
Abstract
Background
Optically pure d-lactic acid (≥ 99%) is an important precursor of polylactic acid. However, there are relatively few studies on d-lactic acid fermentation compared with the extensive investigation of l-lactic acid production. Most lactic acid producers are mesophilic organisms. Optically pure d-lactic acid produced at high temperature not only could reduce the costs of sterilization but also could inhibit the growth of other bacteria, such as l-lactic acid producers.
Results
Thermophilic Bacillus coagulans is an excellent producer of l-lactic acid with capable of growing at 50 °C. In our previous study, the roles of two l-lactic acid dehydrogenases have been demonstrated in B. coagulans DSM1. In this study, the function of another annotated possible l-lactate dehydrogenase gene (ldhL3) was verified to be leucine dehydrogenase with an activity of 0.16 units (μmol/min) per mg protein. Furthermore, the activity of native d-lactate dehydrogenase was too low to support efficient d-lactic acid production, even under the control of strong promoter. Finally, an engineered B. coagulans D-DSM1 strain with the capacity for efficient production of d-lactic acid was constructed by deletion of two l-lactate dehydrogenases genes (ldhL1 and ldhL2) and insertion of the d-lactate dehydrogenase gene (LdldhD) from Lactobacillus delbrueckii subsp. bulgaricus DSM 20081 at the position of ldhL1.
Conclusions
This genetically engineered strain produced only d-lactic acid under non-sterilized condition, and finally 145 g/L of d-lactic acid was produced with an optical purity of 99.9% and a high yield of 0.98 g/g. This is the highest optically pure d-lactic acid titer produced by a thermophilic strain.
Similar content being viewed by others

Background
Technologies for the production of fuels and plastics from renewable biomass are currently being developed to prevent environmental pollution and global warming. Polylactic acid (PLA) is considered as an important bioplastic due to its superior thermal stability, mechanical performance, hydrolysis resistance, and excellent biodegradability [1, 2]. Optically pure l-lactic acid (≥ 99%) is the main precursor of PLA, and addition of optically pure d-lactic acid resulted in high melting point and good appearance. So, the production of d-lactic acid has received increasing focus recently. However, both yield and productivity of d-lactic acid are lower than those of l-lactic acid till now [3, 4]. Chemical synthesis of lactic acid leads to a racemic mixture and environmental pollution, and 70–90% of lactic acid in the world is produced by microbial fermentation in industry [5].
Compared with the extensive investigation of l-lactic acid production, there are relatively few studies on d-lactic acid fermentation [6]. Several strains have been reported to produce d-lactic acid naturally, such as Lactobacillus species, Sporolactobacillus inulinus [4, 6]. Lactic acid is generated from the conversion of pyruvate into l-lactic acid and d-lactic acid by l-lactate dehydrogenases (l-LDHs, encoded by ldhL) and d-lactate dehydrogenases (d-LDHs, encoded by ldhD), respectively [7]. Since d-LDHs is the key gene in d-lactic acid production, some studies reported to obtain engineered d-lactic acid producing strains with the deletion of the native ldhL gene [6]. Then, some metabolically engineered strains, including Saccharomyces cerevisiae, L. plantarum, Corynebacterium glutamicum and Escherichia coli were constructed to produce d-lactic acid [8]. However, all the above strains are mesophilic and the fermentation medium needs to be sterilized, which increases the process cost. Additionally, strains have to reach certain requirements to be used in efficient lactic acid production, such as low-cost nutrition needed, high yield, high optical purity of product, low/no oxygen requirement and short fermentation time [9].
Thermophilic Bacillus coagulans almost meet all the above requirements. They can withstand various harsh conditions, such as low pH values, high temperatures, and no aeration [6]. Moreover, B. coagulans can utilize a broad range of inexpensive carbon resources, and produce optically pure l-lactic acid at 50–55 °C which is expected to minimize contamination during fermentation in industrial scale [7, 10]. Nonsterile fermentation can omit the medium and fermentor sterilization process and largely reduce the production cost [6, 11]. Hence, B. coagulans is expected to be an excellent d-lactic acid producer for industrial uses [12]. Although there are some examples about l-lactic acid production by Bacillus sp. under non-sterilized conditions, no studies about using B. coagulans for optically pure d-lactic acid production under non-sterilized fermentation process are reported until now [10, 11].
In this study, B. coagulans DSM1, an optically pure l-lactic acid producer, was chosen for genetic engineering for d-lactic acid production. The key gene for l-lactic acid production was replaced with LdhD from L. delbrueckii subsp. bulgaricus DSM 20081. This genetically engineered strain produced high optical purity of d-lactic acid under non-sterilized condition.
Methods
Strains, plasmids and culture conditions
Strains and plasmids used in this study are listed in Table 1. The construction of B. coagulans DSM1ΔldhL1ΔldhL2, whose l-lactate dehydrogenase genes (ldhL1 and ldhL2) have been knocked out, was described previously [13]. For genetic manipulation, the BC medium (50 g/L sucrose, 10 g/L yeast extract, 2 g/L (NH4)2HPO4, 3.5 g/L (NH4)2SO4, 10 g/L Bis–Tris, 3 mg/L CaCl2, 5 mg/L MgCl2, 20 μL trace element mixture) was used and the culture condition was 45 °C with 120 rpm. Filter sterilized trance element mixture contained 0.2 mg/L CoCl2·6H2O, 0.01 mg/L CuCl2·2H2O, 0.3 mg/L H3BO3, 0.03 mg/L Na2MoO4·2H2O, 0.02 mg/L NiSO4·6 H2O, 0.03 mg/L MnCl2·4H2O and 0.05 mg/L ZnCl2 [14]. For fermentation, the culture condition was 50 °C with 120 rpm in 513 medium (50 g/L glucose, 10 g/L yeast extract, 30 g/L CaCO3) [10]. Plasmids based on pMH77 were constructed in Lactococcus lactis MG1363, which was grown in GM17 medium (5 g/L soy peptone, 5 g/L tryptone, 2.5 g/L yeast extract, 5 g/L meat extract, 0.5 g/L ascorbic acid, 0.25 g/L MgSO4·7H2O, 5 g/L K2HPO4, 5 g/L glycerol, 10 g/L glucose). Plasmids based on pNW33N were constructed in E. coli TOP10 or BL21 (DE3), which was cultured in Luria–Bertani (LB) medium. When appropriate, 40 μg/mL kanamycin (Kan) and 25 μg/mL chloramphenicol (Cm) were added to E. coli culture, and 7 μg/mL Cm was added to B. coagulans and L. lactis culture.
Function verification of ldhL3
Gene ldhL3 was cloned to pET-28a, and the recombinant plasmid was transformed into E. coli BL21 (DE3). To obtain the protein, isopropyl-β-d-1-thiogalactopyranoside (IPTG) was added to the culture with a final concentration of 0.5 mM when the OD600 value reached 0.6, and a continued culture was followed for 12 h at 16 °C. Cells were harvested (12,000×g for 10 min) and preserved at − 80 °C. After cell sonication, 6× His-Tagged Protein Purification Kit (CWBIO, China) was applied to obtain purified protein. If inclusion body formed, molecular chaperone (Takara, China) was used to help the protein expression.
For lactate dehydrogenase, the in vitro and in vivo enzymatic activities were studied as described previously [7]. For leucine dehydrogenase, reaction was carried out with 4.5 mM 4-methyl-2-oxopentanoate, 0.2 mM NADH and 1 mg purified enzyme in 0.1 M NH4Cl/NH4OH (pH = 9.5) at 45 °C. One unit of leucine dehydrogenase was defined as the amount of enzyme which catalyzed the consumption of 1 μmol of NADH per min under the standard assay conditions. Quantitative real-time (RT)-PCR was employed to determine the transcription levels of ldhL3 in different fermentation periods. Total RNAs of B. coagulans DSM1ΔldhL1ΔldhL2 in logarithmic period, stationary period and decay period were isolated by using an E.Z.N.A bacterial RNA kit (Omega). With the primer pair of RTL3-F/R (Table 2), cDNA copies were synthesized with a FastQuant RT kit (with gDNase) (Tiangen, China) and amplified with SYBR Premix Ex Taq (TaKaRa, China). The threshold cycles (C T ) for each PCR were analyzed according to the Ref. [13].
Functional study of native ldhD in B. coagulans DSM1
The d-lactate dehydrogenase encoding gene from B. coagulans DSM1 (BcldhD) and L. delbrueckii subsp. bulgaricus DSM 20081 (LdldhD) were cloned to pNW33n under the control of the strong promoter of ldhL1 promotor (P ldhL1 , 1182 bp) and the native promoter of BcldhD (P BcldhD ), respectively. In total, four recombinant plasmids in pNW33n, named as P ldhL1 –BcldhD, P ldhL1 –LdldhD, P BcldhD –BcldhD and P BcldhD –LdldhD, were constructed and transformed into B. coagulans DSM1ΔldhL1ΔldhL2. The concentration of d-lactic acid was measured.
Gene insertion in B. coagulans DSM1ΔldhL1ΔldhL2
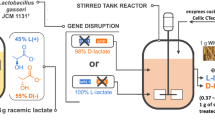
Plasmid pMH77 was used as the primary vehicle to transfer ldhD from L. delbrueckii (LdldhD) to B. coagulans DSM1ΔldhL1ΔldhL2 in the original positon of ldhL1 (Fig. 1). Primers used for gene insertion are listed in Table 2. Homologous arms of the upstream (1000 bp) and downstream (1000 bp) sequence of ldhL1 were PCR amplified with primers L1up-F/R and L1down-F/R, respectively. LdldhD and pMH77 were amplified by primers DS-F/R and 77-F/R correspondingly. Then four fragments were connected by Clone Express® MultiS One Step Cloning Kit (Vazyme Biotech Co., Ltd) resulting in plasmid pMH77–LdldhD.

Insertion of LdldhD in the position of ldhL1 gene of B. coagulans ΔldhL1ΔldhL2. a The upstream and downstream regions of ldhL1 in the chromosome of B. coagulans ΔldhL1ΔldhL2; b The upstream and downstream regions of ldhL1 in the chromosome of B. coagulans d-DSM1
Plasmid pMH77–LdldhD was firstly transformed into L. lactis MG1363 by electroporation. After verifying the correctness by sequencing, pMH77–LdldhD was transformed into B. coagulans DSM1ΔldhL1ΔldhL2 by electroporation. The transformants were cultured in BC medium with Cm at 45 °C for 6–10 h to mid-log phase and then the temperature was shifted to 60 °C for overnight [14]. A dilution series were plated on BC plates with Cm and incubated overnight at 60 °C. Colony PCR analysis was carried out for the first single crossover event. The positive clones were selected to be incubated in BC medium without Cm overnight at 45 °C. The obtained single colonies were firstly streaked onto BC plates with Cm and then without Cm. After incubation overnight at 45 °C, PCR analysis using the primers DS-For/Rev was conducted for the colonies which could grow on BC plates without Cm and could not grow on BC plates with Cm. The positive colonies were sequenced to confirm the gene insertion.
Non-sterilized fermentation of d-lactic acid by B. coagulans D-DSM1
The seed cultures were prepared in 513 medium for 24 h and then inoculated with 10% (v/v) in the fermentation medium. The fermentation medium was maintained at approximately pH 5.5–6.0 with the addition of CaCO3. Fed-batch fermentation was carried out at 50 °C with 50 rpm in a 5-L bioreactor with 2 L fresh medium without sterilization. The initial glucose concentration was 50 g/L. When the glucose concentration decreased to lower than 20 g/L, glucose powder was added directly and repeatedly, leading to the result that the glucose concentration reached to approximately 50 g/L for 4 times.
Analytical methods
Glucose consumption and lactate production were measured by HPLC (Agilent 1260 Series, Hewlett-Packard, Palo Alto, CA, USA) with an organic-acid column (MCI GEL CRS10W). The mobile phase was 6 mM H2SO4 and the flow rate was 0.5 mL/min. Temperature of the column was set to 55 °C. Both UV and differential detector were involved in the detection and the UV detection wavelength was 210 nm. The chiral and optical purity of d-lactic acid were analyzed using HPLC with a chiral column (MCI GEL CRS10W, Tokyo, Japan) [15]. Mobile phase was 2 mM CuSO4 at a flow rate of 0.5 mL/min. Column temperature was set to 25 °C and UV detection wavelength was 254 nm. The optical purity of d-lactic acid was calculated through the following formula: d-lactic acid optical purity = d-lactic acid/(d-lactic acid + l-lactic acid) × 100%. For the growth monitoring, A600 was measured by a 7200 Visible Spectrophotometer (UNICO, Shanghai, China).
Results
l-Lactate dehydrogenase gene (ldhL3) in B. coagulans DSM1ΔldhL1ΔldhL2
Three genes (ldhL1, ldhL2 and ldhL3) related to l-lactic acid production were annotated in the genome of B. coagulans DSM1 (GenBank accession number CP009709). Gene ldhL1 plays a major role in l-lactic acid production, and no L-lactic acid was detected in B. coagulans DSM1ΔldhL1ΔldhL2 [13]. Gene ldhL3 is annotated as leucine dehydrogenase/l-lactate dehydrogenase. To verify the function of ldhL3, l-LDH3 (encoded by ldhL3) was expressed solubly in E. coli with the help of molecular chaperone pGro7 [16]. The purified protein has the activity of leucine dehydrogenase with 0.16 units (μmol/min) per mg protein. RT-PCR and protein mass spectrometry studies showed that ldhL3 expressed at both transcription level and protein level. However, neither d-lactate nor l-lactate was detected in the reaction product of purified l-LDH3 by HPLC. Although purified l-LDH3 could catalyzed the oxidation of NADH, only leucine dehydrogenase activity was detected. So, there is no l-LDH activity in B. coagulans DSM1ΔldhL1ΔldhL2, which makes the strain as a candidate for producing high optical purity of d-lactate.
The activity of the native d-lactic acid dehydrogenase in DSM1ΔldhL1ΔldhL2
A D-LDH-encoding gene BcldhD was annotated in B. coagulans DSM1ΔldhL1ΔldhL2, and our previous study showed that the native d-LDH has an activity of 1.87 ± 0.08 U/mg in vitro [7, 13]. So, we firstly tried to detect the concentration of d-lactic acid produced by its native d-LDH in B. coagulans DSM1ΔldhL1ΔldhL2. However, only trace amount of d-lactic acid (0.26 g/L) was obtained (Additional file 1: Figure S1), and the growth of strain was poor when l-LDH genes deleted [13]. For B. coagulans DSM1, l-LDH deletion did not increase the expression or activity of native d-LDH. To trace the reason, a plasmid containing ldhL1 promoter and native ldhD from B. coagulans DSM1 (BcldhD) was first constructed by using plasmid pNW33N (P ldhL1 –BcldhD) and introduced into DSM1ΔldhL1ΔldhL2. Unfortunately, there was no difference in the concentrations of d-lactic acid between strain DSM1ΔldhL1ΔldhL2 carrying P ldhL1 –BcldhD (0.15 ± 0.03 g/L) and strain DSM1ΔldhL1ΔldhL2 (0.16 ± 0.05 g/L) (Fig. 2). It seems that the enzyme activity of the native D-LHD in DSM1 was very low. L. delbrueckii subsp. bulgaricus DSM20081 is a d-lactic acid producer. The D-LDH (encoded by LdldhD) plays a central role in d-lactic acid production with a high catalytic efficiency (k cat ) of 235.5 ± 1.6 s−1 [7]. To further investigate the mechanism, two strains carrying LdldhD under the control of BcldhD promoter (P Bcldh –LdldhD) and ldhL1 promoter from DSM1 (P ldhL1 –LdldhD) were constructed, respectively. Results showed that 46.14 ± 0.52 and 45.05 ± 0.47 g/L d-lactic acid were obtained, with a yield of 0.98 and 0.96 g/g, respectively (Fig. 2). Same glucose consumption rates and OD600 values were also observed in both strains.

Fermentation profiles of B. coagulans DSM1ΔldhL1ΔldhL2 wild-type and mutant strains. a Glucose consumption; b d-lactic acid production; c A600 value. Each data point represents the average of three replicates, with the error bars representing the standard deviation
B. coagulans D-DSM1 construction by introducing LdldhD
Since the native d-lactate dehydrogenase could not support the efficient d-lactic acid production in the ldhL knockout strain, the ldhD gene from L. delbrueckii subsp. bulgaricus DSM20081 were introduced into B. coagulans DSM1ΔldhL1ΔldhL2 at the position of ldhL1, which is driven by the native promoter of ldhL1 (Fig. 1). Using the primers of DS-For/Rev, a 3002 bp-band was detected by PCR amplification, which means that the gene LdldhD had been inserted into the positon of ldhL1 in B. coagulans DSM1ΔldhL1ΔldhL2 (Additional file 1: Figure S2). The resultant strain ldhL1:: LdldhD was named as B. coagulans d-DSM1.
Strain B. coagulans d-DSM1 produced 48.65 g/L d-lactic acid with an initial glucose concentration of 50 and 117.96 g/L d-lactic acid with an initial glucose concentration of 120 g/L (Table 3). The optical purities were around 99.0–99.6%. To investigate the source of l-lactic acid in the broth, different yeast extraction concentration of 10, 5 and 1 g/L was added to the fermentation medium. At 0 h, 0.6 g/L l-lactic acid was detected in the medium with 10 g/L yeast extract, and the concentration was finally leveling out at 0.6 g/L during fermentation. The optical purity of d-lactate was 99.0, 99.5 and 100% respectively at 24 h. Therefore, it was concludes that the trace amount of l-lactic acid came from yeast extract and not from the endogenous metabolism of d-DSM1. Acetic acid (0.35 ± 0.02 g/L) and pyruvate (0.03 ± 0.00 g/L) were also found in the fermentation broth, and no ethanol, oxaloacetic acid and formic acid were detected (Additional file 1: Figure S3).
Non-sterilized fed-batch fermentation of d-lactic acid
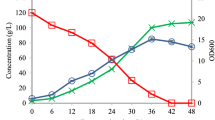
Non-sterilized fermentation of d-lactic acid production was performed in a 5-L bioreactor with fed-batch strategy. The initial glucose concentration was 50 g/L. When residual glucose decreased to approximately 20 g/L, glucose was added to a concentration of 50 g/L. During the first 12 h, the concentration of glucose decreased rapidly from 50 to 20 g/L. The final concentration of d-lactic acid reached 145.23 ± 0.82 g/L with a yield of 0.98 g/g and a productivity of 1.51 ± 0.05 g/L/h (Fig. 3). Since yeast extract contained a small amount of l-lactic acid (0.4 g/L at 0 h), the optical purity of d-lactic acid was just 91.6% at 0 h and increased gradually to 99.2% at 24 h. Interestingly, the optical purity achieved about 100% at the end of fermentation (Fig. 4).

Non-sterilized fed-batch fermentation of B. coagulans d-DSM1. Each data point represents the average of three replicates, with the error bars representing the standard deviation

Optical purity of d-lactic acid in non-sterilized fed-batch fermentation of B. coagulans d-DSM1. Each data point represents the average of three replicates, with the error bars representing the standard deviation
Discussion
Poly-d-lactic acid is an important polymer because it improves the thermostability of poly-l-lactic acid through stereo complex formation. However, fermentation of d-lactic acid monomer has been little studied in comparison with l-lactic acid [17]. Till now, most d-lactic acid producing strains are mesophilic microorganisms, whose growth temperature is between 30 and 42 °C (Table 4) [6]. For example, Yamada et al. reported that 60 g/L d-lactic acid was obtained by genetically manipulated S. cerevisiae at 30 °C [18]. The main drawback of d-lactic acid production by mesophilic microorganisms is the contamination during fermentation, because most species of l-lactic acid producers display mesophilic properties. To overcome such technical shortcomings, strains with thermotolerant characteristics are highly desirable. Tashiro et al. isolated a strain named L. delbrueckii subsp. lactis QU 41, which could produce d-lactic acid at 49–55 °C [4]. B. coagulans strains typically produce l-lactic acid with low or undetectable level of d-lactic acid. Compared to the most of d-lactic acid producers, such as L. bulgaricus, L. delbrueckii and L. plantarum, B. coagulans does not need complex nutrients and require less cooling [19]. B. coagulans could growth under micro aerobic conditions, which do not need oxygen application and high-speed agitation during lactic acid production. So, B. coagulans is an promising host with robustness for d-lactic acid production.
The key enzymes of lactic acid production are lactate dehydrogenases. l-lactate dehydrogenases (l-LDHs) are responsible for l-lactic acid production and d-lactate dehydrogenases (d-LDHs) are responsible for d-isomer production [7]. So many genetic modifications of strains focused on encoding genes of above two enzymes [20–23]. Actually, a d-lactic acid producing strain B. coagulans QZ19 was constructed by deleting the native ldhL and alsS (acetolactate synthase encoding gene) to impede anaerobic growth. Combing the growth-based selection, glycerol dehydrogenase was found to have the ability of d-LDH. Finally, 90 g/L of optically pure d-lactic acid was produced after a long time metabolic evolution in batch fermentation [12]. Low d-lactic acid titer hinders the further use for this strain. B. coagulans DSM1 is a homo-fermentative l-lactic acid producer. Three l-lactate dehydrogenase (l-LDH) encoding genes (ldhL1, ldhL2 and ldhL3) were discerned according to the whole-genome sequence of B. coagulans DSM1. The key roles of ldhL1 and ldhL2 have been systematically studied in our previous paper [13]. Both l-LDH1 (encoded by ldhL1, 5.27 ± 0.14 U/mg) and l-LDH2 (encoded by ldhL2, 4.53 ± 0.26 U/mg) were found to have catalytic activities in vitro. However, no transcription level was detected in ldhL2 in vivo. Deletion of the ldhL2 gene revealed no difference in fermentation profile compared to the wild-type strain, while ldhL1 single deletion or ldhL1ldhL2 double deletion completely blocked l-lactic acid production. Therefore, the genes encoding l-LDH1 (ldhL1) was inferred to contribute to l-lactic acid synthesis in B. coagulans DSM1. Because lactate dehydrogenases drive the consumption of NADH, which is an important step in the metabolism and energy conversion of living cells, ldhL1 deletion may provoke the increased expression of other NADH consumption enzymes, such as l-LDH2 [21]. So, B. coagulans DSM1ΔldhL1ΔldhL2 was used as a candidate in this study.
Because gene ldhL3 is annotated as leucine dehydrogenase/l-lactate dehydrogenase, the enzymatic characterization of l-LDH3 (encoded by ldhL3) was firstly studied. l-LDH3 was confirmed to be leucine dehydrogenase. There is no l-LDHs in B. coagulans DSM1ΔldhL1ΔldhL2. Then we tried to use the native d-LDH encoding gene BcldhD from B. coagulans DSM1 to construct the d-lactic acid producer. However, only trace amount of d-lactic acid was produced in B. coagulans DSM1ΔldhL1ΔldhL2, and the growth of strain was poor when l-LDH encoding genes deleted (Additional file 1: Figure S1) [12]. To trace the reason for low d-lactic acid production, d-lactic acid productions using four recombinants, expressing different d-LDH encoding genes under the control of BcldhD promoter (P BcldhD ) and ldhL1 promoter (P ldhL1 ) respectively, were performed. The introduction of BcldhD under the control of P BcldhD or P ldhL1 led to low production of d-lactic acid and low consumption of glucose. However, the introduction of a d-LDH encoding gene LdldhD from L. delbrueckii subsp. bulgaricus DSM20081 under the control of P BcldhD or P ldhL1 resulted in high production of d-lactic acid and high consumption of glucose (Fig. 2), which suggested that the activity of native promoter of BcldhD functioned enough to drive the gene expression. Even when strong promoter (P ldhL1 ) was introduced to drive the high expression of BcldhD, the enzyme of d-LDH from strain DSM1 did not show sufficient activity. So, the activity of native d-LDH was too low to support the efficient d-lactic acid production.
Lactobacillus delbrueckii subsp. bulgaricus DSM20081 is a d-lactic acid producer. Its key enzyme d-LDH, encoded by LdldhD, exhibited a high catalytic efficiency [7]. Most importantly, d-LDH from strain DSM20081 could work at the temperature at 50 °C, which is consistent with the growth temperature of B. coagulans (Additional file 1: Figure S4). Gene LdldhD from strain DSM20081 was inserted into the original location of ldhL1 using the ldhL1 promoter (Fig. 1). The obtained strain d-DSM1 produced 145 g/L d-lactic acid in fed-batch fermentation, with a yield of 0.98 g/g and an optical purity of 99.9% (Fig. 3), and 118 g/L d-lactic acid with a yield of 0.98 g/g was obtained in batch fermentation (Table 3). Attempts have been made to produce d-lactic acid using Lactic Acid Bacteria and E. coli [2, 26]. Some acid-tolerant strains, such as S. cerevisiae, P. acidilactici, S. inulinus, were also engineered to produce d-lactic acid [18, 24, 25, 27]. However, low d-lactic acid concentration was obtained under non-neutralized condition [25]. Actually, most strains are mesophilic (Table 4). A d-lactic acid producing strain was constructed based on a thermophilic Bacillus strain and 28 g/L d-lactic acid with an optical purity of 99.5% was obtained [23]. For thermophilic organism, the maximal yield of d-lactic acid was 90 g/L in batch fermentation [12]. In this study, besides metabolic engineering, also process optimization was carried out to obtain a thermophilic d-lactic-acid-producing strain. Compared with the existing d-lactic acid producing strains, strain d-DSM1 obtained in this study is more suitable for industry uses with high d-lactic acid titer (145 g/L) and high optical purity (99.9%). Until now, it is the highest d-lactic acid titer produced by thermo-tolerant strains (Table 4).
In conclusion, this present strain d-DSM1, which expresses LdldhD from L. delbrueckii subsp. bulgaricus DSM20081 under the control of the native ldhL1 promoter, produced d-lactic acid with high yield (0.98 g/g) and high optical purity (99.9%). Furthermore, non-sterilized fermentation was conducted which would significantly lower the fermentation costs and nutrition loss, and thus increase the economy of the fermentation process. B. coagulans d-DSM1 could be a promising d-lactic acid producer in industrial settings.
References
Marius M, Philippe D. PLA composites: from production to properties. Adv Drug Deliv Rev. 2016;107:17–46.
Okano K, Hama S, Kihara M, Noda H, Tanaka T, Kondo A. Production of optically pure d-lactic acid from brown rice using metabolically engineered Lactobacillus plantarum. Appl Microbiol Biotechnol. 2017;101:1869–75.
Södergard A, Stolt M. Properties of lactic acid based polymers and their correlation with composition. Prog Polym Sci. 2002;27:1123–63.
Tashiro Y, Kaneko W, Sun Y, Shibata K, Inokuma K, Zendo T, Sonomoto K. Continuous d-lactic acid production by a novel thermotolerant Lactobacillus delbrueckii subsp. lactis QU 41. Appl Microbiol Biotechnol. 2011;89:1741–50.
Ghaffar T, Irshad M, Anwar Z, Aqil T, Zulifqar Z, Tariq A, Kamran M, Ehsan N, Mehmood S. Recent trends in lactic acid biotechnology: a brief review on production to purification. J Radiat Res Appl Sci. 2014;7:222–9.
Wang L, Zhao B, Li F, Xu K, Ma C, Tao F, Li Q, Xu P. Highly efficient production of d-lactate by Sporolactobacillus sp. CASD with simultaneous enzymatic hydrolysis of peanut meal. Appl Microbiol Biotechnol. 2011;89:1009–17.
Wang L, Cai Y, Zhu L, Guo H, Yu B. Major role of NAD-dependent lactate dehydrogenases in high optically pure l-lactic acid production by thermophilic Bacillus coagulans. Appl Environ Microbiol. 2014;80:7134–41.
Zheng L, Liu M, Sun J, Wu B, He B. Sodium ions activated phosphofructokinase leading to enhanced d-lactic acid production by Sporolactobacillus inulinus using sodium hydroxide as a neutralizing agent. Appl Microbiol Biotechnol. 2017. https://doi.org/10.1007/s00253-017-8120-0.
Silvia K, Norman K, Anja K, Ulf P. Biotechnological production of enantiomerically pure d-lactic acid. Appl Microbiol Biotechnol. 2016;100:9423–37.
Wang L, Zhao B, Liu B, Yu B, Ma C, Yang C, Su F, Hua D, Li Q, Ma Y, Xu P. Efficient production of l-lactic acid from corncob molasses, a waste by-product in xylitol production, by a newly isolated xylose utilizing Bacillus sp. strain. Bioresour Technol. 2010;101:7908–15.
Peng L, Wang L, Che C, Yang G, Yu B, Ma Y. Bacillus sp. strain P38: an efficient producer of l-lactate from cellulosic hydrolysate, with high tolerance for 2-furfural. Bioresour Technol. 2013;149:169–76.
Wang Q, Ingram LO, Shanmugam KT. Evolution of d-lactate dehydrogenase activity from glycerol dehydrogenase and its utility for d-lactate production from lignocellulose. PNAS. 2011;108:18920–5.
Sun L, Zhang C, Lyu P, Wang Y, Wang L, Yu B. Contributory roles of two l-lactate dehydrogenases for l-lactic acid production in thermotolerant Bacillus coagulans. Sci Rep. 2016;6:37916. https://doi.org/10.1038/srep37916.
Kovács T, van Hartskamp M, Kuipers O, van Kranenburg R. Genetic tool development for a new host for biotechnology, the thermotolerant bacterium Bacillus coagulans. Appl Environ Microbiol. 2010;76:4085–8.
Zhu L, Xu X, Wang L, Dong H, Yu B. The d-lactate dehydrogenase from Sporolactobacillus inulinus also possessing reversible deamination activity. PLoS ONE. 2015;10:e0139066.
Nishihara K, Kanemori M, Kitagawa M, Yanagi H, Yura T. Chaperone coexpression plasmids: differential and synergistic roles of DnaK–DnaJ–GroE and GroEL–GroES in assisting folding of an allergen of Japanese cedar pollen, Cryj2, in Escherichia coli. Appl Environ Microbiol. 1998;64:1694–9.
Ishida N, Suzuki T, Tokuhiro K, Nagamori E, Onishi T, Saitoh S, Kitamoto K, Takahashi H. d-lactic acid production by metabolically engineered Saccharomyces cerevisiae. J Biosci Bieng. 2006;101:172–7.
Yamada R, Wakita K, Mitsui R, Ogino H. Enhanced d-lactic acid production by recombinant Saccharomyces cerevisiae following optimization of the global metabolic pathway. Biotechnol Bioeng. 2017. https://doi.org/10.1002/bit.26330.
Akita H, Nakashima N, Hoshino T. Production of d-lactate using a pyruvate-producing Escherichia coli strain. Biosci Biotechnol Biochem. 2017;81:1452–5.
Ferain T, Hobbs JN, Richardson J, Bernard N, Garmyn D, Hols P, Allen NE, Delcour J. Knockout of the two ldh genes has a major impact on peptidoglycan precursor synthesis in Lactobacillus plantarum. J Bacteriol. 1996;178:5431–7.
Rico J, Yebra MJ, Pérez-Martínez G, Deutscher J, Monedero V. Analysis of ldh genes in Lactobacillus casei BL23: role on lactic acid production. J Ind Microbiol Biotechnol. 2008;35:579–86.
Liu Y, Gao W, Zhao X, Wang JH, Garza E, Manow R, Zhou SD. Pilot scale demonstration of d-lactic acid fermentation facilitated by Ca(OH)2 using a metabolically engineered Escherichia coli. Bioresour Technol. 2014;169:559–65.
Van Kranenburg R, Van Hartskamp M, Heintz E, Anthonius J, Van Mullekom E, Snelders J. Genetic modification of homolactic thermophilic Bacilli. WO/2007/085443, 2007.
Qiu Z, Gao Q, Bao J. Constructing xylose-assimilating pathways in Pediococcus acidilactici for high titer d-lactic acid fermentation from corn stover feedstock. Bioresour Technol. 2017;245:1369–76.
Baek SH, Kwon EY, Kim YH, Hahn JS. Metabolic engineering and adaptive evolution for efficient production of d-lactic acid in Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2016;100:2737–48.
Zhou S, Yomano LP, Shanmugam KT, Ingram LO. Fermentation of 10% (w/v) sugar to d(−)-lactate by engineered Escherichia coli B. Biotechnol Lett. 2005;27:1891–6.
Zheng H, Gong J, Chen T, Chen X, Zhao X. Strain improvement of Sporolactobacillus inulinus ATCC 15538 for acid tolerance and production of d-lactic acid by genome shuffling. Appl Microbiol Biotechnol. 2010;85:1541–9.
Authors’ contributions
LW and BY designed the project; CLZ and CZ performed experiments; LW, NA and YM analyzed the data; LW wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The work was supported by grants from the National Natural Science Foundation of China (31670045), the Project of Guangxi Provincial Science & Technology Development, China and the Key International Cooperation Project from Chinese Academy of Sciences (155112KYSB20160010-02). The work was partially supported by grant from the Key International Cooperation Project of Chinese Academy of Sciences (155112KYSB20150024).
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data and materials are available.
Consent for publication
All the authors give their consents to the publication.
Ethics approval and consent to participate
Not applicable.
Funding
The work was funded by grants from the National Natural Science Foundation of China (31670045).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, C., Zhou, C., Assavasirijinda, N. et al. Non-sterilized fermentation of high optically pure d-lactic acid by a genetically modified thermophilic Bacillus coagulans strain. Microb Cell Fact 16, 213 (2017). https://doi.org/10.1186/s12934-017-0827-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-017-0827-1