of tumor-derived vascular endothelial growth factor (VEGF) leads to the formation of dysfunctional blood vessels with loose endothelial cell (EC)-EC connections and poor pericyte coverage, which exacerbates hypoxia and acidosis in the TME, thereby impairing the functionality of immune cells. Activated cancer associated fibroblasts (CAFs) lead to excessive extracellular matrix (ECM) deposition, which results in dense and tenacious fibrotic tissue surrounding the tumor mass and an elevated interstitial fluid pressure (IFP). These formidable physical barriers severely hinder immune infiltration and drug perfusion
Tumor cells and tumor-associated immune cells construct an immunosuppressive network
The sophisticated interactions of tumor cells, tumor stroma, and the host immune system construct a highly immunosuppressive TME as a tumor develops. Malignant tumor cells escape from host immunosurveillance by various mechanisms that include downregulating major histocompatibility complex (MHC)-I to escape T cell recognition, expressing immunoinhibitory surface proteins such as programmed cell death protein ligand-1 (PD-L1) to inactivate cytotoxic T cells (CTLs), and secreting immunosuppressive cytokines [e.g. transforming growth factor-β (TGF-β), granulocyte–macrophage colony-stimulating factor (GM-CSF), and interleukin (IL)-10], chemokines [e.g. chemoattractant cytokine ligand (CCL) 20 and CCL17], indoleamine-2,3-dioxygenase, and other secretory factors, thereby inhibiting T cell proliferation, inducing recruitment of immunosuppressive cells such as Tregs and MDSCs, and promoting phenotype conversion of macrophages from anti-tumor M1 to pro-tumor M2-like TAMs [7, 8]. Recruited immunosuppressive cells impair host immunosurveillance through various mechanisms. TAMs promote immunosuppression by producing IL-10 and TGF-β, and recruit Tregs by producing CCL22 [9, 10]. In terms of MDSCs, arginase-1 expressed by MDSCs degrades L-arginine required for T cell proliferation, and T cell receptor signaling is downregulated by MDSCs, which contributes further to T cell inactivation. MDSCs also recruit Tregs by secreting chemokines such as CCL3, CCL4, and CCL5 [11]. Tregs exert a wide range of immunosuppressive effects by secreting immunosuppressive cytokines or acting in a contact-dependent manner, such as inhibiting antigen presentation of DCs, halting early expansion of T cells, downregulating proinflammatory IL-12 signaling by expressing a competitive receptor [12], repressing the expression of T cell-associated cytokines (e.g. IFN-γ, TNF-α, and IL-2), and inhibiting B cells, NK cells, and other immune cells [13, 14]. Collectively, densely distributed negative immune cells and anergic T cells have been recognized as common features of the immunosuppressive TME, which indicate a poor prognosis [15].
Non-immune stromal components and immunosuppression
Tumor-associated vasculature contributes to immunosuppression to a great extent. Rapid progression of a tumor requires tremendous amounts of oxygen and nutrients, which renders the TME relatively hypoxic, resulting in excessive production of proangiogenic factors such as vascular endothelial growth factor (VEGF) during tumorigenesis, this leads to abnormal tumor vasculature with loose EC-EC connections, poor pericyte coverage, a leaky structure, and chaotic organization. Such vasculature with poor delivery functions further exacerbates hypoxia and acidosis as well as elevates interstitial fluid pressure (IFP) of the TME, which create an unfavorable condition for T cell proliferation, infiltration and activation [16]. Apart from contributing to disrupted vasculature, many proangiogenic factors also possess immunosuppressive properties. As an example, VEGF has direct immunosuppressive effects by recruiting immunoinhibitory cells to suppress the activity of T cells and disrupting the maturation and antigen-presenting ability of DCs [17, 18]. Tumor-associated ECs also contribute to T cell inactivation by expressing PD-L1 [19].
In addition to tumor vasculature, other stroma components also contribute to immunosuppression. Activated by tumor-derived molecules such as TGF-β and IL-6, CAFs induce excessive deposition of ECM proteins (mainly collagen) and create a dense and tenacious stroma with elevated IFP, which forms a formidable physical barrier for intratumoral infiltration of immune cells. Additionally, CAF-derived immunosuppressive molecules, such as TGF-β, CXCL2, CCL5, IL-6, and IL-10, contribute to immunosuppression by recruiting immunosuppressive cells such as MDSCs and maintaining their suppressive phenotype. Prevention of T cell infiltration, induction of T cell apoptosis, and impaired cytotoxicity of both NK and T cells have been shown to be mediated by CAFs [20, 21].
Role of the immunosuppressive TME in outcomes of immunotherapy
After the discovery and clinical introduction of ICB and CAR-T cell therapies, there has been a surge in the development of deleveraging the immune system to treat cancer [22,23,24,25]. In ICB therapy, leading checkpoint inhibitors, including anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and anti-programmed cell-death protein 1(PD-1)/PD-ligand (PD-L1) monoclonal antibodies, have reached promising clinical outcomes. Ipilimumab, an anti-CTLA4 antibody, was approved for treatment of melanoma in 2011 [26]. Anti-PD-1/PD-L1 antibodies, such as pembrolizumab, nivolumab, and atezolizumab, have also progressed to clinical applications for various tumors such as non-small cell lung cancer (NSCLC), head and neck cancer, and colorectal cancer [27,28,29]. CAR-T cell therapy is another promising immunotherapy that modifies T cells to be specifically redirected to TAAs on tumor cells. CTL019, an anti-CD19 CAR-T cell product, was approved by the FDA for treatment of refractory B-cell lymphoma [30].
Although desirable therapeutic outcomes have been yielded in some patients, insufficient efficacy and limited responses have been reported for both ICB and CAR-T cell therapies in patients with an immunosuppressive TME phenotype [31]. ICB and CAR-T cell therapies exert anti-tumor effects mainly in a CTL-dependent manner. Preclinical and clinical studies have revealed that the therapeutic efficacy of targeting CTLA4 and PD-1/PD-L1 is closely associated with high infiltration of PD-1 expressing CD8 + T cells, expression of T-helper type 1 genes, and expression of PD-L1 and CTLA4 within tumors at baseline [32,33,34,35,36]. However, the functionality and intratumoral infiltration of CTLs are severely hindered by immunosuppressive and non-immune stromal components that create a non-T cell inflamed TME, characterized by an absence of tumor antigen presentation and sequestered CD8 + T cells around the tumor margin with low expression of cytotoxic markers, such as IFN-γ and granzyme B[37]. This immunogenically “cold” TME is seen in many patients with hypo-immunogenic tumors such as colorectal carcinoma (CRC) and pancreatic ductal adenocarcinoma (PDAC) [37, 38]. Therefore, a rational therapeutic strategy is to introduce a TME-reprogramming agent into immunotherapy. As a versatile genetic engineering platform to reshape the TME, OVs have emerged as a leading candidate to facilitate anti-tumor immunotherapy.
Current development of oncolytic virotherapy
OVs are a class of biological agents with tumor selectivity and replication competence. In the nineteenth century, tumor regression was observed after viral infection, which introduced virotherapy as a therapeutic option to treat cancers [39]. In 1991, the first genetically engineered OV, a herpes simplex virus 1 (HSV-1) mutant with deficient thymidine kinase, was created for treatment of malignant glioma in nude mice [39]. After OV-induced anti-tumor immune activation was revealed by pre-clinical and clinical studies in recent years, oncolytic immunotherapy opened up a new chapter in anti-tumor therapy [40, 41].
OVs are classified into naturally occurring viruses and genetically engineered viruses. The majority of OVs are genetically modified to achieve tumor selectivity and reduce toxicity [5]. Take H101 (Oncorine), an adenovirus recombinant as an example, the coding region responsible for E1B55K protein to inactivate p53 for viral replication in normal cells was deleted, leaving this virus only replicable in malignant cells with aberrant p53 function [42]. In addition, they serve as a powerful transgene-delivering tool by genetically modified with various regulatory molecules to fight against the immunosuppressive TME, such as inserting immunostimulatory factors to potentiate anti-tumor immunity, or ECM-modifying agents to enhance intra-tumoral infiltration of immune cells[5]. The mechanisms and applications of OVs against the TME will be introduced later. To date, a great number of OV agents originate from different species of OVs have been applied in clinical investigations. The biological properties and genetic modifications of the major types of OVs, and clinical applications of some leading OV agents were summarized, as is shown in Table 1.
Herpes simplex virus 1 (HSV-1)
HSV-1 is an enveloped, double-stranded linear DNA virus with a large genome of 152 kb. Owing to the large genome size, HSV allows incorporation of large exogenous DNA, conferring a more versatile transgene capability to HSV. The robust virulence and immunogenicity of HSV-1 provide a “double-edge sword” effect. Potent virulence and immunogenicity enable efficient oncolysis and anti-tumor immune responses. However, rapid immune-mediated viral clearance and potential cytotoxicity result in delivery and safety issues [40].
To achieve tumor selectivity and optimized efficacy, several HSV mutants have been created and applied to many types of tumors. As the first OV approved by the US Food and Drug Administration (FDA), talimogene laherparepvec (T-Vec) was modified to encode exogenous GM-CSF for immune activation another HSV-1 mutant [41].Combination therapy of T-Vec and other therapeutics is currently under intensive clinical evaluations [40]. Another HSV mutant HF10 has also been clinically evaluated in several tumors. A phase I/II clinical trial of HF10 for treatment of patients with solid cutaneous tumors was completed (NCT01017185). Recently, the first intravenous administration of HSV was reported in pediatric patients with cancer, systemic delivery of this HSV mutant called HSV1716 showed good tolerability and produced a similar clinical response comparing to intratumoral HSV1716 [43, 44].
Adenovirus
Adenovirus is a non-enveloped double-stranded DNA virus with a linear genome of 30–38 kb. Considering the well-understood genome structure and life cycle, and the stable physiological properties, genetic manipulation of an adenovirus can be easily achieved [45]. The majority of genetically engineered oncolytic adenoviruses (OAds) are derived from Ad serotype 5, which have been investigated extensively. H101 is the progeny product of ONYX-015, which is the first tumor-specific OAd mutant evaluated in clinic [42]. In 2005, it was approved by the Chinese FDA for the treatment of nasopharyngeal carcinoma in combination with cisplatin or 5-fluorouracil (5-FU), or both [46]. Other OAds that are undergoing clinical evaluations include LOAd703, VCN-01, Telomelysin (OBP-301) and ONCOS-102, clinical responses showed that these OAd recombinants produced potent anti-tumor efficacy and were well-tolerated in patients [47,48,49,50,51].
Vaccinia virus (VV)
Vaccinia virus (VV) is a double-stranded DNA virus with a 180–200 kb genome. The replication, cytotoxicity, and transgene capacity of Lister strain VV is not affected by the hypoxic environment [52]. Its mutant pexastimogene devacirepvec (Pexa-Vec, also known as JX-594) with a thymidine kinase gene mutation and GM-CSF gene insertion is currently under evaluation in a phase III trial of hepatocellular carcinoma (NCT02562755). Another mutant, GL-ONC1 carrying three exogenous genes (green fluorescent protein fusion, β-galactosidase, and β-glucuronidase), is also under clinical evaluation [53, 54]
Reovirus
This non-enveloped RNA virus contains 9–12 segments of linear double-stranded RNA. It has been reported that reovirus maintains its replicating and oncolytic abilities in the hypoxic TME, and downregulation of hypoxia inducible factor 1α (HIF-1α) has been observed during reovirus infection [55]. Moreover, through intravenous injection, reovirus can use peripheral blood mononuclear cells (PBMCs) and dendritic cells (DCs) as a cell-based carrier to evade neutralizing antibodies (NAbs) which mediate potent viral clearance, this biological property of reovirus renders a more efficient viral replication through systemic delivery [56, 57].
Reolysin (also known as pelareorep), a naturally occurring reovirus type 3 Dearing strain, has demonstrated a robust cytotoxic effect in KRAS-activated malignancies [58, 59]. Its combinations with chemotherapy, radiotherapy and immunotherapy have been evaluated in various clinical trials [60].
Measles virus (MV)
MV is a single-stranded RNA virus. CD46 is the receptor that mediates cellular entry of MV, which is often overexpressed on tumor cells. This confers tumor selectivity to MV [61]. MV-NIS is an MV mutant that expresses sodium iodide symporter. Therefore, uptake of I131 by infected tumor cells is increased, which generates radiotoxicity in tumor cells [62, 63]. The safety and efficient viral replication of MV-NIS have been documented in a phase I trial of patients with recurrent multiple myeloma [64].
Coxsackievirus A21 (CVA21 or CAVATAK)
CVA21 is a naturally occurring enterovirus that specifically targets susceptible tumor cells expressing CVA21 cellular receptors, intercellular adhesion molecule-1, and decay-accelerating factor [65]. A phase I clinical trial of CVA21 for treatment of bladder cancer has been reported [66].
There are some other OVs like Newcastle disease virus (NDV), parvovirus and myxoma virus, have been studied for treatment of various tumors [67,68,69,70]. For example, clinical evaluations of parvovirus H-1 (H-1PV or ParvOryx), a naturally occurring parvovirus strain, have undergone in patients with glioblastoma and pancreatic cancer [67, 68].
OVs reverse immunosuppression by modulating TME components
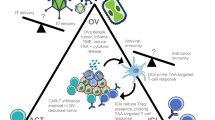
Generally, the multifaceted roles of OVs in tumors are performed through several mechanisms [71] as is shown in Fig. 2. (1) Direct oncolysis: In the context of viral infection, OVs selectively infect and replicate in tumor cells, leading to lysis of tumor cells and subsequent release of viral progeny and tumor cell components. (2) Anti-tumor immunity: Followed by OV-induced immunogenic cell lysis, pathogen-associated molecular pattern molecules (PAMPs), damage-associated molecular pattern molecules (DAMPs), and tumor-associated antigens (TAAs) are released, triggering rapid but unspecific innate immune responses. These reactions enhance tumor and viral antigen presentation by DCs, leading to subsequent T cell priming and activation, and ultimately creating an immunostimulatory microenvironment [72]. By taking advantage of this immune-potentiating ability, enhanced anti-tumor efficacy of OVs is achieved. (3) Vascular pruning: Studies have shown that OVs act as an anti-angiogenic agent by directly targeting tumor-associated ECs and proangiogenic factors through stimulating host immune cells to produce anti-angiogenic factors to reshape the tumor vasculature [4]. (4) Stroma degradation: OVs attract neutrophils that are a potent ECM modifier to decompose the ECM [73]. Additionally, stroma-decomposing agents have been integrated into OV genomes to alleviate fibrotic reactions and facilitate drug spreading and immune cell infiltration within tumors.

Mechanism of oncolytic viruses (OVs) targeting the TME. (i) Direct oncolysis: immunogenic cell death (ICD) induced by OVs leads to the release of numerous molecules, including pathogen-associated molecular pattern molecules (PAMPs), damage-associated molecular pattern molecules (DAMPs), and tumor-associated antigens (TAAs), which enhance activation of antigen presenting cells (APCs) such as dendritic cells (DCs). Simultaneously, infected tumor cells also produce various inflammatory cytokines such as type I interferon (IFN) and chemokines. (ii) Anti-tumor immunity: Inflammatory cytokines and chemokines are produced under OV infection, leading to the recruitment of innate immune cells such as neutrophils and natural killer (NK) cells. Antigen-loaded DCs after OV infection trigger T cell priming and degraded extracellular matrix (ECM) by OVs enhances intratumoral infiltration of T cells. The proinflammatory microenvironment created by OVs includes M2-to-M1 transition of tumor-associated macrophages (TAMs), decreased level of regulatory-T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), and upregulated major histocompatibility complex-I (MHC-I) on tumor cells, which facilitate T cells to overcome immune suppression and complete the final recognition and killing step. Further immunostimulatory effect of OVs was achieved by synergizing with immune checkpoint blockade (ICB) therapy. (iii) Vascular pruning: OVs exert anti-angiogenic effects through direct lysis of tumor-associated endothelial cells (ECs) and reducing the level of vascular endothelial growth factor (VEGF), preventing the immunosuppressive effect from those angiogenic components. (iv) Stroma degradation: various ECM-degrading agents expressed by engineered OVs induce stroma degradation. Concurrently, OV-induced CAF lysis also inhibits excessive ECM production. Alleviated stroma fibrosis subsequently promotes the infiltration of T cells
Next, we focus on the mechanisms and applications of oncolytic immunotherapy in reversing the immunosuppressive TME during the anti-tumor immune cycle.
Activation of innate immunity under OV infection
Soon after viral infection and subsequent oncolysis, non-specific innate immunity is triggered rapidly. Cytokines, such as type-I IFN and tumor necrosis factor-α (TNF-α), are secreted by infected tumor cells, TAAs and viral PAMPs are exposed to the host immune system followed by oncolysis [74]. This virus-induced cell lysis is recognized as immunogenic cell death (ICD) characterized by the release of DMAPs such as ATP, high-mobility group box 1, and calreticulin. DCs are then recruited and convert to the mature phenotype under exposure to DAMPs and TAAs [75, 76]. Toll-like receptors on immune cells are activated by DAMPs and PAMPs, leading to subsequent release of proinflammatory cytokines [e.g. tissue necrosis factor-α (TNF-α), type-I IFN, IL-1β, IL-6, and IL-12] and chemokines, resulting in recruitment and activation of innate immune cells such as neutrophils and NK cells [77, 78].
OV infection facilitates adaptive immune responses
T cell-mediated tumor-specific immune responses are the mainstay of adaptive immunity during OV infection. It has been demonstrated that depletion of T cells leads to absent anti-tumor efficacy despite observing persistent OV replication [79]. However, tumor-infiltrated lymphocytes (TILs) are extensively suppressed by TME components. Therefore, the final goal of oncolytic immunotherapy is to assist T cells in overcoming immunosuppressive barriers and become fully activated to exert tumoricidal activity. According to Twumasi-Boateng et al., both armed and unarmed OVs participate in the whole process of T cell activation, including priming, trafficking, infiltration, activation, and final tumor killing [80].
First, after OV-triggered innate immune responses, antigen-loaded APCs migrate to draining lymph nodes and initiate T cell priming by presenting antigens to naive T cells. Subsequently, production of lymphocyte-recruiting chemokines and proinflammatory cytokines during the virus-induced type-I IFN response leads to the recruitment and activation of T cells. Secreted inflammatory factors such as TNF-α during OV infection also upregulate the expression of selectin in ECs, allowing enhanced extravasation of lymphocytes from the vasculature, which is an EC-dependent process [80,81,82]. Then, during T cell infiltration, OVs alleviate structural barriers in the fibrotic tumor stroma by targeting stromal components and attracting neutrophils that secrete proteases such as matrix metalloproteinases (MMPs) and elastase to degrade the ECM [73, 83]. Increased infiltration of CTLs under OV infection has been observed in both preclinical and clinical studies, which is related to a better prognosis [84,85,86,87]. Finally, during conjugation and the killing step, OVs reverse the immunosuppressive phenotype of immunoinhibitory cells and upregulate MHC-I on the tumor cell surface, so that T cells are able to escape from immunosuppression and achieve efficient tumor recognition and killing [88,89,90].
Apart from localized immune responses, local administration of many OV agents also induces systemic immune responses as demonstrated by tumor regression, increased accumulation of immune cells, and upregulated immune-related gene expression in non-injected sites, which is referred as the “abscopal effect” [91,92,93]. Collectively, this series of immune responses caused by viral infection ultimately induces potent and durable anti-tumor immunity [91, 92].
Despite the immunomodulatory effect of OVs, the immune system acts as an opposing force against OVs. Such obstacles are extremely intractable when OVs are injected intravenously. Virus opsonization mediated by neutralizing antibodies (NAbs), complement proteins, erythrocytes, and other blood components, and the following virus phagocytosis in the liver and spleen impair the effectiveness of OVs to a great extent [94]. Therefore, approaches to induce fully engaged anti-tumor immunity by OVs while minimizing immune-mediated viral clearance require further exploration.
Genetically engineered OVs as a promising immunotherapeutic agent
To further potentiate the anti-tumor immunity of OVs, many genetically engineered OV mutants armed with various therapeutic genes have been investigated intensively.
Expression of immunostimulatory molecules by OVs
To restore the impaired function of APCs in the immunosuppressive TME, OVs have been used as an enhanced cancer vaccine by inserting TAAs into the viral genome to improve T cell priming [95, 96]. Additionally, treatment with OVs armed with proinflammatory cytokines such as GM-CSF, interleukins, and TNF-α, is a common strategy. The feasibility of GM-CSF insertion was confirmed by successful clinical translation of T-VEC [41], and promising clinical outcomes of other types of GM-CSF-inserted OV recombinants are anticipated, such as ONCOS-102 and Pexa-Vec. Genetic insertion of interleukins, such as IL-2, IL-7, and IL-12, has also been applied to enhance activation of lymphocytes and NK cells [97, 98]. For example, synergistic activation of T cells by the combination of IL-12 and IL-7 in vitro has been reported [99]. Consequently, a recent study designed a novel VV recombinant encoding both IL-7 and IL-12. Compared with IL-12 or IL-7 alone, coexpression of IL-12 and IL-7 by the VV caused a significant increase in activated TILs characterized by increased expression of granzyme B and genes related to T cell functions. Additionally, this combination increased tumor susceptibility to ICB treatment [97]. Apart from cytokines, chemokines are also ideal factors to enhance recruitment of immune cells. A recent study enhanced adoptive NK cell transfer therapy by strengthening the CCR5-CCL5 axis that is crucial for NK cell migration to tumors. By delivering CCL5 via a CCL5-encoding lentivirus, recruitment and intratumoral accumulation of modified NK cells overexpressing CCR5 was enhanced significantly. The combination group achieved a superior prognosis with a complete response rate of > 50% in mice [100]. In some cases, cytokines and chemokines have been concomitantly incorporated into OVs for better immunostimulatory effects [101, 102].
Expression of ICB proteins by OVs
Emerging applications of ICB have broadened the application strategies of oncolytic immunotherapy. Because toxicities related to systemic administration of checkpoint inhibitors have been reported in the clinic [103], OVs provide a rational delivery approach by locally expressing checkpoint inhibitors in tumor regions. Recently, a vaccinia virus coexpressing a PD-L1 inhibitor and GM-CSF (VV-iPDL1/GM) was generated and exerted potent systemic anti-tumor effects in several preclinical tumor models. Blockade of PD-L1 expression in both tumor and immune cells, enhanced neoantigen presentation on tumor cells, and improved infiltration and activation of T cells were achieved by VV-iPDL1/GM [104]. Similarly, a myxoma virus mutant expressing a soluble form of PD1 (vPD1) was evaluated in an immunocompetent murine model of B16-F10 melanoma, which showed poor immunogenicity. Compared with the combination of anti-PD1 monoclonal antibodies and the unmodified myxoma virus, vPD1 monotherapy led to more efficient anti-tumor responses and significant survival benefits [105]. Another study using the B16-F10 melanoma mouse model reported that NDV together with radiotherapy-induced significant tumor regression in ICB-treated tumors compared with single treatment. Additionally, an NDV encoding an anti-CTLA4 single-chain variable fragment was created to combine with radiotherapy. The survival outcome was comparable to the combination of radiotherapy and anti-CTLA4 mAbs, which indicated a radiosensitizing effect of NDV and potential in combination with radiotherapy and ICB therapy [106].
Expression of bispecific T-cell engagers by OVs
OVs have also been designed to express bispecific T-cell engagers (BiTEs), which simultaneously bind to TAAs on tumor cells and CD3 on T cells, so that infiltrated T cells are retargeted to tumor cells. For hematopoietic malignancies, BiTEs have progressed to clinical applications. As an example, Blinatumomab is a CD19/CD3 BiTE that has been clinically approved for treating B-cell precursor acute lymphoblastic leukemia [107]. However, BiTEs hardly penetrate into solid tumors because of structural barriers and poor tumor perfusion. Therefore, using OVs as a carrier to achieve local BiTE expression has overcome this physical barrier to a great extent. For example, an MV mutant was generated to deliver BiTEs that target CD3 and CD20 or CEA (VV-BiTE). Durable anti-tumor immunity was achieved through increased infiltration and cytotoxicity of CTLs. Moreover, synergistic effects and prolonged survival were observed in patient-derived colon cancer spheroids when VV-BiTE was combined with human peripheral blood monocytes (PBMCs) [108]. BiTE-armed OVs can also target both tumor and stromal cells such as CAFs. Fibroblast activation protein-α (FAP-α) is overexpressed on CAFs. Therefore, FAP-α-targeted BiTEs (FAP-BiTEs) that mediate binding between T cells and FAP-expressing CAFs were genetically introduced into OAds. In addition to direct oncolysis, T cell activation, cytotoxicity against CAFs, and diminished CAF-associated immunosuppressive factors were achieved by OAds expressing FAP-BiTEs [109, 110]. OVs expressing BiTEs have also been applied to facilitate CAR-T cell therapy [111], which will be introduced later.
Expression of stromal regulators by OVs
To improve intratumoral infiltration of immune cells and the OV itself, strategies of manipulating OVs to alleviate physical barriers in the TME have been considered. As the critical driver of the fibrotic network in the TME, CAFs are considered a common therapeutic target. Apart from the aforementioned FAP-BiTEs expressed by OVs. Urokinase receptor (uPAR) has also been considered as a target because of its overexpression on both tumor and stromal cells. Therefore, a uPAR-retargeted MV has been designed. Virus-mediated regulation of tumor-stroma interactions, including decreased ECs and fibroblasts, and downregulated gene expression associated with stromal components and angiogenesis, have been observed in several tumor models [112, 113]. In terms of targeting the ECM, hyaluronan is a critical ECM component responsible for tissue elasticity and water retention, which is closely related to elevated IFP in tumor stroma [114]. By integrating hyaluronidase into an OV genome to decompose overexpressed hyaluronan, physical barriers of the TME have been alleviated [115,116,117]. Apart from hyaluronidase, MMPs have also been expressed by OVs to enzymatically degrade ECM proteins such as collagen [118, 119]. Tumor-derived TGF-β is a critical signaling molecule that plays essential roles in the phenotypic conversion of CAFs and immunosuppression [120]. OVs have been designed to directly target TGF-β by expressing TGF-β-antagonizing molecules such as decorin [121]. The practicality of this method is supported by solid preclinical evidence including ECM degradation, immune activation, improved intratumoral spreading within tumors, and anti-tumor efficacy of OVs [122,123,124,125].
OVs in combination with immunotherapy
The proinflammatory TME created by OVs renders a tumor more susceptible to immunotherapy. Effective tumor eradication has been achieved in both preclinical and clinical studies that combined OVT with immunotherapy. Next, the applications of OVs in combination with immunotherapy will be introduced, as is shown in Table 2.
Combining OVs with ICB therapy
Immunotherapeutic effects of ICB therapy act in a T cell-dependent manner, while the absence of CTLs is an intractable problem in many tumors with low immunogenicity, which leads to a limited response rate [126, 127]. Both preclinical and clinical studies have revealed that expression of ICB proteins during OV infection, such as PD-L1 and PD-1, was upregulated in both tumor cells and immune cells, and increased T cell trafficking and infiltration into the tumor were observed [85, 128]. Blockade of PD1/PD-L1 and CTLA4 pathways by ICB therapy further enhance activation of recruited CTLs, thereby achieving a synergistic effect.
In an ex vivo melanoma coculture model, upregulated ICB protein, DC maturation, and a decreased TGF-β level were observed after H-1PV infection, and addition of ipilimumab or nivolumab further augmented this immunostimulatory effect characterized by further reduction of the TGF-β level, an increased number of granzyme B+ CD8+ T cells, and increased release of granzyme B, IFNγ, and TNFα [129]. This synergistic effect was further supported by intensive in vivo studies. In an ovarian cancer mouse model, a tumor antigen-armed Maraba virus increased intratumoral CD8+ T cell infiltration, and local T cell suppression via PD-1/PD-L1 axis was subsequently alleviated by anti-PD-1 therapy. Suppressed tumor progression was also achieved by combination therapy [95]. Consistent results were also obtained in a triple-negative breast cancer model using Maraba virus as a neoadjuvant therapy before ICB therapy [130]. Similarly, a study combined VV and PD-L1 blockade in a colon cancer model. Immunological results showed that OV-induced PD-L1+ cells and immunosuppressive cells, such as TAMs, MDSCs, and Tregs, were significantly reduced in the combination group and influx of CTLs with restored cytotoxicity was increased. These synergistic effects collectively alleviated the tumor burden and improved the survival outcome [131]. Inhibition of the leading immunosuppressive factor TFG-β by OVs has also shown synergistic efficacy with ICB therapy. Xu et al., recently designed an OAd encoding a soluble TGFβ receptor II fused with a human IgG Fc fragment (sTGFβRIIFc), which is a TGFβ decoy that inhibits TGFβ signaling. In immunocompetent mouse models of breast and renal cancers, addition of the sTGFβRIIFc-expressing OAd significantly augmented anti-PD-1 and anti-CTLA-4-mediated inhibition of tumor metastasis [132]. In addition to the aforementioned ICB proteins, other ICB proteins are also considered as therapeutic targets. T cell immunoglobulin domain and mucin domain-3 (TIM3) is an ICB protein expressed on immune cells, which correlates with resistance to immunotherapy [133]. A recent study revealed that OVT together with PD-1 blockade was unable to improve T cell activation in a lung cancer mouse model, whereas dual blockade of PD-1 and TIM3 enhanced the viro-immunotherapy. Upregulation of TIM3 on CD8+ T cells and TIM3 ligand on tumor cells induced by OVs contributed to the resistance to PD-1 blockade. This study suggested that targeting multiple ICB pathways is a rational approach to overcome resistance to single ICB treatment and achieve optimal synergy with OVT [134].
With the prevalence of “cocktail” therapy, adding other immunostimulatory agents to this combination regimen has been considered because it minimizes dose-dependent toxicity and drug resistance. For example, colony-stimulating factor 1 (CSF-1) is crucial for the recruitment and differentiation of TAMs, which is highly expressed in tumors. A study reported that combining OAds with anti-PD-1 therapy plus a CSF-1 receptor inhibitor greatly enhanced tumor regression and overall survival, which were achieved through increased functional T cell infiltration and anti-tumor phenotype conversion of TAMs [135]. Additionally, enhanced viral replication, T cell activation, and anti-tumor efficacy in mice with melanoma were achieved by triple combination of T-Vec, anti-PD-1 therapy, and MEK inhibition that targets a BRAF mutation [136].
OV-induced potentiation of ICB therapy has been strongly supported by substantial clinical evidence. In a window-of-opportunity clinical study, nine participants with high-grade glioma were recruited and intravenously administrated with Reolysin. Tumor samples from virus-treated patients showed efficient viral replication and increased CTL infiltration as well as more intensive PD-1 and PD-L1 expression, which were mediated by IFNs. Sequential treatment with Reolysin® prior to PD-1 blockade in a preclinical glioma model led to further immune activation and survival benefits [137]. Notably, a phase Ib clinical study conducted by Ribas et al., reported that in patients with advanced melanoma, the intra-tumoral PD-L1 expression was upregulated after T-Vec injection. Combining T-Vec with anti-PD-1 antibody achieved a high overall response rate (ORR) of 62%. Increased CD8+ T cells infiltration and IFN gene expression in post-treatment tumor samples explained this synergistic efficacy, providing a solid evidence of oncolytic immunotherapy [85]. Subsequently, a phase II clinical trial that combined T-Vec with pembrolizumab (anti-PD-1 mAb) reported an optimal ORR of 30% in 20 patients with advanced sarcoma. Immunological data from patients also confirmed the augmented anti-tumor immunity [138]. Similarly, 198 patients with advanced melanoma were recruited in another phase II trial and the combination of T-Vec and ipilimumab (anti-CTLA4 mAb) showed a superior ORR than ipilimumab alone (39% vs. 18%) [139].
Triple combination of chemotherapy, ICB therapy, and OVT has been applied in the clinic because synergistic efficacy has been reported between OVs and some chemical agents such as gemcitabine and paclitaxel [140, 141]. Recently, this triple combination strategy was applied in a phase Ib trial treating patients with unresectable PDAC. Combination of Reolysin, pembrolizumab, and first-line chemotherapy regimen (gemcitabine, irinotecan, leucovorin, and 5-FU) of PDAC achieved a disease control rate of 27%, and immunological data were highlighted in this study. High clonality and expansion of peripheral T cells and increased expression of inflammatory genes indicated an immune activation in patients with clinical benefits [142].
Currently, extensive clinical trials combining OVs with ICB therapy are under evaluation and promising outcomes are anticipated.
Combining OVs with CAR-T cell therapy
The existence of the TME in solid tumors poses intractable challenges for CAR-T cell therapy, including inadequate CAR-T cell infiltration caused by fibrotic stroma and suppressed CAR-T cell functions due to the immunosuppressive components. To facilitate the recruitment of CAR-T cells, a VV loaded with CXCL11 was designed. Two delivery approaches of CXCL11 were established. CXCL11 was expressed either via CAR T cells (CAR/CXCL11) or vaccinia virus (VV.CXCL11). Compared with CAR/CXCL11, VV.CXCL11 significantly increased intratumoral accumulation of CAR-T cells and potent anti-tumor efficacy was observed, which indicated the therapeutic potential of OVs in assisting CAR T cell therapy [143]. Another study combined CAR-T cells targeting folate receptor alpha with an OAd expressing an epidermal growth factor receptor-targeting BiTE. This combination induced robust T cell activation and proliferation both in vitro and in vivo with improved tumoricidal efficacy and survival compared with single agent treatment [111].
CAR-T cell therapy can be further facilitated by the combination of OVs and ICB therapy. As mentioned previously, local expression of checkpoint inhibitors by OVs alleviate the adverse effects caused by systemic delivery of checkpoint antibodies. Therefore, an OAd encoding both a PD-L1-blocking antibody and IL-12p70 (CAd12_PDL1) was designed to combine with HER2-specific CAR T cell therapy. In a xenograft model of head and neck cancer, compared with the insufficient anti-tumor efficacy of single therapy, combining systemic delivery of CAR T cells and local treatment of CAd12_PDL1 significantly improved the survival outcome. Control of tumor growth in both primary and metastatic sites was also achieved by the combination therapy in the orthotopic model [144]. Consistent results were also reported by Tanoue et al. Compared with the combination of anti-PD-L1 IgG and CAR T-cells, the addition of a OAd expressing anti-PD-L1 mini-antibody had superior therapeutic efficacy in a xenograft model of HER2+ prostate cancer [145].
To date, no clinical investigation of OVs in combination with CAR-T cell therapy has been reported.
Current challenges and future directions
The development of OVT has led to anti-tumor therapy beyond the traditional concept. Although promising results have been obtained by employing OVs in TME-targeted therapy, a multitude of challenges remain to be overcome.
Optimizing preclinical models to simulate the human TME
The abundant and sophisticated components in the TME render traditional preclinical models such as cell-derived tumor models insufficient to gain insights into the TME. Moreover, despite animal models, such as murine and canine models, sharing similarity with humans, species variation is hard to overcome, resulting in discrepant results between preclinical models and clinical patients. Although the patient-derived xenograft (PDX) model has achieved better translation with its well-preserved biological properties, such as mutation profiles and drug sensitivity of the primary tumor and its surroundings [146], the immunocompromised mice used for PDX engraftment are not suitable to evaluate oncolytic immunotherapy [147]. Hence, a humanized mouse model with a human immune system was established by injecting human leucocyte antigen (HLA)-matched PBMCs or cord blood CD34+ hematopoietic stem cells into an immunocompromised mouse. Thus far, the immunomodulatory effect of OVs has been evaluated in several studies using humanized mice [97, 148, 149]. Furthermore, by joining PDX and a humanized mouse model together, a novel preclinical model termed as Hu-PDX with human tumor cells, stroma, and an immune system is considered as the most desirable tool for drug screening or as an “avatar” model to predict clinical responses of patients. Several studies evaluating immunotherapy, especially ICB therapy, in solid tumors using a Hu-PDX model have been reported [150,151,152]. It is conceivable that introducing OVT into Hu-PDX models together with immunotherapy provides a great insight into translational research and precision medicine.
So far, developing Hu-PDX models to investigate the interactions between OVs and host immunity together with immunotherapy still largely lacks in-depth research. Limitations in performing such studies are as follows. (1) The acquirement of a PDX is difficult considering that many patients are diagnosed at an advanced stage with no indication for surgery. (2) The establishment of a Hu-PDX model is time consuming and costly with strict requirements of bringing two sophisticated biological systems together, such as HLA matching and stable engraftment. Therefore, technical progress in this field is urgently needed.
Minimizing immune-mediated viral clearance to achieve systemic delivery of OVs
The therapeutic benefits and safety of OVs have been demonstrated using IT administration, While some additional benefits, such as simplified operation, reaching metastatic sites, and easily triggered systemic immune responses, can be achieved by IV administration [153]. However, in practice, several challenges in IV injection result in limited administration of OVs in solid tumors. Through IV injection, the concentration of OVs is diluted by the bloodstream, and rapid viral clearance is mediated by NAbs and other blood components such as complement proteins and erythrocytes, resulting in only a fraction of OVs accessing tumor sites, leading to the limited infectivity and efficacy [154, 155].
To overcome these limitations, several methods have been employed. Capsid-modified OVs have been developed to prevent OVs from virus neutralization in circulation. For example, a high level of NAbs has been a problem for highly immunogenic HSV. By genetically modifying the envelope glycoproteins on HSVs, evasion from viral neutralization has been achieved [156, 157]. Alternatively, OAds covalently coated with PEG- or poly [N-(2-hydroxypropyl) methacrylamide] (PHPMA)-based polymers have been shown to induce a low titer of Nabs, reduced anti-virus immune responses, and increased circulation time and intratumoral accumulation of OAds [158, 159].
Another promising delivery approach is cell-based carriers. MSCs are considered as the most promising OV carrier based on their inherent tumor-homing and immune-silencing properties [160, 161]. Several preclinical and clinical studies have highlighted the transportation capability of OV-loaded MSCs. For example, delivery of VVs by MSCs augmented viral amplification and delivery by alleviating anti-virus immune responses [162]. An autologous MSC carrying OAds called Celyvir has recently completed its phase I clinical trial [163]. Despite showing promise, challenges including the strict condition for ex vivo infection culture, prevention of premature carrier cell lysis, and allogenic rejections form a formidable barrier to clinical translation considering the current technical restrictions [164].
Conclusions
OVs reverse the predicament of immunotherapy by creating a proinflammatory TME with the prevalence of “cocktail” combination therapy, and combining OVs with immunotherapy and other therapies such as chemotherapy and anti-angiogenic therapy is awaiting to be further explored. Despite some achievements, there are still many limitations in oncolytic immunotherapy. Because of the potent immune-mediated viral clearance in circulation, OVs can hardly access distant metastatic sites, which results in limited synergistic effects with immunotherapy. Methods such as cell carriers have been developed to achieve minimal anti-viral immune responses as well as optimal viral replication and spreading. Additionally, the Hu-PDX model is expected to shed light on virus-immunity interactions in the context of the humanized TME, which is important to achieve successful clinical translation. Collectively, oncolytic immunotherapy has built a solid basis for treating malignancies, a new era of anti-cancer therapy on the basis of oncolytic virotherapy is anticipated.
Availability of data and materials
Not applicable.
Abbreviations
- APC:
-
Antigen presenting cell
- BiTE:
-
Bispecific T-cell engager
- CAF:
-
Cancer-associated fibroblast
- CCL:
-
Chemoattractant cytokine ligand
- CRC:
-
Colorectal carcinoma
- CTLA4:
-
Cytotoxic T lymphocyte-associated antigen 4
- DC:
-
Dendritic cell
- DAMP:
-
Damage-associated molecular pattern molecule
- EC:
-
Endothelial cell
- ECM:
-
Extracellular matrix
- FAP-α:
-
Fibroblast activation protein-α
- FDA:
-
US Food and Drug Administration
- FOLFIRI:
-
Leucovorin and fluorouracil plus irinotecan
- GM-CSF:
-
Granulocyte–macrophage colony-stimulating factor
- HLA:
-
Human leucocyte antigen
- HMGB1:
-
High-mobility group box 1
- HSV:
-
Herpes simplex virus
- H-1PV:
-
Parvovirus H-1
- ICB:
-
Immune checkpoint blockade
- ICD:
-
Immunogenic cell death
- IDO:
-
Indoleamine-2,3-dioxygenase
- IFN:
-
Interferon
- IFP:
-
Interstitial fluid pressure
- IL:
-
Interleukin
- IT:
-
Intratumoral
- IV:
-
Intravenous
- MDSC:
-
Myeloid-derived suppressor cell
- MHC-I:
-
Major histocompatibility complex-I
- MSC:
-
Mesenchymal stem cell
- MMP:
-
Matrix metalloproteinase
- MV:
-
Measle virus
- NAb:
-
Neutralizing antibody
- NDV:
-
Newcastle disease virus
- NK cell:
-
Natural killer cell
- OAd:
-
Oncolytic adenovirus
- ORR:
-
Overall response rate
- OV:
-
Oncolytic virus
- OVT:
-
Oncolytic virotherapy
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PAMP:
-
Pathogen-associated molecular pattern molecule
- PBMC:
-
Peripheral blood mononuclear cell
- PDX:
-
Patient-derived xenograft
- PD-L1:
-
Programmed cell death protein ligand-1
- TAA:
-
Tumor associated-antigen
- TAM:
-
Tumor-associated macrophage
- TGF-β:
-
Transforming growth factor-β
- TIL:
-
Tumor-infiltrated lymphocytes
- TLRs:
-
Toll-like receptors
- TME:
-
Tumor microenvironment
- TNF-α:
-
Tumor necrosis factor-α
- Treg:
-
Regulatory T cell
- MSC:
-
Mesenchymal stem cell
- T-Vec:
-
Talimogene laherparepvec
- uPAR:
-
Urokinase receptor
- VEGF:
-
Vascular endothelial growth factor
- VV:
-
Vaccinia virus
- 5-FU:
-
5-Fluorouracil
References
Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019;79(18):4557–66.
Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16(9):566–81.
Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol. 2020;20(1):25–39.
Berkey SE, Thorne SH, Bartlett DL. Oncolytic virotherapy and the tumor microenvironment. Adv Exp Med Biol. 2017;1036:157–72.
Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci. 2016;107(10):1373–9.
Thomas D, Radhakrishnan P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol Cancer. 2019;18(1):14.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Raja J, et al. Oncolytic virus immunotherapy: future prospects for oncology. J Immunother Cancer. 2018;6(1):140.
Quatromoni JG, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res. 2012;4(4):376–89.
Curiel TJ, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–9.
Fleming V, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. 2018;9:398.
Chappert P, et al. Antigen-specific Treg impair CD8(+) T-cell priming by blocking early T-cell expansion. Eur J Immunol. 2010;40(2):339–50.
Kim JH, Kim BS, Lee SK. Regulatory T cells in tumor microenvironment and approach for anticancer immunotherapy. Immune Netw. 2020;20(1):e4.
Ghiringhelli F, et al. The role of regulatory T cells in the control of natural killer cells: relevance during tumor progression. Immunol Rev. 2006;214:229–38.
Crespo J, et al. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. 2013;25(2):214–21.
Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3(6):401–10.
Voron T, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212(2):139–48.
Gabrilovich DI, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2(10):1096–103.
Fukumura D, et al. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. 2018;15(5):325–40.
Harper J, Sainson RC. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin Cancer Biol. 2014;25:69–77.
Zhao W, et al. The role of hepatic stellate cells in the regulation of T-cell function and the promotion of hepatocellular carcinoma. Int J Oncol. 2012;41(2):457–64.
Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125(9):3335–7.
Topalian SL, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020–30.
Queirolo P, Spagnolo F. Atypical responses in patients with advanced melanoma, lung cancer, renal-cell carcinoma and other solid tumors treated with anti-PD-1 drugs: a systematic review. Cancer Treat Rev. 2017;59:71–8.
Zimmer L, et al. Adjuvant nivolumab plus ipilimumab or nivolumab monotherapy versus placebo in patients with resected stage IV melanoma with no evidence of disease (IMMUNED): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2020;395(10236):1558–68.
Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
Khoja L, et al. Pembrolizumab. J Immunother Cancer. 2015;3:36.
Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8.
Chen R, et al. The efficacy and safety of nivolumab, pembrolizumab, and atezolizumab in treatment of advanced non-small cell lung cancer. Discov Med. 2018;26(143):155–66.
Schuster SJ, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377(26):2545–54.
Filley AC, Henriquez M, Dey M. CART immunotherapy: development, success, and translation to malignant gliomas and other solid tumors. Front Oncol. 2018;8:453.
Agdashian D, et al. The effect of anti-CTLA4 treatment on peripheral and intra-tumoral T cells in patients with hepatocellular carcinoma. Cancer Immunol Immunother. 2019;68(4):599–608.
Spranger S, et al. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2:3.
Taube JM, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20(19):5064–74.
Sun C, et al. Expression of PD-1 and PD-L1 on tumor-infiltrating lymphocytes predicts prognosis in patients with small-cell lung cancer. OncoTargets Ther. 2020;13:6475–83.
Herbst RS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–7.
Binnewies M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–50.
Morrison AH, Byrne KT, Vonderheide RH. Immunotherapy and prevention of pancreatic cancer. Trends Cancer. 2018;4(6):418–28.
Martuza RL, et al. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252(5007):854–6.
Watanabe D, Goshima F. Oncolytic virotherapy by HSV. Adv Exp Med Biol. 2018;1045:63–84.
Andtbacka RH, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–8.
Bischoff JR, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274(5286):373–6.
Streby KA, et al. Intratumoral injection of HSV1716, an oncolytic herpes virus, is safe and shows evidence of immune response and viral replication in young cancer patients. Clin Cancer Res. 2017;23(14):3566–74.
Streby KA, et al. First-in-human intravenous seprehvir in young cancer patients: a phase 1 clinical trial. Mol Ther. 2019;27(11):1930–8.
Niemann J, Kühnel F. Oncolytic viruses: adenoviruses. Virus Genes. 2017;53(5):700–6.
Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst. 2006;98(5):298–300.
Nattress CB, Hallden G. Advances in oncolytic adenovirus therapy for pancreatic cancer. Cancer Lett. 2018;434:56–69.
Pascual-Pasto G, et al. Therapeutic targeting of the RB1 pathway in retinoblastoma with the oncolytic adenovirus VCN-01. Sci Transl Med. 2019. https://doi.org/10.1126/scitranslmed.aat9321.
Rodriguez-Garcia A, et al. Safety and efficacy of VCN-01, an oncolytic adenovirus combining fiber HSG-binding domain replacement with RGD and hyaluronidase expression. Clin Cancer Res. 2015;21(6):1406–18.
Koski A, et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther. 2010;18(10):1874–84.
Nemunaitis J, et al. A phase I study of telomerase-specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Mol Ther. 2010;18(2):429–34.
Hiley CT, et al. Lister strain vaccinia virus, a potential therapeutic vector targeting hypoxic tumours. Gene Ther. 2010;17(2):281–7.
Lauer UM, et al. Phase I study of oncolytic vaccinia virus GL-ONC1 in patients with peritoneal carcinomatosis. Clin Cancer Res. 2018;24(18):4388–98.
Mell LK, et al. Phase I trial of intravenous oncolytic vaccinia virus (GL-ONC1) with cisplatin and radiotherapy in patients with locoregionally advanced head and neck carcinoma. Clin Cancer Res. 2017;23(19):5696–702.
Gupta-Saraf P, Miller CL. HIF-1α downregulation and apoptosis in hypoxic prostate tumor cells infected with oncolytic mammalian orthoreovirus. Oncotarget. 2014;5(2):561–74.
Ilett EJ, et al. Internalization of oncolytic reovirus by human dendritic cell carriers protects the virus from neutralization. Clin Cancer Res. 2011;17(9):2767–76.
Adair RA, et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci Transl Med. 2012;4(138):138–77.
Carew JS, et al. Reolysin is a novel reovirus-based agent that induces endoplasmic reticular stress-mediated apoptosis in pancreatic cancer. Cell Death Dis. 2013;4(7):e728.
Villalona-Calero MA, et al. Oncolytic reovirus in combination with chemotherapy in metastatic or recurrent non-small cell lung cancer patients with KRAS-activated tumors. Cancer. 2016;122(6):875–83.
Zhao X, et al. Strategic combinations: the future of oncolytic virotherapy with reovirus. Mol Cancer Ther. 2016;15(5):767–73.
Aref S, Bailey K, Fielding A. Measles to the rescue: a review of oncolytic measles virus. Viruses. 2016. https://doi.org/10.3390/v8100294.
Opyrchal M, et al. Effective radiovirotherapy for malignant gliomas by using oncolytic measles virus strains encoding the sodium iodide symporter (MV-NIS). Hum Gene Ther. 2012;23(4):419–27.
Penheiter AR, et al. Sodium iodide symporter (NIS)-mediated radiovirotherapy for pancreatic cancer. AJR Am J Roentgenol. 2010;195(2):341–9.
Dispenzieri A, et al. Phase I trial of systemic administration of Edmonston strain of measles virus genetically engineered to express the sodium iodide symporter in patients with recurrent or refractory multiple myeloma. Leukemia. 2017;31(12):2791–8.
Shafren DR, et al. Systemic therapy of malignant human melanoma tumors by a common cold-producing enterovirus, coxsackievirus a21. Clin Cancer Res. 2004;10(1 Pt 1):53–60.
Annels NE, et al. Phase I trial of an ICAM-1-targeted immunotherapeutic-Coxsackievirus A21 (CVA21) as an oncolytic agent against non muscle-invasive bladder cancer. Clin Cancer Res. 2019;25(19):5818–31.
Hajda J, et al. A non-controlled, single arm, open label, phase II study of intravenous and intratumoral administration of ParvOryx in patients with metastatic, inoperable pancreatic cancer: ParvOryx02 protocol. BMC Cancer. 2017;17(1):576.
Geletneky K, et al. Oncolytic H-1 parvovirus shows safety and signs of immunogenic activity in a first phase I/IIa glioblastoma trial. Mol Ther. 2017;25(12):2620–34.
Laurie SA, et al. A phase 1 clinical study of intravenous administration of PV701, an oncolytic virus, using two-step desensitization. Clin Cancer Res. 2006;12(8):2555–62.
Kellish P, et al. Oncolytic virotherapy for small-cell lung cancer induces immune infiltration and prolongs survival. J Clin Invest. 2019;129(6):2279–92.
Gujar S, Bell J, Diallo J-S. SnapShot: cancer immunotherapy with oncolytic viruses. Cell. 2019;176(5):1240-1240.e1.
Achard C, et al. Lighting a fire in the tumor microenvironment using oncolytic immunotherapy. EBioMedicine. 2018;31:17–24.
Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res. 2011;71(7):2411–6.
Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–62.
Zhou J, et al. Immunogenic cell death in cancer therapy: present and emerging inducers. J Cell Mol Med. 2019;23(8):4854–65.
Krysko DV, et al. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12(12):860–75.
Hendrickx R, et al. Innate immunity to adenovirus. Hum Gene Ther. 2014;25(4):265–84.
Chaurasiya S, Chen NG, Fong Y. Oncolytic viruses and immunity. Curr Opin Immunol. 2018;51:83–90.
Li X, et al. The efficacy of oncolytic adenovirus is mediated by T-cell responses against virus and tumor in Syrian hamster model. Clin Cancer Res. 2017;23(1):239–49.
Twumasi-Boateng K, et al. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat Rev Cancer. 2018;18(7):419–32.
Pan J, et al. Tumor necrosis factor-alpha- or lipopolysaccharide-induced expression of the murine P-selectin gene in endothelial cells involves novel kappaB sites and a variant activating transcription factor/cAMP response element. J Biol Chem. 1998;273(16):10068–77.
Kunkel EJ, Jung U, Ley K. TNF-alpha induces selectin-mediated leukocyte rolling in mouse cremaster muscle arterioles. Am J Physiol. 1997;272(3 Pt 2):H1391–400.
Naumenko V, et al. Neutrophils in viral infection. Cell Tissue Res. 2018;371(3):505–16.
Chon HJ, et al. Tumor microenvironment remodeling by intratumoral oncolytic vaccinia virus enhances the efficacy of immune-checkpoint blockade. Clin Cancer Res. 2019;25(5):1612–23.
Ribas A, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170(6):1109-1119 e10.
Garcia-Carbonero R, et al. Phase 1 study of intravenous administration of the chimeric adenovirus enadenotucirev in patients undergoing primary tumor resection. J Immunother Cancer. 2017;5(1):71.
Kleijn A, et al. The in vivo therapeutic efficacy of the oncolytic adenovirus Delta24-RGD is mediated by tumor-specific immunity. PLoS ONE. 2014;9(5):e97495.
Prestwich RJ, et al. Tumor infection by oncolytic reovirus primes adaptive antitumor immunity. Clin Cancer Res. 2008;14(22):7358–66.
Brown MC, et al. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci Transl Med. 2017. https://doi.org/10.1126/scitranslmed.aan4220.
Murphy JP, et al. Therapy-induced MHC I ligands shape neo-antitumor CD8 T cell responses during oncolytic virus-based cancer immunotherapy. J Proteome Res. 2019;18(6):2666–75.
Fend L, et al. Immune checkpoint blockade, immunogenic chemotherapy or IFN-α blockade boost the local and abscopal effects of oncolytic virotherapy. Cancer Res. 2017;77(15):4146–57.
Kaufman HL, et al. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17(3):718–30.
Havunen R, et al. Abscopal effect in non-injected tumors achieved with cytokine-armed oncolytic adenovirus. Mol Ther Oncolytics. 2018;11:109–21.
Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30(7):658–70.
McGray AJR, et al. Oncolytic Maraba virus armed with tumor antigen boosts vaccine priming and reveals diverse therapeutic response patterns when combined with checkpoint blockade in ovarian cancer. J Immunother Cancer. 2019;7(1):189.
de Vries CR, Monken CE, Lattime EC. The addition of recombinant vaccinia HER2/neu to oncolytic vaccinia-GMCSF given into the tumor microenvironment overcomes MDSC-mediated immune escape and systemic anergy. Cancer Gene Ther. 2015;22(3):154–62.
Nakao S, et al. Intratumoral expression of IL-7 and IL-12 using an oncolytic virus increases systemic sensitivity to immune checkpoint blockade. Sci Transl Med. 2020. https://doi.org/10.1126/scitranslmed.aax7992.
Dempe S, et al. Antitumoral activity of parvovirus-mediated IL-2 and MCP-3/CCL7 delivery into human pancreatic cancer: implication of leucocyte recruitment. Cancer Immunol Immunother. 2012;61(11):2113–23.
Mehrotra PT, Grant AJ, Siegel JP. Synergistic effects of IL-7 and IL-12 on human T cell activation. J Immunol. 1995;154(10):5093–102.
Li F, et al. CCL5-armed oncolytic virus augments CCR5-engineered NK cell infiltration and antitumor efficiency. J Immunother Cancer. 2020. https://doi.org/10.1136/jitc-2019-000131.
Li Y, et al. CCL21/IL21-armed oncolytic adenovirus enhances antitumor activity against TERT-positive tumor cells. Virus Res. 2016;220:172–8.
Ye JF, et al. The combination of NK and CD8+T cells with CCL20/IL15-armed oncolytic adenoviruses enhances the growth suppression of TERT-positive tumor cells. Cell Immunol. 2017;318:35–41.
McDermott DF, et al. Atezolizumab, an anti-programmed death-ligand 1 antibody, in metastatic renal cell carcinoma: long-term safety, clinical activity, and immune correlates from a phase Ia study. J Clin Oncol. 2016;34(8):833–42.
Wang G, et al. An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat Commun. 2020;11(1):1395.
Bartee MY, Dunlap KM, Bartee E. Tumor-localized secretion of soluble PD1 enhances oncolytic virotherapy. Cancer Res. 2017;77(11):2952–63.
Vijayakumar G, Palese P, Goff PH. Oncolytic Newcastle disease virus expressing a checkpoint inhibitor as a radioenhancing agent for murine melanoma. EBioMedicine. 2019;49:96–105.
Guo ZS, et al. Bi- and tri-specific T cell engager-armed oncolytic viruses: next-generation cancer immunotherapy. Biomedicines. 2020;8(7):204.
Speck T, et al. Targeted BiTE expression by an oncolytic vector augments therapeutic efficacy against solid tumors. Clin Cancer Res. 2018;24(9):2128–37.
Freedman JD, et al. An oncolytic virus expressing a T-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res. 2018;78(24):6852–65.
de Sostoa J, et al. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J Immunother Cancer. 2019;7(1):19.
Wing A, et al. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol Res. 2018;6(5):605–16.
Jing Y, et al. Molecular effects of stromal-selective targeting by uPAR-retargeted oncolytic virus in breast cancer. Mol Cancer Res. 2017;15(10):1410–20.
Jing Y, et al. In vivo antitumor activity by dual stromal and tumor-targeted oncolytic measles viruses. Cancer Gene Ther. 2020;12:910–22.
Kudo D, Suto A, Hakamada K. The development of a novel therapeutic strategy to target hyaluronan in the extracellular matrix of pancreatic ductal adenocarcinoma. Int J Mol Sci. 2017;18(3):600.
Guedan S, et al. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol Ther. 2010;18(7):1275–83.
Laborda E, et al. A pRb-responsive, RGD-modified, and hyaluronidase-armed canine oncolytic adenovirus for application in veterinary oncology. Mol Ther. 2014;22(5):986–98.
Martinez-Quintanilla J, et al. Encapsulated stem cells loaded with hyaluronidase-expressing oncolytic virus for brain tumor therapy. Mol Ther. 2015;23(1):108–18.
Schäfer S, et al. Vaccinia virus-mediated intra-tumoral expression of matrix metalloproteinase 9 enhances oncolysis of PC-3 xenograft tumors. BMC Cancer. 2012;12:366.
Sette P, et al. GBM-targeted oHSV armed with matrix metalloproteinase 9 enhances anti-tumor activity and animal survival. Mol Ther Oncolytics. 2019;15:214–22.
Caja L, et al. TGF-β and the tissue microenvironment: relevance in fibrosis and cancer. Int J Mol Sci. 2018;19(5):1294.
Choi IK, et al. Effect of decorin on overcoming the extracellular matrix barrier for oncolytic virotherapy. Gene Ther. 2010;17(2):190–201.
Yang Y, et al. An oncolytic adenovirus targeting transforming growth factor β inhibits protumorigenic signals and produces immune activation: a novel approach to enhance anti-PD-1 and anti-CTLA-4 therapy. Hum Gene Ther. 2019;30(9):1117–32.
Seth P. et al. LyP-1-modified oncolytic adenoviruses targeting transforming growth factor β inhibit tumor growth and metastases and augment immune checkpoint inhibitor therapy in breast cancer mouse models. Hum Gene Ther. 2020; 31(15–16):863–80
Zhang W, et al. Efficacy of an oncolytic adenovirus driven by a chimeric promoter and armed with Decorin against renal cell carcinoma. Hum Gene Ther. 2020. https://doi.org/10.1089/hum.2019.352.
Li Y, et al. Potent antitumor effect of tumor microenvironment-targeted oncolytic adenovirus against desmoplastic pancreatic cancer. Int J Cancer. 2018;142(2):392–413.
Saleh R, Elkord E. Treg-mediated acquired resistance to immune checkpoint inhibitors. Cancer Lett. 2019;457:168–79.
Pitt JM, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. 2016;44(6):1255–69.
Woller N, et al. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol Ther. 2015;23(10):1630–40.
Goepfert K, et al. Rational combination of parvovirus H1 with CTLA-4 and PD-1 checkpoint inhibitors dampens the tumor induced immune silencing. Front Oncol. 2019;9:425.
Bourgeois-Daigneault MC, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Transl Med. 2018. https://doi.org/10.1126/scitranslmed.aao1641.
Liu Z, et al. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat Commun. 2017;8:14754.
Xu W, et al. LyP-1-modified oncolytic adenoviruses targeting transforming growth factor β inhibit tumor growth and metastases and augment immune checkpoint inhibitor therapy in breast cancer mouse models. Hum Gene Ther. 2020. https://doi.org/10.1089/hum.2020.078.
Acharya N, Sabatos-Peyton C, Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. J Immunother Cancer. 2020. https://doi.org/10.1136/jitc-2020-000911.
Sun F, et al. Dual but not single PD-1 or TIM-3 blockade enhances oncolytic virotherapy in refractory lung cancer. J Immunother Cancer. 2020. https://doi.org/10.1136/jitc-2019-000294.
Shi G, et al. Modulating the tumor microenvironment via oncolytic viruses and CSF-1R inhibition synergistically enhances anti-PD-1 immunotherapy. Mol Ther. 2019;27(1):244–60.
Bommareddy PK, et al. MEK inhibition enhances oncolytic virus immunotherapy through increased tumor cell killing and T cell activation. Sci Transl Med. 2018;10(471):eaau0417.
Samson A, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. 2018;10(422):eaam7577.
Kelly CM, et al. Objective response rate among patients with locally advanced or metastatic sarcoma treated with talimogene laherparepvec in combination with pembrolizumab: a phase 2 clinical trial. JAMA Oncol. 2020;6(3):402–8.
Chesney J, et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36(17):1658–67.
Hossain E. et al. Advantages of using paclitaxel in combination with oncolytic adenovirus utilizing RNA destabilization mechanism. Cancers (Basel). 2020;12(5):1210.
Angelova AL, et al. Complementary induction of immunogenic cell death by oncolytic parvovirus H-1PV and gemcitabine in pancreatic cancer. J Virol. 2014;88(10):5263–76.
Mahalingam D, et al. Pembrolizumab in combination with the oncolytic virus pelareorep and chemotherapy in patients with advanced pancreatic adenocarcinoma: a phase Ib study. Clin Cancer Res. 2020;26(1):71–81.
Moon EK, et al. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology. 2018;7(3):e1395997.
Rosewell Shaw A, et al. Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol Ther. 2017;25(11):2440–51.
Tanoue K, et al. Armed oncolytic adenovirus-expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res. 2017;77(8):2040–51.
Tentler JJ, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9(6):338–50.
Chen Q, et al. Cancer immunotherapies and humanized mouse drug testing platforms. Transl Oncol. 2019;12(7):987–95.
Zafar S, et al. CD40L coding oncolytic adenovirus allows long-term survival of humanized mice receiving dendritic cell therapy. Oncoimmunology. 2018;7(10):e1490856.
Kuryk L, Møller AW, Jaderberg M. Abscopal effect when combining oncolytic adenovirus and checkpoint inhibitor in a humanized NOG mouse model of melanoma. J Med Virol. 2019;91(9):1702–6.
Meraz IM, et al. An improved patient-derived xenograft humanized mouse model for evaluation of lung cancer immune responses. Cancer Immunol Res. 2019;7(8):1267–79.
Rosato RR, et al. Evaluation of anti-PD-1-based therapy against triple-negative breast cancer patient-derived xenograft tumors engrafted in humanized mouse models. Breast Cancer Res. 2018;20(1):108.
Capasso A, et al. Characterization of immune responses to anti-PD-1 mono and combination immunotherapy in hematopoietic humanized mice implanted with tumor xenografts. J Immunother Cancer. 2019;7(1):37.
Hill C, Carlisle R. Achieving systemic delivery of oncolytic viruses. Expert Opin Drug Deliv. 2019;16(6):607–20.
Evgin L, et al. Complement inhibition prevents oncolytic vaccinia virus neutralization in immune humans and cynomolgus macaques. Mol Ther. 2015;23(6):1066–76.
Filley AC, Dey M. Immune system, friend or foe of oncolytic virotherapy? Front Oncol. 2017;7:106.
Liu XQ, et al. Oncolytic herpes simplex virus tumor targeting and neutralization escape by engineering viral envelope glycoproteins. Drug Deliv. 2018;25(1):1950–62.
Tuzmen C, et al. Point mutations in retargeted gD eliminate the sensitivity of EGFR/EGFRvIII-targeted HSV to key neutralizing antibodies. Mol Ther Methods Clin Dev. 2020;16:145–54.
Kim J, et al. Efficient lung orthotopic tumor-growth suppression of oncolytic adenovirus complexed with RGD-targeted bioreducible polymer. Gene Ther. 2014;21(5):476–83.
Fisher KD, et al. Passive tumour targeting of polymer-coated adenovirus for cancer gene therapy. J Drug Target. 2007;15(7–8):546–51.
Mohammadi M, et al. Mesenchymal stem cell: a new horizon in cancer gene therapy. Cancer Gene Ther. 2016;23(9):285–6.
Hadryś A, et al. Mesenchymal stem cells as carriers for systemic delivery of oncolytic viruses. Eur J Pharmacol. 2020;874:172991.
Draganov DD, et al. Delivery of oncolytic vaccinia virus by matched allogeneic stem cells overcomes critical innate and adaptive immune barriers. J Transl Med. 2019;17(1):100.
Ruano D, et al. First-in-human, first-in-child trial of autologous MSCs carrying the oncolytic virus icovir-5 in patients with advanced tumors. Mol Ther. 2020;28(4):1033–42.
Kim J, et al. Stem cell-based cell carrier for targeted oncolytic virotherapy: translational opportunity and open questions. Viruses. 2015;7(12):6200–17.
Acknowledgements
None.
Funding
This work was financially supported by the National Natural Science Foundation of China (81622049, 81871989); the Shanghai Science and Technology Committee Program (19XD1420900) and Shanghai Education Commission Program (17SG04).
Author information
Authors and Affiliations
Contributions
Yalei Zhang and Ye Li contributed to the conception of this work and manuscript preparation, Peng Wang, Ling Qian and Kun Chen provided constructive suggestions for this work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Y., Li, Y., Chen, K. et al. Oncolytic virotherapy reverses the immunosuppressive tumor microenvironment and its potential in combination with immunotherapy. Cancer Cell Int 21, 262 (2021). https://doi.org/10.1186/s12935-021-01972-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-021-01972-2