
Abstract
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia in humans. Several risk factors promote AF, among which diabetes mellitus has emerged as one of the most important. The growing recognition that obesity, diabetes and AF are closely intertwined disorders has spurred major interest in uncovering their mechanistic links. In this article we provide an update on the growing evidence linking oxidative stress and inflammation to adverse atrial structural and electrical remodeling that leads to the onset and maintenance of AF in the diabetic heart. We then discuss several therapeutic strategies to improve atrial excitability by targeting pathways that control oxidative stress and inflammation.
Similar content being viewed by others
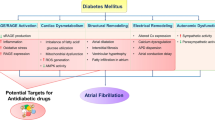

Background
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia. Several risk factors promote AF, among which diabetes mellitus has emerged as one of the most important [1, 2]. A meta-analysis by Huxley et al. [3] revealed that diabetic patients exhibit ~ 40% greater risk of developing AF than their non-diabetic counterparts. In addition, obesity, a major component of the metabolic syndrome that promotes diabetes, is also independently associated with AF [2, 4, 5].
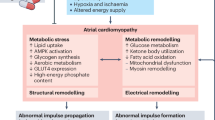
The growing recognition that obesity, diabetes and AF are closely intertwined epidemics has spurred major interest in uncovering their mechanistic links. In recent years, numerous lines of evidence have implicated oxidative stress and inflammation as central mediators of AF in metabolically-stressed hearts [6,7,8]. For one, mitochondria isolated from atrial tissues of diabetic patients [9] and animal models [10] exhibit increased emission of reactive oxygen species (ROS) due, in part, to impaired electron transport and hyperglycemia [11, 12]. Moreover glucose fluctuations which are common in diabetes promote ROS overproduction [13, 14]. The ensuing mitochondrial dysfunction and DNA damage are central to the progression of a host of cardiovascular diseases, including diabetic cardiomyopathy and AF. Other important sources of ROS that are altered in the diabetic heart include Xanthine oxidase, NADPH oxidase, Monoamine oxidase, Protein Kinase C, Nitric oxide synthase (NOS), and Advanced glycation end-products (AGE) [15]. In addition to increased ROS generation, antioxidant defense systems such as glutathione are depleted in the atria of diabetic hearts [11]. Mismatch between ROS scavenging and generation promotes oxidative stress and inflammation. In what follows, we review mechanisms by which these two key factors cause atrial structural and electrical remodeling in the diabetic heart. We then discuss several therapeutic approaches for diabetes-related AF, which have the common feature of ameliorating oxidative stress and reducing inflammation. This article is intended as an update on the evolving link between oxidative stress, inflammation, and AF in the diabetic heart [16]. For a more comprehensive viewpoint, we refer the reader to other excellent reviews on the subject matter [6, 17,18,19,20].
Oxidative stress in diabetes and obesity exacerbates atrial structural remodeling
Structural remodeling (atrial enlargement, increased fat depots, interstitial fibrosis) is a major factor by which obesity and diabetes mellitus promote AF [21]. Clinically, pericardial fat volume which correlates with left atrial enlargement [22,23,24] has been associated with increased risk of AF [22, 25]. There is strong evidence, however, to suggest that this relationship is influenced by race. Specifically, in the Multi-Ethnic Study of Atherosclerosis and the Jackson Heart Study, Hispanics but not African Americans, Whites, or Chinese Americans exhibited significant association between pericardial fat and AF risk after adjusting for body-mass index [26]. Nonetheless, the pathophysiological significance of pericardial fat is illustrated by its ability to predict success of AF ablation procedures [25]. Finally, reversal of atrial fat by weight loss is associated with reduced AF burden and improved cardiac performance [27, 28].
Given its direct apposition to the myocardium, pericardial fat is an active mediator of cardiovascular pathology, including increased AF vulnerability [29,30,31]. Importantly, pericardial fat is a visceral adipose tissue that possesses endocrine and paracrine properties. As such, it expresses both anti-inflammatory adipokines (e.g. adiponectin, omentin, etc.) and pro-inflammatory cytokines (including interleukin (IL)-1β, IL-6, IL-8, TNF-α) that readily influence atrial excitability and structure [32]. In addition to serving as a pro-inflammatory source, direct infiltration of the atrial myocardium by adipocytes disrupts the myocardial architecture causing conduction slowing, a critical factor that promotes the maintenance of AF circuits [33].
Interstitial fibrosis is another prominent feature of atrial structural remodeling in diabetes. It involves the formation of collagen-rich myocardial tissue which also disrupts cell-to-cell coupling, hinders action potential propagation, and promotes reentrant excitation underlying fibrillatory activity. Moreover, fibroblast proliferation and differentiation into myofibroblasts results in adverse heterocellular (myocyte-myofibroblast) interactions through electrical, mechanical and biochemical coupling.
Finally, atrial stretch and enlargement in obese patients promote the onset and maintenance of AF by triggering stretch-activated channels and increasing the pathlength across which multiple reentrant circuits can form. In humans and large animal models, chronic AF requires a structurally remodeled atrial substrate which arises from a multitude of pro-inflammatory processes linked to oxidative stress. Here, we focus on three key pathways that are known for their relevance in obesity and diabetes.
AGE–RAGE axis
Cross-talk between advanced glycation end products (AGE) and their receptors (RAGE) with the dipeptidyl peptidase-4 (DPP-4)-incretin system has been implicated in the pathogenesis of a number of diabetic complications, including retinopathy, nephropathy, and atherosclerosis [34, 35]. AGE levels are elevated in states of hyperglycemia and oxidative stress that arise in the diabetic heart [34]. Kato et al. [36] were among the first to highlight the putative role of the AGE–RAGE axis in diabetes-induced atrial fibrosis. Indeed, they reported markedly elevated levels of atrial fibrosis and RAGE expression in the atria of streptozotocin-induced diabetic rats, a standard model of type-1 diabetes mellitus [36]. Interestingly, fibrosis in this model was partially reversed following treatment with an AGE inhibitor suggesting a causal relationship between AGE levels, atrial fibrosis, and AF [36]. Raposeiras-Roubin et al. [37] reported increased expression of AGE and soluble RAGE in the venous blood of patients in permanent AF compared to those in sinus rhythm. More recently, Begieneman et al. compared AGE levels in the left atrial appendages (a hotspot for AF triggers) of AF patients compared to controls, and found increased N(ε)-(carboxymethyl)lysine which coincided with a marked rise in the number of inflammatory cells [38].
Although the exact mechanism by which the AGE–RAGE axis promotes atrial fibrosis remains unclear, it likely stems from the interaction of AGE with molecules in the basement membrane of the extracellular matrix. Binding of AGE to their receptors increases the expression of inflammatory mediators, namely NF-κB, which, in turn, causes tissue remodeling and damage [39]. Of note, AGE-mediated rise in NF-κB levels increases RAGE expression, causing further ROS elevation in a vicious cycle that exacerbates oxidative stress and inflammation, and therefore promotes disease progression.
Transforming growth factor β1
TGF-β1, a member of the transforming growth factor superfamily of cytokines that mediates cell proliferation and differentiation, is also elevated in diabetes [40]. This, in turn, modulates inflammatory processes in multiple organs, including the heart. The relevance of this pathway to AF has been demonstrated by multiple groups [41,42,43]. For example, Liu et al. [44] demonstrated increased atrial interstitial fibrosis, atrial ion channel remodeling, and AF vulnerability that were accompanied by elevated TGF-β1 levels in alloxan-induced diabetic rabbits. Targeted gene-based reduction of TGF-β1 signaling via constitutive expression of dominant negative TGF-β type II receptor in the posterior left atrium decreased the extent of replacement fibrosis, and in doing so, improved atrial conduction and decreased AF propensity [43]. A comprehensive analysis of the differential regulation of atrial versus ventricular remodeling by TGF-β signaling is required in future studies.
RhoA–ROCK pathway
Another pathway that promotes atrial fibrosis is mediated by the Ras homolog gene family member A (RhoA), a GTPase protein that regulates cell adhesion, smooth muscle contraction, and reorganization of the actin cytoskeleton [45]. The so-called RhoA/ROCK pathway was initially implicated in ventricular fibrosis in a model of type-1 diabetes mellitus [46]. More recently, Chen et al. [47] extended this observation to the atria of diabetic animals. Specifically, in a rat model of type-2 diabetes mellitus that exhibits significant atrial fibrosis, they found increased expression levels of RhoA and its effectors (ROCK1 and ROCK2) at both the mRNA and protein levels. Treatment of these animals with the Rho-kinase inhibitor fasudil hydrochloride hydrate suppressed the pro-fibrotic program by decreasing the expression of RhoA and its effectors [47].
Oxidative stress in diabetes exacerbates atrial electrical remodeling
Atrial electrical remodeling is a cause and consequence of AF. At the tissue level, electrical remodeling comprises effective refractory period shortening, conduction velocity slowing, wavelength reduction, and frequent atrial ectopy caused by calcium (Ca2+)-dependent triggered activity [48,49,50]. While Ca2+-mediated triggers serve as the critical initiators of AF, an appropriate substrate formed through progressive electrical and structural remodeling of the atria is required for the long-term perpetuation of the arrhythmia and its conversion from paroxysmal to chronic forms.
At the molecular level, electrical remodeling arises from altered expression and function of a host of ion channel, Ca2+ cycling, and gap junction proteins. While a comprehensive discussion of mechanisms by which these proteins are regulated by redox signaling is outside the scope of this review article, we focus on three key players, namely the ryanodine receptors (RYR2), sodium (Na) channels, and gap junction proteins owing to their importance to AF initiation and maintenance. In what follows, we highlight key studies linking oxidative stress to the malfunction of these critical targets.
Oxidative stress in diabetes promotes Ca2+-mediated triggers and the initiation of AF
Triggered activity caused by delayed afterdepolarizations (DADs) is typically required for the initiation of AF. DADs arise from increased Ca2+ leak from the sarcoplasmic reticulum (SR) via RYR2. This, in turn, causes diastolic SR Ca2+ release events, which activate the Na+/Ca2+ exchanger (NCX), producing a transient-inward current that causes membrane depolarization. If large enough, these depolarizations trigger propagated wavefronts forming premature atrial beats that can initiate AF. Numerous studies have implicated hyperphosphorylation of RYR2 via calmodulin-dependent protein kinase II (CaMKII) in arrhythmogenic SR Ca2+ leak in ventricular and atrial myocytes of diseased hearts. CAMKII, which typically requires the binding of Ca2+ to calmodulin for its activation, is also triggered by oxidation. Multiple groups have highlighted the role of mitochondria-derived ROS in the oxidation and therefore activation of CAMKII leading to the arrhythmogenic hyperphosphorylation of RYR2. Indeed, mitochondria-derived ROS have been shown to cause AF in multiple transgenic mouse models harboring leaky RYR2 channels [51]. With regards to diabetes, Joseph et al. [52] demonstrated that cardiac lipid overload secondary to peroxisome proliferator-activated receptor gamma (PPAR-γ) overexpression was associated with mitochondrial oxidative stress and increased SR Ca2+ leak via oxidized RyR2 channels. These defects likely resulted in frequent ventricular ectopy, which was reversed by treatment with a mitochondria targeted anti-oxidant [52]. Whether a similar mechanism promotes atrial ectopy and AF in diabetes awaits further study.
Abnormal atrial conduction and the maintenance of AF
In large animals and humans, AF is maintained by reentrant excitation forming stable or meandering rotors, leading circle reentry, or multiple circulating wavelets. Unidirectional conduction block is a prerequisite for reentrant excitation and conduction slowing is a key predisposing factor for conduction block. Conduction slowing causes wavelength shortening, which in turn, promotes the stability of AF circuits. Studies by multiple groups have demonstrated substantial conduction slowing and reduced conduction reserve in the atria of diabetic animals. In general, these changes arise as a consequence of structural remodeling (covered in the previous section), decreased Na channel activity, or altered expression, phosphorylation, and localization of gap junction proteins.
Atrial gap junctions are formed by the assembly of connexin (Cx) proteins, namely Cx40 and Cx43. Downregulation of Cx40 as well as hyperphosphorylation and downregulation of Cx43 have been implicated in the electrical remodeling of the diabetic heart that culminates in conduction slowing and AF [53]. However, these molecular changes have not been corroborated in all studies. For example, Mitasikova et al. [54] demonstrated paradoxical upregulation (not downregulation) of Cx43 with a decrease (not increase) in Cx43 phosphorylation. These seemingly conflicting results may be attributed to the specific sites of phosphorylation on Cx43 or the stage of diabetes. As case in point, we reported stage-dependent discrepancies in Cx43 phosphorylation in ventricular myocardium of rats with pressure overload hypertrophy [55]. Specifically, we found hyperphosphorylation and increased expression of Cx43 at the early (compensated) stage of hypertrophy that were followed by marked downregulation and dephosphorylation of the protein at late stages of remodeling [55]. Notably, conduction slowing was observed at both early and late stages of remodeling but was more severe during the latter [55]. While the molecular mechanisms underlying gap junction remodeling leading to AF are largely unknown, there is substantial evidence that oxidative stress plays a major role. For one, oxidative stress alters the atrial expression of Cx40 and Cx43 as well as the size of atrial gap junctions in a model of intermittent hypoxia mimicking obstructive sleep apnea [56]. Moreover, oxidative modification of tyrosine-mediated signaling plays a key role in Cx43 remodeling during the progression of streptozotocin-induced diabetes [57]. Finally, oxidative stress disrupts Cx43 forward trafficking to the intercalated disk resulting in abnormal gap junction coupling [58].
Na channel activity plays a major role in mediating proper action potential conduction across the heart. In alloxan-induced diabetic rabbits that are prone to AF, Liu et al. [44] demonstrated decreased INa density that was likely caused by the pro-inflammatory rise in NF-κB levels. In addition to its regulation by inflammatory cytokines, INa in ventricular myocytes is highly sensitive to oxidative stress, elevated NADH levels and protein kinase C activation [59]. Remarkably, treatment with a mitochondria-targeted antioxidant reversed this defect in murine models and in ventricular samples from patients with non-ischemic heart failure [59]. Although the role of NADPH-ROS signaling in the modulation of atrial Na channel expression and gating will require direct investigation in models of diabetes, elegant findings by the Dudley group highlight the importance of metabolic pathways in the regulation of impulse formation and conduction via direct effects on INa activity [60].
A master metabolic pathway which is highly relevant in the setting of diabetes is mediated by liver kinase B1 (LKB1), an upstream kinase with multiple downstream effectors. One of those effectors is 5′ adenosine monophosphate-activated protein kinase (AMPK), a critical component of the metabolic stress response of the heart to injury and a target of the anti-diabetic agent Metformin. The relevance of this metabolic pathway in diabetes is underscored by studies demonstrating the cardioprotective efficacy of AMPK activation in Goto Kakizaki type-2 diabetic rats [61] and streptozotocin-induced type-1 diabetic mice [62]. While LKB1 knockdown was shown by multiple groups to cause adverse ventricular remodeling, hypertrophy and AF, the underlying mechanisms by which defective LKB1 signaling promotes atrial arrhythmias remained unclear. Specifically, whether loss of LKB1 per se causes primary atrial electrical remodeling and AF independently of ventricular dysfunction and heart failure remained unknown. Using LKB1 knockout mice, we showed early remodeling of atrial gap junctions and ion channels that preceded ventricular remodeling or the spontaneous onset of permanent AF [63]. Specifically, knockdown of LKB1 led to significant downregulation of atrial Cx40 and INa peak density, causing prolonged intra-atrial depolarization and inter-atrial conduction block [63]. Future studies aimed at investigating the role of this metabolic pathway to diabetes-related AF are needed.
Therapies targeting oxidative stress and Inflammation for treatment of AF in diabetes
Standard pharmacotherapies targeting sarcolemmal ion channels for AF treatment or prevention are associated with limited efficacy and potential toxicity, including risk of pro-arrhythmia. This is largely due to the fact that ion channel ligands typically modulate the activities of atrial and ventricular ion channels alike. As a result, they often disrupt ventricular electrophysiological properties, especially in the context of a chronic disease such as diabetes that causes adverse ventricular remodeling and fibrosis. In light of this major challenge, there is growing interest in developing non-ion channel targeting agents that have the potential to alter the underlying atrial substrate without provoking pro-arrhythmic effects [64,65,66,67,68]. Since oxidative stress and inflammation are critical upstream mediators of adverse atrial structural and electrical remodeling, targeting pro-oxidant and pro-inflammatory factors may hold substantial promise for anti-AF therapies. In what follows, we provide several notable examples. This, however, is by no means intended as a comprehensive listing of promising therapies.
Pioglitazone
Thiazolidinediones (TZDs) are a class of PPAR-γ activators that exhibit potent glucose lowering efficacy, and hence are widely used in patients with insulin resistance and diabetes mellitus. In addition, TZDs which exert a number of pleiotropic effects including decreased inflammation and adiposity, are attractive agents for chronic cardiovascular disorders. A recent meta-analysis by Zhang et al. [69] highlighted the potential for TZDs in conferring protection against AF in patients with diabetes mellitus. Experimentally, pioglitazone, a prominent member of this class of agents, has been shown to inhibit AF by modulating pro-inflammatory and hypertrophic signaling pathways via suppression of TGF-β1, tumor necrosis factor alpha (TNF-α), and phospho-ERK levels [70,71,72,73]. Moreover, administration of pioglitazone results in the depolarization of the inner mitochondrial membrane with a reduction in maximal ROS production [74]. The dual antioxidant and anti-inflammatory effects of pioglitazone are therefore thought to ameliorate atrial electrical and structural remodeling [19, 70, 75,76,77]. PPAR-γ agonists, including pioglitazone, inhibit inducible NOS (iNOS) activity, enhance endothelial NOS bioavailability and reduce NADPH oxidase-dependent superoxide production [19]. Finally, pioglitazone increases soluble RAGE levels while decreasing overall RAGE expression, effects that are consistent with improved structural remodeling and anti-fibrotic action [75, 78, 79]. In light of these encouraging findings, more studies are needed to determine the electrophysiological effects of pioglitazone, and its efficacy in the prevention and treatment of AF in animal models of diabetes.
Polyunsaturated and nitrated fatty acids
Omega-3 polyunsaturated fatty acids (PUFAs) provide beneficial effects in insulin resistance and type-2 diabetes mellitus by enhancing anti-oxidant defense mechanisms. They do so by reducing the accumulation of fatty acid metabolites, providing cytoprotection for pancreatic β-cells, decreasing inhibitor of NF-κB and c-Jun N-terminal kinase (JNK) pathways, activating AMPK stress response signaling, and modulating PPAR-γ activity [80]. With regards to AF, supplementation with PUFAs and antioxidant vitamins decreases NF-κB activation likely due to the attenuation of the inflammatory and pro-oxidant state [81].
Increasing lines of evidence support the notion that PUFA supplementation is cardioprotective and likely to exert anti-arrhythmic effects in the setting of diabetes [82,83,84,85]. Acute administration of eicosapentaenoic acid (EPA) inhibits the formation of noradrenalin-induced DADs and triggered activity [86, 87]. In addition, EPA reduces pulmonary vein firing, an established AF driver, through NO-dependent mechanoelectrical feedback [87]. Finally, Rudolph et al. [88] demonstrated a potent protective effect of nitrated fatty acids against angiotensin II mediated atrial fibrosis and AF. This protection was mediated by suppressing Smad2-dependent myofibroblast differentiation and xanthine oxidase-dependent atrial superoxide levels [88]. Further studies are needed to comprehensively define the electrophysiological effects of fatty acids on atrial excitability and ion channel function in models of diabetes.
Vitamins and anti-oxidants
Since oxidative stress plays a critical role in the pathogenesis of AF, the use of vitamins C and E holds promise as an adjunctive therapeutic strategy. Besides their ROS scavenging capacity, these vitamins exert other modulatory actions including downregulation of NADPH oxidase and up-regulation of endothelial NOS activities. This, in turn, increases NO synthesis, decreases ROS formation and improves overall vascular tone [89]. Studies testing the anti-AF effects of vitamin C have suggested modest benefits [90,91,92,93,94]. Although Carnes et al. [95] found a substantial reduction in the incidence of post-operative AF in patients supplemented with Ascorbate, double-blind, placebo-controlled multi-center clinical trials based on these early studies ultimately showed mixed results [96,97,98]. Development of targeted anti-oxidant approaches that interfere selectively at the level of the defective protein or organelle are likely to produce more favorable outcomes.
One such approach is the use of mitochondria-targeted coenzyme Q (MitoQ), an antioxidant enzyme acting specifically on mitochondria to ameliorate ROS-induced injury. Administration of MitoQ to rodents was found to be safe [99] and effective against oxidative stress caused by ischemia–reperfusion (IR) injury [100], hypertension [101], and kidney damage in type-1 diabetes mellitus [100, 102, 103]. MitoQ also elicited protective effects against doxorubicin-induced cardiotoxicity [104]. Findings by Escribano-Lopez et al. [105] support the notion that the dual antioxidant and anti-inflammatory action of MitoQ is mediated, in large part, by decreased ROS production in the leukocytes of type-2 diabetic patients. In recognition of the ability of MitoQ to improve oxidative stress and inflammation, human trials have been undertaken, primarily for Parkinson’s disease [106] and chronic hepatitis C [107]. Testing of mitochondria-targeted antioxidants in patients with diabetes mellitus is warranted considering the disappointing outcome of trials examining the efficacy of vitamins as vehicles for anti-oxidant treatment [105].
Statins
Antioxidant and anti-inflammatory effects of statins have been extensively described in animal models and humans. Statins (such as Atorvastatin) reduce the risk of myocardial infarction, stroke, and death, by primarily inhibiting ROS levels and inflammation [108]. Underlying mechanisms include enhancement of eNOS activity and NO bioavailability. This, in turn, inhibits the overexpression of adhesion molecules and preserves mitochondrial membrane potential in response to oxidative stress [109, 110].
At the cellular level, Atorvastatin inhibits angiotensin-mediated cell injury by suppressing ROS production in neonatal rat ventricular myocytes [111]. More importantly, Atorvastatin treatment prevents atrial electrical remodeling, including effective refractory period shortening and reduction of the L-type calcium current in a rabbit model of tachycardia-pacing induced AF [112]. Clinically, statins confer their greatest benefit in the prevention of post-operative AF [94, 113, 114]. On the other hand, statins seem to offer limited benefits in the primary prevention of AF. Further studies are needed to determine the efficacy of statins in the prevention or management of obesity and diabetes related AF.
Dipeptidyl peptidase inhibitors
Emerging evidence supports a role for DPP-4 inhibitors in the treatment of AF. DPP-4 is a transmembrane glycoprotein that has two key substrates, namely glucagon like peptide-1(GLP-1) and glucose-dependent insulinotropic peptide (GIP) [115]. The mechanism by which DPP-4 modulates glucose metabolism involves the inhibition of GLP-1 degradation and glucagon secretion, as well as enhancement of beta-cell function, which stimulates insulin secretion [116, 117].
DPP-4 inhibition mediates anti-inflammatory, antioxidant, and anti-fibrotic effects which are critical for the prevention of AF in diabetes and obesity [118,119,120]. Zhang et al. [10] found that Alogliptin, a DPP-4 inhibitor, improved atrial remodeling by decreasing mitochondrial ROS, preventing mitochondrial membrane depolarization, enhancing mitochondrial biogenesis, and alleviating mitochondrial swelling in diabetic rabbits. The safety of this approach was highlighted by Wu et al. [121] who found that multiple DPP-4 inhibitors (alogliptin, linagliptin, saxaglipton, sitagliptin, teneligliptin, and vildagliptin) were associated with less adverse gastrointestinal side-effects compared to GLP-1 receptor agonists, metformin, and α-glucosidase inhibitors.
Summary
Atrial fibrillation, obesity, and diabetes mellitus are intertwined disorders that are linked through oxidative stress and inflammation. Both factors exacerbate atrial electrical and structural remodeling leading to the formation of an adverse substrate that facilitates AF initiation and maintenance. Developing mechanism-based strategies targeting oxidative stress and inflammation will likely generate new, safe, and effective therapeutic opportunities for combatting the growing epidemic of diabetes-related AF.
References
Huxley RR, Alonso A, Lopez FL, Filion KB, Agarwal SK, Loehr LR, Soliman EZ, Pankow JS, Selvin E. Type 2 diabetes, glucose homeostasis and incident atrial fibrillation: the atherosclerosis risk in communities study. Heart. 2012;98:133–8.
Lau DH, Nattel S, Kalman JM, Sanders P. Modifiable risk factors and atrial fibrillation. Circulation. 2017;136:583–96.
Huxley RR, Filion KB, Konety S, Alonso A. Meta-analysis of cohort and case–control studies of type 2 diabetes mellitus and risk of atrial fibrillation. Am J Cardiol. 2011;108:56–62.
Baek YS, Yang PS, Kim TH, Uhm JS, Park J, Pak HN, Lee MH, Joung B. Associations of abdominal obesity and new-onset atrial fibrillation in the general population. J Am Heart Assoc. 2017;6:e004705.
Grundvold I, Bodegard J, Nilsson PM, Svennblad B, Johansson G, Ostgren CJ, Sundstrom J. Body weight and risk of atrial fibrillation in 7,169 patients with newly diagnosed type 2 diabetes; an observational study. Cardiovasc Diabetol. 2015;14:5.
Van Wagoner DR. Oxidative stress and inflammation in atrial fibrillation: role in pathogenesis and potential as a therapeutic target. J Cardiovasc Pharmacol. 2008;52:306–13.
Gutierrez A, Van Wagoner DR. Oxidant and inflammatory mechanisms and targeted therapy in atrial fibrillation: an update. J Cardiovasc Pharmacol. 2015;66:523–9.
da Silva RM. Influence of inflammation and atherosclerosis in atrial fibrillation. Curr Atheroscler Rep. 2017;19:2.
Duicu OM, Lighezan R, Sturza A, Balica R, Vaduva A, Feier H, Gaspar M, Ionac A, Noveanu L, Borza C, Muntean DM, Mornos C. Assessment of mitochondrial dysfunction and monoamine oxidase contribution to oxidative stress in human diabetic hearts. Oxid Med Cell Longev. 2016;2016:8470394.
Zhang X, Zhang Z, Zhao Y, Jiang N, Qiu J, Yang Y, Li J, Liang X, Wang X, Tse G, Li G, Liu T. Alogliptin, a dipeptidyl peptidase-4 inhibitor, alleviates atrial remodeling and improves mitochondrial function and biogenesis in diabetic rabbits. J Am Heart Assoc. 2017;6:e005945.
Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. 2009;54:1891–8.
Peiro C, Romacho T, Azcutia V, Villalobos L, Fernandez E, Bolanos JP, Moncada S, Sanchez-Ferrer CF. Inflammation, glucose, and vascular cell damage: the role of the pentose phosphate pathway. Cardiovasc Diabetol. 2016;15:82.
Saito S, Teshima Y, Fukui A, Kondo H, Nishio S, Nakagawa M, Saikawa T, Takahashi N. Glucose fluctuations increase the incidence of atrial fibrillation in diabetic rats. Cardiovasc Res. 2014;104:5–14.
Wu N, Shen H, Liu H, Wang Y, Bai Y, Han P. Acute blood glucose fluctuation enhances rat aorta endothelial cell apoptosis, oxidative stress and pro-inflammatory cytokine expression in vivo. Cardiovasc Diabetol. 2016;15:109.
Faria A, Persaud SJ. Cardiac oxidative stress in diabetes: mechanisms and therapeutic potential. Pharmacol Ther. 2017;172:50–62.
Odegaard AO, Jacobs DR Jr, Sanchez OA, Goff DC Jr, Reiner AP, Gross MD. Oxidative stress, inflammation, endothelial dysfunction and incidence of type 2 diabetes. Cardiovasc Diabetol. 2016;15:51.
Goudis CA, Korantzopoulos P, Ntalas IV, Kallergis EM, Liu T, Ketikoglou DG. Diabetes mellitus and atrial fibrillation: pathophysiological mechanisms and potential upstream therapies. Int J Cardiol. 2015;184:617–22.
Ziolo MT, Mohler PJ. Defining the role of oxidative stress in atrial fibrillation and diabetes. J Cardiovasc Electrophysiol. 2015;26:223–5.
Zhang Q, Liu T, Ng CY, Li G. Diabetes mellitus and atrial remodeling: mechanisms and potential upstream therapies. Cardiovasc Ther. 2014;32:233–41.
Zakkar M, Ascione R, James AF, Angelini GD, Suleiman MS. Inflammation, oxidative stress and postoperative atrial fibrillation in cardiac surgery. Pharmacol Ther. 2015;154:13–20.
Tadic M, Cuspidi C. The influence of type 2 diabetes on left atrial remodeling. Clin Cardiol. 2015;38:48–55.
Al Chekakie MO, Welles CC, Metoyer R, Ibrahim A, Shapira AR, Cytron J, Santucci P, Wilber DJ, Akar JG. Pericardial fat is independently associated with human atrial fibrillation. J Am Coll Cardiol. 2010;56:784–8.
Al Chekakie MON, Welles CC, Metoyer R, Ibrahim A, Santucci P, Wilber DJ, Archer KR, Akar JG. Pericardial fat predicts human atrial fibrillation. Circulation. 2010;122:A13832.
Batal O, Schoenhagen P, Shao M, Ayyad AE, Van Wagoner DR, Halliburton SS, Tchou PJ, Chung MK. Left atrial epicardial adiposity and atrial fibrillation. Circ Arrhythm Electrophysiol. 2010;3:230–6.
Wong CX, Abed HS, Molaee P, Nelson AJ, Brooks AG, Sharma G, Leong DP, Lau DH, Middeldorp ME, Roberts-Thomson KC, Wittert GA, Abhayaratna WP, Worthley SG, Sanders P. Pericardial fat is associated with atrial fibrillation severity and ablation outcome. J Am Coll Cardiol. 2011;57:1745–51.
Heckbert SR, Wiggins KL, Blackshear C, Yang Y, Ding J, Liu J, McKnight B, Alonso A, Austin TR, Benjamin EJ, Curtis LH, Sotoodehnia N, Correa A. Pericardial fat volume and incident atrial fibrillation in the multi-ethnic study of atherosclerosis and jackson heart study. Obesity. 2017;25:1115–21.
Mahajan R, Kuklik P, Grover S, Brooks AG, Wong CX, Sanders P, Selvanayagam JB. Cardiovascular magnetic resonance of total and atrial pericardial adipose tissue: a validation study and development of a 3 dimensional pericardial adipose tissue model. J Cardiovasc Magn Reson Off J Soc Cardiovasc Magn Reson. 2013;15:73.
Gaborit B, Jacquier A, Kober F, Abdesselam I, Cuisset T, Boullu-Ciocca S, Emungania O, Alessi MC, Clement K, Bernard M, Dutour A. Effects of bariatric surgery on cardiac ectopic fat: lesser decrease in epicardial fat compared to visceral fat loss and no change in myocardial triglyceride content. J Am Coll Cardiol. 2012;60:1381–9.
Mazurek T, Zhang L, Zalewski A, Mannion JD, Diehl JT, Arafat H, Sarov-Blat L, O’Brien S, Keiper EA, Johnson AG, Martin J, Goldstein BJ, Shi Y. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation. 2003;108:2460–6.
Rosito GA, Massaro JM, Hoffmann U, Ruberg FL, Mahabadi AA, Vasan RS, O’Donnell CJ, Fox CS. Pericardial fat, visceral abdominal fat, cardiovascular disease risk factors, and vascular calcification in a community-based sample: the framingham heart study. Circulation. 2008;117:605–13.
Mahabadi AA, Massaro JM, Rosito GA, Levy D, Murabito JM, Wolf PA, O’Donnell CJ, Fox CS, Hoffmann U. Association of pericardial fat, intrathoracic fat, and visceral abdominal fat with cardiovascular disease burden: the framingham heart study. Eur Heart J. 2009;30:850–6.
Levelt E, Pavlides M, Banerjee R, Mahmod M, Kelly C, Sellwood J, Ariga R, Thomas S, Francis J, Rodgers C, Clarke W, Sabharwal N, Antoniades C, Schneider J, Robson M, Clarke K, Karamitsos T, Rider O, Neubauer S. Ectopic and visceral fat deposition in lean and obese patients with type 2 diabetes. J Am Coll Cardiol. 2016;68:53–63.
Munger TM, Dong YX, Masaki M, Oh JK, Mankad SV, Borlaug BA, Asirvatham SJ, Shen WK, Lee HC, Bielinski SJ, Hodge DO, Herges RM, Buescher TL, Wu JH, Ma C, Zhang Y, Chen PS, Packer DL, Cha YM. Electrophysiological and hemodynamic characteristics associated with obesity in patients with atrial fibrillation. J Am Coll Cardiol. 2012;60:851–60.
Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–70.
Yamagishi S, Fukami K, Matsui T. Crosstalk between advanced glycation end products (ages)-receptor rage axis and dipeptidyl peptidase-4-incretin system in diabetic vascular complications. Cardiovasc Diabetol. 2015;14:2.
Kato T, Yamashita T, Sekiguchi A, Tsuneda T, Sagara K, Takamura M, Kaneko S, Aizawa T, Fu LT. Ages-rage system mediates atrial structural remodeling in the diabetic rat. J Cardiovasc Electrophysiol. 2008;19:415–20.
Raposeiras-Roubin S, Rodino-Janeiro BK, Grigorian-Shamagian L, Seoane-Blanco A, Moure-Gonzalez M, Varela-Roman A, Alvarez E, Gonzalez-Juanatey JR. Evidence for a role of advanced glycation end products in atrial fibrillation. Int J Cardiol. 2012;157:397–402.
Begieneman MP, Rijvers L, Kubat B, Paulus WJ, Vonk AB, van Rossum AC, Schalkwijk CG, Stooker W, Niessen HW, Krijnen PA. Atrial fibrillation coincides with the advanced glycation end product n(epsilon)-(carboxymethyl)lysine in the atrium. Am J Pathol. 2015;185:2096–104.
Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605.
Ban CR, Twigg SM. Fibrosis in diabetes complications: pathogenic mechanisms and circulating and urinary markers. Vasc Health Risk Manag. 2008;4:575–96.
Kishima H, Mine T, Takahashi S, Ashida K, Ishihara M, Masuyama T. The impact of transforming growth factor-beta1 level on outcome after catheter ablation in patients with atrial fibrillation. J Cardiovasc Electrophysiol. 2017;28:402–9.
Polejaeva IA, Ranjan R, Davies CJ, Regouski M, Hall J, Olsen AL, Meng Q, Rutigliano HM, Dosdall DJ, Angel NA, Sachse FB, Seidel T, Thomas AJ, Stott R, Panter KE, Lee PM, Stevens JR, Wang Z, Macleod RS, Marrouche NF, White KL. Increased susceptibility to atrial fibrillation secondary to atrial fibrosis in transgenic goats expressing transforming growth factor-beta1. J Cardiovasc Electrophysiol. 2016;27:1220–9.
Kunamalla A, Ng J, Parini V, Yoo S, McGee KA, Tomson TT, Gordon D, Thorp EB, Lomasney J, Zhang Q, Shah S, Browne S, Knight BP, Passman R, Goldberger JJ, Aistrup G, Arora R. Constitutive expression of a dominant-negative tgf-beta type ii receptor in the posterior left atrium leads to beneficial remodeling of atrial fibrillation substrate. Circ Res. 2016;119:69–82.
Liu C, Fu H, Li J, Yang W, Cheng L, Liu T, Li G. Hyperglycemia aggravates atrial interstitial fibrosis, ionic remodeling and vulnerability to atrial fibrillation in diabetic rabbits. Anadolu Kardiyol Derg. 2012;12:543–50.
Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from rho to the actin cytoskeleton through protein kinases rock and lim-kinase. Science. 1999;285:895–8.
Zhou H, Li YJ, Wang M, Zhang LH, Guo BY, Zhao ZS, Meng FL, Deng YG, Wang RY. Involvement of rhoa/rock in myocardial fibrosis in a rat model of type 2 diabetes. Acta Pharmacol Sin. 2011;32:999–1008.
Chen J, Li Q, Dong R, Gao H, Peng H, Wu Y. The effect of the ras homolog gene family (rho), member a/rho associated coiled-coil forming protein kinase pathway in atrial fibrosis of type 2 diabetes in rats. Exp Ther Med. 2014;8:836–40.
Nattel S, Guasch E, Savelieva I, Cosio FG, Valverde I, Halperin JL, Conroy JM, Al-Khatib SM, Hess PL, Kirchhof P, De Bono J, Lip GY, Banerjee A, Ruskin J, Blendea D, Camm AJ. Early management of atrial fibrillation to prevent cardiovascular complications. Eur Heart J. 2014;35:1448–56.
Nattel S. Atrial electrophysiology and mechanisms of atrial fibrillation. J Cardiovasc Pharmacol Ther. 2003;8(Suppl 1):S5–11.
Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res. 2014;114:1483–99.
Shan J, Xie W, Betzenhauser M, Reiken S, Chen BX, Wronska A, Marks AR. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2012;111:708–17.
Joseph LC, Barca E, Subramanyam P, Komrowski M, Pajvani U, Colecraft HM, Hirano M, Morrow JP. Inhibition of napdh oxidase 2 (nox2) prevents oxidative stress and mitochondrial abnormalities caused by saturated fat in cardiomyocytes. PLoS ONE. 2016;11:e0145750.
Lin H, Ogawa K, Imanaga I, Tribulova N. Alterations of connexin 43 in the diabetic rat heart. Adv Cardiol. 2006;42:243–54.
Mitasikova M, Lin H, Soukup T, Imanaga I, Tribulova N. Diabetes and thyroid hormones affect connexin-43 and pkc-epsilon expression in rat heart atria. Physiol Res. 2009;58:211–7.
Jin H, Chemaly ER, Lee A, Kho C, Hadri L, Hajjar RJ, Akar FG. Mechanoelectrical remodeling and arrhythmias during progression of hypertrophy. FASEB J. 2010;24:451–63.
Gemel J, Su Z, Gileles-Hillel A, Khalyfa A, Gozal D, Beyer EC. Intermittent hypoxia causes nox2-dependent remodeling of atrial connexins. BMC Cell Biol. 2017;18:7.
Joshi MS, Mihm MJ, Cook AC, Schanbacher BL, Bauer JA. Alterations in connexin 43 during diabetic cardiomyopathy: competition of tyrosine nitration versus phosphorylation. J Diabetes. 2015;7:250–9.
Smyth JW, Hong TT, Gao D, Vogan JM, Jensen BC, Fong TS, Simpson PC, Stainier DY, Chi NC, Shaw RM. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J Clin Investig. 2010;120:266–79.
Liu M, Gu L, Sulkin MS, Liu H, Jeong EM, Greener I, Xie A, Efimov IR, Dudley SC Jr. Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy. J Mol Cell Cardiol. 2013;54:25–34.
Dudley SC Jr, Hoch NE, McCann LA, Honeycutt C, Diamandopoulos L, Fukai T, Harrison DG, Dikalov SI, Langberg J. Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: role of the nadph and xanthine oxidases. Circulation. 2005;112:1266–73.
Paiva MA, Rutter-Locher Z, Goncalves LM, Providencia LA, Davidson SM, Yellon DM, Mocanu MM. Enhancing ampk activation during ischemia protects the diabetic heart against reperfusion injury. Am J Physiol Heart Circ Physiol. 2011;300:H2123–34.
Zhang Z, Wang S, Zhou S, Yan X, Wang Y, Chen J, Mellen N, Kong M, Gu J, Tan Y, Zheng Y, Cai L. Sulforaphane prevents the development of cardiomyopathy in type 2 diabetic mice probably by reversing oxidative stress-induced inhibition of lkb1/ampk pathway. J Mol Cell Cardiol. 2014;77:42–52.
Kim GE, Ross JL, Xie C, Su KN, Zaha VG, Wu X, Palmeri M, Ashraf M, Akar JG, Russell KS, Akar FG, Young LH. Lkb1 deletion causes early changes in atrial channel expression and electrophysiology prior to atrial fibrillation. Cardiovasc Res. 2015;108:197–208.
Leonardi M, Bissett J. Prevention of atrial fibrillation. Curr Opin Cardiol. 2005;20:417–23.
Lozano HF, Conde CA, Florin T, Lamas GA. Treatment and prevention of atrial fibrillation with non-antiarrhythmic pharmacologic therapy. Heart rhythm. 2005;2:1000–7.
Murray KT, Mace LC, Yang Z. Nonantiarrhythmic drug therapy for atrial fibrillation. Heart rhythm. 2007;4:S88–90.
Musco S, Seltzer J, Kowey PR. Future directions in antiarrhythmic drug therapy for atrial fibrillation. Future Cardiol. 2006;2:545–53.
Page RL, Roden DM. Drug therapy for atrial fibrillation: where do we go from here? Nat Rev Drug Discov. 2005;4:899–910.
Zhang Z, Zhang X, Korantzopoulos P, Letsas KP, Tse G, Gong M, Meng L, Li G, Liu T. Thiazolidinedione use and atrial fibrillation in diabetic patients: a meta-analysis. BMC Cardiovasc Disord. 2017;17:96.
Shimano M, Tsuji Y, Inden Y, Kitamura K, Uchikawa T, Harata S, Nattel S, Murohara T. Pioglitazone, a peroxisome proliferator-activated receptor-gamma activator, attenuates atrial fibrosis and atrial fibrillation promotion in rabbits with congestive heart failure. Heart Rhythm. 2008;5:451–9.
Chen J, Mehta JL. Angiotensin ii-mediated oxidative stress and procollagen-1 expression in cardiac fibroblasts: blockade by pravastatin and pioglitazone. Am J Physiol Heart Circ Physiol. 2006;291:H1738–45.
Xu D, Murakoshi N, Igarashi M, Hirayama A, Ito Y, Seo Y, Tada H, Aonuma K. Ppar-gamma activator pioglitazone prevents age-related atrial fibrillation susceptibility by improving antioxidant capacity and reducing apoptosis in a rat model. J Cardiovasc Electrophysiol. 2012;23:209–17.
Goette A, Staack T, Rocken C, Arndt M, Geller JC, Huth C, Ansorge S, Klein HU, Lendeckel U. Increased expression of extracellular signal-regulated kinase and angiotensin-converting enzyme in human atria during atrial fibrillation. J Am Coll Cardiol. 2000;35:1669–77.
Cabrera JA, Ziemba EA, Colbert R, Kelly RF, Kuskowski M, Arriaga EA, Sluiter W, Duncker DJ, Ward HB, McFalls EO. Uncoupling protein-2 expression and effects on mitochondrial membrane potential and oxidant stress in heart tissue. Transl Res. 2012;159:383–90.
Kume O, Takahashi N, Wakisaka O, Nagano-Torigoe Y, Teshima Y, Nakagawa M, Yufu K, Hara M, Saikawa T, Yoshimatsu H. Pioglitazone attenuates inflammatory atrial fibrosis and vulnerability to atrial fibrillation induced by pressure overload in rats. Heart Rhythm. 2011;8:278–85.
Nakajima T, Iwasawa K, Oonuma H, Imuta H, Hazama H, Asano M, Morita T, Nakamura F, Suzuki J, Suzuki S, Kawakami Y, Omata M, Okuda Y. Troglitazone inhibits voltage-dependent calcium currents in guinea pig cardiac myocytes. Circulation. 1999;99:2942–50.
Liu T, Zhao H, Li J, Korantzopoulos P, Li G. Rosiglitazone attenuates atrial structural remodeling and atrial fibrillation promotion in alloxan-induced diabetic rabbits. Cardiovasc Ther. 2014;32:178–83.
Liu B, Wang J, Wang G. Beneficial effects of pioglitazone on retardation of persistent atrial fibrillation progression in diabetes mellitus patients. Int Heart J. 2014;55:499–505.
Chao TF, Leu HB, Huang CC, Chen JW, Chan WL, Lin SJ, Chen SA. Thiazolidinediones can prevent new onset atrial fibrillation in patients with non-insulin dependent diabetes. Int J Cardiol. 2012;156:199–202.
Thota RN, Acharya SH, Abbott KA, Garg ML. Curcumin and long-chain omega-3 polyunsaturated fatty acids for prevention of type 2 diabetes (cop-d): study protocol for a randomised controlled trial. Trials. 2016;17:565.
Castillo R, Rodrigo R, Perez F, Cereceda M, Asenjo R, Zamorano J, Navarrete R, Villalabeitia E, Sanz J, Baeza C, Aguayo R. Antioxidant therapy reduces oxidative and inflammatory tissue damage in patients subjected to cardiac surgery with extracorporeal circulation. Basic Clin Pharmacol Toxicol. 2011;108:256–62.
Kromhout D, Giltay EJ, Geleijnse JM. N-3 fatty acids and cardiovascular events after myocardial infarction. N Engl J Med. 2010;363:2015–26.
Dijkstra SC, Brouwer IA, van Rooij FJ, Hofman A, Witteman JC, Geleijnse JM. Intake of very long chain n-3 fatty acids from fish and the incidence of heart failure: the rotterdam study. Eur J Heart Fail. 2009;11:922–8.
Belin RJ, Greenland P, Martin L, Oberman A, Tinker L, Robinson J, Larson J, Van Horn L, Lloyd-Jones D. Fish intake and the risk of incident heart failure: the women’s health initiative. Circ Heart Fail. 2011;4:404–13.
Tavazzi L, Maggioni AP, Marchioli R, Barlera S, Franzosi MG, Latini R, Lucci D, Nicolosi GL, Porcu M, Tognoni G. Effect of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the gissi-hf trial): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:1223–30.
Li GR, Sun HY, Zhang XH, Cheng LC, Chiu SW, Tse HF, Lau CP. Omega-3 polyunsaturated fatty acids inhibit transient outward and ultra-rapid delayed rectifier k+ currents and na+ current in human atrial myocytes. Cardiovasc Res. 2009;81:286–93.
Suenari K, Chen YC, Kao YH, Cheng CC, Lin YK, Kihara Y, Chen YJ, Chen SA. Eicosapentaenoic acid reduces the pulmonary vein arrhythmias through nitric oxide. Life Sci. 2011;89:129–36.
Rudolph TK, Ravekes T, Klinke A, Friedrichs K, Mollenhauer M, Pekarova M, Ambrozova G, Martiskova H, Kaur JJ, Matthes B, Schwoerer A, Woodcock SR, Kubala L, Freeman BA, Baldus S, Rudolph V. Nitrated fatty acids suppress angiotensin ii-mediated fibrotic remodelling and atrial fibrillation. Cardiovasc Res. 2016;109:174–84.
Ulker S, McKeown PP, Bayraktutan U. Vitamins reverse endothelial dysfunction through regulation of enos and nad(p)h oxidase activities. Hypertension. 2003;41:534–9.
Hammwohner M, Smid J, Lendeckel U, Goette A. New drugs for atrial fibrillation. J Interv Card Electrophysiol. 2008;23:15–21.
Rodrigo R, Cereceda M, Castillo R, Asenjo R, Zamorano J, Araya J, Castillo-Koch R, Espinoza J, Larrain E. Prevention of atrial fibrillation following cardiac surgery: basis for a novel therapeutic strategy based on non-hypoxic myocardial preconditioning. Pharmacol Ther. 2008;118:104–27.
Korantzopoulos P, Kolettis TM, Kountouris E, Dimitroula V, Karanikis P, Pappa E, Siogas K, Goudevenos JA. Oral vitamin c administration reduces early recurrence rates after electrical cardioversion of persistent atrial fibrillation and attenuates associated inflammation. Int J Cardiol. 2005;102:321–6.
Rodrigo R, Vinay J, Castillo R, Cereceda M, Asenjo R, Zamorano J, Araya J, Castillo-Koch R, Espinoza J, Larrain E. Use of vitamins c and e as a prophylactic therapy to prevent postoperative atrial fibrillation. Int J Cardiol. 2010;138:221–8.
Simon JN, Ziberna K, Casadei B. Compromised redox homeostasis, altered nitroso-redox balance, and therapeutic possibilities in atrial fibrillation. Cardiovasc Res. 2016;109:510–8.
Carnes CA, Chung MK, Nakayama T, Nakayama H, Baliga RS, Piao S, Kanderian A, Pavia S, Hamlin RL, McCarthy PM, Bauer JA, Van Wagoner DR. Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ Res. 2001;89:E32–8.
Eslami M, Badkoubeh RS, Mousavi M, Radmehr H, Salehi M, Tavakoli N, Avadi MR. Oral ascorbic acid in combination with beta-blockers is more effective than beta-blockers alone in the prevention of atrial fibrillation after coronary artery bypass grafting. Tex Heart Inst J. 2007;34:268–74.
Papoulidis P, Ananiadou O, Chalvatzoulis E, Ampatzidou F, Koutsogiannidis C, Karaiskos T, Madesis A, Drossos G. The role of ascorbic acid in the prevention of atrial fibrillation after elective on-pump myocardial revascularization surgery: a single-center experience–a pilot study. Interact Cardiovasc Thorac Surg. 2011;12:121–4.
Bjordahl PM, Helmer SD, Gosnell DJ, Wemmer GE, O’Hara WW, Milfeld DJ. Perioperative supplementation with ascorbic acid does not prevent atrial fibrillation in coronary artery bypass graft patients. Am J Surg. 2012;204:862–7 (discussion 867).
Rodriguez-Cuenca S, Cocheme HM, Logan A, Abakumova I, Prime TA, Rose C, Vidal-Puig A, Smith AC, Rubinsztein DC, Fearnley IM, Jones BA, Pope S, Heales SJ, Lam BY, Neogi SG, McFarlane I, James AM, Smith RA, Murphy MP. Consequences of long-term oral administration of the mitochondria-targeted antioxidant mitoq to wild-type mice. Free Radic Biol Med. 2010;48:161–72.
Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–95.
Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RA, Cocheme HM, Murphy MP, Dominiczak AF. Mitochondria-targeted antioxidant mitoq10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54:322–8.
Chacko BK, Reily C, Srivastava A, Johnson MS, Ye Y, Ulasova E, Agarwal A, Zinn KR, Murphy MP, Kalyanaraman B, Darley-Usmar V. Prevention of diabetic nephropathy in ins2(+/)(−)(akitaj) mice by the mitochondria-targeted therapy mitoq. Biochem J. 2010;432:9–19.
Apostolova N, Victor VM. Molecular strategies for targeting antioxidants to mitochondria: therapeutic implications. Antioxid Redox Signal. 2015;22:686–729.
Chandran K, Aggarwal D, Migrino RQ, Joseph J, McAllister D, Konorev EA, Antholine WE, Zielonka J, Srinivasan S, Avadhani NG, Kalyanaraman B. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: cardioprotection by mito-q. Biophys J. 2009;96:1388–98.
Escribano-Lopez I, Diaz-Morales N, Rovira-Llopis S, de Maranon AM, Orden S, Alvarez A, Banuls C, Rocha M, Murphy MP, Hernandez-Mijares A, Victor VM. The mitochondria-targeted antioxidant mitoq modulates oxidative stress, inflammation and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients. Redox Biol. 2016;10:200–5.
Hwang JM, Wang CJ, Chou FP, Tseng TH, Hsieh YS, Lin WL, Chu CY. Inhibitory effect of berberine on tert-butyl hydroperoxide-induced oxidative damage in rat liver. Arch Toxicol. 2002;76:664–70.
Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RA, Murphy MP. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase ii study of hepatitis c patients. Liver Int. 2010;30:1019–26.
Amarenco P, Bogousslavsky J, Callahan A 3rd, Goldstein LB, Hennerici M, Rudolph AE, Sillesen H, Simunovic L, Szarek M, Welch KM, Zivin JA. Stroke prevention by aggressive reduction in cholesterol levels I. high-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355:549–59.
Jones SP, Teshima Y, Akao M, Marban E. Simvastatin attenuates oxidant-induced mitochondrial dysfunction in cardiac myocytes. Circ Res. 2003;93:697–9.
Mangialardi G, Monopoli A, Ongini E, Spinetti G, Fortunato O, Emanueli C, Madeddu P. Nitric oxide-donating statin improves multiple functions of circulating angiogenic cells. Br J Pharmacol. 2011;164:570–83.
Ma Y, Kong L, Qi S, Wang D. Atorvastatin blocks increased l-type ca2+ current and cell injury elicited by angiotensin ii via inhibiting oxide stress. Acta Biochim Biophys Sin (Shanghai). 2016;48:378–84.
Yang Q, Qi X, Dang Y, Li Y, Song X, Hao X. Effects of atorvastatin on atrial remodeling in a rabbit model of atrial fibrillation produced by rapid atrial pacing. BMC Cardiovasc Disord. 2016;16:142.
Laakso M, Kuusisto J. Diabetes secondary to treatment with statins. Curr Diabetes Rep. 2017;17:10.
Fauchier L, Clementy N, Babuty D. Statin therapy and atrial fibrillation: systematic review and updated meta-analysis of published randomized controlled trials. Curr Opin Cardiol. 2013;28:7–18.
Zhang X, Zhang Z, Li M, Li G, Liu T. Potential role of dipeptidyl peptidase-4 inhibitors in atrial fibrillation. Int J Cardiol. 2016;207:46–7.
Ito R, Fukui T, Hayashi T, Osamura A, Ohara M, Hara N, Higuchi A, Yamamoto T, Hirano T. Teneligliptin, a dipeptidyl peptidase-4 inhibitor, improves early-phase insulin secretion in drug-naive patients with type 2 diabetes. Drugs R&D. 2015;15:245–51.
Fukui K, Kawahito H, Wakana N, Kikai M, Terada K, Yamamoto K, Irie D, Kato T, Miyagawa S, Yamada H. Dipeptidyl peptidase-4 inhibitor sitagliptin improves pancreatic beta-cell function in hypertensive diabetic patients treated with angiotensin receptor blockers. J Renin Angiotensin Aldosterone Syst. 2015;16:1001–9.
Bostick B, Habibi J, Ma L, Aroor A, Rehmer N, Hayden MR, Sowers JR. Dipeptidyl peptidase inhibition prevents diastolic dysfunction and reduces myocardial fibrosis in a mouse model of western diet induced obesity. Metab Clin Exp. 2014;63:1000–11.
Yamamoto T, Shimano M, Inden Y, Takefuji M, Yanagisawa S, Yoshida N, Tsuji Y, Hirai M, Murohara T. Alogliptin, a dipeptidyl peptidase-4 inhibitor, regulates the atrial arrhythmogenic substrate in rabbits. Heart Rhythm. 2015;12:1362–9.
Hayami N, Sekiguchi A, Iwasaki YK, Murakawa Y, Yamashita T. No additional effect of dpp-4 inhibitor on preventing atrial fibrosis in streptozotocin-induced diabetic rat as compared with sulfonylurea. Int Heart J. 2016;57:336–40.
Wu S, Chai S, Yang J, Cai T, Xu Y, Yang Z, Zhang Y, Ji L, Sun F, Zhan S. Gastrointestinal adverse events of dipeptidyl peptidase 4 inhibitors in type 2 diabetes: A systematic review and network meta-analysis. Clin Ther. 2017;39:1780–9.
Authors’ contributions
All authors participated in the writing and editing of this review article. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable. No new datasets were generated for this review article.
Consent for publication
All authors have approved the final manuscript for publication.
Disclosures
All authors report no relevant relationships to the content of this review paper.
Ethics approval and consent to participate
Not applicable for this review article as no new studies were conducted or data generated.
Funding
Supported by Grants from the National Institutes of Health to FGA (R01 HL091923, R21AG054211) and JGA (R01 HL113352).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Karam, B.S., Chavez-Moreno, A., Koh, W. et al. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc Diabetol 16, 120 (2017). https://doi.org/10.1186/s12933-017-0604-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-017-0604-9