
Abstract
Being generally regarded as safe, Kluyveromyces lactis has been widely taken for food, feed, and pharmaceutical applications, owing to its ability to achieve high levels of protein secretion and hence being suitable for industrial production of heterologous proteins. Production platform strains can be created through genetic engineering; while prototrophic cells without chromosomally accumulated antibiotics resistance genes have been generally preferred, arising the need for dominant counterselection. We report here the establishment of a convenient counterselection system based on a Frs2 variant, Frs2v, which is a mutant of the alpha-subunit of phenylalanyl-tRNA synthase capable of preferentially incorporating a toxic analog of phenylalanine, r-chloro-phenylalanine (4-CP), into proteins to bring about cell growth inhibition. We demonstrated that expression of Frs2v from an episomal plasmid in K. lactis could make the host cells sensitive to 2 mM 4-CP, and a Frs2v-expressing plasmid could be efficiently removed from the cells immediately after a single round of cell culturing in a 4-CP-contianing YPD medium. This Frs2v-based counterselection helped us attain scarless gene replacement in K. lactis without any prior engineering of the host cells. More importantly, counterselection with this system was proven to be functionally efficient also in Saccharomyces cerevisiae and Komagataella phaffii, suggestive of a broader application scope of the system in various yeast hosts. Collectively, this work has developed a strategy to enable rapid, convenient, and high-efficiency construction of prototrophic strains of K. lactis and possibly many other yeast species, and provided an important reference for establishing similar methods in other industrially important eukaryotic microbes.
Similar content being viewed by others


Introduction
Kluyveromyces lactis, a non-conventional yeast, was for the first used as a source of lactase in milk products to overcome lactose intolerance of consumers in the early 1950s [1]. Since then, it has been studied and industrially applied for decades. As a host for heterologous protein production, K. lactis exhibits excellent fermentation characterics [1, 2]. For instance, it can be fermented at high cell densities using inexpensive standard yeast culture media. And the great capability of K. lactis in secretory protein production has allowed for reaching high extracellular protein concentrations in large-scale fermentations [1, 3]. These, together with its GRAS (generally regarded as safe) status, have promoted the safe use of K. lactis in various food, feed, and pharmaceutical applications [2].
The basic requirement for heterologous protein expression in K. lactis is the engineering of the cells to harbor an expression cassette of genes of interest. Most often, the expression cassette was introduced into cells in the form of either an episomal plasmid fragment or a chromosomal insert, where an antibiotic resistance marker was usually employed to provide selective pressure. The use of antibiotic resistance genes has been beneficial for initial strain construction and testing, but would not be practical in industrial protein production. Large-scale growth of cells in the presence of antibiotics can be extremely costly, and the addition of antibiotics is unfriendly for the safe use of K. lactis in applications concerning food, feed, or pharmacy. Moreover, the accumulation and subsequent limitation of viable selection markers can be a big problem if engineering of multi-gene pathways were in need. Therefore, it is always good to perform clean genetic manipulations with the help of counterselection for marker recovery.
Counterselection using the Aspergillus nidulans amdS gene has been employed for marker-free gene deletion in K. lactis. Interestingly, this marker can be used in both positive selection and counterselection schemes [4,5,6], thus suggestive of no need for a second selection marker. This can be beneficial for construction of plasmids with relatively smaller sizes. However, it was reported that during the counterselection process, three rounds of cell growth had to be performed to completely remove the chromosomally integrated admS gene, that is, at least 6–9 days were generally required for the isolation of a pure mutant [4]. Several recyclable marker genes, such as URA3 [7,8,9], LYS2 [10], and MET15 [11], did have good performance in counterselection in yeast, but only in the corresponding auxotrophic yeast mutants, hence requiring prior engineering of the wild-type host cells. Although fortunately the development of the bacteriophage-derived Cre/loxP [12] and Flp/FRT [13] recombinase systems have allowed for recycling virtually any prototrophic markers, the repeated use of these systems would cause major chromosomal rearrangements [14]. In recent years, several PheS variants-based counterselection systems have been used in various bacterial species in a host-independent manner [15,16,17,18,19,20,21,22]. In Escherichia coli, where a PheS variant was first studied [23], PheS is the alpha-subunit of phenylalanyl-tRNA synthetase responsible for phenylalanine aminoacylation; whereas its derivative PheS* (PheS with T251A substitution) preferentially catalyzes the incorporation of an analog of phenylalanine, p-chloro-phenylalanine (4-CP), into proteins, resulting in cell death and therein providing robust counterselection pressure [24]. Conveniently, this method can be directly applied in prototrophic strains without any genetic modification.
We envisioned that genes coding for the alpha-subunit of phenylalanyl-tRNA synthetase could be functionally conserved in both prokaryotic and eukaryotic organisms, paving the possibility of establishing similar counterselection in K. lactis. To address the possibility, we attempted developing the FRS2 gene of K. lactis GG799 [25], which encodes the alpha-subunit of phenylalanyl-tRNA synthetase Frs2, a eukaryotic PheS counterpart, as a counterselection marker by converting it to a conditional-lethal variant, Frs2v, mimicking the bacterial PheS* mutant. We experimentally evidenced the capacity of Frs2v for counterselection in K. lactis. With its assistance, rapid plasmid curing and scarless genetic manipulation of K. lactis have been efficiently accomplished, providing a versatile genetic manipulation toolkit for this yeast. Interestingly, we found that the counterselection was also efficiently functional for other yeasts including S. cerevisiae and K. phaffii. Considering that FRS2 genes are present and highly conserved across yeast species, application of Frs2v-based counterselection in a broader host range of yeasts can be envisioned. This work would enable the development and further improvement of many yeasts as ideal production platforms for industrial applications, and has also provided an important reference for developing similar methods in other industrially important eukaryotic microbes.
Materials and methods
Strains, growth conditions and electroporation transformation
Yeast strains used or constructed in this work are listed in Table 1. Yeast cells were grown at 28ºC in a YPD medium (2% yeast extract, 1% bacto peptone, and 2% dextrose). If required, bleomycin was supplemented to the final concentration of 200 µg/mL for yeast and of 50 µg/mL for E. coli. Yeast competent cells were prepared and transformed with plasmids by electroporation using Bio-Rad Gene Pulser (0.1-cm gap cuvettes, 1.6 kV, 200 W, 25 µF) (Bio-Rad, Hercules, CA, USA) following the previously described methods [26]. Electroporated cells were incubated in a YPD medium with 500 mM sorbitol for 2 h at 28ºC prior to plating.
Plasmid construction and DNA manipulation
The E. coli-K. lactis shuttle vector pEKb was generated by ligating three PCR fragments together, including an autonomously replicating sequence originated from K. lactis (panARS) being functional in diverse yeast species [29], the replication origin amplified from the E. coli pUC19 plasmid (pUC ori), and a bleomycin-resistant gene (BleoR) allowing for positive selection in both hosts. The panARS and the pUC ori were assembled together via splicing and overlap extension PCR (SOE-PCR) [30], and then ligated with the BleoR marker after BamHI and HindIII digestions. The FRS2v gene variant was created through the SOE-PCR method [30] and cloned into pEKb using the T5 exonuclease-dependent DNA assembly (TEDA) method [31], giving the pFrs2v plasmid. DNA fragment of the GFP expression cassette was amplified from a previous constructed plasmid [32] and used for generating the pFrs2v-GFP plasmid upon its insertion into pFrs2v.
DNA fragment used to facilitate homologous recombination and hence HAP1 deletion was created by connecting the recombination arms, the gfp gene sequence, and the expression cassettes of the BleoR and the Frs2v markers. The fragments of each recombination arm were amplified from the genome of the wild-type GG799 strain, while that of the gfp gene sequence and the marker cassettes were amplified from the pFrs2v-GFP and pFrs2v plasmids, respectively. These fragments were connected by SOE-PCR [30].
All plasmids are listed in Table 2. All oligonucleotides were synthesized from GenScript (Nanjing, China) and listed in Table S3. Restriction enzymes and T5 exonuclease were purchased from New England Biolabs (Beijing) Ltd (Beijing, China).
4-CP sensitivity assay
Growth inhibition test was conducted for assaying the sensitivity of K. lactis strains to 4-CP. Overnight cultures of K. lactis strains were diluted in fresh YPD medium and growth to OD600 of 0.6. Then, each of the culture was serially 10-fold diluted up to 10− 5, and 5 µL of each dilution was spotted onto YPD4 agar plates where different concentrations of 4-CP were supplemented. The growth of each strain was photographically recorded after 72-hour incubation at 28ºC.
Plasmid curing
Cells of the pFrs2v-GFP transformants were cultured in a liquid YPD medium containing bleomycin (YPDB). Afterwards, the growing cells were spread onto a YPD agar plate with 2.0 mM 4-CP (YPD4) while without bleomycin. Cells that formed colonies were regarded as those lost the plasmid.
FACS analysis and florescence microscopy
Using similar protocols described previously [22, 32,33,34], cells were washed with phosphate buffered saline (PBS) twice, resuspended into PBS to a concentration of 107/cells/mL, and analyzed with a Beckman CytoFLEX FCM (Beckman Coulter, Inc., USA) with PBS as the sheath fluid. The GFP fluorescent intensity of cells was detected with the FITC channel, 525/40 nm band pass. Fluorescent images of cells were obtained using a Zeiss laser-scanning microscope (Zeiss, German) with a 488-nm argon-ion laser.
Construction and screening of HAP1 deletant
The DNA fragment for HAP1 replacement was introduced into yeast cells by electroporation. Electroporated cells were spread on an YPDB agar plate and incubated at 28ºC until colonies were seen. Cells of the transformants were grown up in a liquid YPDB medium, and then spread on a YPD4 agar. Cells that formed colonies were regarded as HAP1 mutant candidates and subjected to colony PCR screening using primers listed in Table S2. The PCR products were analysed by agarose gel electrophoresis.
Results and discussion
Identifying the K. lactis GG799 Frs2 and constructing its Frs2v derivate
The complete genome sequence of K. lactis GG799 has been publicly available [25], yet without annotation. In order to find out the sequence encoding the hypothetical PheS ortholog from the K. lactis GG799 genome, we performed local BLAST search with the NCBI BLAST+ (tblastn) program, taking amino acid sequence of the Phe-tRNA synthetase alpha-subunit (Frs2) from K. lactis strain CBS 2105 (GenBank: QEU58128) as an input. The BLAST result suggested a 1500-bp sequence spanning positions 2,348,421 to 2,349,920 of the chromosome F of K. lactis GG799 (Table 1), forming a complete ORF predictably coding for a 499-aa (amino acid) protein. Strikingly, the sequence of this peptide showed a near-100% identity to that of the Frs2 of K. lactis CBS2105 (Fig. 1A).
We then performed amino acid sequence alignment of the identified Frs2 with several bacterial PheS proteins, looking for the residue counterpart in Frs2 corresponding to the T251 residue of the E. coli PheS [24]. A conserved TEPSxE motif was seen in all the analyzed proteins, in which the T residue corresponds to the T251 of the E. coli PheS and the T411 of the K. lactis Frs2, respectively (Fig. 1B). Alike the T251A substitution previously made to the E. coli PheS [24], a T411A substitution was made to the K. lactis Frs2, giving Frs2v.
Determining the sensitivity of Frs2v-expressing K. lactis cells to 4-CP
Previously, we and others have reported that higher levels expression of the mutated marker genes conferred much better performances in functional competition with the respective originals in respect of 4-CP incorporation and hence cell growth inhibition [20, 22, 35]. Therefore, a Frs2v-expression cassette with the strong constitutive PADH1 promoter [36] was constructed and cloned onto pEKb, an E. coli-K. lactis shuttle vector expressing bleomycin resistance (BleoR), yielding the pFrs2v plasmid. Electroporation of this construct into K. lactis cells obtained as many transformants as that of the pEKb shuttle vector, showing a similar level of transformation efficiencies. From each transformation, three transformants were randomly chosen and individually cultured in liquid YPD media without 4-CP supplementation. No obvious growth retardation was observed for all of them (data not shown), suggestive of no influence of Frs2v expression on cell growth.
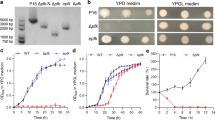
In order to examined the 4-CP sensitivity conferred by Frs2v to the K. lactis cells, we assayed growth inhibition of the pFrs2v transformants using YPDB plates individually containing 0, 0.2, 1.0, 2.0, and 4.8 mM of 4-CP. As shown in Fig. 2A, without 4-CP (0 mM), transformants of both pFrs2v and pEKb (as a reference) were normally and evenly grown up, including the 10− 5 dilutions. As the concentration of 4-CP was gradually increasingly supplemented, the inhibition effect on the growth of the pFrs2v transformants became clearer. Very few cells from the 103-fold diluted pFrs2v transformants could grow up when spotted on YPD agar plates containing 2.0 mM of 4-CP (YPD4), whereas under the same condition the cells carrying pEKb kept growing up normally.

K. lactis Frs2 identification and Frs2v variant construction. (A) Amino acid sequence alignment of Frs2 proteins from two K. lactis strains, GG799 and CSB2105. (B) Amino acid sequence alignment of Frs2 from K. lactis GG799 and PheS from E. coli K-12. The conserved TEPSxE motif was highlighted with a red box, within which the T residual being subject to mutagenesis is indicated with a red arrow
The results supported that Frs2v can be employed as an efficient counterselection marker for K. lactis, to the best of our knowledge, being the first eukaryotic counterpart of PheS for counterselection in yeast. Considering its no requirement for pretreatment of the wild-type host cells at all, it would represent one of currently the most convenient counterselectable systems for yeast.
Attaining efficient plasmid-curing upon Frs2v-mediated counterselection
Next, we performed a plasmid-curing assay to verify the feasibility of Frs2v for counterselection. For a more straightforward observation of the removal of a Frs2v-bearing plasmid, a green fluorescent protein (GFP) expression cassette was inserted in the pFrs2v plasmid, generating pFrs2v-GFP. This would allow us to monitor the plasmid presence/absence by measuring the GFP fluorescence intensity through flow cytometry analysis. After electroporating the pFrs2v-GFP plasmid into K. lactis cells, many transformants appeared on a bleomycin-containing YPD plate (YPDB) without 4-CP supplementation. Cells of a randomly chosen transformant were transferred into a liquid 4-CP-containing medium of YPD (YPD4) or, of YPDB (YPDB4), for 48-hour incubation. They could not grow up in YPDB4 while grew normally in YPD4, and the growing cells could form colonies on agar plate of YPD4 but not YPDB4 (Fig. 2B).

Plasmid curing using the Frs2v as a counterselection. (A) Growth inhibition of Frs2v-expressing strains by 4-CP. Cell cultures of transformants either carrying the shuttle vector pEKb, or the Frs2v-expressing plasmid pFrs2v, were serially 10-fold diluted. Dilutions were spotted onto YPD plates containing 4-CP at the indicated concentrations. (B) Examination of the counterselection effect of 4-CP on plasmid curing from the Frs2v transformants. (C) Detection of GFP signal in the cells of the pEKb transformant, and that of the pFrs2v-GFP transformant before and after plasmid curing via 4-CP counterselection
In addition, signal of green fluorescence was detected in the cells before counterselection, whereas was not detectable in those suffered 4-CP treatment (Fig. 2C). These combined results suggested that the cells have completely discarded the Frs2v-expression plasmid and hence lost the resistance to bleomycin after a single round of 4-CP counterselection, demonstrating the effectiveness of the Frs2v marker for counterselection in K. lactis.
Accomplishing scarless genetic manipulation of K. Lactis via Frs2v-based counterselection
Having constructed the high-efficiency Frs2v counterselection marker, we inferred its usefulness for scarless genetic manipulation of K. lactis and exemplified such by replacing the non-essential HAP1 gene [37] with a gfp gene in an insertion-and-excision manner. As illustrated in Fig. 3A, the method simply included a single DNA fragment consisting of 3 recombination arms, the gfp gene, and a block of selection markers, that is, a gene arm (G-arm) composed of the first 948 bp coding sequences of HAP1, a left arm (L-arm) and a right arm (R-arm) corresponding to sequences immediately upstream and downstream of HAP1, respectively, sandwiching the gfp gene, and a block formed by two expression cassettes of selection markers, BleoR and Frs2v for positive selection and counterselection, respectively.
Upon introducing the DNA fragment into K. lactis cells, bleomycin-resistant transformants, designated as INT strains, were selected by the BleoR marker after integration of the entire DNA donor into the HAP1 locus through recombination between the G-arm and the R-arm. The transformants could be seen on an YPDB agar plate after 2–3 days. Subsequently, the markers were excised via recombination between the homologous L-arms forced by 2.0 mM 4-CP selection via simply culturing the transformants in a liquid YPD4 medium for 2 days, leaving only the gfp gene at the HAP1 locus. This gave the Δhap1::gfp strain in which the transcription of the gfp gene was set to be driven by the native HAP1 promoter. Both the integration (int.) and excision (ex.) were verified by PCR analyses using the primer set of chk_fwd + chk_rev, amplifying products with predicted sizes of 10,782 bp and 2,992 bp, respectively; whereas a 6,034-bp product was expected when the genome DNA of the wild-type cells was taken as a template (Fig. 3B). The fluorescence of the chromosomally incorporated GFP was observed using microscopy in both the strains of INT and Δhap1::gfp (Fig. 3C). Much stronger fluorescence intensity was seen in the Δhap1::gfp cells, presumably due to better cell growth after release of the bleomycin conferred selection pressure. These results demonstrated that an intended mutation could be precisely made to the K. lactis chromosome in a total of only 4–5 days. Conclusively, with the assistance of Frs2v-based counterselection, simplified, convenient, yet highly efficient genetic modifications of K. lactis genome can be readily attainable as designed.

Scarless chromosomal modification with the assistance of Frs2v for counterselection. (A) Schematic showing design of the replacement of HAP1 with gfp. A DNA stretch was designed to contain the bleomycin resistance gene (bleoR), a Frs2v-expression cassette, a gfp gene, and three arms (G-arm, L-arm, and R-arm) for homologous recombination. Transformants each with a DNA insert were selected on bleomycin; while excision of both the bleoR and the FRS2v markers was forced to occur in the form of recombination between two L-arms by 4-CP selection, giving the expected editing outcome of HAP1 replacement by gfp. (B) PCR amplification verifying the strains of INT and Δhap1::gfp using the primer set of chk_fwd and chk_rev indicated in (A). The predicted sizes of PCR products in the INT strain (int.) and the Δhap1::gfp mutant (ex.), as well as the wild-type cells (wt), are shown. M, DNA size marker. (C) Microscopic evaluation of GFP fluorescence
We believe that, equipped with this counterselection system, genetic manipulation toolkits, such as the advanced CRISPR-Cas-based technologies, would generally perform better in yeast genome editing, being beneficial for the development and further improvement of yeast strains. For instance, its capacity in curing of the genome editing plasmid, a process reported to be tedious and time-consuming, can make the host cells be rapidly ready for the next round of editing immediately after the Frs2v-mediated counterselection, thus expediting multi-round genome engineering.
Using the same K. lactis Frs2v for counterselection in yeasts other than K. lactis

Application of the Frs2v from K. lactis for counterselection in S. cerevisiae and K. phaffii. (A) Multiple sequence alignment of Frs2 orthologs derived from yeasts, including K. lactis GG799 (Kla shown in red), S. cerevisiae S288C (Sce), K. phaffii GS115 (Kph), Candida boidinii BVG19_g4589 (Cbo), Eremothecium cymbalariae DBVPG 7215 (Ecy), E. sinecaudum HDL 489Cp (Esi), Hanseniaspora osmophila (Hos), Kazachstania Africana CBS 2517 (Kaf), K. dobzhanskii CBS 2104 (Kdo), K. marxianus DMKU3 1042 (Kma), K. naganishii CBS 8797 (Kna), Lachancea meyersii CBS 8951 (Lme), L. mirantina LAMI_0G08526g1-1 (Lmi), L. thermotolerans CBS 6340 (Lth), Naumovozyma dairenensis CBS 421 (Nda), Saccharomycodes ludwigii SCDLUD_000677 (Slu), Vanderwaltozyma polyspora DSM 70,294 (Vpo), and Zygosaccharomyces parabailii YFL022C (Zpa). The T411 residual of the K. lactis Frs2 and the corresponding T residuals in other Frs2 proteins are indicated with a red box. (B) 4-CP sensitivity of S. cerevisiae and K. phaffii cells with or without a Frs2v-expressing plasmid. Cell cultures of transformants of each strain either harboring the pEKb vector, or the Frs2v-expressing plasmid pFrs2v, were serially 10-fold diluted. Dilutions were spotted onto YPD plates containing 4-CP at the indicated concentrations. (C) Neighbor-joining phylogenetic tree based on amino acid sequences of Frs2 orthologs showing the phylogenetic relationship of the Frs2 from K. lactis GG799 (shown in red fonts) and that from the selected yeast species
Amino acid sequence alignment of Frs2 from K. lactis with several Frs2 homologues from other yeasts was performed, revealing their extremely high overall identity (Fig. 4A). Given this observation, the Frs2v-mediated counterselection might be generally applicable in many yeast species. For initial confirmation, we tested the counterselection capability of the Frs2v in two commonly used yeast strains, i.e. S. cerevisiae S288C and K. phaffii GS115. Results showed that cells of both yeasts expressing Frs2v were highly sensitive to 4-CP, although the lowest concentrations for growth inhibition of each yeast varied (Fig. 4B). Interestingly, according to the constructed phylogenetic tree of the Frs2 proteins, the enzymes from K. lactis GG799 and K. phaffii GS115 fell in distinct clades away apart (Fig. 4C). This was suggestive of that such Frs2v-based counterselection could be applicable in a broader range of yeast hosts.
Conclusions
A counterselectable system based on a variant of the Frs2 protein derived from K. lactis GG799 was established, which not only worked in K. lactis to help attain efficient plasmid curing and scarless chromosomal engineering, but also was practicable in other yeasts such as the commonly applied S. cerevisiae and K. phaffii, providing a versatile tool for genetic manipulations of yeasts. Given the fact that no prior engineering of the host cells was required at all, it would represent one of currently the most convenient broad-host counterselection systems to allow for the speed-up of constructing yeast production platforms.
Availability of data and material
The authors declare that the main data supporting the findings of this work are available within the article and its supplementary information files or from the corresponding authors upon reasonable request.
Data availability
No datasets were generated or analysed during the current study.
References
van Ooyen AJ, Dekker P, Huang M, Olsthoorn MM, Jacobs DI, Colussi PA, Taron CH. Heterologous protein production in the yeast Kluyveromyces Lactis. FEMS Yeast Res. 2006;6:381–92.
De Brabander P, Uitterhaegen E, Delmulle T, De Winter K, Soetaert W. Challenges and progress towards industrial recombinant protein production in yeasts: a review. Biotechnol Adv. 2023;64:108121.
Baghban R, Farajnia S, Rajabibazl M, Ghasemi Y, Mafi A, Hoseinpoor R, Rahbarnia L, Aria M. Yeast expression systems: overview and recent advances. Mol Biotechnol. 2019;61:365–84.
Ganatra MB, Vainauskas S, Hong JM, Taylor TE, Denson JP, Esposito D, Read JD, Schmeisser H, Zoon KC, Hartley JL, Taron CH. A set of aspartyl protease-deficient strains for improved expression of heterologous proteins in Kluyveromyces Lactis. FEMS Yeast Res. 2011;11:168–78.
Read JD, Colussi PA, Ganatra MB, Taron CH. Acetamide selection of Kluyveromyces lactis cells transformed with an integrative vector leads to high-frequency formation of multicopy strains. Appl Environ Microbiol. 2007;73:5088–96.
Colussi PA, Taron CH. Kluyveromyces Lactis LAC4 promoter variants that lack function in bacteria but retain full function in K. lactis. Appl Environ Microbiol. 2005;71:7092–8.
Langle-Rouault F, Jacobs E. A method for performing precise alterations in the yeast genome using a recycable selectable marker. Nucleic Acids Res. 1995;23:3079–81.
Shuster JR, Moyer D, Irvine B. Sequence of the Kluyveromyces lactis URA3 gene. Nucleic Acids Res. 1987;15:8573.
Losberger C, Ernst JF. Sequence and transcript analysis of the C. Albicans URA3 gene encoding orotidine-5’-phosphate decarboxylase. Curr Genet. 1989;16:153–8.
Chattoo BB, Sherman F, Azubalis DA, Fjellstedt TA, Mehnert D, Ogur M. Selection of lys2 mutants of the yeast Saccharomyces cerevisiae by the utilization of alpha-AMINOADIPATE. Genetics. 1979;93:51–65.
Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–32.
Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH. A second set of loxP marker cassettes for cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002;30:e23.
Storici F, Coglievina M, Bruschi CV. A 2-microm DNA-based marker recycling system for multiple gene disruption in the yeast Saccharomyces cerevisiae. Yeast. 1999;15:271–83.
Delneri D, Tomlin GC, Wixon JL, Hutter A, Sefton M, Louis EJ, Oliver SG. Exploring redundancy in the yeast genome: an improved strategy for use of the cre-loxp system. Gene. 2000;252:127–35.
Ishikawa M, Yokoe S, Kato S, Hori K. Efficient counterselection for Methylococcus capsulatus (bath) by using a mutated pheS gene. Appl Environ Microbiol 2018, 84.
Kino Y, Nakayama-Imaohji H, Fujita M, Tada A, Yoneda S, Murakami K, Hashimoto M, Hayashi T, Okazaki K, Kuwahara T. Counterselection employing mutated pheS for markerless genetic deletion in Bacteroides species. Anaerobe. 2016;42:81–8.
Liu Y, He X, Zhu P, Cheng M, Hong Q, Yan X. pheS (AG) based rapid and efficient markerless mutagenesis in Methylotuvimicrobium. Front Microbiol. 2020;11:441.
Xin Y, Guo T, Mu Y, Kong J. Development of a counterselectable seamless mutagenesis system in lactic acid bacteria. Microb Cell Fact. 2017;16:116.
Ran Sapir S, Boichis E, Herskovits AA. Generation of markerless gene deletion mutants in Listeria monocytogenes using a mutated pheS for counterselection. Methods Mol Biol. 2022;2427:3–10.
Gao G, Wei D, Li G, Chen P, Wu L, Liu S, Zhang Y. Highly effective markerless genetic manipulation of streptococcus suis using a mutated PheS-based counterselectable marker. Front Microbiol. 2022;13:947821.
Schuster CF, Howard SA, Grundling A. Use of the counter selectable marker PheS* for genome engineering in Staphylococcus aureus. Microbiol (Reading). 2019;165:572–84.
Zheng Y, Fu H, Chen J, Li J, Bian Y, Hu P, Lei L, Liu Y, Yang J, Peng W. Development of a counterselectable system for rapid and efficient CRISPR-based genome engineering in Zymomonas mobilis. Microb Cell Fact. 2023;22:208.
Kast P, Hennecke H. Amino acid substrate specificity of Escherichia coli phenylalanyl-tRNA synthetase altered by distinct mutations. J Mol Biol. 1991;222:99–124.
Miyazaki K. Molecular engineering of a PheS counterselection marker for improved operating efficiency in Escherichia coli. Biotechniques. 2015;58:86–8.
Chuzel L, Ganatra MB, Schermerhorn KM, Gardner AF, Anton BP, Taron CH. Complete genome sequence of Kluyveromyces lactis strain GG799, a common yeast host for heterologous protein expression. Genome Announc 2017, 5.
Wang TT, Choi YJ, Lee BH. Transformation systems of non-Saccharomyces yeasts. Crit Rev Biotechnol. 2001;21:177–218.
Fisk DG, Ball CA, Dolinski K, Engel SR, Hong EL, Issel-Tarver L, Schwartz K, Sethuraman A, Botstein D, Cherry JM. Saccharomyces Genome Database P: Saccharomyces cerevisiae S288C genome annotation: a working hypothesis. Yeast. 2006;23:857–65.
Barone GD, Emmerstorfer-Augustin A, Biundo A, Pisano I, Coccetti P, Mapelli V, Camattari A. Industrial production of proteins with Pichia pastoris-komagataella phaffii. Biomolecules 2023, 13.
Liachko I, Dunham MJ. An autonomously replicating sequence for use in a wide range of budding yeasts. FEMS Yeast Res. 2014;14:364–7.
Horton RM, Cai Z, Ho SM, Pease LR. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. BioTechniques 8(5):528–535 (November 1990). Biotechniques 2013, 54:129–133.
Xia Y, Li K, Li J, Wang T, Gu L, Xun L. T5 exonuclease-dependent assembly offers a low-cost method for efficient cloning and site-directed mutagenesis. Nucleic Acids Res. 2019;47:e15.
Mei M, Zhai C, Li X, Zhou Y, Peng W, Ma L, Wang Q, Iverson BL, Zhang G, Yi L. Characterization of aromatic residue-controlled protein retention in the endoplasmic reticulum of Saccharomyces cerevisiae. J Biol Chem. 2017;292:20707–19.
Zheng Y, Han J, Wang B, Hu X, Li R, Shen W, Ma X, Ma L, Yi L, Yang S, Peng W. Characterization and repurposing of the endogenous type I-F CRISPR-Cas system of Zymomonas mobilis for genome engineering. Nucleic Acids Res. 2019;47:11461–75.
Li J, Wang BY, Yang Q, Si H, Zhao YT, Zheng YL, Peng WF. Enabling efficient genetic manipulations in a rare actinomycete Pseudonocardia alni Shahu. Front Microbiol 2022, 13.
Argov T, Rabinovich L, Sigal N, Herskovits AA. An effective counterselection system for Listeria monocytogenes and its use to characterize the monocin genomic region of strain 10403S. Appl Environ Microbiol 2017, 83.
Rosa JC, Colombo LT, Alvim MC, Avonce N, Van Dijck P, Passos FM. Metabolic engineering of Kluyveromyces lactis for L-ascorbic acid (vitamin C) biosynthesis. Microb Cell Fact. 2013;12:59.
Yu J, Jiang J, Fang Z, Li Y, Lv H, Liu J. Enhanced expression of heterologous inulinase in Kluyveromyces lactis by disruption of hap1 gene. Biotechnol Lett. 2010;32:507–12.
Funding
This work was financially supported by the Innovation Base for Introducing Talents of Discipline of Hubei Province (2019BJH021). WP and YZ acknowledge the support from the State Key Laboratory of Biocatalysis and Enzyme Engineering.
Author information
Authors and Affiliations
Contributions
WP and YZ designed the research; YZ, YD, PH, SW, JW, SL, JY and LL performed the experiments; WP and YZ wrote the manuscript. All authors contributed to data analyses, read, revised and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zheng, Y., Deng, Y., Hu, P. et al. A convenient broad-host counterselectable system endowing rapid genetic manipulations of Kluyveromyces lactis and other yeast species. Microb Cell Fact 23, 212 (2024). https://doi.org/10.1186/s12934-024-02488-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-024-02488-w