
Abstract
Background
Malate is a C4-dicarboxylic acid widely used as an acidulant in the food and beverage industry. Rational engineering has been performed in the past for the development of microbial strains capable of efficient production of this metabolite. However, as malate can be a precursor for specialty chemicals, such as 2,4-dihydroxybutyric acid, that require additional cofactors NADP(H) and ATP, we set out to reengineer Escherichia coli for Krebs cycle-dependent production of malic acid that can satisfy these requirements.
Results
We found that significant malate production required at least simultaneous deletion of all malic enzymes and dehydrogenases, and concomitant expression of a malate-insensitive PEP carboxylase. Metabolic flux analysis using 13C-labeled glucose indicated that malate-producing strains had a very high flux over the glyoxylate shunt with almost no flux passing through the isocitrate dehydrogenase reaction. The highest malate yield of 0.82 mol/mol was obtained with E. coli Δmdh Δmqo ΔmaeAB ΔiclR ΔarcA which expressed malate-insensitive PEP carboxylase PpcK620S and NADH-insensitive citrate synthase GltAR164L. We also showed that inactivation of the dicarboxylic acid transporter DcuA strongly reduced malate production arguing for a pivotal role of this permease in malate export.
Conclusions
Since more NAD(P)H and ATP cofactors are generated in the Krebs cycle-dependent malate production when compared to pathways which depend on the function of anaplerotic PEP carboxylase or PEP carboxykinase enzymes, the engineered strain developed in this study can serve as a platform to increase biosynthesis of malate-derived metabolites such as 2,4-dihydroxybutyric acid.
Similar content being viewed by others

Background
The four-carbon dicarboxylic acid malate is a Krebs cycle intermediate and a compound of considerable economic interest. It is mainly used as an acidulant in the food and beverage industry, or as a precursor for specialty chemicals [1, 2].
Currently, d/l-malic acid is chemically produced by hydration of maleic anhydride and has an estimated annual market volume of ~ 10 kt/year. However, due to its potential to replace maleic acid as a major building block in the chemical industry, its annual production could rise to ~ 200 kt if a cost-efficient biochemical production process could be developed [2]. Therefore, malic acid has been ranked as one of the 12 top target molecules of biochemical production processes [3], and much research effort has been invested in recent years to optimize the metabolic network and cultivation conditions of malate-producing organisms.
The filamentous fungus Aspergillus flavus was the first identified natural malate-producing organism [4]. After optimization of cultivation conditions, it accumulated 113 g/l malate with a yield and productivity of 1.26 mol/mol and 0.59 g/(lh), respectively [5]. However, since A. flavus produces toxic aflatoxin, this organism was never employed in industrial production processes. During the search for alternative natural malate-producing organisms, the yeast Zygosaccharomyces rouxii and the filamentous fungi Aspergillus oryzae and Penicillium sclerotiorum were shown to accumulate malate to high levels, reaching 92 g/l at a yield of 1.2 mol/mol for the latter organism [6,7,8].
In complementary studies carried out in yeast, bacteria, and fungi, it was tried to further increase malate production by rational optimization of the metabolic network. The oxaloacetate (OAA)-yielding carboxylation of either phosphoenolpyruvate (PEP) or pyruvate, followed by direct reduction of OAA to produce malate represents the most efficient metabolic pathway, which has a theoretical yield of 2 mol malate per mol glucose [9]. Therefore, all metabolic engineering endeavors to increase malate production focused on the optimization of this pathway. The overexpression of pyruvate carboxylase, malate dehydrogenase, and malate permease activities in a pyruvate-overproducing Saccharomyces cerevisiae platform strain enabled the accumulation of 59 g/l malate at a yield of 0.42 mol/mol [9,10,11]. Using a similar strategy, malate production by an A. oryzae strain could be increased to 154 g/l and a yield of 1.38 mol/mol after overexpressing pyruvate carboxylase, and a malate permease [6]. While the optimal function of the processes employing yeast and fungi required aerobic cultivation conditions, anaerobic malate production was demonstrated with Escherichia coli. A strain devoid of all alternative fermentative pathways and evolved for the production of succinate [12, 13] was converted into a malate producer by additionally deleting the genes coding for malic enzyme, fumarate reductase and fumarase activities. The strain accumulated 34 g/l malate with a yield and productivity of 1.42 mol/mol and 0.47 g/(l h), respectively [14]. Malate and succinate production in the evolved strain relied on mutations that conferred increased expression of PEP carboxykinase (Pck) and inactivation of the PTS sugar uptake system [15, 16]. Together, these modifications enabled an ATP-producing carboxylation of PEP and thus increases the energy output of the pathway. The microbial production of malic acid by the use of additional microorganisms and alternative substrates was discussed in detail in two recent reviews [17, 18].
The biosynthesis of malate from glucose via direct reduction of oxaloacetate is redox neutral and produces a maximum of two mole ATP per glucose molecule if PEP is converted into oxaloacetate via the ATP-forming Pck. Thus, in biosyntheses that employ additional reduction steps for the conversion of malate into other target molecules, cofactor supply may become limiting. We recently proposed a synthetic pathway which converts malate into DHB requiring one mol of ATP and two moles of NAD(P)H [19]. These additional cofactors cannot be produced by employing the above-cited conventional pathways for malate biosynthesis. Therefore, we set out to investigate metabolic engineering requirements to enable aerobic malate production via Krebs cycle and glyoxylate shunt, which increases ATP and NADH production at the expense of a decreased maximum malate yield which drops to 1.33 mol/mol [9].
We show that significant malate production through the Krebs cycle could not be achieved by solely deleting all malate dehydrogenases and malic enzymes, but additionally required the over-expression of a malate-insensitive PEP carboxylase mutant. Malate production was further increased by the inactivation of the acetate pathways, or by the overexpression of a NADH-insensitive citrate synthase mutant. Inactivation of the dicarboxylic acid transporter DcuA strongly reduced malate production arguing for a pivotal role of this permease in malate export. The best strain produced malate from glucose at a yield of 0.82 mol/mol.
Methods
Media and cultivation conditions
Strains were grown in Luria–Bertani (LB) broth (containing per liter: 10 g tryptone, 5 g yeast extract, 10 g NaCl, and 50 µg kanamycin sulfate and 25 µg chloramphenicol when necessary) [49] during strain construction and in starter cultures. All growth and pre-cultures were carried out in defined mineral medium. One liter mineral medium contained 10 g glucose, 18 g Na2HPO4 * 12 H2O, 3 g KH2PO4, 0.5 g NaCl, 2 g NH4Cl, 0.5 g MgSO4 * 7 H2O, 0.015 CaCl2 * 2 H2O, 1 ml of 0.06 mol/l FeCl3 stock solution (prepared in 100 times diluted concentrated HCl), 2 ml of 10 mM thiamine HCl stock solution, 20 g MOPS, 50 µg kanamycin sulfate (and 25 µg chloramphenicol when necessary), and 1 ml of trace element solution (containing per liter: 0.4 g Na2EDTA * 2H2O, 1.8 g CoCl2 * 6 H2O, 1.8 g ZnSO4 * 7 H2O, 0.4 g Na2MoO4 * 2 H2O, 0.1 g H3BO3, 1.2 g MnSO4 * H2O, 1.2 g CuCl2 * H2O). Medium was adjusted to pH 7 and filter-sterilized. Pre-cultures were inoculated at OD of ~ 0.5 from starter cultures that were grown overnight in LB. Growth cultures were inoculated at OD of ~ 0.2 from exponentially growing pre-cultures. When OD of the growth cultures reached 0.6, 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) was added. Strains were incubated in 250 ml flasks containing 30 ml medium, and shaken at 200 rpm and 37 °C in a rotary shaker (Infors). Samples were regularly withdrawn to follow growth and metabolite secretion.
Construction of strains and plasmids
Strain construction
All strains were derived from the E. coli K-12 substr. MG1655 wild-type strain. Gene deletions were introduced successively using the phage transduction method adapted from Miller [50]. Strains carrying the desired single deletions were recovered from the Keio collection [51]. Phage lysates of single deletion mutants were prepared by inoculating 10 ml of LB medium containing 50 µg/ml kanamycin, 2 g/l glucose, and 5 mM CaCl2 with 100 µl of overnight precultures. Following an incubation of 1 h at 37 °C, 200 µl of phage lysate prepared from the wild-type E. coli K-12 MG1655 strain were added, and cultures were incubated for another 2–3 h until cell lysis had completed. After addition of 200 µl chloroform, cell preparations were first vigorously vortexed and then centrifuged for 10 min at 4500×g. The clear lysate was recovered and stored at 4 °C. The receptor strain was prepared for phage transduction by an overnight cultivation at 37 °C in LB medium. A volume of 1.5 ml of the preculture was centrifuged at 1500×g for 10 min. The supernatant was discarded and the cell pellet was resuspended in 600 µl of a solution containing 10 mM MgSO4 and 5 mM CaCl2. The transduction was carried out by mixing 100 µl of the solution containing the receptor strain with 100 µl of lysate and incubating this mixture at 30 °C for 30 min. Thereafter, 100 µl of a 1 M sodium citrate solution were added followed by vigorous vortexing. After addition of 1 ml LB medium, the cell suspension was incubated at 37 °C for 1 h before spreading the cells on LB agar dishes containing 50 µg/ml kanamycin. Clones able to grow in presence of the antibiotic were confirmed by colony PCR to contain the desired deletion using the primers listed in Table 3. The resistance gene (FRT-kan-FRT) was subsequently excised from the chromosome using the FLP recombinase-harbouring plasmid pCP20 [52] leaving a scar region containing one FRT site. Kanamycin resistant mutants were transformed with pCP20, and ampicillin-resistant transformants were selected at 30 °C. Transformants were then grown on solid LB medium at 37 °C and tested for loss of all antibiotic resistances. Excision of the FRT-kanamycin cassette was analyzed by colony PCR using crimson taq polymerase and the flanking locus-specific primers (Table 1). All strains used in this study are listed in Table 2.
Plasmid construction
The wild-type ppc gene was PCR amplified from genomic DNA the E. coli MG1655 strain using Phusion polymerase (Thermo-Scientific) and the forward and reverse primers ppc_clon_for and ppc_clon_frev respectively. The resulting DNA fragment was ligated into pACT3 [26] using SmaI and XbaI restriction sites to obtain vector pACT3w-ppcwt. The amino acid exchange Lys620Ser was introduced into ppc by site directed PCR mutagenesis using primers ppc_k620s_for and ppc_k620s_rev. The PCR product was DpnI digested and transformed into NEB 5-alpha competent E. coli cells (NEB). The resulting plasmid, pACT3w-ppcK620S was isolated and verified by DNA sequencing to contain the desired mutation. Subcloning into the pEXT20 vector was achieved using SalI and KpnI restriction sites. To replace the original ribosome-binding site (RBS) of pACT3 in front of the ppcK620S gene by a stronger one, ppcK620S was PCR amplified using primers ppc_sRBS_for and ppc_sRBS_for, respectively, and cloned into the pACT3 and pEXT20 vectors using SmaI and XbaI restriction sites. The wild-type gltA gene was PCR-amplified from genomic DNA of the E. coli MG1655 strain using pfu polymerase primers gltA_clon_for and gltA_clon_rev. The resulting DNA fragment was cloned into the pACT3 vector using SacI and HindIII restriction sites. The amino acid exchange R164L was introduced into the gltA gene by site directed PCR mutagenesis using primers gltA_R164L_for and gltA_R164L_rev. The PCR product was DpnI digested and transformed into NEB 5-alpha competent E. coli cells (NEB). The resulting plasmid, pACT3-gltAR164L was isolated and verified by DNA sequencing to contain the desired mutation. For construction of vector pACT3w-ppcK620S-gltAR164L, the mutant gltAR164L gene was amplified from the pACT3-gltAR164L plasmid using Phusion polymerase (Biolabs) and primers gltA_R164L_for_1 and gltA_R164L_for_2. The PCR product was purified and recombined into the pACT3w-ppcK620S vector that was linearized with HindIII and XbaI, using the In-Fusion® HD Cloning Kit (Clontech).
Analytical techniques
Glucose and extracellular metabolites were quantified on a Dionex Ultimate 3000 HPLC system equipped with a UV and an RI (Shimadzu RID 10A) detector, an Aminex HPX-87H column thermostated at 32 °C, and a “Micro-Guard cation H Refill Cartridge” pre-column. Dilute sulfuric acid (1.25 mM) was used as the mobile phase at an isocratic flow rate of 0.5 ml/min.
Metabolic flux analysis
Culture conditions
The strain was grown in batch culture using a 0.5 l fermenter (Multifors Infors HT, The Netherlands) with a working volume of 0.4 l coupled to a Mass Gas Analyzer (Proline Dycor, Ametek process instrument, USA). Cells were cultivated in modified M9 medium adapted for IC-MS/MS analysis [31] that contained 50 mM glucose (ratio of uniformly labeled to C1-labeled glucose was 1/4), 0.6 mM IPTG and 25 µg/ml chloramphenicol. Temperature, pH, air flow, and stirring speed were maintained at 37 °C, pH 7 (with KOH 1 M), 0.2 l/min and 800 rpm, respectively. Under these conditions, oxygen concentration remained above 50% of saturation throughout the cultivation. The N2, O2, Argon, CO2 concentrations in the bioreactors off-gas were measured on-line using a Mass Gas Analyzer.
Quantification of extracellular metabolites
Culture broth was separated from cells by centrifugation over 0.45 µm pore size membranes (Minisart, Sartorius) and stored at − 20 °C until analysis. To quantify secreted metabolites, clear supernatant was subject to 1H 1D-NMR analysis (Avance 800 MHz spectrometer, Bruker, Germany) carried out at 292 °K, using a 30° pulse and a relaxation delay of 20 s. Trimethylsilyl propionate was used as internal standard.
Quantification of mass isotopologues in intracellular metabolites
Approximately 0.8 ml samples (cells + culture medium) with a cell concentration of 0.4 g/l were manually withdrawn from the bioreactor and directly quenched and extracted by mixing with 3 ml of a mixture of acetonitrile, methanol, and formic acid (0.1 M) (40:40:20 v/v), and incubation at − 20 °C for 1 h. Next, the tubes were moved to − 80 °C until further treatment. Sample evaporation was performed in a rotavapor (Büchi, Switzerland) during approximately 15 h until complete dryness. Afterwards, the samples were dissolved in a 500 μl ultrapure water and stored at − 20 °C until analysis. IC-MS/MS analysis was carried out as described by [34] to quantify the isotopic enrichment of selected metabolites. The analyzed metabolites were PEP, R5P, S7P, 6PG, G6P, FBP, F6P, 23 PG, Cit, Fum, Mal and Suc. Raw peak areas were corrected for the contribution of all naturally abundant isotopes using the IsoCor software [53]. In addition, label incorporation into secreted acetate and malate was measured using 1H 1D-NMR as described above.
Calculation of fluxes
Intracellular carbon fluxes were calculated from data on mass isotopologue enrichments and production/consumption rates of extracellular metabolites using the Influx_s software [21].
Results
Efficient malate production requires inactivation of malic enzymes and dehydrogenases, and appropriate expression of malate-insensitive PEP carboxylase
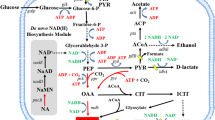
To investigate the Krebs cycle-dependent malate production, we step-wise introduced deletions in competing metabolic pathways (Fig. 1) and analyzed the behavior of the mutants in shake flask cultures using M9 mineral medium which contained 10 g/l glucose as a carbon source. To trigger malate secretion under aerobic conditions we first interrupted the Krebs cycle by deleting the soluble and membrane associated malate dehydrogenases, encoded by mdh and mqo, respectively [20, 21]. However, no significant malate accumulation in the culture medium was observed in the single and double mutants but acetate production strongly increased (Table 1). We therefore additionally deleted the acetate-forming pathway, encoded by ackA-pta [22], and the two malic enzymes, encoded by maeA and maeB. The resulting quintuple mutant Sy162 produced malate at a molar yield of 0.12 (Table 3).

Relevant metabolic pathways during aerobic production of malate in Escherichia coli. Gene names are depicted in blue, names of enzymes that were overexpressed to alleviate allosteric inhibition are depicted in green. Bold lines indicate enzymatic activities that were overexpressed or derepressed, dashed black lines indicate transcriptional repression, and dashed red lines indicate allosteric inhibition. Red crosses indicate deletions of genes and attenuated allosteric inhibitions (Oac oxaloacetate, Cit citrate, Ici isocitrate, 2-OG 2-oxoglutarate, Suc-CoA Succinyl-CoA, Suc succinate, Pyr pyruvate, Ac-CoA acetyl-CoA, Glyox glyoxylate)
It was previously demonstrated that the aerobic production of the Krebs cycle intermediates succinate and fumarate could be improved by increasing expression of the anaplerotic enzymes phosphoenolpyruvate (PEP) carboxylase (Ppc) [23, 24], or pyruvate carboxylase [25]. However, overexpression of the natural Ppc from E. coli did not increase malate production (Table 1). We therefore hypothesized that intracellular accumulation of malate inhibits Ppc [26], thereby preventing production of oxaloacetate and thus synthesis of the first Krebs cycle intermediate citrate. We alleviated malate feedback inhibition by introducing mutation Lys620Ser into the Ppc enzyme [27] and varied the expression level of the resulting mutant using the high and medium copy plasmids, pEXT20 and pACT3 [28], respectively, and strong and weak ribosome binging sites (RBS). Indeed, we found that the expression of the PpcK620S mutant strongly improved malate production, and that malate yield varied by more than threefold depending on the choice of the expression vector and strength of the RBS. Our data does not provide a clear explanation for the observed differences in the production of malate and other metabolic by-products, since they can be due to changes in the activity of the PpcK620S enzyme or due to the altered metabolic burden imposed by different plasmid copy numbers. However, it was previously demonstrated by Lee et al. [29] that efficient production of threonine required moderate over-expression of Ppc and that an excessively high Ppc activity was deleterious for the production of this amino acid. In line with these results, we found that the medium-copy plasmid pACT3w-ppcK620S, which contained a weak RBS and which can therefore be considered the weakest expression system of the four tested vectors, produced the highest malate yield of 0.48 mol/mol (Table 1). Plasmid pACT3w-ppcK620S was therefore used in all further experiments (Table 4).
We then tested the impact of expressing the malate-insensitive PpcK620S mutant in strains that were still wild-type for at least one malic enzyme, or that still contained the functional AckA-Pta-dependent acetate pathway. We found that malate yields strongly decreased in strains expressing at least one malic enzyme and did not exceed 0.15 mol/mol (Additional file 1: Table S1), whereas the presence of the AckA-Pta pathway only had a minor impact on the malate yield which was estimated at 0.4 mol/mol (Table 2). Taken together these results show that inactivation of all malate dehydrogenases and malic enzymes together with appropriate expression of a malate insensitive Ppc mutant are necessary and sufficient to enable significant malate production.
Characterization of carbon flux distribution in a malate-producing strain and further genotype optimization
After having identified the major requirements for aerobic malate production in E. coli, we carried out a detailed analysis on the impact of additional genetic modifications on malate production by this organism. Starting from strain Sy502, which carried deletions in both malate dehydrogenases and malic enzymes (Δmdh Δmqo ΔmaeA ΔmaeB) and which had a malate yield of 0.4 mol/mol, we sequentially deleted the transcriptional repressor of the glyoxylate shunt genes (aceA, aceB), encoded by iclR [30, 31], and the enzymes acetate kinase and pyruvate oxidase, encoded by ackA [32] and poxB [33], respectively. We observed that the transcriptional derepression of the glyoxylate pathway upon deletion of iclR in strain Sy502 giving rise to strain Sy504 did not result in an increased malate yield. In addition, acetate production remained unchanged when compared to strain Sy502 (Table 2), which is at variance to the behavior of an ΔiclR single mutant that is known to have reduced acetate formation [34]. Nevertheless, we continued to work with strains that carried an iclR deletion to assure complete derepression of the glyoxylate shunt. Deletion of ackA (Sy510) increased malate production by 24% and decreased acetate (Table 2). Deleting poxB alone (Sy508) or together with ackA (Sy512) strongly reduced acetate production, but also resulted in an up to 50% drop of malate production compared to strain Sy510, which was mainly due to an augmented production of pyruvate (Table 2). These results showed that the major metabolic by-product acetate could not be channeled into the Krebs cycle by simply deleting the ackA and poxB-dependent acetate pathways. In addition, the strains that carried the ackA deletion alone or in combination with a deletion of poxB (Sy510, Sy512) exhibited strongly extended lag phases (not shown) which was in agreement with previous studies [35, 36]. The strategy of increasing malate production by directly engineering the acetate pathways was therefore dismissed.
To identify alternative metabolic targets to increase malate production without deleting the acetate pathways we carried out a carbon flux analysis on strain Sy504. The strain was cultivated on 13C-labelled glucose in a bioreactor where glucose consumption and the formation of the metabolic end-products biomass, carbon dioxide, and organic acids were monitored. Samples for the quantification of 13C-label enrichment in individual metabolite pools were withdrawn manually during exponential phase and quantified by IC-MS/MS analysis [37]. In addition, the label incorporation into acetate and malate was measured using 1H 1D-NMR.
The carbon flux distribution in strain Sy504 was calculated from these data and is depicted in Fig. 2. In qualitative agreement to the shake flask experiments we found that malate and fumarate were co-secreted into the supernatant and that acetate was the major metabolic by-product in addition to the two C4 compounds. Unexpectedly, we calculated a direct flux from oxaloacetate towards malate of ~ 8 mol%. This result is hard to explain considering that (i) under aerobic conditions this flux normally occurs in the opposite direction, and (ii) malate dehydrogenase was deleted in the analysed strain. A possible explanation for this putative flux is that carbon flux actually passed from oxaloacetate to fumarate and further to malate through aspartate transaminase, aspartase, and fumarase, respectively.

Carbon flux distribution in strain E. coli Δmdh Δmqo ΔmaeA ΔmaeB ΔiclR. Fluxes are indicated in blue as mol percent per consumed glucose. Thickness of the arrows corresponds to amount of carbon flux. Boxed metabolites were directly measured. Grey arrows indicate carbon flux into biomass. The flux values were inferred from two independent experiments (BM biomass, G6P glucose-6P, F6P fructose-6P, F16bP fructose-1,6-bisP, DHAP dihydroxyacetonephosphate, GA3P glyceraldehyde-3P, 3PG 3P-glycerate, PEP phosphoenolpyruvate, Ac-CoA acetyl-CoA, Cit citrate, Ici isocitrate, 2-OG 2-oxoglutarate, Suc-CoA succinyl-CoA, Fumin intracellular fumarate, Malin intracellular malate, 6PG 6P-gluconate, Ribu5P ribulose-5P)
As expected, strong differences in carbon flux repartitioning between wild-type strains [34, 38, 39] and the malate-producing strain Sy504 have been observed regarding the metabolic fate of PEP and the solicitation of the Krebs cycle and glyoxylate shunt reactions. Since oxaloacetate cannot be regenerated from malate in strain Sy504, the net-flux from PEP to oxaloacetate, that is, the flux over PEP carboxylase minus the flux over PEP carboxykinase, is on average 60% higher in this mutant than in the corresponding E. coli MG1655 wild-type strains (Additional file 1: Table S2).
While the flux from pyruvate to acetyl-CoA remained largely the same in all strains, acetyl-CoA was utilized very differently in our strain when compared to wild-type cells. Sy504 produced nearly 50% less acetate, and condensed a particularly large fraction of 30 mol% acetyl-CoA with glyoxylate to yield malate. Even in an ΔiclR single mutant, which has a derepressed glyoxylate shunt [34], nearly two-times less acetyl-CoA is directly converted into malate which indicates a particularly strong solicitation of the glyoxylate shunt in the malate-producing strain. The split ratio of fluxes at the isocitrate lyase (Icl)–isocitrate dehydrogenase (Icd) bifurcation was 5 in strain Sy504 (Additional file 1: Table S2), whereas it was only 0.5 in the ΔiclR mutant [34] during growth on glucose, and 0.4 during growth of wild type cells on acetate [40]. Even more strikingly, no carbon flux was observed between 2-oxoglutarate and succinate in our malate-producing strain (Fig. 2). Our study did not identify the actual physiological reason for this unexpected behaviour. However, since high flux across the glyoxylate shunt is required to attain the maximum theoretical malate yield of 1.33 mol/mol with the this pathway [9], this result of the flux analysis made clear that malate production could not be significantly increased by engineering the Icl/Icd node.
Instead, our attention was attracted by the observation that strain Sy504 produced 50% less acetate and 20% more malate in the bioreactor when compared to shake flask cultivations (Table 1, Fig. 2). We hypothesized that better aeration of the fermenter cultures could explain increased malate production in the bioreactor. Oxygen limitation is known to cause both ArcA-mediated repression of genes implicated in respiratory metabolism and accumulation of NADH [41]. We therefore tested the effect of deleting the transcriptional repressor ArcA, and of overexpressing the NADH-insensitive citrate synthase mutant GltAR164L [42]. We found that the deletion of arcA in strain Sy729 which bears the iclR deletion increased the malate yield to 0.53 mol/mol (Table 2). The additional overexpression of GltAR164L in strain Sy731 resulted in a further improved malate yield which amounted to 0.82 mol/mol. It is of note that this increase of malate yield with strain Sy731 was accompanied by a strongly reduced acetate production which dropped to only 0.28 mol/mol without requiring the deletion of any of the acetate-producing pathways. The additional deletion of aspartase, encoded by aspA, did not increase malate production (not shown).
The dicarboxylic acid transporter DcuA is the major malate-exporting permease in the engineered strain
Previous studies have shown that malate production could be strongly increased by overexpressing malate-exporting permeases [6, 9]. Alternatively, the deletion of malate permease(s) increases intracellular availability of malate which can be expected to be beneficial for the conversion of malate into other value-added products, such as 2,4-dihydroxybutyric acid [19]. We therefore set out to identify the malate transporter in the malate-producing strain E. coli Δmdh Δmqo ΔackA-pta ΔmaeA ΔmaeB pACT3w-ppcK620S (Sy280) by deleting the permease DctA, which is required for the uptake of dicarboxylic acids aerobic conditions [43, 44], and the permeases DcuA and DcuB which are responsible for the uptake of dicarboxylic acids under anaerobic conditions [45]. In addition, we tested the impact of deleting two putative transporter proteins, YeeE and YqhC [46], which were found transcriptionally up-regulated in our malate-producing strain (data not shown). We observed that malate and fumarate secretion decreased by ~ 80% upon deletion of the constitutively expressed dicarboxylic acid transporter, DcuA [47], whereas the deletion of the other permeases did not cause any decrease of malate secretion. Thus, DcuA was identified as the major malate-exporting permease under aerobic conditions (Fig. 3). However, plasmid-born overexpression of this transporter did not increase malate production (data not shown).

Identification of the major malate-exporting permease. Candidate permeases were individually deleted in parent strain E. coli Δmdh Δmqo ΔackA-pta ΔmaeA ΔmaeB expressing pACT3w-ppcK620S (Sy280 = REF)
Discussion
In the present study we investigated the metabolic requirements for Krebs cycle-dependent production of malic acid in E. coli. We found that significant malate production under these conditions required deletion of all malic enzymes and dehydrogenases, and concomitant expression of a malate-insensitive PEP carboxylase. Interestingly, there was almost no gradual increase of malate production upon step-wise introduction of these metabolic modifications, but all genetic modifications had to be present simultaneously to trigger malate production. These results appear to be at variance with data of [48] who reported aerobic production of malate at a yield of 0.56 mol/mol upon expression of a PEP carboxykinase (Pck) from Mannheimia succiniciproducens in an E. coli pta mutant. It is not clear to us how aerobic malate production could be achieved in this metabolic set-up given that (i) reduction of oxaloacetate (OAA) to malate requires high NADH/NAD ratios which are commonly only achieved under oxygen-limited or anaerobic conditions in E. coli, (ii) Pck-dependent synthesis of OAA from PEP requires high carbon dioxide and PEP concentrations [11], which are commonly achieved by imposing either anaerobic conditions or sparging with carbon dioxide [11, 15] and by deleting the PEP-consuming phosphotransferase system or inactivating pyruvate kinase [11, 15, 16]. In addition, (iii) our own results show in agreement with the observations made by Zhang et al. [14] that activity of the malic enzymes prevents significant malate accumulation (Additional file 1: Table S2). The actual reason for these apparent discrepancies remains unclear which may indicate our still incomplete understanding of the metabolic regulation that occurs at the PEP/OAA/malate/pyruvate node in E. coli.
The major metabolic by-product in our aerobic metabolic setup was acetate. Our attempts to reduce acetate secretion by deleting the AckA and/or PoxB-dependent pathways came at the cost of extended lag-phases or even reduced malate production. We therefore set out to engineer strains that produced less acetate despite having functional acetate pathways. Acetate production is mainly regulated by citrate synthase activity. Citrate synthase is allosterically inhibited by high intracellular NADH concentrations, and excess acetyl-CoA is converted to acetate instead of being condensed with oxaloacetate to enter the Krebs cycle in the form of citrate [42, 49, 50]. In agreement with this notion, the deletion of the transcriptional repressor of respiratory enzymes, ArcA, and the overexpression of NADH-insensitive citrate synthase reduced acetate formation by 50% and increased malate production by ~ 90% (when comparing strains Sy504 and Sy731). Together, these results indicated that reducing acetate production via engineering NADH metabolism and citrate synthase was an effective means to improve the performance of malate-producing strains.
We found that malate production by our strains coincided with the accumulation of significant amounts of fumarate. Both compounds accumulated in the supernatant at a molar ratio of approximately 8, which is very similar to the ratio of their intracellular concentrations that was estimated with approximately 12 [51]. Thus, because the malate/fumarate ratio is imposed by the thermodynamic equilibrium of the reversible fumarase reaction, Krebs cycle-dependent malate production will almost inevitably coincide with the formation of fumarate as a co-product. In line with these arguments, we found that the dicarboxylic acid transporter DcuA, which accepts both malate and fumarate as substrates [45], was responsible for malate secretion. Only replacement of this rather unspecific permease by a highly malate-specific transporter could reduce fumarate secretion during Krebs cycle-dependent malate production.
In summary, Krebs-cycle dependent malate production can be achieved at the cost of lower maximum product yields and significant accumulation of fumarate as a metabolic by-product. These results make this metabolic engineering strategy unattractive when compared to Ppc or Pck-dependent pathways if malate is the actual target molecule. However, if malate is an intermediate product which is further processed to yield derivatives such as DHB [19], fumarate will not accumulate and the additional cofactors NAD(P)H and ATP which are generated in the Krebs cycle and respiratory chain can be used to increase product yields. Our study has laid the foundation for these microbial product syntheses.
Our study was carried out in aerobic shake flasks on mineral M9 medium. These cultivation conditions are clearly not representative for a fermentation process which enables cost efficient production of malate which would require to carry out the fermentations in bioreactors using low-cost mineral salt medium [52]. Under such conditions, product titers and yields obtained with the strains described in this work may significantly change. However, at the present stage of our analyses we focused on the fundamental genetic requirements for Krebs cycle-dependent malate production, which represents an intermediate stage on the way to the construction of production strains for biosynthesis of malate-derived value-added chemicals such as DHB.
References
Goldberg I, Rokem JS, Pines O. Organic acids: old metabolites, new themes. J Chem Technol Biotechnol. 2006;81:1601–11.
Sauer M, Porro D, Mattanovich D, Branduardi P. Microbial production of organic acids: expanding the markets. Trends Biotechnol. 2008;26:100–8.
Werpy T, Petersen G. Top value added chemicals from biomass. Results of screening for potential candidates from sugars and synthesis gas. Washington DC; 2004. http://dx.doi.org/10.2172/15008859.
Peleg Y, Stieglitz B, Goldberg I. Malic acid accumulation by Aspergillus flavus. Appl Microbiol Biotechnol. 1988;28:69–75.
Battat E, Peleg Y, Bercovitz A, Rokem JS, Goldberg I. Optimization of l-malic acid production by Aspergillus flavus in a stirred fermentor. Biotechnol Bioeng. 1991;37:1108–16.
Brown SH, Bashkirova L, Berka R, Chandler T, Doty T, McCall K, et al. Metabolic engineering of Aspergillus oryzae NRRL 3488 for increased production of l-malic acid. Appl Microbiol Biotechnol. 2013;97:8903–12.
Taing O, Taing K. Production of malic and succinic acids by sugar-tolerant yeast Zygosaccharomyces rouxii. Eur Food Res Technol. 2007;224:343–7.
Wang Z-P, Wang G-Y, Khan I, Chi Z-M. High-level production of calcium malate from glucose by Penicillium sclerotiorum K302. Bioresour Technol. 2013;143:674–7.
Zelle RM, de Hulster E, van Winden WA, de Waard P, Dijkema C, Winkler AA, et al. Malic acid production by Saccharomyces cerevisiae: engineering of pyruvate carboxylation, oxaloacetate reduction, and malate export. Appl Environ Microbiol. 2008;74:2766–77.
Zelle RM, de Hulster E, Kloezen W, Pronk JT, van Maris AJA. Key process conditions for production of C(4) dicarboxylic acids in bioreactor batch cultures of an engineered Saccharomyces cerevisiae strain. Appl Environ Microbiol. 2010;76:744–50.
Zelle RM, Trueheart J, Harrison JC, Pronk JT, van Maris AJA. Phosphoenolpyruvate carboxykinase as the sole anaplerotic enzyme in Saccharomyces cerevisiae. Appl Environ Microbiol. 2010;76:5383–9.
Jantama K, Haupt MJ, Svoronos SA, Zhang X, Moore JC, Shanmugam KT, et al. Combining metabolic engineering and metabolic evolution to develop nonrecombinant strains of Escherichia coli C that produce succinate and malate. Biotechnol Bioeng. 2008;99:1140–53.
Jantama K, Zhang X, Moore JC, Shanmugam KT, Svoronos SA, Ingram LO. Eliminating side products and increasing succinate yields in engineered strains of Escherichia coli C. Biotechnol Bioeng. 2008;101:881–93.
Zhang X, Wang X, Shanmugam KT, Ingram LO. l-Malate production by metabolically engineered Escherichia coli. Appl Environ Microbiol. 2011;77:427–34.
Zhang X, Jantama K, Moore JC, Jarboe LR, Shanmugam KT, Ingram LO. Metabolic evolution of energy-conserving pathways for succinate production in Escherichia coli. Proc Natl Acad Sci USA. 2009;106:20180–5.
Zhang X, Jantama K, Shanmugam KT, Ingram LO. Reengineering Escherichia coli for succinate production in mineral salts medium. Appl Environ Microbiol. 2009;75:7807–13.
Chi Z, Wang Z-P, Wang G-Y, Khan I, Chi Z-M. Microbial biosynthesis and secretion of l-malic acid and its applications. Crit Rev Biotechnol. 2016;36:99–107.
West TP. Microbial production of malic acid from biofuel-related coproducts and biomass. Fermentation. 2017;3:14.
Walther T, Topham CM, Irague R, Auriol C, Baylac A, Cordier H, et al. Construction of a synthetic metabolic pathway for biosynthesis of the non-natural methionine precursor 2,4-dihydroxybutyric acid. Nat Commun. 2017;8:15828.
Sutherland P, McAlister-Henn L. Isolation and expression of the Escherichia coli gene encoding malate dehydrogenase. J Bacteriol. 1985;163:1074–9.
van der Rest ME, Frank C, Molenaar D. Functions of the membrane-associated and cytoplasmic malate dehydrogenases in the citric acid cycle of Escherichia coli. J Bacteriol. 2000;182:6892–9.
Kakuda H, Hosono K, Shiroishi K, Ichihara S. Identification and characterization of the ackA (acetate kinase A)-pta (phosphotransacetylase) operon and complementation analysis of acetate utilization by an ackA-pta deletion mutant of Escherichia coli. J Biochem (Tokyo). 1994;116:916–22.
Sokol S, Millard P, Portais J-C. influx_s: increasing numerical stability and precision for metabolic flux analysis in isotope labelling experiments. Bioinforma Oxf Engl. 2012;28:687–93.
Song CW, Kim DI, Choi S, Jang JW, Lee SY. Metabolic engineering of Escherichia coli for the production of fumaric acid. Biotechnol Bioeng. 2013;110:2025–34.
Sanchez AM, Bennett GN, San K-Y. Novel pathway engineering design of the anaerobic central metabolic pathway in Escherichia coli to increase succinate yield and productivity. Metab Eng. 2005;7:229–39.
Izui K, Taguchi M, Morikawa M, Katsuki H. Regulation of Escherichia coli phosphoenolpyruvate carboxylase by multiple effectors in vivo. II. Kinetic studies with a reaction system containing physiological concentrations of ligands. J Biochem (Tokyo). 1981;90:1321–31.
Yano M, Izui K. The replacement of Lys620 by serine desensitizes Escherichia coli phosphoenolpyruvate carboxylase to the effects of the feedback inhibitors l-aspartate and l-malate. Eur J Biochem FEBS. 1997;247:74–81.
Dykxhoorn DM, St Pierre R, Linn T. A set of compatible tac promoter expression vectors. Gene. 1996;177:133–6.
Lee KH, Park JH, Kim TY, Kim HU, Lee SY. Systems metabolic engineering of Escherichia coli for l-threonine production. Mol Syst Biol. 2007;3:149.
Cortay JC, Nègre D, Galinier A, Duclos B, Perrière G, Cozzone AJ. Regulation of the acetate operon in Escherichia coli: purification and functional characterization of the IclR repressor. EMBO J. 1991;10:675–9.
Maloy SR, Nunn WD. Genetic regulation of the glyoxylate shunt in Escherichia coli K-12. J Bacteriol. 1982;149:173–80.
Matsuyama A, Yamamoto H, Nakano E. Cloning, expression, and nucleotide sequence of the Escherichia coli K-12 ackA gene. J Bacteriol. 1989;171:577–80.
Chang YY, Cronan JE, Li SJ, Reed K, Vanden Boom T, Wang AY. Locations of the lip, poxB, and ilvBN genes on the physical map of Escherichia coli. J Bacteriol. 1991;173:5258–9.
Waegeman H, Beauprez J, Moens H, Maertens J, De Mey M, Foulquié-Moreno MR, et al. Effect of iclR and arcA knockouts on biomass formation and metabolic fluxes in Escherichia coli K12 and its implications on understanding the metabolism of Escherichia coli BL21 (DE3). BMC Microbiol. 2011;11:70.
Abdel-Hamid AM, Attwood MM, Guest JR. Pyruvate oxidase contributes to the aerobic growth efficiency of Escherichia coli. Microbiol Read Engl. 2001;147:1483–98.
Causey TB, Shanmugam KT, Yomano LP, Ingram LO. Engineering Escherichia coli for efficient conversion of glucose to pyruvate. Proc Natl Acad Sci USA. 2004;101:2235–40.
Kiefer P, Nicolas C, Letisse F, Portais JC. Determination of carbon labeling distribution of intracellular metabolites from single fragment ions by ion chromatography tandem mass spectrometry. Anal Biochem. 2007;360:182–8.
Nicolas C, Kiefer P, Letisse F, Krömer J, Massou S, Soucaille P, et al. Response of the central metabolism of Escherichia coli to modified expression of the gene encoding the glucose-6-phosphate dehydrogenase. FEBS Lett. 2007;581:3771–6.
Sauer U, Canonaco F, Heri S, Perrenoud A, Fischer E. The soluble and membrane-bound transhydrogenases UdhA and PntAB have divergent functions in NADPH metabolism of Escherichia coli. J Biol Chem. 2004;279:6613–9.
Walsh K, Koshland DE. Branch point control by the phosphorylation state of isocitrate dehydrogenase. A quantitative examination of fluxes during a regulatory transition. J Biol Chem. 1985;260:8430–7.
Alexeeva S, Hellingwerf KJ, Teixeira de Mattos MJ. Requirement of ArcA for redox regulation in Escherichia coli under microaerobic but not anaerobic or aerobic conditions. J Bacteriol. 2003;185:204–9.
Stokell DJ, Donald LJ, Maurus R, Nguyen NT, Sadler G, Choudhary K, et al. Probing the roles of key residues in the unique regulatory NADH binding site of type II citrate synthase of Escherichia coli. J Biol Chem. 2003;278:35435–43.
Janausch IG, Zientz E, Tran QH, Kröger A, Unden G. C4-dicarboxylate carriers and sensors in bacteria. Biochim Biophys Acta. 2002;1553:39–56.
Lo TC, Rayman MK, Sanwal BD. Transport of succinate in Escherichia coli. I. Biochemical and genetic studies of transport in whole cells. J Biol Chem. 1972;247:6323–31.
Six S, Andrews SC, Unden G, Guest JR. Escherichia coli possesses two homologous anaerobic C4-dicarboxylate membrane transporters (DcuA and DcuB) distinct from the aerobic dicarboxylate transport system (Dct). J Bacteriol. 1994;176:6470–8.
Keseler IM, Mackie A, Peralta-Gil M, Santos-Zavaleta A, Gama-Castro S, Bonavides-Martínez C, et al. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 2013;41:D605–12.
Golby P, Kelly DJ, Guest JR, Andrews SC. Transcriptional regulation and organization of the dcuA and dcuB genes, encoding homologous anaerobic C4-dicarboxylate transporters in Escherichia coli. J Bacteriol. 1998;180:6586–96.
Moon SY, Hong SH, Kim TY, Lee SY. Metabolic engineering of Escherichia coli for the production of malic acid. Biochem Eng J. 2008;40:312–20.
Nguyen NT, Maurus R, Stokell DJ, Ayed A, Duckworth HW, Brayer GD. Comparative analysis of folding and substrate binding sites between regulated hexameric type II citrate synthases and unregulated dimeric type I enzymes. Biochemistry (Mosc). 2001;40:13177–87.
Vemuri GN, Altman E, Sangurdekar DP, Khodursky AB, Eiteman MA. Overflow metabolism in Escherichia coli during steady-state growth: transcriptional regulation and effect of the redox ratio. Appl Environ Microbiol. 2006;72:3653–61.
Bennett BD, Yuan J, Kimball EH, Rabinowitz JD. Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat Protoc. 2008;3:1299–311.
Martinez A, Grabar TB, Shanmugam KT, Yomano LP, York SW, Ingram LO. Low salt medium for lactate and ethanol production by recombinant Escherichia coli B. Biotechnol Lett. 2007;29:397–404.
Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–5.
Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2006(2):0008.
Authors’ contributions
TW and JMF originally conceived the general structure of the project. DT, AB, CD and RI carried out strain engineering. CA and AB performed fermentation process and physiological analysis. MC-H and SH carried out metabolic flux analysis. TW and JMF wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors are grateful to Amélie Vax, Hélène Serrano-Bataille, Florence Calvayrac, and Julie Fredonnet for their technical assistance. We also acknowledge support from MetaToul (Metabolomics & Fluxomics facilities, Toulouse, France, http://www.metatoul.fr), which is part of the national infrastructure MetaboHUB-ANR-11-INBS-0010 (The French National Infrastructure for Metabolomics and Fluxomics, http://www.metabohub.fr).
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The data and all materials related to this work will be generously provided upon request.
Ethics approval and consent to participate
Not applicable.
Funding
This work was supported by the French National Research Agency (ANR programme d’Investissement d’Avenir) ANR-10-BTBR-05-01.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Table S1.
Fermentation products of E. coli MG1655 mutants in mineral medium with glucose as the carbon source. Table S2. Comparison of carbon fluxes given in mol% relative to glucose uptake measured in different studies for wild-type and mutant E. coli strains.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Trichez, D., Auriol, C., Baylac, A. et al. Engineering of Escherichia coli for Krebs cycle-dependent production of malic acid. Microb Cell Fact 17, 113 (2018). https://doi.org/10.1186/s12934-018-0959-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-018-0959-y