
Abstract
The prevalence of obesity and atrial fibrillation (AF), which are inextricably linked, is rapidly increasing worldwide. Obesity rates are higher among patients with AF than healthy individuals. Some epidemiological data indicated that obese patients were more likely to develop AF, but others reported no significant correlation. Obesity-related hypertension, diabetes, and obstructive sleep apnea are all associated with AF. Additionally, increased epicardial fat, systemic inflammation, and oxidative stress caused by obesity can induce atrial enlargement, inflammatory activation, local myocardial fibrosis, and electrical conduction abnormalities, all of which led to AF and promoted its persistence. Weight loss reduced the risk and reversed natural progression of AF, which may be due to its anti-fibrosis and inflammation effect. However, fluctuations in weight offset the benefits of weight loss. Therefore, the importance of steady weight loss urges clinicians to incorporate weight management interventions in the treatment of patients with AF. In this review, we discuss the epidemiology of obesity and AF, summarize the mechanisms by which obesity triggers AF, and explain how weight loss improves the prognosis of AF.
Similar content being viewed by others


Introduction
Obesity has become a global epidemic. Obesity rates have nearly doubled in the last 30 years in several countries, such as China [1, 2], America [3], the United Kingdom [4], and India [5]. It is estimated that, by 2030, more than half of the world’s population will be obese, with the prevalence of severe obesity reaching 11% [6]. Truly, obesity has become a major public health concern.
Atrial fibrillation (AF) was first detected on electrocardiography by Einthoven and Lewis over 100 years ago [7]; at that time, it was considered a trivial disorder. With increased understanding of AF over the years, we have learned that AF is among the most common life-threatening arrhythmias worldwide. The current overall AF prevalence in the general population is approximately 1–2% [8,9,10,11]. Although striking, these data may underestimate the true AF incidence as patients with paroxysmal AF (pAF: defined by episodes that last < 7 days and terminate spontaneously) were not included in studies, with persistent AF (duration of episodes > 7 days) as the standard[12]. In addition, 5–35% of patients with persistent AF are asymptomatic.
There are a number of epidemiological studies that clearly demonstrate the relationship between obesity and AF (Table 1). For example, Wang TJ et al. found that regardless of gender, for every 1% increase in BMI, there was a 4% increase in AF risk in a multivariate model adjusted for cardiovascular risk factors and temporary myocardial infarction or heart failure, based on the Framingham Heart Study observation cohort of 5282 participants with an average age of 57 years and an average follow-up of 13.7 years [13]. Supportively, a prospective cohort conducted in the Danish Diets, Cancer, and Health Study by Frost L et al., enrolling 47,589 participants with an average age of 56 years, followed up for an average of 5.7 years, found that the adjusted risk ratio for AF or atrial flutter was 1.08 for males and 1.06 for females for every increase in BMI [14]. In addition, a retrospective study on middle-aged men (with an average age of 51.5 years) conducted by Rosengren A et al. revealed that excessive weight during youth and weight gain from the age of 20 to middle age were independently associated with the development of AF [15]. Similar conclusions have been confirmed in studies targeting women conducted by Tedrow UB et al. They analyzed 34,309 medical records from women’s health studies and found that overweight and obesity are associated with an increased risk of AF. Compared with participants who maintain a BMI of < 30 kg/m2, participants who become obese within the first 60 months from the beginning of the investigation have a 41% increased risk of developing AF [16]. Even among young fertile women with low incidence rate of AF (average age of 30.6 years), the risk ratio for AF of young obese women is 2.04, and that of extremely obese people is 3.50 comparing with normal weight women [17]. Therefore, obesity is largely associated with the increased risk of AF. Maintaining a normal weight or losing weight may prevent the onset of AF. Berkovich et al. analyzed the weight data of 18,290 men and women, and investigated the relationship between weight loss and the risk of AF. They found that weight loss was independently associated with a decrease in the risk of developing AF. For every 5 kg of weight loss, the risk of developing AF was significantly reduced by 12% [18].
Obesity is often accompanied by metabolic syndrome, diabetes, hypertension, and OSA [19]. Multiple studies evaluated the impact of these conditions as confounders on obesity-promoted AF development. In a retrospective cohort of 389,321 individuals, Lee et al. reported that metabolically healthy obesity was associated with a 20% increased risk of AF, whereas a 40% increase in the risk of AF was associated with metabolically unhealthy obesity [20]. Grundvold I et al. analyzed 7169 newly diagnosed patients with type 2 diabetes, and found that the risk of AF in overweight or obese patients at baseline was 1.9 times and 2.9 times higher than that in patients with normal BMI, respectively. 14% of patients with subsequent weight gain had a risk of AF of 1.5 times comparing with patients with stable or light weight [21]. Similarly, Kim et al. analyzed 9,797,418 patients who received the national health examination, and found that obese diabetes patients have higher risk (HR = 1.823) as compared with the diabetes patients [22]. Apart from metabolic syndrome and diabetes, Gami et al. demonstrated that obesity predicts AF independently of OSA syndrome by analyzing the factors influencing AF in 3,542 adults from Olmsted County who underwent diagnostic polysomnography between 1987 and 2003 [23]. And the recent Busselton Health Study in Busselton, Western Australia, demonstrated that a high BMI was a risk factor for AF independent of hypertension and more predictive of AF in men [24]. The above studies suggests that obesity is associated with an increased risk of AF regardless of the presence or absence of underlying diseases.
This review summarizes the AF incidence in the obese population, investigates obesity-induced AF pathogenesis and the impact of obesity on ablation, and highlights how weight loss and risk factor control improve AF prognosis.
AF occurrence, maintenance, and progression
AF is a complex cardiac arrhythmia that first develops in a paroxysmal form, then progresses to a persistent form, and finally continues as a long-term persistent form [29]. AF, the final outcome of different pathophysiological processes, presents with significant heterogeneity among patients [30].
AF is caused by focal ectopic-triggering activity that is mainly caused by early afterdepolarization (EAD) and delayed afterdepolarization (DAD). EAD typically occurs in the context of prolonged action potential duration (APD) [31]. In a normal action potential, L-type calcium (Ca2+) channels undergo voltage- and Ca2+-dependent inactivation, thereby limiting Ca2+ influx. APD progression allows L-type Ca2+ channels to recover from inactivation, thereby generating inward currents that lead to EAD [32]. DAD is mainly caused by abnormal sarcoplasmic reticulum (SR) Ca2+ leakage and diastolic SR Ca2+ release events. During diastole, Ca2+ released by SR activates the sodium–calcium exchanger, producing a transient inward current that results in membrane depolarization. In addition, gap junction coupling occurs between fibroblasts and cardiomyocytes through connexin-43 and connexin-45 proteins [33]. Compared to cardiomyocytes, the membrane potential of cardiac fibroblasts is relatively depolarized. The interaction between fibroblasts and cardiomyocytes promotes depolarization of atrial cardiomyocytes, thereby promoting DAD [34]. Any factor that leads to prolonged APD, endoplasmic reticulum (ER) Ca2+ leakage, ER Ca2+ release during diastole, or increased cardiac fibroblasts can induce ectopic-triggering activity and cause AF.
The muscular sleeve within the ostia of the pulmonary veins (PV) is the main source of ectopic-triggering activity that causes AF. The PV muscle sleeve consists of branching fibers with limited lateral coupling and abrupt fiber orientation changes, which provide a structural basis for ectopic triggering of AF [35]. Additionally, compared to cardiomyocytes in other regions of the atrium, the diastolic ER Ca2+ release events in the PV muscle sleeve region are increased [36], and the effective refractory period is shorter, which further increases the possibility of AF triggered by the ectopic-triggering activity in the PV muscle sleeve region.
AF maintenance and progression are based on the electrical and structural remodeling of atrial tissue. Atrial electrical remodeling is mainly caused by the following factors: downregulation of the Ca2+ current leading to shortening of the refractory period [37]; increased outward potassium (K+) current leading to accelerated repolarization and hyperpolarization of atrial cells [38]; and altered expression and localization of connexins that connect atrial myocytes, causing conduction abnormalities [39]. These changes promote reentry and maintenance of atrial activation [40]. Atrial structural remodeling mainly consists of fibrosis, atrial enlargement, and changes in cardiomyocyte ultrastructure. Changes in the cardiomyocyte ultrastructure during AF include myolysis [41], glycogen accumulation [42], gap junction impairment [43], nuclear chromatin changes [44], mitochondrial disruption [45] and redistribution, and SR alterations [46]. These changes reduce the contractility of atrial cardiomyocytes, prolong conduction, and induce and maintain AF. Atrial enlargement is an important determinant in the clinical assessment of the likelihood and prognosis of AF [47]. Greater atrial enlargement reflects a greater stretch of atrial myocytes and atrial damage. Additionally, atrial enlargement is closely associated with fibrosis [48], which can contribute to the maintenance of AF in several ways. First, fibrous tissue physically separates atrial muscle fibers, resulting in the local slowing of conduction. Second, an increased number of fibroblasts increases fibroblast–cardiomyocyte interactions, resulting in slowed conduction, cardiomyocyte depolarization, and a prolonged APD. Fibroblasts also influence the electrical activity of cardiomyocytes via paracrine bioactive substances [49]. AF typically progresses from infrequent to frequent and persistent episodes, followed by persistent AF. Clinical observations, numerous animal studies, and autopsy studies support the concept of “AF begets AF.“ In other words, AF directly induces atrial remodeling, thereby supporting AF maintenance and progression [50].
Mechanisms of obesity-promoting AF
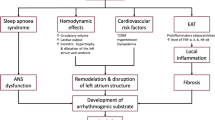
Although epidemiological studies have established the role of obesity in independently predicting the occurrence and progression of AF, the pathophysiological mechanisms associated with AF in obese patients are complex and remain unclear. Obese patients are susceptible to AF, which may be related to systemic changes caused by obesity, such as hemodynamic changes, hypertension, diabetes, and the OSA syndrome. Additionally, in terms of molecular biology, adipose tissue secretes a variety of pro-inflammatory and pro-fibrotic factors that can accelerate the structural and functional remodeling of the left atrium and induce and maintain electrical conduction abnormalities. Oxidative stress induced by adipose tissue and activated autonomic nerves in the ganglion plexus are also involved in the occurrence of AF (Fig. 1).

The arrhythmogenic effects of obesity on cardiomyocytes. Obesity is closely related to systemic hemodynamic changes, hypertension, sleep apnea syndrome, diabetes, and increased local epicardial fat. These systemic and local changes promote inflammation and oxidative stress, as well as activating the ganglionic plexus, leading to structural remodeling including fibrosis, left atrial enlargement, and electrical remodeling including conduction slowing and heterogeneity enhancing, and ultimately cause atrial fibrillation. OSA Obstructive sleep apnea, ROS Reactive oxygen species, NO nitric oxide, d-ROM derivative of reactive oxidative metabolites
Epicardial fat in obesity
Epicardial adipose tissue (EAT) refers to adipose tissue between the visceral pericardium and epicardium [51]. The EAT and myocardium share the same microcirculation with no muscle fascia between them [52]. Thus, atrial pathophysiological changes and localized EAT cumulative infiltration are closely related [53, 54].
Clinical studies have shown that periatrial EAT is associated with AF [55, 56]. Patients with persistent AF had larger EAT volumes (EATV) and higher levels of serum inflammatory biomarkers than those with pAF, which has nothing to do with the presence of obesity, age, sex, cardiovascular diseases, diabetes, dyslipidemia or hypertension [57]. The EATV index (EATVI) [58], high left atrial EAT/total EAT ratio [59], activin A expression in the EAT [60], and EAT content in the isolated left atrial posterior wall isolation line of the posterior left atrium are all strongly associated with the increased prevalence and severity of atrial fibrillation [59, 61, 62].
Atrial electrical remodeling, inflammation, fibrosis, and neurological factors are involved in the process of EAT-promoting AF (Fig. 1). Serum levels of inflammatory markers (MCP-1, IL-1, IL-6, soluble IL-6 receptor, and TNF-α) are directly correlated with the EAT of lower density and higher volume [63]. Regional IL-1β levels in the EAT are independent risk factors for persistent AF (Fig. 1) [64]. Local conduction block and conduction delay caused by localized fibrosis of the atrium underlie the formation of re-entrant arrhythmias [65].Various pro-fibrotic cytokines/chemokines in the EAT, such as YKL-40 [66] and cTGF [67], are positively correlated with total collagen content in the left atrial myocardium (Fig. 1) [68] [69]. Blockade of the adipofibrokine (activin A) by neutralizing antibodies has been shown to reverse atrial fibrosis [70]. In addition to inducing fibroblasts, adipose tissue can also develop fibrosis [55]. The EAT is the site of the ganglionic plexus, which contains sympathetic and vagal fibers that regulate the autonomic nerves of the heart and is closely related to the initiation and maintenance of AF (Fig. 1). In addition to being pro-fibrotic and pro-inflammatory, EAT also contributes to the formation and maintenance of AF by promoting oxidative stress [71] and Ca2+ homeostasis imbalance [72].
Obesity, inflammation and AF
In obese individuals, adipose tissue is infiltrated by a large number of activated macrophages, which is a state associated with systemic inflammation. The degree of macrophage infiltration is proportional to the body weight [73]. When the body weight is reduced, the number of infiltrated macrophages and inflammatory factors decrease [74, 75]. Besides, There are more CD4 + regulatory T cells involved in suppressing pro-inflammatory macrophages in lean mice [76], while obese mice are dominated by CD8 + effector T cells, which recruit and activate pro-inflammatory macrophages and promote the inflammatory cascade [77]. Therefore, the macrophages in the adipocytes of lean mice were dominated by the M2 anti-inflammatory phenotype, whereas the macrophages in the adipose tissue of obese mice were dominated by the M1 type [78].
Patients with AF had higher levels of inflammatory markers, including serum C-reactive protein (CRP), heat-shock protein (HSP) β1 (commonly referred to as HSP27), interleukin (IL)-6, IL-8, and tumor necrosis factor-α (TNF-α), when compared to patients in normal sinus rhythm [79, 80]. The pathology of atrial biopsy specimens from patients with solitary AF refractory to antiarrhythmic therapy showed lymphomonocytic infiltration and necrosis of adjacent myocytes [81], whereas these inflammatory abnormalities were absent in atrial biopsy specimens from patients in sinus rhythm. Large prospective cohort studies suggested that higher CRP levels can predict the risk of AF [82, 83], and CRP levels are reportedly higher in patients with persistent AF than in those with PAF [84]. Medicines with known anti-inflammatory effects, including glucocorticoids, N-3 fatty acids, statins, angiotensin-converting enzyme inhibitors, and angiotensin II receptor blockers, can reduce AF prevalence [85, 86]. Amiodarone’s antiarrhythmic effect may be partially dependent on its anti-inflammatory effect since it inhibits the synthesis of several cytokines, including IL-6 and TNF-α [87].
TNF-α plays a primary role in promoting atrial structural remodeling and affecting atrial ion channel function. TNF-α changes connexin-40 expression in mice and activates myofibroblasts through the transforming growth factor (TGF)-β signaling pathway, thereby inducing atrial fibrosis [88]. In addition, TNF-α can induce the expressions of matrix metalloproteinase-2 and matrix metalloproteinase-9 (MMP9), change the distribution of connexin-43 in the atrial tissue, and promote myocardial remodeling (Fig. 1) [89].The stimulation of PV cardiomyocytes with TNF-α increases the amplitude of DAD and decreases the L-type Ca2+ current [90]. Moreover, TNF-α reduces the Ca2+ content in the SR and increases the intracellular Ca2+ concentration during diastole, thereby inducing AF [91].
Similarly, IL-6 promotes the occurrence of AF mainly by influencing atrial electrical remodeling and fibrosis. Specifically, IL-6 induces early atrial fibrosis by activating pSTAT3/STAT3 signaling pathways[92], and promotes the expression of α-SMA, type I collagen, and type III collagen by inhibiting Tregs function [93]. IL-6 also mediates abnormalities in calcium processing in cardiac myocytes. IL-6 neutralizing antibodies can reverse the prolongation of Ca2 + transient duration and regional heterogeneity, reducing the incidence of discordant alternans, reducing the susceptibility and recurrence frequency of AF [94]. Moreover, IL-6 directly impairs the production of connexin in myocardial cells, including connexin 40 and connexin 43, thereby inducing atrial electrical remodeling and increasing the risk of AF [95].
Obesity, oxidative stress and AF
Accumulation of oxidative stress in adipose tissue is an early event in obesity [96]. Oxidative stress is significantly increased in white adipose tissue in obese animals and humans than in non-obese subjects [97]. Weight gain and obesity in children and adolescents are positively associated with elevated oxidative stress levels [98, 99]. Moreover, maternal obesity leads to increased placental oxidative damage and placental concentrations of ROS and protein carboxyl groups [100]. Reducing body weight and fat accumulation through exercise training [101], dietary restriction [102], increasing the content of high-fiber fruits in the diet [103], or gastrectomy [104] can reduce the formation of ceruloplasmin [105], identifiable reactive oxygen metabolites, and alpha-dicarbonyl compounds; thereby, reducing oxidative stress indicators such as ROS, nitric oxide, and malondialdehyde (Fig. 1) [106].
Oxidative stress is closely related to AF development. For every 10% increase in redox glutathione, the odds of developing AF increased by 30% [107]. The increased AF odds ratios for derivatives of reactive oxidative metabolites and cysteine were 6.1 and 15.9 [108], respectively. NOX-2-dependent ROS production in human right atrial samples is independently associated with postoperative AF[109]. Burst pacing–induced AF was significantly increased in transgenic mice overexpressing NOX-2, whereas the inhibition of mitochondrial oxidative stress by overexpressing mitochondrial catalase reduced the occurrence of AF [110]. Angiotensin II–induced AF is dependent on NADPH-oxidase-dependent ROS production and elevated ox-CaMKII levels [111]. Antioxidants also prevented atrial electrical remodeling in animal models of atrial tachycardia and new-onset AF after cardiac surgery. By correcting the myocardial redox balance, statins, canagliflozin, and allopurinol prevented AF-induced electrical remodeling in AF animal models and, therefore, may reduce the incidence of AF after cardiac surgery [112,113,114].
ROS generated by oxidative stress enhances late Na+ currents and induces early depolarization; thus, triggering activity [115]. Oxidative stress also induces AF by affecting L-type calcium channels and ryanodine receptor 2 (RyR2), thereby prolonging the APD [116]. Additionally, ROS stimulates the proliferation of atrial fibroblasts and promotes the expression of inflammatory and pro-fibrotic factors, such as MMP9, p38, and c-Jun (Fig. 1) [117].
Obesity, systemic diseases and AF
Clinically, hypertension and AF often coexist [118]. More than 60% of patients with AF also have hypertension. The Framingham study showed that high blood pressure increased the risk of AF by 40-50% [119]. BMI is an important cause of high blood pressure [120]. Obese subjects were 3.5 times more likely to develop high blood pressure, and more than 60% of the cases with high blood pressure were attributable to fat accumulation [121]. Across all age groups, the prevalence of hypertension in normal-weight subjects was 34%, whereas in obese subjects, it ranged from 60 to 77% and increased accordingly with the BMI [122].Decreased myocardial systolic-diastolic function caused by hypertension eventually leads to an increase in left atrial pressure, which forces left atrial dilation and forms the basis of AF. The increase in total blood volume in obese patients leads to an increase in cardiac output, which predisposes patients to left ventricular remodeling, hypertrophy, and diastolic dysfunction. Additionally, the potential development of AF has been observed in the myocardium of hypertensive mice, such as impaired Ca2+ transport [123], ultrastructural changes in cardiomyocytes [124], inflammatory infiltration, and activation of fibroblasts (Fig. 1). Moreover, electrophysiological changes were found in the left atrium, such as enhanced conduction heterogeneity, shortened atrial wavelengths, and prolonged AF duration [125, 126], which was detected several weeks after the onset of hypertension.
Multiple prospective cohort studies, retrospective studies, and meta-analyses have demonstrated that diabetes is an independent risk factor for AF [127,128,129,130]. Prediabetes and diabetes increase the risk of AF by 20% and 28%, respectively, and there is a correspondence between elevated blood glucose and AF, with an 11% increased risk of AF for every 20 mg/dL increase in blood glucose [131]. After matching for age and sex, the AF risk in patients with diabetes increased to 35% [132]. With a prolonged duration of diabetes, the risk of AF further increases. Patients with diabetes also had a higher recurrence rate after catheter ablation for AF than patients without diabetes [133]. The recent Dapagliflozin Effect on Cardiovascular Events - Thrombolysis in Myocardial Infarction 58 trial suggests that dapagliflozin reduces the risk of AF/flutter events by 19% in patients with T2DM, further underscoring the role of diabetes in the induction of AF [134, 135]. However, obesity is a major contributor to the development of type 2 diabetes mellitus (T2DM) [136]. Obesity in childhood increases the risk of developing T2DM later in life. More than 80% of people diagnosed with T2DM are obese [137].
Diabetes-induced atrial fibrosis and atrial dilation underlie AF induction. Various stimuli, including inflammation, advanced glycation end products (AGEs), and TGF-β, can promote the development of fibrosis [138, 139]. Pro-inflammatory cytokines and chemokines recruit fibrotic leukocyte subsets to the interstitium. Hyperglycemia-induced accumulation of AGEs transduces fibrotic signals through the receptor for AGE pathway/oxidative stress. TGF-β/Smad signaling activates fibroblasts and induces the deposition of extracellular matrix proteins. Additionally, diabetes-induced prolongation of atrial conduction time, increased dispersion of atrial effective refractory period, and imbalance of sympathetic and parasympathetic nerve activity all increase susceptibility to AF (Fig. 1).
OSA is prevalent in obese individuals and has been identified as an important risk factor for the development and progression of AF [23, 140]. OSA increases the risk of developing AF two-fold. Interestingly, the prevalence of OSA was higher in patients with AF than in those without AF [141]. After matching for age, sex, and other electrophysiological symptoms, patients with AF had a 24% higher prevalence of OSA than those without AF [142]. In addition, OSA reduces the efficacy of antiarrhythmic drugs, electrical cardioversion, and catheter ablation in AF treatment [143].
The relationship between OSA syndrome and AF is complex. Multiple studies have confirmed that OSA is an independent predictor of myocardial diastolic dysfunction, which may lead to left atrial enlargement [144, 145]. Hypoxic episodes caused by OSA activate the sympathetic nerves, leading to tachycardia and increased blood pressure, resulting in relative ischemia of the atrial myocardium [146, 147]. Furthermore, hypoxia reduces atrial conduction velocity and increases its heterogeneity, thus increasing susceptibility to AF [148, 149]. Prolonged apnea also promotes neuronal firing in the ganglionic plexus, near the PV [150]. Additionally, OSA-induced sympathovagal imbalance and atrial fibrosis promote the occurrence and maintenance of AF (Fig. 1) [151].
Effect of weight loss on AF
Multiple studies have shown that obesity management reverses the natural progression of AF [152,153,154]. In the Swedish obese subject study conducted in Sweden, bariatric surgery was associated with sustained weight loss (18% weight loss after 20 years), and in the 19-year follow-up, the risk of AF was reduced by 29% in the bariatric surgery group than it was in the control group (Fig. 2) [25]. Donnellan et al. compared the effects of different bariatric surgeries on AF, and the results showed that the percentage of weight loss significantly correlated with the reversal of AF [155]. Weight loss in obese patients with long-term persistent AF also effectively improved their quality of life, although there was no change in symptom severity or long-term ablation outcomes [156]. Mahajan et al. partially explained the role of weight loss in the development of AF in animal experiments conducted on 30 sheep. After weight loss in obese sheep, the cardiac structure and electrophysiology are inversely reconstructed, which manifest as a reduction in inflammation and fibrosis and an increase in the atrial effective refractory period and conduction velocity (Fig. 2) [157].

Proposed mechanism of benefits of weight loss to reduce AF. Reverse remodeling of cardiac structure and electrophysiology occurs after weight loss, manifested as decreased inflammation and fibrosis, increased atrial effective refractory period, and increased conduction velocity
Sustained and stable weight loss is an essential factor in obesity management to reverse AF, and weight fluctuations offset the benefits of weight loss. The Long-Term Effect of Goal Directed Weight Management on Atrial Fibrillation Cohort: A 5 Year follow-up study showed that when the weight loss was ≥ 10%, the arrhythmic free survival rate, AF burden, and symptom severity were significantly decreased, while weight fluctuation > 5% partially offset this benefit, and the risk of arrhythmia recurrence was doubled [158]. A meta-analysis conducted by Jones et al. showed that weight loss of 5% was not associated with a significant change in AF incidence (HR 1.04) [159]. Johansson et al. conducted a health survey of more than 100,000 people in northern Sweden and found that middle-aged weight loss was not significantly correlated with AF risk, which may be related to fluctuations in body weight [160]. Lee et al. quantified the effects of weight variability on AF. They found that for every 1 standard deviation increase in weight variability, the risk of AF increased by 5%. Except for the extremely obese group (BMI ≥ 30 kg/m2), high body weight variability in all baseline BMI groups was significantly associated with AF occurrence, and this correlation was stronger in subjects with lower body weight [161].
The current mainstream view is that it is beneficial to reduce weight and manage the impact of risk factors on the prognosis of AF [19]. The American Heart Association strongly recommends reducing AF by adjusting lifestyle and controlling risk factors, including obesity, lack of exercise, OSA, diabetes, hypertension, and other modifiable factors [162]. However, a multicenter cross-sectional descriptive study conducted in Belgium found that more than 30% of obese patients did not understand the positive impact of weight loss on AF progression. Patients with low education, hypertension, who are living alone, who have never tried to lose weight, and have low BMI, but are still at high risk of developing AF, lacked the intrinsic motivation to lose weight [163]. Weight management is an effective intervention that does not require marketing and a large amount of financial promotion, such as drug- and device-based therapies, and should be vigorously promoted. Cardiovascular physicians should also include weight management programs for the treatment of obese patients with AF to improve patient-centered treatment outcomes [164].
Conclusion and perspective remarks
Obesity is a major risk factor for AF. Obese individuals are more likely to develop AF than normal-weight individuals. Adipose tissue leads to left atrial enlargement and electrical remodeling through various mechanisms, including inflammation and oxidative stress. This induces AF development and promotes type switching. Weight loss reduces AF development and is associated with a reduced rate of AF recurrence after ablation. Although the relationship between obesity and AF has been well explained, the pathophysiological mechanisms are complex and have not been fully elucidated. For example, from the perspective of metabolism, obesity is categorized into metabolically healthy and unhealthy. Even if metabolically healthy obese patients have a high BMI, the AF risk is low; thus, using BMI alone to identify obesity is not accurate. In addition, adipose tissue is classified as white, beige, and brown fats, which are distributed throughout the body and function differently. Do these different changes in fat correspond to different AF risks? Is it necessary to define the role of fat type in AF? Obesity is a risk factor for the recurrence of PAF after catheter ablation, but not for permanent AF; therefore, further studies are needed to explore the differences in the development of paroxysmal and permanent AF.
Studies have demonstrated the effect of obesity on AF, with obesity in adults as the main target of mechanistic exploration; however, obesity in early life is a better predictor of AF risk throughout adulthood. Further studies are required to explore the mechanisms by which early obesity promotes AF development in adulthood.
Epicardial fat in obese patients plays a key role in left atrial structural and electrical remodeling. The size and thickness of epicardial tissue predict the occurrence, development, and type switching of AF. However, the current imaging techniques used to detect EAT cannot meet the requirements of speed, economy, and accuracy at the same time, and cannot quantify the relationship between epicardial fat volume and AF severity and prognosis. Understanding the importance of EAT in AF development is still in the clinical research stage. Exploration of the possibility of linking EAT with AF diagnosis and treatment may be an important direction in future research.
The current mainstream view is that weight loss is beneficial in reducing the occurrence of AF; however, there is still an “obesity paradox.“ Specifically, weight loss was not associated with a reduced AF risk [165], whereas an increased BMI was independently associated with a reduced risk of stroke and improved survival after AF [166]. Therefore, to clarify the status of weight loss in AF prevention and treatment, it is necessary to explore the reasons for the obesity paradox. The idea that weight loss can reduce the AF burden has not been widely accepted, and most people lack intrinsic motivation to lose weight. Therefore, it is necessary to vigorously promote weight management and make weight management programs mandatory for patients with AF as a part of their daily treatment.
Availability of data and materials
Not applicable.
Abbreviations
- AF:
-
Atrial fibrillation
- AGE:
-
Advanced glycation end product
- Angptl2:
-
Angiopoietin-like protein 2
- ATPase:
-
Adenosine triphosphatase
- BMI:
-
Body mass index
- CRP:
-
C-reactive protein
- DAD:
-
Delayed afterdepolarization
- DIO:
-
Diet-induced obese
- EAD:
-
Early afterdepolarization
- EAT:
-
Epicardial adipose tissue
- EATV:
-
EAT volumes
- EATVI:
-
EATV index
- ER:
-
Endoplasmic reticulum
- HFD:
-
High-fat diet
- HR:
-
Hazard ratio
- HSP:
-
Heat-shock protein
- LV:
-
Left ventricular
- MCP-1:
-
Monocyte chemoattractant protein 1
- MMP9:
-
Matrix metalloproteinase-9
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NE:
-
Norepinephrine
- NOX:
-
NADPH-oxidase
- OSA:
-
Obstructive sleep apnea
- PAF:
-
Paroxysmal AF
- PAT:
-
Paroxysmal atrial tachycardia
- PKA:
-
Protein kinase A
- PV:
-
Pulmonary veins
- ROS:
-
Reactive oxygen species
- RyR2:
-
Ryanodine receptor 2
- SR:
-
Sarcoplasmic reticulum
- T2DM:
-
Type 2 diabetes mellitus
- TNF:
-
Tumor necrosis factor
References
Chu NF. Prevalence of obesity in Taiwan. Obes Rev. 2005;6:271–4.
Chen CM. Overview of obesity in Mainland China. Obes Rev. 2008;9(Suppl 1):14–21.
Baskin ML, Ard J, Franklin F, Allison DB. Prevalence of obesity in the United States. Obes Rev. 2005;6:5–7.
Rennie KL, Jebb SA. Prevalence of obesity in Great Britain. Obes Rev. 2005;6:11–2.
Garg C, Khan SA, Ansari SH, Garg M. Prevalence of obesity in indian women. Obes Rev. 2010;11:105–8.
Finkelstein EA, Khavjou OA, Thompson H, Trogdon JG, Pan L, Sherry B, et al. Obesity and severe obesity forecasts through 2030. Am J Prev Med. 2012;42:563–70.
Pahlm O, Uvelius B. The winner takes it all: Willem Einthoven, Thomas Lewis, and the Nobel prize 1924 for the discovery of the electrocardiogram. J Electrocardiol. 2019;57:122–7.
Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and risk factors in Atrial Fibrillation (ATRIA) Study. JAMA. 2001;285:2370–5.
Naccarelli GV, Varker H, Lin J, Schulman KL. Increasing prevalence of atrial fibrillation and flutter in the United States. Am J Cardiol. 2009;104:1534–9.
Murphy NF, Simpson CR, Jhund PS, Stewart S, Kirkpatrick M, Chalmers J, et al. A national survey of the prevalence, incidence, primary care burden and treatment of atrial fibrillation in Scotland. Heart. 2007;93:606–12.
Wilke T, Groth A, Mueller S, Pfannkuche M, Verheyen F, Linder R, et al. Incidence and prevalence of atrial fibrillation: an analysis based on 8.3 million patients. Europace. 2013;15:486–93.
Nattel S, Dobrev D. Electrophysiological and molecular mechanisms of paroxysmal atrial fibrillation. Nat Rev Cardiol. 2016;13:575–90.
Wang TJ, Parise H, Levy D, D’Agostino RB, Sr., Wolf PA, Vasan RS, et al. Obesity and the risk of new-onset atrial fibrillation. JAMA. 2004;292:2471–7.
Frost L, Hune LJ, Vestergaard P. Overweight and obesity as risk factors for atrial fibrillation or flutter: the danish Diet, Cancer, and Health Study. Am J Med. 2005;118:489–95.
Rosengren A, Hauptman PJ, Lappas G, Olsson L, Wilhelmsen L, Swedberg K. Big men and atrial fibrillation: effects of body size and weight gain on risk of atrial fibrillation in men. Eur Heart J. 2009;30:1113–20.
Tedrow UB, Conen D, Ridker PM, Cook NR, Koplan BA, Manson JE, et al. The long- and short-term impact of elevated body mass index on the risk of new atrial fibrillation the WHS (women’s health study). J Am Coll Cardiol. 2010;55:2319–27.
Karasoy D, Bo Jensen T, Hansen ML, Schmiegelow M, Lamberts M, Gislason GH, et al. Obesity is a risk factor for atrial fibrillation among fertile young women: a nationwide cohort study. Europace. 2013;15:781–6.
Berkovitch A, Kivity S, Klempfner R, Segev S, Milwidsky A, Erez A, et al. Body mass index and the risk of new-onset atrial fibrillation in middle-aged adults. Am Heart J. 2016;173:41–8.
Lavie CJ, Pandey A, Lau DH, Alpert MA, Sanders P. Obesity and Atrial Fibrillation Prevalence, Pathogenesis, and prognosis: Effects of Weight loss and Exercise. J Am Coll Cardiol. 2017;70:2022–35.
Lee H, Choi EK, Lee SH, Han KD, Rhee TM, Park CS, et al. Atrial fibrillation risk in metabolically healthy obesity: a nationwide population-based study. Int J Cardiol. 2017;240:221–7.
Grundvold I, Bodegard J, Nilsson PM, Svennblad B, Johansson G, Östgren CJ, et al. Body weight and risk of atrial fibrillation in 7,169 patients with newly diagnosed type 2 diabetes; an observational study. Cardiovasc Diabetol. 2015;14:5.
Kim YG, Han KD, Choi JI, Boo KY, Kim DY, Oh SK, et al. The impact of body weight and diabetes on new-onset atrial fibrillation: a nationwide population based study. Cardiovasc Diabetol. 2019;18:128.
Gami AS, Hodge DO, Herges RM, Olson EJ, Nykodym J, Kara T, et al. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J Am Coll Cardiol. 2007;49:565–71.
Knuiman M, Briffa T, Divitini M, Chew D, Eikelboom J, McQuillan B, et al. A cohort study examination of established and emerging risk factors for atrial fibrillation: the Busselton Health Study. Eur J Epidemiol. 2014;29:181–90.
Jamaly S, Carlsson L, Peltonen M, Jacobson P, Sjöström L, Karason K. Bariatric surgery and the risk of New-Onset Atrial Fibrillation in Swedish obese subjects. J Am Coll Cardiol. 2016;68:2497–504.
Zacharias A, Schwann TA, Riordan CJ, Durham SJ, Shah AS, Habib RH. Obesity and risk of new-onset atrial fibrillation after cardiac surgery. Circulation. 2005;112:3247–55.
Foy AJ, Mandrola J, Liu G, Naccarelli GV. Relation of obesity to New-Onset Atrial Fibrillation and Atrial Flutter in adults. Am J Cardiol. 2018;121:1072–5.
Lim YM, Yang PS, Jang E, Yu HT, Kim TH, Uhm JS, et al. Body Mass Index variability and long-term risk of New-Onset Atrial Fibrillation in the General Population: a korean Nationwide Cohort Study. Mayo Clin Proc. 2019;94:225–35.
Kallergis EM, Goudis CA, Vardas PE. Atrial fibrillation: a progressive atrial myopathy or a distinct disease? Int J Cardiol. 2014;171:126–33.
Schotten U, Verheule S, Kirchhof P, Goette A. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev. 2011;91:265–325.
Huang X, Song Z, Qu Z. Determinants of early afterdepolarization properties in ventricular myocyte models. PLoS Comput Biol. 2018;14:e1006382.
Chu Z, Yang D, Huang X. Conditions for the genesis of early afterdepolarization in a model of a ventricular myocyte. Chaos. 2020;30:043105.
Rohr S. Myofibroblasts in diseased hearts: new players in cardiac arrhythmias? Heart Rhythm. 2009;6:848–56.
Yue L, Xie J, Nattel S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc Res. 2011;89:744–53.
Nattel S. Paroxysmal atrial fibrillation and pulmonary veins: relationships between clinical forms and automatic versus re-entrant mechanisms. Can J Cardiol. 2013;29:1147–9.
Honjo H, Boyett MR, Niwa R, Inada S, Yamamoto M, Mitsui K, et al. Pacing-induced spontaneous activity in myocardial sleeves of pulmonary veins after treatment with ryanodine. Circulation. 2003;107:1937–43.
Yue L, Feng J, Gaspo R, Li GR, Wang Z, Nattel S. Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ Res. 1997;81:512–25.
Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res. 2014;114:1483–99.
Igarashi T, Finet JE, Takeuchi A, Fujino Y, Strom M, Greener ID, et al. Connexin gene transfer preserves conduction velocity and prevents atrial fibrillation. Circulation. 2012;125:216–25.
Chen PS, Chen LS, Fishbein MC, Lin SF, Nattel S. Role of the autonomic nervous system in atrial fibrillation: pathophysiology and therapy. Circ Res. 2014;114:1500–15.
Allessie M, Ausma J, Schotten U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc Res. 2002;54:230–46.
Embi AA, Scherlag BJ. An endocrine hypothesis for the genesis of atrial fibrillation: the hypothalamic-pituitary-adrenal axis response to stress and glycogen accumulation in atrial tissues. N Am J Med Sci. 2014;6:586–90.
Ueda N, Yamamoto M, Honjo H, Kodama I, Kamiya K. The role of gap junctions in stretch-induced atrial fibrillation. Cardiovasc Res. 2014;104:364–70.
Qiu D, Peng L, Ghista DN, Wong KKL. Left Atrial Remodeling Mechanisms Associated with Atrial Fibrillation. Cardiovasc Eng Technol. 2021;12:361–72.
Yu LM, Dong X, Xu YL, Zhou ZJ, Huang YT, Zhao JK, et al. Icariin attenuates excessive alcohol consumption-induced susceptibility to atrial fibrillation through SIRT3 signaling. Biochim Biophys Acta Mol Basis Dis. 2022;1868:166483.
Ai X. SR calcium handling dysfunction, stress-response signaling pathways, and atrial fibrillation. Front Physiol. 2015;6:46.
Tsioufis C, Konstantinidis D, Nikolakopoulos I, Vemmou E, Kalos T, Georgiopoulos G, et al. Biomarkers of Atrial Fibrillation in Hypertension. Curr Med Chem. 2019;26:888–97.
Platonov PG, Mitrofanova LB, Orshanskaya V, Ho SY. Structural abnormalities in atrial walls are associated with presence and persistency of atrial fibrillation but not with age. J Am Coll Cardiol. 2011;58:2225–32.
Sohns C, Marrouche NF. Atrial fibrillation and cardiac fibrosis. Eur Heart J. 2020;41:1123–31.
Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995;92:1954–68.
Gaborit B, Venteclef N, Ancel P, Pelloux V, Gariboldi V, Leprince P, et al. Human epicardial adipose tissue has a specific transcriptomic signature depending on its anatomical peri-atrial, peri-ventricular, or peri-coronary location. Cardiovasc Res. 2015;108:62–73.
Monti CB, Codari M, De Cecco CN, Secchi F, Sardanelli F, Stillman AE. Novel imaging biomarkers: epicardial adipose tissue evaluation. Br J Radiol. 2020;93:20190770.
Marchington JM, Pond CM. Site-specific properties of pericardial and epicardial adipose tissue: the effects of insulin and high-fat feeding on lipogenesis and the incorporation of fatty acids in vitro. Int J Obes. 1990;14:1013–22.
Iacobellis G, di Gioia CR, Di Vito M, Petramala L, Cotesta D, De Santis V, et al. Epicardial adipose tissue and intracoronary adrenomedullin levels in coronary artery disease. Horm Metab Res. 2009;41:855–60.
Haemers P, Hamdi H, Guedj K, Suffee N, Farahmand P, Popovic N, et al. Atrial fibrillation is associated with the fibrotic remodelling of adipose tissue in the subepicardium of human and sheep atria. Eur Heart J. 2017;38:53–61.
Kusayama T, Furusho H, Kashiwagi H, Kato T, Murai H, Usui S, et al. Inflammation of left atrial epicardial adipose tissue is associated with paroxysmal atrial fibrillation. J Cardiol. 2016;68:406–11.
Chen PS, Turker I. Epicardial adipose tissue and neural mechanisms of atrial fibrillation. Circ Arrhythm Electrophysiol. 2012;5:618–20.
Zhou Y, Yu M, Cui J, Hu F, Yang Z, Yuan J, et al. The predictive value of epicardial adipose tissue volume assessed by cardiac magnetic resonance for atrial fibrillation in patients with hypertrophic obstructive cardiomyopathy. Int J Cardiovasc Imaging. 2021;37:1383–93.
Kogo H, Sezai A, Osaka S, Shiono M, Tanaka M. Does Epicardial Adipose tissue influence postoperative atrial fibrillation? Ann Thorac Cardiovasc Surg. 2019;25:149–57.
Wang Q, Min J, Jia L, Xi W, Gao Y, Diao Z, et al. Human epicardial adipose tissue activin a expression predicts occurrence of postoperative atrial fibrillation in patients receiving cardiac surgery. Heart Lung Circ. 2019;28:1697–705.
Nakatani Y, Sakamoto T, Yamaguchi Y, Tsujino Y, Kinugawa K. Epicardial adipose tissue affects the efficacy of left atrial posterior wall isolation for persistent atrial fibrillation. J Arrhythm. 2020;36:652–9.
Maeda M, Oba K, Yamaguchi S, Arasaki O, Sata M, Masuzaki H, et al. Usefulness of Epicardial Adipose tissue volume to Predict Recurrent Atrial Fibrillation after Radiofrequency catheter ablation. Am J Cardiol. 2018;122:1694–700.
Goeller M, Achenbach S, Marwan M, Doris MK, Cadet S, Commandeur F, et al. Epicardial adipose tissue density and volume are related to subclinical atherosclerosis, inflammation and major adverse cardiac events in asymptomatic subjects. J Cardiovasc Comput Tomogr. 2018;12:67–73.
Liu Q, Zhang F, Yang M, Zhong J. Increasing level of Interleukin-1β in Epicardial Adipose tissue is Associated with Persistent Atrial Fibrillation. J Interferon Cytokine Res. 2020;40:64–9.
Everett THt, Olgin JE. Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm. 2007;4:24–7.
Wang Q, Shen H, Min J, Gao Y, Liu K, Xi W, et al. YKL-40 is highly expressed in the epicardial adipose tissue of patients with atrial fibrillation and associated with atrial fibrosis. J Transl Med. 2018;16:229.
Wang Q, Xi W, Yin L, Wang J, Shen H, Gao Y, et al. Human epicardial adipose tissue cTGF expression is an independent risk factor for Atrial Fibrillation and highly Associated with Atrial Fibrosis. Sci Rep. 2018;8:3585.
Abe I, Teshima Y, Kondo H, Kaku H, Kira S, Ikebe Y, et al. Association of fibrotic remodeling and cytokines/chemokines content in epicardial adipose tissue with atrial myocardial fibrosis in patients with atrial fibrillation. Heart Rhythm. 2018;15:1717–27.
Shaihov-Teper O, Ram E, Ballan N, Brzezinski RY, Naftali-Shani N, Masoud R, et al. Extracellular vesicles from Epicardial Fat facilitate Atrial Fibrillation. Circulation. 2021;143:2475–93.
Venteclef N, Guglielmi V, Balse E, Gaborit B, Cotillard A, Atassi F, et al. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur Heart J. 2015;36:795–805a.
Kim YM, Guzik TJ, Zhang YH, Zhang MH, Kattach H, Ratnatunga C, et al. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ Res. 2005;97:629–36.
Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, et al. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation. 2014;129:145–56.
Cancello R, Henegar C, Viguerie N, Taleb S, Poitou C, Rouault C, et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes. 2005;54:2277–86.
Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–30.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808.
Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–9.
Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8 + effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–20.
Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604.
Chang G, Chen Y, Liu Z, Wang Y, Ge W, Kang Y, et al. The PD-1 with PD-L1 Axis is pertinent with the Immune Modulation of Atrial Fibrillation by regulating T cell excitation and promoting the secretion of inflammatory factors. J Immunol Res. 2022;2022:3647817.
Guo Y, Lip GY, Apostolakis S. Inflammation in atrial fibrillation. J Am Coll Cardiol. 2012;60:2263–70.
Frustaci A, Chimenti C, Bellocci F, Morgante E, Russo MA, Maseri A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation. 1997;96:1180–4.
Aviles RJ, Martin DO, Apperson-Hansen C, Houghtaling PL, Rautaharju P, Kronmal RA, et al. Inflammation as a risk factor for atrial fibrillation. Circulation. 2003;108:3006–10.
Conen D, Ridker PM, Everett BM, Tedrow UB, Rose L, Cook NR, et al. A multimarker approach to assess the influence of inflammation on the incidence of atrial fibrillation in women. Eur Heart J. 2010;31:1730–6.
Dernellis J, Panaretou M. C-reactive protein and paroxysmal atrial fibrillation: evidence of the implication of an inflammatory process in paroxysmal atrial fibrillation. Acta Cardiol. 2001;56:375–80.
Healey JS, Baranchuk A, Crystal E, Morillo CA, Garfinkle M, Yusuf S, et al. Prevention of atrial fibrillation with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: a meta-analysis. J Am Coll Cardiol. 2005;45:1832–9.
Siu CW, Lau CP, Tse HF. Prevention of atrial fibrillation recurrence by statin therapy in patients with lone atrial fibrillation after successful cardioversion. Am J Cardiol. 2003;92:1343–5.
Scridon A, Dobreanu D, Chevalier P, Şerban RC. Inflammation, a link between obesity and atrial fibrillation. Inflamm Res. 2015;64:383–93.
Liew R, Khairunnisa K, Gu Y, Tee N, Yin NO, Naylynn TM, et al. Role of tumor necrosis factor-α in the pathogenesis of atrial fibrosis and development of an arrhythmogenic substrate. Circ J. 2013;77:1171–9.
Sawaya SE, Rajawat YS, Rami TG, Szalai G, Price RL, Sivasubramanian N, et al. Downregulation of connexin40 and increased prevalence of atrial arrhythmias in transgenic mice with cardiac-restricted overexpression of tumor necrosis factor. Am J Physiol Heart Circ Physiol. 2007;292:H1561–7.
Lee SH, Chen YC, Chen YJ, Chang SL, Tai CT, Wongcharoen W, et al. Tumor necrosis factor-alpha alters calcium handling and increases arrhythmogenesis of pulmonary vein cardiomyocytes. Life Sci. 2007;80:1806–15.
Kao YH, Chen YC, Cheng CC, Lee TI, Chen YJ, Chen SA. Tumor necrosis factor-alpha decreases sarcoplasmic reticulum Ca2+-ATPase expressions via the promoter methylation in cardiomyocytes. Crit Care Med. 2010;38:217–22.
Liu Y, Wu F, Wu Y, Elliott M, Zhou W, Deng Y, et al. Mechanism of IL-6-related spontaneous atrial fibrillation after coronary artery grafting surgery: IL-6 knockout mouse study and human observation. Transl Res. 2021;233:16–31.
Chen Y, Chang G, Chen X, Li Y, Li H, Cheng D, et al. IL-6-miR-210 suppresses Regulatory T cell function and promotes Atrial Fibrosis by Targeting Foxp3. Mol Cells. 2020;43:438–47.
Liao J, Zhang S, Yang S, Lu Y, Lu K, Wu Y, et al. Interleukin-6-Mediated-Ca(2+) handling abnormalities contributes to Atrial Fibrillation in sterile pericarditis rats. Front Immunol. 2021;12:758157.
Lazzerini PE, Laghi-Pasini F, Acampa M, Srivastava U, Bertolozzi I, Giabbani B, et al. Systemic inflammation rapidly induces reversible Atrial Electrical remodeling: the role of Interleukin-6-Mediated changes in Connexin expression. J Am Heart Assoc. 2019;8:e011006.
Suzuki K, Ito Y, Ochiai J, Kusuhara Y, Hashimoto S, Tokudome S, et al. Relationship between obesity and serum markers of oxidative stress and inflammation in japanese. Asian Pac J Cancer Prev. 2003;4:259–66.
Sun X, Li X, Jia H, Wang H, Shui G, Qin Y, et al. Nuclear factor E2-Related factor 2 mediates oxidative Stress-Induced lipid Accumulation in Adipocytes by increasing adipogenesis and decreasing Lipolysis. Antioxid Redox Signal. 2020;32:173–92.
Warolin J, Coenen KR, Kantor JL, Whitaker LE, Wang L, Acra SA, et al. The relationship of oxidative stress, adiposity and metabolic risk factors in healthy black and white american youth. Pediatr Obes. 2014;9:43–52.
Tran B, Oliver S, Rosa J, Galassetti P. Aspects of inflammation and oxidative stress in pediatric obesity and type 1 diabetes: an overview of ten years of studies. Exp Diabetes Res. 2012;2012:683680.
Hu C, Yan Y, Ji F, Zhou H. Maternal obesity increases oxidative stress in Placenta and it is Associated with Intestinal Microbiota. Front Cell Infect Microbiol. 2021;11:671347.
Nono Nankam PA, Mendham AE, De Smidt MF, Keswell D, Olsson T, Blüher M, et al. Changes in systemic and subcutaneous adipose tissue inflammation and oxidative stress in response to exercise training in obese black african women. J Physiol. 2020;598:503–15.
Dandona P, Mohanty P, Ghanim H, Aljada A, Browne R, Hamouda W, et al. The suppressive effect of dietary restriction and weight loss in the obese on the generation of reactive oxygen species by leukocytes, lipid peroxidation, and protein carbonylation. J Clin Endocrinol Metab. 2001;86:355–62.
Crujeiras AB, Parra MD, Rodríguez MC, Martínez de Morentin BE, Martínez JA. A role for fruit content in energy-restricted diets in improving antioxidant status in obese women during weight loss. Nutrition. 2006;22:593–9.
Wang M, Xiong Y, Zhu W, Ruze R, Xu Q, Yan Z, et al. Sleeve gastrectomy ameliorates diabetes-related spleen damage by improving oxidative stress status in Diabetic obese rats. Obes Surg. 2021;31:1183–95.
Hariri N, Darafshi Ghahroudi S, Jahangiri S, Borumandnia N, Narmaki E, Saidpour A. The beneficial effects of sumac (Rhus coriaria L.) supplementation along with restricted calorie diet on anthropometric indices, oxidative stress, and inflammation in overweight or obese women with depression: a randomized clinical trial. Phytother Res. 2020;34:3041–51.
Vîrgolici B, Mohora M, Stoian I, Lixandru D, Găman L, Paveliu F. A comparative oxidative stress study–obesity with and without diabetes mellitus. Rom J Intern Med. 2005;43:261–8.
Samman Tahhan A, Sandesara PB, Hayek SS, Alkhoder A, Chivukula K, Hammadah M, et al. Association between oxidative stress and atrial fibrillation. Heart Rhythm. 2017;14:1849–55.
Neuman RB, Bloom HL, Shukrullah I, Darrow LA, Kleinbaum D, Jones DP, et al. Oxidative stress markers are associated with persistent atrial fibrillation. Clin Chem. 2007;53:1652–7.
Okada A, Kashima Y, Tomita T, Takeuchi T, Aizawa K, Takahashi M, et al. Characterization of cardiac oxidative stress levels in patients with atrial fibrillation. Heart Vessels. 2016;31:80–7.
Avula UMR, Dridi H, Chen BX, Yuan Q, Katchman AN, Reiken SR, et al. Attenuating persistent sodium current-induced atrial myopathy and fibrillation by preventing mitochondrial oxidative stress. JCI Insight. 2021. https://doi.org/10.1172/jci.insight.147371.
Yang KC, Dudley SC Jr. Oxidative stress and atrial fibrillation: finding a missing piece to the puzzle. Circulation. 2013;128:1724–6.
Nishinarita R, Niwano S, Niwano H, Nakamura H, Saito D, Sato T, et al. Canagliflozin suppresses atrial remodeling in a Canine Atrial Fibrillation Model. J Am Heart Assoc. 2021;10:e017483.
Yang Y, Zhao J, Qiu J, Li J, Liang X, Zhang Z, et al. Xanthine oxidase inhibitor Allopurinol prevents oxidative stress-mediated atrial remodeling in Alloxan-Induced Diabetes Mellitus rabbits. J Am Heart Assoc. 2018. https://doi.org/10.1161/JAHA.118.008807.
Pinho-Gomes AC, Reilly S, Brandes RP, Casadei B. Targeting inflammation and oxidative stress in atrial fibrillation: role of 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibition with statins. Antioxid Redox Signal. 2014;20:1268–85.
Pezhouman A, Cao H, Fishbein MC, Belardinelli L, Weiss JN, Karagueuzian HS. Atrial Fibrillation initiated by early afterdepolarization-mediated triggered activity during Acute oxidative stress: efficacy of late Sodium Current Blockade. J Heart Health. 2018. https://doi.org/10.16966/2379-769X.146.
Yoo S, Aistrup G, Shiferaw Y, Ng J, Mohler PJ, Hund TJ, et al. Oxidative stress creates a unique, CaMKII-mediated substrate for atrial fibrillation in heart failure. JCI Insight. 2018. https://doi.org/10.1172/jci.insight.120728.
Liang X, Zhang Q, Wang X, Yuan M, Zhang Y, Xu Z, et al. Reactive oxygen species mediated oxidative stress links diabetes and atrial fibrillation. Mol Med Rep. 2018;17:4933–40.
Kallistratos MS, Poulimenos LE, Manolis AJ. Atrial fibrillation and arterial hypertension. Pharmacol Res. 2018;128:322–6.
Mills KT, Bundy JD, Kelly TN, Reed JE, Kearney PM, Reynolds K, et al. Global disparities of hypertension prevalence and control: a systematic analysis of Population-Based Studies from 90 countries. Circulation. 2016;134:441–50.
Shihab HM, Meoni LA, Chu AY, Wang NY, Ford DE, Liang KY, et al. Body mass index and risk of incident hypertension over the life course: the Johns Hopkins Precursors Study. Circulation. 2012;126:2983–9.
Landsberg L, Aronne LJ, Beilin LJ, Burke V, Igel LI, Lloyd-Jones D, et al. Obesity-related hypertension: pathogenesis, cardiovascular risk, and treatment–a position paper of the the obesity society and the American Society of Hypertension. Obes (Silver Spring). 2013;21:8–24.
Bramlage P, Pittrow D, Wittchen HU, Kirch W, Boehler S, Lehnert H, et al. Hypertension in overweight and obese primary care patients is highly prevalent and poorly controlled. Am J Hypertens. 2004;17:904–10.
Pluteanu F, Heß J, Plackic J, Nikonova Y, Preisenberger J, Bukowska A, et al. Early subcellular Ca2 + remodelling and increased propensity for Ca2 + alternans in left atrial myocytes from hypertensive rats. Cardiovasc Res. 2015;106:87–97.
Fialová M, Dlugošová K, Okruhlicová L, Kristek F, Manoach M, Tribulová N. Adaptation of the heart to hypertension is associated with maladaptive gap junction connexin-43 remodeling. Physiol Res. 2008;57:7–11.
Choisy SC, Arberry LA, Hancox JC, James AF. Increased susceptibility to atrial tachyarrhythmia in spontaneously hypertensive rat hearts. Hypertension. 2007;49:498–505.
Lau DH, Shipp NJ, Kelly DJ, Thanigaimani S, Neo M, Kuklik P, et al. Atrial arrhythmia in ageing spontaneously hypertensive rats: unraveling the substrate in hypertension and ageing. PLoS ONE. 2013;8:e72416.
Pallisgaard JL, Schjerning AM, Lindhardt TB, Procida K, Hansen ML, Torp-Pedersen C, et al. Risk of atrial fibrillation in diabetes mellitus: a nationwide cohort study. Eur J Prev Cardiol. 2016;23:621–7.
Bisson A, Bodin A, Fauchier G, Herbert J, Angoulvant D, Ducluzeau PH, et al. Sex, age, type of diabetes and incidence of atrial fibrillation in patients with diabetes mellitus: a nationwide analysis. Cardiovasc Diabetol. 2021;20:24.
Higa S, Maesato A, Ishigaki S, Suenari K, Chen YJ, Chen SA. Diabetes and Endocrine Disorders (Hyperthyroidism/Hypothyroidism) as risk factors for Atrial Fibrillation. Card Electrophysiol Clin. 2021;13:63–75.
Mujović N, Potpara TS. Predicting incident atrial fibrillation in patients with diabetes mellitus. Int J Cardiol. 2018;269:194–5.
Aune D, Feng T, Schlesinger S, Janszky I, Norat T, Riboli E. Diabetes mellitus, blood glucose and the risk of atrial fibrillation: a systematic review and meta-analysis of cohort studies. J Diabetes Complications. 2018;32:501–11.
Seyed Ahmadi S, Svensson AM, Pivodic A, Rosengren A, Lind M. Risk of atrial fibrillation in persons with type 2 diabetes and the excess risk in relation to glycaemic control and renal function: a swedish cohort study. Cardiovasc Diabetol. 2020;19:9.
Anselmino M, Matta M, D’Ascenzo F, Pappone C, Santinelli V, Bunch TJ, et al. Catheter ablation of atrial fibrillation in patients with diabetes mellitus: a systematic review and meta-analysis. Europace. 2015;17:1518–25.
Zelniker TA, Bonaca MP, Furtado RHM, Mosenzon O, Kuder JF, Murphy SA, et al. Effect of Dapagliflozin on Atrial Fibrillation in patients with type 2 diabetes Mellitus: insights from the DECLARE-TIMI 58 Trial. Circulation. 2020;141:1227–34.
Li WJ, Chen XQ, Xu LL, Li YQ, Luo BH. SGLT2 inhibitors and atrial fibrillation in type 2 diabetes: a systematic review with meta-analysis of 16 randomized controlled trials. Cardiovasc Diabetol. 2020;19:130.
Schiller JS, Lucas JW, Ward BW, Peregoy JA. Summary health statistics for US adults: National Health interview Survey, 2010. Vital Health Stat. 2012;10:1–207.
Tanamas SK, Reddy SP, Chambers MA, Clark EJ, Dunnigan DL, Hanson RL, et al. Effect of severe obesity in childhood and adolescence on risk of type 2 diabetes in youth and early adulthood in an american indian population. Pediatr Diabetes. 2018;19:622–9.
Karam BS, Chavez-Moreno A, Koh W, Akar JG, Akar FG. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc Diabetol. 2017;16:120.
Yue Y, Meng K, Pu Y, Zhang X. Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res Clin Pract. 2017;133:124–30.
Kwon Y, Koene RJ, Johnson AR, Lin GM, Ferguson JD. Sleep, sleep apnea and atrial fibrillation: questions and answers. Sleep Med Rev. 2018;39:134–42.
Gami AS, Pressman G, Caples SM, Kanagala R, Gard JJ, Davison DE, et al. Association of atrial fibrillation and obstructive sleep apnea. Circulation. 2004;110:364–7.
Stevenson IH, Teichtahl H, Cunnington D, Ciavarella S, Gordon I, Kalman JM. Prevalence of sleep disordered breathing in paroxysmal and persistent atrial fibrillation patients with normal left ventricular function. Eur Heart J. 2008;29:1662–9.
Linz D, McEvoy RD, Cowie MR, Somers VK, Nattel S, Lévy P, et al. Associations of Obstructive Sleep Apnea with Atrial Fibrillation and continuous positive airway pressure treatment: a review. JAMA Cardiol. 2018;3:532–40.
Fung JW, Li TS, Choy DK, Yip GW, Ko FW, Sanderson JE, et al. Severe obstructive sleep apnea is associated with left ventricular diastolic dysfunction. Chest. 2002;121:422–9.
Alchanatis M, Paradellis G, Pini H, Tourkohoriti G, Jordanoglou J. Left ventricular function in patients with obstructive sleep apnoea syndrome before and after treatment with nasal continuous positive airway pressure. Respiration. 2000;67:367–71.
Skalidis EI, Hamilos MI, Karalis IK, Chlouverakis G, Kochiadakis GE, Vardas PE. Isolated atrial microvascular dysfunction in patients with lone recurrent atrial fibrillation. J Am Coll Cardiol. 2008;51:2053–7.
Lin YK, Lai MS, Chen YC, Cheng CC, Huang JH, Chen SA, et al. Hypoxia and reoxygenation modulate the arrhythmogenic activity of the pulmonary vein and atrium. Clin Sci (Lond). 2012;122:121–32.
Linz D, Mahfoud F, Schotten U, Ukena C, Neuberger HR, Wirth K, et al. Renal sympathetic denervation suppresses postapneic blood pressure rises and atrial fibrillation in a model for sleep apnea. Hypertension. 2012;60:172–8.
Linz D, Schotten U, Neuberger HR, Böhm M, Wirth K. Negative tracheal pressure during obstructive respiratory events promotes atrial fibrillation by vagal activation. Heart Rhythm. 2011;8:1436–43.
Ghias M, Scherlag BJ, Lu Z, Niu G, Moers A, Jackman WM, et al. The role of ganglionated plexi in apnea-related atrial fibrillation. J Am Coll Cardiol. 2009;54:2075–83.
Li M, Yang G, Xie B, Babu K, Huang C. Changes in matrix metalloproteinase-9 levels during progression of atrial fibrillation. J Int Med Res. 2014;42:224–30.
Middeldorp ME, Pathak RK, Meredith M, Mehta AB, Elliott AD, Mahajan R, et al. PREVEntion and regReSsive effect of weight-loss and risk factor modification on Atrial Fibrillation: the REVERSE-AF study. Europace. 2018;20:1929–35.
Lynch KT, Mehaffey JH, Hawkins RB, Hassinger TE, Hallowell PT, Kirby JL. Bariatric surgery reduces incidence of atrial fibrillation: a propensity score-matched analysis. Surg Obes Relat Dis. 2019;15:279–85.
Peigh G, Wasserlauf J, Vogel K, Kaplan RM, Pfenniger A, Marks D, et al. Impact of pre-ablation weight loss on the success of catheter ablation for atrial fibrillation. J Cardiovasc Electrophysiol. 2021;32:2097–104.
Donnellan E, Wazni OM, Elshazly M, Kanj M, Hussein AA, Baranowski B, et al. Impact of bariatric surgery on Atrial Fibrillation type. Circ Arrhythm Electrophysiol. 2020;13:e007626.
Mohanty S, Mohanty P, Natale V, Trivedi C, Gianni C, Burkhardt JD, et al. Impact of weight loss on ablation outcome in obese patients with longstanding persistent atrial fibrillation. J Cardiovasc Electrophysiol. 2018;29:246–53.
Mahajan R, Lau DH, Brooks AG, Shipp NJ, Wood JPM, Manavis J, et al. Atrial fibrillation and obesity: reverse remodeling of atrial substrate with weight reduction. JACC Clin Electrophysiol. 2021;7:630–41.
Pathak RK, Middeldorp ME, Meredith M, Mehta AB, Mahajan R, Wong CX, et al. Long-term effect of Goal-Directed Weight Management in an Atrial Fibrillation Cohort: a long-term Follow-Up study (LEGACY). J Am Coll Cardiol. 2015;65:2159–69.
Jones NR, Taylor KS, Taylor CJ, Aveyard P. Weight change and the risk of incident atrial fibrillation: a systematic review and meta-analysis. Heart. 2019;105:1799–805.
Johansson C, Lind MM, Eriksson M, Johansson L. Weight, height, weight change, and risk of incident atrial fibrillation in middle-aged men and women. J Arrhythm. 2020;36:974–81.
Lee HJ, Choi EK, Han KD, Lee E, Moon I, Lee SR, et al. Bodyweight fluctuation is associated with increased risk of incident atrial fibrillation. Heart Rhythm. 2020;17:365–71.
Chung MK, Eckhardt LL, Chen LY, Ahmed HM, Gopinathannair R, Joglar JA, et al. Lifestyle and risk factor modification for reduction of Atrial Fibrillation: A Scientific Statement from the American Heart Association. Circulation. 2020;141:e750–e72.
Delesie M, Desteghe L, Bertels M, Gerets N, Van Belleghem F, Meyvis J, et al. Motivation of overweight patients with atrial fibrillation to lose weight or to follow a weight loss management program: a cross-sectional study. Acta Cardiol. 2021;76:494–503.
Atreya AR, Giugliano GR. The heavy LEGACY: Should weight management be part of every atrial fibrillation clinic? Glob Cardiol Sci Pract. 2015;2015:57.
Alonso A, Bahnson JL, Gaussoin SA, Bertoni AG, Johnson KC, Lewis CE, et al. Effect of an intensive lifestyle intervention on atrial fibrillation risk in individuals with type 2 diabetes: the look AHEAD randomized trial. Am Heart J. 2015;170:770–7e5.
Boriani G, Ruff CT, Kuder JF, Shi M, Lanz HJ, Rutman H, et al. Relationship between body mass index and outcomes in patients with atrial fibrillation treated with edoxaban or warfarin in the ENGAGE AF-TIMI 48 trial. Eur Heart J. 2019;40:1541–50.
Acknowledgements
The authors would like to acknowledge https://app.editage.cn/ for providing editorial support.
Funding
This work was supported by grants from the National Natural Science Foundation of China (no. 82070316, no. 82200317, no. 81700357).
Author information
Authors and Affiliations
Contributions
Authors HYS, JC, YZP, NL, ZXZ, JLN, WY, DWW, and NZ contributed to the conception, drafting and revision of the manuscript, approved submission of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shu, H., Cheng, J., Li, N. et al. Obesity and atrial fibrillation: a narrative review from arrhythmogenic mechanisms to clinical significance. Cardiovasc Diabetol 22, 192 (2023). https://doi.org/10.1186/s12933-023-01913-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-023-01913-5