
Abstract
In the era of precision medicine, treatments that target specific modifiable characteristics of high-risk patients have the potential to lower further the residual risk of atherosclerotic cardiovascular events. Correction of atherogenic dyslipidemia, however, remains a major unmet clinical need. Elevated plasma triglycerides, with or without low levels of high-density lipoprotein cholesterol (HDL-C), offer a key modifiable component of this common dyslipidemia, especially in insulin resistant conditions such as type 2 diabetes mellitus. The development of selective peroxisome proliferator-activated receptor alpha modulators (SPPARMα) offers an approach to address this treatment gap. This Joint Consensus Panel appraised evidence for the first SPPARMα agonist and concluded that this agent represents a novel therapeutic class, distinct from fibrates, based on pharmacological activity, and, importantly, a safe hepatic and renal profile. The ongoing PROMINENT cardiovascular outcomes trial is testing in 10,000 patients with type 2 diabetes mellitus, elevated triglycerides, and low levels of HDL-C whether treatment with this SPPARMα agonist safely reduces residual cardiovascular risk.
Similar content being viewed by others

Preamble: why we need this consensus
Atherosclerotic cardiovascular disease (ASCVD) presents a growing global health challenge. Over the last 20 years, chronic lifestyle-related diseases such as visceral obesity, type 2 diabetes mellitus (T2DM), and non-alcoholic fatty liver disease (NAFLD) have exacerbated the burden of death and disability due to ASCVD. While this burden affects all regions, it presents a particular threat in low- and middle-income countries, which have the largest populations affected by obesity and diabetes [1,2,3]. Furthermore, escalation in the prevalence of NAFLD in these regions, particularly the Middle East and Latin America, has contributed to this increasing ASCVD burden independent of traditional risk factors [4, 5].
Atherogenic dyslipidemia, however, remains a major unmet clinical need in such populations. Elevated plasma triglycerides (TG), with or without low levels of high-density lipoprotein cholesterol (HDL-C), offer a key modifiable component of this common pattern of dyslipidemia, especially in those with insulin resistant conditions such as T2DM. After statins, guidelines recommend peroxisome proliferator-activated receptor alpha (PPARα) agonists—fibrates—for management of hypertriglyceridemia [6]. However, these agents have limitations, most importantly due to pharmacokinetic interactions, such as increased risk of myopathy with statins for gemfibrozil [7], or side effects, which include reversible elevation in serum creatinine (with fenofibrate), as well as liver enzyme elevation [8,9,10]. Hence, there is a clear need for new therapeutic options.
Is it possible to selectively modify the pharmacological characteristics of a PPARα agonist to improve the profile of beneficial effects and address known safety issues associated with fibrate treatment? And, if this is feasible, would this represent a novel therapeutic class? This Joint Consensus Panel from the International Atherosclerosis Society (IAS) and the Residual Risk Reduction Initiative (R3i) evaluated these questions in the context of evidence for the first of the selective peroxisome proliferator-activated receptor alpha modulators (SPPARMα). Box 1 delineates the search strategy and selection criteria for studies that informed this statement.
Residual vascular risk: a key therapeutic concept
Despite guideline-recommended treatment of ASCVD risk, including antihypertensive and high-intensity statin therapy, or antiaggregant agents, high-risk patients, especially those with established ASCVD, continue to experience cardiovascular events [11, 12]. This residual vascular risk, particularly in T2DM, includes both macrovascular disease as well as the microvascular changes that predispose to diabetic nephropathy, retinopathy, and neuropathy [13], for which there are limited effective medical therapies beyond strict glycemic control, an approach that entails risk of hypoglycemia and perhaps aggravated macrovascular outcomes, as well as blood pressure control.
Addressing this residual cardiovascular risk has advanced considerably. Further lowering of low-density lipoprotein cholesterol (LDL-C), a major component of modifiable ASCVD risk [14], by inhibiting proprotein convertase subtilisin/kexin type 9 (against a background of intense statin therapy) provides incremental reduction in risk [15, 16], with greatest benefit in individuals at highest absolute risk, including those with diabetes or peripheral artery disease [15,16,17,18]. Beyond lipids, the CANTOS trial (Canakinumab Antiinflammatory Thrombosis Outcome Study) established that targeting inflammation in patients with high residual risk despite well-controlled LDL-C levels on statin therapy reduces the incidence of major adverse cardiovascular events (MACE) [19], paving the way for exploration of further anti-inflammatory therapies. Additionally, the COMPASS trial (Cardiovascular Outcomes for People using Anticoagulation Strategies) demonstrated that low dose rivaroxaban plus aspirin significantly reduced MACE and major adverse limb events (albeit with a small increase in bleeding events) in patients with stable ASCVD [20].
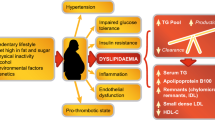
Together these findings underpin the concept of deploying ‘precision medicine’ to optimize ASCVD prevention. This approach involves focusing on specific modifiable residual cardiovascular risk targets (i.e. lipids, inflammation, or coagulation), according to patient characteristics [21]. Targeting only one component, however, does not eliminate residual cardiovascular risk. The changing landscape of cardiovascular risk drivers provides one explanation, in particular the increasing prevalence of visceral obesity. Visceral obesity and ectopic fat accumulation, particularly in the liver, associate with metabolic diseases and adverse cardiovascular outcomes [5]. Deposition of fat within the liver accompanies a plethora of associated metabolic abnormalities including elevated lipids and blood pressure, insulin resistance, as well as prothrombotic and proinflammatory states [22]. Despite observational association of liver fat content and NAFLD with ASCVD, genetic evidence shows that NAFLD is not causal for ASCVD [23, 24].
Insulin resistant cardiometabolic disease often entails an ‘atherogenic dyslipidemia’, characterized by elevated plasma TG, low HDL-C levels, a preponderance of small, dense LDL particles, and elevated apolipoproteins (apo) B (apoB 100 and apoB48) and C-III concentrations [25, 26]. This dyslipidemic profile is common, especially in low- and middle-income regions where obesity is prevalent, such as Latin America, where it affects nearly 20% of the general adult population [27]. Among high- and very high-risk patients, up to 35% have elevated TG and 10–15% have atherogenic dyslipidemia (Table 1) [26, 28,29,30,31,32]. Atherogenic dyslipidemia therefore offers a particularly attractive target for new therapies to mitigate residual ASCVD risk.
Atherogenic dyslipidemia and cardiometabolic risk
Recognition of the contribution of atherogenic dyslipidemia to ASCVD risk is not new [11, 33]. Consistent epidemiological data associate low HDL-C with risk for ASCVD [34]. Genetic studies, however, do not support a protective role of HDL-C in humans [35], and clinical outcomes trials using different therapeutic approaches to target low HDL-C also failed to meet their primary endpoints [36,37,38,39,40]. Together, these findings imply that low HDL-C is a marker of risk and not a therapeutic target.
In contrast to the situation with HDL-C, the case for elevated TG as a biomarker for causal risk has grown in strength [41, 42]. Before discussing the evidence, it merits mention that most of the studies that have evaluated the association between TG and ASCVD risk have measured fasting levels, due to previous concerns that non-fasting samples may overestimate plasma TG. As current evidence does not support this view, either fasting or nonfasting TG concentrations can serve as a marker of increased risk of cardiovascular events and death in both men and women [43,44,45,46].
In the PROVE IT-TIMI 22 trial, on-treatment TG < 1.7 mmol/L associated independently with a lower risk of recurrent coronary events in acute coronary syndrome (ACS) patients at LDL-C goal [47]. Pooled analysis of the TNT (Treating to New Targets) and IDEAL (Incremental Decrease in Endpoints Through Aggressive Lipid lowering) trials showed a trend for association between lowering TG levels and reduction in ASCVD events [48, 49]. Elevated TG also predicted recurrent ischemic events in ACS patients treated with statins, as well as progression of coronary atherosclerosis in patients with stable coronary heart disease [50, 51]. Furthermore, long-term (> 20 years) follow-up of the BIP (Bezafibrate Infarction Prevention) Study showed an association between elevated TG and all-cause mortality [52].
In patients with T2DM treated with statin therapy in the ACCORD (Action to Control Cardiovascular Risk in Diabetes) Lipid study, the presence of atherogenic dyslipidemia (TG ≥ 2.3 mmol/L and HDL-C levels ≤ 0.88 mmol/L) associated with an increase in cardiovascular event rates [53]. These findings derive support from real-world data in statin-treated diabetes patients with elevated TG (2.3–5.6 mmol/L), which showed higher rates for non-fatal myocardial infarction (MI, by 30%) compared with patients with lower TG [54]. This result undoubtedly translates to greater healthcare costs associated with management of these complications [55]. Furthermore, as previously noted, high TG and low HDL-C associate with diabetic microangiopathy, in particular nephropathy, as supported mainly by evidence from observational studies, especially in individuals with LDL-C at goal [13].
Which is the risk factor: triglycerides or triglyceride-rich lipoproteins?
The atherogenic entities of particular interest are, however, TG-rich lipoproteins and their remnants, for which circulating TG levels serve as a biomarker. TG-rich lipoproteins encompass a mixture of chylomicrons (synthesized in the intestine) and very low-density lipoprotein (VLDL) particles (synthesized in the liver) (Fig. 1) [56]. Under fasting conditions, the liver secretes both VLDL1 and VLDL2 containing apo B100; the larger form, VLDL1, carries most of the TG and associates with NAFLD [57]. Lipoprotein lipase (LpL) subsequently hydrolyzes both VLDL classes to form smaller and denser lipoprotein particles. The action of LpL on VLDL can generate endogenous, natural PPARα ligands, resulting in anti-inflammatory and anti-atherosclerotic responses [58]. Consistent with this finding, overexpression of LpL in settings where it is not usually found, e.g. cardiac myocytes, induces expression of PPARα target genes [59, 60]. In the post-prandial phase, the intestine secretes chylomicrons containing apo B48, which subsequently undergo hydrolysis by LpL, with release of free fatty acids and formation of chylomicron remnants. LpL activity undergoes both pre- and post-transcriptional regulation mediated by free fatty acids, apo C-II, apo C-III, apo A-V, angiopoietin-like members 3, 4, 8 (ANGPTL 3, 4, 8), and glycosylphosphatidylinositol anchored HDL binding protein 1 (GPIHBP1) [56, 61, 62].

Remnant lipoproteins accumulate in the arterial wall where they elicit inflammation. This provides a mechanistic basis for a causal role in atherosclerosis. Adapted from Nordestgaard and Varbo [56] with permission. FFA, free fatty acids; LDL, low-density lipoproteins; LPL, lipoprotein lipase
Insulin resistance drives dysregulation of the metabolism of TG-rich lipoproteins by two mechanisms. On the one hand, excess flow of free fatty acids to the liver, compensatory hyperinsulinemia, together with concomitant activation of enzymes involved in hepatic de novo lipogenesis (DNL) contribute to overproduction of VLDL1 particles. DNL generates malonyl-CoA that inhibits carnitine palmitoyl transferase I, resulting in reduced uptake of long-chain fatty acyl groups into mitochondria and hence reduced beta-oxidation. On the other hand, increased secretion of apo C-III mediates impaired clearance of VLDL1-TG. The combination of hepatic TG-rich lipoprotein overproduction and inefficient clearance increases the residence time of circulating TG-rich lipoproteins [63]. This delayed clearance enhances the exchange of components such as cholesteryl ester, TG, and apolipoproteins between lipoproteins, and further remodeling by hepatic lipase results in cholesterol-enriched remnants, small dense LDL particles and low plasma HDL-C levels [63].
Triglyceride-rich lipoproteins, remnants and ASCVD
TG-rich lipoproteins and their remnants contain both TG and cholesterol. As all cells in the body readily degrade TG, the enhanced ASCVD risk likely results from the cholesterol component of TG-rich lipoproteins and their remnants (referred to as ‘remnant cholesterol’ and estimated in clinical practice as total cholesterol − [LDL-C + HDL-C]). Indeed, with the exception of very large particles such as chylomicrons, these lipoproteins and their remnants can enter the arterial wall, ultimately depositing their cholesterol load in the atherosclerotic plaque (Fig. 1) [56, 64, 65]. Post hoc analysis of the TNT study also showed that TG-rich lipoprotein cholesterol concentration was an independent marker of residual ASCVD risk [66].
Insights from Mendelian randomization and genetic studies
Mendelian randomization studies strongly support the causality of remnant cholesterol carried by TG-rich lipoproteins in ASCVD. Elevated levels of remnant cholesterol associate with both increased observational and genetic risk for ASCVD, independent of HDL-C levels [67, 68]. Furthermore, while elevated nonfasting remnant and LDL-C levels each associate with increased risk of ischemic heart disease and MI, only elevated remnant cholesterol concentration associates with increased risk of all-cause mortality [69]. Elevated nonfasting remnant cholesterol may also contribute to the residual risk of all-cause mortality in individuals with established ischemic heart disease [70]. These findings reinforce the long-held view that postprandial lipemia contributes to atherogenesis, as during an ordinary day, individuals spend more time in the nonfasting than fasting state [71, 72]. Mechanistically, the atherogenicity of elevated remnant cholesterol may involve inflammation [73], as the Copenhagen studies show that elevated plasma C-reactive protein levels (> 2 mg/dL), a marker of inflammation, commonly accompany elevated TG levels (≥ 1.7 mmol/L) [74].
Genetic studies which investigated the impact of mutations in genes involved in TG-rich lipoprotein metabolism have strengthened evidence for a link between TG-rich lipoproteins, their remnants and ASCVD risk (Fig. 2). Loss-of-function (LOF) variants in genes encoding apo AV and LpL associate with lifelong higher plasma TG levels and an increased risk of coronary artery disease [75,76,77,78], whereas LOF mutations in APOC3 and ANGPTL4 associate with lifelong decreased plasma TG levels and reduction in the risk of coronary artery disease [79,80,81,82]. These data are highly consistent with the action of LpL releasing endogenous PPARα ligands that limit atherosclerosis [60]. Evidence also implicates ANGPTL3 in control of TG and promotion of coronary risk [83]. Thus, mutations in all five genes that regulate TG-rich lipoprotein metabolism impact the subsequent risk for ASCVD.

Genetic studies suggest novel approaches for the management of hypertriglyceridemia focused on key targets involved in the regulation of triglyceride-rich lipoprotein metabolism: apolipoprotein C-III (encoded by APOC3), angiopoietin-like proteins (ANGPTL) 3 and 4, apolipoprotein A V (apo A V) and lipoprotein lipase (LPL)0 [75,76,77,78,79,80,81,82,83]. IDL, intermediate-density lipoproteins; TG, triglycerides; VLDL, very low-density lipoproteins
The potential of apo C-III as a therapeutic target merits emphasis. Clinical evidence has already established apo C-III as a cardiovascular risk predictor independent of TG levels [84]. Accumulating preclinical studies also suggest that apo C-III exerts lipid-independent pro-inflammatory effects [85]. Individuals with diabetes mellitus have elevated apo C-III concentrations, in part mediated via effects on the functionality of the β-cell, affecting intracellular calcium handling and insulin sensitivity [86]. Therefore, targeting apo C-III may offer benefits beyond TG lowering in patients with diabetes.
Insights from trials of TG-lowering therapies
Guidelines recommend fibrates (PPARα agonists) and omega-3 fatty acids for the management of hypertriglyceridemia, usually as an add-on to primary statin treatment [6, 87,88,89]. Cardiovascular outcomes studies with these agents have, however, yielded mixed results. In the case of the major fibrate trials, none recruited selectively patients with high TG levels. For example, the ACCORD Lipid study, which aimed to examine the benefit of adding a fibrate to statin therapy in patients with T2DM, had no TG entry criteria, and the median TG was only 1.8 mmol/L (interquartile range 1.3 to 2.6 mmol/L). The study showed no significant benefit of add-on fenofibrate treatment on residual cardiovascular risk [53]. Despite these shortcomings, post hoc analyses of the major fibrate trials did indicate benefit in individuals with atherogenic dyslipidemia [90]. Moreover, long-term follow-up of patients in ACCORD Lipid showed continued benefit from fenofibrate in this subgroup [91]. Reinforcing the relevance of elevated TG-rich lipoproteins to ASCVD risk, regression analysis including data from the major fibrate trials showed a 54% (95% confidence interval 5 to 78%) reduction in cardiovascular events per 1 mmol/L reduction in TG levels [56]. As with any treatment, however, these agents have limitations, predominantly due to drug–drug interactions (in particular, between gemfibrozil and statins), or effects on renal function (notably with fenofibrate, reversible elevation in serum creatinine), or hepatic safety [7,8,9,10].
Whether omega-3 fatty acids reduce cardiovascular events has engendered debate. While the JELIS (Japan EPA Lipid Intervention Study) trial reported a 19% reduction in major coronary events [92], other studies were inconclusive, perhaps because they used lower doses of omega-3 fatty acids than required clinically to lower TG substantially [93]. Recently, however, REDUCE–IT (Reduction of Cardiovascular Events with Icosapent Ethyl–Intervention Trial) showed that treatment with high dose (4 g) eicosapentaenoic acid ethyl ester in high-risk individuals (58% with diabetes) with elevated TG (median 2.4 mmol/L [interquartile range 2.0–3.1 mmol/L]) resulted in relative reductions of 25% in the incidence of MACE and 20% in cardiovascular mortality against a background of well-controlled LDL-C levels on statin treatment [94]. Although the cardiovascular outcomes benefit exceeded that anticipated by the magnitude of TG lowering (18.3%), suggesting the involvement of other mechanism(s), selection of an appropriate patient population, including both primary (30%) and secondary prevention groups with elevated TG, supports REDUCE-IT as a landmark trial supporting the concept of targeting elevated TG to reduce residual ASCVD risk. The question is, can application of a precision medicine approach to improve the clinical profile of fibrates (PPARα agents), also offer potential to mitigate residual ASCVD risk?
PPARα: the nuclear receptor ‘hub’ for TG-rich lipoprotein metabolism
Understanding the role of PPARα in lipid metabolism is fundamental to defining the SPPARMα concept. PPAR belongs to the extended family of nuclear receptors, ligand-dependent transcriptional regulators—‘hubs’—that control key metabolic processes involved in development, reproduction, metabolism, and inflammation. The PPAR subfamily of nuclear receptors comprises three isotypes: PPARα, PPARβ/δ and PPARγ, each encoded by separate genes and with a unique albeit overlapping tissue distribution. These three isotypes share a common structural organization, namely, a variable N-terminal domain with a ligand-independent activation function, a conserved DNA binding domain, and a C-terminal ligand-binding domain, which contains the ligand-dependent activation function 2 (AF2) (Fig. 3) [95]. Attention has focused on PPARα given that (1) it is highly prevalent in metabolically active tissues such as the liver, kidney, heart, muscle, brown adipose, and macrophages, and (2) has a key role in transcriptional regulation of lipoprotein metabolism, specifically fatty acid transport and beta-oxidation, as well as vascular inflammation [95]. Hepatic PPARα agonism accounts for most of these effects. Under circumstances of diminished hepatic PPARα function, PPARα-dependent regulation of fatty acid oxidation in peripheral tissues may also become relevant [96].

Structural organization of nuclear receptors. The ligand binding domain of PPARα includes the ligand dependent activation function 2 interface. PPRE, peroxisome proliferator response element
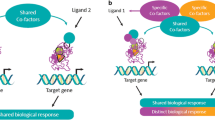
The ligand binding domain, which accommodates the lipophilic ligands and also harbors a transcriptional activation function at the C-terminus, has a critical role [97]. Binding of an agonist to the ligand binding domain triggers a conformational change. The activated nuclear receptor then binds to a specific DNA sequence in the promoter region of the target gene, resulting in activation of gene transcription (a process referred to as transactivation). The nuclear receptor may also bind to a repressor protein that prevents transcription of other genes (referred to as transrepression) [98]. For PPARα, transcriptional activation is a three-step process (Fig. 4) [95, 99]. Binding of an endogenous ligand (e.g. prostaglandins, leukotrienes, and medium-long-chain free fatty acids, especially when released by LpL) or a synthetic PPARα agonist (e.g. a fibrate) to PPARα triggers a conformational change which stabilizes the ligand binding domain and facilitates the recruitment of a specific profile of coactivators and/or the release of corepressors [100]. Of the 320 known cofactors that bind to nuclear receptors, 38 bind to PPAR. Such PPAR cofactors include PGC-1α (peroxisome proliferator-activated receptor-γ coactivator-1α), SRC1 (steroid receptor coactivator 1), and NcoR1 (nuclear receptor co-repressor 1). The ligand-activated PPARα forms a heterodimeric complex with another ligand-activated nuclear receptor, the Retinoid X Receptor (RXR), and binds to a specific DNA sequence in the promotor region of target genes referred to as a peroxisome proliferator response element (PPRE) [101]. Activation by a coactivator-acetyl transferase complex results in the expression of key genes involved in lipid metabolism, including those encoding apo A-I, A-II, A-V and C-III, LpL, scavenger receptor BI, adenosine triphosphate-binding cassette transporter A1 (ABCA1), ATP binding cassette subfamily G member 1 (ABCG1), and acyl CoA synthase. Thus, the net effects of PPARα activation on lipid metabolism include increases in HDL production, VLDL clearance, and LDL particle size, with downstream decreases in VLDL production, and LDL particle concentration [95, 102]. PPARα can also compete for co-activators of the cytokine-activated nuclear factor-κB, inhibiting the expression of pro-inflammatory genes, resulting in reduced vascular inflammation [95]. Indeed, studies demonstrated the anti-inflammatory activity of PPARα more than 20 years ago [103,104,105,106]. More recent findings have shown that PPARα activation in mouse liver reduces the CCAAT/enhancer binding protein (C/EBPβ), as well as nuclear factor-κB protein expression, resulting in lower levels of C-reactive protein, interleukin-6 and prostaglandins [107].

Transcriptional activation of PPARα is a three-part process
PPARα may also regulate glucose homeostasis and reduce thrombogenesis [95, 108]. Activation of PPARα may shift the balance of glucose versus fatty acid as the major energy source for intracellular metabolism. In the metabolically challenged liver in a glucose-rich environment, suppression of the tricarboxylic acid cycle in the mitochondria leads to an increase in acetyl co-A levels in the cytoplasm, impairing cellular homeostasis (for example, a decrease in transcription of autophagy-related genes, and an increase in oxidative stress) [109]. Similar phenomena may occur in activated macrophages, cells which contribute to the pathogenesis of ASCVD [110]. Instead, PPARα activation may promote beta oxidation, and the tricarboxylic acid cycle, triggering starvation signaling-like responses and ameliorating intracellular dysmetabolism. Taken together, these findings suggest that PPARα has the potential for addressing multiple contributors to residual cardiovascular risk.
In summary, PPARα is the nuclear receptor ‘hub’ for transcriptional regulation of lipoprotein metabolism and vascular inflammation. Conformational changes induced by binding of a ligand (either endogenous or synthetic) to PPARα facilitate the recruitment of a specific profile of cofactors, which either promote or repress expression of target genes involved in key metabolic pathways. |
Defining the SPPARMα concept
Development of selective estrogen receptor modulators (SERMs) provides an analogy for the SPPARMα concept. Depending on the tissue, SERMs can act as either agonists or antagonists of the estrogen receptor, with the cofactor milieu and structure of the bound receptor-ligand complex influencing tissue-specific cellular transcriptional activity and the subsequent profile of physiological effects [111]. Modulation of the estrogen receptor activity of the ligand permitted promotion of specific beneficial effects (in breast tissue) and avoidance of adverse effects (such as uterotropic effects) [112]. SERMs therefore suggest a ‘blueprint’ for modulating the ligand binding profile of PPARα, to improve potency and selectivity and potentially, limit tolerability issues seen with fibrates. This rationale underpins the SPPARMα concept [113].
The PPARα receptor has a large ligand binding pocket which can bind a range of endogenous and synthetic ligands, each capable of triggering specific conformational changes, resulting in a characteristic cofactor binding pattern. Different transcriptional responses seen between endogenous LpL-released fatty acids, prescription omega 3 fatty acids and different pharmacologic forms of fibrates are strongly supportive of the SPPARMα concept [60]. Modulation of the receptor–cofactor binding profile of the PPARα ligand tuned tissue- and gene-selective effects and, thus physiological responses [113]. LY-518674 was among the first SPPARMα agonists evaluated. Its higher potency than fenofibrate in vitro did not translate to superior efficacy in lowering TG and raising HDL-C in patients with atherogenic dyslipidemia. Additionally, there were safety concerns, notably an increase in serum creatinine (similar to that observed with fenofibrate) in clinical studies [114]. A subsequent search for a novel SPPARMα involved the synthesis and screening of over 1300 compounds before identification of one compound, K-877 (subsequently named pemafibrate), with potential SPPARMα activity.
In summary, binding interactions between the ligand and the PPARα receptor modulate the receptor–cofactor binding profile; this rationale underpins the SPPARMα concept. |
Differentiating SPPARMα and PPARα agonists: pharmacology
As in the case of SERMs, structural features allow differentiation of this SPPARMα agonist from PPARα agonists [115]. Specifically, the addition of unique benzoxazole and phenoxyalkyl side-chains confer a Y-shape to the SPPARMα agonist pemafibrate, contrasting with the linear structure of PPARα agonists such as fenofibrate (Fig. 5). In silico computer simulation, which enables coupling of information relating to structure and sequence, demonstrated that this SPPARMα agonist binds to the entire Y-shaped ligand binding pocket with an enhanced induced fit compared with PPARα agonists such as fenofibrate (Fig. 5, Additional files 1, 2). Changes in PPARα conformation form a new interface which binds to PGC-1α, a transcriptional coactivator, resulting in complete activation of PPARα [116]. Quantitative evaluation of ligand docking using computer-linked fragment molecular orbit analysis predicted which amino acids mediate binding to the SPPARMα agonist, as confirmed by mutation experiments. Identification of the key role of PGC-1α in binding is important, given that it regulates metabolic adaptation, and thus influences the development of systemic insulin resistance, glucose intolerance and insulin deficiency [117]. This SPPARMα agonist exhibited greatly enhanced PPARα potency and selectivity in cell-based transactivation assays, > 2500-fold more potent than fenofibric acid, the active metabolite of fenofibrate, and > 5000-fold more specific for human PPARα than either PPARγ or δ [118].

Transcriptome analysis showed that while 11 of the main 20 genes induced by pemafibrate or fenofibrate participate in carbohydrate and lipid metabolism, there were differences in the magnitude of effect. For example, in human hepatocytes this SPPARMα agonist further induced key target genes such as VLDLR and ABCA1 at 10-fold lower concentration than fenofibrate (10 μM vs. 100 μM) [119]. SPPARMα agonism predominantly induced mitochondrial genes encoding 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) synthase 2, fatty acid-binding protein 1 (FABP1), and pyruvate dehydrogenase kinase isozyme 4 (PDK4), involved in maintaining glucose homeostasis and increasing ketone body utilization. This SPPARMα agonist (but not fenofibric acid) also augmented the expression of fibroblast growth factor 21 (FGF21) [119], a metabolic regulator with favourable effects on glucose and lipid metabolism [120]. Experimentally, FGF21 induces fatty acid oxidation, ketogenesis and gluconeogenesis, as well as suppresses lipogenesis; [121] some reports have also shown this effect with fibrates [122]. In addition, there was increased expression of genes involved in the regulation of the innate immune system (mannose-binding lectin 2 [MBL2]), inflammation, blood pressure (glutamyl aminopeptidase [ENPEP]), and glucose and energy homeostasis, implying the potential for effects beyond lipid modification [119]. Moreover, this SPPARMα agonist had no effect on peroxisome biogenesis genes in human hepatocytes, suggesting that it does not stimulate peroxisome proliferation, and thus avoids hepatic adverse effects in humans [119].
SPPARMα in pre-clinical studies
Multiple preclinical studies investigated the pharmacological profile of this novel SPPARMα agonist (reviewed in reference 118 and summarized in Fig. 6). Compared with fenofibrate, pemafibrate resulted in greater TG-lowering and elevation in HDL-C in animals with hypertriglyceridemia [118, 123], and in C57BL/6J mice fed a high-fat diet, attenuated postprandial hypertriglyceridemia more effectively, by suppressing the postprandial increase in chylomicrons and accumulation of chylomicron remnants [124]. This SPPARMα agonist produced similar lipid modulating actions in the liver and intestine [125].

Differentiation of the pharmacological profile of a SPPARMα (pemafibrate) based on available data. ALT, alanine aminotransferase; apo apolipoprotein; AST, aspartate aminotransferase; C, cholesterol; FGF21, fibroblast growth factor 21; HDL, high-density lipoprotein; TG, triglycerides
Beyond lipid effects, this SPPARMα agonist also promoted potent anti-inflammatory effects, increased macrophage cholesterol efflux to HDL, inhibited lipid deposition in the aorta, and attenuated atherosclerotic lesion development in animals [126, 127]. Evidence from obese mice also suggests that this SPPARMα agonist ameliorates visceral obesity-induced hyperglycemia and elevated TG-rich lipoproteins, possibly mediated by an increase in circulating FGF21 levels, as well as enhanced expression of genes involved in thermogenesis and fatty acid oxidation in both white and brown adipose tissue [128]. In rodents with non-alcoholic steatohepatitis (NASH), pemafibrate improved liver dysfunction by modulation of hepatic lipid turnover and energy metabolism [129]. Finally, this SPPARMα agonist may produce beneficial microvascular benefits, with evidence of reduction of diabetic nephropathy in diabetic db/db mice, attributed, at least partly, to inhibition of renal lipid content and oxidative stress [130].
In summary, preclinical studies have revealed that enhanced potency, selectivity and cofactor binding profile differentiate this novel SPPARMα agent from traditional non-selective PPARα agonists. Clinically relevant genes regulated by this SPPARMα agonist include those involved in regulation of lipoprotein metabolism, such as VLDLR and ABCA1, inflammation, the innate immune system (MBL2) and energy metabolism (FGF21). In preclinical studies, this SPPARMα activator had lipid modifying and anti-inflammatory effects, as well as regulatory effects in glucose homeostasis and liver dysfunction. |
Differentiating SPPARMα and PPARα agonists: clinical trial evidence
Efficacy
Thus, the pharmacological profile of this SPPARMα agonist suggests benefit in the management of atherogenic dyslipidemia, in particular elevated TG-rich lipoproteins and remnant cholesterol common in overweight patients with T2DM [131]. A phase II dose-ranging trial (oral pemafibrate 0.025–0.2 mg twice daily) in Japanese patients with elevated TG (≥ 2.3 mmol/L) and low HDL-C (< 1.3 mmol/L in men and < 1.4 mmol/L in women) defined the clinically relevant dose range for this SPPARMα agonist [132]. After 12 weeks, this agent produced dose-related reductions from baseline in TG (by 30.9% to 42.7%), VLDL-cholesterol (by 24.3% to 48.4%), remnant-cholesterol (by 32.3% to 50.1%), apo B48 (by 28.4% to 55.9%), and apo C-III (by 2.2% to 34.6%), as well as an increase in HDL-C (by 11.9% to 21.0%), compared with both placebo and micronized fenofibrate 100 mg once daily, with maximal effects at a dose of 0.2–0.4 mg daily (Table 2). Treatment with this SPPARMα agent also ameliorated the atherogenic lipoprotein profile, reducing the proportion of small and very small LDL particles, and increasing small and very small HDL particles. Reduction in non-HDL-C and apo B100 was less pronounced (~ 10%) during pemafibrate therapy [132]. In another study in Japanese patients with high TG and low HDL–C, pemafibrate 0.2 mg or 0.4 mg daily was significantly more effective than a low dose of fenofibrate (solid dispersion tablet 106.6 mg, equivalent to micronized fenofibrate 134 mg daily) and non-inferior to fenofibrate 200 mg daily [133]. Subsequent phase II/III trials in Japanese and European patients with elevated TG with or without T2DM confirmed the lipid-modifying activity of this SPPARMα agonist, in particular robust and sustained lowering of remnant cholesterol (by up to 80%), and TG and apo C-III (by ~ 50%) [134,135,136,137,138,139]. As this SPPARMα agent depends predominantly on excretion by the liver [140], the TG-lowering response with pemafibrate does not vary with baseline estimated glomerular filtration rate (eGFR) [141]. Table 2 summarizes clinical trials with this SPPARMα agonist.
Subsequent studies showed that treatment with this SPPARMα agonist significantly reduced the postprandial area under the curve for TG, apoB 48, and remnant cholesterol for patients with and without T2DM [136, 139]. In patients with atherogenic dyslipidemia, treatment with pemafibrate not only significantly increased HDL-C, apo A-I, and apo A-II levels, but also improved indices related to HDL function, as shown by increases in prebeta-HDL, smaller HDL particles (HDL3 and HDL2), and macrophage cholesterol efflux capacity, a marker of the ability of HDL to mediate reverse cholesterol transport [139]. Some evidence also suggested non-lipid effects with pemafibrate 0.2 to 0.4 mg daily, including beneficial effects on glycemic control and insulin sensitivity in patients with and without T2DM [132, 136, 142]. In a hyperinsulinemic–euglycemic clamp study in patients with elevated TG (mean 3.3 mmol/L [standard deviation 1.10 mmol/L]) and insulin resistance, pemafibrate 0.4 mg daily for 12 weeks significantly increased splanchnic glucose uptake, although there was no change in peripheral glucose uptake rates compared with placebo [143]. Treatment with pemafibrate also significantly increased FGF21 to a greater extent than 100 mg micronized fenofibrate [132, 134,135,136, 139], and lowered biomarkers of inflammation (C-reactive protein and serum amyloid A) [139].
Safety
As with all novel therapies, clinicians and patients alike share concerns regarding benefits versus risks. Across all trials, this SPPARMα agonist was generally well tolerated, particularly with respect to renal and hepatic safety signals. The incidence of adverse events with pemafibrate resembled that of placebo (or statin alone in pemafibrate combination treatment trials) and showed no association with pemafibrate dose. Moreover, there were fewer adverse effects relating to renal or hepatic function with this SPPARMα agonist than with fenofibrate 200 mg daily [118, 142]. Pooled analyses of phase II/III studies showed significant improvement in liver function tests (alanine aminotransferase, gamma glutamyl transferase, and bilirubin) with this SPPARMα agonist administered over 12–24 weeks [143]. Importantly, and in contrast to studies with fenofibrate which showed reversible increases in serum creatinine and a decline in eGFR [8, 9], no pemafibrate dose studied elevated serum creatinine over up to 52 weeks in patients with or without pre-existing renal dysfunction [142]. In addition, while both pemafibrate and fenofibrate (solid-dispersion tablet 106.6 mg daily) increased serum homocysteine, the effect was less with pemafibrate [134].
In summary, the sum of evidence from clinical studies provides further support for the SPPARMα concept. Briefly, treatment with this SPPARMα agonist resulted in robust and sustained lowering of TG-rich lipoproteins, remnant cholesterol, and apo C-III, together with improvement in the atherogenic lipoprotein profile, as well as attenuation of postprandial hyperlipidemia in patients with and without T2DM. Pemafibrate also favourably affected glycemia, FGF21, and inflammatory markers. The safety data for this SPPARMα agonist are encouraging over the relatively short duration of exposure in clinical trials so far, especially for renal and hepatic safety, with no evidence of elevation in serum creatinine during treatment. There remain, however, a number of outstanding questions. Chief among them is whether translation of the SPPARMα concept to the clinic will reduce residual cardiovascular risk and prove safe during long-term treatment.
Clinical trials support the SPPARMα concept, showing robust and sustained reduction of TG-rich lipoproteins in patients with atherogenic dyslipidemia, with or without T2DM. The risk versus benefit profile so far is also encouraging, especially the lack of any effect on serum creatinine during treatment, although longer-term safety data are needed. |
Unanswered questions: SPPARMα, residual vascular risk and NAFLD
A number of lines of evidence suggest that treatment with this SPPARMα agonist could limit atherosclerotic lesion progression. In preclinical studies, pemafibrate promoted macrophage cholesterol efflux to HDL and attenuated atherosclerotic lesion development [126, 127] and, in patients with atherogenic dyslipidemia, pemafibrate treatment improved macrophage cholesterol efflux capacity [139]. Moreover, this SPPARMα agent robustly reduces TG, and exerts potent anti-inflammatory effects. Therefore, pemafibrate may offer a novel approach to target residual cardiovascular risk in high-risk patients with atherogenic dyslipidemia, especially those with T2DM. The data so far support testing the SPPARMα concept to determine whether therapeutic lowering of TG-rich lipoproteins with pemafibrate, on a background of best evidence-based treatment including statin therapy, will reduce incident ASCVD events and exhibit long-term safety.
The PROMINENT study (Pemafibrate to Reduce cardiovascular OutcoMes by reducing triglycerides IN diabetic patiENTs) addresses these critical questions. PROMINENT aims to recruit 10,000 T2DM patients with atherogenic dyslipidemia (TG ≥ 2.3 mmol/L and < 5.6 mmol/L, and low HDL-C) despite statin therapy, with or without established ASCVD (Fig. 7) [144]. Thus, unlike the previous fibrate trials, PROMINENT has specifically targeted the hypertriglyceridemic patient population. The primary endpoint is a four-point MACE of nonfatal MI, nonfatal ischemic stroke, cardiovascular death, or unstable angina requiring unplanned revascularization [144]. The trial is event-driven, requiring 1092 events (at least 200 events in female patients), and is powered to detect an 18% relative risk reduction. Allowing for a placebo event rate of 3.7 per 100 person-years, the trial should take 4–5 years. Within PROMINENT, a prospective nested substudy will investigate whether this SPPARMα agonist slows the progression of diabetic retinopathy in patients with non-proliferative diabetic retinopathy at study enrolment [145]. This substudy follows on evidence of fenofibrate limiting progression of diabetic retinopathy in the FIELD (Fenofibrate Intervention and Event Lowering in Diabetes) and ACCORD studies [146, 147].

Design of the PROMINENT study with pemafibrate. Adapted from Pradhan et al. [144] with permission. BID, twice daily; HDL, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; PAD, peripheral artery disease; TG, triglycerides
Beyond reduction in residual cardiovascular risk, other effects may differentiate this SPPARMα agonist from current fibrates. Notably, pemafibrate can benefit experimental NASH [129], which suggests that this SPPARMα agent can impact progression of complications such as fibrosis, cirrhosis, hepatocellular carcinoma and liver failure [148]. These pathophysiological consequences also extend beyond the liver, contributing to ASCVD burden [149]. Ongoing studies are investigating the effects of this SPPARMα agonist in the setting of NAFLD [150]. Furthermore, combination with a sodium-glucose cotransporter-2 inhibitor may merit exploration, with evidence of favourable effects on weight gain, TG, and glucose levels, and pathogenesis in animals that develop NASH and have heightened risk of hepatocellular carcinoma [151].
Conclusion
The pandemic of visceral obesity poses enormous socioeconomic challenges in managing the associated cardiometabolic comorbidities of T2DM, NAFLD, and ASCVD. Atherogenic dyslipidemia, chiefly elevated TG-rich lipoproteins and remnant cholesterol (often accompanied by low HDL-C), likely drive this association. There is an unmet clinical need for treatments that effectively reduce residual cardiovascular risk associated with atherogenic dyslipidemia. Realization of the SPPARMα concept and translation to the clinic offers a precision medicine approach to this challenge. On the basis of evidence from preclinical and clinical studies, this Joint Consensus Panel concludes that this SPPARMα represents a new therapeutic class, differentiated from fibrates by its profile of activity, especially improved renal and hepatic safety, as well as lipid-independent anti-inflammatory effects. Consistent with this, the Japanese Atherosclerosis Society has recently ratified SPPARMα as a new therapeutic class, on the basis of these criteria. PROMINENT is testing whether these SPPARMα characteristics translate to reduction in cardiovascular events in T2DM patients with atherogenic dyslipidemia. This study aims to validate SPPARMα as a novel therapeutic class for managing residual vascular risk driven by visceral obesity and T2DM.
Availability of data and materials
The datasets used and/or analyzed during the current study (as defined in Box 1) are available from the corresponding author on reasonable request.
References
Joseph P, Leong D, McKee M, Anand SS, Schwalm JD, Teo K, et al. Reducing the global burden of cardiovascular disease. Part 1: the epidemiology and risk factors. Circ Res. 2017;121:677–94.
World Health Organization. Fact sheet. Obesity and overweight. http://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. Accessed 21 Jan 2019.
NCD Countdown 2030 collaborators. NCD Countdown 2030: worldwide trends in non-communicable disease mortality and progress towards Sustainable Development Goal target 3.4. Lancet. 2018;392:1072–88.
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84.
Olubamwo OO, Virtanen JK, Voutilainen A, Kauhanen J, Pihlajamäki J, Tuomainen TP. Association of fatty liver index with the risk of incident cardiovascular disease and acute myocardial infarction. Eur J Gastroenterol Hepatol. 2018;30:1047–54.
Piepoli MF, Hoes AW, Agewall S, Albus C, Brotons C, Catapano AL, et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: the Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J. 2016;37:2315–81.
Davidson MH. Statin/fibrate combination in patients with metabolic syndrome or diabetes: evaluating the risks of pharmacokinetic drug interactions. Expert Opin Drug Saf. 2006;5:145–56.
Mychaleckyj JC, Craven T, Nayak U, Buse J, Crouse JR, Elam M, et al. Reversibility of fenofibrate therapy-induced renal function impairment in ACCORD type 2 diabetic participants. Diabetes Care. 2012;35:1008–14.
Davis TM, Ting R, Best JD, Donoghoe MW, Drury PL, Sullivan DR, et al. Effects of fenofibrate on renal function in patients with type 2 diabetes mellitus: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study. Diabetologia. 2011;54:280–90.
Hedrington MS, Davis SN. Peroxisome proliferator-activated receptor alpha-mediated drug toxicity in the liver. Expert Opin Drug Metab Toxicol. 2018;14:671–7.
Fruchart JC, Sacks F, Hermans MP, Assmann G, Brown WV, Ceska R, et al. The Residual Risk Reduction Initiative: a call to action to reduce residual vascular risk in patients with dyslipidemia. Am J Cardiol. 2008;102(Suppl 10):1K–34K.
Jernberg T, Hasvold P, Henriksson M, Hjelm H, Thuresson M, Janzon M. Cardiovascular risk in post-myocardial infarction patients: nationwide real-world data demonstrate the importance of a long-term perspective. Eur Heart J. 2015;36:1163–70.
Sacks FM, Hermans MP, Fioretto P, Valensi P, Davis T, Horton E, et al. Association between plasma triglycerides and high-density lipoprotein cholesterol and microvascular kidney disease and retinopathy in type 2 diabetes mellitus: a global case–control study in 13 countries. Circulation. 2014;129:999–1008.
Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38:2459–572.
Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–22.
Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379:2097–107.
Bonaca MP, Nault P, Giugliano RP, Keech AC, Pineda AL, Kanevsky E, et al. Low-density lipoprotein cholesterol lowering with evolocumab and outcomes in patients with peripheral artery disease: insights from the FOURIER Trial (Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects With Elevated Risk). Circulation. 2018;137:338–50.
Sabatine MS, Leiter LA, Wiviott SD, Giugliano RP, Deedwania P, De Ferrari GM, et al. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: a prespecified analysis of the FOURIER randomised controlled trial. Lancet Diabetes Endocrinol. 2017;5:941–50.
Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–31.
Anand SS, Bosch J, Eikelboom JW, Connolly SJ, Diaz R, Widimsky P, et al. Rivaroxaban with or without aspirin in patients with stable peripheral or carotid artery disease: an international, randomised, double-blind, placebo-controlled trial. Lancet. 2018;391:219–29.
Patel KV, Pandey A, de Lemos JA. Conceptual framework for addressing residual atherosclerotic cardiovascular disease risk in the era of precision medicine. Circulation. 2018;137:2551–3.
Piché ME, Poirier P, Lemieux I, Després JP. Overview of epidemiology and contribution of obesity and body fat distribution to cardiovascular disease: an update. Prog Cardiovasc Dis. 2018;61:103–13.
Lauridsen BK, Stender S, Kristensen TS, Kofoed KF, Køber L, Nordestgaard BG, et al. Liver fat content, non-alcoholic fatty liver disease, and ischaemic heart disease: mendelian randomization and meta-analysis of 279 013 individuals. Eur Heart J. 2018;39:385–93.
Santos RD, Valenti L, Romeo S. Does nonalcoholic fatty liver disease cause cardiovascular disease? Current knowledge and gaps. Atherosclerosis. 2019;282:110–20.
Taskinen M-R, Boren J. Why is apolipoprotein CIII emerging as a novel therapeutic target to reduce the burden of cardiovascular disease? Curr Atheroscler Rep. 2016;18:59.
Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Borén J, Catapano AL, et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32:1345–61.
Ponte-Negretti CI, Isea-Perez JE, Lorenzatti AJ, Lopez-Jaramillo P, Wyss-Q FS, Pintó X, et al. Atherogenic dyslipidemia in Latin America: prevalence, causes and treatment: expert’s position paper made by The Latin American Academy for the Study of Lipids (ALALIP) Endorsed by the Inter-American Society of Cardiology (IASC), the South American Society of Cardiology (SSC), the Pan-American College of Endothelium (PACE), and the International Atherosclerosis Society (IAS). Int J Cardiol. 2017;243:516–22.
Fan W, Philip S, Granowitz C, Toth PP, Wong ND. Hypertriglyceridemia in statin-treated US adults: the National Health and Nutrition Examination Survey. J Clin Lipidol. 2018. https://doi.org/10.1016/j.jacl.2018.11.008 (Epub ahead of print).
Reiner Ž, De Bacquer D, Kotseva K, Prugger C, De Backer G, Wood D, et al. Treatment potential for dyslipidaemia management in patients with coronary heart disease across Europe: findings from the EUROASPIRE III survey. Atherosclerosis. 2013;231:300–7.
Tóth PP, Potter D, Ming EE. Prevalence of lipid abnormalities in the United States: the National Health and Nutrition Examination Survey 2003–2006. J Clin Lipidol. 2012;6:325–30.
Halcox JP, Banegas JR, Roy C, Dallongeville J, De Backer G, Guallar E, et al. Prevalence and treatment of atherogenic dyslipidemia in the primary prevention of cardiovascular disease in Europe: EURIKA, a cross-sectional observational study. BMC Cardiovasc Disord. 2017;17:160.
Nussbaumerová B, Rosolová H, Mayer O, Filipovský J, Cífková R, Bruthans J. Residual cardiovascular risk in patients with stable coronary heart disease over the last 16 years (Czech part of the EUROASPIRE I–IV surveys). Cor et Vasa. 2014;56:e98–104.
Brunzell JD, Davidson M, Furberg CD, Goldberg RB, Howard BV, Stein JH, et al. Lipoprotein management in patients with cardiometabolic risk: consensus statement from the American Diabetes Association and the American College of Cardiology Foundation. J Am Coll Cardiol. 2008;31:811–22.
The Emerging Risk Factors Collaboration. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000.
Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–80.
Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22.
AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67.
Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–99.
HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J. 2013;34:1279–91.
Lincoff AM, Nicholls SJ, Riesmeyer JS, Barter PJ, Brewer HB, Fox KAA, et al. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N Engl J Med. 2017;376:1933–42.
Miller M, Stone NJ, Ballantyne C, Bittner V, Criqui MH, Ginsberg HN, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123:2292–333.
Libby P. Triglycerides on the rise: should we swap seats on the seesaw? Eur Heart J. 2015;36:774–6.
Nordestgaard BG, Langsted A, Mora S, Kolovou G, Baum H, Bruckert E, et al. Fasting is not routinely required for determination of a lipid profile: clinical and laboratory implications including flagging at desirable concentration cut-points-a joint consensus statement from the European Atherosclerosis Society and European Federation of Clinical Chemistry and Laboratory Medicine. Eur Heart J. 2016;37:1944–58.
Austin MA, McKnight B, Edwards KL, Bradley CM, McNeely MJ, Psaty BM, et al. Cardiovascular disease mortality in familial forms of hypertriglyceridemia: a 20-year prospective study. Circulation. 2000;101:2777–82.
Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298:299–308.
Bansal S, Buring JE, Rifai N, Mora S, Sacks FM, Ridker PM. Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. JAMA. 2007;298:309–16.
Miller M, Cannon CP, Murphy SA, Qin J, Ray KK, Braunwald E, PROVE IT-TIMI 22 Investigators. Impact of triglyceride levels beyond low-density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 trial. J Am Coll Cardiol. 2008;51:724–30.
Faergeman O, Holme I, Fayyad R, Bhatia S, Grundy SM, Kastelein JJ, et al. Plasma triglycerides and cardiovascular events in the treating to new targets and incremental decrease in end-points through aggressive lipid lowering trials of statins in patients with coronary artery disease. Am J Cardiol. 2009;104:459–63.
Kastelein JJ, van der Steeg WA, Holme I, Gaffney M, Cater NB, Barter P, et al. Lipids, apolipoproteins, and their ratios in relation to cardiovascular events with statin treatment. Circulation. 2008;117:3002–9.
Schwartz GG, Abt M, Bao W, DeMicco D, Kallend D, Miller M, et al. Fasting triglycerides predict recurrent ischemic events in patients with acute coronary syndrome treated with statins. J Am Coll Cardiol. 2015;65:2267–75.
Puri R, Nissen SE, Shao M, Elshazly MB, Kataoka Y, Kapadia SR, et al. Non-HDL cholesterol and triglycerides: implications for coronary atheroma progression and clinical events. Arterioscler Thromb Vasc Biol. 2016;36:2220–8.
Klempfner R, Erez A, Sagit BZ, Goldenberg I, Fisman E, Kopel E, et al. Elevated triglyceride level is independently associated with increased all-cause mortality in patients with established coronary heart disease: twenty-two-year follow-up of the bezafibrate infarction prevention study and registry. Circ Cardiovasc Qual Outcomes. 2016;9:100–8.
ACCORD Study Group, Ginsberg HN, Elam MB, Lovato LC, Crouse JR 3rd, Leiter LA, Linz P, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Eng J Med. 2010;362:1563–74.
Nichols GA, Philip S, Reynolds K, Granowitz CB, Fazio S. Increased residual cardiovascular risk in patients with diabetes and high versus normal triglycerides despite statin-controlled LDL cholesterol. Diabetes Obes Metab. 2019;21:366–71.
Toth PP, Granowitz C, Hull M, Liassou D, Anderson A, Philip S. High triglycerides are associated with increased cardiovascular events, medical costs, and resource use: a real-world administrative claims analysis of statin-treated patients with high residual cardiovascular risk. J Am Heart Assoc. 2018;7:e008740.
Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. Lancet. 2014;384:626–35.
Adiels M, Taskinen MR, Packard C, Caslake MJ, Soro-Paavonen A, Westerbacka J, et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia. 2006;49:755–65.
Ziouzenkova O, Perrey S, Asatryan L, Hwang J, MacNaul KL, Moller DE, et al. Lipolysis of triglyceride-rich lipoproteins generates PPAR ligands: evidence for an antiinflammatory role for lipoprotein lipase. Proc Natl Acad Sci USA. 2003;100:2730–5.
Augustus A, Yagyu H, Haemmerle G, Bensadoun A, Vikramadithyan RK, Park SY, et al. Cardiac-specific knock-out of lipoprotein lipase alters plasma lipoprotein triglyceride metabolism and cardiac gene expression. J Biol Chem. 2004;279:25050–7.
Brown JD, Plutzky J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation. 2007;115:518–33.
Tikka A, Jauhiainen M. The role of ANGPTL3 in controlling lipoprotein metabolism. Endocrine. 2016;52:187–93.
Kersten S. Angiopoietin-like 3 in lipoprotein metabolism. Nat Rev Endocrinol. 2017;13:731–9.
Borén J, Watts GF, Adiels M, Söderlund S, Chan DC, Hakkarainen A, et al. Kinetic and related determinants of plasma triglyceride concentration in abdominal obesity: multicenter tracer kinetic study. Arterioscler Thromb Vasc Biol. 2015;35:2218–24.
Nordestgaard BG, Wootton R, Lewis B. Selective retention of VLDL, IDL, and LDL in the arterial intima of genetically hyperlipidemic rabbits in vivo. Molecular size as a determinant of fractional loss from the intima-inner media. Arterioscler Thromb Vasc Biol. 1995;15:534–42.
Nordestgaard BG, Stender S, Kjeldsen K. Reduced atherogenesis in cholesterol-fed diabetic rabbits. Giant lipoproteins do not enter the arterial wall. Arteriosclerosis. 1988;8:421–8.
Vallejo-Vaz AJ, Fayyad R, Boekholdt SM, Hovingh GK, Kastelein JJ, Melamed S, et al. Triglyceride-rich lipoprotein cholesterol and risk of cardiovascular events among patients receiving statin therapy in the TNT Trial. Circulation. 2018;138:770–81.
Varbo A, Benn M, Nordestgaard BG. Remnant cholesterol as a cause of ischemic heart disease: evidence, definition, measurement, atherogenicity, high risk patients, and present and future treatment. Pharmacol Ther. 2014;141:358–67.
Varbo A, Benn M, Tybjærg-Hansen A, Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61:427–36.
Varbo A, Freiberg JJ, Nordestgaard BG. Extreme nonfasting remnant cholesterol vs extreme LDL cholesterol as contributors to cardiovascular disease and all-cause mortality in 90000 individuals from the general population. Clin Chem. 2015;61:533–43.
Jepsen AM, Langsted A, Varbo A, Bang LE, Kamstrup PR, Nordestgaard BG. Increased remnant cholesterol explains part of residual risk of all-cause mortality in 5414 patients with ischemic heart disease. Clin Chem. 2016;62:593–604.
Steinberg D, Carew TE, Fielding C, Fogelman AM, Mahley RW, Sniderman AD, et al. Lipoproteins and the pathogenesis of atherosclerosis. Circulation. 1989;80:719–23.
Zilversmit DB. Atherogenesis: a postprandial phenomenon. Circulation. 1979;60:473–85.
Bernelot Moens SJ, Verweij SL, Schnitzler JG, Stiekema LCA, Bos M, Langsted A, et al. Remnant cholesterol elicits arterial wall inflammation and a multilevel cellular immune response in humans. Arterioscler Thromb Vasc Biol. 2017;37:969–75.
Hansen SEJ, Madsen CM, Varbo A, Nordestgaard BG. Low-grade inflammation in the association between mild-to-moderate hypertriglyceridemia and risk of acute pancreatitis: a study of more than 115000 individuals from the general population. Clin Chem. 2018. https://doi.org/10.1373/clinchem.2018.294926 (Epub ahead of print).
Pennacchio LA, Rubin EM. Apolipoprotein A5, a newly identified gene that affects plasma triglyceride levels in humans and mice. Arterioscler Thromb Vasc Biol. 2003;23:529–34.
Nilsson SK, Heeren J, Olivecrona G, Merkel M. Apolipoprotein A–V; a potent triglyceride reducer. Atherosclerosis. 2011;219:15–21.
Khera AV, Won HH, Peloso GM, O’Dushlaine C, Liu D, Stitziel NO, Natarajan P, et al. Association of rare and common variation in the lipoprotein lipase gene with coronary artery disease. JAMA. 2017;317:937–46.
Nordestgaard BG, Abildgaard S, Wittrup HH, Steffensen R, Jensen G, Tybjaerg-Hansen A. Heterozygous lipoprotein lipase deficiency: frequency in the general population, effect on plasma lipid levels, and risk of ischemic heart disease. Circulation. 1997;96:1737–44.
Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32–41.
TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute, Crosby J, Peloso GM, Auer PL, Crosslin DR, Stitziel NO, Lange LA, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31.
Dewey FE, Gusarova V, O’Dushlaine C, Gottesman O, Trejos J, Hunt C, et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N Engl J Med. 2016;374:1123–33.
Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators, Stitziel NO, Stirrups KE, Masca NG, Erdmann J, Ferrario PG, König IR, et al. Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N Engl J Med. 2016;374:1134–44.
Stitziel NO, Khera AV, Wang X, Bierhals AJ, Vourakis AC, Sperry AE, et al. ANGPTL3 deficiency and protection against coronary artery disease. J Am Coll Cardiol. 2017;69:2054–63.
Sacks FM, Alaupovic P, Moye LA, Cole TG, Sussex B, Stampfer MJ, et al. VLDL, apolipoproteins B, CIII, and E, and risk of recurrent coronary events in the Cholesterol and Recurrent Events (CARE) trial. Circulation. 2000;102:1886–92.
Zheng C, Azcutia V, Aikawa E, Figueiredo JL, Croce K, Sonoki H, et al. Statins suppress apolipoprotein CIII-induced vascular endothelial cell activation and monocyte adhesion. Eur Heart J. 2013;34:615–24.
Juntti-Berggren L, Berggren PO. Apolipoprotein CIII is a new player in diabetes. Curr Opin Lipidol. 2017;28:27–31.
Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2018. https://doi.org/10.1016/j.jacc.2018.11.003 (Epub ahead of print).
Handelsman Y, Bloomgarden ZT, Grunberger G, Umpierrez G, Zimmerman RS, Bailey TS, et al. American association of clinical endocrinologists and american college of endocrinology—clinical practice guidelines for developing a diabetes mellitus comprehensive care plan—2015. Endocr Pract. 2015;21(Suppl 1):1–87.
Catapano AL, Graham I, De Backer G, Wiklund O, Chapman MJ, Drexel H, et al. ESC/EAS guidelines for the management of dyslipidaemias: the task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS) developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J. 2016;37:2999–3058.
Sacks FM, Carey VJ, Fruchart JC. Combination lipid therapy in type 2 diabetes. N Engl J Med. 2010;363:692–4.
Elam MB, Ginsberg HN, Lovato LC, Corson M, Largay J, Leiter LA, et al. Association of fenofibrate therapy with long-term cardiovascular risk in statin-treated patients with type 2 diabetes. JAMA Cardiol. 2017;2:370–80.
Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y, Saito Y, Ishikawa Y, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet. 2007;369:1090–8.
Aung T, Halsey J, Kromhout D, Gerstein HC, Marchioli R, Tavazzi L, et al. Associations of omega-3 fatty acid supplement use with cardiovascular disease risks: meta-analysis of 10 trials Involving 77 917 individuals. JAMA Cardiol. 2018;3:225–34.
Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med. 2018;380:11–22.
Fruchart JC. Peroxisome proliferator-activated receptor-alpha (PPARalpha): at the crossroads of obesity, diabetes and cardiovascular disease. Atherosclerosis. 2009;205:1–8.
Brocker CN, Patel DP, Velenosi TJ, Kim D, Yan T, Yue J, et al. Extrahepatic PPARα modulates fatty acid oxidation and attenuates fasting-induced hepatosteatosis in mice. J Lipid Res. 2018;59:2140–52.
Gronemeyer H, Laudet V. Transcription factors 3: nuclear receptors. Protein Profile. 1995;2:1173–308.
Tugwood JD, Issemann I, Anderson RG, Bundell KR, McPheat WL, Green S. The mouse peroxisome proliferator activated receptor recognizes a response element in the 50-flanking sequence of the rat acyl CoA oxidase gene. EMBO J. 1992;11:433–9.
Marx N, Duez H, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors and atherogenesis: regulators of gene expression in vascular cells. Circ Res. 2004;94:1168–78.
Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, et al. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem. 1999;274:32048–54.
Pineda Torra I, Jamshidi Y, Flavell DM, Fruchart JC, Staels B. Characterization of the human PPARalpha promoter: identification of a functional nuclear receptor response element. Mol Endocrinol. 2002;16:1013–28.
Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B, et al. Molecular actions of PPARα in lipid metabolism and inflammation. Endocr Rev. 2018;39:760–802.
Marx N, Sukhova GK, Collins T, Libby P, Plutzky J. PPARalpha activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 1999;99:3125–31.
Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature. 1996;384:39–43.
Neve BP, Corseaux D, Chinetti G, Zawadzki C, Fruchart JC, Duriez P, et al. PPARalpha agonists inhibit tissue factor expression in human monocytes and macrophages. Circulation. 2001;103:207–12.
Marx N, Mackman N, Schoenbeck U, Yilmaz N, Hombach V, Libby P, et al. PPARα activators inhibit tissue factor expression and activity in human monocytes. Circulation. 2001;103:213–9.
Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab. 2012;23:351–63.
Staels B, Maes M, Zambon A. Fibrates and future PPARalpha agonists in the treatment of cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2008;5:542–53.
Shi L, Tu BP. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol. 2015;33:125–31.
Angajala A, Lim S, Phillips JB, Kim JH, Yates C, You Z, et al. Diverse roles of mitochondria in immune responses: novel insights into immuno-metabolism. Front Immunol. 2018;9:1605.
Krishnan V, Heath H, Bryant HU. Mechanism of action of estrogens and selective estrogen receptor modulators. Vitam Horm. 2000;60:123–47.
Lewis JS, Jordan VC. Selective estrogen receptor modulators (SERMs): mechanisms of anticarcinogenesis and drug resistance. Mutat Res. 2005;591:247–63.
Fruchart JC. Selective peroxisome proliferator-activated receptor α modulators (SPPARMα): the next generation of peroxisome proliferator-activated receptor α-agonists. Cardiovasc Diabetol. 2013;12:82.
Nissen SE, Nicholls SJ, Wolski K, Howey DC, McErlean E, Wang MD, et al. Effects of a potent and selective PPAR-alpha agonist in patients with atherogenic dyslipidemia or hypercholesterolemia: two randomized controlled trials. JAMA. 2007;297:1362–73.
Yamazaki Y, Abe K, Toma T, Nishikawa M, Ozawa H, Okuda A, et al. Design and synthesis of highly potent and selective human peroxisome proliferator-activated receptor alpha agonists. Bioorg Med Chem Lett. 2007;17:4689–93.
Yamamoto Y, Takei K, Arulmozhiraja S, Sladek V, Matsuo N, Han SI, et al. Molecular association model of PPARα and its new specific and efficient ligand, pemafibrate: structural basis for SPPARMα. Biochem Biophys Res Commun. 2018;499:239–45.
Besseiche A, Riveline JP, Gautier JF, Bréant B, Blondeau B. Metabolic roles of PGC-1α and its implications for type 2 diabetes. Diabetes Metab. 2015;41:347–57.
Fruchart JC. Pemafibrate (K-877), a novel selective peroxisome proliferator-activated receptor alpha modulator for management of atherogenic dyslipidaemia. Cardiovasc Diabetol. 2017;16:124.
Raza-Iqbal S, Tanaka T, Anai M, Matsumura Y, Ikeda K, Taguchi A, et al. Transcriptome analysis of K-877 (a novel Selective PPARα Modulator (SPPARMα))-regulated genes in primary human hepatocytes and the mouse liver. J Atheroscler Thromb. 2015;22:754–72.
Kharitonenkov A, DiMarchi R. FGF21 Revolutions: recent advances illuminating FGF21 biology and medicinal properties. Trends Endocrinol Metab. 2015;26:608–17.
Schlein C, Talukdar S, Heine M, Fischer AW, Krott LM, Nilsson SK, et al. FGF21 lowers plasma triglycerides by accelerating lipoprotein catabolism in white and brown adipose tissues. Cell Metab. 2016;23:441–53.
Ong KL, O’Connell R, Januszewski AS, Jenkins AJ, Xu A, Sullivan DR, et al. Baseline circulating FGF21 concentrations and increase after fenofibrate treatment predict more rapid glycemic progression in type 2 diabetes: results from the FIELD Study. Clin Chem. 2017;63:1261–70.
Takei K, Han SI, Murayama Y, Satoh A, Oikawa F, Ohno H, et al. Selective peroxisome proliferator-activated receptor-α modulator K-877 efficiently activates the peroxisome proliferator-activated receptor-α pathway and improves lipid metabolism in mice. J Diabetes Investig. 2017;8:446–52.
Sairyo M, Kobayashi T, Masuda D, Kanno K, Zhu Y, Okada T, et al. A novel selective PPAR Modulator (SPPARM), K-877 (pemafibrate), attenuates postprandial hypertriglyceridemia in mice. J Atheroscler Thromb. 2018;25:142–52.
Takei K, Nakagawa Y, Wang Y, Han SI, Satoh A, Sekiya M, et al. Effects of K-877, a novel selective PPARα modulator, on small intestine contribute to the amelioration of hyperlipidemia in low-density lipoprotein receptor knockout mice. J Pharmacol Sci. 2017;133:214–22.
Hennuyer N, Duplan I, Paquet C, Vanhoutte J, Woitrain E, Touche V, et al. The novel selective PPARα modulator (SPPARMα) pemafibrate improves dyslipidemia, enhances reverse cholesterol transport and decreases inflammation and atherosclerosis. Atherosclerosis. 2016;249:200–8.
Iwata H, Murakami K, Ricchiuto P, Singh S, Libby P, Aikawa E. The novel PPARα selective agonist K-877 suppresses pro-inflammatory pathways and experimental arterial lesion formation. Circ Res. 2014;115:e86–93 (Abstract 24160).
Araki M, Nakagawa Y, Oishi A, Han SI, Wang Y, Kumagai K, et al. The peroxisome proliferator-activated receptor α (PPARα) agonist pemafibrate protects against diet-induced obesity in mice. Int J Mol Sci. 2018;19:E2148.
Honda Y, Kessoku T, Ogawa Y, Tomeno W, Imajo K, Fujita K, et al. Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci Rep. 2017;7:42477.
Maki T, Maeda Y, Sonoda N, Makimura H, Kimura S, Maeno S, et al. Renoprotective effect of a novel selective PPARα modulator K-877 in db/db mice: a role of diacylglycerol-protein kinase C-NAD(P)H oxidase pathway. Metabolism. 2017;71:33–45.
Varbo A, Freiberg JJ, Nordestgaard BG. Remnant cholesterol and myocardial infarction in normal weight, overweight, and obese individuals from the Copenhagen General Population Study. Clin Chem. 2018;64:219–30.
Ishibashi S, Yamashita S, Arai H, Araki E, Yokote K, Suganami H, et al. Effects of K-877, a novel selective PPARα modulator (SPPARMα), in dyslipidaemic patients: a randomized, double blind, active- and placebo-controlled, phase 2 trial. Atherosclerosis. 2016;249:36–43.
Arai H, Yamashita S, Yokote K, Araki E, Suganami H, Ishibashi S, K-877 Study Group. Efficacy and safety of pemafibrate versus fenofibrate in patients with high triglyceride and low HDL cholesterol levels: a multicenter, placebo-controlled, double-blind, randomized trial. J Atheroscler Thromb. 2018;25:521–38.
Ishibashi S, Arai H, Yokote K, Araki E, Suganami H, Yamashita S, K-877 Study Group. Efficacy and safety of pemafibrate (K-877), a selective peroxisome proliferator-activated receptor α modulator, in patients with dyslipidemia: results from a 24-week, randomized, double blind, active-controlled, phase 3 trial. J Clin Lipidol. 2018;12:173–84.
Arai H, Yamashita S, Yokote K, Araki E, Suganami H, Ishibashi S, K-877 Study Group. Efficacy and safety of K-877, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα), in combination with statin treatment: two randomised, double-blind, placebo-controlled clinical trials in patients with dyslipidaemia. Atherosclerosis. 2017;261:144–52.
Araki E, Yamashita S, Arai H, Yokote K, Satoh J, Inoguchi T, et al. Effects of pemafibrate, a novel selective PPARα modulator, on lipid and glucose metabolism in patients with type 2 diabetes and hypertriglyceridemia: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. 2018;41:538–46.
Kastelein JJP, Senko Y, Hounslow N. K-877, a selective PPAR alpha modulator (SPPARM alpha), improves dyslipidaemia in statin-treated patients with type 2 diabetes mellitus. Eur Heart J. 2015;36:1048.
Kastelein JJP, Senko Y, Hounslow N, Hovingh GK, Ginsberg HN. K-877, a selective PPAR alpha modulator (SPPARM alpha), ameliorates dyslipidaemia in patients with well-controlled LDL Cholesterol levels on statin therapy, without increases in serum creatinine. Eur Heart J. 2015;36:1048.
Yamashita S, Arai H, Yokote K, Araki E, Suganami H, Ishibashi S, K-877 Study Group. Effects of pemafibrate (K-877) on cholesterol efflux capacity and postprandial hyperlipidemia in patients with atherogenic dyslipidemia. J Clin Lipidol. 2018;12:1267–79.
Hounslow N, Mair S, Suganami H, Nakamura M. Pemafibrate has high bioavailability and is principally excreted via the liver. Atheroscler Suppl. 2018;32:155.
Yokote K, Yamashita S, Arai H, Araki E, Suganami H, Ishibashi S. Long-term efficacy and safety of pemafibrate, a novel selective peroxisome proliferator-activated receptor-α modulator (SPPARMα), in dyslipidemic patients with renal impairment. Int J Mol Sci. 2019;20:E706.
Yokote K, Yamashita S, Arai H, Araki E, Suganami H, Ishibashi S. A pooled analysis of pemafibrate Phase II/III clinical trials indicated significant improvement in glycemic and liver function-related parameters. Atheroscler Suppl. 2018;32:155.
Matsuba I, Matsuba R, Ishibashi S, Yamashita S, Arai H, Yokote K, et al. Effects of a novel selective peroxisome proliferator-activated receptor-α modulator, pemafibrate, on hepatic and peripheral glucose uptake in patients with hypertriglyceridemia and insulin resistance. J Diabetes Investig. 2018. https://doi.org/10.1111/jdi.12845.
Pradhan AD, Paynter NP, Everett BM, Glynn RJ, Amarenco P, Elam M, et al. Rationale and design of the Pemafibrate to Reduce Cardiovascular Outcomes by Reducing Triglycerides in Patients with Diabetes (PROMINENT) study. Am Heart J. 2018;206:80–93.
PROMINENT-Eye Ancillary Study (Protocol AD). ClinicalTrials.gov Identifier NCT03345901. https://clinicaltrials.gov/ct2/show/NCT03345901. Accessed 7 Aug 2018.
Keech AC, Mitchell P, Summanen PA, O’Day J, Davis TM, Moffitt MS, et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007;370:1687–97.
ACCORD Study Group; ACCORD Eye Study Group, Chew EY, Ambrosius WT, Davis MD, Danis RP, Gangaputra S, Greven CM, et al. Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med. 2010;363:233–44.
Doycheva I, Issa D, Watt KD, Lopez R, Rifai G, Alkhouri N. Nonalcoholic steatohepatitis is the most rapidly increasing indication for liver transplantation in young adults in the United States. J Clin Gastroenterol. 2018;52:339–46.
Adams LA, Anstee QM, Tilg H, Targher G. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut. 2017;66:1138–53.
A study of pemafibrate in patients with nonalcoholic fatty liver disease (NAFLD). ClinicalTrials.gov Identifier. https://clinicaltrials.gov/ct2/show/NCT03350165. Accessed 21 Jan 2019.
Sasaki Y, Asahiyama M, Kamiya W, Sakai J, Kodama T, Tanaka T. Tofogliflozin and pemafibrate combination therapy prevents nonalcoholic steatohepatitis (NASH) development of novel metabolic syndrome-related NASH model mice. Diabetes. 2018;67:1. https://doi.org/10.2337/db18-1153-p.
Acknowledgements
Not applicable.
Funding
The Consensus Panel members met at a closed expert meeting in Chantilly, France in November 2018 where evidence relating to SPPARMα was critically appraised and discussed. Support for travel was provided by an academic Grant from the R3i. There were no other sources of funding.
Author information
Authors and Affiliations
Contributions
JCF and RDS conceived this consensus statement and prepared the initial manuscript together with JKS. CAS, MA, AC, JPD, ME, MF, HNG, MPH, SI, MK, PL, AJL, SL, RM, BGN, CJP, CIP, ZR, PMR, HS, AS, MRT, AT, PV, WW, GFW, SY all provided critical writing and interpretation of the data. All authors critically reviewed drafts and approved the final version of this manuscript before submission. The corresponding author had full access to all data and final responsibility for the decision to submit for publication. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The following authors report disclosures outside the submitted work. These relate to grants and personal fees (honoraria for consultancy, lectures and/or advisory bureaux). J-C Fruchart reports personal fees from Kowa Company. RD Santos reports personal fees from Amgen, AstraZeneca, Merck, Akcea, Sanofi/Regeneron, Biolab, Esperion, Kowa, and Novo-Nordisk. M Aikawa reports grants from Kowa Company, Ltd., Pfizer, Inc., and Sanofi, Inc. P Amarenco reports personal fees from Kowa, grants and personal fees from Pfizer, Sanofi, Bristol Myers Squibb, Boston Scientific and AstraZeneca, grants from Merck, and personal fees from Amgen, GSK, Fibrogen and Shin Poong. PJ Barter reports personal fees from Amgen, Sanofi, and Pfizer. A Corsini reports personal fees from AstraZeneca, Amgen, Sanofi, Recordati, Novartis, MSD, Mediolanum, DOC, Mylan and Pfizer. RH Eckel reports grants from NINDS, NIA, NHLBI, Ionis Pharmaceutical, Uniqure Biopharma, CTSA Pilot Project, Endece, LLC, personal fees from Cardiometabolic Health Congress, Prime, Medtelligence, Medscape, Medical Education Resources, Vox Media, Sanofi/Regeneron, Merck, Novo Nordisk, and Kowa. M Farnier reports grants, consulting fees and/or honoraria for delivering lectures for Abbott, Akcea/Ionis, Amarin, Amgen, AstraZeneca, Daiichi-Sankyo, Eli Lilly, Kowa, Merck and Co, Mylan, Pfizer, Sanofi/Regeneron and Servier. HN Ginsberg reports and consults for Kowa, a pharmaceutical company that has a SPPARMalpha in clinical trials for the treatment of dyslipidemia and prevention of cardiovascular disease. MP Hermans reports personal fees from Amgen, AstraZeneca, MSD, Mylan, Sanofi-Aventis, Teva, and Tilman. S Ishibashi reports personal fees from Kowa, grants from Astellas Pharma, Boehringer Ingelheim, Daiichi Sankyo, Ono Pnharma, Shionogi, and Teijin Pharma. T Kodama is the recipient of a research grant from Kowa Company. W Koenig reports personal fees from AstraZeneca, Novartis, Pfizer, The Medicines Company, GSK, DalCor, Sanofi, Berlin-Chemie, Kowa, Amgen, grants and non-financial support from Roche Diagnostics, Beckmann, Singulex, and Abbott. M Krempf reports personal fees from Amgen, Sanofi Regeneron, MSD, Novartis, Abbott, Servier, AstraZeneca and grants from Pfizer. P Libby reports a research grant from Novartis and honoraria as a scientific advisory board member for Dalcor Pharmaceuticals. He also provides unpaid consultancy for Amgen, AstraZeneca, Ionis Pharmaceuticals, Kowa Pharmaceuticals, Pfizer, Sanofi-Regeneron, XBiotech Inc., Corvidia Therapeutics, IFM Therapeutics, Olatec Therapeutics, Medimmune and Esperion Therapeutics. AJ Lorenzatti reports grants and personal fees from Amgen, Kowa, personal fees from Sanofi, Pfizer, and grants from Resverlogix. BG Nordestgaard reports personal fees from AstraZeneca, Sanofi, Regeneron, Ionis, Akcea, Amgen, Kowa, Denka Seiken, Amarin and Kowa. CJ Packard reports grants from MSD, and personal fees from Sanofi/Regeneron, Amgen, and Daiichi-Sankyo. A Pradhan reports grants from Kowa Research Institute. KK Ray reports grants from Sanofi, Regeneron, Pfizer, Amgen, MSD, and honoraria for lectures/speaker’s bureau or consultancy from Sanofi, Amgen, Regeneron, Lilly, Medicines Company, Astra Zeneca, Pfizer, Kowa, Algorithm, IONIS, Esperion, Novo Nordisk, Takeda, Boehringer Ingelheim, Resverlogix, and Abbvie. Z Reiner reports personal fees from Sanofi Aventis and Akcea. PM Ridker reports grants from Kowa, Inc, Novartis, and Pfizer. H Shimano reports grants and personal fees from Kowa Company Ltd., Kowa Pharmaceutical Co. Ltd., MSD K.K., DAIICHI SANKYO Co., Ltd., Takeda Pharmaceutical Co.,Ltd., Astellas Pharma Inc., AstraZeneca K.K., ONO PHARMACEUTICAL CO., LTD., Nippon Boehringer lngelheim Co., Ltd., Eli Lilly Japan, Mitsubishi Tanabe Pharma Corporation, Sanofi K.K., MOCHIDA PHARMACEUTICAL CO., LTD., Novartis Pharma K.K., Bayer Yakuhin, Ltd., SHIONOGI & CO., LTD., and Taisho Toyama Pharmaceutical Co., Ltd. A Susekov reports personal fees from AstraZeneca, Pfizer, Sanofi, KRKA, Teva and Kowa. MR Taskinen reports grants and personal fees from Amgen, NovoNordisk, and personal fees from Sanofi and Akcea. LS Tokgözoğlu reports personal fees from Abbott, Merck, Amgen, Astra, Novartis, Daiichi Sankyo, Pfizer, Actelion, Servier, Sanofi, Menarini, Mylan, Recordati and Novonordisk. B Tomlinson reports grants from Amgen, Merck Sharp and Dohme, Pfizer, Roche, and personal fees from Amgen, Dr. Reddy’s Laboratories Ltd, Merck Serono and Sanofi. P Valensi reports personal fees from Abbott, AstraZeneca, Merck-Sharp Dohme, Novo-Nordisk, and Sanofi-Aventis. M Vrablík reports personal fees from Abbott, Actavis, AstraZeneca, Amgen, BMS, Genzyme, KRKA, MSD Idea, Novartis, Pfizer and Sanofi-Regeneron. GF Watts reports support from Kowa for a lecture in Japan; honoraria (for advisory boards and lectures) and research grants from Amgen, Sanofi, and Regeneron; and honoraria (for advisory board) from Gemphire. S Yamashita reports grants and personal fees from Kowa Company, Ltd., Otsuka Pharmaceutical Co., Ltd., Shionogi & Co., Ltd., Bayer Yakuhin, Ltd., MSD K.K., Takeda Pharmaceutical Company, Ltd., Sanwa Kagaku Kenkyusho Co., Ltd., Astellas Pharma Inc., Daiichi-Sankyo Company, Ltd., Astra Zeneka K.K., Kaken Pharmaceutical Co., Ltd., grants from Nippon Boehringer Ingelheim Co., Ltd., National Institute of Biomedical Innovation, Kyowa Medex Co., Ltd., Mochida Pharmaceutical Company, Ltd., Hayashibara Co., Ltd., Teijin Pharma Limited and Kissei; and personal fees from Ono Pharmaceutical Company, Ltd., Skylight Biotec, Inc., Pfizer, Astellas Amgen, Sanofi, and Aegerion In addition, S Yamashita has a patent PCT/JP2016/074402 (Assisting Method for the Diagnosis of Type III Hyperlipidemia) pending to Fujirebio & Osaka University, a patent PCT/JP2017/038766 (Method for Selecting Subject Needing Treatment for Dyslipidemia and Reagent for Such Selection) pending to Osaka University & Kyowa Medex Co., Ltd., and a patent PCT/JP2017/038715 (Method for Measuring Oxidized High-Density Lipoprotein) pending to Osaka University & Kyowa Medex Co., Ltd.. A Zambon reports honoraria for lectures from Abbott, Amgen, Sanofi and Mylan. K Yokote reports grants and personal fees from Kowa Pharmaceutical Co, Astellas, AstraZeneca, MSD, Sanofi, Takeda, Pfizer, Mochida, and personal fees from Kowa Company, Astellas-Amgen Biopharm, and Bayer. C Aguilar-Salinas, K Al Rasadi, R Ceska, JP Després, P Duriez, MV Ezhov, F Karpe, S Lim, R McPherson, J Millan Nuñez-Cortes, H Ogawa, CI Ponte-Negretti, J Plutzky, M Ruscica, S Sadikot, P Sritara, JK Stock, T-C Su, A Tartar, A Tenenbaum, A Tybjærg-Hansen, W Wahli have no competing interests in relation to this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1.
Interaction with the selective peroxisome proliferator-activated receptor alpha modulator (pemafibrate, K-877).
Additional file 2.
Interaction with a PPARα agonist (fenofibrate).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fruchart, JC., Santos, R.D., Aguilar-Salinas, C. et al. The selective peroxisome proliferator-activated receptor alpha modulator (SPPARMα) paradigm: conceptual framework and therapeutic potential. Cardiovasc Diabetol 18, 71 (2019). https://doi.org/10.1186/s12933-019-0864-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-019-0864-7