
Abstract
Background
1,2-propanediol (1,2-PDO) is widely used in the cosmetic, food, and drug industries with a worldwide consumption of over 1.5 million metric tons per year. Although efforts have been made to engineer microbial hosts such as Corynebacterium glutamicum to produce 1,2-PDO from renewable resources, the performance of such strains is still improvable to be competitive with existing petrochemical production routes.
Results
In this study, we enabled 1,2-PDO production in the genome-reduced strain C. glutamicum PC2 by introducing previously described modifications. The resulting strain showed reduced product formation but secreted 50 ± 1 mM d-lactate as byproduct. C. glutamicum PC2 lacks the d-lactate dehydrogenase which pointed to a yet unknown pathway relevant for 1,2-PDO production. Further analysis indicated that in C. glutamicum methylglyoxal, the precursor for 1,2-PDO synthesis, is detoxified with the antioxidant native mycothiol (MSH) by a glyoxalase-like system to lactoylmycothiol and converted to d-lactate which is rerouted into the central carbon metabolism at the level of pyruvate. Metabolomics of cell extracts of the empty vector-carrying wildtype, a 1,2-PDO producer and its derivative with inactive d-lactate dehydrogenase identified major mass peaks characteristic for lactoylmycothiol and its precursors MSH and glucosaminyl-myo-inositol, whereas the respective mass peaks were absent in a production strain with inactivated MSH synthesis. Deletion of mshA, encoding MSH synthase, in the 1,2-PDO producing strain C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) improved the product yield by 56% to 0.53 ± 0.01 mM1,2−PDO mMglucose−1 which is the highest value for C. glutamicum reported so far.
Conclusions
Genome reduced-strains are a useful basis to unravel metabolic constraints for strain engineering and disclosed in this study the pathway to detoxify methylglyoxal which represents a precursor for 1,2-PDO production. Subsequent inactivation of the competing pathway significantly improved the 1,2-PDO yield.
Similar content being viewed by others

Introduction
The C3-diols 1,2- and 1,3-propanediol (PDO) are important building blocks and are widely used in the polymer, food, cosmetic and drug industry [1, 2]. For 1,3-PDO and 1,2-PDO a global market size of around 1.4 billion and 0.4 billion US dollars is expected in the next years [2, 3]. Both diols are mainly produced from fossil fuels, but bio-based production processes, utilizing renewable resources, are favorable to tackle the concerns of climate change and the limited availability of fossil resources. Notably, first commercial microbial processes for 1,3-PDO production are readily available [4]. Although several natural 1,2-PDO producers are known and established microbial systems have been extensively engineered for its production, a sustainable process at the industrial level is still missing [2].
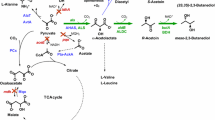
Corynebacterium glutamicum is a facultative anaerobic Gram-positive soil bacterium which is generally recognized as safe (GRAS), robust, and grows with several sugars, organic acids and phenolic compounds as single or combined carbon and energy sources [5,6,7,8,9,10]. Ample knowledge about the physiology, metabolic and regulatory networks has been gathered and a versatile toolbox for genetic engineering is available [6, 11,12,13,14]. This bacterium is known as industrial powerhouse for the production of amino acids such as l-glutamate and l-lysine at a scale of 6 million tons per year [5]. Moreover, sophisticated metabolic engineering approaches extended the product portfolio rapidly [15], also to diols such as 2,3-butanediol [16,17,18,19], 1,3-PDO [20] and 1,2-PDO [21]. Production of 1,2-PDO was initially achieved by heterologous expression of the Escherichia coli gene mgsA (encoding methylglyoxal synthase) and overexpression of a putative aldo-keto reductase (probably functioning as a methylglyoxal reductase) (Fig. 1; [21]). Biosynthesis was further improved by additional heterologous expression of the E. coli genes gldA (encoding glycerol dehydrogenase) and yqhD (encoding aldehyde reductase). Deletions of hdpA (encoding dihydroxyacetone phosphate phosphatase) and ldh (encoding l-lactate dehydrogenase) avoided the secretion of the side-products lactate and glycerol and improved 1,2-PDO production further (Fig. 1; [22]).
Methylglyoxal is a key intermediate in the 1,2-PDO production route, but also represents a potent cytotoxic compound which reacts with arginine, lysine, and cysteine residues and might lead to protein inactivation [23, 24]. Consequently, methylglyoxal is in many organisms detoxified to d-lactate by a GloAB-mediated glyoxalase system with the tripeptide glutathione (GSH). d-lactate is further oxidized to pyruvate which is subsequently channeled back into the central carbon metabolism (Fig. 1) [23, 24]. A characteristic of Actinobacteria, such as C. glutamicum, is the absence of GSH biosynthesis. However, in these organisms the low molecular weight thiol and non-enzymatic antioxidant mycothiol (MSH) is synthesized for detoxification instead [25]. MSH is synthezised from the intermediates myo-inositol and N-acetyl-glucosamine via glucosaminyl-myo-inositol by five enzymatic steps encoded by the genes mshA, mshA2, mshB, mshC and mshD [26]. However, no GloAB homologs have been identified in C. glutamicum and the mechanism for detoxification of methylglyoxal is elusive so far. Besides MSH as antioxidant, the transcriptional regulator OxyR plays an important role in the oxidative stress response in C. glutamicum. Under unstressed conditions this master regulator represses genes encoding enzymes such as catalase. As a result, an oxyR deletion mutant shows increased resistance to H2O2 [27].
The concept of genome reduction to improve physiological characteristics and create optimized hosts for the industrial environment has been applied to many organisms [28] as well as to C. glutamicum [29, 30]. In a systematic top-down approach all genes of the C. glutamicum genome were ranked by their relevance for growth in minimal medium with glucose as sole carbon and energy source. The proposed gene clusters were individually deleted and the genome-reduced strains (GRS) were screened for their growth phenotype [29]. In a follow-up study the deletions of the most promising genomic regions were combined in a stepwise manner, leading to the pre-chassis strains PC1 and PC2, with 8.5% and 12.6% reduced genomes, respectively. While both of these strains grew like the wildtype, the finally engineered chassis strain C1 (13.4% genome-reduction) showed a growth deficit on acetate due to an unwanted mutation in the promoter of the ramA gene, which was repaired yielding the final genome-reduced derivative C1* [30]. Recently, some intermediate strains of the aforementioned genome-reduction approach were utilized to screen for improved heterologous cutinase secretion in C. glutamicum [31].
In this study, we introduced known genetic modifications to enable 1,2-PDO production [22] into the genome-reduced strain C. glutamicum PC2 [30] and its parental strain GRS [29] (in later studies also called CR099). Only in the PC2 genetic background, we observed the formation of d-lactate as byproduct, which was attributed to the lack of the respective d-lactate dehydrogenase and pointed to a yet unknown pathway relevant for 1,2-PDO production. Further analysis indicated that in C. glutamicum methylglyoxal, the precursor for 1,2-PDO synthesis, is converted with MSH by a glyoxalase-like system to lactoylmycothiol and further to d-lactate which is channeled back into the central carbon metabolism on the level of pyruvate (Fig. 1).

Overview of the 1,2-PDO pathway introduced into C. glutamicum [22], including the proposed bypass from methylglyoxal to d-lactate and pyruvate via lactoylmycothiol in blue. Black arrows represent native pathways; dotted arrows indicate more than one reaction; green and disrupted arrows represent heterologously expressed proteins and deletions of gene sequences of mentioned proteins, respectively. Abbreviations: TCA, tricarboxylic acid cycle; DHA, dihydroxyacetone; DHAP, dihydroxyacetone phosphate; GAP, glyceraldehyde 3-phosphate; UDP-GlcNAc, uridine diphosphate-N-acetylglucosamine; 1-l-Ins-1-P, 1-l-myo-inositol 1-phosphate; GldA, glycerol dehydrogenase (from E. coli); HdpA, dihydroxyacetone phosphate phosphatase; MgsA, methylglyoxal synthase (from E. coli); Dld, quinone-dependent d-lactate dehydrogenase; LldD, quinone-dependent l-lactate dehydrogenase; Ldh, NAD-dependent l-lactate dehydrogenase; YqhD, aldehyde reductase (from E. coli); MshA, glycosyltransferase; “GloA”, proposed lactoylglutathione lyase homolog in C. glutamicum; “GloB”, proposed hydroxyacylglutathione hydrolase homolog in C. glutamicum
Results
Reconstruction of the 1,2-PDO production pathway in the C. glutamicum strains GRS and PC2
In a former study, it was shown that the deletion of hdpA and ldh and heterologous expression of the E. coli genes mgsA, yqhD and gldA led to the highest production of 1,2-PDO with C. glutamicum reported yet [22]. To analyze the production capacity of the genome-reduced strains C. glutamicum PC2 [30] and its parental strain GRS [29], the named genetic modifications were introduced into both strains. The resulting strains C. glutamicum GRSΔhdpAΔldh and PC2ΔhdpAΔldh, each carrying the plasmid pEKEx3-mgsA-yqhD-gldA were cultivated in modified CGXII minimal medium with glucose as sole carbon and energy source [22]. Compared to GRS, the strain with PC2 genetic background showed a similar growth rate (0.20 ± 0.01 vs. 0.22 ± 0.01 h-1), accumulated less biomass (max. CDW 6.60 ± 0.38 g L-1 vs. 8.25 ± 0.36) and exhibited a decreased glucose consumption (178 mM ± 7 vs. 126 ± 1 mM consumed) (Fig. 2A). While C. glutamicum GRSΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) produced up to 44 ± 3 mM 1,2-PDO (YP/S = 0.25 ± 0.01 mol mol-1), only 10 ± 2 mM (YP/S = 0.08 ± 0.02 mol mol-1) were secreted by C. glutamicum PC2ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) (Fig. 2B). Interestingly, the PC2-based strain produced up to 50 ± 1 mM lactate whereas for the GRS-derivative no lactate was determined in the supernatant (Fig. 2B). With the applied HPLC method it was not possible to distinguish between l- and d-lactate. However, since in both strains the ldh gene encoding l-lactate dehydrogenase is deleted, we speculated that d-lactate is secreted by C. glutamicum PC2ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA). Notably, compared to the GRS strain, the genome-reduced variant PC2 additionally lacks the dld gene encoding a quinone-dependent d-lactate dehydrogenase [32], which might be a reasonable explanation for the observed phenotype.

(A) Growth (black) and glucose consumption (green) and (B) 1,2-PDO (blue) and lactate (grey) accumulation of the strains C. glutamicum GRSΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) (GRS) and PC2ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) (PC2) in shaking flasks with modified CGXII minimal medium. Error bars represent the standard deviation of the mean values of three biological replicates
Impact of dld on the production of 1,2-propanediol in C. glutamicum
To clarify if the dld gene has a yet unknown effect on 1,2-PDO production in C. glutamicum, we deleted the gene in C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA). The resulting strain C. glutamicum ΔhdpAΔldhΔdld(pEKEx3-mgsA-yqhD-gldA) and its parental strain were cultivated in shaking flasks with modified CGXII medium and glucose as sole carbon and energy source. The additional deletion of dld led to a slightly decreased biomass concentration and reduced glucose consumption (Fig. 3A and B). Further, C. glutamicum ΔhdpAΔldhΔdld(pEKEx3-mgsA-yqhD-gldA) secreted up to 24 ± 1 mM lactate and produced three times less 1,2-PDO compared to the parental strain (59 ± 12 mM as compared to 20 ± 7 mM; Fig. 3C and D). The lactate concentrations in the culture supernatants determined via HPLC were confirmed and identified to be d-lactate by a specific enzyme assay (Figure S1). These findings are in agreement with the results obtained with the chassis strain PC2, confirming the relevance of d-lactate dehydrogenase for 1,2-PDO production and hint towards a not yet known pathway in C. glutamicum which causes a degradation of the main product or an intermediate of the production pathway.

(A) Growth (black), (B) glucose consumption (green), (C) 1,2-PDO (blue) and (D) lactate (grey) accumulation of the strains C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) (-) and C. glutamicum ΔhdpAΔldhΔdld(pEKEx3-mgsA-yqhD-gldA) (Δdld) in shaking flasks with modified CGXII minimal medium. Error bars represent the standard deviation of the mean values of three biological replicates
Inactivation of mycothiol biosynthesis improves 1,2-PDO production
We speculated that methylglyoxal might be converted to d-lactate by a MSH-dependent system similar to the glyoxalase system acting with GSH. To investigate this, we identified several targets potentially involved in the detoxification of methylglyoxal in C. glutamicum: deletion of the mshA gene encoding a glycosyltransferase to avoid MSH biosynthesis [25]; inactivation of the Xi-class mycothiol S-transferase [33] by deletion of the gene cg1426 (NCgl1216/mstX); deletion of the putative gloA and gloB homologs cg1073 as well as cg0071, cg1482 and cg1856, which were identified by annotations in the databases CoryneRegNet 7 [34] and Kyoto Encyclopedia of Genes and Genomes (KEGG) [35]. These deletions were introduced individually into C. glutamicum ΔhdpAΔldh, followed by the transformation of the newly constructed strains with the plasmid pEKEx3-mgsA-yqhD-gldA. The resulting strains were cultivated in shaking flasks with modified CGXII minimal medium and glucose as sole carbon and energy source. Neither inactivation of the genes putatively encoding GloAB homologs nor MstX led to a significant change in formed biomass, glucose consumption or product yield (p > 0.1) compared to the parental strain C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) which showed a YP/S of 0.34 ± 0.06 mM1,2−PDO mMglucose−1 after 72 h of fermentation (Fig. 4 and S2). In contrast, deletion of the mshA gene resulted in a 20% reduced biomass formation after 72 h and impaired glucose consumption, however, improved the YP/S significantly (p < 0.01) (Fig. 4 and S2). C. glutamicum ΔhdpAΔldhΔmshA(pEKEx3-mgsA-yqhD-gldA) showed a YP/S of 0.53 ± 0.01 mM1,2−PDO mMglucose−1 which is 56% higher compared to C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) (Fig. 4), while no lactate was formed. Because the mshA deletion abolishes MSH biosynthesis (see below), the improved YP/S of C. glutamicum ΔhdpAΔldhΔmshA(pEKEx3-mgsA-yqhD-gldA) might be a result of increased metylglyoxal availability and/or also of an overall elevated oxidative stress level. To investigate this effect, we deleted and overexpressed oxyR, encoding the master regulator of the oxidative stress response [27], in C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) and analyzed the impact on growth and 1,2-PDO production. Overexpression of oxyR, increasing the sensitivity to oxidative stress [27], led to a strongly decreased YP/Sof 0.05 ± 0.01 mM1,2−PDO mMglucose−1 whereas deletion of oxyR did not significantly impact 1,2-PDO production after 72 h (p > 0.1 regarding the yield), but inhibited growth and glucose uptake (Figure S3). Since the deletion of mshA increased the YP/S strongly, these results may indicate that in C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) a significant portion of methylglyoxal is rerouted to the central carbon metabolism with MSH as reaction partner.

Glucose-specific 1,2-PDO yields of C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) (-), C. glutamicum ΔhdpAΔldhΔmshA(pEKEx3-mgsA-yqhD-gldA) (ΔmshA), C. glutamicum ΔhdpAΔldhΔcg1426(pEKEx3-mgsA-yqhD-gldA) (Δcg1426), C. glutamicum ΔhdpAΔldhΔcg1073(pEKEx3-mgsA-yqhD-gldA) (Δcg1073), C. glutamicum ΔhdpAΔldhΔcg0071(pEKEx3-mgsA-yqhD-gldA) (Δcg0071), C. glutamicum ΔhdpAΔldhΔcg1482(pEKEx3-mgsA-yqhD-gldA) (Δcg1482) and C. glutamicum ΔhdpAΔldhΔcg1856(pEKEx3-mgsA-yqhD-gldA) (Δcg1856) cultivated in shaking flasks with modified CGXII minimal medium and glucose as sole carbon and energy source. Error bars represent the standard deviation of the mean values of three biological replicates
The metabolome of C. glutamicum comprises lactoylmycothiol
We expected that the detoxification of methylglyoxal with MSH would yield lactoylmycothiol as intermediate and therefore applied high-resolution LC-MS-Qtof metabolomics to analyze cell extracts of C. glutamicum WT(pEKEx3), C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA), C. glutamicum ΔhdpAΔldhΔdld(pEKEx3-mgsA-yqhD-gldA) and C. glutamicum ΔhdpAΔldhΔmshA(pEKEx3-mgsA-yqhD-gldA) from the production phase. In the analysis at negative ionization, for extracts of the WT(pEKEx3), C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) and its derivative carrying the dld deletion, accumulation of a metabolite at Rt 11.1 min was observed. Major mass peaks at m/z = 593.1429 and m/z = 557.1657 and respective expected 13C isotope peaks were detected. These correspond with high accuracy (δ 0.7 ppm and δ 0.2 ppm, respectively) to (C20H34N2O14S + 35Cl)− and (C20H33N2O14S)−. C20H34N2O14S is the sum formula of lactoylmycothiol. In a ms/ms-analysis the (C20H33N2O14S)− ion was fragmented. Based on the accurate masses of major fragment ions, we propose that the observed C20H34N2O14S metabolite is lactoylmycothiol (Figure S4). In contrast to extracts of C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA), we neither detected MSH, mycothione, and the MSH precursor glucosaminyl-myo-inositol nor lactoylmycothiol in extracts of C. glutamicum ΔhdpAΔldhΔmshA(pEKEx3-mgsA-yqhD-gldA) (Figure S5).
Discussion
Genome-reduced strains have been engineered to improve properties of microbes for industrial application, to study the physiological role of gene sets and to indentify essential genes to eventually design minimal genomes with streamlined functions [28, 36]. This concept has also been applied to the industrially relevant bacterium C. glutamicum which yielded a strain genealogy varying in the degree of genome reduction [30]. This genome-reduced strain library was recently utilized to screen for strains with improved heterologous secretion of cutinase [31]. In this study, we harnessed the genome-reduced derivative C. glutamicum PC2 [30] and its parental strain GRS [29] to unravel metabolic bottlenecks for the production of the industrial relevant bulk chemical 1,2-PDO.
The reconstruction of the previously described genetic modifications [22] to enable 1,2-PDO production in C. glutamicum PC2 led to reduced product formation accompanied by the accumulation of lactate. In C. glutamicum three enzymes are known, which are responsible for synthesis and utilization of lactate. The NADH-dependent l-lactate dehydrogenase LdhA, encoded by ldh, which is mainly responsible for l-lactate formation under excess of NADH [22, 37]. Further, C. glutamicum harbors quinone-dependent l- and d-lactate dehydrogenases (LldD and Dld), which are specific for the respective enantiomer and essential for its utilization [32, 38]. C. glutamicum PC2ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) lacks LdhA and due to the genome reduction Dld, as well. The applied enzyme assay identified accumulating lactate to be the d-enantiomer. These results suggested the presence of a metabolic route relevant for 1,2-PDO synthesis and with d-lactate as intermediate which was interrupted by the inactive Dld in the PC2 background. Further, no other genes encoding proteins obviously related to the described production pathway could be identified in the PC2 background (Table S2). However, the PC2-derivative showed higher accumulation of lactate and less propanediol compared to the wildtype background harboring the same genetic modification in addition to dld deletion (50 ± 1 mM vs. 10 ± 2 mM lactate and 24 ± 1 mM vs. 20 ± 7 mM propanediol; Figs. 2B and 3C and D).
Methylglyoxal is a precursor for 1,2-PDO synthesis but it also represents a cytotoxic intermediate leading to protein inactivation and oxidative stress [23, 24]. In E. coli and other organisms methylglyoxal is detoxified with GSH, forming the intermediate S-lactoylglutathione, which is further metabolized via the glyoxalase system GloAB to d-lactate [23, 24, 39]. In actinobacteria, such as C. glutamicum, the low molecular weight thiol MSH is present instead of GSH [25], however, a glyoxalase-like system based on MSH has not been identified so far. Recently, in the actinobacterium Streptomyces coelicolor a Ni2+-activated and MSH-dependent glyoxalase I enzyme was described [40]. However, BLASTx analysis [41] did not identify a homolog in C. glutamicum (data not shown). Also the deletion of the annotated gloA and gloB homologs cg1073 as well as cg0071, cg1482 and cg1856 in C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) did not influence growth, glucose consumption and product formation. Therefore, the identification of the glyoxylase system remains elusive and has to be elucidated in future studies. It was shown that overexpression of mshA, encoding a glycosyltransferase catalyzing the first step of MSH synthesis, and deletion of the gene mstX, encoding a MSH transferase, affected the robustness of C. glutamicum towards methylglyoxal [33, 42]. Taken together, these results indicate that, in analogy to GSH-dependent systems, methylglyoxal in C. glutamicum is detoxified with MSH via S-lactoylmycothiol by an unknown glyoxalase-like system yielding d-lactate. Notably, LC-MS-Qtof-based analysis identified major mass peaks characteristic for lactoylmycothiol, MSH and glucosaminyl-myo-inositol in cell extracts of the wildtype, C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) and its derivative with inactivated d-lactate dehydrogenase. For lactoylmycothiol, the exact mass and isotope pattern matches the expected sum formula C20H34N2O14S, and, although alternative structures cannot be excluded, the observed fragmentation pattern is in accordance with the proposed lactoylmycothiol structure. Moreover, in C. glutamicum ΔhdpAΔldhΔmshA(pEKEx3-mgsA-yqhD-gldA), with inactive MSH synthase, lactoylmycothiol and both precursors were not detectable. These findings support the presence of the MSH-dependent detoxification system converting methylglyoxal with MSH to d-lactate in C. glutamicum.
The secretion of up to 24 ± 1 mM of d-lactate by C. glutamicum ΔhdpAΔldhΔdld(pEKEx3-mgsA-yqhD-gldA) indicated an increased rerouting of methylglyoxal back into the central carbon metabolism in the parental strain with active Dld. Consequently, to improve the precursor availability, we inactivated MshA which significantly improved 1,2-PDO production. C. glutamicum ΔhdpAΔldhΔmshA(pEKEx3-mgsA-yqhD-gldA) showed a YP/S of 0.53 ± 0.01 mM1,2−PDO mMglucose−1 which is 56% higher compared to C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) and represents the highest reported value for C. glutamicum. Also in E. coli inactivation of the methylglyoxal detoxification system by deletion of gloA proved to be beneficial for 1,2-PDO production from glucose [43].
It should be noted, that the strain lacking mshA showed reduced growth and glucose consumption (Figure S2) which might indicate an impaired stress tolerance. In the closely related Mycobacterium tuberculosis MSH is essential for survival whereas for M. smegmatis it is not [44]. For E. coli GSH is dispensable, however, GSH-deficiency results in an increased sensitivity towards oxidative stress [45]. Also in C. glutamicum MSH synthesis is not essential but its absence was shown to increase the sensitivity against oxidative stress and some toxic compounds indicated by reduced growth and cell viability [26, 42, 46]. Notably, a MSH-deficient C. glutamicum strain, lacking mshC, possessed impaired growth in bioreactor cultivations at a pO2 of 30%, whereas at a pO2 level of 20% this growth defect was abolished [46]. The relevance of a proper functioning oxidative stress response for 1,2-PDO production is also indicated by the result that overexpression of oxyR in C. glutamicum ΔhdpAΔldh(pEKEx3-mgsA-yqhD-gldA) strongly diminished 1,2-PDO production. OxyR is the master regulator of the oxidative stress response in C. glutamicum [27] and under unstressed conditions this transcriptional regulator is acting as a repressor of its target genes (e.g. katA encoding catalase). Deletion of oxyR resulted in an increased resistance of the C. glutamicum mutant to hydrogen peroxide whereas its overexpression completely inhibited growth in the presence of H2O2 [47]. Therefore, a carefully adjusted oxygen transfer into the culture broth might be crucial for scale-up of fermentation processes based on MSH-deficient 1,2-PDO production strains.
Conclusion
Libraries with genome-reduced strains provide a valuable tool to identify novel targets for metabolic engineering. In this study, the genetic background of the genome-reduced C. glutamicum PC2 strain was the basis to disclose the pathway for detoxification of methylglyoxal with MSH to d-lactate and its relevance for 1,2-PDO production. Subsequent inactivation of this competing pathway significantly improved 1,2-PDO production in C. glutamicum. Regarding the detoxification of methylglyoxal, the low molecular weight thiol MSH in C. glutamicum, seems to play a role comparable to GSH in other organism, such as E. coli. However, the enzymatic machinery catalyzing the detoxification process with MSH has to be identified in future studies.
Materials and methods
Microorganisms, media and cultivation conditions
All C. glutamicum strains and plasmids used in this work are listed in Table 1. Escherichia coli DH5α [48] was used as host for plasmid construction and was grown in lysogeny broth (LB) or in 2x TY complex medium [49] at 37 °C and 120 rpm. For precultivation of C. glutamicum, LB medium with 2% (w/v) glucose was applied and the main cultivations were carried out in modified CGXII medium [22]. C. glutamicum was cultivated aerobically in 500 mL baffled shaking flasks with 50 mL medium at 30 °C on a rotary shaker at 120 rpm. For induction of gene expression, 1 mM isopropyl-β-D-thiogalactopyranosid (IPTG) and, if appropriate, kanamycin (25 or 50 µg mL− 1) and spectinomycin (100 µg mL− 1) were supplemented to the medium. For solid medium plates 18 g agar-agar L− 1 were added to the liquid medium.
Recombinant DNA work and construction of C. glutamicum deletion mutants
Oligonucleotides (Table S1) were purchased either from Eurofins MWG Operon (Ebersberg, Germany), metabion international AG (Planegg, Germany) or Sigma-Aldrich Chemie GmbH (Steinheim, Germany). The construction of plasmids (Table 1) was carried out as described in [9]. Purification and isolation of PCR products, plasmids and genomic DNA was conducted via “NucleoSpin® Gel and PCR Clean-up”, “NucleoSpin® Plasmid” and “NucleoSpin® Microbial DNA”, respectively, as recommended by MACHEREY-NAGEL GmbH & Co. KG (Düren, Germany). Enzymes were all purchased from New England Biolabs GmbH [(NEB), Frankfurt am Main, Germany] and used as recommended by the manufacturer. For cloning of pVWEx1-oxyR, pVWEx1 [52] was linearized via BamHI and the oxyR gene was PCR-amplified with primers #16 and #17 from genomic C. glutamicum DNA by Phusion® High-Fidelity DNA Polymerase. pVWEx1 and the insert were fused via Gibson assembly [53]. To construct pK19mobsacB [50] derivatives the plasmid was linearized via SmaI and the two homologous flanks for recombination were amplified via PCR, respectively. E.g. for the deletion of the mshA gene the primer pairs #19 + 20 and #21 + 22 (Table S1) were used. Eventually, the flanks and linearized pK19mobsacB were fused via Gibson assembly [53]. For subsequent transformation of E. coli DH5α the calcium chloride method was conducted [49]. Positive clones were identified by colony PCR applying Quick-Load R® Taq 2X Master Mix, validated via restriction digestion with appropriate enzymes and eventually the correct sequence of the inserts was confirmed via Sanger sequencing by Microsynth Seqlab GmbH (Göttingen, Germany).
For transformation and markerless gene deletions in C. glutamicum GRS and PC2 derivatives the procedure of [22] was applied: Electrocompetent cells [54] were transformed via electroporation [6] and, in case of gene deletions via pK19mobsacB, a subsequent two-step homologous recombination procedure followed [6]. All WTΔhdpAΔldh derivatives were engineered as described in [9]: Electrocompetent cells [55] were transformed via electroporation [56] and the subsequent two-step homologous recombination was carried out as described previously [50]. Successful gene deletions were confirmed via colony PCR as described above with the respective primer pairs listed in Table S1.
Analytical methods
Determination of biomass
OD600 (optical density at 600 nm) was measured with a spectrophotometer (either V-1200 of VWR International, Darmstadt, Germany or ULTROSPEC® 10 of Biochrom, Holliston, MA, United States) by diluting the samples into an OD600 range of 0.05 to 0.3. Cell dry weight (CDW) was calculated via a correlation factor of either 0.25 g L− 1 (V-1200) or 0.23 g L− 1 (ULTROSPEC® 10) per OD600 of 1. The growth rates (h− 1) were determined by linear regression in a semi-logarithmic blot and maximizing the coefficient of determination (R2) in the exponential growth phase. At the given time points 1 mL samples were taken, centrifuged (10 min, 16,000 or 21,300 x g) and the resulting supernatants were stored at -20 °C for further analysis.
GC-MS and HPLC analysis
The GC-MS-based quantification of 1,2-PDO and the HPLC measurement of glucose and lactate shown in Fig. 1 was conducted as described before [22].
Elsewhere, glucose, 1,2-PDO, lactate and acetol were determined via a modified HPLC method as described in [9], employing a 1260 Infinity II system (Agilent Technologies, Waldbronn, Germany), equipped with a Hi-Plex H column (7.7 × 300 mm, 8 μm) and a Hi-Plex H guard cartridge (3 × 5 mm, 8 μm) kept at 30 °C. As mobile phase 5 mM sulfuric acid (H2SO4) in water was applied with a flow rate of 0.4 mL min− 1, while signal acquisition was conducted via refractive index detector (RID) hold at 30 °C.
Metabolomics via LC-MS
For LC-MS analyses cell extracts were prepared as previously described for C. glutamicum [57]. From cultures grown as described above for HPLC analysis, cells were harvested via centrifugation (5 min, 3,000 x g, 4 °C), washed once with 0.5 volumes of saline (9 g L− 1 sodium chloride in water) and again centrifuged. The cell pellet was resuspended with boiling water to adjust a concentration of approximately 125 g CDW L− 1. This suspension was incubated three times in a bath with boiling water for 2 min with intermittent vortexing of 5 s. The cell extracts were collected by centrifugation (10 min, 20,000 x g, 4 °C), aliquoted and stored at -80 °C for further analysis.
LC-MS analyses were performed on a bio-inert 1290 series UHPLC system which was interfaced to a Q-TOF mass spectrometer (G6546AA, Agilent Technologies) via a dual Agilent jet stream electrospray ion source. MassHunter LC/MS Data Acquisition (version 10.1) and Qualitative Analysis software (version 10.0) from Agilent Technologies was used for data acquisition and data evaluation, respectively. The mass spectrometer was operated in low mass range (m/z 1700), Gas temp 325 °C, VCap 4000 V, Nozzle Voltage 2000 V. The instrument was auto tuned and calibrated according to manufacturer´s recommendations using ESI-L tuning mix (Agilent Technologies).
Extracts (5 µL injection volume) were separated on a InfinityLab Poroshell 120 HILIC-Z (150 × 2.1 mm, 2.7 μm particle size) using acetonitrile /10 mM CH3COONH4 in milli-Q H2O (pH 9.2), 9:1, v/v and acetonitrile /10 mM CH3COONH4 in milli-Q H2O (pH 9.2), 1:9, v/v, as eluents A and B, respectively. The following binary gradient program at a flow rate of 200 µL min− 1 was applied: 0–1 min isocratic 0% B, 1–31 min linear from 0 to 75% B, 31–35 min linear from 75 to 100% B, 35–40 min isocratic 100% B; 40–50 min linear from 100 to 0% B, 50–65 min, isocratic, 0% B. The column temperature was maintained at 40 °C and the autosampler temperature at 4 °C. Eluting compounds were detected from m/z 50-1700 in negative ion mode. Mass spectra were acquired in centroid mode using an acquisition rate of 1.4 spectra per second.
Collision-induced dissociation (CID) mass spectra were acquired in targeted MS/MS mode. Precursor ions were isolated by the quadrupole using an isolation width of m/z 4 and fragmented inside the collision cell by applying collision energies in the range of 10, 20, and 30 V and nitrogen as collision gas. Fragment ions were detected from m/z 100–1000. For reference mass correction a solution of purine (5 µM) and hexakis-(2,2,3,3-tetrafluoropropoxy)phosphazine (2.5 µM) in acetonitrile/water, 95/5 (v/v) % was continuously introduced through the second sprayer of the dual ion source at a flow rate of 10 µL min-1 using an isocratic HPLC pump (G7110B, Agilent Technologies) equipped with a 1:100 splitting.
Metabolites were annotated according to high resolution mass: Lactoylmycothiol, C20H34N2O14S, Rt = 11.1 min, (m-H)−: 557.1657 (expect: 557.1658, δ 0.2 ppm), m-35Cl−: 593.1429 (expect: 593.1425, δ 0.7 ppm), (m-C3H6O3-H)−: 467.1338 (co-eluting, putative in-source fragment, expect: 467.1341), MSH C17H30N2O12S, Rt = 11.4 min, (m-H)−: 485.1450 (expect: 485.1447, δ 0.7 ppm), mycothione, Rt = 16.1 min C34H57N4O24S2, (m-H)−: 969.2807 (expect: 969.2809, δ 0.2 ppm), glucosaminyl-myo-inositol C12H22NO10 Rt = 14.7 min (m-H)−: 400.1459 (expect: 400.1460, δ 0.3 ppm).
Enzyme assay specific for d-lactate
For the specific, enzymatic determination of d-lactate the “D-LACTIC ACID (D-LACTATE) (Rapid) Assay Kit (K-DATE)” from Megazyme Ltd. (Wicklow, Ireland) was purchased and applied as recommended by the manufacturer. The assay was conducted in a 96-well format with a Tecan Spark and SparkControl V 2.3 (Tecan Group Ltd., Männedorf, Switzerland), while for quantification an external calibration curve ranging from 0 to 0.15 g L− 1 was prepared with a d-lactic acid standard solution.
Determination of significance
The significance (p-value) was calculated via “two-sample t-test assuming equal variances” in Excel 2016 (Microsoft Corporation, Redmond, Washington, USA).
Data availability
No datasets were generated or analysed during the current study.
References
Liu H, Xu Y, Zheng Z, Liu D. 1,3-Propanediol and its copolymers: research, development and industrialization. Biotechnol J. 2010;5:1137–48. https://doi.org/10.1002/biot.201000140.
Tao Y-M, Bu C-Y, Zou L-H, Hu Y-L, Zheng Z-J, Ouyang J. A comprehensive review on microbial production of 1,2-propanediol: micro-organisms, metabolic pathways, and metabolic engineering. Biotechnol Biofuels. 2021;14:216. https://doi.org/10.1186/s13068-021-02067-w.
Zhu Y, Wang Y, Gao H, Wang H, Wan Z, Jiang Y et al. Current advances in microbial production of 1,3-propanediol. Biofuels, Bioproducts and Biorefining. 2021;15:1566–83. https://doi.org/10.1002/bbb.2254.
Ewing TA, Nouse N, van Lint M, van Haveren J, Hugenholtz J, van Es DS. Fermentation for the production of biobased chemicals in a circular economy: a perspective for the period 2022–2050. Green Chem. 2022;24:6373–405. https://doi.org/10.1039/d1gc04758b.
Becker J, Wittmann C. A field of dreams: lignin valorization into chemicals, materials, fuels, and health-care products. Biotechnol Adv. 2019;37:107360. https://doi.org/10.1016/j.biotechadv.2019.02.016.
Eggeling L, Bott M. Handbook of Corynebacterium glutamicum. CRC; 2005.
Merkens H, Beckers G, Wirtz A, Burkovski A. Vanillate metabolism in Corynebacterium glutamicum. Curr Microbiol. 2005;51:59–65. https://doi.org/10.1007/s00284-005-4531-8.
Nishimura T, Vertès AA, Shinoda Y, Inui M, Yukawa H. Anaerobic growth of Corynebacterium glutamicum using nitrate as a terminal electron acceptor. Appl Microbiol Biotechnol. 2007;75:889–97. https://doi.org/10.1007/s00253-007-0879-y.
Siebert D, Altenbuchner J, Blombach B. A timed off-switch for dynamic control of Gene expression in Corynebacterium glutamicum. Front Bioeng Biotechnol. 2021;9:704681. https://doi.org/10.3389/fbioe.2021.704681.
Takeno S, Ohnishi J, Komatsu T, Masaki T, Sen K, Ikeda M. Anaerobic growth and potential for amino acid production by nitrate respiration in Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2007;75:1173–82. https://doi.org/10.1007/s00253-007-0926-8.
Burkovski A. Corynebacteria: Genomics and Molecular Biology. Norfolk: Caister Academic; 2008.
Yukawa H, Inui M. Corynebacterium glutamicum. Berlin, Heidelberg: Springer Berlin Heidelberg; 2013.
Inui M, Toyoda K. Corynebacterium glutamicum. Cham: Springer International Publishing; 2020.
Wang Q, Zhang J, Al Makishah NH, Sun X, Wen Z, Jiang Y, Yang S. Advances and perspectives for genome editing tools of Corynebacterium glutamicum. Front Microbiol. 2021;12:654058. https://doi.org/10.3389/fmicb.2021.654058.
Becker J, Rohles CM, Wittmann C. Metabolically engineered Corynebacterium glutamicum for bio-based production of chemicals, fuels, materials, and healthcare products. Metab Eng. 2018;50:122–41. https://doi.org/10.1016/j.ymben.2018.07.008.
Yang J, Kim B, Kim H, Kweon Y, Lee S, Lee J. Industrial production of 2,3-butanediol from the engineered Corynebacterium glutamicum. Appl Biochem Biotechnol. 2015;176:2303–13. https://doi.org/10.1007/s12010-015-1719-7.
Radoš D, Carvalho AL, Wieschalka S, Neves AR, Blombach B, Eikmanns BJ, Santos H. Engineering Corynebacterium glutamicum for the production of 2,3-butanediol. Microb Cell Fact. 2015;14:171. https://doi.org/10.1186/s12934-015-0362-x.
Radoš D, Turner DL, Catarino T, Hoffart E, Neves AR, Eikmanns BJ, et al. Stereospecificity of Corynebacterium glutamicum 2,3-butanediol dehydrogenase and implications for the stereochemical purity of bioproduced 2,3-butanediol. Appl Microbiol Biotechnol. 2016;100:10573–83. https://doi.org/10.1007/s00253-016-7860-6.
Kou M, Cui Z, Fu J, Dai W, Wang Z, Chen T. Metabolic engineering of Corynebacterium glutamicum for efficient production of optically pure (2R,3R)-2,3-butanediol. Microb Cell Fact. 2022;21:150. https://doi.org/10.1186/s12934-022-01875-5.
Li Z, Dong Y, Liu Y, Cen X, Liu D, Chen Z. Systems metabolic engineering of Corynebacterium glutamicum for high-level production of 1,3-propanediol from glucose and xylose. Metab Eng. 2022;70:79–88. https://doi.org/10.1016/j.ymben.2022.01.006.
Niimi S, Suzuki N, Inui M, Yukawa H. Metabolic engineering of 1,2-propanediol pathways in Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2011;90:1721–9. https://doi.org/10.1007/s00253-011-3190-x.
Siebert D, Wendisch VF. Metabolic pathway engineering for production of 1,2-propanediol and 1-propanol by Corynebacterium glutamicum. Biotechnol Biofuels. 2015;8:91. https://doi.org/10.1186/s13068-015-0269-0.
Allaman I, Bélanger M, Magistretti PJ. Methylglyoxal, the dark side of glycolysis. Front Neurosci. 2015;9:23. https://doi.org/10.3389/fnins.2015.00023.
Anaya-Sanchez A, Feng Y, Berude JC, Portnoy DA. Detoxification of methylglyoxal by the glyoxalase system is required for glutathione availability and virulence activation in Listeria monocytogenes. PLoS Pathog. 2021;17:e1009819. https://doi.org/10.1371/journal.ppat.1009819.
Vetting MW, Frantom PA, Blanchard JS. Structural and enzymatic analysis of MshA from Corynebacterium glutamicum: substrate-assisted catalysis. J Biol Chem. 2008;283:15834–44. https://doi.org/10.1074/jbc.M801017200.
Liu Y-B, Long M-X, Yin Y-J, Si M-R, Zhang L, Lu Z-Q, et al. Physiological roles of mycothiol in detoxification and tolerance to multiple poisonous chemicals in Corynebacterium glutamicum. Arch Microbiol. 2013;195:419–29. https://doi.org/10.1007/s00203-013-0889-3.
Milse J, Petri K, Rückert C, Kalinowski J. Transcriptional response of Corynebacterium glutamicum ATCC 13032 to hydrogen peroxide stress and characterization of the OxyR regulon. J Biotechnol. 2014;190:40–54. https://doi.org/10.1016/j.jbiotec.2014.07.452.
LeBlanc N, Charles TC. Bacterial genome reductions: tools, applications, and challenges. Front Genome Ed. 2022;4:957289. https://doi.org/10.3389/fgeed.2022.957289.
Unthan S, Baumgart M, Radek A, Herbst M, Siebert D, Brühl N, et al. Chassis organism from Corynebacterium glutamicum–a top-down approach to identify and delete irrelevant gene clusters. Biotechnol J. 2015;10:290–301. https://doi.org/10.1002/biot.201400041.
Baumgart M, Unthan S, Kloß R, Radek A, Polen T, Tenhaef N, et al. Corynebacterium glutamicum chassis C1*: building and testing a novel platform host for synthetic biology and industrial biotechnology. ACS Synth Biol. 2018;7:132–44. https://doi.org/10.1021/acssynbio.7b00261.
Hemmerich J, Labib M, Steffens C, Reich SJ, Weiske M, Baumgart M, et al. Screening of a genome-reduced Corynebacterium glutamicum strain library for improved heterologous cutinase secretion. Microb Biotechnol. 2020;13:2020–31. https://doi.org/10.1111/1751-7915.13660.
Kato O, Youn J-W, Stansen KC, Matsui D, Oikawa T, Wendisch VF. Quinone-dependent D-lactate dehydrogenase dld (Cg1027) is essential for growth of Corynebacterium glutamicum on D-lactate. BMC Microbiol. 2010;10:321. https://doi.org/10.1186/1471-2180-10-321.
Si M, Che C, Li G, Li X, Gong Z, Liu J, et al. Characterization of Xi-class mycothiol S-transferase from Corynebacterium glutamicum and its protective effects in oxidative stress. Microb Cell Fact. 2019;18:182. https://doi.org/10.1186/s12934-019-1232-8.
Parise MTD, Parise D, Kato RB, Pauling JK, Tauch A, Azevedo VAdC, Baumbach J. CoryneRegNet 7, the reference database and analysis platform for corynebacterial gene regulatory networks. Sci Data. 2020;7:142. https://doi.org/10.1038/s41597-020-0484-9.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. https://doi.org/10.1093/nar/28.1.27.
Ma S, Su T, Lu X, Qi Q. Bacterial genome reduction for optimal chassis of synthetic biology: a review. Crit Rev Biotechnol. 2023:1–14. https://doi.org/10.1080/07388551.2023.2208285.
Inui M, Murakami S, Okino S, Kawaguchi H, Vertès AA, Yukawa H. Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under oxygen deprivation conditions. J Mol Microbiol Biotechnol. 2004;7:182–96. https://doi.org/10.1159/000079827.
Stansen C, Uy D, Delaunay S, Eggeling L, Goergen J-L, Wendisch VF. Characterization of a Corynebacterium glutamicum lactate utilization operon induced during temperature-triggered glutamate production. Appl Environ Microbiol. 2005;71:5920–8. https://doi.org/10.1128/AEM.71.10.5920-5928.2005.
Ozyamak E, Black SS, Walker CA, Maclean MJ, Bartlett W, Miller S, Booth IR. The critical role of S-lactoylglutathione formation during methylglyoxal detoxification in Escherichia coli. Mol Microbiol. 2010;78:1577–90. https://doi.org/10.1111/j.1365-2958.2010.07426.x.
Suttisansanee U, Honek JF. Preliminary characterization of a Ni2+-activated and mycothiol-dependent glyoxalase I enzyme from Streptomyces coelicolor. Inorganics. 2019;7:99. https://doi.org/10.3390/inorganics7080099.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. https://doi.org/10.1093/nar/25.17.3389.
Liu Y-B, Chen C, Chaudhry MT, Si M-R, Zhang L, Wang Y, Shen X-H. Enhancing Corynebacterium glutamicum robustness by over-expressing a gene, mshA, for mycothiol glycosyltransferase. Biotechnol Lett. 2014;36:1453–9. https://doi.org/10.1007/s10529-014-1501-x.
Jain R, Sun X, Yuan Q, Yan Y. Systematically engineering Escherichia coli for enhanced production of 1,2-propanediol and 1-propanol. ACS Synth Biol. 2015;4:746–56. https://doi.org/10.1021/sb500345t.
Jothivasan VK, Hamilton CJ. Mycothiol: synthesis, biosynthesis and biological functions of the major low molecular weight thiol in actinomycetes. Nat Prod Rep. 2008;25:1091–117. https://doi.org/10.1039/B616489G.
Carmel-Harel O, Storz G. Roles of the glutathione- and thioredoxin-dependent reduction systems in the Escherichia coli and Saccharomyces cerevisiae responses to oxidative stress. Annu Rev Microbiol. 2000;54:439–61. https://doi.org/10.1146/annurev.micro.54.1.439.
Hartmann FSF, Clermont L, Tung QN, Antelmann H, Seibold GM. The industrial organism Corynebacterium glutamicum requires mycothiol as antioxidant to resist against oxidative stress in bioreactor cultivations. Antioxidants. 2020. https://doi.org/10.3390/antiox9100969.
Teramoto H, Inui M, Yukawa H. OxyR acts as a transcriptional repressor of hydrogen peroxide-inducible antioxidant genes in Corynebacterium glutamicum R. FEBS J. 2013;280:3298–312. https://doi.org/10.1111/febs.12312.
Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166:557–80. https://doi.org/10.1016/S0022-2836(83)80284-8.
Green MR, Sambrook J. Molecular cloning: a laboratory manual. 4th ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2012.
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994;145:69–73. https://doi.org/10.1016/0378-1119(94)90324-7.
Blombach B, Riester T, Wieschalka S, Ziert C, Youn J-W, Wendisch VF, Eikmanns BJ. Corynebacterium glutamicum tailored for efficient isobutanol production. Appl Environ Microbiol. 2011;77:3300–10. https://doi.org/10.1128/AEM.02972-10.
Peters-Wendisch PG, Schiel B, Wendisch VF, Katsoulidis E, Möckel B, Sahm H, Eikmanns BJ. Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J Mol Microbiol Biotechnol. 2001;3:295–300.
Gibson DG. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol. 2011;498:349–61. https://doi.org/10.1016/B978-0-12-385120-8.00015-2.
Follettie MT, Peoples OP, Agoropoulou C, Sinskey AJ. Gene structure and expression of the Corynebacterium flavum N13 ask-asd operon. J Bacteriol. 1993;175:4096–103. https://doi.org/10.1128/jb.175.13.4096-4103.1993.
Tauch A, Kirchner O, Löffler B, Götker S, Pühler A, Kalinowski J. Efficient electrotransformation of Corynebacterium diphtheriae with a mini-replicon derived from the Corynebacterium glutamicum plasmid pGA1. Curr Microbiol. 2002;45:362–7. https://doi.org/10.1007/s00284-002-3728-3.
van der Rest ME, Lange C, Molenaar D. A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogeneic plasmid DNA. Appl Microbiol Biotechnol. 1999;52:541–5. https://doi.org/10.1007/s002530051557.
Feith A, Teleki A, Graf M, Favilli L, Takors R. HILIC-enabled 13 C metabolomics strategies: comparing quantitative precision and spectral accuracy of QTOF High- and QQQ low-resolution mass spectrometry. Metabolites. 2019. https://doi.org/10.3390/metabo9040063.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the Deutsche Forschungsgemeinschaft (project number 409172861).
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
B.B. acquired funding of the project. D.S., V.F.W. and B.B. conceived and designed the experiments. D.S., E.G., M.W., M.V., A.S., A.W., E.S. and D.M. performed the experiments and analyzed the data. D.S. and B.B. drafted the manuscript. All authors reviewed and agreed with the final manuscript.
Corresponding author
Ethics declarations
Competing interest
The authors declare that there are no competing interests associated with this work.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This paper is dedicated to Professor Dr. Bernhard Eikmanns on the occasion of his retirement and his significant contributions to the field of microbial biotechnology.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Siebert, D., Glawischnig, E., Wirth, MT. et al. A genome-reduced Corynebacterium glutamicum derivative discloses a hidden pathway relevant for 1,2-propanediol production. Microb Cell Fact 23, 62 (2024). https://doi.org/10.1186/s12934-024-02337-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-024-02337-w